

Abstract
The kidney requires a large number of mitochondria to remove waste from the blood and regulate fluid and electrolyte balance. Mitochondria provide the energy to drive these important functions and can adapt to different metabolic conditions through a number of signalling pathways (for example, mechanistic target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) pathways) that activate the transcriptional co-activator peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α), and by balancing mitochondrial dynamics and energetics to maintain mitochondrial homeostasis. Mitochondrial dysfunction leads to a decrease in ATP production, alterations in cellular functions and structure, and the loss of renal function. Persistent mitochondrial dysfunction has a role in the early stages and progression of renal diseases, such as acute kidney injury (AKI) and diabetic nephropathy, as it disrupts mitochondrial homeostasis and thus normal kidney function. Improving mitochondrial homeostasis and function has the potential to restore renal function, and administering compounds that stimulate mitochondrial biogenesis can restore mitochondrial and renal function in mouse models of AKI and diabetes mellitus. Furthermore, inhibiting the fission protein dynamin 1-like protein (DRP1) might ameliorate ischaemic renal injury by blocking mitochondrial fission.
The kidney is one of the most energy-demanding organs in the human body. A study measuring the resting energy expenditure of various organs in healthy adults, ranging from 21 to 73 years of age, found that the kidney and heart have the highest resting metabolic rates1. The kidney has the second highest mitochondrial content and oxygen consumption after the heart2,3. The resting metabolic rate for the kidney is high because the kidney requires an abundance of mitochondria to provide sufficient energy to enable it to remove waste from the blood, reabsorb nutrients, regulate the balance of electrolytes and fluid, maintain acid–base homeostasis, and regulate blood pressure. These tasks, especially the reabsorption of glucose, ions and nutrients through channels and transporters, are driven by ion gradients.
Mitochondria provide energy to the Na+–K+-ATPase to generate ion gradients across the cellular membrane4. In the kidney, the proximal tubule, the loop of Henle, the distal tubule and the collecting duct all require active transport to reabsorb ions4. By contrast, glomerular filtration is a passive process that is dependent on the maintainence of hydrostatic pressure in the glomeruli5. Proximal tubules require more active transport mechanisms than other renal cell types because they reabsorb 80% of the filtrate that passes through the glomerulus, including glucose, ions, and nutrients. As such, they contain more mitochondria than any other structure in the kidney. The ability of mitochondria to sense and respond to changes in nutrient availability and energy demand by maintaining mitochondrial homeostasis is critical to the proper functioning of the proximal tubule. In this Review, we describe the processes involved in maintaining mitochondrial homeostasis and discuss how these processes provide and maintain sufficient energy to support renal function. We also explore how disease states, such as acute kidney injury (AKI) and diabetic nephropathy, alter mitochondrial function, and how mitochondrial energetics might be targeted as a treatment for these diseases.
Mitochondrial function
Mitochondria are a network of plastic organelles that together maintain a variety of cellular functions and processes, such as the level of reactive oxygen species (ROS), cytosolic calcium and apoptosis6. Most importantly, mitochondria produce ATP, thereby supplying the energy source for basal cell functions as well as cellular repair and regeneration. To accomplish this feat, a population of healthy and functional mitochondria is vital.
ATP production
Aerobic respiration involves the consumption of oxygen to produce ATP, water and carbon dioxide (CO2). Most of the ATP generated by aerobic respiration is produced by the flux of electrons through the electron transport chain (ETC) in a process called oxidative phosphory lation (FIG. 1a). Aerobic respiration begins with the production of pyruvate from glucose via glycolysis7. Pyruvate is converted to acetyl-CoA (via pyruvate dehydro genase in the mitochondrial matrix), which fuels the tricarboxylic acid (TCA) cycle to produce six NADH, four FADH2, and six CO2 per molecule of glucose7. Electrons from NADH and FADH2 are transferred to complex I and complex II, respectively, of the ETC in the mitochondrial inner membrane. Electrons then travel through the ETC to complex IV, where they are accepted by oxygen. Note that the haem protein cytochrome c, which is located in the mitochondrial inner membrane, facilitates the transfer of electrons from complex III to complex IV. Ultimately, protons, which are actively pumped into the intermembrane space as electrons move through complexes I, III, and IV, flow through ATP synthase (also known as complex V) to drive the conversion of ADP to ATP7.
Figure 1. ATP production in the kidney.
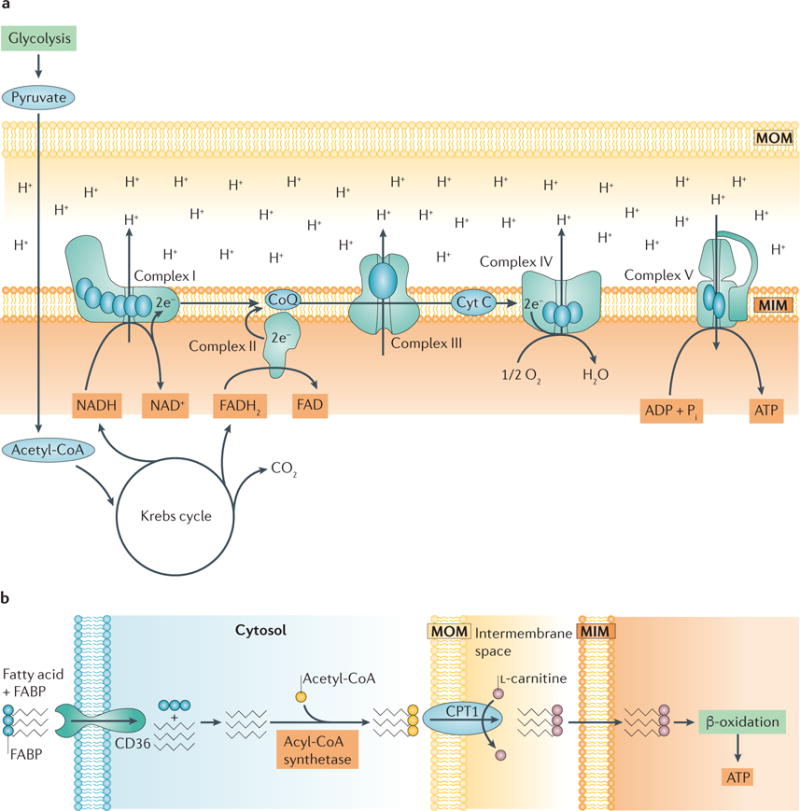
a | The electron transport chain (ETC). A functioning ETC transforms reducing equivalents from NADH and FADH2 to produce NAD+ and FAD+, respectively. The electrons (e−)that are produced travel through the complexes of the ETC and are ultimately accepted by oxygen at complex IV. As electrons are transferred from complex to complex, protons (H+) are actively pumped out from complexes I, III, and IV into the intermembrane space, maintaining the membrane potential and driving the production of ATP by ATP synthase (also known as complex V). b | Fatty acid transport and activation in renal proximal tubule cells. Proximal tubules require large amounts of ATP to drive ion transport and therefore rely on aerobic respiration, the most efficient mechanism for producing ATP. Fatty acids are a main source of energy for proximal tubules because more ATP can be produced from one molecule of palmitate than from one molecule of glucose18. Fatty acids bound to fatty acid-binding proteins (FABP) are transported into the proximal tubule cell via platelet glycoprotein 4 (also known as CD36) and activated by the addition of acetyl-CoA in the cytosol via acyl-CoA synthetase. Activated fatty acids are transported into mitochondria via carnitine O-palmitoyltransferase 1 (CPT1), which exchanges their acyl-CoA group for L-carnitine, whereupon they undergo β-oxidation to produce ATP. CoQ, coenzyme Q; Cyt C, cytochrome c; MIM, mitochondrial inner membrane; MOM, mitochondrial outer membrane; Pi, inorganic phosphate.
In general, all cell types in the kidney need ATP to maintain cellular functions; however, the mechanism by which ATP is produced is cell type-dependent. For example, in the renal cortex, proximal tubules depend on the efficiency of oxidative phosphorylation to produce ATP that drives the active transport of glucose, ions and nutrients8. By contrast, glomerular cells, including podocytes, endothelial cells and mesengial cells, have lower oxidative capacity because their function is to filter blood to remove small molecules (namely, glucose, urea, water and salts) while retaining large proteins, such as haemoglobin9. This passive process does not directly require ATP and, therefore, glomerular cells have the ability to perform aerobic and anaerobic respiration to produce ATP for basal cell processes10–13. Anaerobic respiration, like aerobic respiration, begins with glycolysis, producing pyruvate from glucose, but is characterized by the subsequent production of lactate from pyruvate14. Anaerobic respiration produces two molecules of ATP and is an efficient mechanism for cell types that have a lower O2 supply10. This process is important, as glycolysis frequently occurs in cell types other than proximal tubules and can utilize alternative energy sources, such as amino acids, in the absence of glucose15,16. For example, pyruvate can be generated via the oxidation of amino acids to fuel both anaerobic and aerobic mechanisms of ATP production.
Due to the high energy demand of proximal tubules, aerobic respiration is their primary mechanism of ATP production. Proximal tubules utilize non- esterified fatty acids, such as palmitate, via β-oxidation for maximal ATP production. A single molecule of palmitate produces 106 molecules of ATP, whereas the oxidation of glucose only yields 36 molecules of ATP17,18. Fatty acids are taken up by proximal tubule cells via transport proteins, such as platelet glycoprotein 4 (also known as CD36), or synthesized in the cytoplasm, where they are activated by coA before being transported into mitochondria through the carnitine shuttle19 (FIG. 1b). Specifically, carnitine O-palmitoyltransferase 1 (CPT1) exchanges the coA group on fatty acids with l- carnitine, allowing the transfer of fatty acids across the mitochondrial inner membrane space through the carnitine shuttle. Fatty acids are then broken down for energy via β-oxidation in the mitochondrial matrix. Although β-oxidation is the most efficient mechanism for producing ATP in proximal tubules, it is important to note that due to the high consumption of oxygen by proximal tubules, they are more susceptible than other cell types to changes in oxygen levels20,21. A decrease in oxygen levels can lead to impaired β-oxidation and a reduction in ATP production (see below).
A balance of catabolic and anabolic nutrient-sensing pathways regulates the optimum concentration of fatty acids in a cell (see below). Disease states and different metabolic conditions in the kidney alter this balance and can adversely affect mitochondrial energetics. For example, the accumulation of fatty acids in AKI and diabetic nephropathy can negatively impact ATP production by decreasing β-oxidation in the mitochondria and increasing the formation of lipid droplets inside the cell18. An inverse correlation exists between lipogenesis that is induced by the accumulation of fatty acids and the transcription of genes that are involved in fatty acid oxidation22,23. Fatty acids can also trigger apoptosis and, more importantly, create a toxic environment inside the cell that hinders mitochondrial function24,25. Fatty acid metabolism in disease states, such as AKI and diabetic nephropathy, will be discussed below.
Antioxidant defences
As discussed, mitochondria produce ATP via the ETC. At steady state, when electrons are passed through the ETC to molecular oxygen, a low concentration of superoxide anions is generated from complex I and complex III. Although a low level of ROS, such as superoxide anions, is important for cell function, high concentrations are toxic to mitochondria and the cell26–28 (FIG. 2). For example, under oxidative stress, increased levels of ROS can cause breaks in mitochondrial DNA (mtDNA) that cause mutations in the next generation of mitochondria; breaks in mtDNA also negatively affect the efficiency of the ETC, causing a decrease in ATP production and damaging proteins and lipids29. ROS can also trigger apoptosis in the cell by causing the release of cytochrome c, leading to mitochondrial dysfunction29. Therefore, mitochondria have antioxidant defence systems to counteract the excessive formation of additional ROS. Superoxide dismutase 2 (SOD2), which converts superoxide anions to hydrogen peroxide and oxygen, is specific for mitochondria30. Moreover, the transcription of genes encoding antioxidant enzymes, such as SOD2, catalase and glutathione peroxidase, is activated by nuclear factor erythroid 2-related factor 2 (NRF2) in response to oxidative stress, providing a mechanism to prevent excessive ROS production31. The importance of these antioxidant systems is to maintain optimal ATP production and sustain mitochondrial function.
Figure 2. Oxidative stress and the antioxidant defence system.
