

Abstract
Background
Small cell carcinoma of the bladder (SCCB) is a rare and aggressive neuroendocrine tumor with a dismal prognosis and limited treatment options. As SCCB is histologically indistinguishable from small cell lung cancer, a shared pathogenesis and cell of origin has been proposed. The aim of this study is to determine whether SCCBs arise from a pre-existent urothelial carcinoma or share a molecular pathogenesis in common with small cell lung cancer.
Results
We performed an integrative analysis of 61 SCCB tumors to identify histology- and organ-specific similarities and differences. SCCB has a high somatic mutational burden driven predominantly by an APOBEC-mediated mutational process. TP53, RB1, and TERT promoter mutations were present in nearly all samples. While these events appeared to arise early in all affected tumors and likely reflect an evolutionary branch point that may have driven small cell lineage differentiation, they were unlikely the founding transforming event, as they were often preceded by diverse and less common driver mutations, many of which are common in bladder urothelial cancers but not small cell lung tumors. Most patient tumors (72%) also underwent genome doubling (GD). While arising at different chronological points in the evolution of the disease, GD was often preceded by biallelic mutations in TP53 with retention of two intact copies.
Conclusions
Our findings indicate that small cell cancers of the bladder and lung have a convergent but distinct pathogenesis with SCCBs arising from a cell of origin shared with urothelial bladder cancer.
Introduction
Bladder cancer is the second most common urinary tract malignancy, responsible for over 165,000 deaths per year worldwide(1). While urothelial carcinomas predominate, several histologically distinct subtypes are observed including squamous cell, adenocarcinoma, sarcomatoid, plasmacytoid, and small cell/neuroendocrine tumors(2). Though histology-specific pathognomonic genetic lesions exist(3), little is known about the distinguishing genomic features of most bladder cancer histologies. Additionally, a comprehensive comparison of the genomic profiles of these histologies as an entry point for understanding their diverse clinical and therapeutic differences is lacking. Small cell carcinoma of the bladder (SCCB) is highly aggressive neuroendocrine tumor(4, 5) often associated with a urothelial component. The therapeutic management of SCCB has, to date, been driven by the clinical experience in small cell lung cancers(6, 7) as these diseases are histologically indistinguishable and share many clinicopathological characteristics(8). We sought to understand the molecular etiology of SCCB in the context of small cell lung cancers as well as diverse bladder histologies(2, 3) using genome-wide data from 61 patients compared with comprehensive data from both urothelial tumors and small cell lung cancers. We identify genetic lesions that arise early in SCCB pathogenesis, and through histology- and organ-specific comparisons, reveal differences in mutational patterns and potential therapeutic targets.
Materials and Methods
Patient samples
All specimens and clinical annotation were obtained from patients providing informed consent and in accordance with Institutional Review Board approval at Memorial Sloan Kettering Cancer Center. Tumor samples were obtained from surgical specimens (either transurethral resection or cystectomy specimens). All tumors were reviewed and histopathologically confirmed to be small cell carcinoma of the bladder (H.A.A. and X.H.). Representative formalin-fixed, paraffin- embedded (FFPE) sections (on average, 10 curls of 10 microns) from each sample were selected for analysis. In a subset of cases, macro-dissection or micro-dissection was performed to enrich for tumor content and minimize stromal tissue contamination. Matched normal tissue for germline DNA consisted of blood and/or normal tissues (benign lymph nodes procured at the time of cystectomy). Histologically distinct regions of tumors from patients diagnosed with small cell bladder cancers presenting with mixed histology disease were further macrodissected and sequenced separately. Immunohistochemistry for RB was performed on representative tumor sections using an Rb mouse monoclonal antibody (13A10, Leica Biosystems, Newcastle Ltd) on a Ventana Discovery XT platform at 1:50 dilution. Overall, 15 patients underwent WES and two patients underwent WGS, of which both also possessed higher depth of coverage MSK-IMPACT sequencing, and one also had RNA sequencing data. In total, 56 patients had tumor specimens sequenced with MSK-IMPACT using either a 281-gene version (n=17) or a 341-gene version (n=40; one patient with two tumors, each of which was sequenced on each assay version). Finally, 12 patients had RNA sequencing of which WES was available for 10 and MSK-IMPACT was available for the remaining two. In total, 13 patients had multiple spatially or temporally distinct tumor specimens (two or three) sequenced. Full details of the cohort including clinical data and sequencing platforms utilized are available in Supplementary Tables 1 and 3.
Sequencing and analysis
Exome, transcriptome, and targeted sequencing were performed in the Center for Molecular Oncology at MSKCC. DNA extraction was performed from either FFPE or frozen tumors and matched normal specimens with the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) according to the manufacturer’s modified protocol including the replacement of the AW2 buffer with 80% Ethanol. DNA was eluted in Nuclease free water. In total, 13 tumors were subjected to whole exome sequencing and 500ng of genomic DNA was captured by hybridization using the SureSelect XT Human All Exon V4 (Agilent Technologies). Samples were prepared according to the manufacturer’s instructions. PCR amplification of the libraries was carried out for 6 cycles in the pre-capture step and for 7 cycles post capture. Samples were barcoded and run on a HiSeq 2500 in a 75bp paired end run using the TruSeq SBS Kit v3 (Illumina). The average number of read pairs per sample was 133 and 110 million for tumor and normal samples respectively, the average duplication rate was 4.3%, and 3.2%. Read processing, alignment, and recalibration as well as somatic mutation (substitutions and small insertions and deletions) calling were performed as previously described(3).
