

Abstract
Nicotinamide adenine dinucleotide (NAD+) and related metabolites are central mediators of fuel oxidation and bioenergetics within cardiomyocytes. Additionally, NAD+ is required for the activity of multifunctional enzymes, including sirtuins and poly(ADP-ribose) polymerases that regulate posttranslational modifications, DNA damage responses, and Ca2+ signaling. Recent research has indicated that NAD+ participates in a multitude of processes dysregulated in cardiovascular diseases. Therefore, supplementation of NAD+ precursors, including nicotinamide riboside that boosts or repletes the NAD+ metabolome, may be cardioprotective. This review examines the molecular physiology and preclinical data with respect to NAD+ precursors in heart failure-related cardiac remodeling, ischemic-reperfusion injury, and arrhythmias. In addition, alternative NAD+-boosting strategies and potential systemic effects of NAD+ supplementation with implications on cardiovascular health and disease are surveyed.
Keywords: cardiovascular diseases, ischemia-reperfusion, nicotinamide adenine dinucleotide, oxidation-reduction (redox)
INTRODUCTION
Nicotinamide adenine dinucleotide (NAD+) is the central regulator of metabolism (8). In fuel oxidation, NAD+ accepts hydride groups to form the reduced cofactor NADH. NADH is reoxidized at the inner mitochondrial membrane to drive aerobic production of ATP in the process of oxidative phosphorylation (OXPHOS) (Fig. 1). Within normal cardiac physiological conditions, OXPHOS is responsible for ~95% of the ATP generated, with fatty acids as the predominant substrate (103). Additionally, NADP+, the phosphorylated form of NAD+, is required in the pentose phosphate pathway for generation of NADPH and ribose 5-phosphate. NADPH, the reduced form of NADP+, provides primary protection against reactive oxygen species (ROS) damage and serves as the hydride donor for lipid and steroid hormone biosynthesis. In addition to the classical roles for NAD+ coenzymes in redox biochemistry, NAD+ can be consumed in important regulatory reactions. This review will examine the emerging use of NAD+ precursors in the prevention of cardiovascular disease with a focus on the inhibition of adverse cardiac remodeling in heart failure, attenuation of ischemia-reperfusion (I/R) injury, and mitigation of arrhythmic susceptibility. The implications of NAD+ metabolism in vascular biology and dysfunction will not be covered, since they have been reviewed elsewhere (87, 101). However, the effects of increasing NAD+ in other organ systems that have implications in cardiac health are examined.
Fig. 1.

Nicotinamide adenine dinucleotide (NAD+) in cardiac metabolism. NAD+ is used in fuel metabolism within the heart. The predominant substrate for energy production in the cardiomyocyte is fatty acids. Through fatty acid β-oxidation (FAO), NAD+ is reduced to NADH. NADH is used and oxidized to NAD+ in oxidative phosphorylation to produce ATP. Within conditions of mitochondrial dysfunction and ischemia, the heart relies on carbohydrates and glycolysis for the generation of ATP. Glycolysis reduces NAD+ to NADH. Subsequently, pyruvate is converted to lactate by lactate dehydrogenase to aid in the regeneration of the oxidized NAD+. Glucose 6-phosphate from the glycolytic pathway can be shunted to the pentose phosphate shuttle where the phosphorylated form of NAD+ (NADP+) is reduced to NADPH. Acetyl-CoA can be generated from pyruvate, ketone bodies, and amino acids to participate in the tricarboxylic acid (TCA) cycle. The generation of α-ketoglutarate (α-KG) from amino acids can also contribute in energy production within the TCA cycle. Metabolic reprogramming in failing hearts away from FAO and toward glycolytic and ketone body oxidation is predicted to decrease the NAD+-to-NADH ratio, having major implications in cellular processes. Furthermore, decreasing NAD+ content by downregulation of biosynthesis or upregulation of consumption may limit metabolic processes.
Biosynthesis and Metabolism of NAD+
NAD+ content is regulated by its synthesis and consumption. Depending on which biosynthetic pathways are expressed in a particular tissue, synthesis of NAD+ can be initiated from tryptophan in the de novo pathway or from the following three salvageable precursor vitamins: nicotinic acid (NA), nicotinamide (NAM), or nicotinamide riboside (NR) (Fig. 2) (12). The de novo synthesis pathway of NAD+ primarily occurs in the liver and relies on the kynurenine metabolic pathway. Interestingly, the biosynthetic rate of the de novo pathway at which tryptophan is converted to NAD+ appears to be independent of the presence of NAM and activity of the salvage pathway (29). NAM and NR are converted to nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (NAMPT) and nicotinamide riboside kinase (NMRK) isozymes, respectively (10). Alternatively, NAD+ is synthesized from NA via the Preiss-Handler pathway through nicotinic acid mononucleotide and nicotinic acid adenine dinucleotide. The ability of NA, NAM, and NR to stimulate NAD+ biosynthesis varies (19, 93). Recent animal studies have indicated that equimolar oral NR is superior to NA and NAM in elevating NAD+ content in the liver (93).
Fig. 2.

Biosynthesis and consumption of NAD+. NAD+ is produced from the following 4 precursors: tryptophan, nicotinic acid (NA), nicotinamide riboside (NR), and nicotinamide (NAM). Through the de novo pathway, tryptophan is converted to quinolate through the kynurenine pathway. Quinolate is converted to nicotinic acid mononucleotide (NAMN) by quinolate phosphoribosyltransferase (QPRT). In addition, nicotinic acid is converted to NAMN by nicotinic acid phosphotransferase (NAPRT1). NAMN is converted to nicotinic acid adenine dinucleotide (NAAD) by nicotinamide mononucleotide adenylate transferase. Nicotinamide riboside and nicotinamide are converted to nicotinamide mononucleotide (NMN) by nicotinamide riboside kinases (NMRKs) and nicotinamide phosphotransferase (NAMPT), respectively. In addition, NR can be degraded to NAM by purine nucleoside phosphorylase (PNP). NMN is converted to NAD+ by NMN adenlyltransferase (NMNAT). NAD+ can be consumed by the following 3 classes of enzymes: sirtuins, poly(ADP-ribose) polymerases, and cADP-ribose synthetases. A byproduct of consumption is NAM, which can be recycled back to produce NAD+.
In addition to biosynthesis, the uptake of NAD+ precursors is important in its regulation. NAD+ has reported to enter cells through connexin (Cx)43 hemichannels (11, 13, 76). However, more recent data are more consistent, with Cx43-mediated NAD+ export and cADP-ribose (cADPR) import (86). Given the expression of Cx43 hemichannels within cardiomyocytes, NAD+ export and cADPR import through this mode may be more important in the heart than other organ systems.
Because of bioavailability, preclinical studies have used NMN, NR, or NAM to elevate intracellular NAD+. These metabolites enter the cell as either NR or NAM (84). NAD+ and NMN are nucleotides that are converted to NR extracellularly by CD73 (32). Upon entry in the cell, NR is then phosphorylated to NMN by NMRK isozymes NMRK1 and NMRK2 within the cytoplasm (28, 84). NMN, through an ATP-dependent reaction, is converted to NAD+ by NMN adenlyltransferase. It has been estimated that >80% of cellular NAD+ in cardiomyocytes is found in mitochondria (3, 24). This enrichment is explained by the high density of mitochondria within cardiomyocytes and the high energy demand of the heart.
Biosynthesis of NAD+ is counteracted by NAD+ consumption and degradation. The following three classes of enzymes consume NAD+: sirtuins (SIRTs), poly(ADP-ribose) polymerases (PARPs), and cADPR synthetases (8). SIRTs, the mammalian orthologs of the yeast silent information regulator 2, are a family of NAD+-dependent deacylases that remove acetyl and other acyl groups from protein lysine residues. In so doing, SIRTs consume NAD+ and produce the nonmodified protein substrate plus NAM and an acylated ADP-ribose (ADPR) product. PARPs and other ADP-ribosyl transferases modify target proteins by the addition of an ADP-ribosyl group on glutamic acid, aspartic acid, and lysine residues. PARPs have been shown to alter protein-protein and protein-RNA interactions, modulate protein localization, and signal for ubiquitination (31). Finally, cADPR synthetases, including the ectoenzymes CD38 and CD157, use NAD+ to produce ADPR, cADPR, and nicotinic acid adenine dinucleotide phosphate (NAADP). cADPR and NAADP are important second messengers in Ca2+ mobilization and excitation-contraction coupling (36, 44, 47).
NA and NAM were first used as a therapy and for prevention of pellagra, a nutritional deficiency. Characterized by dermatitis, diarrhea, dementia, and potentially death, pellagra was first described in the 18th century by the Spanish physician Gasper Casal in a poor farming population with a corn-based diet lacking fresh meat (82). At high doses, NA, also known as niacin, increases high-density lipoprotein-cholesterol while decreasing triglycerides and low-density lipoprotein-cholesterol. However, use of NA as a therapy for dyslipidemia is limited by its high rate of side effects, including painful flushing of the face and chest, evoked by the release of PGD2 (39). NAM does not have the same beneficial effect as NA on lipid management (69). Over the past two decades, additional therapeutic applications of NAD+-related metabolites have been discovered based on our further understanding of redox biology and NAD+ signaling. Significantly, NR has been discovered as a third vitamin precursor of NAD+ that does not cause flushing and that functions as a SIRT-activating compound (9, 10). NR has shown to be beneficial in resisting weight gain and improving circulating and hepatic lipids in high-fat diet-fed animal models (14, 94). Now produced under Good Manufacturing Practice conditions, NR is safe for human use as an orally available agent that boosts the NAD+ metabolome (93).
