

Abstract
DOC2B is a high-affinity Ca2+ sensor that translocates from the cytosol to the plasma membrane (PM) and promotes vesicle priming and fusion. However, the molecular mechanism underlying its translocation and targeting to the PM in living cells is not completely understood. DOC2B interacts in vitro with the PM components phosphatidylserine, phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] and target SNAREs (t-SNAREs). Here we show that PI(4,5)P2 hydrolysis at the PM of living cells abolishes DOC2B translocation, whereas manipulations of t-SNAREs and other phosphoinositides have no effect. Moreover, we were able to redirect DOC2B to intracellular membranes by synthesizing PI(4,5)P2 in those membranes. Molecular dynamics simulations and mutagenesis in the calcium and PI(4,5)P2-binding sites strengthened our findings, demonstrating that both calcium and PI(4,5)P2 are required for the DOC2B–PM association and revealing multiple PI(4,5)P2–C2B interactions. In addition, we show that DOC2B translocation to the PM is ATP-independent, and occurs in a diffusion-like manner. Our data suggest that the Ca2+-triggered translocation of DOC2B is diffusion-driven and aimed at PI(4,5)P2-containing membranes.
Keywords: DOC2B; translocation; PM targeting; PI(4,5)P2; Ca2+ sensor
Graphical abstract
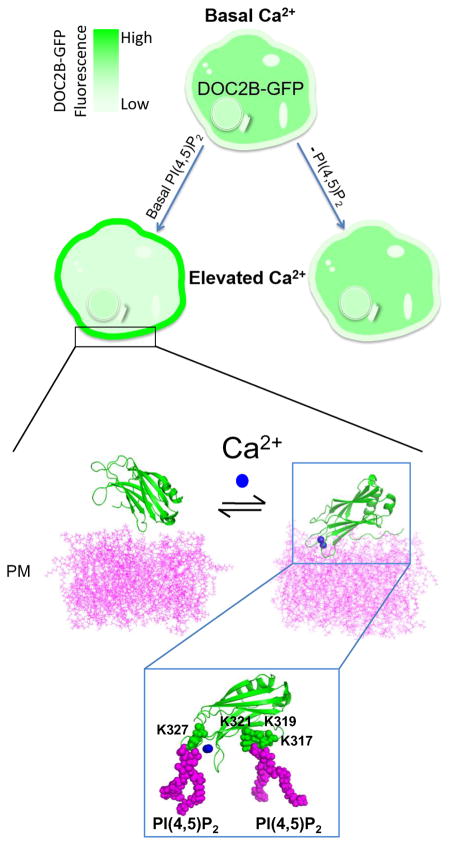
Introduction
Double C2 domain protein B (DOC2B) is a Ca2+ sensor that enhances spontaneous and asynchronous neurotransmitter release 1-3. It is a soluble protein with cytoplasmic distribution under basal intracellular Ca2+ concentrations ([Ca2+]i). When [Ca2+]i rises, DOC2B translocates to the plasma membrane (PM) and supports vesicle priming and fusion. As its name suggests, DOC2B contains two sequential C2 domains which comprise most (70%) of the protein, termed C2A and C2B 1,4-8.
C2 domains are abundant Ca2+-binding modules present in many proteins and involved in cell signaling and the secretory pathway. They are composed of ∼130 residues organized in a sandwich of two four-stranded anti-parallel β-sheets connected by loops. Ca2+ binds to five conserved aspartates located in the interconnecting loops with an affinity dictated by the surrounding sequence 9-12. C2 domains were first discovered in classical Protein Kinase C (PKC) where they were shown to be responsible for Ca2+-dependent membrane binding 13,14.
DOC2B C2 domains are also responsible for DOC2B's Ca2+-dependent ability to bind to membranes in vitro and in living cells. In vitro, DOC2B C2 domains bind the membranal components—phosphatidylserine (PS), phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] and the target SNAREs (t-SNAREs) syntaxin (syx) and SNAP25 when bound together in a binary complex 1,3-5. However, which of these components targets DOC2B specifically to the PM (and not to intracellular membranes), as well as the exact mechanism underlying DOC2B's translocation, remain elusive.
