

Abstract
Alternate frame folding (AFF) is a protein engineering methodology the purpose of which is to convert an ordinary binding protein into a molecular switch. The AFF modification entails duplicating an amino- or carboxy-terminal segment of the protein and appending it to the opposite end of the molecule. This duplication allows the protein to interconvert, in a ligand-dependent fashion, between two mutually exclusive native folds: the wild-type structure and a circularly permuted form. The fold shift can be detected by placement of extrinsic fluorophores at sites sensitive to the engineered conformational change. Here, we apply the AFF mechanism to create several ribose-sensing proteins derived from Thermoanaerobacter tengcongensis ribose binding protein. Our purpose is to systematically explore the parameters of the AFF design. These considerations include the site of circular permutation, the length and location of the duplicated segment, thermodynamic and kinetic optimization of the switching mechanism, and placement of extrinsic fluorophores. Three of the four AFF variants created here undergo the expected conformational shift and exhibit a ribose-dependent fluorescence change. The fourth construct fails to switch folds upon addition of ribose, likely because the circularly permuted form folds much more slowly than the nonpermuted form. This disparity apparently introduces a kinetic barrier that partitions the refolding molecules to the nonpermuted structure. The results of this study serve as a guideline for applying the AFF modification to other proteins of biomedical, diagnostic, and industrial interest.
Graphical abstract
Protein conformational switches are defined by their ability to change structure in response to a stimulus. Switchable proteins are particularly effective platforms for biosensor design because they can convert a molecular recognition event into an output signal via conformational change. Conventional biosensors typically achieve signal transduction by attaching a biological receptor to an electronic, optical, or mechanical device. For example, surface plasmon resonance detects binding by a change in the index of refraction that occurs when the target ligand interacts with a surface-immobilized recognition element. The power of conformational switching is that it allows recognition and reporting functions to be integrated in a single protein molecule, thereby offering significant opportunity for miniaturization, simplification, and economization. Fluorescence detection is advantageous in this regard because it is sensitive, potentially ratiometric, and amenable to quantification by a variety of instruments ranging from simple desktop fluorimeters to sophisticated single-molecule apparatus. Moreover, fluorescence makes it possible to genetically encode the entire sensor by using fluorescent proteins as reporter groups.
Periplasmic binding proteins (PBPs) are a platform for testing the concepts described above. PBPs are a superfamily of nonenzymatic receptors whose members are found in all kingdoms of life. They are useful starting points for biosensor development because they bind a number of small molecules, including sugars, amino acids, metals, and cofactors, with high specificity and dissociation constants (Kd) in the nanomolar to micromolar range.1 PBPs undergo a rigid-body domain movement upon engaging their targets, as shown in Figure 1 for ribose binding protein (RBP). Ribose binds in a cleft at the hinge between the amino- and carboxy-terminal lobes of RBP, causing them to close by ∼30° and almost completely occlude the substrate. Strategic placement of environmentally sensitive fluorophores, either singly2,3 or in donor–acceptor pairs [for Förster resonance energy transfer (FRET)],4–6 has resulted in fluorescent biosensors for ribose and several other target ligands.
Figure 1.
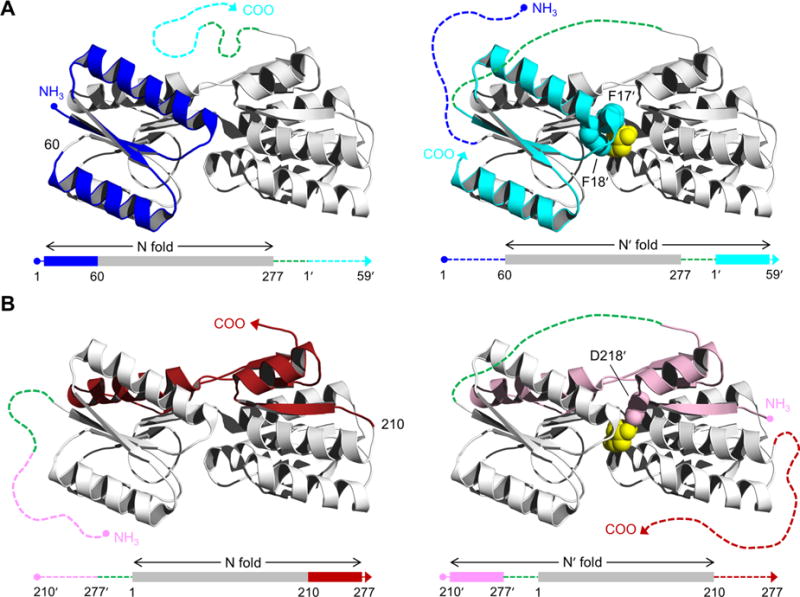
Structure of RBP and schematic of the AFF design. RBP binds ribose (yellow spheres) in the cleft between amino- and carboxy-terminal domains. AFF variants are generated by duplicating either (A) an amino-terminal segment (blue) and fusing it to the carboxy terminus (cyan), as exemplified by AFF60, or (B) a carboxy-terminal segment (red) and appending it to the amino terminus (pink), as demonstrated by AFF210. The N and N′ conformations are shown at THE left and right, respectively. In the amino acid sequences, depicted below each structure, rectangles indicate folded structure and dashed lines represent unfolded/unstable structure. The dashed green line is a 30-amino acid linker. The copy of the duplicated segment that is orphaned in each conformation is depicted as a dashed line extending from one of the termini. Critical ribose contacts are established by Phe17 and Phe18 (cyan spheres in panel A) and by Asp218 (pink spheres in panel B). Substrate binding to the N conformations of AFF60 and AFF210 is abolished by introducing the F17A/F18A double mutation and the D218S mutation into the blue and red regions, respectively. Addition of ribose, and the binding energy provided by Phe17′ and Phe18′ (AFF60) or Asp218′ (AFF210) in the remodeled binding pocket, drive the shift from the N fold to the N′ fold. The folded blue segment of AFF60 becomes an amino-terminal tail, and the cyan carboxy-terminal tail becomes part of the folded protein. The analogous event happens with the red and pink regions of AFF210 except the locations of the tails are reversed. To detect this rearrangement, fluorophores are placed at the site of permutation (position 60 in AFF60, position 210 in AFF210, etc.) and at the carboxy termini of AFF60 and AFF70 or the amino termini of AFF186 and AFF210.
We recently developed the alternate frame folding (AFF) methodology for introducing a binding-dependent conformational change into a protein where none existed previously.7 Like the class of intrinsically disordered proteins (IDPs) found in nature, AFF links binding to folding. The unique aspect of AFF, however, is that folding of one portion of the molecule is coupled to unfolding of another region.8 This folding–unfolding reaction constitutes a conformational change that can, in principle, be engineered into many different proteins. The goals of this study are to test this hypothesis using RBP as a model protein and to explore the parameters that go into the AFF design.
Figure 1 illustrates the steps for converting a binding protein into a switch by the AFF method. First, a segment from the amino terminus of the protein is duplicated and appended to the carboxy terminus (Figure 1A), or a segment from the carboxy terminus is copied and fused to the amino terminus (Figure 1B). The duplicated amino acids are indicated by prime superscripts but are otherwise numbered in a manner identical to that of the parent sequence. In both cases, the linkages are made via a flexible peptide that is long enough to bridge the termini of the wild-type (WT) protein. The twinned segments allow the molecule to adopt one of two native folds: that of the WT protein (N) or that of a circular permutant (N′). Circular permutation generates termini at a new location, usually chosen to be a former surface loop, and creates a new surface loop connecting the original termini. Permutation thus changes the topology of a protein but frequently not its structure or function. The AFF-modified protein cannot exist simultaneously in both forms. Consequently, it interconverts between N and N′ as dictated by their relative thermodynamic stabilities and the kinetic barriers that separate them.9
Here, ligand binding energy is used to drive the N ⇆ N′ conversion. The directionality of the fold shift is established by mutating one or more critical binding residues in one of the duplicate segments (F17 and F18 in Figure 1A and D218 in Figure 1B). One typically chooses to knock out binding of the more stable of the two conformations. In this way, the majority of molecules will exist in the binding-incompetent state and ligand binding can shift the populations to favor the binding-competent form. The key aspect of this design as it pertains to biosensors is that it creates tails (consisting of the “orphaned” segment) that extend from the amino or carboxy terminus of N or N′ (Figure 1). These tails exchange positions as well as potentially fold and unfold when the protein switches from N to N′ and vice versa. This rearrangement forms the basis for detection by distance-sensitive fluorophores.