Current research has shown that NAD+ supplementation using NAD+ precursors increases or protects against diminishing NAD+ content within the heart in various in vivo models (67, 76, 93, 107). Furthermore, in pathological models of cardiac dysfunction, NAD+ supplementation normalizes the redox NAD+-to-NADH ratio, which has downstream effects on cellular processes. However, much remains unknown about how NAD+ supplementation influences various NAD+ pools and associated metabolites in pathological conditions. The effects of NR and other NAD+ precursors have only started to be realized within cardiac health and disease (68).
Emerging benefits of NAD+ in the pathological cardiac remodeling of heart failure.
Oxidative stress and ROS are common mediators of pathological stimuli leading to cardiac remodeling. ROS activate a multitude of transcription factors and kinases to promote prohypertrophic signaling pathways that stimulate extracellular matrix remodeling and myocyte growth (90, 95). The initial study (20) examining the beneficial effects of NAD+ in adverse cardiac remodeling was in a rat model of chronic volume overload by intrarenal arteriovenous fistula. In this model, supplementation of NAM in the drinking water resulted in improved cardiac function by decreasing oxidative stress and inhibiting downstream prohypertrophic signaling and matrix metalloproteinases (20). Further studies have shown that NAD+ supplementation of neonatal rat cardiomyocytes prevented phenylephrine-induced ROS production and the hypertrophic response. In mice, peritoneal infusion of NAD+ prevented the hypertrophic response induced by angiotensin II by inhibiting the prohypertrophic Akt1 signaling pathway and supporting the antihypertrophic AMP-activated protein kinase (AMPK) signaling pathway through SIRT3-dependent activation of liver kinase B1 (LKB1) (76). In a mouse model of obesity and type 2 diabetes, hepatic NADPH was depressed, which may allow for the production of systemic ROS. NR prevented multiple sequelae of obesity and diabetes mediated by this systemic ROS and normalized the NAD+ metabolome (94). These studies have demonstrated that NAD+ supplementation has the potential to protect against adverse cardiac remodeling by decreasing oxidative stress and ROS.
Perturbations of the metabolic state because of mitochondrial impairment from ischemia or other causes is central in the development of hypertrophy and heart failure. During development of heart failure, a shift from fatty acid oxidation (FAO) and OXPHOS to other forms of substrate metabolism (glycolysis and ketone oxidation) occurs (5, 50). Within this reprogramming, the NAD+-to-NADH ratio reportedly decreases (50). This change in the oxidative-reductive capacity increases cardiac susceptibility to stress and protein hyperacetylation. This protein hyperacetylation is driven by decreasing NAD+-dependent deacetylation, as seen in mouse models of pressure overload-induced hypertrophy and in patients with heart failure from ischemic or dilated cardiomyopathy (42, 49). NAD+ precursor supplementation with NMN improved cardiac function and prevented adverse remodeling by normalizing the NAD+-to-NADH ratio and attenuating mitochondrial protein hyperacetylation in a mouse model of pressure overload by transverse aortic constriction (49). Additionally, a cardiac-specific mitochondrial complex-1 protein knockout (Ndufs4−/−) mouse model of mitochondrial dysfunction revealed hyperacetylation of proteins within the heart and increased susceptibility to cardiac stress (42). Increasing NAD+ by NMN supplementation in Ndufs4−/− mice prevented hyperacetylation and protected against cardiac dysfunction after transverse aortic constriction. Alternatively, increasing biosynthesis of NAD+ by overexpression of NAMPT resulted in protection against isoproterenol-induced cardiac dysfunction and adverse remodeling in these Ndufs4−/− mice (49).
Supplementation with NR demonstrates similar protective effects to NMN in adverse cardiac remodeling by normalizing the myocardial NAD+-to-NADH ratio in mice with transverse aortic constriction (23). Interestingly, long-term supplementation of NR increased nucleocytoplasmic protein acetylation by stimulating acetyl-CoA metabolism through mitochondrial citrate synthase and cytosolic ATP citrate lyase (23). Of note, current analyses that rely on Western blot quantification of steady-state protein acetylation levels do not adequately account for the dynamic rates of acetylation and deacetylation reactions. Much still remains to be known about the steady-state balance and subcellular compartmentation of protein acetylation during the evolution of cardiac dysfunction.
SIRTs are emerging as protective mediators in pathological remodeling of the heart. SIRT1, the most extensively studied member of the SIRT family, has been shown to be important in mediating cardiac remodeling in the development of heart failure. Cardiac-specific SIRT1 knockout mice develop a mild cardiomyopathy with cardiac dysfunction (100). Activation and overexpression of SIRT1 in neonatal rat cardiomyocytes protected against metabolic dysregulation by inhibiting the downregulation of FAO genes in response to phenylephrine-induced hypertrophy (77). Furthermore, in vivo activation of SIRT1 protected against cardiac hypertrophy, metabolic dysregulation, and cardiac inflammation in a mouse model of isoproterenol-induced cardiac hypertrophy. Numerous additional studies have shown a protective effect of SIRT1 activation in other models of cardiac dysfunction (4, 17, 85, 105). However, overexpression of SIRT1 by >7.5-fold in transgenic mice resulted in cardiac dysfunction by inducing oxidative stress and mitochondrial dysfunction (4, 75).
SIRT2 and SIRT6 have also emerged as prominent cardioprotective SIRTs (25, 91). Overexpression of SIRT2 repressed angiotensin II-induced cardiac hypertrophy by deacetylating LKB1 and maintaining AMPK signaling, whereas deficiency of SIRT2 aggravated cardiac hypertrophy in aged mice and mice stressed with angiotensin II (91). Alternatively, loss of SIRT6 in mice resulted in the development of cardiac hypertrophy and heart failure by inducing insulin growth factor-Akt signaling (89). The importance of SIRT6 was further demonstrated in a mouse model of transverse aortic constriction-induced heart failure in which SIRT6 overexpression resulted in increased survival, diminished cardiac dysfunction, and attenuation of the fibrotic and inflammatory responses (51, 89). This has been shown to be, in part, the result of suppression of both the prohypertrophic STAT3 pathway and stress-responsive transcription factor c-Jun (89, 113). In another model of isoproterenol-induced cardiac hypertrophy, SIRT6 was cardioprotective by activating autophagy (62).
The activity of mitochondrial SIRTs, including SIRT3 and SIRT4, has implications for cardiac remodeling and the development of heart failure. SIRT3 appears to be required for maintaining cardiac function (37). Deficiency of SIRT3 in mice worsened dysfunction in the heart after transverse aortic constriction by impairing cellular bioenergetics through increased acetylation of mitochondrial proteins (18, 45). Overexpression of SIRT3 blocked the angiotensin II-induced hypertrophic response by repressing hypertrophic signaling, increasing synthesis of antioxidants, and upregulating key metabolic enzymes required for FAO (88). Additionally, in a mouse model of Friedreich’s ataxia cardiomyopathy, NAD+ supplementation by intraperitoneal injections of NMN restored cardiac function through a SIRT3-dependent improvement in cardiac bioenergetics (67). Furthermore, it has been proposed that SIRT3 activity is required to protect against the hyperacetylation of mitochondrial proteomes after overnutrition-induced formation of high levels of acetyl-CoA (30). On the other hand, increased SIRT4 activity may have detrimental effects within the heart. Cardiac-specific overexpression of SIRT4 in mice accentuated development of heart failure by increasing the degree of cardiac dysfunction, hypertrophy, and fibrosis in response to angiotensin II (65). This study (65) demonstrated that SIRT4 overexpression increased oxidative stress by inhibiting SIRT3 binding and deacetylation of manganese SOD, an important mediator in scavenging ROS. In addition, suppression of SIRT4 by miRNA-497 overexpression diminished the adverse hypertrophic response induced by transverse aortic constriction in mice (106). Furthermore, knockdown of SIRT4 was shown to improve metabolic functions by upregulating FAO metabolism in myocytes and hepatocytes, further suggesting that SIRT4 may be detrimental to the heart (48, 72). The differential role of SIRTs and the interplay with NAD+ supplementation need to be further elucidated.
Another hallmark of heart failure is dysregulation of Ca2+ homeostasis (64). Ca2+ handling is disturbed in the presence of oxidative stress, in part because of the effects of reduced NAD+-to-NADH ratio on the Na+/Ca2+ exchanger (NCX). NCX, an antiporter that is responsible for the principal Ca2+ efflux pathway within the cardiomyocyte, is inhibited by increased cytosolic NADH (59). NADH-mediated NCX inhibition is facilitated through the accumulation of cytosolic ROS by an unidentified ROS-generating NADH-driven flavoprotein oxidase. However, it remains unknown how ROS directly acts on NCX. The inhibition of NCX would translate into increased intracellular Ca2+, propagating excitation-contraction coupling abnormalities. In a mouse model of pressure overload hypertrophy by transverse aortic constriction, NCX current density was significantly diminished in isolated hypertrophied cardiomyocytes despite increased NCX transcript and proteins levels (102). Upon inhibition of the hypertrophic signaling by the calcineurin inhibitor cyclosporine, the effect of pressure overload on the heart toward NCX activity and expression was blunted (102). These studies illustrate that NCX is interdependent on the cellular redox state and hypertrophic signaling pathways. The ability to normalize the NAD+-to-NADH ratio by boosting the NAD+ metabolome has the potential to improve NCX function, Ca2+ handling, and contractile function of the heart.
Previous studies have illustrated that knockdown of cADPR synthetase and NADase CD38 significantly increases NAD+ content in multiple tissues, including the heart (2). CD38 knockdown-mediated increase in cellular NAD+ content protected against angiotensin II-induced cardiac hypertrophy and fibrosis, providing an additional therapeutic approach for boosting NAD+ levels (33).