It has been repeatedly demonstrated in vitro that the C2 domains of DOC2B bind PS in a Ca2+-dependent manner 1,3,6, and that the C2A of DOC2B binds PS-containing liposomes at submicromolar [Ca2+] 5. However, isolated DOC2B C2A fails to translocate to the PM in living cells 6, suggesting that PS binding is insufficient to induce translocation in cells. In addition, PS is present in many intracellular membranes other than the PM 15,16, but neither full-length DOC2B nor its isolated domains are targeted to them. Thus, even though PS has been shown to interact with DOC2B C2 and homologous C2 domains, it is unlikely to target DOC2B C2 domains to the PM.
In contrast, syx•SNAP heterodimers are relatively restricted to the PM 17, and PI(4,5)P2 is exclusively localized to the PM 18-20, making them potential targeting molecules for Ca2+-bound DOC2B. Specifically, PI(4,5)P2 has been proposed to function via recruitment of proteins to their sites of action 21,22. Indeed, PI(4,5)P2 was found to be crucial for Ca2+-dependent translocation of the PKC C2 domain 23,24. In addition, specific interactions of PI(4,5)P2 with C2 domains of synaptotagmin (syt) 1 and rabphilin 3A, which present the highest homology to DOC2B C2 domains, have been suggested by nuclear magnetic resonance and in vitro assays 25-27. Other biochemical studies have established that PI(4,5)P2 enhances the Ca2+-dependent and independent interactions of syt1 and DOC2B C2 domains with liposomes 1,25. Another study found a strong correlation between the ability of C2 domains from syt 1–9 to inhibit fusion in cracked rat adrenal pheochromocytoma (PC12) cells, and the ability to bind PI(4,5)P2 and t-SNAREs but not PS 28.
Mutagenesis studies in many C2 domains have revealed a conserved basic patch of amino acids that interact with PI(4,5)P2 and t-SNAREs. Mutating this region in the C2B of syt1 and DOC2B severely impaired the domain's Ca2+-independent PI(4,5)P2 binding in vitro 1,25. In DOC2A, severe impairment of the basic patch in C2B substantially decreased Ca+2-induced PM translocation of the full-length protein 29, and similar mutations in the C2C domain of extended syt1 led to loss of its peripheral distribution 30. In the tandem C2AB of DOC2B, a double mutation in specific lysines in both C2 domains (K237,319E) decreased t-SNARE binding and PI(4,5)P2 Ca2+-independent interactions, and further impaired the domains' Ca2+-dependent ability to enhance fusion 1. However, knowledge is limited regarding the contribution of PI(4,5)P2 and t-SNARE binding to DOC2B's Ca2+-dependent PM translocation in living cells. Understanding this constitutes a key step in the broader question regarding the mechanism of DOC2B in promoting asynchronous and spontaneous release.
To learn more about the significance of PI(4,5)P2 and t-SNARE binding to DOC2B's Ca2+-dependent PM translocation in living cells, and to reveal the molecule that targets DOC2B specifically to the PM, we implemented various molecular, cellular, pharmacological and modeling techniques. We concluded that PI(4,5)P2 is a critical factor for DOC2B translocation to the PM and that its arrival to the PM is driven by diffusion. These findings expand our understanding of how PM composition spatially controls exocytotic events.
Results
Screening for potential targeting molecules
The C2 domains of DOC2B can bind several membranous components 1,3,4,7, but it is unclear which of these components targets DOC2B specifically to the PM under elevated [Ca2+]i. To address this, we set three main requirements for the potential DOC2B target: (1) its distribution pattern must match that of the Ca2+-bound DOC2B; (2) manipulations that affect DOC2B's interaction with the target will affect DOC2B translocation; (3) inducing a change in the target's distribution pattern (i.e., enriching the target in other cellular organelles) should change DOC2B's translocation destination according to the new pattern. We therefore systematically examined each of the known DOC2B-interacting partners at the PM, as well as DOC2B mutants, for these requirements, while using a cell-based system to maintain a physiological environment.