To this exome cohort, we added 3 additional small cell bladder cancers that were profiled by TCGA, but were ultimately excluded from the TCGA bladder cancer study cohort(9) due to their non-urothelial histology. Mutations and CNAs in either 281 or 341 genes were also profiled (in 17 and 41 additional patients respectively) using a solution-phase hybridization-based exon capture and deep sequencing assay as previously described (Supplementary Tables 1–2)(25). Whole-genome sequencing was performed and analyzed, all as previously described(26), on two small cell bladder tumors and their matched normal specimens, whose DNA was extracted as described above. The results of all sequencing data is available on the publicly accessible cBioPortal for Cancer Genomics(27, 28).
RNA sequencing was performed for 12 small cell bladder tumors. These transcriptome data were utilized for fusion detection, mutation cross-validation, and exploring RB1 dysfunction in presumed RB1-wildtype tumors. Briefly, RNA from frozen tissue was extracted using RNeasy mini kit (Qiagen; Valencia, CA) according to manufacturer’s instructions. After ribogreen quantification and quality control with an Agilent BioAnalyzer, 2ug of total RNA (6.5<RIN<8.1) underwent polyA selection and Truseq library (TruSeq™ RNA Sample Prep Kit v2) preparation according to a modified protocol to enhance fusion transcript discovery. Briefly, samples were fragmented for 2 minutes at 94C before undergoing first strand and second strand cDNA synthesis. Libraries were amplified with 10 cycles of PCR and size-selected for fragments between 400 and 550 bp with a Pippin prep instrument (Sage Science). Samples were barcoded and run on a Hiseq 2500 in a 100bp paired end run, using the TruSeq SBS Kit v3 (Illumina). An average of 32 million paired reads were generated per sample. To these we added the RNA-seq data of the three histologically confirmed TCGA small cell bladder cancers (H.A.A.), converting aligned reads to FASTQ for merging with sequenced samples prior to analysis. Transcriptome reads were mapped to the human genome (hg19) using rnaStar ver2.5.0a(29) to map reads genomically and resolve reads across splice junctions. Reads were mapped in a two-pass method(30), the first pass mapping to known annotated junctions from Ensembl, and the second pass is completed on both known and novel junctions found in the first pass. After mapping, expression count matrices were generated from mapped reads using HTSeq ver 0.5.3 using Gencode ver18 gene models and normalized using DESeq. Candidate fusions were identified using chimerascan 0.4.5, defuse 0.6.2, and fusioncatcher v0.99.3e. Pairs of genes identified by more than one algorithm were inspected. Subsequent heuristic filtering and manual inspection confirmed all putative fusion calls were false positives.
Retrospective and prospective data
Results from this study cohort (SCCB) were compared to those of four additional cohorts. For urothelial bladder cancer, we combined data from 131 histologically confirmed tumors of the TCGA project(9) with 172 tumors prospectively sequenced as part of an ongoing clinical sequencing initiative at our institution with the CLIA-certified MSK-IMPACT assay (Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets) that screens 410 cancer-associated genes using the same technology as the targeting sequencing performed above(25). Corresponding aligned read data from the whole-exome sequencing of the TCGA samples were downloaded from CGHub and re-analyzed with the copy number pipeline described below. For small cell lung cancers, we combined the somatic mutational data from a recent whole-genome sequencing study of 110 patients(11) with data from 39 tumors prospectively sequenced as described above. Prospective sequencing analysis was performed as previously described and extensively validated for clinical use(25).
Mutational signatures analysis
Mutational signature decomposition analysis(12, 31) was performed for all tumor samples with 10 or more somatic mutations (n=61)(31). From the somatic mutations in an individual tumor sample, we infer contributions from known mutational signatures, which are probability distributions over the nucleotide change and flanking 5′ and 3′ nucleotide context of each mutation. Each mutation in the sample is viewed as a random draw from the following random process: first a mutational signature is chosen at random according to its contribution, or degree of exposure, then a mutation type is chosen at random according to that signature. This gives a mixture model, a weighted combination of signatures where the weights are unknown and sum to one, with each weight indicating the proportion of mutations in the sample attributed to that signature. These weights are inferred using an optimization algorithm that maximizes a log-likelihood function derived from this random model. As the APOBEC-mediated mutational process has been attributed to two distinct signatures (signatures 2 and 13)(12), for visualization purposes we show the sum of the fractions of both signatures.