Transcription factors are integral in physiology and cellular processing within the heart. Two transcription factors, Kruppel-like factor 4 (KLF4) and serum response factor (SRF), have been identified as playing a critical role in the heart. KLF4, described as a zinc finger transcription factor, was found to be important in maintaining cardiac mitochondrial homeostasis (52). Cardiac-specific KLF4 knockout mice were sensitive to pressure overload stress by transverse aortic constriction, exhibiting decreased ventricular contractile function compared with control mice. NMN supplementation rescued this stress-induced sensitivity to cardiac failure by rescuing the mitochondrial dysfunction (112). In another model, inducible cardiac-specific SRF knockout (SRFHKO) mice develop a dilated cardiomyopathy (23). Strikingly, NAD+ content within the hearts of SRFHKO mice was reduced by ~30% after 15 days of SRF inactivation. Of note, the expression of NMRK2, the enzyme converting NR to NMN, was strongly upregulated in hearts of SRFHKO mice through a transcriptional pathway involving AMPK and peroxisome proliferator-activated receptor-α. The upregulation of the NMRK2 pathway for NAD+ synthesis was coupled with downregulation of the NAMPT pathway. This shift in NAD+ synthetic pathways is seen in many models of heart failure and reflects an energy-sparing adaptation that may be beneficial in the failing heart (23). Furthermore, this adaptation suggests that NR is the preferred precursor for NAD+ restoration in models of heart failure. When supplemented with dietary NR, cardiac structure and function in SRFHKO mice were preserved with prevention of the adverse remodeling and deterioration of ejection fraction seen in SRFHKO mice on a standard diet. In addition, NR-enriched diets potentially aid in the detoxification of ROS by increasing expression of the antioxidant regulator transcription factor Nfel2 and its targets in SRFHKO mice (23).
Collectively, NAD+ precursor supplementation holds the potential to protect against adverse cardiac remodeling by reducing oxidative stress, activating SIRTs, maintaining Ca2+ homeostasis, and normalizing the NAD+ metabolome.
Emerging benefits of NAD+ in I/R injury.
Coronary artery disease, a major cause of heart failure, provokes I/R injury. I/R injury is the damage induced within the myocardium by prolonged hypoxia from decreased blood flow and subsequent reoxygenation of the tissue with the return of blood flow. During ischemia, the cardiomyocyte relies on glycolysis to produce ATP. This shift in metabolism is unsustainable and results in a cascade that includes the depletion of NAD+, reduction of available ATP, and decrease in intracellular pH. Together, this causes inactivation of ATPases, overload of intracellular Ca2+, dysfunction of mitochondria, and activation of intracellular proteases (40). Upon reperfusion, rapid introduction of oxygen and recovery of intracellular pH result in the production of ROS and further increases in intracellular Ca2+. Thus, I/R leads to substantial oxidative stress in cardiomyocytes, resulting in disruption of cellular metabolism.
In the heart, hypoxia has been shown to upregulate apoptotic pathways, including increasing expression of proapoptotic protein p53 (61). SIRT1 regulates p53 activity by removing acetyl groups at several target lysines. In addition, NAMPT, the rate-limiting enzyme that converts NAM to NMN in the NAD+ salvage synthesis pathway, is downregulated at the mRNA and protein levels within the heart in models of ischemia and I/R injury, similar to models of heart failure. The depletion of NAMPT under ischemic conditions results in a decrease of NAD+ synthesis. Therefore, NAD+ depletion and the inability to regenerate NAD+ by NAMPT promote apoptotic signaling of p53 through decreased SIRT1 activity. In addition, SIRT1 has been shown to reduce oxidative stress by activating catalase and manganese SOD through deacetylation of proliferator-activated receptor-γ coactivator-1α and forkhead box O (FOXO) transcription factors (25). Elevation of NAD+ content by either NR supplementation or upregulation of NAMPT could constitute therapeutic strategies to decrease oxidative stress and cell death in I/R injury.
Several studies have demonstrated the protective role of NAD+ in I/R injury. Supplementation of NAD+ in rat cardiac myoblasts increased survival in a dose-dependent manner after hypoxia and reoxygenation by driving SIRT1 activity on p53 (54). These in vitro findings were confirmed in vivo with intraperitoneal injections of NMN before and during ischemia that were protective against I/R injury, as measured by reduced infarct size and increased SIRT1 activity (107). In another study (34), exogenous NAD+ by intraperitoneal injection was found to decrease infarct size in response to left anterior descending coronary artery ligation in wild-type mice. Deficiency of CD38 had a similar protective effect in this model by increasing NAD+ availability. In an in vitro model of hypoxia/reoxygenation, deficiency of CD38 was protective against apoptosis through activation of the SIRT1/FOXO3 signaling pathway and reduction of ROS by upregulating SOD2 and downregulating ROS-generating NADPH oxidase 4 (34).
In addition to SIRT1, SIRT3 appears to play an important role in the response to I/R injury. Deficiency of SIRT3 increased the susceptibility to I/R injury, since adult mouse hearts halpoinsufficent for SIRT3 had decreased functional recovery and increased infarct size in a Langendorff model of simulated I/R injury (78). In addition, SIRT3 has been implicated in mediating the cardioprotective effects in response to I/R injury of metformin (26).
This beneficial effect of NAD+ supplementation was further illustrated by intravenous infusion of NAD+, which resulted in reduced infarction size, decreased troponin I levels, and attenuation of apoptotic signaling and apoptosis in a rat model of I/R injury (114). Although this study did not demonstrate that intravenous NAD+ increased intracellular NAD+ content, previous reports have shown exogenous application of the metabolite prevents the loss of NAD+ in conditions of depletion (76). Similar to CD38 deficiency, NAD+ supplementation displayed protective antioxidant properties against ROS by increasing SOD activity through upregulation of mitochondrial SOD2 expression. Similarly, in a mouse model of sudden cardiac arrest, NAD+ repletion by intravenous NAM administration was protective in short-term survival after an 8-min cardiac arrest protocol (115). In summary, there is strong in vitro and in vivo animal model evidence that NAD+-boosting strategies have therapeutic benefits in protecting against I/R injury. Based on an understanding that intracellular activities of NAD+ and NMN depend on the NMRK pathway because of repression of the NAMPT pathway, the preclinical data appear sufficient to justify early phase evaluation of NAD+ precursor supplementation using NR in I/R injury in humans (84).
Emerging benefits of NAD+ in arrhythmic conditions.
Fatal inherited or acquired arrhythmias are the result of an imbalance of depolarizing and repolarizing currents and may arise from diverse pathological mechanisms. A combination of ion channels is responsible for the depolarizing and repolarizing currents that generate the cardiac action potential. Although NAD+ metabolism and content have not been directly examined in arrhythmic states, recent studies have established that the functions of these ion channels are tightly linked to the metabolic state of the heart (7). Conditions like ischemia that alter cardiac metabolism and reduce the NAD+-to-NADH ratio serve as a substrate for arrhythmic events. Furthermore, arrhythmic events and conditions have been shown to regulate the metabolic profile in shifting from OXPHOS to glycolysis on a transcriptional and proteomic level (6). This interdependence between metabolism and arrhythmias has established a role of NAD+ in cardiovascular electrophysiology. Current research has demonstrated the potential therapeutic effects of NAD+ repletion on ion channels in vitro, the action potential ex vivo, and arrhythmogenesis in vivo using animal models. In addition, downstream effects of NAD+ on Ca2+ signaling are being evaluated.
The initial discovery of glycerol-3-phosphate dehydrogenase 1-like (GPD1L) as a modifier of the primary Na+ channel (Nav1.5) responsible for the depolarization phase of the cardiac action potential indicated an important role for NAD+ in cardiac electrophysiology (60). Sharing homology and activity with the cytoplasmic component of the mitochondrial glycerol phosphate shuttle glycerol 3-phosphate dehydrogenase 1 (GPD1), GPD1L interconverts glycerol 3-phosphate (G3P) and dihydroxyacetone phosphate using the redox coenzymes NAD+ and NADH. Possibly by altering cellular NAD+ and/or NADH metabolism, mutations that disrupt GPD1L activity decrease surface expression of Nav1.5, manifesting in fatal arrhythmias observed in Brugada syndrome and Sudden Infant Death Syndrome (Fig. 3) (60, 99). Common polymorphisms in GPD1L were also found to be associated with the risk of sudden cardiac death in patients with coronary artery disease (104). Subsequent studies within this GPD1L paradigm established a potential mechanism in which a decreased NAD+-to-NADH ratio results in elevated G3P and the downstream metabolic byproduct diacylglycerol, an activator of PKC. PKC, in turn, phosphorylates Nav1.5 at S1503 to decrease channel surface expression (98). Other studies, however, have demonstrated that NADH-mediated PKC phosphorylation at S1503 modulates single channel conductance (58). In addition to NADH directly activating PKC by increasing phospholipase D activity and diacylglycerol levels, NAD+ has been shown to increase Nav1.5 activity through PKA-mediated phosphorylation (57, 58). Interestingly, in hearts isolated from mice that are haploinsufficient for Nav1.5, perfusion of NAD+ for 20 min was protective against arrhythmogenesis assessed by programmed electrical stimulation. Further studies in left ventricular wedge preparations from human failing hearts demonstrated that application of NAD+ increased conduction velocity at multiple pacing cycle lengths (55). The signaling mechanisms underlying the effect of extracellular NAD+ on intracellular signaling remain unknown; however, unpublished work suggests that CD38 may, in part, mediate this signaling of extracellular NAD+ by the production of cADPR in the regulation of Nav1.5 (55). Together, these studies have suggested that the cellular oxidative state and NAD+ signaling within the heart can modify its electrical activity (57).
Fig. 3.

