

Abstract
Rhodium metalloinsertors are a unique set of metal complexes that bind specifically to DNA base pair mismatches in vitro and kill mismatch repair (MMR)-deficient cells at lower concentrations than their MMR-proficient counterparts. A family of metalloinsertors containing rhodium-oxygen ligand coordination, termed “Rh–O” metalloinsertors, has been prepared and shown to have a significant increase in both overall potency and selectivity towards MMR-deficient cells regardless of structural changes in the ancillary ligands. Here we describe DNA-binding and cellular studies with the second generation of Rh–O metalloinsertors in which an ancillary ligand is varied in both steric bulk and lipophilicity. These complexes, of the form [Rh(L)(chrysi)(PPO)]2+, all include the O-containing PPO ligand (PPO = 2-(pyridine-2-yl)propan-2-ol) and the aromatic inserting ligand chrysi (5,6-chrysene quinone diimine) but differ in the identity of their ancillary ligand L, where L is a phenanthroline or bipyridyl derivative. The Rh–O metalloinsertors in this family all show micromolar binding affinities for a 29-mer DNA hairpin containing a single CC mismatch. The complexes display comparable lipophilic tendencies and pKa values of 8.1–9.1 for dissociation of an imine proton on the chrysi ligand. In cellular proliferation and cytotoxicity assays with MMR-deficient cells (HCT116O) and MMR-proficient cells (HCT116N), the complexes containing the phenanthroline-derived ligands show highly selective cytotoxic preference for the MMR-deficient cells at nanomolar concentrations. Using mass spectral analyses, it is shown that the complexes are taken into cells through a passive mechanism and exhibit low accumulation in mitochondria, an off-target organelle that, when targeted by parent metalloinsertors, can lead to non-selective cytotoxicity. Overall, these Rh–O metalloinsertors have distinct and improved behavior compared to previous generations of parent metalloinsertors, making them ideal candidates for further therapeutic assessment.
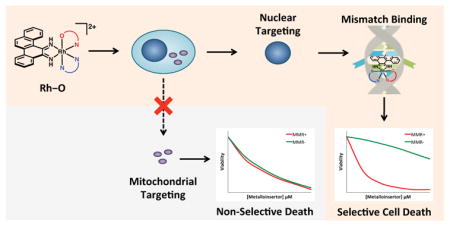
Graphical Abstarct
INTRODUCTION
Over the past 70 years, DNA and its associated metabolic processes have proven to be fruitful targets for the design of new therapeutic agents.1 Many of the most common FDA-approved chemotherapeutics work by binding DNA, such as the DNA-crosslinking agent cisplatin and the DNA-intercalating agent doxorubicin.2–5 Despite the prevalence of these drugs in the clinic, there are many drawbacks to their design and mechanisms of action. In many cases, the drugs target a generic DNA structure that is common to both healthy and cancerous cells. The incidental targeting of healthy tissue can result in dramatic and often dose-limiting side effects, such as emesis and nephrotoxicity.6 To circumvent these off-target effects, it is essential to identify new therapeutic targets that are almost exclusively found within cancerous tissues and cells.
In our research, we focus on one such target: DNA base pair mismatches. Mismatches occur regularly in cells due to polymerase errors or interaction with exogenous compounds.7 In healthy cells, these errors are corrected by the mismatch repair (MMR) machinery of the cell. However, in many solid tumors or tumors of Lynch syndrome patients, mutations in MMR proteins severely down-regulate or completely inactivate repair.8,9 As a result, these cancers contain a relative abundance of DNA base pair mismatches compared to healthy cells, making mismatches a potential biomarker for selective cancer therapy.
Mismatched base pairs have been targeted through the design of metal complexes, called rhodium metalloinsertors, which selectively and non-covalently bind these lesions.10 Rhodium metalloinsertors contain a sterically expansive aromatic chrysi (5,6-chrysenequinone diimine) ligand that is capable of π-stacking with DNA bases. Due to steric bulk, however, the chrysi ligand is unable to easily intercalate into well-matched DNA, and instead primarily interacts with DNA at thermodynamically destabilized sites, such as mismatches or abasic sites.11 The ability of a prototypical metalloinsertor, [Rh(bpy)2(chrysi)]3+ (bpy = 2,2′-bipyridine), to selectively bind DNA mismatches has been verified using both in vitro binding assays and crystallographic studies.12–15 Crystallographic and NMR studies show that this complex binds DNA mismatches via metalloinsertion, a non-covalent binding mode in which the complex inserts into DNA at the mismatched site from the minor groove, ejects the mismatched DNA bases, and π-stacks with the flanking well-matched base pairs.14 This mismatch-targeting ability has also been seen in human cell culture experiments, with metalloinsertors exhibiting enhanced cytotoxicity in MMR-deficient cell lines relative to their MMR-proficient counterparts.15,16 This result is in stark contrast to most DNA-targeting therapeutics, such as the aforementioned cisplatin and doxorubicin, which are selective towards MMR-proficient cell lines over MMR-deficient cell lines, leading to the development of resistance in MMR-deficient tumors following treatment.17,18
Several generations of metalloinsertors have been synthesized since [Rh(bpy)2(chrysi)]3+, which has led to the recent discovery of a potent and selective family of rhodium metalloinsertors containing a pyridyl-alcohol ligand and unique Rh–O ligand coordination (Figure 1).19 This Rh–O ligand coordination is structurally distinct from earlier generations of parent metalloinsertors, which contained solely Rh–N coordination.20 Furthermore, these Rh–O metalloinsertors were found to have improved potency and selectivity towards MMR-deficient cancer cells over MMR-proficient cancer cells. Surprisingly, this high potency and cell selectivity was seen across a variety of metalloinsertors containing O-coordinated ligands that differed significantly in size and structure (spanning methyl, pyridyl, phenyl, and hexyl functionalization), suggesting the biological activities of Rh–O metalloinsertors are not perturbed by ligand substitution off of the O-containing site.
Figure 1.

General structure of the newest generation of Rh−O metalloinsertors (left), which show improved potency and selectivity over parent metalloinsertors that have exclusively Rh–N coordination (right). R has been varied between methyl, phenyl, pyridyl, and hexyl groups and N—N has been varied between several bpy, phen, and HDPA derivatives.
Here, a family of rhodium metalloinsertors was designed and synthesized as variations of the Rh–O metalloinsertor [Rh(phen)(chrysi)(PPO)]2+ (phen = 1,10-phenanthroline). These complexes, of the form [Rh(L)(chrysi)(PPO)]2+, all include the O-containing PPO ligand but differ in the identity of their ancillary ligand, L, where L= bpy, HDPA (2,2′-dipyridylamine), 4,7-DMP (4,7-dimethyl-1,10-phenanthroline), 5,6-DMP (5,6-dimethyl-1,10-phenanthroline), and DIP (4,7-diphenyl-1,10-phenanthroline) (Figure 2). The ancillary ligand substitution alters the steric bulk and lipophilicity of these complexes, which can ultimately affect DNA-binding properties and biological activity.20,21 Each complex described, even the most lipophilic and sterically bulky, shows biological selectivity towards MMR-deficient cell lines, further demonstrating that the Rh–O ligand framework is amenable to a wide array of functionalization. To better understand the trends in biological activity of these complexes, each metalloinsertor was examined for binding affinity to mismatched DNA, pKa, lipophilicity, whole cell uptake, and subcellular localization into the nucleus and mitochondria. The results indicate that minimizing uptake of the complexes into the mitochondria may be a key factor in ensuring high biological selectivity and support that these Rh–O complexes exhibit distinct differences in metalloinsertor-DNA binding and cell activation compared to parent metalloinsertors.
Figure 2.

Chemical structures of [Rh(L)(chrysi)(PPO)]2+ family of rhodium metalloinsertors, with the PPO ligand shown in red and the changing ancillary ligand shown in blue.
EXPERIMENTAL METHODS
Materials
Commercially available chemicals were used as received. All reagents and Sephadex ion-exchange resin were obtained from Sigma-Aldrich with the following exceptions. RhCl3 was purchased from Pressure Chemical, Inc. Dowex ion-exchange beads were purchased from Acros Organics. Analytical standards for Rb and transition metals were purchased from Analytical West and Ultra Scientific, respectively. MTT and ELISA assay kits were obtained from Roche. Pierce BCA assay kit and NP40 were purchased from Thermo Scientific. Sep-pak C18 solid-phase extraction (SPE) cartridges were purchased from Waters Chemical Co. Cell culture media and supplements were purchased from Invitrogen. Tissue culture flasks and plates were obtained from Corning. 32P labeled ATP was purchased from Perkin Elmer. UreaGel supplies were purchased from National Diagnostics. Microbiospin columns were purchased from BioRad.
Synthesis and Characterization of Metal Complexes
[Rh(phen)(chrysi)(PPO)]Cl2 and [Rh(bpy)2(chrysi)]Cl3 were synthesized following the published protocols.19,22 New metal complexes were synthesized in a similar manner to published procedures.19,20,23 A description of the general synthetic procedures are below. Complete synthetic details for each complex, including specific amounts (masses, volumes, and ratios) as well as slight deviations from the synthetic scheme below, can be found in the SI.
Synthesis of [Rh(L)Cl4][K or H3O] complexes
For L = bpy, 4,7-DMP, 5,6-DMP, and DIP: RhCl3•3H2O (1 equiv.) and KCl (1 equiv.) were refluxed in methanol for 2 h at 98 °C. Ligand (L, 1 equiv.) was added in a minimum volume of methanol and refluxed for 4 h, during which the deep red solution turned to golden-brown precipitate. The solution was filtered over a medium fritted filter, rinsed with methanol, and dried under vacuum to produce [Rh(L)Cl4]K. Crude yield: 84% (bpy), 86% (4,7-DMP), 91% (5,6-DMP), 95% (DIP). For L = HDPA: RhCl3•3H2O (1 equiv.) was refluxed in concentrated HCl (38% w/v) for 3 h at 98 °C. Ligand (L, 2 equiv.) was added in a minimum volume of HCl, followed immediately by boiling water. The solution was refluxed for 16 h, then cooled to 4 °C. The golden precipitate was filtered over a Buchner funnel and dried under vacuum to produce [Rh(L)Cl4][H3O]. Crude yield: 100% (HDPA).