Copy number, clonality, and evolutionary analyses
We determined total, allele-specific, and integer DNA copy number genome-wide using the FACETS algorithm(32) in all tumors (n=81), independent of sequencing platform (targeted, exome, or genome). Briefly, FACETS simultaneously segments total and allele-specific DNA copy number from the coverage and genotypes of polymorphic SNPs genome-wide. Allele-specific segmentation is based on the log odds-ratio of allele fractions at SNPs identified as heterozygous in the normal sample. A fit is applied to the resulting segments, identifying in each sample the 1) log ratio corresponding to diploidy, 2) purity, and 3) average ploidy. Major and minor integer copy number is then assigned to each segment by maximum likelihood. Allelic imbalance (including tumor-specific loss-of-heterozygosity) is determined from a change in the zygosity of heterozygous SNPs. We then defined the presence of genome doubling (GD) in samples for which the majority of the genome (≥50%) contains multiple copies from the same parent/allele. Gene-level copy number (Supplementary Table 7) was assigned from spanning segments of integer copy number data in each tumor. Homozygous deletion was determined from regions of total copy number of zero. Amplifications were those regions of total integer copy number greater than 5 or 6 in diploid and GD cases respectively. Partial deletions (with intragenic breakpoints) were called whereas partial amplifications were not. This same FACETS-based analytical pipeline was run on all retrospective and prospective cohorts (including both targeted and WES data) to allow for direct comparison of gene-level copy number calls.
The purity and integer copy number results from FACETS analysis, along with coverage levels and allele frequencies, were used to estimate the fraction of cancer cells harboring each mutation (cancer cell fraction, CCF) in all evaluable specimens (n=77). For mutations in regions of genomic gain, two CCFs were calculated, assuming the minimum and maximum possible number of copies(33). For each CCF, confidence intervals were estimated as the full-width half-maximum of the posterior probability distribution(34). The timing of emergence of GD relative to somatic mutations was estimated by applying a Gaussian mixture model to the allele fractions of somatic mutations in genomic regions of balanced tetraploidy in 10 tumors for which a sufficient number of such mutations were present (≥20). Mutations that could be confidently assigned membership in the mixture model were assumed to have arisen before or after GD. The number of copies of each mutation per cancer cell, and therefore timing relative to GD, was estimated when there was sufficient separation between component allele fraction distributions per sample. In general, somatic CNAs can also be timed, however, the burden of CNAs in small cell bladder cancer is so high that often the majority of a genome has been affected by multiple independent CNA events. As a result, only a few events can be timed unambiguously.
In the two patients in our cohort with mixed histology disease, histology-specific regions were macro-dissected, so we therefore expected that a minimal amount of cross-contamination among the populations may be present. We corrected for this analytically in the following manner. As the estimated tumor purity was similar, for all mutations shared between the two cell populations in regions that lacked CNAs, we determined the mode of the distribution of the ratio of allele frequencies between the small cell and other histologic population. Mutations in the non-small-cell population with allele frequencies less than or equal to the positive full width at half maximum of this distribution of ratios between the two populations were considered to be arising from a minor contaminating population of small cell present in the second histology and were excluded from comparisons.
Results
We analyzed 87 tumor and matched normal specimens from 61 SCCB patients with a combination of whole-exome, -genome, and -transcriptome sequencing along with targeted deep sequencing of hundreds of key cancer-associated genes (see Methods and Supplementary Tables 1–2). This cohort is comprised predominantly of men (79%), largely muscle-invasive (stage ≥2) disease at diagnosis (89%), were pre-treated tumor specimens, and had a median overall survival of 45 months (95% confidence interval, 31-60) (Supplementary Table 3). To explore bladder histology-, cell lineage-, and organ-specific differences, we compared these results with genomic data from 452 retrospectively and prospectively sequenced high-grade urothelial bladder(9, 10) or small cell lung cancers(11) (303 and 149 respectively, see Methods). Whole-exome and/or genome sequencing of the tumor and matched normal specimens from 17 patients revealed a high somatic mutational burden (median of 10.7 mutations per million bases (Mb) sequenced) that was significantly greater than other genitourinary cancers (Fig. 1A). Despite the prevalence of a past history of smoking (73% were current or former smokers; Supplementary Table 3), mutational signature decomposition analysis(12) in these patients revealed that APOBEC, rather than tobacco-associated mutagenesis predominated. Indeed, 95% of these patients harbored evidence of an APOBEC-mediated mutational process that accounted for a median of 60 ± 23.7% of all somatic mutations in each patient (Fig. 1B). This APOBEC-driven mutational signature (predominantly C>G or C>T mutations at the TCW trinucleotide context) was observed to a lesser degree in bladder urothelial carcinoma (UC), but was largely absent from small cell lung cancers, despite a shared risk factor of past smoking history in all three cancer types(4, 13–15) (Fig. 1B, bottom). Endogenous mutational processes other than APOBEC were present in a subset of samples including two patients with a mutational signature associated with polymerase η/activation-induced cytidine deaminase (AID) defects. Taken together, this suggests risk of smoking-associated bladder cancers is driven by a pathogenic mechanism different than the mutagenesis observed in small cell lung cancers.
Fig. 1. Mutational burden of SCCB.