Regulation of Nav1.5 membrane expression by NAD+ and NADH. The III−IV intracellular domain of Nav1.5, historically known for its involvement in channel gating, has more recently been identified as a critical site for posttranslational modifications (PTMs) that regulate channel surface expression. Glycerol-3-phosphate dehydrogenase 1-like (GPD1L) interacts with Nav1.5 and modulates these PTMs. GPD1L interconverts glycerol 3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP) using NAD+ and NADH as coenzymes. NAD+ increases channel membrane expression by activating PKA phosphorylation on the I-II intracellular linking domain in addition to boosting sirtuin (SIRT)1 deacetylation of lysine residue K1479 within the III−IV intracellular linking domain. NADH has been shown to decrease channel membrane expression and single channel conductance by activating PKC, which phosphorylates S1503 within the III−IV linking domain. PKC is also activated by a downstream metabolite of G3P, diacylglycerol (DAG). In a catalytically inactive mutant of GPD1L, a known A280V mutation among others predispose individuals to fatal arrhythmias and Brugada Syndrome, this regulation is disturbed.
Increased oxidative stress within the heart has been established as an arrhythmogenic substrate by observations in models of heart failure. Elevated levels of ROS resulting from mitochondrial dysfunction have been shown to directly inhibit Nav1.5 activity (56). As previously stated, increased oxidative stress in heart failure facilitates hyperacetylation of proteins, including that of Nav1.5 at a specific lysine residue, K1479 (100). This hyperacetylation of Nav1.5 can be reversed by SIRT1, resulting in increased Nav1.5 activity by enhanced surface localization of the channel (100). Mice with cardiac-specific knockout of SIRT1 in mice display increased Nav1.5 acetylation, decreased Nav1.5 current density, and increased arrhythmic events, including bradyarrhythmias, tachyarrhythmias, and conduction abnormalities. Potential therapeutic strategies in arrhythmias derived by Nav1.5 dysfunction could include elevating intracellular NAD+ levels to increase SIRT1 activity to subsequently promote Nav1.5 channel localization and activity (9).
Additionally, K+ voltage-gated channel subfamily D member 2 (Kv4.2), a voltage-gated K+ channel that contributes to the transient outward potassium repolarization current, is subject to modulation by NAD+/NADH through its pyridine-sensing Kvβ1.1 β-subunit (96). In the presence of Kvβ1.1, Kv4.2 demonstrates an inactivation in heterologous cell systems that would mitigate the repolarization current and prolong the action potential. This inactivation is further perpetuated by NADH and prevented by intracellular delivery of NAD+ (92). Kvβ1.1 knockout (Kvβ1.1−/−) mice have electrophysiological changes at baseline, including prolongation of the QTc and reduction in P wave duration and PR interval compared with wild-type mice. Unlike wild-type mice, Kvβ1.1−/− mice do not appear to experience further QTc prolongation after isoproterenol-mediated hypertrophy, a model known to decrease the NAD+-to-NADH ratio (96, 97). This further suggests that Kvβ1.1 senses metabolic changes and modulates Kv4.2.
Several studies have examined the influence of acutely elevating extracellular NAD+ on the electrophysiological properties of the heart through purine receptor signaling. Although extracellular perfusion of NAD+ in mouse heart Langendorff preparations had no effect on action potential duration (APD) at 90% repolarization (57), an acute 5-min extracellular NAD+ application decreased myocardial APD in both an isolated rat heart and guinea pig right atrial appendage preparation (79, 81). An additional study (46) observed a differential effect of NAD+ on atrial APD with an acute transient increase followed by a sustained decrease that is possibly explained by NAD+ acting differentially on both P2X and P2Y purine receptors. Furthermore, the influence of NAD+ on the ventricular conduction system was examined using Purkinje fibers isolated from rabbits and showed that APD at 50% and 90% repolarization was significantly reduced (80). However, NAD+ had no effect on APD in spontaneously active Purkinje fibers, suggestive of distinct effects on repolarizing ionic currents. In addition to effects in the myocardium, NAD+ supplementation increased action potential upstroke velocity and the rate of slow diastolic depolarization within primary sinoatrial pacemaker cells in guinea pigs (79). However, the mechanistic interplay between the electrophysiological effects of extracellular NAD+ and purine receptor signaling remains unclear.
The NAD+-derived Ca2+ signaling second messengers cADPR and NAADP have also been implicated in arrhythmias. Produced by cADPR synthetases, cADPR and NAADP potentiate Ca2+ release from the sarcoplasmic reticulum (SR). The exact mechanisms by which these metabolites modulate SR Ca2+ release are unclear. However, a body of research has shown that cADPR potentiates Ca2+ release by activating ryanodine receptor type 2 (RyR2), whereas NAADP may be acting on more of variety of Ca2+ stores, including microsomes, acidic stores, the endoplasmic reticulum, RyR2, or two-pore domain channels at the lysosomal-SR junction (16, 71, 73, 74). Additional research has shown that cADPR may increase Ca2+ uptake within the SR by increasing the activity of the SR Ca2+ pump, which, in turn, would enhance RyR2-mediated Ca2+ release (63, 108, 109).
cADPR increases the magnitude of Ca2+ transients and the frequency of spontaneous Ca2+ release events or “sparks” in cardiomyocytes from rats and guinea pigs (21, 38). Ca2+ sparks increase the intracellular Ca2+ levels, resulting in NCX activation among modulating other ionic currents (27). To remove excess Ca2+ from the cytoplasm, NCX mediates an influx of Na+ (at an ion ratio of 1 Ca2+:3 Na+). This electrogenic influx promotes depolarization of the cardiomyocyte and may lead to focal or propagated arrhythmias by delayed afterdepolarizations (Fig. 4) (47). This arrhythmogenic potential of cADPR was further exhibited when ventricular myocytes demonstrated the generation of spontaneous action potentials when exposed to cADPR intracellularly (83). Competitive antagonists of cADPR inhibited this effect. In addition, prevention of the formation of cADPR through inhibition of cADPR synthetases has been shown to be effective in the prevention of ouabain-induced arrhythmias in vivo (41). Furthermore, wild-type hearts pretreated with SAN-4825, a selective inhibitor of cADPR synthetases, and hearts from CD38 knockout mice had a significant reduction in β-adrenergic stimulation-induced arrhythmias during isoproterenol application (53).
Fig. 4.
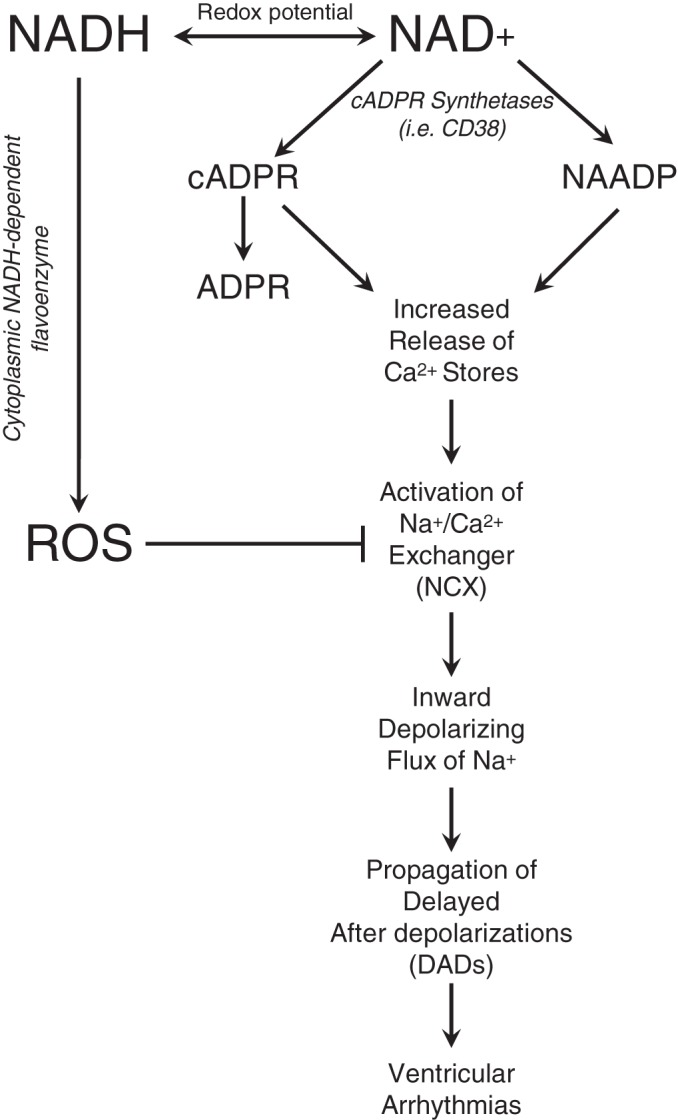
NAD+ and Ca2+ signaling in arrhythmogenesis. NAD+ can be converted to cADP-ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP) by cADPR synthetases, including CD38, which can serve as both a cyclase and hydrolase for the subsequent conversion of cADPR to acylated ADP-ribose (ADPR). cADPR and NAADP trigger a cascade of events, including the activation of the Na+/Ca2+ exchanger (NCX), that may lead to the propagation of ventricular arrhythmias. NADH-dependent reactive oxygen species (ROS) accumulation by an unidentified cytoplasmic flavoprotein oxidase has been shown to inhibit NCX. Collectively, redox regulation and NAD+ metabolism hold the ability to regulate Ca2+ handling and electrical activity in the heart.
Similarly, NAADP increases Ca2+ transients in addition to increasing magnitude and frequency of Ca2+ sparks in cardiomyocytes (66). An antagonist of NAADP action, BZ-194, reduced arrhythmic events induced by isoproterenol in awake mice (73). This research suggests that NAD+ supplementation and possible downstream formation of cADPR and NAADP may be arrthymogenic. The role of NAD+ supplementation and Ca2+ signaling-mediated arrhythmogenesis warrants further exploration.