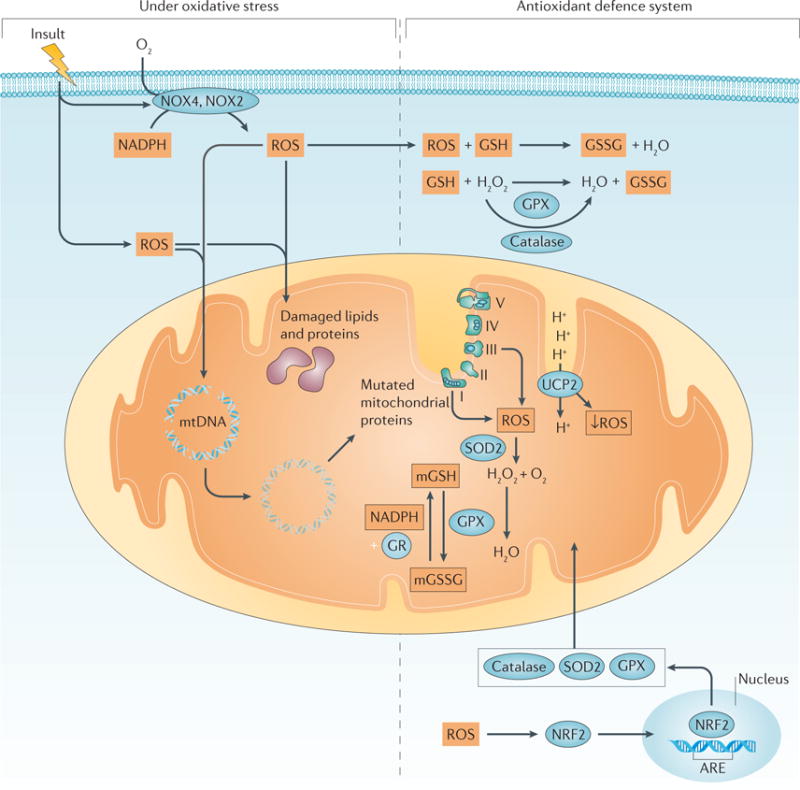
Insults can increase the production of reactive oxygen species (ROS) in the cytosol and mitochondria. NADPH oxidase 2 (NOX2) and NOX4 can also contribute to the production of ROS222. The production of ROS can cause breaks in mitochondrial DNA (mtDNA) and damage lipids and proteins. Damaged mtDNA can produce aberrant mitochondrial proteins and prevent mitochondrial protein synthesis, whereas damaged lipids and proteins result in impaired mitochondrial function, leading to further increases in mitochondrial ROS. ROS also activate nuclear factor erythroid 2-related factor 2 (NRF2), which translocates to the nucleus and binds to antioxidant-responsive elements (AREs) to activate the transcription of genes encoding oxidant-neutralizing enzymes, such as mitochondrial superoxide dismutase 2 (SOD2), glutathione peroxidase (GPX) and catalase. SOD2 reduces superoxide anions to hydrogen peroxide (H2O2) and oxygen (O2). Catalase, found in the cytoplasm, and GPX, located in the cytoplasm and mitochondria, reduce H2O2 to water (H2O)223. GPX also oxidizes glutathione (GSH), resulting in glutathione disulfide (GSSG) as a byproduct of reducing hydrogen peroxide to water. GSSG in mitochondria (mGSSG) is converted back to GSH by glutathione reductase (GR) in a process that requires the presence of NADPH. The activity of the mitochondrial uncoupling protein 2 (UCP2) is increased, dissipating the proton motive force and decreasing ROS production. mGSH, mitochondrial GSH. The electron transport chain complexes I–V are indicated as I, II, III, IV and V.
Another important antioxidant defence mechanism involves glutathione. Glutathione is a tripeptide (γ-glutamyl-cysteinal-glycine) nucleophile that can exist in a reduced form (GSH), or in an oxidized form as glutathione disulfide (GSSG). Mitochondria contain their own pool of glutathione, mitochondrial glutathione (mGSH), which not only helps to decrease excessive ROS levels but also prevents the release of cytochrome c from the inner membrane32. mGSH directly interacts with superoxide anions and becomes oxidized to GSSG33. Glutathione peroxidase (GPX) is located in both the cytoplasm and the mitochondria and uses GSH to reduce hydrogen peroxide to water, resulting in GSSG as a by-product34. GSSG cannot exit the mitochondria and is converted back to mGSH by glutathione reductase, for reuse or for elimination from the mitochondria33. The conversion of GSSG to mGSH requires NADPH, allowing crosstalk between the mechanism that maintains mGSH levels and the pentose phosphate pathway that produces NADPH. Together, these mechanisms have a major role in preventing excessive levels of ROS, and sustaining mitochondrial function.
Uncoupling proteins are a family of mitochondrial transport proteins that are located in the mitochondrial inner membrane35,36. They transport protons across the inner membrane to the mitochondrial matrix. Mitochondrial uncoupling protein 2 (UCP2) is expressed in the kidney and is activated by mitochondrial ROS and other stimuli. An increase in ROS formation in the mitochondria activates UCP2, dissipating the proton motive force as heat and, as a result, reducing ROS production36,37. As ROS production contributes to mitochondrial dysfunction in AKI and diabetic nephropathy, UCP2 has been explored in the kidney and in these disease states38. Studies investigating the role of UCP2 polymorphisms in the kidney that exacerbate disease in patients with diabetic nephro pathy reveal that UCP2 is a potential target for treatment39. Lack of UCP2 has also been shown to worsen tubular injury after induction of experimental AKI in mice38. These studies show the importance of UCP2 in the kidney as well as its role in attenuating excessive ROS production.
Mechanisms also exist to sustain mitochondrial function under hypoxic conditions. The lack of oxygen under hypoxic conditions decreases ATP production and causes cell death. In normoxic conditions, hypoxia-inducible factor 1α (HIF1α) is degraded in the presence of oxygen and α-ketoglutarate, an intermediate of the TCA cycle40. However, under hypoxic conditions, HIF1α heterodimerizes with HIF1β to form a transcription factor that binds to a hypoxia response element (HRE) present in genes that encode glycolytic enzymes and glucose transporters in the kidney41. Hypoxic conditions also alter the composition of complex IV of the ETC in which, at physiological conditions, the regulatory subunit 1 predominates in the ETC; during hypoxia, regulatory subunit 2 predominates in complex IV, which increases the efficiency of the ETC42. Several studies have shown that increasing the efficiency of the ETC increases the production of mitochondrial ROS under hypoxic conditions, although the mechanism by which this occurs is still unclear43–45. The effects of oxidative stress and hypoxia on mitochondrial morphology and energetics are discussed below.
Nutrient-sensing pathways in the kidney
Nutrient-sensing pathways can directly affect mitochondrial energetics in response to external stimuli, such as hypoxia, oxidative stress and energy depletion. Two signalling pathways in particular have been extensively explored in the kidney, namely the mechanistic target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) signalling pathways46,47. Both signalling pathways also have a role in regulating mitochondrial bio-genesis — that is, the production of new and functional mitochondria — to help maintain a healthy population of mitochondria (FIG. 3).
Figure 3. Crosstalk between two nutrient-sensing pathways.
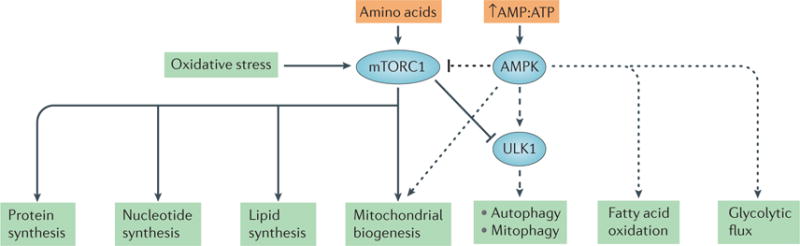
Mechanistic target of rapamycin complex 1 (mTORC1) and AMP-activated protein kinase (AMPK) have key roles in regulating mitochondrial biogenesis and mitophagy. mTORC1 is responsible for triggering anabolic pathways, such as the synthesis of proteins, nucleotides and lipids, as well as mitochondrial biogenesis. AMPK activates catabolic pathways, including autophagy, mitophagy, fatty acid oxidation and glycolysis. AMPK can stimulate mitochondrial biogenesis (dotted arrow). However, in response to stimuli such as nutrient deprivation, AMPK can inhibit mTORC1 (dotted inhibitory line) and phosphorylate ULK1 to activate mitophagy (dashed arrow). Together these two signalling pathways maintain cell function and sustain mitochondrial energetics in response to stimuli such as hypoxia, oxidative stress and energy depletion.
mTOR is a serine/threonine kinase complex that comprises a number of proteins. Two distinct mTOR complexes exist: mTOR complex 1 (mTORC1) and mTORC2, each of which contain their own unique subunits and substrates. mTORC1, which is a complex of mTOR, regulatory-associated protein of mTOR (Raptor) and several other proteins, regulates cell growth and proliferation and inhibits autophagy by stimulating anabolic processes. mTORC2, which is a complex of mTOR, rapamycin- insensitive companion of mTOR (Rictor) and several other proteins, is thought to regulate potassium and sodium levels in the kidney48,49. mTORC1 is considered a nutrient sensor because it can be activated by growth factors, nutrients such as amino acids and glucose, and oxidative stress, triggering pathways that lead to protein synthesis, nucleotide synthesis, lipid synthesis and mitochondrial biogenesis by activating the transcriptional repressor yin and yang 1 (YY1)46,50. In the case of mitochondrial biogenesis, YY1 acts as a transcription factor and co-activator of the master regulator of mitochondrial biogenesis — the transcriptional co-activator peroxi-some proliferator-activated receptor-γ co-activator 1α (PGC1α) — resulting in the transcription of mitochondrial genes50. mTORC1-deficiency specifically in renal proximal tubules of mice decreased the protein levels of PGC1α in vivo51. Of note, the mTOR pathway can be inhibited by hypoxia and AMPK.
AMPK is another nutrient sensor in the kidney that stimulates catabolic processes. When the AMP:ATP ratio in the cell is high in the presence of low oxygen levels, AMPK is activated52. AMPK targets a number of proteins, the phosphorylation of which leads to the production of antioxidant enzymes, the induction of mitochondrial biogenesis, and an increase in glycolytic flux, fatty acid oxidation and glucose transport; all of these events contribute to cell growth and an increase in cellular metabolism53. AMPK can induce mitochondrial biogenesis by stimulating the transcription of the gene encoding PGC1α (PPARGC1A) and by phosphory lating PGC1α at Thr177 and Ser539 to increase its activity54. AMPK stimulates the production of energy and inhibits energy-consuming pathways by inhibiting mTORC1. Under conditions of nutrient deprivation, crosstalk exists between mTORC1 and AMPK (FIG. 3) so that AMPK can inhibit mTORC1 while activating autophagy by phosphorylating the serine/threonine protein kinase ULK1 (REF. 55). Due to the presence of AMPK targets in kidney cells, AMPK is a novel drug target for several renal diseases (see below).
Maintaining mitochondrial homeostasis
Mitochondrial homeostasis requires a balance between mitochondrial biogenesis, fission and fusion, and mitophagy — the selective removal of non-functional and damaged mitochondria from cells by autophagy. All of these processes work together to maintain mitochondrial energetics, that is, the optimal production of ATP in normoxic conditions and in altered metabolic conditions.
Mitochondrial biogenesis
Mitochondrial biogenesis, which produces new and functional mitochondria, increases ATP production in response to increasing energy demands. Mitochondrial biogenesis is regulated by a range of transcriptional co-activators and co-repressors56,57. One study has shown that PGC1α is a prominent regulator, at the transcriptional level, of oxidative phosphorylation, the TCA cycle and fatty acid metabolism in the kidney58. In that study, the investigators performed gene expression profiling of kidneys from control mice and nephron-specific inducible PPARGC1A-knockout (NiPKO) mice that had been fed a chow diet or high-fat diet (HFD). Using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, they analysed transcripts from all four groups of mice, and found a decrease in transcripts related to oxidative phosphorylation, TCA cycle and glycolysis in chow-fed NiPKO mice and in HFD-fed NiPKO mice. This finding supports the idea that inactivation of PGC1α in the kidney reduces mitochondrial function and metabolism and subsequently decreases mitochondrial biogenesis.
Overexpression of PGC1α can also mitigate mitochondrial dysfunction in vitro after oxidant exposure, further supporting a role for mitochondrial biogenesis in mitochondrial homeostasis59. The activation of peroxisome proliferator-activated receptors (PPARs) and oestrogen-related receptors (ERRs) also contributes to the regulation of mitochondrial biogenesis, sometimes by these receptors directly interacting with PGC1α60 (FIG. 4). PPARs and ERRs are nuclear receptors that can be activated by fatty acids and steroid hormones such as oestrogen, and they elicit a response by binding to specific DNA response elements through their DNA-binding domains61. PGC1α can directly bind to these nuclear receptors and co-activate the transcription of genes, the protein products of which are involved in oxidative phosphorylation and fatty acid oxidation62,63. PGC1α activation results in its translocation from the cytoplasm to the nucleus, allowing it to upregulate the transcription of genes that are important for mitochondrial homeostasis and ATP production64. Transcription programmes downstream of PGC1α include nuclear and mitochondrial genes, as well as those involved in signalling pathways that regulate mitochondrial biogenesis (reviewed elsewhere65–67).
Figure 4. Activation and regulation of mitochondrial biogenesis.