We began by examining the distribution patterns of t-SNAREs (syx1A and SNAP25), PI(4,5)P2 and PS, which are known to interact with DOC2B in vitro 1,3,4 and reside on the PM. We examined PI(4,5)P2 and PS distribution by overexpressing the phospholipid (PL) biosensors PLCδ-PH-GFP 19 and Lact-C2-GFP 16, respectively, in PC12 cells. The distribution patterns of t-SNAREs were also observed in PC12 cells using overexpression of fluorescently tagged syx1A or SNAP25. In accordance to previous well-established data 15-17,19, we observed that only PI(4,5)P2 presents clear PM staining resembling that of DOC2B•Ca2+ (Figure S1M). The distribution of mRFP-syx1A was not restricted to the PM as it was also clearly observed in intracellular organelles (Figure S1). However, SNAP25-YFP distribution was relatively restricted to the PM, and SNAP25•syx1A heterodimers were primarily localized to the PM in PC12 cells 17. As DOC2B only binds to SNAP25•syx1A in their heterodimeric state 4, we have to consider t-SNAREs as potential DOC2B-targeting molecules. Finally, PS was enriched in both intracellular membranes and the PM, in a pattern that was distinct from that of Ca2+-activated DOC2B (Figure S1) 6,8,16. Thus, even though it has been repeatedly shown that DOC2B can bind PS in vitro 1,3,5,6, it is less likely to target Ca2+-bound DOC2B to its destination in living cells. We therefore focused on PI(4,5)P2 and the t-SNAREs, whose distribution patterns resembled that of DOC2B•Ca2+.
PI(4,5)P2 targets DOC2B to the PM
We next examined the distribution and translocation of the DOC2B mutant DOC2BK237,319E following Ca2+ elevation 1. This mutation disrupts the basic sequences in the DOC2B C2A and C2B domains responsible for binding to PI(4,5)P2 and t-SNAREs, but not to PS 1. DOC2BK237,319E-GFP was expressed in PC12 cells and its PM translocation following Ca2+ elevation induced by KCl was examined using conventional microscopy and quantified using total internal reflection fluorescent microscopy (TIRFM), which is more sensitive for the detection of membrane fluorescence. Indeed, while DOC2BWT-GFP showed typical PM translocation, DOC2BK237,319E-GFP PM translocation was severely impaired (Figure 1A,B). [Ca2+]i increase in the cells was verified using the red-shifted calcium indicator R-GECO 31 (Figure 1C).
Figure 1.
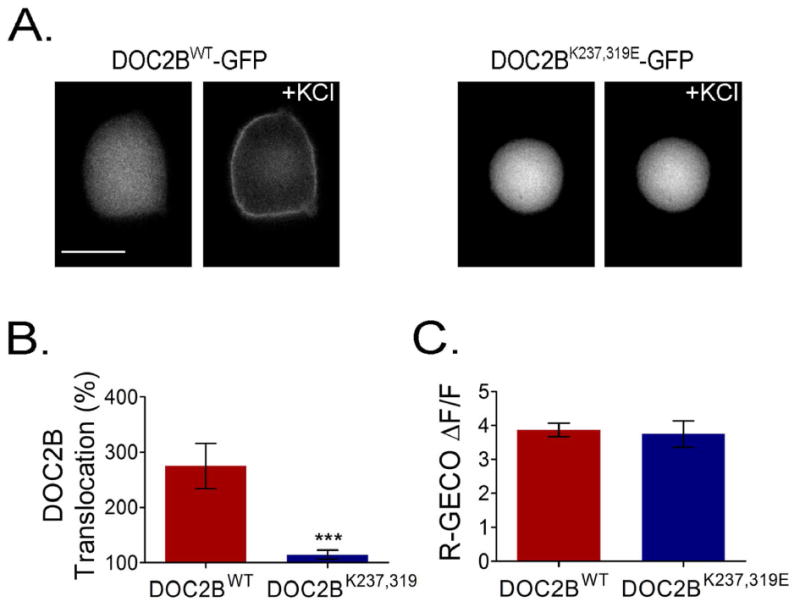
The basic patch in DOC2B is crucial for its PM translocation. (A) DOC2BK237,319E-GFP, deficient in PI(4,5)P2 and t-SNARE binding, fails to translocate to the PM following KCl depolarization, despite Ca2+ elevation as evidenced by R-GECO (n = 11 for both mutant and WT DOC2B; t-test P > 0.05) (C). (B) TIRFM quantification of KCl-induced translocation of DOC2BK237,319E-GFP and DOC2BWT-GFP (n = 12 for mutant and 9 for WT DOC2B; Mann–Whitney P < 0.005). Data presented as mean ± SEM. Scale bar = 10 μm.