Synthesis of [Rh(L)(NH3)4][OTf]3
[Rh(L)Cl4][K or H3O] (1 equiv.) was added to an oven-dried 25 mL Schlenk flask and degassed under argon. Neat triflic acid (HOTf, 10 g, excess) was added to the flask under positive argon pressure, producing a deep red solution. The flask was purged to remove newly formed HCl gas and stirred for 16 h. The solution was then added dropwise to cold, stirring ether at −78 °C to produce a yellow-brown precipitate. The precipitate was filtered over a medium fritted filter and rinsed with additional cold ether. The product, [Rh(L)(OTf)4][K or H3O], was combined with NH4OH (28% w/v) and stirred at 40 °C for 1 h. The solvent was removed under vacuum and the product was suspended in minimal ethanol, precipitated with ether, filtered over a medium fritted filter, and dried further under vacuum to produce [Rh(L)(NH3)4][OTf]3. Crude yields of 42% (bpy), 10% (HDPA), 15% (4,7-DMP), 77% (5,6-DMP), 72% (DIP).
Synthesis of [Rh(L)(chrysi)(NH3)2][OTf]3
[Rh(L)(NH3)4][OTf]3 (1 equiv.) was combined with 5,6-chrysene-quinone (1 equiv.) and a mixture of acetonitrile, water, and NaOH, and stirred for 1–12 h at ambient temperature. The solution changed from bright orange (the color of free ligand) to red-brown (for L = bpy, HDPA, 5,6-DMP, and DIP) or green-brown (for L=4,7-DMP) with no precipitate. The reaction was quenched with HCl, producing a deep red solution, and the solvent was removed under vacuum. The products from L=bpy, HDPA, and DIP were purified using a C18 SepPak, pre-equilibrated with 0.1% TFA (aq, TFA = trifluoroacetic acid) and eluted with 1:3 MeCN:0.1% TFA (aq). The products from L=4,7-DMP and 5,6-DMP were purified by HPLC using a reverse phase C18 column with gradient elution from 15:85 MeCN:0.1% TFA (aq) to 95:5 MeCN:0.1% TFA (aq) over 30 min. Products were in the form of [Rh(L)(chrysi)(NH3)2][TFA]3. Crude yields of 33% (bpy), 51% (HDPA), 46% (4,7-DMP), 62% (5,6-DMP), 100% (DIP).
Synthesis of [Rh(L)(chrysi)(PPO)]Cl2
[Rh(L)(chrysi)(NH3)2][TFA]3 (1 equiv.) was combined with PPO in a mixture of ethanol and water and refluxed 16 h (for L = bpy, 4,7-DMP, 5,6-DMP, and DIP) or 7 days (for L=HDPA). The solvent was removed under vacuum and the product was purified by HPLC using the method described above for L = bpy, HDPA, 4,7-DMP, and DIP. For L = 5,6-DMP, an isocratic method of 30:70 MeCN:0.1% TFA (aq) was used. For L = bpy, HDPA, and 4,7-DMP, the purified product was converted to the chloride salt using Sephadex QAE resin charged with MgCl2. For L = 5,6-DMP and DIP, the purified product was converted to the chloride salt using Dowex 1×2 500-100 mesh ion exchange resin. Purified yields of 30% (bpy), 10% (HDPA), 10% (4,7-DMP), 23% (5,6-DMP), 33% (DIP).
Characterization of [Rh(bpy)(chrysi)(PPO)](TFA)2
LCQ-MS (cation): m/z calc. 650.1 (M-1H+), 325.6 (M2+); obs. 650.0, 325.8. UV-Vis (H2O): 259nm (59,800 M−1 cm−1), 287nm (43,100 M−1 cm−1), 298nm (37,100 M−1 cm−1), 312nm (32,000 M−1 cm−1), 435nm (10,000 M−1 cm−1). 1H NMR (500 MHz, Acetonitrile-d3) δ 13.44 (br s, 1.2H), 11.89 (br s, 2H), 9.45 (d, J = 5.6 Hz, 1H), 9.36 (d, J = 5.7 Hz, 0.6H), 8.80 (d, J = 8.0, 1.4 Hz, 1H), 8.71 (d, J = 5.3 Hz, 0.6H), 8.62 (d, J = 8.2 Hz, 0.6H), 8.60-8.54 (m, 2.6H), 8.43-8.26 (m, 8H), 8.26-8.21 (m, 1H), 8.14 (d, J = 8.2, 1.5 Hz, 0.6H), 8.06-7.89 (m, 4.8H), 7.85-7.78 (m, 1,6H), 7.77-7.68 (m, 3.2H), 7.68-7.61 (m, 2.2H), 7.60-7.52 (m, 2.6H), 7.31 (d, 0.6H), 7.29-7.21 (m, 2.6H), 1.91 (s, 3H), 1.87 (s, 1.8H), 1.58 (s, 4.8H), purified as a 1:0.6 mixture of diastereomers.
Characterization of [Rh(HDPA)(chrysi)(PPO)](TFA)2
LCQ-MS (cation): m/z calc. 665.2 (M-1H+), 333.1 (M2+); obs. 665.3, 333.3. UV-Vis (H2O): 259nm (60,400 M−1 cm−1), 283nm (45,900 M−1 cm−1), 326nm (18,600 M−1 cm−1), 440nm (8,500 M−1 cm−1). 1H NMR (500 MHz, Acetonitrile-d3) δ 12.49 (br s, 1H), 12.04 (br s, 1H), 8.72 (dd, J = 8.0, 1.3 Hz, 1H), 8.50 (d, J = 6.0 Hz, 1H), 8.38-8.31 (m, 3H), 8.31-8.23 (m, 2H), 8.20-8.13 (m, 2H), 8.08-8.00 (m, 2H), 7.98 (td, J = 8.6, 1.6 Hz, 1H), 7.94-7.81 (m, 4H), 7.69 (m, 3H), 7.51 (ddd, J = 7.6, 6.0, 1.4 Hz, 1H), 7.23 (ddd, J = 7.4, 6.1, 1.3 Hz, 1H), 7.17 (ddd, J = 7.4, 6.2, 1.4 Hz, 1H), 1.78 (s, 3H), 1.56 (s, 3H), purified as a single diastereomer.
Characterization of [Rh(4,7-DMP)(chrysi)(PPO)](TFA)2
LCQ-MS (cation): m/z calc. 702.2 (M-1H+), 351.6 (M2+); obs. 702,3, 351.8. UV-Vis (H2O): 269nm (106,800 M−1 cm−1), 440nm (11,400 M−1 cm−1). 1H NMR (500 MHz, Acetonitrile-d3) δ 13.31 (br s, 0.8H), 11.75 (br s, 2H), 9.50 (d, J = 5.4 Hz, 1H), 9.42 (d, J = 5.4 Hz, 0.4H), 8.86 (dd, J = 5.5, 0.9 Hz, 1H), 8.83 (dd, J = 8.0, 1.3 Hz, 1H), 8.73 (d, J = 5.4 Hz, 0.4H), 8.47 (d, J = 2.5 Hz, 0.4H), 8.46-8.35 (m, 4.2H), 8.34 (d, J = 8.2 Hz, 0.4H), 8.27 (d, J = 8.8 Hz, 1H), 8.21-8.17 (m, 1.4H), 8.16 (d, J = 8.1 Hz, 0.4H), 8.08 (dd, J = 5.4, 1.0 Hz, 1H), 8.04 (d, J = 5.4 Hz, 0.4H), 8.00 (dd, J = 7.5, 1.7 Hz, 1H), 7.97-7.92 (m, 2.4H), 7.84 (m, 1.8H), 7.77 (m, 1.4H), 7.61-7.51 (m, 5.2H), 7.19-7.15 (m, 0.4H), 7.10-7.03 (m, 2.8H), 3.05 (s, 3H), 3.04 (s, 1.2H), 3.02 (s, 1.2H), 2.99 (s, 3H), 1.95 (s, 3H), 1.92 (s, 1.2H), 1.62 (s, 3H), 1.61 (s, 1.2H), purified as a 1:0.4 mixture of diastereomers.
Characterization of [Rh(5,6-DMP)(chrysi)(PPO)](TFA)2
LCQ-MS (cation): m/z calc. 702.2 (M-1H+), 351.6 (M2+); obs. 702.3, 351.8. UV-Vis (H2O): 267nm (80,600 M−1 cm−1), 280nm (81,700 M−1 cm−1), 438nm (10,500 M−1 cm−1). 1H NMR (500 MHz, Acetonitrile- d3) δ 13.40 (br s, 0.3H), 11.77 (br s, 1H), 9.68 (d, J = 5.2 Hz, 1H), 9.59 (d, J = 5.1 Hz, 0.3H), 9.06-8.97 (m, 3.9H), 8.84-8.89 (m, 1.3H), 8.43-8.37 (m, 2.6H), 8.34 (d, J = 8.2 Hz, 0.3H), 8.29-8.14 (m, 5.2H), 8.02-7.97 (m, 2.3H), 7.96-7.89 (m, 2.6H), 7.83-7.73 (m, 1.6H), 7.57 (td, J = 7.4, 1.4 Hz, 2H), 7.55-7.50 (m, 1H), 7.17 (d, J = 5.7 Hz, 0.3H), 7.10-7.02 (m, 2.3H), 2.91 (s, 0.9H), 2.90 (s, 0.9H), 2.89 (s, 3H), 2.87 (s, 3H), 1.93 (s, 3H), 1.90 (s, 0.9H), 1.58 (s, 3.9H), purified as a 1:0.3 mixture of diastereomers.
Characterization of [Rh(DIP)(chrysi)(PPO)]Cl2
LCQ-MS (cation): m/z calc. 826.2 (M-1H+); obs. 826.3. UV-Vis (H2O): 267nm (103,000 M−1 cm−1). 1H NMR (500 MHz, Methanol-d4) δ 9.74 (dd, J = 5.5, 0.9 Hz, 1H), 9.70 (dd, J = 5.5, 0.8 Hz, 0.5H), 8.89 (m, 1.5H), 8.76 (m, 1.5H), 8.58-8.46 (m, 4.5H), 8.40-8.28 (m, 6H), 8.14-7.98 (m, 4.5H), 7.81-7.59 (m, 15H), 7.56-7.49 (m, 1.5H), 7.41-7.33 (m, 6H), 7.34-7.23 (m, 3H), 2.07 (s, 3H), 2.02 (s, 1.5H), 1.70 (s, 1.5H), 1.69 (s, 3H), purified as a 1:0.5 mixture of diastereomers.
Enantiomeric Separation of [Rh(phen)(chrysi)(PPO)]Cl2
Purified [Rh(phen)(chrysi)(PPO)][TFA]2 was dissolved in 1:1 ethanol:water and HPLC purified on an Astec CYCLOBOND chiral column using an isocratic elution method of 40:60 ACN:0.1 M KPF6 (aq) over 37 min. The column was periodically rinsed with 40:60 MeCN:H2O to remove KPF6 buildup. Separated enantiomers were collected and exchanged to the chloride salt using Sephadex QAE resin pre-equilibrated with MgCl2. The enantiomeric nature of the collected fractions was verified using circular dichroism (CD) as follows: 200 μM solutions of Δ- and Λ-[Rh(phen)(chrysi)(PPO)]Cl2 were made in aqueous solution and their CD spectra recorded in 1 nm increments on an Aviv 62DS spectropolarimeter under a N2 atmosphere at ambient temperature. The spectra were recorded a second time 30 d later to assess decomposition or racemization of the sample, and none was observed.