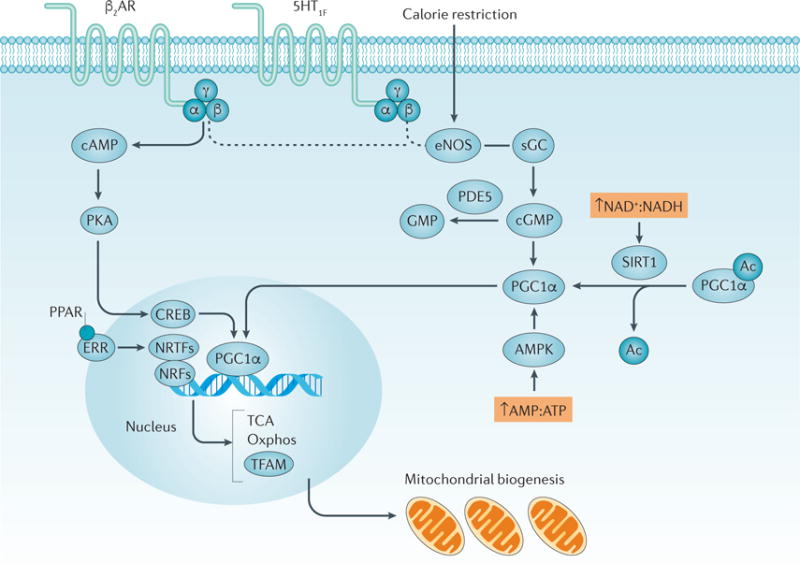
A complex network of pathways regulate mitochondrial biogenesis. Activation of peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α) in the cytosol causes its translocation to the nucleus and the transcription of genes (including that encoding mitochondrial tricarboxylic acid (TCA) cycle and mitochondrial biogenesis. TFAM aids in the transcription of genes that are encoded by mitochondrial DNA224–226. The activation of G protein-coupled receptors (GPCRs), such as the β2 adrenergic receptors (β2AR) and 5-hydroxytryptamine receptor 1F (5-HT1F), leads to the dissociation of heterotrimeric G proteins composed of Gα, Gβ and Gγ subunits and the subsequent activation of protein kinase A and endothelial nitric oxide synthase (eNOS)66. The pathway from GPCRs to eNOS is still under investigation, as indicated by the dashed line. eNOS stimulates soluble guanylyl cyclase (sGC) to form cyclic guanosine monophosphate (cGMP), which in turn activates PGC1α. A number of compounds can activate nuclear receptors such as peroxisome proliferator-activated receptors (PPARs) and oestrogen- related receptors (ERRs) and induce mitochondrial biogenesis. Once activated, these nuclear receptors can act as transcriptional co-activators (labelled in the figure as nuclear receptor transcription factors (NRTFs)), with PGC1α to stimulate mitochondrial biogenesis. Other transcription factors, including nuclear respiratory factor 1 (NRF1) and NRF2, can also directly bind to PGC1α to induce mitochondrial biogenesis227. Stimuli, such as caloric restriction, can activate eNOS, increasing the production of cGMP and leading to the activation of PGC1α. The activity of sirtuin 1 (SIRT1) is increased in the presence of a high ratio of NAD+ to NADH concentrations, leading to the activation of PGC1α. High AMP:ATP ratios also activate AMP-activated protein kinase (AMPK), activating PGC1α by phosphorylation. In all of these cases, the activation of PGC1α stimulates mitochondrial biogenesis. Ac, acetyl; PDE5, cGMP-specific 3ʹ,5ʹ-cyclic phosphodiesterase; PKA, protein kinase A; sGC, soluble guanylyl cyclase.
As the activation or suppression of PGC1α is regulated by external stimuli and post-translational modifications, it can be considered to be a nutrient sensor in the kidney. The expression and regulation of PGC1α in the kidney is still being explored. However, much of what is known about the regulation of PGC1α was discovered in the injured kidney as a result of disease states, such as diabetic nephropathy, ischaemia–reperfusion injury (IRI), sepsis, and cisplatin-induced AKI. Findings in these disease states support a role for PGC1α in the recovery phase from these diseases and in restoring mitochondrial function, highlighting PGC1α as a therapeutic target. Exercise and insulin stimulate an increase in PPARGC1A expression in skeletal muscle and in the heart, whereas fasting increases PPARGC1A expression in the liver65,68. In brown fat and muscle cells, cold exposure activates PGC1α65. In cases of oxidative stress or nutrient depletion, the activation of mitochondrial biogenesis helps rescue mitochondria from apoptosis69,70. In general, if the cell is in need of more energy, PGC1α is activated by deacetylation, whereas PGC1α is inactivated by acetylation when energy levels are high65.
In addition to AMPK and mTOR, other energy sensing pathways that stimulate mitochondrial bio genesis include those involving sirtuins, cAMP and cyclic guanosine monophosphate (cGMP) (FIG. 4). Sirtuin 1 (SIRT1) and SIRT3 are protein deacetylases that have a role in a variety of mitochondrial processes, including the ETC, TCA cycle, fatty acid oxidation, redox homeostasis and mitochondrial biogenesis71. SIRT1 activity is activated by NAD+, after which it activates downstream targets such as PGC1α64. SIRT3 is mitochondria- specific and can be activated to stimulate mitochondrial biogenesis72. The stimulation of adenylyl cyclase results in an increase in cAMP, which activates protein kinase A (PKA) that in turn phosphorylates cyclic AMP-responsive element-binding protein (CREB)65,73. CREB is also a transcriptional activator of PGC1α and can therefore also stimulate mitochondrial biogenesis73. Finally, increased levels of cGMP induced by caloric restriction and the inhibition of phosphodiesterases can stimulate PGC1α activation and mitochondrial bio-genesis in vivo74–76. Several of these pathways are being targeted to increase mitochondrial biogenesis to correct mitochondrial defects.
Mitochondrial dynamics and energetics
Correct mitochondrial morphology must be maintained for maximal ATP production. The processes of fission, fusion and mitophagy drive mitochondrial dynamics as they directly affect mitochondrial structure and morphology. Fission and fusion complement each other under different metabolic conditions to maintain mitochondrial morphology, whereas mitophagy removes damaged mitochondria from the network77. Sustaining mitochondrial dynamics is important for the appropriate maintenance of mitochondrial energetics.
Fission and fusion
Fission, the splitting of mitochondria into two, and fusion, the combining of two mitochondria, are complementary processes that are necessary for mitochondrial homeostasis. At steady state there is a balance between these processes (FIG. 5). The genetic deletion of genes, the protein products of which are involved in fission or fusion, causes human disease. For example, dominant optic atrophy is characterized by a loss of visual acuity owing to mutations in the gene encoding the fusion protein dynamin-like 120 kDa protein (also known as OPA1), and mutations in the gene encoding the fission protein dynamin 1-like protein (DRP1), are lethal78–83. Although exceptions exist, in general, studies have shown that oxidative phosphorylation increases with fusion and decreases with fission to match the energy demands of the cells84,85. Excessive fusion, like excessive fission, can be associated with disease states, as seen in neurodegenerative diseases86. However, some cell types do not adhere to this trend, such as adult cardiomyocytes and senescent cells. Mitochondria in adult cardiomyocytes have a fragmented morphology but maintain oxidative capacity, whereas mitochondria in senescent cells remain elongated, which is characteristic of increased fusion87. Senescent cells in this elongated state have decreased bioenergetic capacity88,89.
Figure 5. Mitochondrial dynamics: fission, fusion and mitophagy.
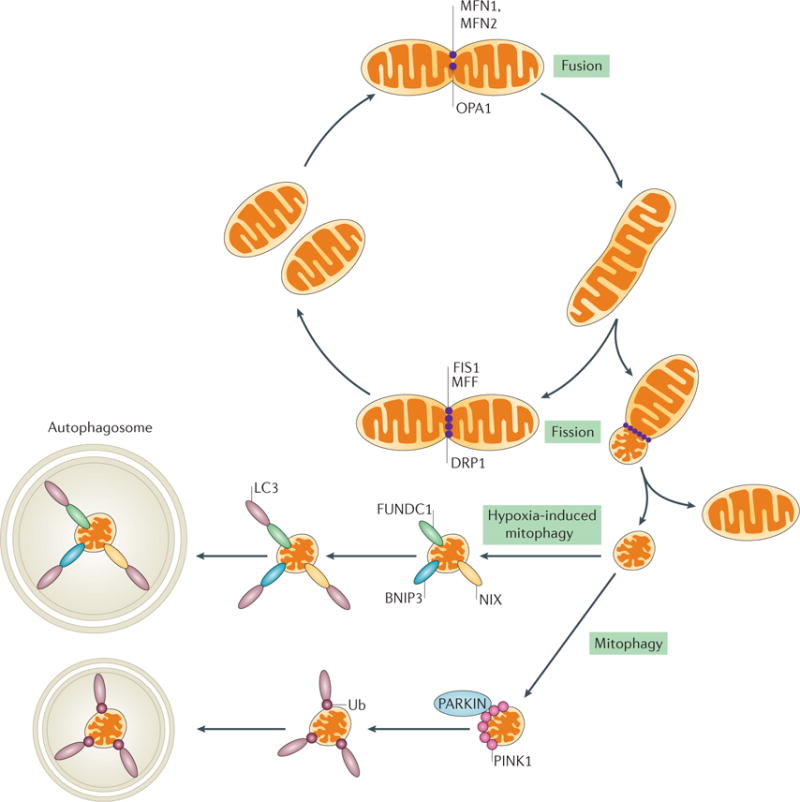
Mitochondria are dynamic organelles that need to maintain their morphology for the optimal production of ATP under different metabolic conditions and as part of a healthy network of mitochondria. Fission and fusion are two processes that are necessary for the maintenance of mitochondria morphology. Mitochondria fuse together via mitofusin 1 (MFN1) and MFN2 (outer membrane fusion) and the activation of dynamin-like 120 kDa (OPA1) (inner membrane fusion). Fusion can occur to maintain ATP production or to redistribute mitochondrial proteins. Fission can isolate depolarized mitochondrion that might not contribute to the healthy network of mitochondria. The activation of fission causes the oligomerization of dynamin 1-like protein (DRP1) on the mitochondrial outer membrane, where it is bound to receptors (namely mitochondrial fission 1 (FIS1) and mitochondrial fission factor (MFF)), forming a ring-like structure that mediates the separation of mitochondria. The network also isolates dysfunctional mitochondria for degradation by mitophagy via a well-studied PTEN-induced putative kinase 1 (PINK1)– PARKIN mechanism. Under adverse conditions such as hypoxia, however, mitochondria will be removed by a FUN14 domain-containing protein 1 (FUNDC1) or BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) and NIP3-like protein (NIX)-dependent mechanism of mitophagy. LC3, microtubule-associated protein 1 light chain 3; Ub, ubiquitin.
Fusion is a two-part process that involves fusion of the outer mitochondrial membrane and, subsequently, the inner mitochondrial membrane of two mitochondria. GTPases of the dynamin superfamily — mitofusin 1 (MFN1), MFN2 and OPA1 — are key players in fusion. MFN1 and MFN2 are located on the outer mitochondrial membrane and are necessary for outer membrane fusion, whereas OPA1 resides in the inner membrane and is important for inner membrane fusion. Fusion leads to the elongation of mitochondria under physiological conditions, which can help to maintain oxidative phosphorylation90. These GTPases have a role in mitochondrial energetics. For example, deletion of MFN2 in mice causes deficiency in coenzyme Q, an electron carrier in complex III, which leads to ETC dysfunction and a decrease in ATP production91. Activation of these mitofusins and the cleavage of OPA1 can be regulated by changes in metabolism (see below).
Mitochondrial outer membranes are tethered by dimerization of MFN1 and MFN2, and external stimuli, such as oxidative stress, can enhance outer membrane fusion92. The activation of inner membrane fusion can be regulated by changes in metabolism at sites of proteolytic cleavage of OPA1 (REF. 93). OPA1 usually exists in a soluble long form and can be cleaved by the ATP-dependent zinc metalloproteinase YME1L or by the metalloendo-peptidase OMA1, which is activated in response to a loss in membrane potential, to yield a soluble short form85. The soluble long and soluble short forms of OPA1 are necessary for fusion to occur. During steady state, both forms can coexist to induce minor structural remodelling of mitochondria94,95. The activation of cleaved OPA1 requires the presence of GTP, and the availability of GTP to activate OPA1 correlates with ATP levels in the cell96,97. The exact mechanism by which outer membrane and inner membrane fusion events are coordinated is still under investigation.