To discriminate the individual contribution of t-SNAREs and PI(4,5)P2 to the PM localization of Ca2+-bound DOC2B, we manipulated each of these factors separately at the PM and examined DOC2B translocation. To substantially reduce t-SNARE levels, we cleaved syx and SNAP25 by expressing the light chains of botulinum toxin C (BoNT/C) and botulinum toxin E (BoNT/E) for 48 h 32-35. We verified t-SNARE cleavage using cells overexpressing syx1A or SNAP25 fused to fluorophores positioned on the side of the protein that is destined for cleavage. As expected, SNAP25-YFP and mRFP-syx1A, which localize to the PM under control conditions, were localized to the cytosol when co-expressed with the toxins, indicating their successful cleavage (Figure 2A). We then measured DOC2B-GFP translocation in the presence or absence of the toxins and found it to be similar under both conditions (Figure 2B,C). To confirm that the toxins do not change Ca2+ elevation in the cells, we also measured R-GECO fluorescence in cells expressing the toxins and in naive cells, and found similar Ca2+ dynamics in both (Figure 2D).
Figure 2.
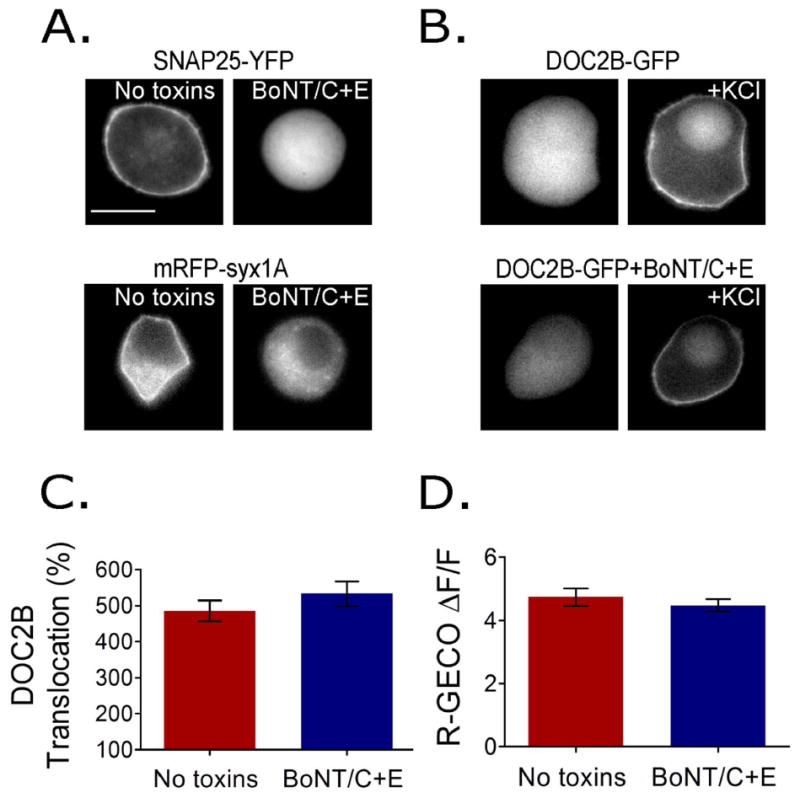
SNARE cleavage using BoNT/C and BoNT/E does not affect DOC2B translocation. (A) Cells co-expressing BoNT/C and BoNT/E (BoNT/C+E) together with SNAP25-YFP or mRFP-syx1A. Fluorophores were placed at the site of the t-SNARE protein that is detached from the membrane and enters the cytosol following cleavage. Images indicate full cleavage by the toxins. (B) Representative epifluorescence images and (C) TIRFM quantification of KCl-induced DOC2B-GFP translocation in the presence or absence of BoNT/C+E (n = 28 in the no toxin group and 34 in the toxin group; t-test P > 0.1). (D) R-GECO change in fluorescence indicates that Ca2+ dynamics induced by KCl remains similar following SNARE cleavage (n = 20 in each group; t-test P > 0.1). Data presented as mean ± SEM. Scale bar = 10 μm.