These mechanistic findings and insights provide evidence that manipulating the NAD+ metabolome has the potential to modulate Nav1.5, Kv4.2, and Ca2+ handling in ways that could prevent arrhythmias and sudden death. It remains unclear how extracellular NAD+-related signaling integrates with intracellular NAD+-related processes to regulate cardiac electrophysiology. In humans, extracellular NAD+ content in plasma can vary between 50 and 100 nM (35). Other NAD+ metabolites have previously been shown to be present and sensitive to NAD+ precursor supplementation in the extracellular milieu (93). It is unclear how extracellular NAD+ and related metabolites mediate cellular signaling and physiology of the heart in vivo. In addition, downstream byproducts of NAD+ precursor supplementation such as cADPR and NAADP may be arrhythmogenic. Thus, further studies are warranted to determine the potential effects of NAD+ and NAD+ precursors in the prevention and propagation of arrhythmias.
Beneficial systemic effects of NAD+ supplementation to cardiac health.
In addition to the benefits for cardiac disease, NAD+ repletion has been shown to be therapeutically beneficial in other organ systems and diseases that have implications in cardiac health (43). Supplementation of the NAD+ precursors was shown to prevent weight gain, improve muscle function, and reverse vascular dysfunction in aged mice (22, 70, 111). Furthermore, NAD+ supplementation has been shown to improve multiple facets and sequelae of obesity and type II diabetes, including protection of high-fat diet-associated weight gain (14, 94, 110). The potential beneficial features of NAD+ precursor supplementation on cardiac health are more expansive than direct effects on the heart.
Future Directions
NAD+ and associated metabolites hold great influence on cardiac physiology in health and disease. NAD+ supplementation has emerged as potential therapy (Table 1) for pathological cardiac remodeling, I/R injury, and arrhythmogenesis. Increasing cellular NAD+ may be achieved by increasing the biosynthesis of NAD+ by supplementation of precursors; tryptophan, NA, NAM, NMN, and NR all hold the ability to increase NAD+ content, although they differ in efficiency (15). Additional research is necessary to examine the differential effects, if any, of these various NAD+ supplements. NR has emerged recently has a leading candidate because of its bioavailability, safety profile, and superiority in raising NAD+ content relative to other precursors (93).
Table 1.
Preclinical evidence supporting NAD+-related metabolites as a therapy for cardiovascular disease
| Disease | Model | NAD+ Therapy | Dose and Route of Administration | Key Findings | Mechanistic Findings | Reference |
|---|---|---|---|---|---|---|
| Heart failure | Rat model of chronic volume overload by intrarenal arteriovenous fistula | NAM | 0.67 mg/ml in drinking water, 2 days presurgeries and 4 wk postsurgeries | Preservation of cardiac function Prevention of adverse remodeling | Decreased oxidative stress Decreased activation of matrix metalloproteinases | 20 |
| Angiotensin-II-induced mouse model of cardiac hypertrophy | NAD+ | 1 mg·kg−1·day−1 for 2 wk (entire duration of ANG II ip infusion) | Preservation of cardiac function Prevention of adverse remodeling | Decreased prohypertrophic signaling via Akt1 Increased antihypertrophic signaling via SIRT3-LKB1-AMPK | 76 | |
| Pressure overload mouse model of hypertrophy: transverse aortic constriction | NMN | 500 mg/kg ip every 3 days, 5 days before and until 4 wk postsurgery | Preservation of cardiac function Prevention of adverse remodeling | Normalization of NAD+/NADH Decreased protein hyperacetylation | 49 | |
| NR | NR-enriched diet with target dose of 400 mg·kg body wt−1·day−1 (starting 2 days postsurgery) | Partial preservation of cardiac function | Partially protected against dysregulation of NAD+ metabolome | 23 | ||
| Mouse model of mitochondrial dysfunction (cardiac-specific Ndufs4−/−) with transverse aortic constriction | NMN | 500 mg/kg ip every 3 days, 5 days before and until 4 wk postsurgery | Preservation of cardiac function | Decreased protein hyperacetylation | 49 | |
| Prevention of adverse remodeling | ||||||
| Mouse model of Friedreich's ataxia cardiomyopathy | NMN | 500 mg/kg ip, 2 times weekly for 4–5 wk | Restored cardiac function | Protection by NMN is mediated by SIRT3 | 67 | |
| Improvement of cardiac and extracardiac metabolic bioenergetics | ||||||
| Mouse model of mitochondrial dysfunction (cardiac-specific KLF4−/− mice) with pressure overload cardiomyopathy | NMN | 500 mg·kg−1·day−1 ip, every day (starting 1 day presurgery to 3–5 days postsurgery) | Restored cardiac contractility Reduction in cardiac cell death Decreased expression of inflammatory markers | Improvement of mitochondrial bioenergetics and ultrastructure | 112 | |
| Mouse model of dilated cardiomyopathy (cardiac-specific SRFHKO mice) | NR | NR-enriched diet with target dose of 400 mg·kg body wt−1·day−1 (starting 5 days post-tamoxifen-induced SRF inactivation) | Preservation of cardiac function Prevention of adverse remodeling | Protected against dysregulation of NAD+ metabolome Increased expression of antioxidant regulator Nfel2 | 23 | |
| I/R injury | Mouse model of I/R injury | NMN | 500 mg/kg ip 30 min before ischemia | Reduced infarct size Improved cardiac function | Increased SIRT1 activity | 107 |
| Mouse model of I/R injury | NAD+ | 200 mg/kg ip | Reduced infarct size | 34 | ||
| Mouse model of cardiac arrest | NAM | 500 mg/kg iv postcardiac arrest protocol | Increased sudden cardiac arrest survival | 115 | ||
| Rat model of I/R injury | NAD+ | 10–20 mg/kg iv before ischemia | Reduced infarct size Improved cardiac function | Decreased troponin I levels Reduction in apoptotic signaling Decreased oxidative stress by upregulation of SOD2 | 114 | |
| Arrhythmias | Nav1.5 haploinsufficient mouse model of Brugada Syndrome: ex vivo assesement of arrhythmic susceptibility | NAD+ | 100 µM perfusion, 20 min | Reduction in arrhythmic susceptibility | Activation of PKA | 57 |
| Nonischemic cardiomyopathic mouse model: ex vivo assessment of Nav1.5 current | NAD+ | 100 mg/kg, 2 injections before euthanasia (24 h, 1 h) | Increased INa in isolated cardiomyocytes | Normalization of NAD+/NADH Decreased PKC phosphorylation of Nav1.5 Effect mediated through CD38 activity | 55 | |
| Human cardiomyopathic left ventricle tissue: ex vivo assessment of conduction velocity | NAD+ | 500 µM (30 min) | Improved conduction velocity | 55 |
NAD+, nicotinamide adenine dinucleotide; KLF−/−, Kruppel-like factor 4 knockout; SRFHKO, cardiac-specific serum response factor knockout; I/R, ischemia-reperfusion; NAM, nicotinamide; NMN, nicotinamide mononucleotide; NR, nicotinamide riboside; SRF, serum response factor; INa, Na+ current; SIRT3, sirtuin 3; LKB1, liver kinase B1; AMPK, AMP-activated protein kinase; SIRT1, sirtuin 1; SOD2, superoxide dismutase 2; PKA, protein kinase A; PKC, protein kinase C.
This review outlined the evidence for beneficial effects of NAD+ supplementation with NAD+, NAM, NMN, and NR in a variety of preclinical disease models. In addition, several mechanisms that highlight the importance of NAD+ in regulating cellular processes were described (Fig. 5). Further understanding of the mechanisms and effects of NAD+ repletion in health and disease will be necessary to fully delineate individual and concerted effects on NAD+-consuming enzymes. For example, activation of SIRT1, SIRT2, SIRT3, and SIRT6 has been implicated in mediating cardioprotective effects of NAD+ supplementation; however, activation of SIRT4 and overactivation of SIRT1 may aid in the development of cardiac dysfunction. It is unknown how NAD+-boosting strategies influence these SIRTs in concert. Furthermore, consumption of NAD+ by CD38 and CD157 to yield cADPR and NAADP may have detrimental effects within the heart because of the role of these metabolites in Ca2+ signaling and arrhythmogenesis. Additional research will need to examine the effects of NAD+ repletion on the multitude of cellular processes involved in NAD+ consumption, signaling, and regulation.
Fig. 5.

Effects of boosting NAD+ in the heart. NAD+ precursor vitamins boost the cardiac NAD+ metabolome, resulting in 1) increased sirtuin activity, 2) increased NAD+/NADH redox balance, and 3) elevated NADPH. Pharmacological NAM is a sirtuin (SIRT) inhibitor, and its conversion to NMN through NAMPT is downregulated in pathological cardiac stress, including models of ischemia/reperfusion (I/R) injury and pressure overload hypertrophy. However, NMRK2 has been shown to be upregulated in the presence of cardiac stress, including a mouse model of dilated cardiomyopathy, suggesting that NR is the preferred agent for cardioprotection. Although SIRT1, SIRT2, SIRT3, and SIRT6 are generally regarded as cardioprotective, the mitochondrial SIRT4 appears to induce cardiac stress and damage.
The translation of these cardioprotective benefits observed in preclinical studies to human patients and disease will yield an exciting period for NAD+ supplementation research. Initial human studies with NR have shown that NR by oral administration is effective in increasing NAD+ metabolism without adverse effects (1, 93). As a natural product and vitamin, NR is undergoing clinical testing for longer-term safety and efficacy. Other therapeutic strategies to increase NAD+ content such as inhibition of NAD+-consuming enzymes or delivery of drugs like NR-releasing compositions offer additional promise.
Conclusions
NAD+-related metabolites have promising therapeutic effects in cardiovascular disease. Dysregulation of the NAD+ metabolome is developing as a focal point of the pathogenesis of adverse effects in many disease processes, including cardiac remodeling, response to I/R, and arrhythmic susceptibility. Stimulation of NAD+ biosynthesis by supplementation of NAD+ precursors may prove to be therapeutically beneficial. Insights on the differential effects of these NAD+ precursors will be critical, since further research is necessary to evaluate the potential of this therapeutic strategy in humans.