Fission is necessary to isolate damaged mitochondria from the mitochondrial network. If the resulting daughter mitochondria are unbalanced and depolarized, they are targeted for mitophagy98 to sustain a population of healthy mitochondria. However, excessive fission, as seen in diseases such as diabetic nephropathy and AKI, can have harmful effects on mitochondrial homeostasis in the long term99. In vitro studies to elucidate the mechanisms that trigger mitochondrial fission have shown that cells that are exposed to an excess of nutrients or oxidative stress have fragmented mitochondria99. Fission is induced by the translocation of DRP1 from the cytosol to the mitochondrial outer membrane as a result of a loss in mitochondrial membrane potential. If the membrane potential is not restored, mitochondria are degraded via mitophagy99. DRP1 oligomerizes on the outer membrane to form a ring-like structure around the mitochondria, which can cause scission of the membrane100. DRP1 can bind to several different receptors, such as mitochondrial fission 1 (FIS1), the mitochondrial dynamics proteins MID49 and MID51, and mitochondrial fission factor (MFF), which reside on the mitochondrial outer membrane81. DRP1 accumulates on the outer mitochondrial membrane by binding to these receptors and mediates the scission of mitochondria, which is dependent on GTP101. MID51 contains a cytosolic domain that has affinity for ADP and GDP, and can therefore act as a metabolic sensor102,103. DRP1 activity can be regulated by post-translational modifications, such as phosphorylation, ubiquitylation, and sumoylation104, and several signalling pathways have been shown to regulate the phosphorylation of DRP1 (REF. 105). For example, phosphorylation of DRP1 at Ser637 by PKA inhibits its GTPase activity and thus inactivates fission81,106. By contrast, dephosphorylation of DRP1 at Ser637 by calcium and calmodulin-dependent serine/threonine protein phosphatase 2B catalytic subunit α isoform or calcineurin (CaN) activates DRP1 and promotes fission107,108. The balance between fission and fusion to maintain a functional population of mitochondria is an intricate process and is still under investigation. Mitochondria that disrupt this balance between fission and fusion, such as damaged mitochondria, are however removed from the network via mitophagy.
Mitophagy
Mitophagy in most cell types is regulated by a PTEN-induced putative kinase 1 (PINK1)– PARKIN mechanism that tags mitochondria for degradation109. PINK1, a kinase that is located in the cytosol, is imported into the mitochondria and then degraded under physiological conditions110. As protein import is dependent on the mitochondrial membrane potential, mitochondrial depolarization results in an accumulation of PINK1 on the outer membrane; the PINK1-mediated phosphorylation of certain proteins on the outer membrane mediates recruitment of the E3 ligase, PARKIN111–114, to the outer membrane. PARKIN ubiquitylates lysine residues in the N-termini of mitochondrial outer membrane proteins, such as MFN1 and MFN2, thereby targeting the mitochondria for degradation by autophagosomes115–119.
Several pathways regulate mitophagy (FIG. 5). Proteins that are important for autophagy, such as ULK1 and ULK2, can mediate mitophagy under different stimuli120. For example, when nutrients are sufficient, AMPK is inhibited and mTOR inhibits ULK1, suppressing mitophagy121. During nutrient deprivation, AMPK is activated and inhibits mTOR, facilitating ULK1 activation and mitophagy120 (FIG. 3). Under oxidative stress, AMPK can be activated and inhibit mTOR, again stimulating mitophagy55,121. A more direct role for AMPK in the activation of mitophagy has also been suggested122, whereby AMPK directly phosphorylates MFF on Ser155 and Ser172, triggering fission and, subsequently, mitophagy123. However, external stimuli that trigger this pathway are unknown and more research is needed.
Other stimuli, such as hypoxia, cause the Ser/Thr protein phosphatase phosphoglycerate mutase family member 5 (PGAM5) to dephosphorylate its substrate, the mitophagy receptor FUN14 domain-containing protein 1 (FUNDC1)124. FUNDC1 then interacts with microtubule-associated protein 1 light chain 3 (LC3), which mediates the formation of an autophagic membrane124,125. Alternatively, hypoxia can induce mitophagy through the actions of BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) and NIP3-like protein X (NIX; also known as BNIP3L) via a mechanism involving HIF1α126,127. HIF1α can directly induce the transcription of BNIP3 and NIX by binding to the promoter of BNIP3 and by recruiting other co-activator proteins to NIX. NIX and BNIP3 are transmembrane proteins located in the mitochondrial outer membrane and can activate mitophagy by dissipating the mitochondrial membrane potential and interacting with LC3 to deliver mitochondria to the autophagosome127–130. BNIP3 and NIX are also apoptotic regulators that can induce cell death or autophagy by increasing the production of ROS, by binding to pro-apoptotic proteins of the BCL-2 family, or by inhibiting the GTP-binding protein RHEB, an upstream activator of mTOR131–133. Previous studies suggest that crosstalk exists between both of the mechanisms that can regulate mitophagy127,134,135, although the mechanisms of this proposed crosstalk are unclear and additional studies are needed to determine the mechanisms that regulate mitophagy in renal disease.
Mitochondria and renal diseases
Diseases such as AKI and diabetic nephropathy can cause an imbalance in mitochondrial homeostasis, negatively impacting mitochondrial energetics and the production of ATP. Much research supports a role for mitochondrial dysfunction in a number of renal diseases136. We focus on AKI and diabetic nephropathy as examples of how mitochondrial dysfunction can negatively affect mitochondrial energetics to contribute to disease progression.
Acute kidney injury
The outcome of AKI is renal dysfunction, as indicated by an increase in blood urea nitrogen (BUN) and serum creatinine level, and/or reduced urinary output137. Current treatment for AKI is lacking owing to its complex pathogenesis138,139. Over the past two decades, the incidence of AKI has increased; furthermore, the mortality rate for patients requiring renal replacement therapy is >60%137,140–143. Ultimately, unresolved AKI can cause long-term damage to the kidney, increasing the risk of chronic kidney disease (CKD)144. AKI can be categorized as prerenal, postrenal or intrinsic139, and can result from sepsis, IRI, exposure to nephrotoxic reagents, trauma145 or in response to decreased cardiovascular function146,147. One of the main sites of injury in AKI is the proximal tubules, where injury is characterized by disrupted brush borders and tight junctions, cell sloughing, apoptosis, necrosis and the subsequent backleak of filtrate across injured proximal tubular cells148.
Much research has been conducted on mitochondrial dysfunction as an initiator of and contributor to AKI and as a therapeutic target149. Histologically, mitochondrial swelling and fragmentation are observed after diverse insults to the kidney150. A decrease in ATP production, an increase in ROS production, the release of cytochrome c, and the disruption of mitochondrial cristae are also observed, supporting a role for mitochondria in AKI150. A decrease in ATP production and mitochondrial dysfunction has been documented in many animal models of AKI, including sepsis, and these outcomes result from the loss of mitochondrial respiratory proteins in proximal tubules151–153. Furthermore, the loss of ETC proteins is persistent in the damaged kidney and might contribute to the slow recovery of renal function after AKI151.
A number of factors in the ischaemic kidney disrupt the oxidation and transport of fatty acids, causing an accumulation of fatty acids in the cytoplasm and contributing to the decrease in ATP production and mitochondrial energetics154,18,150,155,156. For example, cofactors, such as NAD+, are necessary for fatty acid oxidation, but a dysfunctional ETC is not able to regenerate NAD+ (REF. 157). IRI also decreases the activity of CPT1 (REFS 18,158), the rate-limiting enzyme in the carnitine shuttle that transports fatty acids from the cytoplasm into the mitochondria158, which decreases the transport of fatty acids into the mitochondria and reduces β-oxidation158.
Increased levels of lactate and pyruvate and of hexokinase activity in the kidney have been reported after IRI, suggesting that an increase in glycolysis occurs after injury159,160. Increased levels of glycolytic enzymes have also been detected in injured renal tubules after IRI161,162, suggesting that the kidney can respond to injury by altering its metabolic substrates to maintain function163. Further studies are needed to explore how this increase in glycolysis affects mitochondrial function in the kidney and if this change in metabolism contributes to long-term recovery following IRI.
Changes in mitochondrial dynamics also contribute to the decrease in mitochondrial energetics following AKI164 (FIG. 6). The translocation of DRP1 into the mitochondrial outer membrane occurs shortly after kidney injury151,165, and activation of DRP1 in ischaemic kidneys promotes mitochondrial fragmentation and apoptosis166. Loss of cristae structure is also observed in AKI, which dissipates the mitochondrial membrane potential and halts ATP production150. Administration of a pharmacological inhibitor of DRP1, mdivi-1, protected kidneys from AKI by inhibiting mitochondrial fragmentation, supporting a role for altered mitochondrial dynamics in AKI165.
Figure 6. Changes in mitochondrial morphology lead to tubular damage in acute kidney injury.
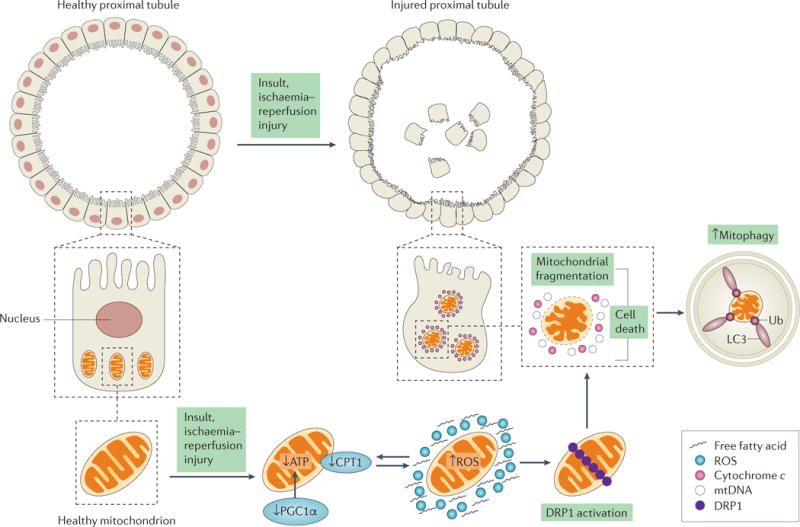
A healthy proximal tubule consists of an intact brush border with tight junctions and contains a network of mitochondria to maintain its function. After ischaemia–reperfusion injury (IRI), changes in mitochondrial function and morphology lead to mitochondrial dysfunction, and eventually to injured proximal tubules. In the early stages of acute kidney injury (AKI), a number of events may happen concurrently to cause a decrease in ATP production. These events include a decrease in the expression of carnitine O-palmitoyltransferase 1 (CPT1) (causing fatty acid accumulation and decreasing β-oxidation for ATP production), a decrease in the expression of peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α) and an increase in the production of reactive oxygen species (ROS) (bidirectional arrows). Together, these events can trigger the activation and accumulation of dynamin 1-like protein (DRP1) on the mitochondrial outer membrane, promoting mitochondrial fragmentation and eventually cell death. The release of cytochrome c and mitochondrial DNA (mtDNA) from dysfunctional mitochondria causes an increase in mitophagy. Mitochondrial dysfunction can induce cell death in injured proximal tubules, resulting in the loss of nuclei and tight junctions and in disrupted brush borders. Apoptotic or necrotic tubules can lead to cell sloughing, as seen in the centre of the tubule.
Mitophagy is also activated after ischaemic AKI. In mice from which the genes encoding the autophagy regulators autophagy-related protein 7 (ATG7) and ATG5 were specifically knocked out in renal proximal tubules, mitochondrial dysfunction was greater in renal proximal tubules in response to IRI, as characterized by severe morphological changes, increased ROS production and apoptosis167–169. Activation of NIX and BNIP3 causes the release of ROS and the pro-apoptotic proteins BAX and BAK, in hypoxic conditions116,170. Deletion of BAX and BAK in mouse kidneys not only protected mice from ischaemic AKI but also suppressed mitochondrial fragmentation and the release of cytochrome c, preserving mitochondrial integrity171. A lack of ATG7 also exacerbated cisplatin-induced AKI in mice134,167. These studies suggest that crosstalk occurs between components of the cell death machinery and the autophagy machinery in the activation of mitophagy.
In mouse models of AKI, the transcription and protein expression of PGC1α are persistently suppressed, but are eventually restored to basal levels with recovery151. As PGC1α can regulate the transcription of mitochondrial proteins, the level of these proteins is also decreased after AKI151,172. In a model of septic AKI, global PPARGC1A-knockout mice showed a greater increase in BUN and creatinine levels than did wild-type mice152. Renal-specific PPARGC1A-knockout mice exhibited persistent AKI in response to sepsis152. By contrast, overexpression of PGC1α in renal proximal tubule cells attenuated oxidant injury in vitro59. Together, these studies show that PGC1α is necessary for the recovery of renal function in AKI.