Next, we manipulated PI(4,5)P2 levels at the PM using the rapamycin system 20. This technique is based on the rapamycin-dependent dimerization of two protein fragments: FKBP and FRB, where FRB is fused to a PM anchor, Lyn11, and FKBP is cloned into a fluorescently tagged 5′ phosphatase, CFP-Inp54p. Upon addition of rapamycin, CFP-FKBP-Inp54p (CF-Inp) translocates to its PM anchor, Lyn11-FRB, and PI(4,5)P2 is dephosphorylated to PI4P. We used the PI(4,5)P2 biosensor PLCδ-PH-GFP to monitor PI(4,5)P2 levels at the PM and found that it detaches from the PM to the cytosol within 2 min of rapamycin addition (Figure 3A). An inactive variant of the phosphatase, CFP-FKBP-Inp54D281A (CF-InpD281A), was used as a control, and under this condition, PLCδ-PH-GFP did not detach from the PM (Figure 3B). We then examined the effect of PI(4,5)P2 hydrolysis on DOC2B translocation and found it to be severely impaired. Fluorescently tagged DOC2B failed to translocate to the PM following high KCl stimulation when CF-Inp was expressed, but translocated to the PM when the inactive phosphatase CF-InpD281A was expressed (Figure 3C,D) despite unchanged calcium dynamics, measured by R-GECO (Figure 3E). This suggested that PI(4,5)P2 is crucial for DOC2B translocation to the PM.
Figure 3.
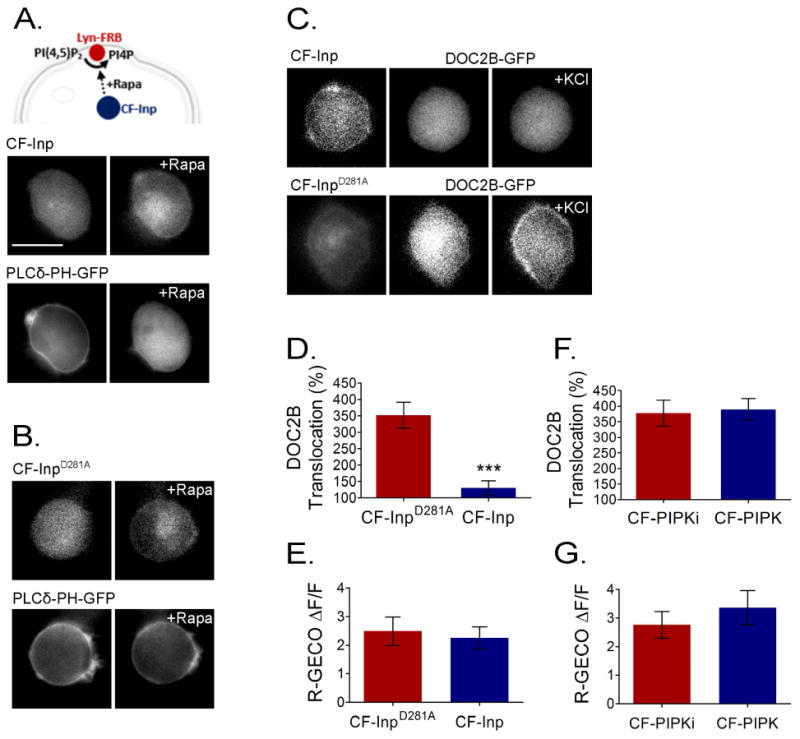
PI(4,5)P2 is crucial for DOC2B translocation to the PM. (A) Top: schematic representation of rapam ycin (Rapa)-induced PI(4,5)P2 hydrolysis at the PM. Bottom: rapamycin-induced translocation of CF-Inp to its Lyn11-FRB PM anchor, thus initiating PI(4,5)P2 hydrolysis reported as PLCδ-PH-GFP dissociation from the PM. (B) This did not occur with the inactive CF-InpD281A variant, which served as a control. (C) When rapamycin induced translocation of CF-Inp to the PM to induce PI(4,5)P2 hydrolysis, DOC2B-GFP translocation was abolished. However, when rapamycin induced the translocation of inactive CF-InpD281A, DOC2B translocation persisted. (D) TIRFM quantification of DOC2B-mRFP translocation under PI(4,5)P2 depletion (CF-Inp) and control (CF-InpD281A) conditions (n = 10 for CF-Inp and 11 for CF-InpD281A; t-test P < 0.005). (F) TIRFM quantification of DOC2B-mRFP translocation in cells co-expressing Lyn11 and CF-PIPK [to induce PI(4,5)P2 synthesis at the PM] or an inactive CF-PIPKi variant following incubation with rapamycin (n = 10 in each group; t-test P > 0.05). (E,G) Change in R-GECO fluorescence indicating that Ca2+ dynamics induced by KCl remains similar under the conditions tested (n = 10 for CF-Inp, 12 for CF-InpD281A and 9 each for PIPK and PIPKi; t-test P > 0.05 in both E and G). Data presented as mean ± SEM. Scale bar = 10 μm.