When implementing the AFF modification, one must first determine the location and length of the segment to duplicate. What factors determine this choice? The primary requirement is that the duplication must encompass at least one residue that, when mutated, eliminates substrate binding. Beyond that criterion, there are two other considerations. (i) The circularly permuted N′ form must be functional and at least moderately stable. Accordingly, conventional wisdom suggests that the duplication site should be in a surface loop/turn, rather than in a secondary structure element or in the core of the protein, and not be overly close to the binding pocket. (ii) The orphaned tails should not cause the protein to misfold, aggregate, or become degraded. One strategy is to make them very short. Duplicating as few amino acids as possible might reduce the statistical probability of misfolding, as well as increase the likelihood that the tails will be unstructured and flexible (which may maximize the distance-dependent fluorescence change resulting from fold switching). The risk of short, disordered tails is that they may foster proteolytic degradation. The second tactic is to make the tails longer so that they retain residual, perhaps nativelike structure. Residual structure may confer resistance to degradation, but it may physically hinder the fold shift. Arguments can be made for either strategy minimizing aggregation.
The second step of the AFF design is to adjust the relative stabilities of N and N′, if necessary, to optimize the signal change (gain) of the sensor and in some cases its affinity. Switch performance is typically maximized when the ratio of binding-incompetent to binding-competent forms is ∼10:1 in the absence of ligand. This ratio balances high gain (ligand binding will induce ≥90% of molecules to switch conformations) and high affinity (a portion of the binding energy is used to drive the unfavorable conformational change, thus lowering intrinsic affinity). If a higher Kd is desired, e.g., to match it to the concentration of the analyte to be detected, then the ratio can be adjusted to >10:1. Stability tuning is achieved by introducing stabilizing or destabilizing mutations into one of the duplicated segments.
The final step in the AFF design is to harness the conformational change to a fluorescent output. The mechanism of N ⇆ N′ switching lends itself to the natural placement of distance-sensitive fluorescent donor–acceptor pairs. For amino-terminal duplications (Figure 1A), the donor and acceptor are attached to the carboxy terminus and to the loop containing the permutation site (position 60). The groups will always be proximal in N′ because they are fixed by structure that corresponds to a loop of the WT protein. They will likely be farther apart in N, depending to an extent on the flexibility and/or residual structure of the amino-terminal tail. Likewise, placing fluorophores at the permutation loop (position 210 in Figure 1B) and the amino terminus is an analogous strategy for the AFF constructs made by duplicating carboxy-terminal segments.
Herein, we apply the AFF design to Thermoanaerobacter tengcongensis RBP in an effort to convert it to a fluorescent ribose sensor. Our goal is to systematically explore the AFF design parameters described above. RBP (277 amino acids) is substantially larger and more structurally complex than the two proteins to which we have previously applied the AFF modification [calbindin D9k (75 amino acids)7–9 and barnase (110 amino acids)10]. This study therefore addresses the generality of AFF as a technique for converting binding proteins to biosensors and will help to define guidelines for that conversion.
EXPERIMENTAL PROCEDURES
Gene Construction and Protein Purification
T. tengcongensis RBP was a gift from H. W. Hellinga (Duke University, Durham, NC). The gene did not contain the amino-terminal signal sequence; the coding sequence begins with Lys at position 37 and ends with a His8 tag connected to the carboxy terminus by a Asp-Leu-Glu linker. The single Cys residue (Cys101) was mutated to Ser in all variants. Circular permutants were created by ligating two halves of the gene using polymerase chain reaction methods. The sequence of the peptide used to link the original amino and carboxy termini of all permutants is GGAASGGAAGGSSAAASSGAGAAGGSGAGG. For the N analogues, a single Cys residue was introduced between residues 59 and 60 (N60), 69 and 70 (N70), 185 and 186 (N186), and 209 and 210 (N210). For N′60 and N′70, Cys was added at the amino terminus and the carboxy-terminal His8 tag linker sequence was changed from Asp-Leu-Glu to Cys-Leu-Glu. For N′186 and N′210, a His6 tag sequence was connected to the amino terminus via an Ala-Ser-Cys linker, and a second Cys was introduced at the carboxy terminus. AFF variants were constructed by ligating appropriate segments of the N and N′ analogue genes described above. All genes were sequenced and verified in their entirety.
Plasmids were transformed into Escherichia coli BL21(DE3), and proteins were expressed by being induced with 40 mg/L isopropyl β-thiogalactopyranoside and shaken for 12–18 h at 20 °C. Proteins were purified using nickel nitrilotriacetic acid affinity resin under denaturing conditions [6 M guanidine hydrochloride (GdnHCl)] to remove bound ribose, refolded by being dialyzed against double-distilled water, and lyophilized. Samples were judged to be >95% pure by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) with Coomassie brilliant blue staining. Protein yields were typically ≥80 mg/L of starting culture.
Analytical Ultracentrifugation
Samples were prepared by reducing the proteins with 3.3 mM tris(2-carboxyethyl)-phosphine (TCEP) in 0.35 M GdnHCl, blocking Cys residues with 6.67 mM N-ethylmaleimide, and desalting the proteins into 20 mM sodium phosphate (pH 7.0), 0.15 mM NaCl, and 4 mM TCEP using a 10DG column (Bio-Rad). Experiments were performed using a Beckman Coulter ProteomeLab XL-A analytical ultracentrifuge equipped with a four-hole An-60 Ti rotor. Sedimentation velocity data were collected at 60000 rpm (262000g) using a 12 mm, two-sector Epon centerpiece with absorbance detection at 230 nm; 300 scans were recorded for each sample with the time interval between scans set to zero. Sedimentation boundaries were analyzed by the continuous distribution method using SEDFIT.11
Circular Dichroism
Wavelength scan and thermal denaturation experiments were performed on a model 202 or model 420 spectropolarimeter (Aviv Biomedical). For wavelength scans, 5 μM protein was incubated in 10 mM sodium phosphate (pH 7.0), 0.1 M NaCl, and 0.5 mM TCEP for 3 h at 50 °C, then transferred to the CD instrument, and allowed to equilibrate at 37 °C for at least 10 min before data were collected. Scans were collected in a 0.2 cm path length cuvette. For thermal melts, the protein concentration was lowered to 1 μM and the cuvette path length was increased to 1 cm. The temperature was increased at a rate of 2 °C/min, and proteins were equilibrated for 60 s prior to each 60 s data collection time.
Isothermal Titration Calorimetry
ITC experiments were performed on a MicroCal VP-ITC instrument. Proteins were dialyzed against the same buffer used in CD experiments, and the protein concentration was adjusted to 35–50 μM. Data were collected at 37 °C with a 2 min interval between ribose injections. Data were fit to the one-site binding model using the Origin software package that was bundled with the MicroCal instrument.
Fluorescence Labeling and FRET Assays
Lyophilized proteins were dissolved in binding buffer [10 mM sodium phosphate (pH 7.0) and 0.10 M NaCl] with 0.35 M GdnHCl and reduced via addition of 20 mM DTT. After 1 h, DTT was removed by passing the sample through a 10DG desalting column equilibrated in the buffer described above. A 1.5-fold excess (relative to Cys groups) of BODIPY-FL N-(2-aminoethyl)maleimide (Invitrogen) was immediately added to the desalted protein and allowed to react for 2 h at room temperature. Samples were then desalted as described above to remove unbound dye and GdnHCl.
Equilibrium FRET assays were performed by incubating 1 μM protein with the indicated concentrations of ribose in binding buffer for 1 h at 50 °C. Samples were then cooled to room temperature and scanned on a Horiba Fluoromax-4 fluorimeter (490 nm excitation, 500–600 nm emission, 1 nm bandpass on each setting). For kinetic FRET experiments, RBP samples were equilibrated at 50 °C for at least 1 h prior to measurements. Samples were then placed in the 50 °C-jacketed cuvette holder, and a 50-fold dilution of ribose was added to the final concentrations indicated in the figure. Baseline curves were generated by adding buffer instead of ribose.
Limited Trypsin Digestion
The same BODIPY-labeled proteins prepared above were used in proteolysis assays. Protein samples (5 μM in 50 μL aliquots) with or without 0.5 mM ribose were incubated at 50 °C for 1 h and then cooled to 20 °C. After 15 min, 0.1 μg of Trypsin Gold (Promega) was added. Digestion proceeded for 60 min at 20 °C, after which it was quenched by addition of formic acid to a final concentration of 1%. Samples were run on 13% SDS–polyacrylamide gels. Gels were scanned for BODIPY fluorescence using a Typhoon 9410 imager (GE Healthcare). For Western blotting, proteins were transferred to Immobilon-P membranes (Millipore) and probed with tetra-His antibody (Qiagen). Immunodetection was performed with a goat anti-mouse HRP-conjugated antibody (Bio-Rad) following the manufacturer’s protocol. Intensities of gel bands were measured using ImageJ (National Institutes of Health, Bethesda, MD).