a) The somatic mutational burden of SCCBs, other genitourinary cancers, and both pulmonary non-small cell adenocarcinomas and small cell carcinomas. Box represents interquartile range (IQR); upper whisker extends from hinge to largest value ≤1.5x IQR b) Mutational signatures in SCCB patients with WES or WGS data (includes three TCGA small cell bladder samples, see Methods), urothelial tumors, and small cell lung cancers (bottom). Patients 06 and 23 possessed a polymerase η-associated AID mutational signature, as indicated. c) Pattern of lesions in TP53, RB1, the TERT promoter, and other effectors of cell cycle regulation in SCCBs and urothelial carcinomas (as labeled, UC inferred from TCGA data). d) Commonly mutated genes in SCCB, UC, and small cell lung cancers (dark blue, light blue, and red respectively) are grouped based on their alteration frequency being predominantly associated with either histology, organ, or cancer type alteration patterns (asterisk, nominal p-value < 0.05; Fisher exact test, error bars represent one standard deviation using the binomial distribution).
TP53 and RB1 were the most frequently altered genes in SCCB, each arising in 90% of patients (Fig. 1C, Supplementary Table 4–5). Mutations in these genes co-occurred in 80% of all tumors, a pattern that is consistent with small cell lung cancers(11) and underscores the importance of G1- to S-phase cell-cycle dysregulation in small cell cancers independent of their organ of origin. Moreover, transcriptome sequencing in 12 patients, while not identifying recurrent gene fusions in SCCBs, did reveal loss of RB1 expression in an SCCB that lacked an RB1 mutation. We confirmed with immunohistochemistry in two additional RB1-wildtype patients that these tumors did not express the retinoblastoma protein (RB) (Supplementary Figure 1), suggesting the presence of occult lesions or epigenetic silencing as the basis for RB1 inactivation in such cases. SCCBs also had a much higher rate of biallelic mutation in these genes compared to urothelial tumors (Supplementary Figure 2). However, 12% of histologically confirmed urothelial bladder cancers also harbored co-occurring alterations in TP53 and RB1(9) suggesting that mutations in one or both of these genes are necessary but not sufficient for the development of the small cell phenotype. Targeted sequencing of 341 key cancer-associated genes at high depth of coverage in 46 additional SCCB confirmed these coincident mutations and further revealed that 95% of SCCB harbor TERT promoter mutations that are also present less frequently in 70% of urothelial tumors (prospective cohort, see Methods; p-value < 1.6×10−4 Fisher exact test), but that are absent from other small cell cancer types including small cell lung cancers(16). Combining the unbiased and targeted sequencing cohorts, we found that recurrent mutations in diverse epigenetic modifiers (KDM6A, ARID1A, CREBBP, EP300, KMT2A/C/D) were present in most SCCB patients (74%, n=45 of 61), a mutational frequency of these genes similar to that observed in UC. Mutations in these chromatin modifying genes were, however, uncommon in small cell lung cancers (p-value < 10−6, Fisher exact test; Fig. 1D) indicating that SCCB likely arise from a UC precursor and have a pathogenesis distinct from that of small cell lung cancers.
Like the genomes of small cell lung cancer, SCCB genomes were complex, although in distinct ways (Supplementary Figure 3). On average, 63% of the SCCB genome harbored DNA copy number alterations (CNAs), similar to UC with TP53 mutations but higher than other epithelial tumors. Focal homozygous and heterozygous deletions of the RB1 and TP53 loci were the most frequent CNAs, contributing to biallelic alterations. Focal CDKN2A deletions and CCND1 amplifications, while common in UC, were absent from SCCB (p-values = 0.02 and 0.0005, Fisher exact test). Organ-specific E2F3 focal amplifications were present in 17% of SCCBs, a frequency similar to that observed in UC but significantly higher than in small cell lung cancers (Fig. 1D). While many of these complex amplicons also span SOX4, another previously hypothesized target of this CNA, whole-genome sequencing resolved the structure of one such event in an affected patient, indicating that E2F3 is the likely target (Supplementary Figure 4). Moreover, the presence of E2F3 amplifications in RB1-null tumors indicates that the former may confer an additional growth advantage or that these effectors have organ-, rather than cell-type-specific non-redundant roles, despite their shared regulation of the G1 to S-phase transition of the cell cycle. Beyond focal CNAs, recurrent broad gains and losses were common, but the frequent 3p losses present in small cell lung cancers were largely absent in SCCB. Notably, while 5p gains were the most common broad CNA in SCCB (10%), these events did not exclusively target the mutant allele of the TERT promoter (5p15). Indeed, 5p genomic gains targeted the wildtype allele of TERT in four tumors, indicating that there is a selective pressure for 5p gains beyond elevating expression of mutant TERT, perhaps targeting another oncogene on the chromosome arm.
Given the chromosomal instability in SCCB, we investigated the presence of genome doubling (GD), which has been previously associated with poor prognosis in individual cancer types(17). Using inferences of ploidy from genome-wide allele-specific copy number (Fig. 2A), we identified GD in 72% of 58 evaluable tumors (Fig. 2B, Supplementary Table 6), a frequency similar to that observed in UC, NOS tumors(18). Whereas GD has been associated with TP53 mutations in other tumor types(17, 18), GD was more common in SCCBs with missense rather than loss-of-function TP53 mutations (nonsense, frameshift, splice site, and homozygous deletions; p-value < 10−4, Fisher exact test; Fig. 2C). Moreover, there appeared to be selection for biallelic alteration of TP53 missense-mutant GD-positive tumors. Among tumors with TP53 biallelic mutation, a single mutation followed by copy-neutral loss-of-heterozygosity (CN-LOH) predominated (Fig. 2C and Supplementary Figure 5). Notably, in GD-positive tumors from three patients in which the two independent TP53 mutations could be phased, we confirmed they were present in trans (Fig. 2D).