Investigations into the mechanisms by which PGC1α regulates the recovery from AKI revealed a role for PGC1α in NAD biosynthesis. The levels of nicotinamide, a precursor for NAD, were decreased after AKI in PPARGC1A-knockout mice, and supplementation with nicotinamide reversed ischaemic AKI173. We have reported that drugs or chemicals can upregulate mitochondrial biogenesis by increasing the expression of PGC1α in the recovery phase following IRI through two G-protein coupled receptors (GPCRs): the β2 adrenergic receptor and the 5-hydroxytryptamine 1F receptor174,175 (see below).
The role of SIRT3 in cisplatin-induced AKI has also been explored. SIRT3 is a mitochondrial-specific protein deacetylase with an active role in mitochondrial function and integrity176. An in vitro study using cisplatin-injured human renal proximal tubules showed that the over-expression of SIRT3 decreased the translocation of DRP1 from the cytosol to the mitochondrial outer membrane and thus decreased mitochondrial fission, supporting a role for SIRT3 in regulating mitochondrial dynamics after AKI176. Deletion of SIRT3 exacerbates injury in a cisplatin-induced AKI mouse model, supporting its role in recovery from AKI176.
Diabetic nephropathy
Diabetic nephropathy is the leading cause of end-stage renal disease (ESRD) in the USA177,178. It is characterized by hyperglycaemia, albuminuria, the accumulation of extracellular matrix proteins, and glomerular and tubular epithelial hypertrophy, as well as a reduced glomerular filtration rate following an initial period of hyperfiltration179. Mitochondrial energetics are altered in diabetic nephropathy owing to increased ROS and hyperglycae-mia180, both of which induce changes in the ETC that cause a decrease in ATP production and an increase in apoptosis180. In line with these observations, increased fission, mitochondrial fragmentation and reduced levels of PGC1α are all observed in the early stages of diabetes mellitus181,182. Structural changes in mitochondria correlate with the observed changes in mitochondrial energetics182.
Hyperglycaemia is the main factor that contributes to the development of diabetic nephropathy (FIG. 7). Hyperglycaemia increases the production of NADH and FADH2 by the TCA cycle, fueling the ETC183. ROS released from the ETC can damage mtDNA, hindering the production of mitochondrial proteins183. The hyperglycaemic state was originally thought to cause mitochondrial dysfunction by stimulating the development of hyperpolarized mitochondria, which produce more ATP and release higher levels of superoxide from complexes I and III than healthy mitochondria180,184,185. Administration of antioxidants such as vitamin E and vitamin A did not, however, attenuate the complications of patients with diabetes mellitus, suggesting that mitochondrial ROS might not be the primary mediator of mitochondrial dysfunction in diabetic nephropathy186. Hyperglycaemia can also increase the level of advanced glycation end products (AGEs), and the activity of the protein kinase C (PKC) and hexosamine pathways, which can contribute to mitochondrial dysfunction187. All three events cause deleterious effects that include increased fibrosis, thrombosis, oxidative damage and abnormalities in the vasculature and in blood flow187.
Figure 7. Factors contributing to mitochondrial dysfunction in diabetic nephropathy.
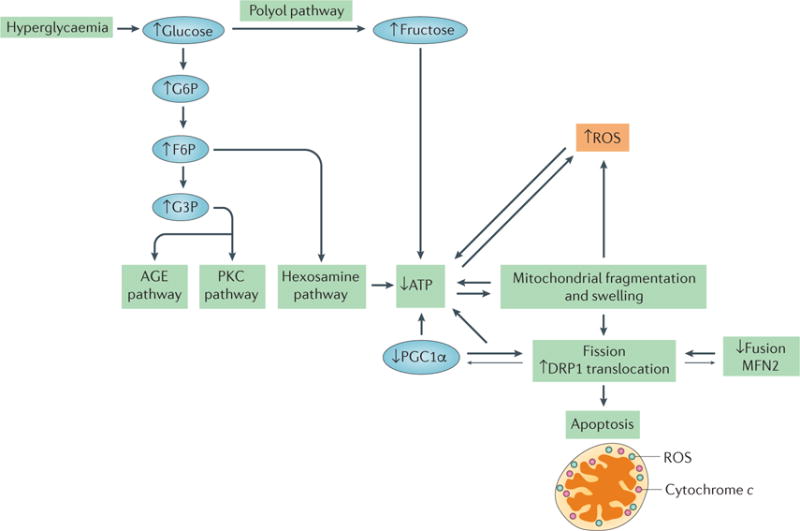
Hyperglycaemia is the primary contributing factor to mitochondrial dysfunction in diabetic nephropathy. An increase in glucose level results in an increase in glycolysis, in turn activating the advanced glycation end product (AGE) pathway, the protein kinase C (PKC) pathway and the hexosamine pathway, which results in a decrease in ATP levels. Hyperglycaemia also activates the polyol pathway, which increases fructose levels and, consequently, decreases ATP levels. Mitochondrial fragmentation and swelling is observed in early diabetic nephropathy, leading to an increase in fission and the production of reactive oxygen species (ROS). The correlations between increased mitochondrial fragmentation and decreased ATP, and between ROS production and decreased ATP, are interdependent. Whether one causes the other is unclear, as depicted by the bidirectional arrows. Decreases in the levels of mitofusin 2 (MFN2) and peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α) correlate with, and might contribute to, the increase in mitochondrial fission observed in diabetic nephropathy, as indicated by the larger arrows pointing towards increased mitochondrial fission. Decreases in mitochondrial energetics that are caused by changes in mitochondrial morphology and hyperglycaemia lead to apoptosis in diabetic nephropathy. F6P, fructose-6-phosphate; G6P, glucose-6-phosphate; G3P, glyceraldehyde-3-phosphate.
Hyperglycaemia also stimulates the conversion of glucose to fructose via the polyol pathway in proximal tubules, leading to ATP depletion188. A role for endogenous fructose metabolism in the regulation of diabetic nephropathy was suggested by a study showing that deleting the gene that encodes ketohexokinase (KHK; also known as hepatic fructokinase) — the enzyme responsible for the conversion of fructose to fructose-1-phosphate — protected mice from streptozotocin-induced diabetic nephropathy189. Proximal tubules are a major site of ketohexokinase expression188,190 and ATP levels were increased and tubular morphology was improved in diabetic Khk−/− mice compared with that of diabetic wild-type mice, suggesting a role for fructose metabolism in the pathogenesis of diabetic nephropathy189.
Mitochondrial fragmentation has been observed in proximal tubules in the early stages of diabetes mellitus181, although the mechanisms that drive changes in mitochondrial dynamics in diabetes are not yet clear. Fission dissipates the mitochondrial membrane potential, decreasing the production of ATP and promoting apoptosis191. Several studies have suggested a role for RHO-associated protein kinase 1 (ROCK1) signalling in activating fission in the diabetic kidney192. ROCK1 promotes the translocation of DRP1 to the mitochondria and triggers fission by phosphorylating DRP1 (REF. 192). Deletion of ROCK1 in mice with streptozotocin-induced diabetes prevents mitochondrial fission, attenuates the increase in ROS production and restores bioenergetic function in the kidney192.
Patients with diabetes mellitus have reduced levels of the fusion protein MFN2193. In line with this finding, kidney-specific overexpression of MFN2 protects rats from streptozotocin-induced diabetic nephropathy193. MFN2 overexpression decreased ROS production, decreased kidney volume and attenuated the pathological changes seen in the diabetic kidney193. Induced in high glucose 1 (IHG1; also known as THG1L) is another protein that is involved in mitochondrial fusion and has been shown to regulate mitochondrial dynamics and biogenesis in the diabetic kidney194. IHG1 can enhance the ability of MFN2 to bind to GTP and interacts directly with MFN2 to mediate fusion194. Inhibition of IHG1 reduces ATP production and hinders fusion in vitro194. IHG1 also stabilizes PGC1α activation195.
Reduced levels of PGC1α have also been observed in diabetic rat kidneys196. The overexpression of PGC1α in mesangial cells in vitro attenuated the pathophysiological changes induced by hyperglycaemic conditions196. The decrease in mitochondrial biogenesis in diabetic rat kidneys is consistent with the translocation of DRP1 to the mitochondrial outer membrane and an increase in mitochondrial fragmentation196. The levels of PGC1α mRNA and protein were also reduced in podocytes that were cultured under hyperglycaemic conditions compared with the levels in podocytes that were cultured under normal glucose conditions, indicating a decrease in mitochondrial biogenesis197.
Another study has described an important role for pyruvate kinase M2 (PKM2) in diabetic nephropathy. The expression and activity of PKM2 is upregulated in patients with long-term diabetes mellitus who have not developed diabetic nephropathy but not in patients with diabetic nephropathy198. Podocytes from PKM2-knockdown mice have decreased PPARGC1A mRNA and mitochondrial mass, whereas activation of PKM2 attenuated the decrease in mitochondrial function and glycolytic flux in podocytes in vitro. In vivo studies showed that activation of PKM2 in mice attenuated the diabetes-induced decrease in PPARGC1A mRNA and increased the expression of OPA1, increasing mitochondrial fusion198. Activation of PKM2 can therefore reverse mitochondrial dysfunction and renal abnormalities associ ated with diabetes mellitus. These studies highlight the need for further research in this area, as targeting the balance between mitochondrial biogenesis and dynamics could be a potential therapeutic approach for diabetic nephropathy.
Mitochondrial energetics and therapy
Targeting AMPK signalling
AMPK signalling has been implicated as a target for correcting metabolism and mitochondrial function, especially in the kidney. As mentioned above, AMPK is a metabolic sensor of ATP in the cell. A high AMP:ATP ratio activates AMPK to stimulate cell growth and cellular metabolism. The AMPK activator 5-aminoimidazole-4-carboxamide-1-β-D-riboside (AICAR), prevents glomerulopathy and tubulointerstitial fibrosis in mice by stimulating fatty acid oxidation199 (TABLE 1). AICAR also has a therapeutic effect in mouse renal IRI and can improve glucose utilization in obese, insulin-resistant rats200,201. The activation of AMPK by AICAR increased the level of PGC1α and mitochondrial proteins while reducing ROS production in a diabetic mouse model202.
Table 1.
Approaches to correct abnormal mitochondrial function in AKI and diabetic nephropathy
| Agent | Mechanism of action | In vivo and clinical studies* |
|---|---|---|
| Acute kidney injury | ||
| AICAR (an AMPK activator) |
|
|
| Formoterol (a β2AR agonist) | Binds to β2AR and induces mitochondrial biogenesis | Formoterol restored mitochondrial and renal function in mice with IRI within 6 days (2014)174 |
| LY344864 (a 5-HT1F receptor agonist) | Binds to 5-HT1F and induces mitochondrial biogenesis | LY344864 restores renal function in mice with IRI within 6 days (2014)175 |
| Elamipretide (a Szeto–Schiller peptide (specifically SS-31)) | Prevents the peroxidation of cardiolipin by cytochrome c |
|
| Diabetic nephropathy | ||
| AICAR (an AMPK activator) | Increases glucose utilization | AICAR decreased blood glucose levels in db/db diabetic mice and ob/ob obese mice (2002)228 |
| Fenofibrate (a PPARα agonist) |
|
|
5-HT1F, 5-hydroxytryptamine receptor 1F; β2AR, β2 adrenergic receptor; AICAR, 5-aminoimidazole-4-carboxamide-1-β-D-riboside; AKI, acute kidney injury; AMPK, AMP-activated protein kinase; BUN, blood urea nitrogen; ETC, electron transport chain; IRI, ischaemia–reperfusion injury; PPARα, peroxisome proliferator-activated receptor-α; SIRT3, sirtuin 3.
The year of the clinical study is given in parentheses.
Several studies have suggested that crosstalk exists between AMPK and SIRT3 signalling203,204. SIRT1 and SIRT3 are activated by NAD+ (REF.205). Cisplatin-treated mice have decreased expression of Sirt3 and lower SIRT3 protein levels, increased tubular damage, and decreased levels of phosphorylated AMPK compared with that of saline-treated control mice206. Administration of AICAR to cisplatin-treated mice attenuated the decrease in SIRT3 expression, phosphorylated AMPK level, and tubular damage206. These studies provide a therapeutic rationale for targeting AMPK signalling in the kidney to improve outcomes in AKI and diabetic nephropathy.
Targeting PPARs
PPARs can regulate cellular metabolism, mitochondrial function, mitochondrial biogenesis, fatty acid oxidation and glucose homeostasis; thus, targeting them could be beneficial for patients with renal disease related to mitochondrial dysfunction.
Activation of PPARs can ameliorate ischaemic AKI207–209. As discussed above, an accumulation of fatty acids and increased ROS production can decrease the efficiency of the ETC. Defects in fatty acid oxidation have been attributed to the downregulation of PPARs during renal ischaemia18. Fenofibrate, which is used to treat dyslipidaemia, activates PPARα210 (TABLE 1). Activation of PPARα leads to activation of lipoprotein lipase, which hydrolyses triglycerides into glycerol and free fatty acids for metabolism210. PPARs can also stimulate mitochondrial biogenesis; for example, compounds such as bardoxolone increase the level of PPARG (encoding PPARγ) and NFE2L2 (encoding NRF2) mRNA, leading to mitochondrial biogenesis211. However, the use of bardoxolone in clinical trials for patients with type 2 diabetes mellitus and stage 4 CKD showed adverse effects in patients, including an increase in the rate of heart failure events, resulting in termination of the trial212.