Determination of Extinction Coefficients
Aqueous solutions of each [Rh(L)(chrysi)(PPO)]Cl2 complex were made and a UV-Visible spectrum was recorded for each. The solutions were diluted 50x, 100x, 500x, and 1000x in 2% HNO3. The dilutions were analyzed for Rh content via ICP-MS (inductively coupled plasma mass spectrometry) and the concentration was determined by comparison to a standard curve. Extinction coefficients were determined from the UV-Visible absorbance measurement of the initial solution and the Rh concentration of the dilutions following Beer’s law (A=εlc). L = DIP was observed to significantly adsorb onto plastics, therefore PTFE (polytetrafluoroethylene) and PFA (perfluoroalkoxy alkane) coated materials were used in the workup and analysis of its extinction coefficient.
Partition Coefficient Determination
One-octanol and 10 mM Tris-HCl, pH 7.4 were pre-equilibrated with each other by vigorously shaking the phases together. A solution of each metalloinsertor was made in octanol and the UV-Visible spectrum of the solution recorded. Each solution was combined with an equal volume of aqueous buffer and shaken using a foam insert on a Vortex-Genie 2 running at maximum speed for 16 h. The samples were centrifuged to separate the aqueous and octanol phases and a UV-Visible spectrum of each octanol fraction was recorded. The baseline value obtained at 800 nm was used to normalize the spectra to a common zero point. The absorbance of the ~260 nm peak in the final spectrum was compared to the initial spectrum to determine the partition coefficient following the literature.24 The partition coefficients from three experiments were measured for each [Rh(L)(chrysi)(PPO)]Cl2 complex and averaged to give the partition coefficient.
pKa Determination of Metalloinsertors
A ~25 μM solution of each metalloinsertor was made in 100 mM NaCl. The pH of the sample was adjusted to 4.5 using HCl (10 mM). NaOH (10 mM) was titrated into the solution, with stirring. The pH and UV-Visible spectrum were recorded after each base addition, up to a pH of 10.5. A back titration to pH 6 was performed to check for decomposition, and none was observed. Spectra were corrected for baseline and volume changes. The absorbance of the ~430 nm peak was plotted against pH and fit to a sigmoidal curve in OriginPro v8.5, and the pKa was determined as the inflection point of the curve. Three pKa titrations were performed for each [Rh(L)(chrysi)(PPO)]Cl2 complex and averaged to give an average pKa value.
Binding Constant Determination
A DNA hairpin (5′-GGCAGGXATGGCTTTTTGCCATYCCTGCC-3′, where XY=CG or CC for a well-matched or mismatched hairpin, respectively) was radiolabeled with γ-32P ATP and prepared following the literature.10,19,22 Full details of DNA preparation and purification can be found in the SI. A 4 μM solution of the photocleaving metalloinsertor [Rh(bpy)2(chrysi)]Cl3 and solutions containing 0–400 μM of a competing metalloinsertor, [Rh(L)(chrysi)(PPO)]Cl2 (which does not photocleave DNA), were made in MilliQ water. Five μL of the [Rh(bpy)2(chrysi)]Cl3 solution, 5 μL of the competing metalloinsertor, and 10 μL of the hairpin DNA were combined to create a solution containing 1 μM [Rh(bpy)2(chrysi)]Cl3, 0–100 μM competing metalloinsertor, and 1 μM DNA. The samples were irradiated with an Oriel 1000 W Hg/Xe solar simulator (340–440 nm) for 20 min. After irradiation, solvent was removed from the samples and the samples were counted on a scintillation counter to determine the necessary exposure time (with 300,000 cpm needing a 1 hour exposure) and they were suspended in a denaturing formamide loading dye. Samples were electrophoresed on a 20% denaturing polyacrylamide urea gel.
A phosphor screen was exposed to the polyacrylamide gel and imaged using a Typhoon FLA 9000 biomolecular imager. The ratio of photocleaved to uncleaved DNA was quantified using ImageQuant TL software. The ratio was plotted against the concentration of [Rh(L)(chrysi)(PPO)]Cl2 and fit to a sigmoidal curve in OriginPro v8.5 to determine the inflection point of the fit. The binding affinity of the competing metalloinsertor was calculated in Mathematica 9.0 by solving simultaneous equilibria involving DNA, [Rh(bpy)2(chrysi)]Cl3, and [Rh(L)(chrysi)(PPO)]Cl2. Three photocleavage titrations were performed for each [Rh(L)(chrysi)(PPO)]Cl2 complex and averaged to give the binding affinity.
Melting Temperature Analysis
Melting temperature analysis was performed on a Beckman DU 7400 spectrophotometer equipped with a Tm Analysis Accessory. The short oligomer, 5′-CGGACTCCG-3′ (underline denotes mismatch), was purchased from IDT DNA and purified by HPLC. Samples containing 11 μM ssDNA (ultimately 5.5 μM dsDNA and mismatches) and 6 μM of [Rh(phen)(chrysi)(PPO)]Cl2, [Rh(bpy)2(chrysi)]Cl3 or no metal complex were prepared in phosphate buffer (5 mM phosphate, 50 mM NaCl, pH 7.0). Samples were heated at a rate of 0.5 °C/min and absorbance was measured at 260 nm every 0.5 °C between 10 °C and 50 °C. Data from three experiments was combined and fit to a sigmoidal curve in OriginPro v8.5 and the Tm was taken as the inflection point of the curve.
Cell Culture
HCT116N and HCT116O cells were grown in RPMI (Roswell Park Memorial Institute) 1640 media supplemented with 10% FBS (fetal bovine serum), 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 100 units/mL penicillin and streptomycin, and 100 μg/mL Geneticin (G418). The cells were incubated in tissue culture flasks or plates at 37 °C in a 5% CO2 atmosphere. All cell studies were performed with the chloride salt of each metalloinsertor.
Cell Proliferation ELISA
Cell proliferation ELISA (enzyme-linked immunosorbent assay) was performed following the manufacturers instructions. Briefly, 2×103 HCT116N or HCT116O cells in 100 μL media were plated into each well of a 96-well plate. The cells were allowed to adhere for 24 h before the addition of 100 μL of media containing various concentrations of rhodium metalloinsertor. The plates were incubated for an additional 48 h before the rhodium-containing media was replaced with fresh media, with which the cells were allowed to grow for the remainder of a 72 h period. Cells were then treated with an excess of the unnatural nucleic acid, BrdU (bromodeoxyuridine), for 24 h during which time it could be incorporated into newly synthesized DNA. Cells were then fixed, labeled with a BrdU antibody, and quantified using a colorimetric substrate solution and stop solution. Absorbance was measured at 450 nm (background subtracted at 690 nm). Decrease in cellular proliferation was determined for each metalloinsertor concentration through comparison to untreated cells. Outliers were removed using a modified Thompson Tau test. An additional variation of this assay was performed in which the cells were treated with rhodium metalloinsertor for 24 h, then directly treated with BrdU in fresh media.
MTT Cytotoxicity Assay
Cell proliferation MTT (MTT = 2-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltatrazolium bromide) assays were performed following the manufacturers instructions. Briefly, 5×104 HCT116N or HCT116O cells in 100 μL media were plated into each well of a 96-well plate. Various concentrations of a rhodium metalloinsertor were added to each well. The cells were allowed to incubate for 72 h before treatment with MTT for 4 h, during which time MTT could be converted into formazan by metabolically active cells. The formazan crystals were solubilized and quantified by absorbance at 570 nm (background subtracted at 690 nm). Viability was determined for each metalloinsertor concentration through comparison to untreated cells. Outliers were removed using a modified Thompson Tau test. An additional variation of this assay was performed in which the cells were allowed to adhere to the 96-well plate overnight before treated with rhodium metalloinsertor for 24 h, followed by MTT treatment.
Uptake and Localization Experiments
Whole-cell uptake, mitochondrial localization, and nuclear localization of metalloinsertors were determined following published methods.25 Prior to whole-cell, mitochondrial, and nuclear rhodium determination, 24-hour ELISA and MTT assays were performed to determine a metalloinsertor concentration that would not result in significant cell death by MTT but showed some anti-proliferative effect by ELISA. The concentrations used in the uptake and localization studies of the [Rh(L)(chrysi)(PPO)]Cl2 family were 0.2 μM for L=DIP, 0.5 μM for L=phen, bpy, HDPA, 4,7-DMP, and 5,6-DMP, and 10 μM for [Rh(bpy)2(chrysi)]Cl3, which was included as a control.
Assay for Whole-Cell Rhodium Concentration
Whole-cell uptake experiments were performed following published protocols.20 Briefly, 1×106 HCT116N or HCT116O cells were plated into 6-well tissue culture treated plates and allowed to adhere for 24 h. Media was aspirated from the cells and fresh media containing a metalloinsertor was added to each well. Cells were allowed to incubate for an additional 0.5–24 h with the Rh-containing media. After incubation, media was aspirated and the cells were rinsed with PBS (phosphate buffered saline, pH 7.4) to remove surface rhodium. Cells were lysed directly in the well using 1 mL of 1% SDS solution. These samples were transferred to microcentrifuge tubes and sonicated for 10 s at 20% amplitude on a Qsonica Ultrasonic sonicator. Cell lysate was combined with an equal volume 2% HNO3. This solution was analyzed for Rh content on an Agilent 8800 Triple Quadrupole ICP-MS and the concentration of Rh in each sample was determined by comparison to a standard curve (ranging from 1–100 ppb Rh) and normalized using the protein content of each sample. The protein content of each sample was determined using a Pierce BCA assay, following the manufacturer’s instructions.