Fig. 2. Genome doubling as a function of TP53 aberrations.
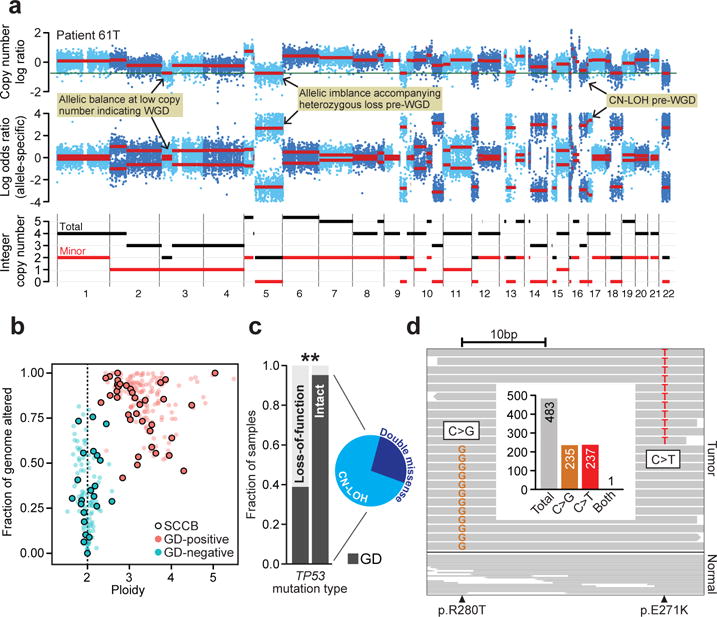
a) Total, allele-specific, and integer copy number segmentation inferred from whole-exome sequencing (chromosomes 1-22; top, middle, bottom) of a representative SCCB genome is shown (patient 61T). Hallmark lesions are indicated (yellow). b) The overall burden of CNAs as a function of tumor ploidy in both SCCBs and urothelial carcinomas (points are individual tumors, SCCBs are those with black border). GD-positive tumors are in red. GD-positive tumors have both higher ploidy and elevated levels of CNAs genome-wide. c) GD as a function of TP53 mutational and zygosity status (biallelic missense with inset representing underlying mechanism; asterisk, p-value < 10−4, Fisher exact test). d) A representative tumor possessing two independent TP53 mutations (patient 70, R280T and E271K) that could be phased, reads spanning both mutant sites indicate that the mutations were in trans. Inset, targeting sequencing coverage of reads spanning both mutations are shown indicating the near absence of reads harboring both mutant alleles.
To better understand how such cardinal events and GD evolved in SCCB, we sought to time the emergence of specific genomic alterations in the chronology of disease pathogenesis from individual tumor specimens. In 10 tumors harboring sufficient somatic mutations (≥20) in regions of balanced tetraploidy for the timing analysis, we found that GD arose at various points in molecular time relative to other somatic mutations (Fig. 3A). The co-incident RB1, TP53, and TERT promoter mutations, however, arose prior to GD in all tumors, emphasizing their early role in SCCB pathogenesis. In some patients, typified by the paired primary and metastatic tumors of patient 37, only a small minority of the total somatic mutational burden (<5%) arose before GD, indicating GD itself was an early event, perhaps arising shortly after TP53 alteration(s), while in other patients, most somatic mutations including the cardinal co-incident lesions arose before GD, as was the case for patients 7 and 61. Notably, even the dominant mutational process varied with time. For instance, the APOBEC-associated mutational signature so pervasive in SCCBs was observed only after GD in patient 59, despite 28% of its somatic mutational burden arising prior to GD (Fig. 3B). In other patients, the APOBEC-associated mutational process produced most somatic mutations early, but ebbed as the tumor evolved after GD. Integrating these chronological analyses in a representative SCCB (patient 61), we found that the cardinal TP53, RB1, and TERT promoter mutations all arose very early in molecular time. RB1 and PTEN biallelic inactivation evolved from truncating mutations after which heterozygous losses targeted the wildtype allele occurred. Conversely, a TP53 E285K missense mutation was followed by CN-LOH resulting in two copies of the mutant allele after which GD arose, increasing the mutant allele burden of all four of these genes (Fig. 3C). While most other predominantly APOBEC-associated somatic mutations arose prior to GD, indicating it was a later event in this patient, several broad CNAs appeared both before and after GD. In the patient in whom we profiled a primary diagnostic tumor and matched adrenal metastasis, the vast majority of somatic mutations were clonal in both, indicating both tumors emerged from a shared antecedent clone after which linear clonal evolution was evident with only a few metastasis-specific CNAs arising late.