RESULTS
RBP Constructs and Nomenclature
Three categories of RBP constructs were created for this study: AFF variants, N frame analogues, and N′ frame analogues. The nomenclature and sequence connectivities of these variants are listed in Table 1. AFF variants are named according to the position at which the sequence was duplicated (Figure 1). For example, AFF60 was made by copying residues 1–59 and appending them to the carboxy terminus, and AFF210 was generated by duplicating residues 210–277 and fusing them to the amino terminus. N analogues (designated N60, N210, etc.) consist of the WT sequence with the addition of the binding-knockdown mutations and Cys insertions described below and in Table 1. N′ analogues (N′60, N′210, etc.) are circular permutants of RBP in which the sequence was permuted at the position indicated and Cys groups were inserted at the designated locations. All variants in this study, including WT RBP, contain the C101S mutation in which the single naturally occurring Cys residue was removed to facilitate fluorescence labeling at Cys residues placed elsewhere in the sequence.
Table 1.
Partial List of RBP Constructs Created for This Study
| variant | amino acid connectivity | additional mutationsa |
|---|---|---|
| WT | 1–277 | none |
| N60 | 1–277 | F17A and F18A (binding), Cys60* (labeling) |
| N70 | 1–277 | F17A and F18A (binding), Cys70* (labeling) |
| N186 | 1–277 | D218S (binding), Cys186* (labeling) |
| N210 | 1–277 | D218S (binding), Cys210* (labeling) |
| N′60 | (60–277)–linker–(1–59) | Cys inserted at amino and carboxy termini (labeling) |
| N′70 | (70–277)–linker–(1–69) | Cys inserted at amino and carboxy termini (labeling) |
| N′186 | (186–277)–linker–(1–185) | Cys inserted at amino and carboxy termini (labeling) |
| N′210 | (210–277)–linker–(1–209) | Cys inserted at amino and carboxy termini (labeling) |
| AFF60 | (1–277)–linker–(1′–59′) | F17A and F18A (binding), Cys60* and Cys inserted at the carboxy terminus (labeling) |
| AFF70 | (1–277)–linker–(1′–69′) | F17A and F18A (binding), Cys70* and Cys inserted at the carboxy terminus (labeling) |
| AFF186 | (186′–277′)–linker–(1–277) | D218S (binding), Cys186* and Cys inserted at the amino terminus (labeling) |
| AFF210 | (210′–277′)–linker–(1–277) | D218S (binding), Cys210* and Cys inserted at the amino terminus (labeling) |
| AFF60rev, AFF70rev, AFF186rev, AFF210rev | same as AFF60, AFF70, AFF186, AFF210 | same as AFF60, AFF70, AFF186, AFF210 except binding mutations transferred to the N′ frame |
| N60rev, N70rev, N186rev, N210rev | same as N60, N70, N186, N210 | same Cys* labeling mutations as N60, N70, N186, N210 |
| N′60rev, N′70rev, N′186rev, N′210rev | same as N′60, N′70, N′186, N′210 | F17′A and F18′A (binding; N′60rev and N′70rev), D218′S (binding; N′186rev and N′210rev), same Cys* labeling mutations as N′60, N′70, N′186, N′210 |
All variants, including WT, contain the C101S mutation. All variants except for WT contain the L131D kinetic tuning mutation. An asterisk indicates an insertion rather than a substitution mutation; e.g., Cys60* is inserted between residues 59 and 60.
Choice of Circular Permutants and AFF Design
We first determined which circular permutants of RBP are stable and capable of binding ribose. RBP contains 16 surface loops and turns. Six of these loops are ruled out as permutation sites because they are either in contact with ribose or at the interface of the amino and carboxy domains. An additional consideration is that, because the duplicated segment must contain at least one substrate binding residue, one such amino acid should reside in the shorter of the two polypeptides bisected by the permutation site. This allows one to duplicate the short segment rather than the long one when making the AFF construct. Several studies of E. coli RBP have identified mutations that significantly diminish the level of ribose binding.2,12,13 In T. tengcongensis RBP, the sequence of which is 57% identical and 76% similar to that of the E. coli protein,14 these mutations correspond to N15A, F17A, F18A, N66A, D91A, F167A, N193A, F217A, and D218S. We therefore excluded two additional permutation sites at loop positions 228 and 260. The remaining eight candidate loops are centered at positions 60, 70, 85, 97, 125, 137, 186, and 210.
We expressed, purified, and characterized all eight of the circular permutants. These permutants are designated N′60WT, N′70WT, N′85WT, N′97WT, N′125WT, N′137WT, N′186WT, and N′210WT (WT indicates that they do not contain the additional mutations described in Table 1 that were added to subsequent constructs). We first assessed stability of the permutants by thermal denaturation. WT RBP denatures above 100 °C; therefore, thermal unfolding experiments were performed in the presence of 0.34 M GdnHCl. Under these conditions, WT unfolds reversibly with an apparent melting temperature (Tm) near 90 °C (Figure S1A of the Supporting Information). All curves in Figure S1A of the Supporting Information show significant hysteresis on cooling, suggesting that the proteins fold and/or unfold slowly. Melting temperatures are therefore apparent figures that likely depend on the rate of heating. Tm values of the permutants range from 73 to 83 °C (Figure S1A of the Supporting Information). Addition of ribose increases the Tm values of the permutants by 11–15 °C and increases the Tm of WT RBP above 100 °C (Figure S1B of the Supporting Information). All eight of the permutants are therefore folded, stable, and functional.
We selected N′60WT, N′70WT, N′186WT, and N′210WT for further characterization. The reason is that we elected to build four AFF variants: two by duplicating amino-terminal segments (Figure 1A) and two by duplicating carboxy-terminal segments (Figure 1B). Choosing permutation sites closest to the amino terminus (positions 60 and 70) and closest to the carboxy terminus (positions 186 and 210) results in AFF constructs with the shortest possible duplications.
Kinetic Tuning
The hystereses observed in thermal denaturation curves suggest that folding and/or unfolding rates of RBP are slow. This can potentially be problematic for AFF switching because the N ⇆ N′ transition can be at least partially rate-limited by an unfolding step.9 We denatured WT RBP [bearing the F17A and F18A ribose binding mutations (see below)] in various concentrations of GdnHCl (20 °C) and extrapolated the fitted rate constants to estimate unfolding halftimes in the absence of denaturant (tunf). Unfolding is complex; two kinetic phases are observed below 6 M GdnHCl, and the logarithm of the slower rate exhibits curvature when plotted against GdnHCl concentration (Figure S2 of the Supporting Information). Extrapolating the most linear portion of the slower rate yields an upper limit of 3.9 × 10−10 s−1 (tunf ≥ 55 years) in the absence of denaturant.
The strategy for decreasing tunf, and potentially increasing the rate of N ⇆ N′ switching, is to use site-directed mutagenesis to destabilize the N state or the N′ state (or both) and rely on the commonly observed outcome that such mutations destabilize the transition state ensemble for unfolding as well.9,15 If the equilibrium population of N or N′ needs to be adjusted, then kinetic optimization can go hand in hand with stability tuning. Here, however, we placed destabilizing mutations into the region of the protein shared by N and N′ (colored gray in Figure 1). These mutations will in theory destabilize N and N′ to comparable extents, thereby increasing their unfolding rates while maintaining the equilibrium position of the switch. We chose to introduce the hydrophobic-to-charged mutations V52E, I77D, I90D, and L131D individually into the same F17A/F18A construct described above. The first three decrease tunf but not enough: values remain in the regime of days (not shown). The L131D mutation decreases tunf to 2.2 h (Figure S2 of the Supporting Information). We therefore incorporated it into all AFF constructs and analogues from this point forward (Table 1).
Stability Tuning
The structures of the WT and circular permuted forms of a protein approximate only those of the N and N′ states that the molecule takes on in its AFF-modified incarnation. Nevertheless, the relative stabilities of the analogues have been shown to predict the population distribution of N and N′ reasonably accurately.7 Accordingly, the goal of this step is to adjust the folding free energies of the analogues such that the N analogues are more stable than the N′ analogues in the absence of ligand, and the situation is reversed in the presence of ligand. To match the sequences of the analogues as closely as possible to those of the eventual AFF constructs, we introduced Cys residues and ribose binding mutations into the positions indicated in Table 1.