Assay for Mitochondrial Rhodium Concentration
Mitochondrial uptake experiments were performed following published protocols.20,26 Briefly, 1.5×107 HCT116N and HCT116O cells were plated in T75 tissue culture treated flasks. The cells were allowed to adhere for 24 h, after which media was aspirated from each flask and restored with 20 mL media containing a rhodium metalloinsertor. The cells were allowed to grow in the presence of Rh-containing media for 24 h, then harvested using 0.05% trypsin over 5 minutes. Cells were pelleted by centrifugation at 1200 rpm for 5 min. The pellet was rinsed and suspended in PBS, then pelleted again and the PBS removed. The cell pellet was suspended in 500 μL mitochondrial extraction buffer (200 mM mannitol, 68 mM sucrose, 50 mM PIPES, 50 mM KCl, 5 mM EGTA, 2 mM MgCl2, 1 mM DTT added just before use, and protease inhibitors added just before use) and incubated on ice for 20 min. Each sample was homogenized by 35 passes thorough a 21-gauge needle and syringe. The resultant solution was centrifuged for 5 min at 750 rpm. The supernatant of each sample was transferred to a 1.5 mL microcentrifuge tube and centrifuged for 10 min at 14,000 g. The supernatant was decanted and the resulting pellet was the mitochondrial fraction. SDS (800 μL of a 1% solution) was added to the pellet and sonicated for 10 s at 40% amplitude on a Qsonica Ultrasonic sonicator. Mitochondrial lysate was combined with an equal volume of 2% nitric acid. This solution was analyzed for Rh content on an Agilent 8800 Triple Quadrupole ICP-MS and the concentration of Rh in each sample was determined by comparison to a standard curve (ranging from 1–100 ppb Rh) and normalized using the protein content of each sample. The protein content of each sample was determined using a Pierce BCA assay, following the manufacturer’s instructions.
Assay for Nuclear Rhodium Concentration
Nuclear uptake experiments were performed following published protocols.20 Briefly, 1×107 HCT116N and HCT116O cells were plated in T75 tissue culture treated flasks. The cells were allowed to adhere for 24 h before the media was aspirated and restored with 20 mL media containing a rhodium metalloinsertor. The cells were allowed to grow in the presence of Rh-containing media for 24 h, then harvested using 0.05% trypsin over 5 minutes. Cells were pelleted by centrifugation at 1200 rpm for 5 min. The pellet was rinsed and suspended in PBS, then pelleted and the PBS removed. Each cell pellet was suspended in 1 mL hypotonic buffer (20 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2), transferred to a microcentrifuge tube, and incubated on ice for 15 min. NP-40 (50 μL of a 10% solution) was added to each sample, vortexed for 10 s at the highest setting, and centrifuged at 3000 g for 10 min. The supernatant was decanted and the resulting pellet was the nuclear fraction. SDS (800 μL of a 1% solution) was added to the pellet and then sonicated for 10 s at 40% amplitude on a Qsonica Ultrasonic sonicator. Nuclear lysate was combined with an equal volume of 2% HNO3. This solution was analyzed for Rh content on an Agilent 8800 Triple Quadrupole ICP-MS and the concentration of Rh in each sample was determined by comparison to a standard curve (ranging from 1–100 ppb Rh) and normalized using the protein content of each sample. The protein content of each sample was determined using a Pierce BCA assay, following the manufacturer’s instructions.
Assay for Uptake of Metalloinsertors
Mechanism of uptake experiments were adapted from published protocols.27 RbCl and [Ru(DIP)(dppz)]Cl2 were used as positive and negative controls, respectively. Briefly, 1×106 HCT116N or HCT116O cells were plated into 6-well tissue culture treated plates and allowed to adhere for 24 h. Metabolic inhibitors (5 μM oligomycin in ethanol and 50 mM 2-deoxy-D-glucose) or control solutions (5 mM glucose and ethanol) were added to the cell culture media and samples were incubated for 1 h. Media was removed by aspiration and each well was washed with PBS. Media (3 mL) containing the Rh–O metalloinsertor [Rh(phen)(chrysi)(PPO)]Cl2 (0.5 μM), the parent metalloinsertor [Rh(bpy)2(chrysi)]Cl3 (10 μM), [Ru(DIP)(dppz)]Cl2 (2 μM), or RbCl (25 μM) was then added to each well and incubated for 1 h. Media was aspirated and cells were rinsed with PBS to remove surface rhodium, ruthenium, or rubidium. Cells were lysed directly in the well using 1 mL of 1% SDS solution. Samples were transferred to microcentrifuge tubes and sonicated for 10 s at 20% amplitude on a Qsonica Ultrasonic sonicator. Cell lysate was combined with an equal volume of 2% HNO3 and analyzed for Rh, Ru, and Rb content on an Agilent 8800 Triple Quadrupole ICP-MS, and the concentration of Rh, Ru, or Rb in each sample was determined by comparison to a standard curve (ranging from 1–100 ppb) and normalized using the protein content of each sample. The protein content of each sample was determined using a Pierce BCA assay, following the manufacturer’s instructions.
RESULTS
Establishing the Enantiomeric Activity of [Rh(phen)(chrysi)(PPO)]2+
Enantiomeric separation was performed for the complex [Rh(phen)(chrysi)(PPO)]2+ to establish the interaction of its Δ- and Λ-enantiomers with DNA in vitro and in MMR-deficient or -proficient cells in culture. The Δ- and Λ- enantiomers of [Rh(phen)(chrysi)(PPO)]2+ were isolated with >90% and >95% enantiomeric excess, respectively (Supplementary Figure S1). Circular dichroism experiments confirmed the enantiomeric nature of the isolated complexes, and no racemization was observed at ambient temperature over 1 month (Supplementary Figure S1). Competition titrations between [Rh(phen)(chrysi)(PPO)]2+ and the photocleaving metalloinsertor [Rh(bpy)2(chrysi)]3+ in the presence of 32P-radiolabeled DNA containing a CC mismatch revealed both enantiomers are capable of binding mismatched DNA base pairs with similar affinity (106 M−1, Table 1).10 Furthermore, both enantiomers were found to have selective cytotoxic effects towards MMR-deficient cells over MMR-proficient cells in MTT experiments (Supplementary Figure S2). These studies confirm that both enantiomers of the PPO-containing metalloinsertor, [Rh(phen)(chrysi)(PPO)]2+, exhibit binding properties towards mismatched DNA that are consistent with a previous generation of Rh–O metalloinsertors. These Rh–O complexes show no enantiomeric preference in binding DNA, unlike parent metalloinsertors, which show a high enantiomeric preference for the Δ-isomer in binding DNA.15,19
Table 1.
Binding affinity, pKa, and Log P values for each metalloinsertor
| Metalloinsertor | Binding Constant (× 106 M−1)a | pKa (2+ to 1+) | Log P |
|---|---|---|---|
| Δ-[Rh(phen)(chrysi)(PPO)]2+ | 6.6 | – | – |
| Λ-[Rh(phen)(chrysi)(PPO)]2+ | 9.2 | – | – |
| rac-[Rh(phen)(chrysi)(PPO)]2+ | 5.5b | 8.3 ± 0.3a | 1.4 ± 0.1 |
| rac-[Rh(bpy)(chrysi)(PPO)]2+ | 7.2 | 8.9 ± 0.1 | 0.68 ± 0.07 |
| rac-[Rh(HDPA)(chrysi)(PPO)]2+ | 3.0 | 9.1 ± 0.1 | 0.69 ± 0.08 |
| rac-[Rh(4,7-DMP)(chrysi)(PPO)]2+ | 1.5 | 9.1 ± 0.1 | 1.1 ± 0.1 |
| rac-Rh[(5,6-DMP)(chrysi)(PPO)]2+ | 2.3 | 9.0 ± 0.3 | 0.71 ± 0.01 |
| rac-[Rh(DIP)(chrysi)(PPO)]2+ | 1.6 | 8.1 ± 0.1 | > 2.0c |
binding affinities measured using the DNA hairpin 5′-GGCAGGCATGGCTTTTTGCCATCCCTGCC-3′ (underline denotes mismatch) in 100 mM NaCl, 20 mM NaPi, pH 7.1 buffer. Competition titrations were performed against the photocleaving metalloinsertor [Rh(bpy)2(chrysi)]Cl3.
Values from reference 19
The change in absorbance in the [Rh(DIP)(chrysi)(PPO)]2+-containing 1-octanol phase before and after equilibration with the aqueous phase was too small to accurately and consistently measure.
Binding of Metalloinsertors to a Single Base Pair Mismatch
The binding affinities of [Rh(L)(chrysi)(PPO)]2+ metalloinsertors to DNA containing a single CC mismatch were determined. The [Rh(L)(chrysi)(PPO)]2+ complexes do not photocleave DNA upon irradiation, so their binding affinities were assayed via a competition titration with [Rh(bpy)2(chrysi)]3+, a complex known to photocleave DNA selectively upon mismatch binding and irradiation.22 A CC mismatch was used as it is highly destabilized relative to other mismatches and therefore undergoes significant photocleavage in the presence of [Rh(bpy)2(chrysi)]3+. A constant concentration of [Rh(bpy)2(chrysi)]3+ and varying concentrations of the competing [Rh(L)(chrysi)(PPO)]2+ metalloinsertor were incubated with a DNA hairpin containing a single CC mismatch, irradiated, and the DNA photocleavage products were separated on a denaturing gel. The ratio of photocleaved DNA to intact DNA was plotted against the log of the rhodium concentration and fit to a sigmoidal curve (Figure S3). The inflection point of the sigmoidal fit was used to determine the binding affinity of the competing [Rh(L)(chrysi)(PPO)]2+ metalloinsertor by solving simultaneous equilibria equations using the known binding affinity of [Rh(bpy)2(chrysi)]3+. The binding affinities of these complexes are shown in Table 1. All complexes were tested as racemic mixtures and exhibit binding affinities in the range of 2.4 to 7.2 × 106 M−1 (Table 1). Despite differences in ligand steric bulk, all Rh–O metalloinsertors tested have binding affinities within one order of magnitude of each other, and thus bind DNA with comparable affinity.