The efficacy of PPAR agonists in animal models suggests these agents could show promise for the treatment of diabetic nephropathy. Treatment of db/db diabetic mice with fenofibrate led to decreased hyperglycaemia and insulin resistance, potentially by correcting glucose homeostasis213. Studies have also shown that treatment of diabetic mice with fenofibrate leads to a decrease in fatty acids in the kidney, supporting its potential as a therapeutic for diabetic nephropathy214–216. These in vivo studies provide evidence that fenofibrate might be suitable for the treatment of patients with diabetic nephropathy. Indeed, fenofibrate decreased dyslipidaemia and albuminuria in patients with type 2 diabetes mellitus and reduced the risk of further cardiovascular events217. Taken together, these studies confirm that PPARs have a role in diabetic nephropathy and are a therapeutic target.
Targeting G protein-coupled receptors
Although a wide variety of GPCRs are expressed in the kidney, few studies correlate GPCRs with mitochondrial function in the kidney and other organs. We proposed that compounds that target two different GPCRs — β2 adrenergic receptor (β2AR) and 5-hydroxytryptamine receptor 1F (5-HT1F) — can induce mitochondrial bio-genesis, restore mitochondrial function and stimulate the recovery of renal function following IRI. Formoterol, a β2AR agonist used to treat asthma and chronic obstructive pulmonary disease, stimulates mitochondrial biogenesis and the expression of PGC1α in renal proximal tubular cells in mice174. The administration of formoterol in a model of IRI accelerated the recovery of mitochondrial and renal function by 6 days174. LY344864 is a potent 5-HT1F agonist; it induced mitochondrial biogenesis in naive mice and accelerated the recovery of mitochondrial biogenesis and renal function in the same AKI model175. Several GPCR ligands, such as atrasentan, are currently in clinical trials of diabetic nephropathy; however, whether they act by influencing mitochondrial energetics is unknown and requires further research. These studies provide a foundation for pursuing the targeting of GPCRs, particularly β2AR and 5-HT1F, as a treatment for mitochondrial dysfunction in renal diseases.
Using mitochondrial peptides
A 2014 study described a family of peptides, called Szeto–Schiller peptides (SS peptides), which specifically target cytochrome c activity in the ETC, enhancing its efficiency and increasing ‘state 3 respiration’ — that is, ATP production in the presence of excess substrates and ADP218. SS peptides are highly polar, water-soluble tetrapeptides that can cross the blood–brain barrier and specifically target the inner mitochondrial membrane. The SS peptides do not cause mitochondrial depolarization upon entry, making these compounds highly promising for treatment. SS peptides prevent the peroxidation of cardiolipin, a phospholipid that is important for maintaining cristae formation, by cytochrome c218. Cytochrome c binds to and oxidizes cardiolipin, disrupting cristae formation and detaching cytochrome c from the inner mitochondrial membrane219,220. The SS-31 peptide (also known as elamipretide) has been shown, in a variety of animal disease models, especially in AKI, to promote ATP recovery and cristae formation218. Pretreatment of rats with SS-31 in vivo maintained cristae formation and prevented mitochondrial swelling of renal tubular epithelial cells218. Due to the success in animal models, SS-31 is currently in clinical trials for the treatment of impaired renal function221 (TABLE 1).
Conclusions
Mitochondrial homeostasis involves a network of cellular processes that regulate ATP production; the disruption of these processes can result in mitochondrial dysfunction and organ damage. Although much is known about mitophagy and mitochondrial fission, fusion and biogenesis, the precise role of these processes in renal disease remains to be determined. It is clear, however, that mitochondrial dysfunction is common and occurs early in AKI and diabetic nephropathy. Furthermore, the absence of recovery of mitochondrial function after diverse insults might lead to the continued impairment of renal function, leading to CKD. As renal cell repair and the recovery of renal function is dependent on the ability of mitochondria to produce ATP, restoring mitochondrial function might reverse cellular injury and restore renal function, particularly for diseases such as AKI and diabetic nephropathy. Collectively, the available studies corroborate the need to target mitochondrial homeostasis to restore mitochondrial function and stimulate organ repair or prevent further declines in organ function.
Key points.
Mitochondrial homeostasis requires a fine-tuned balance between mitochondrial dynamics and mitochondrial energetics, and ensures the maintenance of properly functioning mitochondria
Mitochondria can adapt to different metabolic conditions via the regulation of mechanistic target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) nutrient sensing pathways, to maintain a healthy population of mitochondria
External stimuli can augment mitochondrial processes, such as mitophagy, fission and fusion, and mitochondrial biogenesis to attenuate irregular levels of ATP production
The disruption of mitochondrial homeostasis in the early stages of acute kidney injury is an important factor that drives tubular injury and persistent renal dysfunction
Hyperglycaemia-induced ATP depletion triggers changes in mitochondrial morphology that lead to the onset of diabetic nephropathy in diabetes mellitus
Correcting abnormal electron transport chain function directly, and/or by targeting the pathways that regulate mitochondrial biogenesis, is likely to improve renal outcomes by restoring mitochondrial function and stimulating organ repair
Glossary
- Carnitine shuttle
Enzymes in the mitochondrial membrane that transport long-chain fatty acids from the cytosol to the mitochondrial matrix by replacing their coA group with carnitine
- Mitochondrial cristae
Folds in the mitochondrial inner membrane that increase the surface area for mitochondrial respiration to take place
- Streptozotocin
A glucosamine-nitrosourea that is used to induce experimental diabetes in animals by specifically targeting and damaging beta cells
- Dyslipidaemia
Abnormalities in lipoprotein metabolism, resulting in elevated or deficient levels of lipids and/or lipoproteins in the body
Footnotes
Author contributions
Both authors researched data for the article, made substantial contributions to discussions of the content, wrote the article and reviewed and/or edited the manuscript before submission.
Competing interests statement
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
DATABASES
Kyoto Encyclopedia of Genes and Genomes (KEGG) database: http://www.genome.jp/kegg/
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Wang ZM, et al. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr. 2010;92:1369–1377. doi: 10.3945/ajcn.2010.29885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pagliarini DJ, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Connor PM. Renal oxygen delivery: matching delivery to metabolic demand. Clin Exp Pharmacol Physiol. 2006;33:961–967. doi: 10.1111/j.1440-1681.2006.04475.x. [DOI] [PubMed] [Google Scholar]
- 4.Soltoff SP. ATP and the regulation of renal cell function. Annu Rev Physiol. 1986;48:9–31. doi: 10.1146/annurev.ph.48.030186.000301. [DOI] [PubMed] [Google Scholar]
- 5.Holechek MJ, et al. Glomerular filtration: an overview. Nephrol Nurs J. 2003;30:285–290. quiz 281–282. [PubMed] [Google Scholar]
- 6.Dimmer KS, Scorrano L. (De)constructing mitochondria: what for? Physiol (Bethesda) 2006;21:233–241. doi: 10.1152/physiol.00010.2006. [DOI] [PubMed] [Google Scholar]
- 7.Lodish H, et al. Molecular Cell Biology. W. H. Freeman and Company; 2000. [Google Scholar]
- 8.Weinberg JM, et al. Anaerobic and aerobic pathways for salvage of proximal tubules from hypoxia-induced mitochondrial injury. Am J Physiol Renal Physiol. 2000;279:F927–F943. doi: 10.1152/ajprenal.2000.279.5.F927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollak MR, Quaggin SE, Hoenig MP, Dworkin LD. The glomerulus: the sphere of influence. Clin J Am Soc Nephrol. 2014;9:1461–1469. doi: 10.2215/CJN.09400913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Fry BC, Layton AT. Modeling glucose metabolism and lactate production in the kidney. Math Biosci. 2017;289:116–129. doi: 10.1016/j.mbs.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med. 2010;27:136–142. doi: 10.1111/j.1464-5491.2009.02894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas SR. Inner medullary lactate production and accumulation: a vasa recta model. Am J Physiol Renal Physiol. 2000;279:F468–F481. doi: 10.1152/ajprenal.2000.279.3.F468. [DOI] [PubMed] [Google Scholar]
- 13.Ross BD, Espinal J, Silva P. Glucose metabolism in renal tubular function. Kidney Int. 1986;29:54–67. doi: 10.1038/ki.1986.8. [DOI] [PubMed] [Google Scholar]
- 14.Scott C. Misconceptions about aerobic and anaerobic energy expenditure. J Int Soc Sports Nutr. 2005;2:32. doi: 10.1186/1550-2783-2-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wirthensohn G, Guder WG. Renal substrate metabolism. Physiol Rev. 1986;66:469–497. doi: 10.1152/physrev.1986.66.2.469. [DOI] [PubMed] [Google Scholar]
- 16.Guder WG, Ross BD. Enzyme distribution along the nephron. Kidney Int. 1984;26:101–111. doi: 10.1038/ki.1984.143. [DOI] [PubMed] [Google Scholar]
- 17.Lewy PR, Quintanilla A, Levin NW, Kessler RH. Renal energy metabolism and sodium reabsorption. Annu Rev Med. 1973;24:365–384. doi: 10.1146/annurev.me.24.020173.002053. [DOI] [PubMed] [Google Scholar]
- 18.Simon N, Hertig A. Alteration of fatty acid oxidation in tubular epithelial cells: from acute kidney injury to renal fibrogenesis. Front Med (Lausanne) 2015;2:52. doi: 10.3389/fmed.2015.00052. This review discusses the mechanisms behind fatty acid transport and oxidation in proximal tubules and how therapeutic agents restore β-oxidation in renal diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwao Y, et al. CD36 is one of important receptors promoting renal tubular injury by advanced oxidation protein products. Am J Physiol Renal Physiol. 2008;295:F1871–F1880. doi: 10.1152/ajprenal.00013.2008. [DOI] [PubMed] [Google Scholar]
- 20.Sabbahy ME, Vaidya VS. Ischemic kidney injury and mechanisms of tissue repair. Wiley Interdiscip Rev Syst Biol Med. 2011;3:606–618. doi: 10.1002/wsbm.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forbes JM. Mitochondria-power players in kidney function? Trends Endocrinol Metab. 2016;27:441–442. doi: 10.1016/j.tem.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Bobulescu IA. Renal lipid metabolism and lipotoxicity. Curr Opin Nephrol Hypertens. 2010;19:393–402. doi: 10.1097/MNH.0b013e32833aa4ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Proctor G, et al. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes. 2006;55:2502–2509. doi: 10.2337/db05-0603. [DOI] [PubMed] [Google Scholar]
- 24.Arici M, Chana R, Lewington A, Brown J, Brunskill NJ. Stimulation of proximal tubular cell apoptosis by albumin-bound fatty acids mediated by peroxisome proliferator activated receptor-γ. J Am Soc Nephrol. 2003;14:17–27. doi: 10.1097/01.asn.0000042167.66685.ea. [DOI] [PubMed] [Google Scholar]
- 25.Ruggiero C, et al. Albumin-bound fatty acids but not albumin itself alter redox balance in tubular epithelial cells and induce a peroxide-mediated redox-sensitive apoptosis. Am J Physiol Renal Physiol. 2014;306:F896–F906. doi: 10.1152/ajprenal.00484.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gutteridge JMC, Halliwell B. Invited review free radicals in disease processes: a compilation of cause and consequence. Free Radic Res Commun. 1993;19:141–158. doi: 10.3109/10715769309111598. [DOI] [PubMed] [Google Scholar]
- 27.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 29.Ruiz S. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic. Kidney Int. 2013;83:1029–1041. doi: 10.1038/ki.2012.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weisiger RA, Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J Biol Chem. 1973;248:4793–4796. [PubMed] [Google Scholar]
- 31.Teruya R, et al. Expression of oxidative stress and antioxidant defense genes in the kidney of inbred mice after intestinal ischemia and reperfusion. Acta Cir Bras. 2013;28:848–855. doi: 10.1590/s0102-86502013001200007. [DOI] [PubMed] [Google Scholar]
- 32.Ribas V, García-Ruiz C, Fernández-Checa JC. Glutathione and mitochondria. Front Pharmacol. 2014;5:151. doi: 10.3389/fphar.2014.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. 2012;2012:26. doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Handy DE, et al. Glutathione peroxidase-1 regulates mitochondrial function to modulate. J Biol Chem. 2009;284:11913–11921. doi: 10.1074/jbc.M900392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell. 2012;151:400–413. doi: 10.1016/j.cell.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005;2:85–93. doi: 10.1016/j.cmet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Brand MD, et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 38.Zhou Y, et al. UCP2 attenuates apoptosis of tubular epithelial cells in renal ischemia/reperfusion injury. Am J Physiol Renal Physiol. 2017 doi: 10.1152/ajprenal.00118.2017. http://dx.doi.org/10.1152/ajprenal.00118.2017. This study suggests a role for UCP2 in restoring tubular function after AKI by reducing tubular cell apoptosis. [DOI] [PubMed]