Fig. 3. Heterogeneous evolutionary histories.
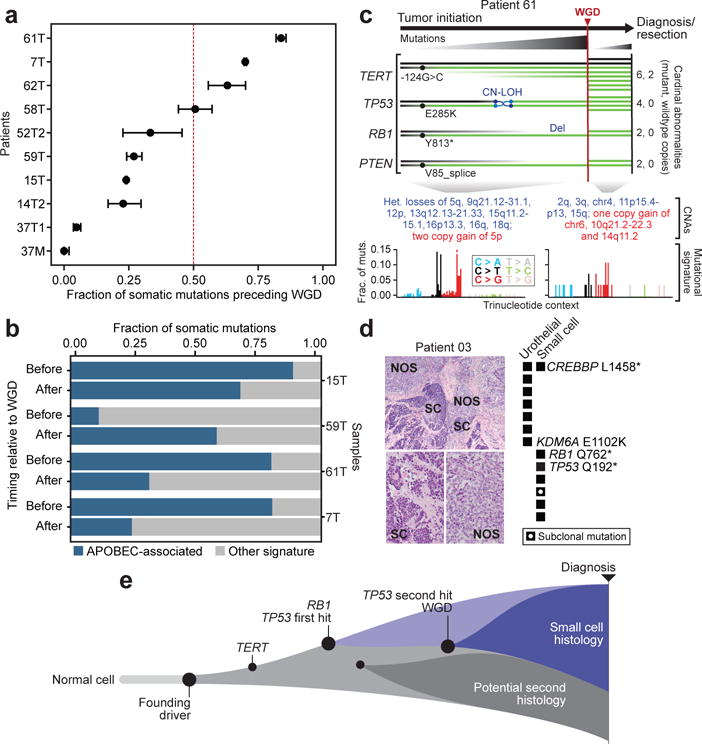
a) Genome doubling (GD) can arise at different points in the chronology of tumor development as indicated by the fraction of somatic mutations present at diagnosis that arose before and after GD (error bars represent one standard deviation using the binomial distribution; 10 patients shown are those with ≥20 somatic mutations in regions of balanced tetraploidy). b) While in some tumors, APOBEC-associated mutagenesis is constant before and after GD, in others it either accelerates or ebbs after a genome doubling indicating the pressure of a given mutagenic process is not constant in SCCB evolution. c) Integrated analysis of mutational event timing in a typical SCCB (patient 61) indicates that multiple mutant copies of cardinal lesions are present after a late GD event that is preceded by most the APOBEC-induced somatic mutational burden. d) Coincident urothelial (not otherwise specified – NOS) and small cell (SC) histologies from a single patient with mixed histology disease (left) and the corresponding lesion-specific somatic mutations (right). e) Schematic of the evolution of SCCBs in which a founding lesion that initiates cellular transformation precedes the cardinal RB1/TP53 lesions that define a branch point of the small cell histology that may or may not co-exist with a second minor population of a distinct histology with its own lesions.
To explore how cardinal events such as RB1, TP53, and TERT promoter mutations arise and contribute to the conversion of a preceding urothelial carcinoma to predominant small cell histology, we sampled and deeply sequenced histologically distinct regions of tumors with clear and spatially separated regions of both small cell and urothelial carcinoma, NOS from the same patients. By comparing the mutations common among, or specific to, multiple histologically distinct regions of mixed-histology tumors presumably originating from a single common ancestor, we can draw inferences on the timing of their emergence and their phylogenetic origins. In two such patients, each with two histologically distinct tumor specimens sequenced by MSK-IMPACT, a pattern of branching evolution was apparent. The branch point, defined by specific somatic mutations, represented cellular differentiation between histologies. In both illustrative cases, one or more truncal mutations were clonal in both cell populations. In one patient, a single CREBBP L1458* nonsense mutation was clonal in both the small cell and urothelial cell populations (Fig. 3D), whereas RB1 and TP53 mutations were present and clonal in only the small cell component. Conversely, several mutations including KDM6A E1102K were present exclusively in the urothelial component, a finding that is consistent with the increased frequency of KDM6A mutations in UC overall compared to SCCB (Fig. 1D). In the other patient, both a TERT promoter mutation (−145/C>T) and a PIK3CA Q546P hotspot mutation, along with several others, were clonal in both the small cell and urothelial (papillary) tumor components arising prior to the cellular differentiation program that defined the two histologies (Supplementary Figure 6). On the other hand, the small cell component again exclusively possessed RB1 and TP53 mutations, whereas the papillary population possessed a clonal activating ERBB2 L755S mutation. These results reaffirm the nearly obligate emergence of RB1 and TP53 mutations in SCCB, but also indicate that these two cardinal events are not the founder mutations necessary for initial transformation and clonal outgrowth of a histologically distinct bladder tumor cell population (Fig. 3E).