Figure 2 superimposes thermal denaturation curves of the N and N′ analogues. We first note that the L131D kinetic tuning mutation destabilizes RBP, but all variants remain relatively stable (Tm > 45 °C). Neither the L131D mutation nor insertion of Cys residues appears to adversely affect the ability of the permutants to bind ligand, as demonstrated by the ribose-induced increase in Tm values of N′60, N′70, N′186, and N′210. Melting curves of N60, N70, N186, and N210 are identical in the presence and absence of ribose (not shown), indicating that binding is abolished by the F17A and F18A mutations (N60 and N70) and the D218S mutation (N186 and N210).
Figure 2.
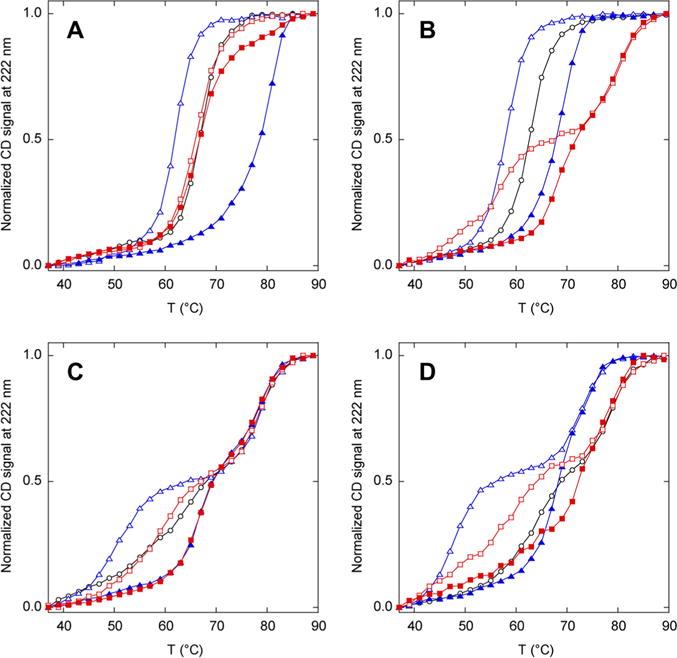
Relative stabilities of N analogues (black), N′ analogues (blue), and AFF constructs (red) determined by thermal denaturation. Empty and filled symbols indicate the absence and presence of 1 mM ribose, respectively. Lines are drawn to guide the eye only: (A) N60, N′60, and AFF60, (B) N70, N′70, and AFF70, (C) N186, N′186, and AFF186, and (D) N210, N′210, and AFF210.
The melting temperature of N′60 is lower than that of N60 in the absence of ribose and is higher than that of N60 in the presence of ribose (Figure 2A). The same is true for N′70 and N70 (Figure 2B). Further stability tuning is therefore not necessary for AFF60 and AFF70. Unexpectedly, N186 and N210 denature in two transitions (Figure 2C,D). This result is surprising because the only differences between N186 and N210 and between N60 and N70 (which denature in an apparent two-state fashion) are the ribose binding mutations and the placement of a single Cys residue in different surface loops. The first transitions exhibit slightly lower Tm values compared to those of N60 and N70, whereas the midpoints of the second transitions occur at temperatures beyond which N60 and N70 are already unfolded. This result indicates that the D218S mutation and/or Cys insertions cause a partially folded intermediate to be populated at high temperatures. The unusually high thermal stability of the intermediate suggests that it may be an associated state. N186′ and N210′ denature in an even more pronounced three-state fashion because of a shift in the first transition to lower temperatures (Figure 2C,D). Addition of ribose increases the Tm of the first transitions of N186′ and N210′ but not those of the second transitions, consistent with the idea that the first transition corresponds to unfolding of native RBP to the intermediate state. If this is true, then no additional thermodynamic tuning is necessary for AFF186 and AFF210 because Tm values of the lower-temperature transitions of N′186 and N′210, in the presence and absence of ribose, bracket those of their respective N state analogues.
Monomer and Oligomer Tests
To assess the oligomerization state of AFF variants, and to establish whether CD and fluorescence data could be explained by a change in the monomer–oligomer distribution rather than a fold shift, we determined apparent molecular masses by analytical ultra-centrifugation (AUC) and size exclusion chromatography (SEC). In sedimentation velocity experiments performed at 10 μM in the absence of ribose, AFF70, AFF186, and AFF210 sediment predominantly as monomers with apparent molecular masses that are slightly lower than the theoretical values (Table 2 and Figure S3 of the Supporting Information). The difference in molecular mass suggests that the proteins may contain disordered or extended regions, possibly corresponding to the orphaned segments, which are known to increase the frictional coefficient.16 A faster-sedimenting minor species of approximately twice the monomer molecular mass is observed in all samples. This peak likely corresponds to a small population of a disulfide-bonded dimer that is seen in nonreducing PAGE of the same samples (not shown). In SEC experiments conducted at the same protein concentration, all AFF constructs elute in a single major peak with an apparent molecular mass of 91–110 kDa (Figure S4A of the Supporting Information). In light of the AUC results, the most likely interpretation is that the proteins are monomeric and contain regions of disorder. As a rule of thumb, a compact IDP elutes with an apparent molecular mass roughly double that of the actual molecular mass, with the discrepancy increasing to 4–6-fold for a more extended IDP.17 Addition of ribose does not affect the AUC sedimentation (Figure S3 of the Supporting Information and Table 2) or SEC elution (Figure S4B of the Supporting Information) of any protein examined. We therefore conclude that AFF variants are monomeric and that the ribose-induced changes in CD, fluorescence, and proteolytic digestion patterns observed below are not caused by a change in the oligomerization state.
Table 2.
Analytical Ultracentrifugation Parameters of AFF Variantsa
| variant |
s value (S) |
% total peak area | apparent molecular mass (kDa) |
theoretical molecular mass (kDa) |
|---|---|---|---|---|
| AFF70 | 1.93 | 82 | 35.1 | 40.6 |
| AFF70 | 3.02 | 13 | 68.0 | 81.3 (dimer) |
| AFF186 | 1.85 | 82 | 39.5 | 42.6 |
| AFF186 | 2.68 | 13 | 68.8 | 85.2 (dimer) |
| AFF210 | 1.87 | 89 | 33.7 | 40.1 |
| AFF210 | 2.71 | 6.2 | 58.8 | 80.1 (dimer) |
| AFF70 with ribose | 1.89 | 87 | 38.2 | 40.8 |
| AFF70 with ribose | 2.99 | 11 | 76.5 | 81.5 (dimer) |
| AFF186 with ribose | 1.85 | 88 | 38.5 | 42.7 |
| AFF186 with ribose | 2.74 | 10 | 69.3 | 85.5 (dimer) |
| AFF210 with ribose | 1.87 | 90 | 33.3 | 40.2 |
| AFF210 with ribose | 2.79 | 4.4 | 60.6 | 80.4 (dimer) |
The protein concentration was 10 μM and the temperature 10 °C.
Fold Shift Test: Tm Change
Thermal denaturation of AFF variants provides an initial test for the ribose-induced fold shift (Figure 2). AFF60 denatures in a single transition that is unperturbed by the addition of ribose. AFF70, AFF186, and AFF210 unfold in two transitions of which the first shifts in the presence of ribose. These data demonstrate that AFF70, AFF186, and AFF210 bind ribose and suggest that AFF60 does not. The results do not, however, prove that ribose binding induces the N to N′ fold shift. It is instead possible that these variants already exist in the N′ conformation in the absence of ribose, despite the finding that the N′ analogues are less stable than the corresponding N analogues (Figure 2).
Fold Shift Test: Limited Proteolysis
To address that question, we sought to test the fold shift mechanism of AFF by partial trypsin digestion. Trypsin preferentially attacks Lys and Arg residues in disordered, weakly stable, or otherwise exposed regions. In AFF variants, these regions are hypothesized to consist of the orphaned tails (Figure 1). The major proteolytic product is thus predicted to be the N analogue (in the absence of ribose) or the N′ analogue (in the presence of ribose), with each species made longer by the fragment of the tail that terminates at the first accessible Lys or Arg residue. In particular, the (His)6–8 purification tags (HT) that were placed at the carboxy termini of AFF60 and AFF70 and at the amino termini of AFF186 and AFF210 are expected to be cleaved off in N and remain attached in N′ (Figure 3). The test is to use an anti-HT Western blot to determine whether a band is absent in the ribose-free lane and apparent in the ribose-containing lane. HTs were also appended to the amino termini of N′186 and N′210 and to the carboxy termini of N′60, N′70, and all N analogues. We digested the identical BODIPY-labeled proteins that were prepared for FRET studies. Fluorescence scans of the same gels further identify the products, as the BODIPY groups reveal fragments that do not contain HT (Figure 3).