- 39.Souza BMD, et al. Polymorphisms of the UCP2 gene are associated with glomerular filtration rate in type 2 diabetic patients and with decreased UCP2 gene expression in human kidney. PLoS ONE. 2015;10:e0132938. doi: 10.1371/journal.pone.0132938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 41.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol. 2006;291:F271–F281. doi: 10.1152/ajprenal.00071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 43.Chandel NS, et al. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chandel NS, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 45.Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–666. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- 46.Fantus D, Rogers NM, Grahammer F, Huber TB, Thomson AW. Roles of mTOR complexes in the kidney: implications for renal disease and transplantation. Nat Rev Nephrol. 2016;12:587–609. doi: 10.1038/nrneph.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim Y, Park CW. Adenosine monophosphate-activated protein kinase in diabetic nephropathy. Kidney Res Clin Pract. 2016;35:69–77. doi: 10.1016/j.krcp.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grahammer F, et al. mTORC2 critically regulates renal potassium handling. J Clin Invest. 2016;126:1773–1782. doi: 10.1172/JCI80304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gleason CE, et al. mTORC2 regulates renal tubule sodium uptake by promoting ENaC activity. J Clin Invest. 2015;125:117–128. doi: 10.1172/JCI73935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cunningham JT, et al. mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 51.Grahammer F, et al. mTORC1 maintains renal tubular homeostasis and is essential in response to ischemic stress. Proc Natl Acad Sci USA. 2014;111:E2817–E2826. doi: 10.1073/pnas.1402352111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mihaylova MM, Shaw RJ. The AMP-activated protein kinase (AMPK) signaling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Melser S, et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013;17:719–730. doi: 10.1016/j.cmet.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 56.Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. 2011;1813:1269–1278. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23:459–466. doi: 10.1016/j.tem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Svensson K, Schnyder S, Cardel B, Handschin C. Loss of renal tubular PGC-1α exacerbates diet-induced renal steatosis and age-related urinary sodium excretion in mice. PLoS ONE. 2016;11:e0158716. doi: 10.1371/journal.pone.0158716. This study shows the importance of PGC1α in basic renal physiology, further supporting PGC1α as a therapeutic target for renal diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rasbach KA, Schnellmann RG. PGC-1α over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem Biophys Res Commun. 2007;355:734–739. doi: 10.1016/j.bbrc.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 60.Fan W, Evans R. PPARs and ERRs: molecular mediators of mitochondrial metabolism. Curr Opin Cell Biol. 2015;33:49–54. doi: 10.1016/j.ceb.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol. 2010;72:247–272. doi: 10.1146/annurev-physiol-021909-135917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1α (PGC-1α) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-α and -γ. Identification of novel leucine-rich interaction motif within PGC-1α. J Biol Chem. 2002;277:40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- 64.Whitaker RM, Corum D, Beeson CC, Schnellmann RG. Mitochondrial biogenesis as a pharmacological target: A new approach to acute and chronic diseases. Annu Rev Pharmacol Toxicol. 2016;56:229–249. doi: 10.1146/annurev-pharmtox-010715-103155. [DOI] [PubMed] [Google Scholar]
- 65.Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. 2011;93:884S–890S. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cameron RB, Beeson CC, Schnellmann RG. Development of therapeutics that induce mitochondrial biogenesis for the treatment of acute and chronic degenerative diseases. J Med Chem. 2016;59:10411–10434. doi: 10.1021/acs.jmedchem.6b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015;282:647–672. doi: 10.1111/febs.13175. [DOI] [PubMed] [Google Scholar]
- 68.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Palikaras K, Tavernarakis N. Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol. 2014;56:182–188. doi: 10.1016/j.exger.2014.01.021. [DOI] [PubMed] [Google Scholar]
- 70.Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol. 2005;37:822–834. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 71.Ahn BH, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kong X, et al. Sirtuin 3, a new target of PGC-1α, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE. 2010 doi: 10.1371/journal.pone.0011707. http://dx.doi.org/10.1371/journal.pone.0011707. [DOI] [PMC free article] [PubMed]
- 73.Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. Proc Natl Acad Sci USA. 2003;100:7111–7116. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nisoli E, et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci USA. 2004;101:16507–16512. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nisoli E, et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 76.Whitaker RM, Wills LP, Stallons LJ, Schnellmann R. G cGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J Pharmacol Exp Ther. 2013;347:626–634. doi: 10.1124/jpet.113.208017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuven Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 78.Alexander C, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 79.Delettre C, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 80.Delettre C, Lenaers G, Pelloquin L, Belenguer P, Hamel CP. OPA1 (Kjer type) dominant optic atrophy: a novel mitochondrial disease. Mol Genet Metab. 2002;75:97–107. doi: 10.1006/mgme.2001.3278. [DOI] [PubMed] [Google Scholar]
- 81.Labbe K, Murley A, Nunnari J. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol. 2014;30:357–391. doi: 10.1146/annurev-cellbio-101011-155756. [DOI] [PubMed] [Google Scholar]
- 82.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 83.Waterham HR, et al. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- 84.Rossignol R, et al. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- 85.Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19:630–641. doi: 10.1016/j.cmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu B. Mitochondrial dynamics and neurodegeneration. Curr Neurol Neurosci Rep. 2009;9:212–219. doi: 10.1007/s11910-009-0032-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Song M, Dorn GW. II. Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 2015;21:195–205. doi: 10.1016/j.cmet.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ziegler DV, Wiley CD, Velarde MC. Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell. 2015;14:1–7. doi: 10.1111/acel.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yoon YS, et al. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J Cell Physiol. 2006;209:468–480. doi: 10.1002/jcp.20753. [DOI] [PubMed] [Google Scholar]
- 90.Romanello V, Sandri M. Mitochondrial quality control and muscle mass maintenance. Front Physiol. 2015 doi: 10.3389/fphys.2015.00422. http://dx.doi.org/10.3389/fphys.2015.00422. [DOI] [PMC free article] [PubMed]
- 91.Mourier A, et al. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J Cell Biol. 2015;208:429–442. doi: 10.1083/jcb.201411100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shutt T, Geoffrion M, Milne R, McBride HM. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012;13:909–915. doi: 10.1038/embor.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Anand R, et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. 2014;204:919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frezza C, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 96.Boissan M, et al. Nucleoside diphosphate kinases fuel dynamin superfamily proteins with GTP for membrane remodeling. Science. 2014;344:1510–1515. doi: 10.1126/science.1253768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212:379–387. doi: 10.1083/jcb.201511036. This review summarizes recent studies and mechanisms that relate metabolism and mitochondrial energetics to mitochondrial dynamics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mears JA, et al. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol. 2011;18:20–26. doi: 10.1038/nsmb.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256–1268. doi: 10.1016/j.bbamcr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 102.Loson OC, et al. The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure. 2014;22:367–377. doi: 10.1016/j.str.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Richter V, et al. Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J Cell Biol. 2014;204:477–486. doi: 10.1083/jcb.201311014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013 doi: 10.1101/cshperspect.a011072. http://dx.doi.org/10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed]
- 105.Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann NY Acad Sci. 2010;1201:34–39. doi: 10.1111/j.1749-6632.2010.05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282:21583–21587. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 107.Slupe AM, et al. A calcineurin docking motif (LXVP) in dynamin-related protein 1 contributes to mitochondrial fragmentation and ischemic neuronal injury. J Biol Chem. 2013;288:12353–12365. doi: 10.1074/jbc.M113.459677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cereghetti GM, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eiyama A, Okamoto K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol. 2015;33:95–101. doi: 10.1016/j.ceb.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 110.Greene AW, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Matsuda N, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Narendra DP, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Okatsu K, et al. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol. 2015;209:111–128. doi: 10.1083/jcb.201410050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vives-Bauza C, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tanaka A, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe. 2014;15:403–411. doi: 10.1016/j.chom.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sarraf SA, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chan NC, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Groenewoud MJ, Zwartkruis FJ. Rheb and mammalian target of rapamycin in mitochondrial homoeostasis. Open Biol. 2013;3:130185. doi: 10.1098/rsob.130185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Toyama EQ, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang CS, Lin S-C. AMPK promotes autophagy by facilitating mitochondrial fission. Cell Metab. 2016;23:399–401. doi: 10.1016/j.cmet.2016.02.017. This study suggests a direct role for AMPK in mitophagy by phosphorylating MFF, a mitophagy receptor on the outer mitochondrial membrane, to initiate fission and therefore mitophagy. [DOI] [PubMed] [Google Scholar]
- 124.Chen G, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54:362–377. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 125.Liu L, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 126.Novak I, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thomas RL, Kubli DA, Gustafsson AB. Bnip3-mediated defects in oxidative phosphorylation promote mitophagy. Autophagy. 2011;7:775–777. doi: 10.4161/auto.7.7.15536. [DOI] [PubMed] [Google Scholar]
- 129.Hanna RA, et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kanki T. Nix, a receptor protein for mitophagy in mammals. Autophagy. 2010;6:433–435. doi: 10.4161/auto.6.3.11420. [DOI] [PubMed] [Google Scholar]
- 131.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 132.Li Y, et al. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem. 2007;282:35803–35813. doi: 10.1074/jbc.M705231200. [DOI] [PubMed] [Google Scholar]
- 133.Maiuri MC, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ishihara M, et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol. 2013;305:F495–F509. doi: 10.1152/ajprenal.00642.2012. [DOI] [PubMed] [Google Scholar]
- 135.Tang C, He L, Liu J, Dong Z. Mitophagy: Basic Mechanism and Potential Role in Kidney Diseases. Kidney Diseases. 2015;1:71–79. doi: 10.1159/000381510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol - Renal Physiol. 2014;306:F367–F378. doi: 10.1152/ajprenal.00571.2013. [DOI] [PubMed] [Google Scholar]
- 137.Yang Y, et al. Renoprotective approaches and strategies in acute kidney injury. Pharmacol Ther. 2016;163:58–73. doi: 10.1016/j.pharmthera.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shusterman N, et al. Risk factors and outcome of hospital-acquired acute renal failure. Clinical epidemiologic study. Am J Med. 1987;83:65–71. doi: 10.1016/0002-9343(87)90498-0. [DOI] [PubMed] [Google Scholar]
- 139.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334:1448–1460. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 140.Kelly KJ, Molitoris BA. Acute renal failure in the new millennium: time to consider combination therapy. Semin Nephrol. 2000;20:4–19. [PubMed] [Google Scholar]
- 141.Murugan R, Kellum JA. Acute kidney injury: what’s the prognosis? Nat Rev Nephrol. 2011;7:209–217. doi: 10.1038/nrneph.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Waikar SS, Liu KD, Chertow GM. Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol. 2008;3:844–861. doi: 10.2215/CJN.05191107. [DOI] [PubMed] [Google Scholar]
- 143.Doyle JF, Forni LG. Acute kidney injury: short-term and long-term effects. Crit Care. 2016;20:188. doi: 10.1186/s13054-016-1353-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hsu C, Liu KD. Cardiovascular events after AKI: a new dimension. J Am Soc Nephrol. 2014;25:425–427. doi: 10.1681/ASN.2013121276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Selewski DT, Symons JM. Acute kidney injury. Pediatr Rev. 2014;35:30–41. doi: 10.1542/pir.35-1-30. [DOI] [PubMed] [Google Scholar]
- 146.Paraskevas KI, Mikhailidis DP. Contrast-induced acute kidney injury in patients undergoing carotid artery stenting: an underestimated issue. Angiology. 2016 doi: 10.1177/0003319716668934. http://dx.doi.org/10.1177/0003319716668934. [DOI] [PubMed]
- 147.Schefold JC, Filippatos G, Hasenfuss G, Anker SD, von Haehling S. Heart failure and kidney dysfunction: epidemiology, mechanisms and management. Nat Rev Nephrol. 2016;12:610–623. doi: 10.1038/nrneph.2016.113. [DOI] [PubMed] [Google Scholar]
- 148.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol. 2012;2:1303–1353. doi: 10.1002/cphy.c110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ishimoto Y, Inagi R. Mitochondria: a therapeutic target in acute kidney injury. Nephrol Dial Transplant. 2016;31:1062–1069. doi: 10.1093/ndt/gfv317. [DOI] [PubMed] [Google Scholar]