We then examined the effect of PI(4,5)P2 synthesis at the PM. We again used the rapamycin system, this time to induce the dimerization of CFP-FKBP-PIPK (CF-PIPK) to Lyn11-FRB at the PM. CF-PIPK, a PIPK 5 kinase, phosphorylates PI4P to PI(4,5)P2. In the control group, CF-PIPK was replaced by the inactive variant CF-PIPKi, and we verified that Ca2+ dynamics does not differ between the two experimental conditions (Figure 3G). DOC2B-mRFP translocation was not affected by this manipulation (Figure 3F). This was not surprising considering the fact that the PI(4,5)P2 biosensor, PLCδ-PH, is also unaffected by this manipulation 20.
Previous studies have found that certain mutations in the C2A Ca2+-binding loops, specifically neutralization of the negative charge in aspartates 218 and 220, mimic the Ca2+-bound form of the protein, resulting in constitutive localization of the mutant at the PM under basal [Ca2+]i 5,8,36. However, the nature of this interaction is not clear. We therefore examined the previously described DOC2BD218,220N 4,8 for its requirement of PI(4,5)P2 for constitutive PM distribution. DOC2BD218,220N completely relocalized to the cytosol following PI(4,5)P2 hydrolysis by CF-Inp (Figure 4A), but not under control conditions (Figure 4B). The relocalization was not due to changes in basal [Ca2+]i as this was unchanged following this manipulation [R-GECO ΔF/F = -0.02 ± 0.05 for the PI(4,5)P2 depletion group and -0.02 ± 0.02 for controls; n = 10 and 9, accordingly; t-test P > 0.05]. Since DOC2BD218,220N closely mimics the distribution and function of Ca2+-activated DOC2B 4,36, this experiment supported the requirement of PI(4,5)P2 for DOC2B•Ca2+ PM localization.
Figure 4.
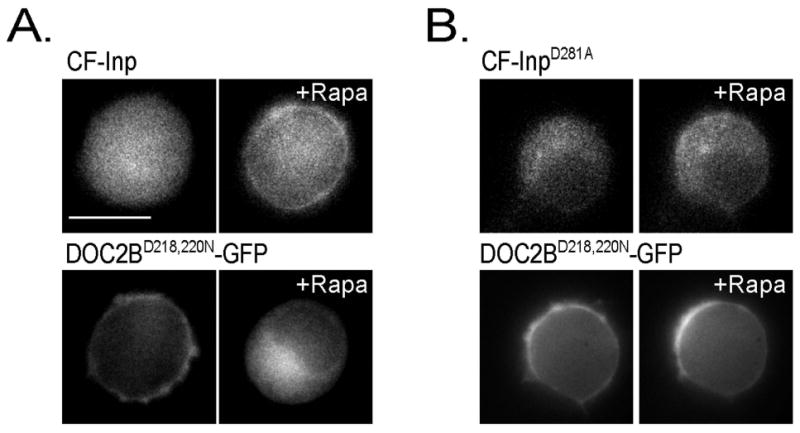
PI(4,5)P2 is crucial for DOC2BD218,220N-GFP PM localization. (A) DOC2BD218,220N-GFP relocalizes from the PM to the cytosol following PI(4,5)P2 hydrolysis induced by rapamycin (Rapa) treatment in the presence of the active CF-Inp unit. (B) DOC2BD218,220N-GFP remains on the PM following rapamycin treatment in the presence of the inactive CF-InpD281A variant which leaves PI(4,5)P2 levels unchanged (n >10 for each condition). Scale bar = 10 μm.