GRANTS
This work is supported by National Institutes of Health (NIH) Grant R01-HL-115955. D. S. Matasic is supported by American Heart Association Predoctoral Fellowship 17PRE33410450 and the NIH-supported Medical Scientist Training Program at the University of Iowa (NIH Grant T32-GM-007337).
DISCLOSURES
Conflicts of interest: DSM: none, CB: a stockholder and Chief Scientific Adviser of ChromaDex, which manufactures and sells nicotinamide riboside as a nutritional supplement and is developing NAD+-boosting therapeutics, BL: none.
AUTHOR CONTRIBUTIONS
D.S.M. prepared figures; D.S.M. drafted manuscript; D.S.M., C.B., and B.L. edited and revised manuscript; D.S.M., C.B., and B.L. approved final version of manuscript.
REFERENCES
- 1.Airhart SE, Shireman LM, Risler LJ, Anderson GD, Nagana Gowda GA, Raftery D, Tian R, Shen DD, O’Brien KD. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One 12: e0186459, 2017. doi: 10.1371/journal.pone.0186459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun 345: 1386–1392, 2006. doi: 10.1016/j.bbrc.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 3.Alano CC, Tran A, Tao R, Ying W, Karliner JS, Swanson RA. Differences among cell types in NAD+ compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J Neurosci Res 85: 3378–3385, 2007. doi: 10.1002/jnr.21479. [DOI] [PubMed] [Google Scholar]
- 4.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res 100: 1512–1521, 2007. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 5.Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Krüger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP. The failing heart relies on ketone bodies as a fuel. Circulation 133: 698–705, 2016. doi: 10.1161/CIRCULATIONAHA.115.017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barth AS, Merk S, Arnoldi E, Zwermann L, Kloos P, Gebauer M, Steinmeyer K, Bleich M, Kääb S, Hinterseer M, Kartmann H, Kreuzer E, Dugas M, Steinbeck G, Nabauer M. Reprogramming of the human atrial transcriptome in permanent atrial fibrillation: expression of a ventricular-like genomic signature. Circ Res 96: 1022–1029, 2005. doi: 10.1161/01.RES.0000165480.82737.33. [DOI] [PubMed] [Google Scholar]
- 7.Barth AS, Tomaselli GF. Cardiac metabolism and arrhythmias. Circ Arrhythm Electrophysiol 2: 327–335, 2009. doi: 10.1161/CIRCEP.108.817320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci 32: 12–19, 2007. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 129: 473–484, 2007. doi: 10.1016/j.cell.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 10.Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117: 495–502, 2004. doi: 10.1016/S0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- 11.Billington RA, Travelli C, Ercolano E, Galli U, Roman CB, Grolla AA, Canonico PL, Condorelli F, Genazzani AA. Characterization of NAD uptake in mammalian cells. J Biol Chem 283: 6367–6374, 2008. doi: 10.1074/jbc.M706204200. [DOI] [PubMed] [Google Scholar]
- 12.Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr 28: 115–130, 2008. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- 13.Bruzzone S, Guida L, Zocchi E, Franco L, De Flora A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J 15: 10–12, 2001. doi: 10.1096/fj.00-0566fje. [DOI] [PubMed] [Google Scholar]
- 14.Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, Auwerx J. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 15: 838–847, 2012. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cantó C, Menzies KJ, Auwerx J. NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab 22: 31–53, 2015. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capel RA, Bolton EL, Lin WK, Aston D, Wang Y, Liu W, Wang X, Burton RA, Bloor-Young D, Shade KT, Ruas M, Parrington J, Churchill GC, Lei M, Galione A, Terrar DA. Two-pore channels (TPC2s) and nicotinic acid adenine dinucleotide phosphate (NAADP) at lysosomal-sarcoplasmic reticular junctions contribute to acute and chronic β-adrenoceptor signaling in the heart. J Biol Chem 290: 30087–30098, 2015. doi: 10.1074/jbc.M115.684076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cappetta D, Esposito G, Piegari E, Russo R, Ciuffreda LP, Rivellino A, Berrino L, Rossi F, De Angelis A, Urbanek K. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int J Cardiol 205: 99–110, 2016. doi: 10.1016/j.ijcard.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Chen T, Liu J, Li N, Wang S, Liu H, Li J, Zhang Y, Bu P. Mouse SIRT3 attenuates hypertrophy-related lipid accumulation in the heart through the deacetylation of LCAD. PLoS One 10: e0118909, 2015. doi: 10.1371/journal.pone.0118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins PB, Chaykin S. The management of nicotinamide and nicotinic acid in the mouse. J Biol Chem 247: 778–783, 1972. [PubMed] [Google Scholar]
- 20.Cox MJ, Sood HS, Hunt MJ, Chandler D, Henegar JR, Aru GM, Tyagi SC. Apoptosis in the left ventricle of chronic volume overload causes endocardial endothelial dysfunction in rats. Am J Physiol Heart Circ Physiol 282: H1197–H1205, 2002. doi: 10.1152/ajpheart.00483.2001. [DOI] [PubMed] [Google Scholar]
- 21.Cui Y, Galione A, Terrar DA. Effects of photoreleased cADP-ribose on calcium transients and calcium sparks in myocytes isolated from guinea-pig and rat ventricle. Biochem J 342: 269–273, 1999. doi: 10.1042/bj3420269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, Imai S, Seals DR. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15: 522–530, 2016. doi: 10.1111/acel.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, Gouge A, Gressette M, Manoury B, Blanc J, Breton M, Decaux JF, Lavery G, Baczkó I, Zoll J, Garnier A, Li Z, Brenner C, Mericskay M. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation In press. doi: 10.1161/CIRCULATIONAHA.116.026099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Lisa F, Menabò R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 276: 2571–2575, 2001. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 25.D’Onofrio N, Servillo L, Balestrieri ML. SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid Redox Signal 28: 711–732, 2018. doi: 10.1089/ars.2017.7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du Y, Zhang J, Fang F, Wei X, Zhang H, Tan H, Zhang J. Metformin ameliorates hypoxia/reoxygenation-induced cardiomyocyte apoptosis based on the SIRT3 signaling pathway. Gene 626: 182–188, 2017. doi: 10.1016/j.gene.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 27.Fan X, Ma J, Wan W, Zhang P, Wang C, Wu L. Increased intracellular calcium concentration causes electrical turbulence in guinea pig ventricular myocytes. Sci China Life Sci 54: 240–247, 2011. doi: 10.1007/s11427-011-4146-1. [DOI] [PubMed] [Google Scholar]
- 28.Fletcher RS, Ratajczak J, Doig CL, Oakey LA, Callingham R, Da Silva Xavier G, Garten A, Elhassan YS, Redpath P, Migaud ME, Philp A, Brenner C, Canto C, Lavery GG. Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Mol Metab 6: 819–832, 2017. doi: 10.1016/j.molmet.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukuwatari T, Shibata K. Effect of nicotinamide administration on the tryptophan-nicotinamide pathway in humans. Int J Vitam Nutr Res 77: 255–262, 2007. doi: 10.1024/0300-9831.77.4.255. [DOI] [PubMed] [Google Scholar]
- 30.Ghanta S, Grossmann RE, Brenner C. Mitochondrial protein acetylation as a cell-intrinsic, evolutionary driver of fat storage: chemical and metabolic logic of acetyl-lysine modifications. Crit Rev Biochem Mol Biol 48: 561–574, 2013. doi: 10.3109/10409238.2013.838204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13: 411–424, 2012. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 32.Grozio A, Sociali G, Sturla L, Caffa I, Soncini D, Salis A, Raffaelli N, De Flora A, Nencioni A, Bruzzone S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J Biol Chem 288: 25938–25949, 2013. doi: 10.1074/jbc.M113.470435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guan XH, Hong X, Zhao N, Liu XH, Xiao YF, Chen TT, Deng LB, Wang XL, Wang JB, Ji GJ, Fu M, Deng KY, Xin HB. CD38 promotes angiotensin II-induced cardiac hypertrophy. J Cell Mol Med 21: 1492–1502, 2017. doi: 10.1111/jcmm.13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guan XH, Liu XH, Hong X, Zhao N, Xiao YF, Wang LF, Tang L, Jiang K, Qian YS, Deng KY, Ji G, Fu M, Xin HB. CD38 deficiency protects the heart from ischemia/reperfusion injury through activating SIRT1/FOXOs-mediated antioxidative stress pathway. Oxid Med Cell Longev 2016: 7410257, 2016. doi: 10.1155/2016/7410257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guida L, Bruzzone S, Sturla L, Franco L, Zocchi E, De Flora A. Equilibrative and concentrative nucleoside transporters mediate influx of extracellular cyclic ADP-ribose into 3T3 murine fibroblasts. J Biol Chem 277: 47097–47105, 2002. doi: 10.1074/jbc.M207793200. [DOI] [PubMed] [Google Scholar]
- 36.Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, Walseth TF, Lee HC. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 262: 1056–1059, 1993. doi: 10.1126/science.8235624. [DOI] [PubMed] [Google Scholar]
- 37.Hu DX, Liu XB, Song WC, Wang JA. Roles of SIRT3 in heart failure: from bench to bedside. J Zhejiang Univ Sci B 17: 821–830, 2016. doi: 10.1631/jzus.B1600253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iino S, Cui Y, Galione A, Terrar DA. Actions of cADP-ribose and its antagonists on contraction in guinea pig isolated ventricular myocytes. Influence of temperature. Circ Res 81: 879–884, 1997. doi: 10.1161/01.RES.81.5.879. [DOI] [PubMed] [Google Scholar]