Figure 3.
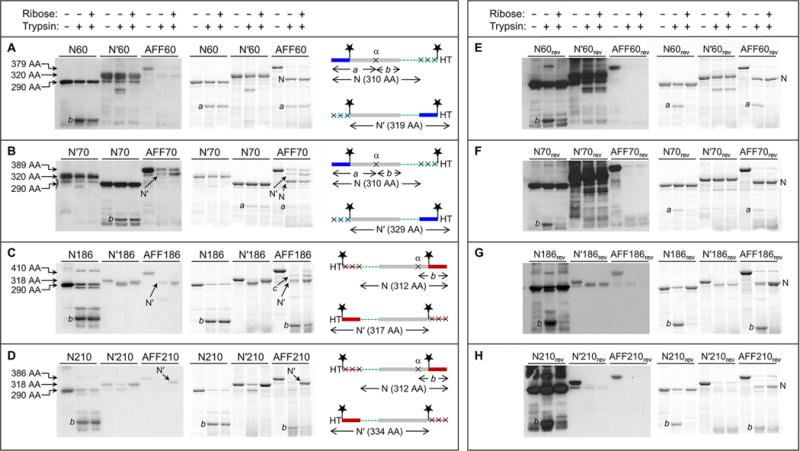
Fold shift test by partial trypsin digestion. Gels at the left are anti-HT Western blots. Gels at the right are BODIPY fluorescence scans of the same gels at the left. Amino acid sequences are shown at the far right, schematized to highlight the N and N′ conformations and the positions of the HTs, BODIPY groups (stars), and trypsin cleavage sites (×). The same color scheme as in Figure 1 is used, as well as same the rectangle and line designation for folded and unfolded structure, respectively. Amino acid lengths of undigested N analogues, N′ analogues, and AFF constructs are labeled to the left of the gels. These lengths, as well as the amino acid sequence diagrams, are omitted from panels E–H because they are identical to those in panels A–D, respectively. Note that the loading order of N70 and N′70 is switched compared to that of gels of the other proteins.
We first consider AFF60 (Figure 3A). A Western blot does not show any bands in the presence or absence of ribose, suggesting that the protein adopts the N fold under both conditions. Two bands are, however, observed by BODIPY fluorescence. The larger is consistent with the theoretical N fragment (cleavage at Lys1′). The smaller (fragment a) is produced by incomplete cleavage of the N fragment at a single position (site α), the approximate location of which is shown in Figure 3A. This deduction is supported by the observation that all N analogues are partially cleaved at site α, generating the same BODIPY-labeled fragment a plus fragment b, which contains HT but not BODIPY groups. Fragment b is expected to be present in the AFF60 lanes, but it is undetectable because it lacks both BODIPY and HT. These results confirm the previous conclusion that AFF60 does not bind substrate because it is unable to convert to the N′ fold (Figure 2A).
Trypsinization of AFF70 theoretically produces N and N′ fragments of 310 amino acids (cleavage at Lys1′) and 329 amino acids (cleavage at Lys59), respectively (Figure 3B). The Western blot shows a single band indicative of the N′ fragment. This band is faint in the absence of ribose and gains intensity in its presence. BODIPY fluorescence detects both N and N′ fragments. Comparing the densities of these bands estimates the N:N′ ratio to be ∼10:1 in the ribose-free lane and ∼1:1 in the ribose-containing lane. This finding suggests that AFF70 shifts from N to N′ in response to ribose but that the conversion is not complete within the 1 h incubation time.
Digestion of AFF186 is anticipated to yield N and N′ fragments of 312 amino acids (cleavage at Lys273′) and 317 amino acids (cleavage at Lys185) (Figure 3C). These species are likely to run very closely on gels, but the Western blot resolves the N′ fragment and finds that its intensity increases upon addition of ribose in a manner similar to that of AFF70. N186 shows extensive cleavage at site α; consequently, fragment b serves as a marker of the N conformation (fragment a cannot be observed because it lacks BODIPY and HT). Band b is dark in the ribose-free lane of AFF186 and lighter in the ribose-containing lane. Substrate binding thus appears to induce the N to N′ shift. In the absence of ribose, N′186 is cleaved at an internal position near the carboxy terminus (the amino-terminal His tag is retained), but this site is not attacked in ribose-free AFF186 as the N′ fragment is of the expected size. A larger, unexplained band (fragment c) is observed. Lacking the amino-terminal HT and BODIPY groups but containing BODIPY at position 186*, fragment c is generated by truncation from the amino and possibly carboxy termini. Although its identity remains uncertain, one intriguing possibility is that it arises from a second permuted structure in which only a subset of the duplicated residues are used to fold. For example, if residues 210–277 swap with residues 210′–277′, then the N′ conformation of AFF186 would be that of AFF210 with short tails protruding from both termini (consisting of residues 186′–209′ and 186–209). Cleavage of these tails at the anticipated sites (Lys202′ and Lys225) would produce a 331-amino acid product consistent with the size of band c.
Limited proteolysis of AFF210 theoretically generates an N fragment of 312 amino acids (cleavage at Lys273′) and an N′ fragment of 334 amino acids (cleavage at Lys225) (Figure 3D). The digestion patterns of AFF210 resemble those of AFF70 and AFF186, and similar conclusions are drawn for all three. The N′ fragment is absent in ribose-free AFF210 and appears as a strong band in the ribose-containing sample. Band b, the marker for the N conformation, changes intensity in the opposite manner. In summary, AFF70, AFF186, and AFF210 all show evidence of the ribose-induced shift from N to N′, whereas AFF60 appears to remain stuck in the N conformation.
Reverse Binding Mutants
As a final test of the fold shift mechanism, we created a set of AFF variants (designated AFF60rev, AFF70rev, AFF186rev, and AFF210rev) in which the substrate binding mutations were moved from N to N′. Transferring the binding mutations reverses the position of the switch so that the ribose-bound form is N. Moreover, because this transfer also stabilizes the Nrev analogues relative to the N′rev analogues (Figure S5 of the Supporting Information), it is predicted to reduce the minor population of N′ that appeared to be present in the ribose-free states of AFF70, AFF186, and AFF210. Western blots of tryptic digests detect either no N′ fragment [AFF60rev, AFF70rev, and AFF210rev (panels E–H, respectively, of Figure 3)] or a faint N′ band in the ribose-free lane only [AFF186rev (Figure 3G)]. By contrast, BODIPY scans reveal robust N fragment and/or fragment a/b bands in the presence and absence of ribose. These bands correspond to those generated by cleavage of the Nrev analogues under both conditions.
FRET Biosensors for Ribose
To visualize the fold shift, we monitored quenching of BODIPY groups placed at the Cys positions indicated in Table 1 and Figure 3. The transfer of energy between BODIPY groups occurs with a Förster radius of 57 Å. As controls, we labeled N′ analogues with BODIPY at positions equivalent to those in the parent AFF molecules. N analogues do not bind ribose and were not included in FRET experiments. The average labeling efficiency was approximately 50%. Consequently, only ∼25% of molecules are labeled at both positions, whereas ∼50% are singly labeled; the maximal theoretical FRET response is reduced accordingly.
FRET results agree with those of the thermal denaturation and limited proteolysis experiments. The fluorescence of AFF60 does not change significantly upon addition of ribose, confirming that it does not bind substrate (Figure 4A). Emission of AFF70 (Figure 4B), AFF186 (Figure 4C), and AFF210 (Figure 4D), by contrast, is quenched in the expected manner when ribose is added. Fitting the data to the one-site binding equation yields Kd values of 4.2 μM (AFF70 and AFF210) and 12.3 μM (AFF186). Taken together, these findings indicate that (i) substrate binding induces AFF70, AFF186, and AFF210 to undergo the N to N′ fold shift and (ii) the fold shift can be consistently detected by placement of donor and acceptor fluorophores at the positions specified by the AFF design (Figure 1).
Figure 4.