Binding was assessed further via melting temperature analysis. A short, palindromic DNA sequence containing a central CC mismatch was incubated in the presence of the parent metalloinsertor, [Rh(bpy)2(chrysi)]Cl3, or the Rh–O metalloinsertor, [Rh(phen)(chrysi)(PPO)]Cl2. The chosen DNA sequence has a low Tm and therefore exists as ssDNA at room temperature.13 In the presence of metalloinsertor, however, the DNA anneals and the melting temperature increases dramatically to 44.9 ± 0.6 and 41.3 ± 0.5 °C for [Rh(bpy)2(chrysi)]Cl3 and [Rh(phen)(chrysi)(PPO)]Cl2, respectively (Supplementary Figure S3B). These results are in good agreement with the results of the DNA binding assay describe above and corroborate the result that parent and Rh–O metalloinsertors have comparable binding affinities to mismatches in DNA, with [Rh(phen)(chrysi)(PPO)]Cl2 stabilizing DNA to a slightly lesser extent than [Rh(bpy)2(chrysi)]Cl3.
pKa Determination of Metalloinsertors
The pKa values of [Rh(L)(chrysi)(PPO)]2+ metalloinsertors were assessed via spectroscopic pH titrations (Table 1, Supplementary Figures S4–S8). The absorbance of a 435–440 nm peak, which corresponds to a charge transfer located on the chrysi ligand, was plotted against the pH of the solution for each complex.28 Data were fit to a sigmoidal curve and the inflection point was taken as the pKa of the complex, specifically of the imine proton on the chrysi ligand. All Rh–O metalloinsertors exhibited pKa values in the range of 8.1 to 9.1, which are above physiological pH (Table 1), indicating that the chrysi ligands of these complexes remain protonated in cell culture media or within cells. It has been shown previously that fully protonated chrysi ligands, which are seen with Rh–O metalloinsertors, buckle in contrast to the deprotonated chrysi ligands of the parent metalloinsertors, which are completely flat and thus easy to stack with the DNA base pairs once inserted.19
Partition Coefficient and Lipophilicity of Metalloinsertors
The [Rh(L)(chrysi)(PPO)]Cl2 family of metalloinsertors was designed to vary in lipophilicity, and the partition coefficients of each [Rh(L)(chrysi)(PPO)]2+ metalloinsertor were determined between aqueous buffer (10 mM Tris-HCl, pH 7.4) and 1-octanol according to literature methods.24 Absorbance measurements at the ~260 nm peak were made in the 1-octanol phase before and after equilibration with the aqueous phase. These absorbance values were compared to determine the partition coefficient, log P (Table 1, Supplementary Figures S9–S12). The log P values followed the expected trend with the least bulky complexes ([Rh(bpy)(chrysi)(PPO)]2+ and [Rh(HDPA)(chrysi)(PPO)]2+) having the lowest log P values and the bulkiest complex ([Rh(DIP)(chrysi)(PPO)]2+) having the greatest log P value. Surprisingly, despite their cationic nature, under these conditions the [Rh(L)(chrysi)(PPO)]2+ metalloinsertors are all lipophilic and have partition coefficients favoring octanol over water, ranging from 0.68 to >2.0.
Cytotoxic and Anti-Proliferative Effects in MMR-Deficient and -Proficient Cells
The ability of metalloinsertors to selectively kill or impair growth of MMR-deficient cells is a critical factor in their potential value as chemotherapeutic agents.19,29 In this structure-activity relationship study, we used ELISA and MTT assays to determine the effect of ligand substitution on biological activity in MMR-deficient and -proficient cells. The ELISA was used to determine the inhibitory effects on DNA synthesis and the MTT assay was performed to establish levels of cytotoxicity. For the ELISA, each metalloinsertor was incubated with HCT116N (MMR-proficient) or HCT116O (MMR-deficient) cells at various concentrations before treatment with the unnatural nucleic acid BrdU. Colorimetric antibody treatment allowed the relative BrdU incorporation into DNA to be quantified, and cellular proliferation was then determined as the ratio of BrdU incorporation between metalloinsertor-treated cells and untreated control cells. The results of the 48-hour metalloinsertor treatment are shown in Figure 3, and the results of a 24-hour treatment are shown in Supplemental Figure S15. All [Rh(L)(chrysi)(PPO)]2+ metalloinsertors exhibit anti-proliferative activity with selectivity towards the MMR-deficient cell line. The maximum proliferation difference (referred to as selectivity) between the cell lines and the concentration at which this selectivity occurs (referred to as potency) are as follows: 77 ± 10% at 400 nM for [Rh(phen)(chrysi)(PPO)]2+, 78 ± 18% at 2 μM for [Rh(bpy)(chrysi)(PPO)]2+, 47 ± 10% at 25 μM for [Rh(HDPA)(chrysi)(PPO)]2+, 66 ± 6% at 400 nM for [Rh(4,7-DMP)(chrysi)(PPO)]2+, 67 ± 5% at 400 nM for [Rh(5,6-DMP)(chrysi)(PPO)]2+, and 70 ± 23% at 160 nM for [Rh(DIP)(chrysi)(PPO)]2+.
Figure 3.

Cellular proliferation ELISA for the [Rh(L)(chrysi)(PPO)]2+ metalloinsertors in MMR-deficient (HCT116O, red circles) and MMR-proficient (HCT116N, blue squares) cells. Cells were incubated with various concentrations of metalloinsertor for 48 h before treatment with BrdU. Cell proliferation is shown as %BrdU incorporated into DNA compared to untreated control cells. Error is shown as the standard deviation of 5 replicates.
For the MTT assay, each metalloinsertor was incubated with HCT116N (MMR-proficient) or HCT116O (MMR-deficient) cells at various concentrations before the addition of MTT, which can be converted into formazan by mitochondrial reductase activity in a functioning cell. Colorimetric measurements of formazan allow the relative viability to be quantified, and cellular viability is then determined as the ratio of formazan produced between metalloinsertor-treated cells and untreated control cells. The results of the 72-hour treatment are shown in Figure 4 and the results of the 24-hour treatment are shown in Supplemental Figure S16. All [Rh(L)(chrysi)(PPO)]2+ metalloinsertors exhibit cytotoxic activity with selectivity towards the MMR-deficient cell line. The maximum proliferation difference between the cell lines and the concentration at which this difference occurs are as follows: 52 ± 5% at 300 nM for [Rh(phen)(chrysi)(PPO)]2+, 30 ± 7% at 2 μM for [Rh(bpy)(chrysi)(PPO)]2+, 13 ± 11% at 32 μM for [Rh(HDPA)(chrysi)(PPO)]2+, 46 ± 8% at 600 nM for [Rh(4,7-DMP)(chrysi)(PPO)]2+, 49 ± 3% at 600 nM for [Rh(5,6-DMP)(chrysi)(PPO)]2+, and 39 ± 6% at 640 nM for [Rh(DIP)(chrysi)(PPO)]2+.
Figure 4.

Cellular viability MTT assay for the [Rh(L)(chrysi)(PPO)]2+ metalloinsertors in MMR-deficient (HCT116O, red circles) and MMR-proficient (HCT116N, blue squares) cells. Cells were incubated with various concentrations of metalloinsertor for 72 h before treatment with MTT. Cell proliferation is shown as % viability from MTT metabolism, compared to untreated control cells. Error is shown as the standard deviation of 5 replicates.
Whole-Cell Uptake, Mechanism of Uptake, and Organelle Localization
To better understand the range of biological activities of these complexes, cellular uptake and mechanism of uptake were examined via ICP-MS based assays. 24-hour ELISA and MTT assays were performed to determine a suitable concentration for uptake and localization studies (which were performed over a 24-hour timescale). To minimize cell death in this assay, a factor which can complicate data interpretation, suitable dosing was determined to be at a concentration at which there was noticeable anti-proliferative effects in the HCT116O cells via ELISA but no significant cytotoxicity via MTT assay. Whole cell uptake studies were performed with each [Rh(L)(chrysi)(PPO)]2+ complex at 0.5 μM with the exception of [Rh(DIP)(chrysi)(PPO)]2+, which was performed at 0.2 μM due to its high cytotoxicity at 0.5 μM. For whole cell uptake studies, cells were incubated with metalloinsertors for 24 h before they were lysed and analyzed for rhodium content via ICP-MS, with rhodium concentrations normalized to the protein content of each sample. The whole cell uptakes of each metalloinsertor in HCT116O cells are shown in Figure 5 (results in HCT116N cells are similar and shown in Supplementary Figure S17). Overall, all [Rh(L)(chrysi)(PPO)]2+ complexes exhibit uptake into cells at concentrations within one order of magnitude of each other. The uptake of these complexes correlates generally with their lipophilicity values, with the least lipophilic complexes ([Rh(HDPA)(chrysi)(PPO)]2+ and [Rh(bpy)(chrysi)(PPO)]2+) having the poorest uptake and the most lipophilic complex ([Rh(DIP)(chrysi)(PPO)]2+) having the highest uptake. Lipophilicity has long been correlated with an increase in cellular uptake and a resultant increase in drug potency.30,31
Figure 5.

Whole-cell rhodium uptake assays. (left) Rhodium accumulation in HCT116O was measured by ICP-MS analysis after a 24 hour incubation with [Rh(L)(chrysi)(PPO)]2+ metalloinsertors (where L = phen, bpy, HDPA, 4,7-DMP, 5,6-DMP, or DIP). (right) Rhodium accumulation over time in HCT116O cells was measured by ICP-MS for three metalloinsertors, [Rh(phen)(chrysi)(PPO)]2+ (phen), [Rh(DIP)(chrysi)(PPO)]2+ (DIP), and the parent metalloinsertor [Rh(bpy)2(chrysi)]3+ (Rh-BC). Rhodium content was normalized to protein content determined by BCA assay, and the results of four independent trials were averaged with error shown as the standard deviation.
In addition to examining whole cell uptake of the [Rh(L)(chrysi)(PPO)]2+ metalloinsertors, the uptake over time and the mechanism of uptake were also examined. In the former experiment, cells were incubated with a metalloinsertor for 0.5, 1, 3, 6, 9, or 24 h before being lysed and analyzed for rhodium content by ICP-MS. The whole-cell uptake over time of these metalloinsertors in HCT116O cells is shown in Figure 5 (results in HCT116N cells are similar and shown in Supplementary Figure S17). The complexes appear to show significant increases in uptake over the first 3–6 h of incubation with cells, followed by plateau with no evidence of significant efflux during a 24-hour period. These results are consistent with previous studies on metalloinsertors.20
A metabolic inhibition assay was performed to better understand the mechanism of cellular uptake of [Rh(L)(chrysi)(PPO)]2+ metalloinsertors. HCT116N and HCT116O cells were pre-treated with the metabolic inhibitors oligomycin A, an inhibitor of oxidative phosphorylation, and 2-deoxy-D-glucose, an inhibitor of glycolysis.27 Metabolic inhibition depletes cellular ATP (adenosine triphosphate), so any compound that is taken into the cell via an active, ATP-dependent mechanism should have reduced uptake in metabolically depleted cells. Conversely, complexes taken into the cell via a passive mechanism, such as passive diffusion, are not affected by metabolic inhibition and therefore the drug should accumulate in inhibited and uninhibited cells at similar concentrations. [Rh(phen)(chrysi)(PPO)]2+ and the parent metalloinsertor, [Rh(bpy)2(chrysi)]3+, were studied to determine if the mechanism of metalloinsertor uptake was ATP-dependent. The compounds RbCl and [Ru(dppz)(DIP)2]2+ were included as positive and negative controls, respectively. The rubidium ion of RbCl is transported into the cell by Na,K-ATPase, an ATP-dependent ion pump, while [Ru(dppz)(DIP)2]2+ has previously been shown to enter the cell via passive diffusion.27, 32 Cells were treated with each compound for 1 h before they were lysed and analyzed by ICP-MS for metal content. As rubidium, ruthenium, and rhodium are not naturally present in cells or cell culture reagents, all three elements can be analyzed as low-background analytes by ICP-MS. The results of each compound in HCT116O cells are shown in Figure 6 (results in HCT116N cells are similar and shown in Supplementary Figure S18). As expected, RbCl showed a significant decrease in uptake when pre-treated with metabolic inhibitors and [Ru(dppz)(DIP)2]2+ was unaffected by inhibitor pre-treatment. Similar to [Ru(dppz)(DIP)2]2+, [Rh(phen)(chrysi)(PPO)]2+ and [Rh(bpy)2(chrysi)]2+ were also unaffected by inhibitor pre-treatment, suggesting these complexes are also taken into the cell via an ATP-independent mechanism, such as passive diffusion. Since these complexes are all lipophilic and cationic, passive diffusion is a reasonable uptake mechanism, with the negative membrane potential driving diffusion and relatively high lipophilicity facilitating the process as the molecules can more readily partition into the cellular membranes.33
Figure 6.