- 39.Julius U, Fischer S. Nicotinic acid as a lipid-modifying drug--a review. Atheroscler Suppl 14: 7–13, 2013. doi: 10.1016/j.atherosclerosissup.2012.10.036. [DOI] [PubMed] [Google Scholar]
- 40.Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 298: 229–317, 2012. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kannt A, Sicka K, Kroll K, Kadereit D, Gögelein H. Selective inhibitors of cardiac ADPR cyclase as novel anti-arrhythmic compounds. Naunyn Schmiedebergs Arch Pharmacol 385: 717–727, 2012. doi: 10.1007/s00210-012-0750-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18: 239–250, 2013. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katsyuba E, Auwerx J. Modulating NAD+metabolism, from bench to bedside. EMBO J 36: 2670–2683, 2017. doi: 10.15252/embj.201797135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim H, Jacobson EL, Jacobson MK. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science 261: 1330–1333, 1993. doi: 10.1126/science.8395705. [DOI] [PubMed] [Google Scholar]
- 45.Koentges C, Pfeil K, Schnick T, Wiese S, Dahlbock R, Cimolai MC, Meyer-Steenbuck M, Cenkerova K, Hoffmann MM, Jaeger C, Odening KE, Kammerer B, Hein L, Bode C, Bugger H. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol 110: 36, 2015. doi: 10.1007/s00395-015-0493-6. [DOI] [PubMed] [Google Scholar]
- 46.Kuzmin VS, Pustovit KB, Abramochkin DV. Effects of exogenous nicotinamide adenine dinucleotide (NAD+) in the rat heart are mediated by P2 purine receptors. J Biomed Sci 23: 50, 2016. doi: 10.1186/s12929-016-0267-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Landstrom AP, Dobrev D, Wehrens XHT. Calcium signaling and cardiac arrhythmias. Circ Res 120: 1969–1993, 2017. doi: 10.1161/CIRCRESAHA.117.310083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laurent G, de Boer VC, Finley LW, Sweeney M, Lu H, Schug TT, Cen Y, Jeong SM, Li X, Sauve AA, Haigis MC. SIRT4 represses peroxisome proliferator-activated receptor α activity to suppress hepatic fat oxidation. Mol Cell Biol 33: 4552–4561, 2013. doi: 10.1128/MCB.00087-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE, Tian R. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation 134: 883–894, 2016. doi: 10.1161/CIRCULATIONAHA.116.022495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee CF, Tian R. Mitochondrion as a target for heart failure therapy- role of protein lysine acetylation. Circ J 79: 1863–1870, 2015. doi: 10.1253/circj.CJ-15-0742. [DOI] [PubMed] [Google Scholar]
- 51.Li Y, Meng X, Wang W, Liu F, Hao Z, Yang Y, Zhao J, Yin W, Xu L, Zhao R, Hu J. Cardioprotective effects of SIRT6 in a mouse model of transverse aortic constriction-induced heart failure. Front Physiol 8: 394, 2017. doi: 10.3389/fphys.2017.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao X, Zhang R, Lu Y, Prosdocimo DA, Sangwung P, Zhang L, Zhou G, Anand P, Lai L, Leone TC, Fujioka H, Ye F, Rosca MG, Hoppel CL, Schulze PC, Abel ED, Stamler JS, Kelly DP, Jain MK. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J Clin Invest 125: 3461–3476, 2015. doi: 10.1172/JCI79964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin WK, Bolton EL, Cortopassi WA, Wang Y, O’Brien F, Maciejewska M, Jacobson MP, Garnham C, Ruas M, Parrington J, Lei M, Sitsapesan R, Galione A, Terrar DA. Synthesis of the Ca2+-mobilizing messengers NAADP and cADPR by intracellular CD38 enzyme in the mouse heart: Role in β-adrenoceptor signaling. J Biol Chem 292: 13243–13257, 2017. doi: 10.1074/jbc.M117.789347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu L, Wang P, Liu X, He D, Liang C, Yu Y. Exogenous NAD+ supplementation protects H9c2 cardiac myoblasts against hypoxia/reoxygenation injury via Sirt1-p53 pathway. Fundam Clin Pharmacol 28: 180–189, 2014. doi: 10.1111/fcp.12016. [DOI] [PubMed] [Google Scholar]
- 55.Liu M, Gu L, Sulkin MS, Liu H, Jeong EM, Greener I, Xie A, Efimov IR, Dudley SC Jr. Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol 54: 25–34, 2013. doi: 10.1016/j.yjmcc.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu M, Liu H, Dudley SC Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res 107: 967–974, 2010. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu M, Sanyal S, Gao G, Gurung IS, Zhu X, Gaconnet G, Kerchner LJ, Shang LL, Huang CL, Grace A, London B, Dudley SC Jr. Cardiac Na+ current regulation by pyridine nucleotides. Circ Res 105: 737–745, 2009. doi: 10.1161/CIRCRESAHA.109.197277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu M, Shi G, Yang KC, Gu L, Kanthasamy AG, Anantharam V, Dudley SC Jr. Role of protein kinase C in metabolic regulation of the cardiac Na+channel. Heart Rhythm 14: 440–447, 2017. doi: 10.1016/j.hrthm.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu T, O’Rourke B. Regulation of the Na+/Ca2+ exchanger by pyridine nucleotide redox potential in ventricular myocytes. J Biol Chem 288: 31984–31992, 2013. doi: 10.1074/jbc.M113.496588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC Jr. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116: 2260–2268, 2007. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Long X, Boluyt MO, Hipolito ML, Lundberg MS, Zheng JS, O’Neill L, Cirielli C, Lakatta EG, Crow MT. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J Clin Invest 99: 2635–2643, 1997. doi: 10.1172/JCI119452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu J, Sun D, Liu Z, Li M, Hong H, Liu C, Gao S, Li H, Cai Y, Chen S, Li Z, Ye J, Liu P.. SIRT6 suppresses isoproterenol-induced cardiac hypertrophy through activation of autophagy. Transl Res 172: 96–112 e6, 2016. doi: 10.1016/j.trsl.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 63.Lukyanenko V, Györke I, Wiesner TF, Györke S. Potentiation of Ca2+ release by cADP-ribose in the heart is mediated by enhanced SR Ca2+ uptake into the sarcoplasmic reticulum. Circ Res 89: 614–622, 2001. doi: 10.1161/hh1901.098066. [DOI] [PubMed] [Google Scholar]
- 64.Luo M, Anderson ME. Mechanisms of altered Ca2+ handling in heart failure. Circ Res 113: 690–708, 2013. doi: 10.1161/CIRCRESAHA.113.301651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo YX, Tang X, An XZ, Xie XM, Chen XF, Zhao X, Hao DL, Chen HZ, Liu DP. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur Heart J 38: 1389–1398, 2017. doi: 10.1093/eurheartj/ehw138. [DOI] [PubMed] [Google Scholar]
- 66.Macgregor A, Yamasaki M, Rakovic S, Sanders L, Parkesh R, Churchill GC, Galione A, Terrar DA. NAADP controls cross-talk between distinct Ca2+ stores in the heart. J Biol Chem 282: 15302–15311, 2007. doi: 10.1074/jbc.M611167200. [DOI] [PubMed] [Google Scholar]
- 67.Martin AS, Abraham DM, Hershberger KA, Bhatt DP, Mao L, Cui H, Liu J, Liu X, Muehlbauer MJ, Grimsrud PA, Locasale JW, Payne RM, Hirschey MD. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich's ataxia cardiomyopathy model. JCI Insight 2: 93885, 2017. doi: 10.1172/jci.insight.93885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mericskay M. Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: pathophysiological implications and therapeutic potential. Arch Cardiovasc Dis 109: 207–215, 2016. doi: 10.1016/j.acvd.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 69.Miller ON, Hamilton JG, Goldsmith GA. Studies on the mechanism of effects of large doses of nicotinic acid and nicotinamide on serum lipids of hypercholesterolemic patients. Circulation 18: 489–490, 1958. [Google Scholar]
- 70.Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, Yoshino J, Imai SI. Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab 24: 795–806, 2016. doi: 10.1016/j.cmet.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mojzisová A, Krizanová O, Záciková L, Komínková V, Ondrias K. Effect of nicotinic acid adenine dinucleotide phosphate on ryanodine calcium release channel in heart. Pflugers Arch 441: 674–677, 2001. doi: 10.1007/s004240000465. [DOI] [PubMed] [Google Scholar]
- 72.Nasrin N, Wu X, Fortier E, Feng Y, Bare’ OC, Chen S, Ren X, Wu Z, Streeper RS, Bordone L. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J Biol Chem 285: 31995–32002, 2010. doi: 10.1074/jbc.M110.124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nebel M, Schwoerer AP, Warszta D, Siebrands CC, Limbrock AC, Swarbrick JM, Fliegert R, Weber K, Bruhn S, Hohenegger M, Geisler A, Herich L, Schlegel S, Carrier L, Eschenhagen T, Potter BV, Ehmke H, Guse AH. Nicotinic acid adenine dinucleotide phosphate (NAADP)-mediated calcium signaling and arrhythmias in the heart evoked by β-adrenergic stimulation. J Biol Chem 288: 16017–16030, 2013. doi: 10.1074/jbc.M112.441246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ogunbayo OA, Zhu Y, Rossi D, Sorrentino V, Ma J, Zhu MX, Evans AM. Cyclic adenosine diphosphate ribose activates ryanodine receptors, whereas NAADP activates two-pore domain channels. J Biol Chem 286: 9136–9140, 2011. doi: 10.1074/jbc.M110.202002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, Sadoshima J. PPARα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab 14: 598–611, 2011. doi: 10.1016/j.cmet.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem 285: 3133–3144, 2010. doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Planavila A, Iglesias R, Giralt M, Villarroya F. Sirt1 acts in association with PPARα to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res 90: 276–284, 2011. doi: 10.1093/cvr/cvq376. [DOI] [PubMed] [Google Scholar]