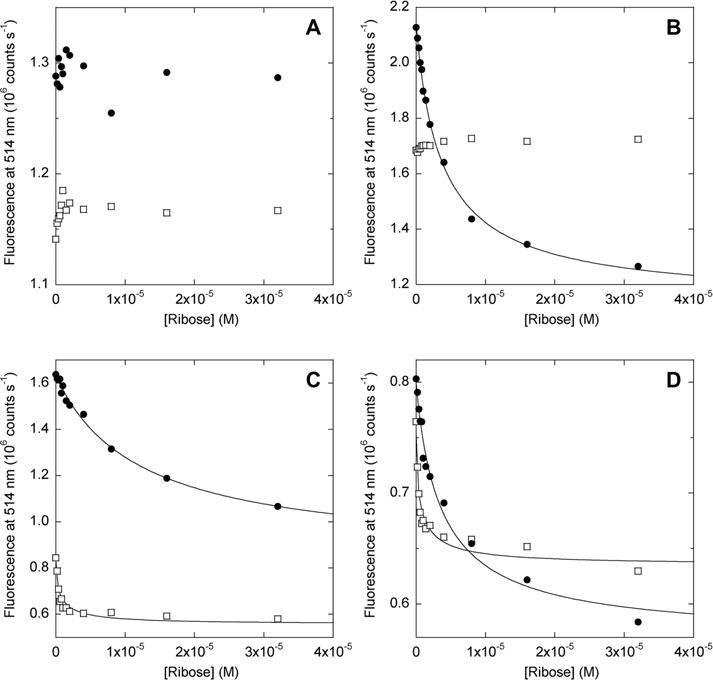
Ribose binding of AFF biosensors monitored by FRET. Filled circles show data for AFF variants; empty squares show data for N′ analogues. Lines indicate best fits to the single-site binding equation. Samples in each panel and their fitted dissociation constants (where applicable) are as follows: (A) AFF60, N′60; (B) AFF70 (Kd = 4.2 μM), N′70; (C) AFF186 (Kd = 12.3 μM), N′186; (D) AFF210 (Kd = 4.2 μM), N′210. Kd values of N′186 and N′210 are not accurately determined because they are significantly below the protein concentration employed (1 μM). The temperature was 50 °C.
Surprisingly, the fluorescence of two of the N′ analogues (N′186 and N′210) is also quenched by ribose (Figure 4C,D). This result is striking because the distance between BODIPY groups, situated at the termini, is expected to be much shorter than the Förster radius and not change appreciably as a result of domain closure. The fluorescence change nevertheless appears to result from ribose binding, as the data fit to the one-site binding equation and yield Kd values in the submicromolar range, consistent with those measured by isothermal titration calorimetry (ITC) for N′ analogues (vide infra). Kd values below 1 μM cannot be determined accurately from panels C and D of Figure 4 because of stoichiometric binding. The amplitudes of the fluorescence changes observed for N′186 and N′210, while significant, are less than those recorded for AFF186 and AFF210, respectively. The data suggest that the FRET changes observed for AFF186 and AFF210 arise from a combination of fold switching and more subtle perturbations within the N′ structure, both of which contribute to the overall response of these switches.
To further explore the extent to which fluorescence changes might reflect minor environmental perturbations rather than fold shifts, we repeated the FRET experiments with the reverse binding mutants. The reverse mutants bind ribose (Figure S5 of the Supporting Information) but remain in conformation N after binding (Figure 3). As expected, no ribose-dependent fluorescence change is seen for any of the AFFrev constructs (Figure S6 of the Supporting Information). These data indicate that the BODIPY groups do not report on domain closure when the switches are in the N state.
Switching Kinetics
In addition to establishing the response times of the AFF biosensors, ribose binding kinetics also provide insight into their mechanisms of conformational change. Binding of substrates to PBPs is rapid, diffusion-limited in many cases, and association rates (kon) are concentration-dependent.18 The mechanism by which AFF variants bind substrate involves a major conformational rearrangement. On rates are hence expected to be slower and independent of substrate concentration.
FRET-monitored kinetic data confirm these predictions. Association kinetics for AFF70 fit to single-exponential decays, of which the fitted rates do not vary significantly from 8 to 100 μM ribose [average kon = 0.065 ± 0.0076 min−1 (Figure 5A)]. On rates for AFF186 and AFF210 (Figure 5B) are likewise independent of ribose concentration (not shown). Data for AFF210 fit adequately to a single-exponential function (kon = 0.092 min−1), whereas AFF186 requires a double exponential, with major (86%) and minor (14%) phases yielding kon values of 0.018 and 2.1 min−1, respectively. AFF70 and AFF210 show evidence of a minor fast phase as well, but this reaction occurs too quickly to characterize. Limited proteolysis data suggest that the N′ conformations of AFF186 (Figure 3C) and AFF210 (Figure 3D) are populated in the absence of ribose, and it is possible that the faster phase reflects binding to this subpopulation. The same speculation does not seem to apply to AFF70, however, because binding of ribose to N′70 does not result in a FRET change (Figure 4B).
Figure 5.
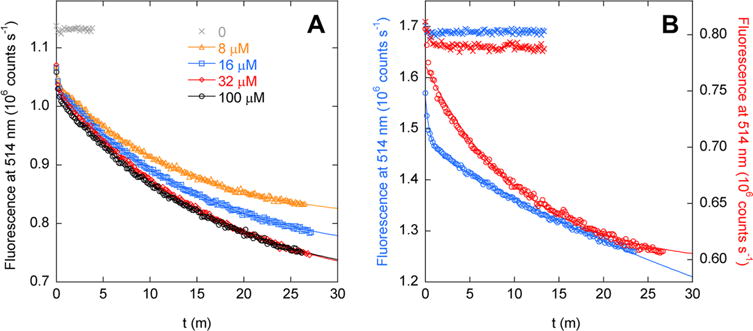
Kinetics of binding of ribose to (A) AFF70 and (B) AFF186 (blue symbols, left axis) and AFF210 (red symbols, right axis), determined by FRET. Lines are best fits to a single-exponential function (AFF70 and AFF 210) or a double-exponential function (AFF186). Crosses indicate the fluorescence baseline reading in the absence of ribose. Panel A shows that rates of binding of ribose to AFF70 are independent of substrate concentration. Fitted rate constants are (in order of increasing ribose concentration) 0.075, 0.066, 0.058, and 0.062 min−1. In panel B, the ribose concentration is 100 μM and fitted rates are 2.1 min−1 (14% total amplitude) and 0.018 min−1 (86% total amplitude) for AFF186 and 0.092 min−1 for AFF210. The temperature was 50 °C.
Isothermal Titration Calorimetry
The major FRET-detected binding event in Figure 4 occurs too slowly to be monitored by ITC. ITC can, however, give thermodynamic information about the rapid binding phase. Table 3 lists binding parameters for AFF variants and their N′ analogues. N′ analogues bind ribose with Kd values that are similar to each other (21–38 nM) except for N′70, which binds 100-fold less tightly (Kd = 2.7 μM). One possible explanation for the diminished affinity is that the site of permutation, while relatively distant (∼15 Å) from the bound ribose, is located in the crevice between the amino and carboxy domains of RBP. Permutation sites of the other N′ analogues are on the opposite sides of the two domains, facing outward in the structures in Figure 1.
Table 3.
Thermodynamic Parameters of Ribose Binding Determined by ITCa
| variant | Kd (nM) | −ΔH (kcal/mol) | n |
|---|---|---|---|
| N′60 | 21.3 ± 1.1 | 16.4 ± 0.25 | 0.79 ± 0.032 |
| N′70 | 2740 ± 240 | 17.5 ± 0.15 | 0.85 ± 0.010 |
| N′186 | 38.1 ± 0.40 | 17.5 ± 0.21 | 0.78 ± 0.021 |
| N′210 | 21.6 ± 1.4 | 18.3 ± 0.15 | 0.68 ± 0.012 |
| AFF60 | no binding detected | no binding detected | no binding detected |
| AFF70 | 46.6 ± 5.5 | 18.4 ± 0.15 | 0.75 ± 0.012 |
| AFF186 | 58.8 ± 0.17 | 22.5 ± 2.1 | 0.44 ± 0.035 |
| AFF210 | 26.1 ± 1.5 | 18.9 ± 0.71 | 0.17 ± 0.010 |
Errors are the standard deviations of three measurements. The temperature was 37 °C.
Enthalpy changes of AFF variants (with the exception of AFF60 for which no binding is detected) are identical within error to those of their N′ analogues, suggesting that binding involves similar interactions and conformational changes. Binding affinities of AFF186 and AFF210 are close to those of N′186 and N′210, respectively. Curiously, AFF70 binds ribose 60-fold more tightly than N′70. We envision that the major structural difference between N′70 and the N′ conformation of AFF70 is that the amino and carboxy termini are at position 70 in the former, whereas only the carboxy terminus is located at that equivalent position in the latter (see Figure 1A for the analogous situation in AFF60). Removal of the positive charge associated with the α-amino group may be responsible for the increased ribose affinity of AFF70.