Despite the evolutionary heterogeneity of SCCB, most driver mutations of potential clinical actionability were clonal at diagnosis. Exploring the landscape of potentially actionable lesions in SCCB, we found that 46% of patients (n=28 of 61) harbored a lesion of potential therapeutic significance defined as either an FDA-approved or NCCN compendium-listed biomarker or prior clinical evidence associating it as a biomarker of drug response in this or another indication (Supplementary Figure 7). In total, 13% of patients (8 of 61) harbored mutations in PIK3CA (Supplementary Figure 7A). Unlike in UC, SCCBs lack ERBB2 amplifications(9, 19), but 14 patients harbored likely activating ERBB2 mutations including hotspot mutations in both the extracellular (S310) and kinase domains (L755) (Supplementary Figure 7B). Among other RTKs, ERBB3 mutations affected 15% of patients. FGFR3 hotspot mutations (S249C) were much less common in SCCB than in UC, likely owing to their mutual exclusivity with RB1 loss, reaffirming the highly RB1-dependent G1/S checkpoint dysfunction in these tumors. Beyond mutations in ERCC2, which correlate with sensitivity to platinum-based therapy in muscle-invasive bladder cancers(20), there were also mutations in other effectors of DNA repair signaling (Supplementary Figure 7C). Overall, the presence of therapeutically actionable lesions within SCCBs revealed specific clinical hypotheses that can be tested as part of broader early-phase ‘basket’ studies that are uniquely suited to study such rare tumor types.
Discussion
Our findings indicate that small cell carcinomas of the bladder and lung have a convergent but distinct pathogenesis. Consistent with small cell lung cancer, we observed obligate likely early-arising lesions in RB1 and TP53. We also identified bladder-specific mutations in the TERT promoter and in chromatin modifying genes, among others. A substantial subset of TP53 mutations were biallelic missense rather than loss-of-function, a setting in which GD arose preferentially suggesting that GD is more strongly associated with TP53 neomorphism rather than conventional loss of function in SCCB. Also, while evolutionarily diverse, we demonstrated that there are truncal mutations in tumors with mixed histology(21), but histology-specific lesions in RB1 and TP53 determine the small cell phenotype and appear to arise early in molecular time, likely shortly after the founding driver. This indicates that small cell and urothelial bladder cancers have a shared cellular origin, with the former representing a de-differentiation from urothelial carcinoma, quite unlike small cell histologies in other organ types (Fig. 3E)(22, 23). Of course, a small percentage of UC also harbor alterations in RB1 and TP53, which can be detected in even non-invasive precursor lesions (unpublished data). Therefore, while these lesions are necessary, they alone are likely insufficient to drive small cell differentiation. Moreover, unlike in prostate cancers progressing on anti-androgen therapy(24), transdifferentiation in bladder cancers has been difficult to identify. Therefore, future studies should explore whether epigenomic or transcriptional events interact with the loss of RB1 and TP53 to confer the small cell phenotype. Taken together, however, these findings suggest that the undifferentiated neuroendocrine features present in small cell carcinoma are likely associated with the specific alterations present in the affected cell rather than prior mutagen exposure, tissue of origin, or mutational burden. Overall, aside from RB1 and TP53 alterations, genomic alterations present in SCCB more closely resemble UC than small cell lung cancers, indicating that most alterations contribute to oncogenesis in an organ-specific manner rather than cell type-specific manner.
Supplementary Material
Statement of Translational Relevance.
Beyond their cardinal lesions and distinct pathogenesis, nearly half of small cell bladder cancers (46%) harbor a potentially therapeutically actionable lesion that may serve as a rationale for clinical hypothesis testing as part of broader early-phase basket studies uniquely suited to the study of such rare tumor types.
Acknowledgments
We would like to thank the members of the Marie-Josée and Henry R. Kravis Center for Molecular Oncology for their assistance.
Financial support: This work was supported in part through National Institutes of Health awards T32 GM007175 (M.T.C), P30 CA008748, UL1 TR024996, and R01 CA204749 (B.S.T); the Sontag Foundation (B.S.T.); the Robertson Foundation and Prostate Cancer Foundation (N.S. and B.S.T.); and Cycle for Survival (H.A., D.B.S., and B.S.T.).
Footnotes
Conflict of interest disclosure: The authors declare no competing financial interests.
Data availability
All somatic mutational calls and CNAs along with accompanying clinical data are available for analysis and visualization in the cBioPortal for Cancer Genomics, http://cbioportal.org/. Raw sequencing data have been deposited in the Database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap/) under the accession number phsXXXXXX.