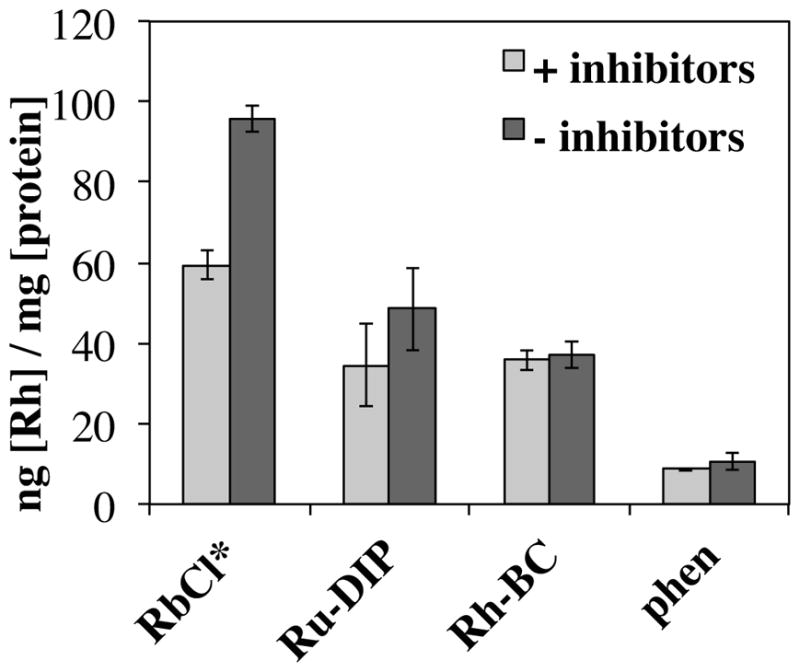
Uptake assay for rhodium metalloinsertors and controls. [Rh(phen)(chrysi)(PPO)]2+ (phen), [Rh(bpy)2(chrysi)]3+ (Rh-BC), [Ru(DIP)2(chrysi)]2+ (Ru- DIP), and RbCl accumulation in HCT116O cells was measured by ICP-MS analysis after treatment with or without metabolic inhibitors (oligomycin and 2-deoxy-D-glucose). Rhodium, ruthenium, and rubidium contents were normalized to protein content determined by BCA assay. Each experiment was performed in triplicate and averaged, with error shown as the standard deviation. Each experiment was repeated three times with similar outcomes (not shown). RbCl* indicates that Rb concentrations for RbCl have been lowered by a factor of 500 in this graphic.
Subcellular localization into the nucleus (the on-target organelle) and mitochondria (a major off-target organelle) were also examined by an ICP-MS assay. Localization studies were performed with each [Rh(L)(chrysi)(PPO)]2+ metalloinsertor at 0.5 μM with the exception of [Rh(DIP)(chrysi)(PPO)]2+, which was performed at 0.2 μM. For localization studies, cells were incubated with metalloinsertors for 24 h before they were lysed and analyzed for rhodium content via ICP-MS, with rhodium concentrations normalized to the protein content of each sample. The whole cell uptakes of each metalloinsertor in HCT116O cells are shown in Figure 7 (results in HCT116N cells are similar and shown in Supplementary Figure S19). Overall, all [Rh(L)(chrysi)(PPO)]2+ complexes have comparable nuclear uptakes and mitochondrial uptakes to one another with the exception of [Rh(DIP)(chrysi)(PPO)]2+, which has nuclear and mitochondrial uptakes that are 2–3 times higher than other complexes despite being dosed at a lower concentration. All complexes appear to enter the nucleus at high enough concentrations to bind DNA mismatches, with a significant enrichment in nuclear concentration over the extracellular concentration of rhodium (Supplementary Table S1).
Figure 7.
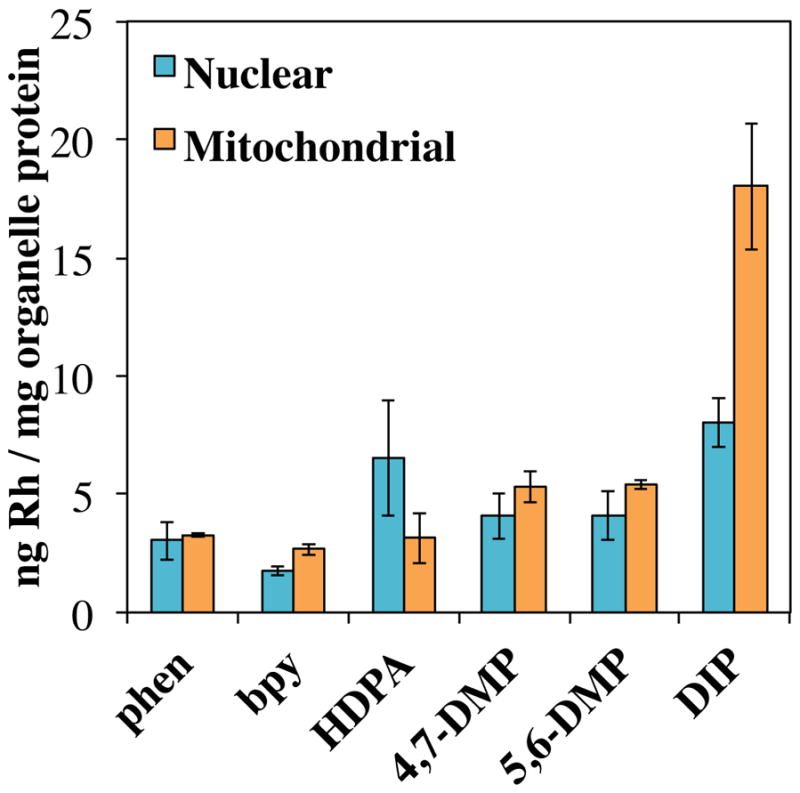
ICP-MS assay for nuclear and mitochondrial uptake of rhodium metalloinsertors. Rhodium accumulation in HCT116O cells was measured by ICP-MS analysis after a 24 hour incubation with [Rh(L)(chrysi)(PPO)]2+ (where L = phen, bpy, HDPA, 4,7-DMP, 5,6-DMP or DIP). Rhodium content was normalized to protein content of each organelle fraction determined by BCA assay. The results of 4 independent studies were averaged with error shown as the standard deviation.
DISCUSSION
Early generations of rhodium metalloinsertors, which exclusively contain Rh–N ligand coordination, are a richly studied family of metal complexes that can selectively bind to DNA base pair mismatches and lead to selective cell death in MMR-deficient cells. Across multiple studies, these metalloinsertors were determined to have several characteristic and consistent behaviors. Through in vitro experiments, we have observed that only the Δ- enantiomer of these Rh–N coordinated complexes is capable of binding mismatches in B-form DNA.34 In cellular studies, these metalloinsertors have been observed to selectively kill cells in concentration ranges of 5–40 μM.20,21 In one structure-activity relationship study, the steric bulk of the ancillary ligands on a metalloinsertor was seen to influence DNA binding properties and, ultimately, alter cellular selectivity.21 In another structure-activity relationship study, the lipophilicity of the ancillary ligands on a metalloinsertor was seen to dramatically influence its subcellular localization within a cell and, again, alter cellular selectivity.20
While the above trends seem to ring true across parent metalloinsertors containing exclusively Rh–N ligand coordination, the recent emergence of a new family of metalloinsertors that contain Rh–O ligand coordination has challenged many of these characteristics and behaviors.19 For instance, both enantiomers of Rh–O metalloinsertors are capable of binding DNA mismatches in vitro, and are furthermore capable of inducing selective cellular toxicity at nanomolar concentrations. Additionally, changes in lipophilicity and steric bulk of the O-containing ligand seemed to have little, if any, effect on DNA binding affinity and cellular selectivity. This remarkable shift in metalloinsertor activity revealed that these Rh–O complexes have distinct in vitro characteristics and biological properties from their parent metalloinsertor complexes. As such, a new family of Rh–O metalloinsertors has been synthesized, characterized, and investigated for biological activity. In contrast to the first generation of Rh–O metalloinsertors in which the O-containing ligand was varied, in this new family an ancillary ligand was varied and the O-containing ligand was kept constant. This family is of the form [Rh(L)(chrysi)(PPO)]2+, where L = bpy, phen, HDPA, 4,7-DMP, 5,6-DMP, and DIP. This ligand variation influences many features of the metalloinsertor, including steric bulk and lipophilicity, both of which have previously been seen to affect DNA binding and cellular activity of the parent metalloinsertors.20,21 In studying this family of complexes, we aimed to test the unique biological activity of metalloinsertors containing the Rh–O ligand framework and begin to understand the high potency and improved selectivity exhibited by these metalloinsertors over parent metalloinsertors and other DNA-binding complexes.
Robustness of Biological Activity of the Rh–O Ligand Framework
A primary aim of this structure-activity relationship study was to determine if altering the ancillary ligand of Rh–O metalloinsertors would significantly affect the biological activity of these complexes. Biological activity was assessed through both ELISA and MTT assays in two cell lines, HCT116N and HCT116O. These cells are derived from the same colorectal carcinoma cell line but differ primarily in that HCT116N cells are MMR-proficient whereas HCT116O cells are MMR-deficient.35 For this reason, HCT116O cells have a higher relative abundance of DNA mismatches over HCT116N cells and therefore should be more sensitive to mismatch-targeting metalloinsertors.36
Indeed, all complexes prepared showed highly selective anti-proliferative or cytotoxic effects toward the MMR-deficient cells over the MMR-proficient cells in both ELISA (Figure 3) and MTT assays (Figure 4), with the exception of [Rh(HDPA)(chrysi)(PPO)]2+, which only shows activity in the ELISA. While selectivity was seen for all complexes, the effective concentrations varied by two orders of magnitude across the family. For instance, [Rh(HDPA)(chrysi)(PPO)]2+ has very low potency and little selectivity compared to other Rh–O metalloinsertors. Although it does appear to interfere selectively with DNA synthesis via ELISA, this biological interaction does not appear significant enough to produce cytotoxic effects in the MTT assay, even at high drug concentrations (Figure 4). HDPA is the only ligand containing a labile proton and the only ligand that forms a 6-ring chelate with the metal, and it seems possible that these structural features ultimately influence the biological activity of the [Rh(HDPA)(chrysi)(PPO)]2+. It is possible that the 6-member chelate could cause structural aberrations and the proton on HDPA could cause hydrogen-bonding interactions that ultimately alter DNA-binding or DNA-processing by proteins, which could cause a decrease in toxicity. [Rh(bpy)(chrysi)(PPO)]2+ has the second lowest potency of this new family, though remarkably this complex still shows higher potency than the parent metalloinsertors containing only Rh–N coordination.20 The phenanthroline-derived metalloinsertors, [Rh(phen)(chrysi)(PPO)]2+, [Rh(4,7-DMP)(chrysi)(PPO)]2+, and [Rh(5,6-DMP)(chrysi)(PPO)]2+, all show comparable nanomolar potencies and selectivities in the ELISA and MTT assays.