Ribose binding stoichiometries (n) of AFF70, AFF186, and AFF210 are significantly lower than those of their N′ analogues (Table 3). The N → N′ conformational change occurs slowly on the time scale of the 2 min ITC injections; therefore, n likely reflects binding to the subpopulation of molecules that already exist in N′. This finding is in broad agreement with proteolysis results, although the N′ population appears to be somewhat smaller in the latter experiment.
Structural Characterization by Circular Dichroism
We used circular dichroism (CD) to ask (i) whether circular permutation has an effect on secondary structure, (ii) the extent to which the duplicated region is folded or unfolded in AFF variants, and (iii) whether substrate binding changes the secondary structure content of AFF constructs (or that of their N′ analogues). All variants exhibit dual minima at 222 and 208 nm characteristic of predominantly helical proteins (Figure 6). N′60 (Figure 6A), N′70 (Figure 6B), and N′186 (Figure 6C) possess slightly lower per-residue helical contents than their respective N analogues. The magnitude of the decrease is consistent with the presence of the presumably disordered 30-amino acid linker in the circular permutants. N′210, however, appears to be much less helical than N210 (Figure 6D). Loss of this amount of structure would seem likely to compromise stability and/or function; however, N′210 is only marginally less stable than N′186 [as gauged by Tm values (Figure 2C,D)], and N′210 binds ribose at least as tightly as the other permutants (Table 3). The basis for the anomalously weak CD signal of N′210 is unclear.
Figure 6.
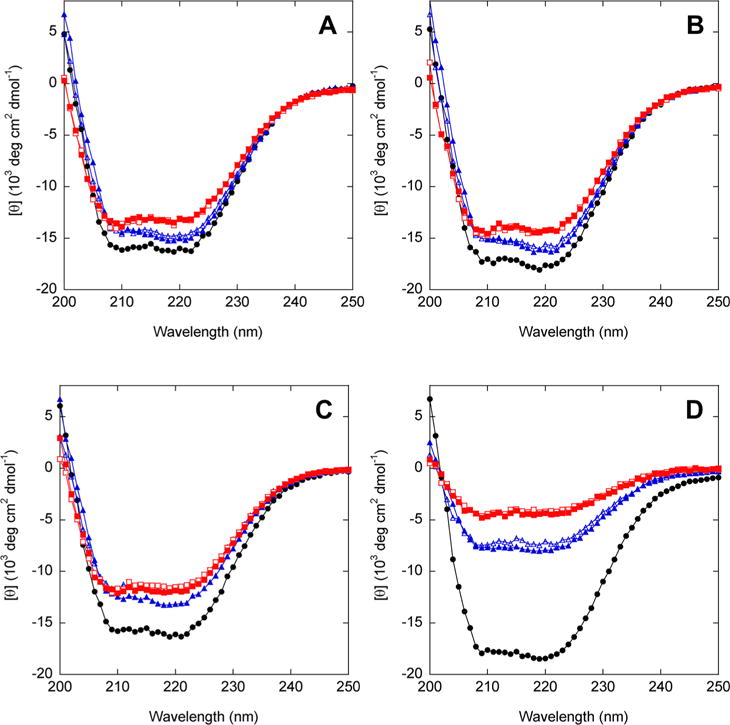
CD spectra of AFF variants (red), N analogues (black), and N′ analogues (blue). Empty and filled symbols indicate the absence and presence of 1 mM ribose, respectively. The temperature was 37 °C.
Upon comparing empty and filled blue symbols in Figure 6, we can conclude that ribose binding does not substantially alter the secondary structure content of any of the N′ analogues. This result is expected because the binding-dependent conformational change involves rigid-body hinge movement without gain or loss of secondary structure.
If the duplicated segments of AFF variants retain nativelike structure when orphaned, then CD spectra of AFF variants will be comparable to those of their N′ analogues (both species contain the 30-amino acid linker). On the other hand, if the duplicated regions are completely disordered, then CD spectra of AFF constructs will be considerably less intense, as these segments comprise 16–22% of the total sequence lengths. [θ]222 values of AFF60 (Figure 6A), AFF70 (Figure 6B), and AFF186 (Figure 6C) are 3–10% less intense than those of their N′ analogues in the absence of ribose, implying that these duplicated regions retain significant structure when orphaned. For AFF210, the decrease in [θ]222 (43%) is larger than the maximal expected value of 17%. This result suggests that residues 210–277 are predominantly disordered when orphaned, although the anomalously weak CD spectrum of AFF210 (as well as that of N′210) remains unexplained.
The duplicated segments within each AFF construct are identical with the exception of the ribose binding mutation(s). One may therefore conjecture that the CD spectra will not change as a result of the ribose-induced fold shift. Figure 6 confirms this prediction for all AFF variants. In the case of AFF210, however, CD spectra of the N and N′ analogues are substantially different, and one could alternatively argue that addition of ribose should shift the CD signal. That outcome is not observed.
DISCUSSION
The major objective of this work is to establish the extent to which a relatively large, structurally complex binding protein can be transformed to a molecular switch for biosensing. We draw the following conclusions for RBP with the expectation that they may be applicable to the larger goal of converting a generic binding protein into a fluorescent biosensor.
Viable Permutants Were Chosen with a High Success Rate, but Many Were Destabilized
The rules for selecting optimal permutation sites remain obscure. Perhaps contrary to structural intuition, creating new termini at surface loops does not always produce the most stable permutants, and breaking the chain within secondary structure elements sometimes generates viable permutants.19 Nevertheless, the simple strategy of targeting surface loops distant from the active site proved to be adequate in the case of RBP. All eight permutants tested were functional and stable, albeit less stable than WT RBP. It is therefore desirable to choose a parent protein of high intrinsic stability. Indeed, we began this study using E. coli RBP [Tm = 55 °C (Figure S7 of the Supporting Information)]. Permuting E. coli RBP at positions 57 and 70 resulted in proteins that bound ribose but were too unstable in their apo forms to be useful (Tm = 47 and 45 °C, respectively). It may be feasible to increase stability by optimizing the length and/or composition of the linker sequence, as demonstrated by others.20
The Response Time of the Switch Can Be Adjusted by Kinetic Tuning Mutations
The potential downside of a highly stable starting protein is that its unfolding rate can be slow. Proteins with high Tm values, particularly those from thermophilic organisms, often unfold unusually slowly at ambient temperature. T. tengcongensis (optimal growth temperature of 75 °C) RBP is an extreme example; the extrapolated half-time (55 years) of the slow unfolding phase is among the longest we were able to find in the literature. Our previous study showed that an unfolding step at least partially limits the rate of the N ⇆ N′ fold shift for the calbindin D9k Ca2+ sensor (calbindin-AFF).9 For the work presented here, we created a second set of AFF60, AFF70, AFF186, and AFF210 constructs that did not contain the L131D kinetic tuning mutation. None of them exhibited a ribose-dependent shift in melting temperature (data not shown), consistent with the notion that the fold shift is too slow to measure. Introducing hydrophobic-to-charged core mutations in the shared sequence proved to be effective, as the L131D substitution increased the unfolding rate of RBP by >105-fold and rendered the fold shift of the AFF variants observable. The FRET-detected ribose binding rate of AFF70 at 50 °C (Figure 5A) is 12.5-fold faster than the extrapolated unfolding rate (at 25 °C) of a variant that closely approximates N70 (Figure S2 of the Supporting Information). The similarity of these values, especially considering that the unfolding rate is more rapid at 50 °C, suggests that the fold shift rate is limited by global unfolding of the N conformation. If this hypothesis is correct, then the switching mechanism would differ from that of calbindin-AFF, which does not require whole-molecule unfolding.9
Relative Stabilities of N and N′ Analogues Predict the Gain and Sensitivity of the Switch
Gain (e.g., FRET change between on and off states) is determined by the extent to which the populations of N and N′ change as a result of ligand binding. Ideally, either N or N′ should predominate over the other species in the absence of ligand, and the populations should invert in the presence of ligand. We were able to achieve this condition by taking advantage of two axioms of protein stability. First, permuting a protein almost always destabilizes it. In the rare instance in which it does not (e.g., calbindin D9k7), one can simply shorten the linker to introduce conformational strain.21 For a generic AFF construct, the default expectation is that N will prevail over N′, and N will thus be chosen as the binding-incompetent conformation. Depending on available binding energy, it may be necessary to destabilize N (relative to N′) to facilitate the fold shift. The second adage, that it is easy to destabilize a protein by mutation, is invoked for this purpose. Here, the binding-knockdown substitutions served as stability tuning mutations as well, although for other proteins it will likely be necessary to introduce them separately.