Finally, to further demonstrate the ability of PI(4,5)P2 to target DOC2B to the PM, we used a novel approach of protein targeting in living cells by examining whether a change in the distribution pattern of PI(4,5)P2 will retarget DOC2B translocation accordingly. In this experiment, we induced the synthesis of PI(4,5)P2 in intracellular membranes 37 by co-expressing DOC2B-mRFP and PLCδ-PH-GFP together with CF-PIPK (or inactive CF-PIPKi as a control) and Rab7-FRB. The latter was used as an anchor for late endosomes/lysosomes. We performed these experiments in COS7 cells as they provide a better platform for the multiple transfections required in this particular experiment. Rapamycin induced the dimerization of CF-PIPK and Rab7-FRB, as expressed by intracellular CFP staining. This resulted in the appearance of PLCδ-PH-GFP fluorescence at the intracellular membranes, indicating PI(4,5)P2 synthesis (Figure 5A). At this stage, DOC2B-mRFP was still observed primarily in the cytosol. We then elevated [Ca2+]i and this induced robust translocation of DOC2B-mRFP to the intracellular membranes in addition to the PM, in a manner that perfectly matched the new PI(4,5)P2 pattern reported by PLCδ-PH-GFP (Figure 5A): intracellular organelles that expressed PI(4,5)P2 also showed DOC2B-mRFP staining following calcium elevation. In the control cells, rapamycin induced dimerization of the inactive PIPK (CF-PIPKi) with the Rab7-FRB, as observed by CFP fluorescence in intracellular membranes. However, PI(4,5)P2 was not formed, and DOC2B-mRFP calcium-induced translocation was limited to the PM (Figure 5B). We also used this system to test the PI(4,5)P2-targeting of C2B isolated from DOC2B. C2B is the primary Ca2+ sensor in DOC2B, capable of independent translocation to the PM 6. Similar to the full-length DOC2B, isolated C2B translocated to intracellular membranes containing PI(4,5)P2 following rapamycin and ionomycin treatments (Figure S2A). Furthermore, C2BK319E, previously shown to be impaired in PI(4,5)P2 binding1 and comprising one of the mutations in the non-translocating DOC2BK237,319E (Figure 1), did not translocate to the PM (Figure 7A,B) or to intracellular membranes following PI(4,5)P2 synthesis in these organelles (Figure S2B). In summary, under all of the conditions tested, Ca2+-bound DOC2B and isolated C2B followed PLCδ-PH as reported by their prominent co-localization (Figure 5A,B and Figure S2, respectively). This places PI(4,5)P2 as a crucial targeting element for Ca2+-bound DOC2B, which spatially controls its cellular localization.
Figure 5.
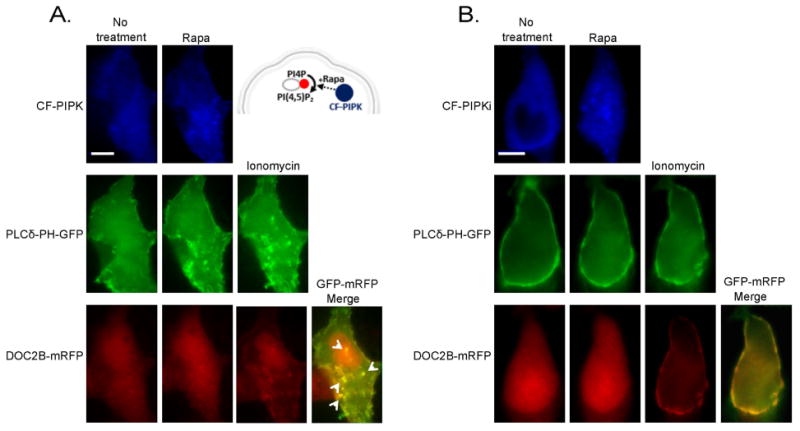
Synthesis of PI(4,5)P2 in intracellular membranes targets DOC2B to those membranes. (A) The scheme (top-right) describes the rapamycin (Rapa)-induced PI(4,5)P2 synthesis in intracellular membranes. Rapamycin induce dimerization of CF-PIPK and the endosomal anchor, FRB-Rab7, and this initiates phosphorylation of PI4P to PI(4,5)P2 in the intracellular membranes. Images show COS7 cells co-expressing CF-PIPK together with FRB-Rab7, PLCδ-PH-GFP and DOC2B-mRFP. PLCδ-PH-GFP translocated to the intracellular membranes following addition of rapamycin (Rapa), indicating synthesis of PI(4,5)P2 from PI4P in those membranes. Following the addition of ionomycin, DOC2B-mRFP co-localized to PLCδ-PH-GFP in the PM and in the new intracellular organelles (arrowheads). (B) This did not occur when the inactive CF-PIPKi variant was used; n > 5 for both conditions. Scale bar = 10 μm.
Figure 7.