Perhaps the most surprising biological activity is seen with [Rh(DIP)(chrysi)(PPO)]2+. Historically, metalloinsertors containing the bulky DIP ligand have shown no selectivity for the MMR-deficient cell line.21 This lack of selectivity was attributed to substantially lower mismatch binding affinities (104 M−1 for [Rh(DIP)2(chrysi)]3+) owing to ancillary bulk, as well as off-target localization into the mitochondria, a property that is common with lipophilic cations.20,37 [Rh(DIP)(chrysi)(PPO)]2+, however, does exhibit selective cytotoxicity towards MMR-deficient cells over proficient cells in both the ELISA and MTT assays. In fact, [Rh(DIP)(chrysi)(PPO)]2+ displays a similar selectivity and ~2-fold higher potency than [Rh(phen)(chrysi)(PPO)]2+ when measured by ELISA (Figure 3).
Overall, these results confirm that Rh–O metalloinsertor biological selectivity is minimally influenced by substitution at the ancillary ligand.19 Thus far, all of the Rh–O metalloinsertors, derivatized at the O-containing ligand or ancillary ligand, have exhibited selectivity in ELISA and/or MTT assays, regardless of steric bulk or lipophilicity, factors that had heavily influenced (and sometimes abolished) the selectivity of parent metalloinsertors. It is noteworthy that this selectivity profile, wherein the Rh–O metalloinsertors selectively kill MMR-deficient cells, is shared with the parent complexes and is in stark contrast to what is seen with all other DNA-targeting therapeutics, which preferentially kill MMR-proficient cells.17,18 Although parent and Rh–O metalloinsertors share this unique selectivity profile and have similar in vitro binding properties, suggesting they should interact with DNA in a similar way, the Rh–O metalloinsertors are dramatically more potent than the parent metalloinsertors, with nearly all Rh–O complexes (with the sole exception being [Rh(HDPA)(chrysi)(PPO)]2+) having greater cytotoxicity in MMR-deficient cells than any of the parent metalloinsertors. It stands to reason, then, that the high potency and selectivity of these Rh–O complexes does not reflect a difference in DNA binding affinity from the parent complexes, but rather it must instead reflect a difference in structure associated with the DNA-metalloinsertor lesion. That is, if the frequency of DNA binding is comparable between the Rh–O and parent metalloinsertors, the lesion formed by Rh–O metalloinsertors must activate a cellular response at lower concentrations.
Uptake Characteristics
Although the [Rh(L)(chrysi)(PPO)]2+ family shows consistent activity towards MMR-deficient cells, the selectivities and potencies of these complexes vary significantly across the family from 160 nM to 25 μM. It was initially hypothesized that these differences in biological activity could be due to differences in cellular uptake. In particular, it seemed possible that the least potent complexes, [Rh(HDPA)(chrysi)(PPO)]2+ (which has almost no cytotoxic properties at 40 μM) and [Rh(bpy)(chrysi)(PPO)]2+ (which has nearly 10-fold lower potency than [Rh(phen)(chrysi)(PPO)]2+), could be less effective due to low uptake. Similarly, it was proposed that increased uptake could be responsible for the high potency of [Rh(DIP)(chrysi)(PPO)]2+. Indeed, it does seem possible that uptake may explain some of the observed potency trends: despite being dosed at 0.2 μM, [Rh(DIP)(chrysi)(PPO)]2+ exhibits similar uptake to [Rh(phen)(chrysi)(PPO)]2+, which was dosed at 0.5 μM. The finding suggests that [Rh(DIP)(chrysi)(PPO)]2+ may induce biological effects at half the concentration of [Rh(phen)(chrysi)(PPO)]2+ as a result of complexes exhibiting similar uptakes at these concentrations. However, uptake alone appears insufficient to explain the potencies of other complexes. For instance, [Rh(HDPA)(chrysi)(PPO)]2+ and [Rh(bpy)(chrysi)(PPO)]2+ have comparably low uptake into the cell despite a >10-fold difference in activity.
Organelle-specific uptake is also worthy of consideration when examining the activity of these complexes. Studies on previous generations of parent metalloinsertors bearing solely Rh–N ligand coordination showed that off-target mitochondrial uptake is strongly influenced by ligand lipophilicity, with the most lipophilic parent metalloinsertors having high mitochondrial uptake and low selectivity for MMR-deficient cells.20,38 Surprisingly, all Rh–O metalloinsertors studied here are more lipophilic than any of the parent metalloinsertors described above, yet all Rh–O complexes exhibit selective cytotoxicity towards MMR-deficient cells, making their selectivity patterns distinct from trends followed by the parent metalloinsertors. To better understand this marked change in trends, on-target nuclear localization and off-target mitochondrial localization experiments were performed to assess the biological activity of [Rh(L)(chrysi)(PPO)]+2 complexes, particularly DIP, which shows selectivity despite its very high lipophilicity.
As indicated, all [Rh(L)(chrysi)(PPO)]2+ metalloinsertors enter the nuclei to a similar extent and at high enough concentrations to bind DNA mismatches (Figure 7 and Supplementary Table S1). Similarly, all [Rh(L)(chrysi)(PPO)]2+ metalloinsertors enter the mitochondria to a comparable extent. Although nuclear and mitochondrial uptake cannot be compared directly (since each is normalized to the total protein in the organelle), the localization patterns of Rh–O versus parent metalloinsertors can be compared (See Supplementary Figure S20). This comparison shows that, unlike their Rh–N coordinated predecessors, Rh–O metalloinsertor localization into the mitochondria is not significantly influenced by lipophilicity. In fact, despite being lipophilic, Rh–O complexes exhibit uptake profiles that are comparable to hydrophilic parent metalloinsertors (which have low mitochondrial uptake) and are distinct from lipophilic parent metalloinsertors (which have high mitochondrial uptake). This trend in localization is consistent with the biological activity we observed; similar to the hydrophilic parent metalloinsertors, Rh–O complexes are highly selective and show little off-target cytotoxicity. Overall, these data indicate that Rh–O metalloinsertors are able to maintain their high selectivity and potency because the ligand substitutions do not strongly influence their subcellular localization. Since these complexes exhibit low mitochondrial uptake, off-target mitochondria-induced toxicity does not overwhelm the biological response, and the selective nuclear- and mismatch-mediated response can prevail.
It is also interesting to note that both MMR-proficient HCT116N cells and MMR-deficient HCT116O cells had comparable levels of uptake and similar localization profiles, showing that metalloinsertors enter HCT116N and HCT116O cells at the same rate, through the same passive mechanism, and to the same extent (Figure 5 and 6 and Supplementary Figures S17 and S18). These details support the idea that the biological selectivity seen in these cells is not a feature of different cellular uptake or elimination properties. Furthermore, the nuclear uptake into the MMR-deficient and proficient cells are comparable (Figure 7 and Supplementary Figure S19). Therefore, with similar concentrations of metalloinsertors entering the nuclei and similar mismatch binding affinities, any DNA-mediated cytotoxicity must result from a difference in how the drugs interact with the DNA. Rationally, this difference must depend upon an increased mismatch targeting in MMR-deficient cells, where DNA base pair mismatches are more abundant.36
Source of Potency for the Rh–O Metalloinsertors
Although MMR-deficient cells have a relative abundance of mismatches compared to MMR-proficient cells, the total number of mismatches formed during each cellular replication is ultimately small due to the high fidelity and proofreading abilities of polymerases. It is clear, therefore, that the lesion formed by parent metalloinsertors must be significantly potent such that even a small number of metalloinsertor-DNA lesions can result in selective cell death. Moreover, despite their similar mismatch binding affinities, the Rh–O metalloinsertors are even more potent than parent metalloinsertors, and therefore these Rh–O metalloinsertors must produce a unique lesion structure at the mismatched site that can activate a response at even lower concentrations (and therefore fewer metalloinsertor-DNA lesions) than parent metalloinsertors.
Does the increase in potency depend upon a difference in how these Rh–O metalloinsertors bind to DNA within the cell?19 As discussed above, both the Δ- and Λ-enantiomers of [Rh(phen)(chrysi)(PPO)]2+ can bind to DNA mismatches in vitro and selectively kill MMR-deficient cells in culture. This behavior is distinct from parent metalloinsertors, for which only the Δ-enantiomer can bind mismatches and produce biological effects.15 The ability of both enantiomers of Rh–O metalloinsertors to bind mismatched DNA suggests the binding interaction must be fundamentally distinct from that of the parent metalloinsertors; these new Rh–O metalloinsertors must bind DNA in a way that can accommodate the Λ-enantiomer.
Furthermore, some evidence suggests that even the DNA-binding ability of the Δ-enantiomer may be altered in these Rh–O metalloinsertors. Previously, it was observed that bulky parent metalloinsertors, such as [Rh(DIP)2(chrysi)]3+, exhibited poor binding affinities (104 M−1) and could not easily be modeled to fit into a mismatched DNA lesion due to significant steric clashing between the DIP ligands and the DNA backbone.21 In contrast, significant differences in ancillary ligand steric bulk have minimal effect on the binding affinities of Rh–O metalloinsertors, which all bind to DNA with micromolar affinity. Even the most sterically bulky complex, [Rh(DIP)(chrysi)(PPO)]2+, has a relatively high affinity for mismatched DNA (106 M−1) despite containing the bulky DIP ligand. It therefore seems that the inclusion of the DIP ligand is not sufficient to preclude DNA binding, and perhaps this dramatic increase in binding affinity of a DIP-containing metalloinsertor may indicate that a new binding interaction exists that can accommodate the steric bulk of these Rh–O metalloinsertors.