- 150.Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol. 2016;12:267–280. doi: 10.1038/nrneph.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Funk JA, Schnellmann RG. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol. 2012;302:F853–F864. doi: 10.1152/ajprenal.00035.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tran M, et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121:4003–4014. doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Parikh SM. Therapeutic targeting of the mitochondrial dysfunction in septic acute kidney injury. Curr Opin Crit Care. 2013;19:554–559. doi: 10.1097/MCC.0000000000000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ruidera E, et al. Fatty acid metabolism in renal ischemia. Lipids. 1988;23:882–884. doi: 10.1007/BF02536209. [DOI] [PubMed] [Google Scholar]
- 155.Johnson AC, Stahl A, Zager RA. Triglyceride accumulation in injured renal tubular cells: alterations in both synthetic and catabolic pathways. Kidney Int. 2005;67:2196–2209. doi: 10.1111/j.1523-1755.2005.00325.x. [DOI] [PubMed] [Google Scholar]
- 156.Zager RA, Johnson AC, Hanson SY. Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int. 2005;67:111–121. doi: 10.1111/j.1523-1755.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- 157.Portilla D. Role of fatty acid beta-oxidation and calcium-independent phospholipase A2 in ischemic acute renal failure. Curr Opin Nephrol Hypertens. 1999;8:473–477. doi: 10.1097/00041552-199907000-00012. [DOI] [PubMed] [Google Scholar]
- 158.Idrovo JP, Yang WL, Nicastro J, Coppa GF, Wang P. Stimulation of carnitine palmitoyltransferase 1 improves renal function and attenuates tissue damage after ischemia/reperfusion. J Surg Res. 2012;177:157–164. doi: 10.1016/j.jss.2012.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Smith JA, Stallons LJ, Schnellmann RG. Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am J Physiol Renal Physiol. 2014;307:F435–F444. doi: 10.1152/ajprenal.00271.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zager RA, Johnson AC, Becker K. Renal cortical pyruvate depletion during AKI. J Am Soc Nephrol. 2014;25:998–1012. doi: 10.1681/ASN.2013070791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lan R, et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol. 2016;27:3356–3367. doi: 10.1681/ASN.2015020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol. 2015;26:1765–1776. doi: 10.1681/ASN.2015010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Eklund T, Wahlberg J, Ungerstedt U, Hillered L. Interstitial lactate, inosine and hypoxanthine in rat kidney during normothermic ischaemia and recirculation. Acta Physiol Scand. 1991;143:279–286. doi: 10.1111/j.1748-1716.1991.tb09233.x. [DOI] [PubMed] [Google Scholar]
- 164.Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83:568–581. doi: 10.1038/ki.2012.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cho SG, Du Q, Huang S, Dong Z. Drp1 dephosphorylation in ATP depletion-induced mitochondrial injury and tubular cell apoptosis. Am J Physiol Renal Physiol. 2010;299:F199–F206. doi: 10.1152/ajprenal.00716.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jiang M, et al. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–1283. doi: 10.1038/ki.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liu S, et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 2012;8:826–837. doi: 10.4161/auto.19419. [DOI] [PubMed] [Google Scholar]
- 169.Kimura T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22:902–913. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Duann P, Lianos EA, Ma J, Lin PH. Autophagy, innate immunity and tissue repair in acute kidney injury. Int J Mol Sci. 2016;17:662. doi: 10.3390/ijms17050662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wei Q, Dong G, Chen JK, Ramesh G, Dong Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013;84:138–148. doi: 10.1038/ki.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Stallons LJ, Whitaker RM, Schnellmann RG. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol Lett. 2014;224:326–332. doi: 10.1016/j.toxlet.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tran MT, et al. PGC1α-dependent NAD biosynthesis links oxidative metabolism to renal protection. Nature. 2016;531:528–532. doi: 10.1038/nature17184. This investigation shows the importance of NAD biosynthesis in the recovery phase of AKI and of PGC1α as an important regulator of NAD biosynthesis, highlighting this pathway as a therapeutic target for AKI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jesinkey SR, et al. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol. 2014;25:1157–1162. doi: 10.1681/ASN.2013090952. This study provides the first proof of principle that stimulation of mitochondrial biogenesis after AKI can restore mitochondrial function and renal function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Garrett SM, Whitaker RM, Beeson CC, Schnellmann RG. Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J Pharmacol Exp Ther. 2014;350:257–264. doi: 10.1124/jpet.114.214700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Perico L, Morigi M, Benigni A. Mitochondrial sirtuin 3 and renal diseases. Nephron. 2016;134:14–19. doi: 10.1159/000444370. [DOI] [PubMed] [Google Scholar]
- 177.Maahs DM, Rewers M. Mortality and renal disease in type 1 diabetes mellitus—progress made, more to be done. J Clin Endocrinol Metab. 2006;91:3757–3759. doi: 10.1210/jc.2006-1730. [DOI] [PubMed] [Google Scholar]
- 178.Collins AJ, et al. US Renal Data System 2011 annual data report. Am J Kidney Dis. 2012;59:A7. doi: 10.1053/j.ajkd.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 179.Miranda-Diaz AG, Pazarin-Villasenor L, Yanowsky-Escatell FG, Andrade-Sierra J. Oxidative stress in diabetic nephropathy with early chronic kidney disease. J Diabetes Res. 2016;2016:7047238. doi: 10.1155/2016/7047238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Flemming NB, Gallo LA, Ward MS, Forbes JM. Tapping into mitochondria to find novel targets for diabetes complications. Curr Drug Targets. 2016;17:1341–1349. doi: 10.2174/1389450116666150727114410. This review summarizes the role of mitochondrial ROS production in diabetes, which is a controversial contributor to the development and progression of diabetes. [DOI] [PubMed] [Google Scholar]
- 181.Coughlan MT, et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin Sci (Lond) 2016;130:711–720. doi: 10.1042/CS20150838. This study showed that diabetes-induced changes in mitochondrial morphology and energetics occur prior to renal lesions, suggesting that mitochondrial dysfunction is a primary cause of diabetes rather than a contributor. [DOI] [PubMed] [Google Scholar]
- 182.Higgins GC, Coughlan MT. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol. 2014;171:1917–1942. doi: 10.1111/bph.12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Coughlan MT, Sharma K. Challenging the dogma of mitochondrial reactive oxygen species overproduction in diabetic kidney disease. Kidney Int. 2016;90:272–279. doi: 10.1016/j.kint.2016.02.043. [DOI] [PubMed] [Google Scholar]
- 184.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 186.Lonn E, et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293:1338–1347. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 187.Hallan S, Sharma K. The role of mitochondria in diabetic kidney disease. Curr Diab Rep. 2016;16:61. doi: 10.1007/s11892-016-0748-0. [DOI] [PubMed] [Google Scholar]
- 188.Burch HB, et al. Metabolic effects of large fructose loads in different parts of the rat nephron. J Biol Chem. 1980;255:8239–8244. [PubMed] [Google Scholar]
- 189.Lanaspa MA, et al. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J Am Soc Nephrol. 2014;25:2526–2538. doi: 10.1681/ASN.2013080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Diggle CP, et al. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem. 2009;57:763–774. doi: 10.1369/jhc.2009.953190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 192.Wang W, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15:186–200. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tang WX, Wu WH, Zeng XX, Bo H, Huang SM. Early protective effect of mitofusion 2 overexpression in STZ-induced diabetic rat kidney. Endocr. 2012;41:236–247. doi: 10.1007/s12020-011-9555-1. [DOI] [PubMed] [Google Scholar]
- 194.Hickey FB, et al. IHG-1 increases mitochondrial fusion and bioenergetic function. Diabetes. 2014;63:4314–4325. doi: 10.2337/db13-1256. [DOI] [PubMed] [Google Scholar]
- 195.Hickey FB, et al. IHG-1 promotes mitochondrial biogenesis by stabilizing PGC-1α. J Am Soc Nephrol. 2011;22:1475–1485. doi: 10.1681/ASN.2010111154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Guo K, et al. Protective role of PGC-1α in diabetic nephropathy is associated with the inhibition of ROS through mitochondrial dynamic remodeling. PLoS ONE. 2015;10:e0125176. doi: 10.1371/journal.pone.0125176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Imasawa T, et al. High glucose repatterns human podocyte energy metabolism during differentiation and diabetic nephropathy. FASEB J. 2017;31:294–307. doi: 10.1096/fj.201600293R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Qi W, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. 2017;23:753–762. doi: 10.1038/nm.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Szeto HH, et al. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016;90:997–1011. doi: 10.1016/j.kint.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 200.Lempiainen J, Finckenberg P, Levijoki J, Mervaala E. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br J Pharmacol. 2012;166:1905–1915. doi: 10.1111/j.1476-5381.2012.01895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123:2764–2772. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dugan LL, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123:4888–4899. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pillai VB, et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. 2010;285:3133–3144. doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Palacios OM, et al. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1α in skeletal muscle. Aging (Albany NY) 2009;1:771–783. doi: 10.18632/aging.100075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Nogueiras R, et al. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev. 2012;92:1479–1514. doi: 10.1152/physrev.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Morigi M, et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. 2015;125:715–726. doi: 10.1172/JCI77632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Singh JP, Singh AP, Bhatti R. Explicit role of peroxisome proliferator-activated receptor γ in gallic acid-mediated protection against ischemia-reperfusion-induced acute kidney injury in rats. J Surg Res. 2014;187:631–639. doi: 10.1016/j.jss.2013.11.1088. [DOI] [PubMed] [Google Scholar]
- 208.Chung BH, et al. Protective effect of peroxisome proliferator activated receptor γ agonists on diabetic and non-diabetic renal diseases. Nephrol (Carlton, Vic) 2005;10(Suppl):S40–S43. doi: 10.1111/j.1440-1797.2005.00456.x. [DOI] [PubMed] [Google Scholar]
- 209.Sivarajah A, et al. Agonists of peroxisome-proliferator activated receptor-γ reduce renal ischemia/reperfusion injury. Am J Nephrol. 2003;23:267–276. doi: 10.1159/000072088. [DOI] [PubMed] [Google Scholar]
- 210.Staels B, et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98:2088–2093. doi: 10.1161/01.cir.98.19.2088. [DOI] [PubMed] [Google Scholar]
- 211.Wu QQ, et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARγ, and HO-1. Am J Physiol Renal Physiol. 2011;300:F1180–F1192. doi: 10.1152/ajprenal.00353.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.de Zeeuw D, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369:2492–2503. doi: 10.1056/NEJMoa1306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Park CW, et al. PPARα agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. 2006;69:1511–1517. doi: 10.1038/sj.ki.5000209. [DOI] [PubMed] [Google Scholar]
- 214.Stadler K, Goldberg IJ, Susztak K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr Diabetes Rep. 2015;15:40. doi: 10.1007/s11892-015-0611-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Al-Rasheed NM, et al. Fenofibrate attenuates diabetic nephropathy in experimental diabetic rat’s model via suppression of augmented TGF-β1/Smad3 signaling pathway. Arch Physiol Biochem. 2016;122:186–194. doi: 10.3109/13813455.2016.1164186. [DOI] [PubMed] [Google Scholar]
- 216.Hong YA, et al. Fenofibrate improves renal lipotoxicity through activation of AMPK-PGC-1α in db/db mice. PLoS ONE. 2014;9:e96147. doi: 10.1371/journal.pone.0096147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kawanami D, Matoba K, Utsunomiya K. Dyslipidemia in diabetic nephropathy. Ren Replace Ther. 2016;2:16. [Google Scholar]
- 218.Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther. 2014;96:672–683. doi: 10.1038/clpt.2014.174. The Szeto peptides described in this study are novel, as they prevent the peroxidation of cardiolipin and therefore preserve mitochondrial function, demonstrating that they are renoprotective. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hanske J, et al. Conformational properties of cardiolipin-bound cytochrome c. Proc Natl Acad Sci USA. 2012;109:125–130. doi: 10.1073/pnas.1112312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Basova LV, et al. Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry. 2007;46:3423–3434. doi: 10.1021/bi061854k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.US National Library of Medicine. ClinicalTrials.gov. 2015 https://clinicaltrials.gov/ct2/show/NCT02436447.
- 222.Sedeek M, Nasrallah R, Touyz RM, Hebert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol. 2013;24:1512–1518. doi: 10.1681/ASN.2012111112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bai J, Cederbaum AI. Mitochondrial catalase and oxidative injury. Biol Signals Recept. 2001;10:189–199. doi: 10.1159/000046887. [DOI] [PubMed] [Google Scholar]
- 224.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 225.Kaufman BA, et al. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Biol Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci USA. 1994;91:1309–1313. doi: 10.1073/pnas.91.4.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wu Z, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 228.Halseth AE, Ensor NJ, White TA, Ross SA, Gulve EA. Acute and chronic treatment of ob/ob and db/db mice with AICAR decreases blood glucose concentrations. Biochem Biophys Res Commun. 2002;294:798–805. doi: 10.1016/S0006-291X(02)00557-0. [DOI] [PubMed] [Google Scholar]
- 229.Keech A, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–1861. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]