AFF70, AFF186, and AFF210 exhibited fluorescence changes of 1.9-, 1.9-, and 1.4-fold, respectively (Figure 4). These modest values are likely attributed to a combination of partial labeling, incomplete switching [because of minor populations of N′ in the free state and/or N in the bound state (Figure 3)], and residual structure of the orphaned tails. The first two issues can be addressed by improved labeling methods and thermodynamic fine-tuning. We discuss the orphaned tails in the next section.
The sensitivity of the switch (how much ligand is required to change folds) is also established by the relative stabilities of N and N′. The observed dissociation constant (Kd) is weaker than the intrinsic dissociation constant of binding to the N′ state according to the following relationship
where and are the equilibrium constants for folding of the N and N′ conformations (from the globally unfolded state), respectively.9 When , this equation simplifies to . FRET-detected Kd values are approximately 500-fold greater than the ITC-measured values, suggesting that the N conformation is more stable than the N′ conformation by 3.5–4.0 kcal/mol. This figure is reasonable based on the melting temperatures of the N and N′ analogues (Figure 2).
Orphaned Tails Exhibit Varying Degrees of Residual Structure
CD spectra in Figure 6 indicate that residues 210–277 are largely unstructured when they extend from the termini of AFF210. Increasing the length of the tail by adding residues 186–277 seems to induce it to adopt significant nativelike structure. Residues 1–59 and 1–69 likewise appear to be substantially structured. All of the tails, however, were preferentially attacked by trypsin, implying that any existing structure is relatively unstable. The sloping native baselines seen in thermal melts of AFF70 (Figure 2B), AFF186 (Figure 2C), and AFF210 (Figure 2D) may arise from noncooperative unfolding of the tails. The presence of structure in the tails is expected to draw the BODIPY-FL groups closer when the proteins are in conformation N, reducing the observed change in FRET. Surprisingly, all AFF variants were soluble at >500 μM (not shown). The orphaned tails, regardless of their structure, did not induce aggregation or misfolding.
Why Did AFF60 Fail To Switch?
AFF60 met all of the initial criteria for the AFF design, and none of its characteristics stood out from those of its three successful cousins; however, it is stuck in conformation N. To understand why, we measured folding rates of N60 and N′60 (Figure S8 of the Supporting Information). The extrapolated folding rate of N60 is 4.77 min−1 (25 °C). N′60 folds too slowly to measure at 25 °C, but the rate is 0.25 min−1 at 32 °C. Thus, the N state of AFF60 is predicted to fold at least 20 times faster than the N′ state. If the fold shift involves whole-molecule unfolding, as our data suggest, then most of the refolding molecules will partition into N regardless of whether ribose is present. The origin of the high kinetic barrier in N′60 folding is unknown, but it is less pronounced in N′70. N′70 folds 2.7 times faster than N′60 (Figure S8 of the Supporting Information), and AFF70 undergoes normal switching behavior.
CONCLUSIONS
We have tested the steps by which a medium-sized (31 kDa) binding protein with mixed α/β structure can be converted to a fluorescent biosensor according to the AFF design. All of the steps, circular permutation, kinetic and thermodynamic tuning, and fluorophore placement, are implemented using proven principles of protein structure, stability, and folding kinetics. Three of four AFF variants exhibited the expected ribose-dependent fold shift. The fourth failed because of a kinetic partitioning event that is likely exacerbated by the unusually slow rate at which T. tengcongensis RBP cycles between folded and unfolded states. It may be possible to transfer the modifications described herein to other members of the PBP superfamily to generate a class of related biosensors for a variety of targets. More significantly, this study provides a guideline for converting an arbitrary binding protein into a fluorescent switch for biosensing.
Supplementary Material
Acknowledgments
We thank Xiaowen Wang for assistance with Western blots.
Funding
This work was supported by National Institutes Health Grant GM069755 to S.N.L.
Footnotes
Supporting Information
Figures S1–S8 as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Dwyer MA, Hellinga HW. Periplasmic binding proteins: A versatile superfamily for protein engineering. Curr Opin Struct Biol. 2004;14:495–504. doi: 10.1016/j.sbi.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Vercillo NC, Herald KJ, Fox JM, Der BS, Dattelbaum JD. Analysis of ligand binding to a ribose biosensor using site-directed mutagenesis and fluorescence spectroscopy. Protein Sci. 2007;16:362–368. doi: 10.1110/ps.062595707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Lorimer RM, Smith JJ, Dwyer MA, Looger LL, Sali KM, Paavola CD, Rizk SS, Sadigov S, Conrad DW, Loew L, Hellinga HW. Construction of a fluorescent biosensor family. Protein Sci. 2002;11:2655–2675. doi: 10.1110/ps.021860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alicea I, Marvin JS, Miklos AE, Ellington AD, Looger LL, Schreiter ER. Structure of the Escherichia coli Phosphonate Binding Protein PhnD and Rationally Optimized Phosphonate Biosensors. J Mol Biol. 2011;414:356–369. doi: 10.1016/j.jmb.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fehr M, Frommer WB, Lalonde S. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proc Natl Acad Sci USA. 2002;99:9846–9851. doi: 10.1073/pnas.142089199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fehr M, Lalonde S, Lager I, Wolff MW, Frommer WB. In vivo imaging of the dynamics of glucose uptake in the cytosol of COS-7 cells by fluorescent nanosensors. J Biol Chem. 2003;278:19127–19133. doi: 10.1074/jbc.M301333200. [DOI] [PubMed] [Google Scholar]
- 7.Stratton MM, Mitrea DM, Loh SN. A Ca2+-sensing molecular switch based on alternate frame protein folding. ACS Chem Biol. 2008;3:723–732. doi: 10.1021/cb800177f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stratton MM, McClendon S, Eliezer D, Loh SN. Structural characterization of two alternate conformations in a calbindin D9k-based molecular switch. Biochemistry. 2011;50:5583–5589. doi: 10.1021/bi102040g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stratton MM, Loh SN. On the mechanism of protein fold-switching by a molecular sensor. Proteins: Struct, Funct, Bioinf. 2010;78:3260–3269. doi: 10.1002/prot.22833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitrea DM, Parsons L, Loh SN. Engineering an artificial zymogen by alternate frame protein folding. Proc Natl Acad Sci USA. 2010;107:2824–2829. doi: 10.1073/pnas.0907668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boas FE, Harbury PB. Design of protein-ligand binding based on the molecular-mechanics energy model. J Mol Biol. 2008;380:415–424. doi: 10.1016/j.jmb.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson JM, Church GM. Predicting ligand-binding function in families of bacterial receptors. Proc Natl Acad Sci USA. 2000;97:3965–3970. doi: 10.1073/pnas.050580897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuneo MJ, Tian Y, Allert M, Hellinga HW. The backbone structure of the thermophilic Thermoanaerobacter tengcongensis ribose binding protein is essentially identical to its mesophilic E. coli homolog. BMC Struct Biol. 2008;8:20. doi: 10.1186/1472-6807-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fersht AR, Sato S. ϕ-value analysis and the nature of protein-folding transition states. Proc Natl Acad Sci USA. 2004;101:7976–7981. doi: 10.1073/pnas.0402684101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott DJ, Harding SE, Rowe AJ, editors. Analytical Ultracentrifugation: Techniques and Methods. Royal Society of Chemistry; Cambridge, U.K: 2005. [Google Scholar]
- 17.Tompa P, Fersht AR. Structure and Function of Intrinsically Disordered Proteins. Vol. 1. Chapman and Hall/CRC Press; 2009. [Google Scholar]
- 18.Miller DM, III, Olson JS, Pflugrath JW, Quiocho FA. Rates of ligand binding to periplasmic proteins involved in bacterial transport and chemotaxis. J Biol Chem. 1983;258:13665–13672. [PubMed] [Google Scholar]
- 19.Iwakura M, Nakamura T, Yamane C, Maki K. Systematic circular permutation of an entire protein reveals essential folding elements. Nat Struct Biol. 2000;7:580–585. doi: 10.1038/76811. [DOI] [PubMed] [Google Scholar]
- 20.Iwakura M, Nakamura T. Effects of the length of a glycine linker connecting the N- and C-termini of a circularly permuted dihydrofolate reductase. Protein Eng. 1998;11:707–713. doi: 10.1093/protein/11.8.707. [DOI] [PubMed] [Google Scholar]
- 21.Butler JS, Mitrea DM, Mitrousis G, Cingolani G, Loh SN. Structural and thermodynamic analysis of a conformationally-strained circular permutant of barnase. Biochemistry. 2009;48:3497–3507. doi: 10.1021/bi900039e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.