Another consideration is the conformation of the chrysi ligand of these new Rh–O metalloinsertors. All Rh–O complexes have chrysi imine pKa values above physiological pH, indicating that they remain protonated in the intracellular environment. This protonation results in steric clashing between the imine proton and an aromatic proton in the chrysi system and, as a result, the chrysi ligand becomes buckled relative to the rhodium center to relieve the steric strain.19 This is in stark contrast to parent metalloinsertors which deprotonate at cellular pH and therefore do not exhibit steric clashing between the imine and aromatic protons. As a result, the chrysi ligand lays planar in these parent metalloinsertors. Distortion of the chrysi ligand, the ligand that interacts most intimately with the DNA, likely disrupts the overall metalloinsertor-DNA binding interaction, further suggesting there is likely a difference in how Rh–O and parent metalloinsertors bind to DNA.19
Lastly, the Rh–O complexes reported here are lipophilic (log P > 0), whereas comparable parent metalloinsertors are hydrophilic (log P < 0).38 This change in lipophilicity could alter the way Rh–O complexes interact with the hydrophobic bases of DNA or even DNA-processing proteins that may be responsible for recognizing the DNA-metalloinsertor lesion. Overall, these results suggest that the Rh–O metalloinsertors bind to DNA differently than parent metalloinsertors. While these complexes still appear to undergo metalloinsertion, as evidenced by their ability to bind mismatched DNA with high affinity, it is unclear how their binding might be distinct from parent metalloinsertors. It seems possible that a subtle difference in the extent or orientation of mismatched base ejection or in the unwinding of the DNA helix by the metalloinsertor could ultimately result in a difference in how that lesion is recognized or processed within the cell, which could lead to overall cellular response and increased potency. Crystallographic studies of Rh–O metalloinsertors with DNA are currently underway to investigate the potential difference between parent and Rh–O metalloinsertor binding.
Implications for Future Metalloinsertor Design
The [Rh(L)(chrysi)(PPO)]2+ family of metalloinsertors described herein display biological selectivity and potency that are maintained across various ligand frameworks varying in size and lipophilicity. When compared with other Rh–O metalloinsertors in which the PPO-type ligand is varied, metalloinsertors containing the Rh–O motif are consistent in their biological selectivity (and, to a large extent, potency) for MMR-deficient cells regardless of significant alterations to their ancillary ligands. It has previously been shown that the metalloinsertors with DIP ligands and PPO-type ligands cannot be easily modeled into a mismatched DNA lesion due to steric clashes with the DNA structure.19,21 Despite steric bulk, Rh–O metalloinsertors have comparable binding affinities to parent metalloinsertors and significantly improved biological activity. Furthermore, these complexes show little enantioselectivity; both isomers bind DNA and show high potency, further supporting that their metalloinsertion binding interaction markedly differs from parent metalloinsertors. Taken together, these observations show that the Rh–O metalloinsertor framework has great potential for the design of new therapeutics and for the attachment of new payloads, while maintaining biological selectivity.39–43 The consistently high potency and cell selectivity of these complexes is unique and provides the basis for new generations of metalloinsertors and metalloinsertor conjugates.
Supplementary Material
Acknowledgments
Financial support for this work from the NIH (GM33309) and AMGEN is gratefully acknowledged. We thank the Department of Defense for supporting K.M.B. through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program. We also thank Paige Gannon for assisting in uptake and localization studies of the complexes. This project benefitted from the use of instrumentation made available by the Caltech Environmental Analysis Center and we gratefully acknowledge guidance from Dr. Nathan Dalleska. We also thank the Moore Foundation Center for the Chemistry of Cellular Signaling. The Caltech Center for Catalysis and Chemical Synthesis and Dr. Scott Virgil are gratefully acknowledged for assistance with enantiomeric separation.
Footnotes
Associated Content
Some experimental methods, representative binding affinity determination, pH titrations, enantiomeric characterization, further DNA binding characterization, and cytotoxicity of the two enantiomers as well as cellular uptake data. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes: The authors declare no competing financial interest.
References
- 1.Cheung-Ong K, Giaever G, Nislow C. Chem Biol. 2013;20:648–659. doi: 10.1016/j.chembiol.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Wang D, Lippard SJ. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 3.Kelland L. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 4.Nitiss JL. Nat Rev Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waring MJ, editor. Chemical Biology. 2. The Royal Society of Chemistry; 2018. DNA-targeting Molecules as Therapeutic Agents. [Google Scholar]
- 6.Shoshan MC, Linder S. Expert Opin Drug Metab Toxicol. 2008;4:273–280. doi: 10.1517/17425255.4.3.273. [DOI] [PubMed] [Google Scholar]
- 7.Tham KC, Kanaar R, Lebbink JHG. DNA Repair (Amst) 2016;38:75–83. doi: 10.1016/j.dnarep.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Arzimanoglou II, Gilbert F, Barber HRK. Cancer. 1998;82:1808–1820. doi: 10.1002/(sici)1097-0142(19980515)82:10<1808::aid-cncr2>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 9.Pino MS, Mino-Kenudson M, Wildemore BM, Ganguly A, Batten J, Sperduti I, Iafrate AJ, Chung DC. J Mol Diagnostics. 2009;11:238–247. doi: 10.2353/jmoldx.2009.080142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Jackson BA, Barton JK. J Am Chem Soc. 1997;119:12986–12987. [Google Scholar]; (b) Jackson BA, Barton Biochemistry. 2000;39:6176–6182. doi: 10.1021/bi9927033. [DOI] [PubMed] [Google Scholar]
- 11.Zeglis BM, Boland JA, Barton JK. J Am Chem Soc. 2008;130:7530–7531. doi: 10.1021/ja801479y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson BA, Alekseyev VY, Barton JK. Biochemistry. 1999;38:4655–4662. doi: 10.1021/bi990255t. [DOI] [PubMed] [Google Scholar]
- 13.Cordier C, Pierre VC, Barton JK. J Am Chem Soc. 2007;129:12287–12295. doi: 10.1021/ja0739436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Pierre VC, Kaiser JT, Barton JK. Proc Natl Acad Sci. 2007;104:429–434. doi: 10.1073/pnas.0610170104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zeglis BM, Pierre VC, Kaiser JT, Barton JK. Biochem. 2009;48:4247–4253. doi: 10.1021/bi900194e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hart JR, Glebov O, Ernst RJ, Kirsch IR, Barton JK. Proc Natl Acad Sci. 2006;103:15359–15363. doi: 10.1073/pnas.0607576103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bailis JM, Gordon ML, Gurgel JL, Komor AC, Barton JK, Kirsch IR. PLoS One. 2013;8:e78726. doi: 10.1371/journal.pone.0078726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fink D, Nebel S, Aebi S, Zheng H, Cenm B, Nehmã A, Christen D, Howell SB. Cancer Res. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 18.Fink D, Nebel S, Norris PS, Aebi S, Kim HK, Haas M, Howell SB. Br J Cancer. 1998;77:703–708. doi: 10.1038/bjc.1998.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komor AC, Barton JK. J Am Chem Soc. 2014;136:14160–14172. doi: 10.1021/ja5072064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komor AC, Schneider CJ, Weidmann AG, Barton JK. J Am Chem Soc. 2012;134:19223–19233. doi: 10.1021/ja3090687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ernst RJ, Song H, Barton JK. J Am Chem Soc. 2009;131:2359–2366. doi: 10.1021/ja8081044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeglis BM, Barton JK. Nat Protoc. 2007;2:357–371. doi: 10.1038/nprot.2007.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JR, Lung JW. J Chinese Chem Soc. 2003;50:227–232. [Google Scholar]
- 24.Sangster J. J Phys Chem Ref Data. 1989;18:1111–1229. [Google Scholar]
- 25.Komor AC, Schneider CJ, Weidmann AG, Barton JK. J Am Chem Soc. 2012;134:19223–19233. doi: 10.1021/ja3090687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmad KA, Iskandar KB, Hirpara JL, Clement MV, Pervaiz S. Cancer Res. 2004;64:7867–7878. doi: 10.1158/0008-5472.CAN-04-0648. [DOI] [PubMed] [Google Scholar]
- 27.Puckett CA, Barton JK. Biochemistry. 2008;47:11711–11716. doi: 10.1021/bi800856t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sitlani A, Long EC, Pyle AM, Barton JK. J Am Chem Soc. 1992;114:2303–2312. [Google Scholar]
- 29.Bailis JM, Weidmann AG, Mariano NF, Barton JK. Proc Natl Acad Sci. 2017;114:6948–6953. doi: 10.1073/pnas.1706665114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reithofer MR, Bytzek AK, Valiahdi SM, Kowol CR, Groessl M, Hartinger CG, Jakupec MA, Galanski M, Keppler BK. J Inorg Biochem. 2011;105:46–51. doi: 10.1016/j.jinorgbio.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 31.Waring MJ, Johnstone C. Bioorganic Med Chem Lett. 2007;17:1759–1764. doi: 10.1016/j.bmcl.2006.12.061. [DOI] [PubMed] [Google Scholar]
- 32.Landero Figueroa JA, Stiner CA, Radzyukevich TL, Heiny JA. Sci Rep. 2016;6:20551. doi: 10.1038/srep20551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X, Testa B, Fahr A. Pharm Res. 2011;28:962–977. doi: 10.1007/s11095-010-0303-7. [DOI] [PubMed] [Google Scholar]
- 34.Hart JR, Glebov O, Ernst RJ, Kirsch IR, Barton JK. Proc Natl Acad Sci. 2006;103:15359–15363. doi: 10.1073/pnas.0607576103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- 36.Glaab WE, Tindall KR. Carcinogenesis. 1997;18:1–8. doi: 10.1093/carcin/18.1.1. [DOI] [PubMed] [Google Scholar]
- 37.Modica-Napolitano JS, Aprille JR. Adv Drug Deliv Rev. 2001;49:63–70. doi: 10.1016/s0169-409x(01)00125-9. [DOI] [PubMed] [Google Scholar]
- 38.Weidmann AG, Komor AC, Barton JK. Philisophocal Trans R Soc. 2013;371:20120117. doi: 10.1098/rsta.2012.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weidmann AG, Barton JK. Inorg Chem. 2015;54:9626–9636. doi: 10.1021/acs.inorgchem.5b01722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petitjean A, Barton JK. J Am Chem Soc. 2004;126:14728–14729. doi: 10.1021/ja047235l. [DOI] [PubMed] [Google Scholar]
- 41.Chari RVJ. Acc Chem Res. 2008;41:98–107. doi: 10.1021/ar700108g. [DOI] [PubMed] [Google Scholar]
- 42.Zeglis BM, Barton JK. J Am Chem Soc. 2006;128:5654–5655. doi: 10.1021/ja061409c. [DOI] [PubMed] [Google Scholar]
- 43.Nano A, Boynton AN, Barton JK. J Am Chem Soc. 2017;139:17301–17304. doi: 10.1021/jacs.7b10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.