- 78.Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM. SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol 306: H1602–H1609, 2014. doi: 10.1152/ajpheart.00027.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pustovit KB, Abramochkin DV. Effects of nicotinamide adenine dinucleotide (NAD+) and diadenosine tetraphosphate (Ap4A) on electrical activity of working and pacemaker atrial myocardium in guinea pigs. Bull Exp Biol Med 160: 733–736, 2016. doi: 10.1007/s10517-016-3297-2. [DOI] [PubMed] [Google Scholar]
- 80.Pustovit KB, Kuz’min VS, Sukhova GS. Effect of exogenous extracellular nicotinamide adenine dinucleotide (NAD+) on bioelectric activity of the pacemaker and conduction system of the heart. Bull Exp Biol Med 159: 188–191, 2015. doi: 10.1007/s10517-015-2919-4. [DOI] [PubMed] [Google Scholar]
- 81.Pustovit KB, Kuz’min VS, Sukhova GS. [Influence exogenous nicotinamide adenine dinucleotide (NAD+) on contractile and bioelectric activity of the rat heart]. Ross Fiziol Zh Im I M Sechenova 100: 445–457, 2014. [PubMed] [Google Scholar]
- 82.Rajakumar K. Pellagra in the United States: a historical perspective. South Med J 93: 272–277, 2000. doi: 10.1097/00007611-200093030-00005. [DOI] [PubMed] [Google Scholar]
- 83.Rakovic S, Cui Y, Iino S, Galione A, Ashamu GA, Potter BV, Terrar DA. An antagonist of cADP-ribose inhibits arrhythmogenic oscillations of intracellular Ca2+ in heart cells. J Biol Chem 274: 17820–17827, 1999. doi: 10.1074/jbc.274.25.17820. [DOI] [PubMed] [Google Scholar]
- 84.Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, Kulkarni SS, Rodrigues M, Redpath P, Migaud ME, Auwerx J, Yanes O, Brenner C, Cantó C. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun 7: 13103, 2016. doi: 10.1038/ncomms13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shen T, Ding L, Ruan Y, Qin W, Lin Y, Xi C, Lu Y, Dou L, Zhu Y, Cao Y, Man Y, Bian Y, Wang S, Xiao C, Li J. SIRT1 functions as an important regulator of estrogen-mediated cardiomyocyte protection in angiotensin II-induced heart hypertrophy. Oxid Med Cell Longev 2014: 713894, 2014. doi: 10.1155/2014/713894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Song EK, Rah SY, Lee YR, Yoo CH, Kim YR, Yeom JH, Park KH, Kim JS, Kim UH, Han MK. Connexin-43 hemichannels mediate cyclic ADP-ribose generation and its Ca2+-mobilizing activity by NAD+/cyclic ADP-ribose transport. J Biol Chem 286: 44480–44490, 2011. doi: 10.1074/jbc.M111.307645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sosnowska B, Mazidi M, Penson P, Gluba-Brzózka A, Rysz J, Banach M. The sirtuin family members SIRT1, SIRT3 and SIRT6: their role in vascular biology and atherogenesis. Atherosclerosis 265: 275–282, 2017. doi: 10.1016/j.atherosclerosis.2017.08.027. [DOI] [PubMed] [Google Scholar]
- 88.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest 119: 2758–2771, 2009. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, Cunningham JM, Deng CX, Lombard DB, Mostoslavsky R, Gupta MP. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med 18: 1643–1650, 2012. doi: 10.1038/nm.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 49: 241–248, 2007. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 91.Tang X, Chen XF, Wang NY, Wang XM, Liang ST, Zheng W, Lu YB, Zhao X, Hao DL, Zhang ZQ, Zou MH, Liu DP, Chen HZ. SIRT2 acts as a cardioprotective deacetylase in pathological cardiac hypertrophy. Circulation 136: 2051–2067, 2017. doi: 10.1161/CIRCULATIONAHA.117.028728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tipparaju SM, Saxena N, Liu SQ, Kumar R, Bhatnagar A. Differential regulation of voltage-gated K+ channels by oxidized and reduced pyridine nucleotide coenzymes. Am J Physiol Cell Physiol 288: C366–C376, 2005. doi: 10.1152/ajpcell.00354.2004. [DOI] [PubMed] [Google Scholar]
- 93.Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME, Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun 7: 12948, 2016. doi: 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trammell SA, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, Obrosov A, Kardon RH, Yorek MA, Brenner C. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci Rep 6: 26933, 2016. doi: 10.1038/srep26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 301: H2181–H2190, 2011. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 96.Tur J, Chapalamadugu KC, Katnik C, Cuevas J, Bhatnagar A, Tipparaju SM. Kvβ1.1 (AKR6A8) senses pyridine nucleotide changes in the mouse heart and modulates cardiac electrical activity. Am J Physiol Heart Circ Physiol 312: H571–H583, 2017. doi: 10.1152/ajpheart.00281.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tur J, Chapalamadugu KC, Padawer T, Badole SL, Kilfoil PJ II, Bhatnagar A, Tipparaju SM. Deletion of Kvβ1.1 subunit leads to electrical and haemodynamic changes causing cardiac hypertrophy in female murine hearts. Exp Physiol 101: 494–508, 2016. doi: 10.1113/EP085405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol 297: H1446–H1452, 2009. doi: 10.1152/ajpheart.00513.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, Ackerman MJ. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation 116: 2253–2259, 2007. doi: 10.1161/CIRCULATIONAHA.107.704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vikram A, Lewarchik CM, Yoon JY, Naqvi A, Kumar S, Morgan GM, Jacobs JS, Li Q, Kim YR, Kassan M, Liu J, Gabani M, Kumar A, Mehdi H, Zhu X, Guan X, Kutschke W, Zhang X, Boudreau RL, Dai S, Matasic DS, Jung SB, Margulies KB, Kumar V, Bachschmid MM, London B, Irani K. Sirtuin 1 regulates cardiac electrical activity by deacetylating the cardiac sodium channel. Nat Med 23: 361–367, 2017. doi: 10.1038/nm.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang P, Li WL, Liu JM, Miao CY. NAMPT and NAMPT-controlled NAD metabolism in vascular repair. J Cardiovasc Pharmacol 67: 474–481, 2016. doi: 10.1097/FJC.0000000000000332. [DOI] [PubMed] [Google Scholar]
- 102.Wang Z, Nolan B, Kutschke W, Hill JA. Na+-Ca2+ exchanger remodeling in pressure overload cardiac hypertrophy. J Biol Chem 276: 17706–17711, 2001. doi: 10.1074/jbc.M100544200. [DOI] [PubMed] [Google Scholar]
- 103.Weiss RG, Maslov M. Normal myocardial metabolism: fueling cardiac contraction. Advan Stud Med 4: S457–S463, 2004. [Google Scholar]
- 104.Westaway SK, Reinier K, Huertas-Vazquez A, Evanado A, Teodorescu C, Navarro J, Sinner MF, Gunson K, Jui J, Spooner P, Kaab S, Chugh SS. Common variants in CASQ2, GPD1L, and NOS1AP are significantly associated with risk of sudden death in patients with coronary artery disease. Circ Cardiovasc Genet 4: 397–402, 2011. doi: 10.1161/CIRCGENETICS.111.959916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Winnik S, Auwerx J, Sinclair DA, Matter CM. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J 36: 3404–3412, 2015. doi: 10.1093/eurheartj/ehv290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xiao Y, Zhang X, Fan S, Cui G, Shen Z. MicroRNA-497 inhibits cardiac hypertrophy by targeting Sirt4. PLoS One 11: e0168078, 2016. doi: 10.1371/journal.pone.0168078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S, Sadoshima J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS One 9: e98972, 2014. doi: 10.1371/journal.pone.0098972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yamasaki-Mann M, Demuro A, Parker I. cADPR stimulates SERCA activity in Xenopus oocytes. Cell Calcium 45: 293–299, 2009. doi: 10.1016/j.ceca.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yamasaki-Mann M, Demuro A, Parker I. Modulation of endoplasmic reticulum Ca2+ store filling by cyclic ADP-ribose promotes inositol trisphosphate (IP3)-evoked Ca2+ signals. J Biol Chem 285: 25053–25061, 2010. doi: 10.1074/jbc.M109.095257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab 14: 528–536, 2011. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ, Auwerx J. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352: 1436–1443, 2016. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- 112.Zhang R, Shen Y, Zhou L, Sangwung P, Fujioka H, Zhang L, Liao X. Short-term administration of nicotinamide mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J Mol Cell Cardiol 112: 64–73, 2017. doi: 10.1016/j.yjmcc.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang X, Li W, Shen P, Feng X, Yue Z, Lu J, You J, Li J, Gao H, Fang S, Li Z, Liu P. STAT3 suppression is involved in the protective effect of SIRT6 against cardiomyocyte hypertrophy. J Cardiovasc Pharmacol 68: 204–214, 2016. doi: 10.1097/FJC.0000000000000404. [DOI] [PubMed] [Google Scholar]
- 114.Zhang Y, Wang B, Fu X, Guan S, Han W, Zhang J, Gan Q, Fang W, Ying W, Qu X. Exogenous NAD+ administration significantly protects against myocardial ischemia/reperfusion injury in rat model. Am J Transl Res 8: 3342–3350, 2016. [PMC free article] [PubMed] [Google Scholar]
- 115.Zhu XD, Li J, Wang HS, Lee C, Vanden Hoek TV. Nicotinamide restores tissue Nad+ and improves outcomes in a murine model of cardiac arrest (Abstract). Circulation 134: A12474, 2016. [Google Scholar]