Author Contributions
D.B.S., H.A.A., and B.S.T. conceived the study. N.B.D., E.K.C., X.H., V.E.R., C.M.R., B.H.B., J.E.R., D.F.B., G.I., and H.A.A. assisted with specimen collection, clinical data, and pathology. M.T.C., A.P., N.D.S., R.S., V.E.S., R.K., A.A., A.V., N.S., M.F.B., and G.I. assisted with sequencing and data analysis. M.T.C., A.P., D.B.S., H.A.A., and B.S.T. wrote the manuscript with input from all authors.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15:25–41. doi: 10.1038/nrc3817. [DOI] [PubMed] [Google Scholar]
- 3.Al-Ahmadie HA, Iyer G, Lee BH, Scott SN, Mehra R, Bagrodia A, et al. Frequent somatic CDH1 loss-of-function mutations in plasmacytoid variant bladder cancer. Nat Genet. 2016;48:356–8. doi: 10.1038/ng.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng L, Pan CX, Yang XJ, Lopez-Beltran A, MacLennan GT, Lin H, et al. Small cell carcinoma of the urinary bladder: a clinicopathologic analysis of 64 patients. Cancer. 2004;101:957–62. doi: 10.1002/cncr.20456. [DOI] [PubMed] [Google Scholar]
- 5.Quek ML, Nichols PW, Yamzon J, Daneshmand S, Miranda G, Cai J, et al. Radical cystectomy for primary neuroendocrine tumors of the bladder: the university of southern california experience. J Urol. 2005;174:93–6. doi: 10.1097/01.ju.0000162085.20043.1f. [DOI] [PubMed] [Google Scholar]
- 6.Mukesh M, Cook N, Hollingdale AE, Ainsworth NL, Russell SG. Small cell carcinoma of the urinary bladder: a 15-year retrospective review of treatment and survival in the Anglian Cancer Network. BJU Int. 2009;103:747–52. doi: 10.1111/j.1464-410X.2008.08241.x. [DOI] [PubMed] [Google Scholar]
- 7.Siefker-Radtke AO, Kamat AM, Grossman HB, Williams DL, Qiao W, Thall PF, et al. Phase II clinical trial of neoadjuvant alternating doublet chemotherapy with ifosfamide/doxorubicin and etoposide/cisplatin in small-cell urothelial cancer. J Clin Oncol. 2009;27:2592–7. doi: 10.1200/JCO.2008.19.0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abbas F, Civantos F, Benedetto P, Soloway MS. Small cell carcinoma of the bladder and prostate. Urology. 1995;46:617–30. doi: 10.1016/S0090-4295(99)80290-8. [DOI] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–22. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim PH, Cha EK, Sfakianos JP, Iyer G, Zabor EC, Scott SN, et al. Genomic predictors of survival in patients with high-grade urothelial carcinoma of the bladder. Eur Urol. 2015;67:198–201. doi: 10.1016/j.eururo.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. doi: 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cumberbatch MG, Rota M, Catto JW, La Vecchia C. The Role of Tobacco Smoke in Bladder and Kidney Carcinogenesis: A Comparison of Exposures and Meta-analysis of Incidence and Mortality Risks. Eur Urol. 2015 doi: 10.1016/j.eururo.2015.06.042. [DOI] [PubMed] [Google Scholar]
- 14.Freedman ND, Silverman DT, Hollenbeck AR, Schatzkin A, Abnet CC. Association between smoking and risk of bladder cancer among men and women. JAMA. 2011;306:737–45. doi: 10.1001/jama.2011.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kenfield SA, Wei EK, Stampfer MJ, Rosner BA, Colditz GA. Comparison of aspects of smoking among the four histological types of lung cancer. Tob Control. 2008;17:198–204. doi: 10.1136/tc.2007.022582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng X, Zhuge J, Bezerra SM, Faraj SF, Munari E, Fallon JT, 3rd, et al. High frequency of TERT promoter mutation in small cell carcinoma of bladder, but not in small cell carcinoma of other origins. J Hematol Oncol. 2014;7:47. doi: 10.1186/s13045-014-0047-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014;4:175–85. doi: 10.1158/2159-8290.CD-13-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–40. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iyer G, Al-Ahmadie H, Schultz N, Hanrahan AJ, Ostrovnaya I, Balar AV, et al. Prevalence and co-occurrence of actionable genomic alterations in high-grade bladder cancer. J Clin Oncol. 2013;31:3133–40. doi: 10.1200/JCO.2012.46.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Allen EM, Mouw KW, Kim P, Iyer G, Wagle N, Al-Ahmadie H, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014;4:1140–53. doi: 10.1158/2159-8290.CD-14-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thota S, Kistangari G, Daw H, Spiro T. A clinical review of small-cell carcinoma of the urinary bladder. Clin Genitourin Cancer. 2013;11:73–7. doi: 10.1016/j.clgc.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Fine SW. Neuroendocrine lesions of the genitourinary tract. Adv Anat Pathol. 2007;14:286–96. doi: 10.1097/PAP.0b013e3180ca8a89. [DOI] [PubMed] [Google Scholar]
- 23.Wick MR, Marchevsky AM. Neuroendocrine neoplasms of the lung: Concepts and terminology. Semin Diagn Pathol. 2015;32:445–55. doi: 10.1053/j.semdp.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 24.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–64. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Ahmadie H, Iyer G, Hohl M, Asthana S, Inagaki A, Schultz N, et al. Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer Discov. 2014;4:1014–21. doi: 10.1158/2159-8290.CD-14-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engstrom PG, Steijger T, Sipos B, Grant GR, Kahles A, Ratsch G, et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods. 2013;10:1185–91. doi: 10.1038/nmeth.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–59. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenman CD, Pleasance ED, Newman S, Yang F, Fu B, Nik-Zainal S, et al. Estimation of rearrangement phylogeny for cancer genomes. Genome Res. 2012;22:346–61. doi: 10.1101/gr.118414.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7:283ra54. doi: 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.