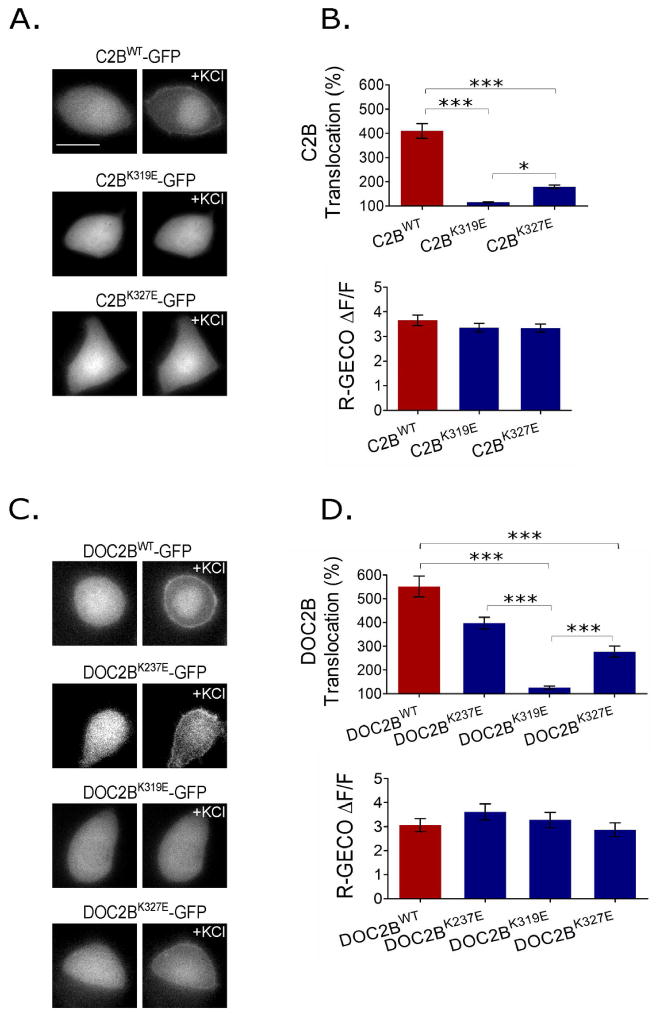
Single mutations in basic residues that bind PI(4,5)P2 impair translocation. (A) Representative epifluorescence images of isolated DOC2B C2B-GFP domain and the mutants C2BK319E-GFP and C2BK327E-GFP, suggested by the MD simulation. (B) Top: TIRFM quantification of the KCl-induced translocation of isolated C2BWT-GFP and mutants. C2BWT-GFP translocation is severally impaired by the K319E and K327 mutations (n = 12 in all groups; one-way ANOVA P < 0.05; Tukey P < 0.05 for all pairs). Bottom: Change in R-GECO fluorescence (n = 13 for WT and 10, 11 for C2BK319E, C2BK327E respectively; one-way ANOVA P > 0.05). (C) Representative epifluorescence images of the KCl-induced translocation of DOC2BWT-GFP and single DOC2B mutants-GFP as indicated. (D) Top: TIRFM quantification of DOC2BWT-GFP and DOC2B mutants-GFP translocation (n = 35 for DOC2BWT and DOC2BK237E, 37 for DOC2BK319E and 36 for DOC2BK327E; Kruskal-Wallis P < 0.05; Dunns P < 0.05 for all pairs except DOC2BK237E-GFP and DOC2BWT-GFP, and DOC2BK237E-GFP and DOC2BK327E-GFP). Bottom: Change in R-GECO fluorescence indicating that Ca2+ dynamics was similar between groups under the conditions tested (n = 13 for DOC2BWT, 10 for DOC2BK237E, 12 for DOC2BK319E and 15 for DOC2BK327E; one-way ANOVA P > 0.05). Data presented as mean ± SEM. Scale bar = 10 μm.
Molecular dynamics (MD) simulations reveal multiple PI(4,5)P2 –C2B interactions
The fact that isolated C2B presents behavior similar to that of the full-length DOC2B in terms of translocation and requirement of PI(4,5)P2 for translocation makes it attractive for modeling and further examining the PM association. To investigate lipid penetration by the C2B domain of DOC2B, we performed prolonged MD simulations of the C2B domain in Ca2+-free and Ca2+-bound forms, interacting with the POPC/POPS/PI(4,5)P2 bilayer (75:20:5). In the absence of Ca2+, the protein remained on the surface of the lipid bilayer and did not show any deep penetration (Figure 6A,B). The energy of Van der Waals (VdW) interactions between the protein and lipids (Figure 6A, black line) fluctuated around zero, demonstrating a lack of strong close-range interactions. Consistent with this, only a few contacts between the protein and the lipid bilayer were observed in the protein region distal to the Ca2+-binding loops (Figure 6B, right) at the final point of the trajectory.
Figure 6.
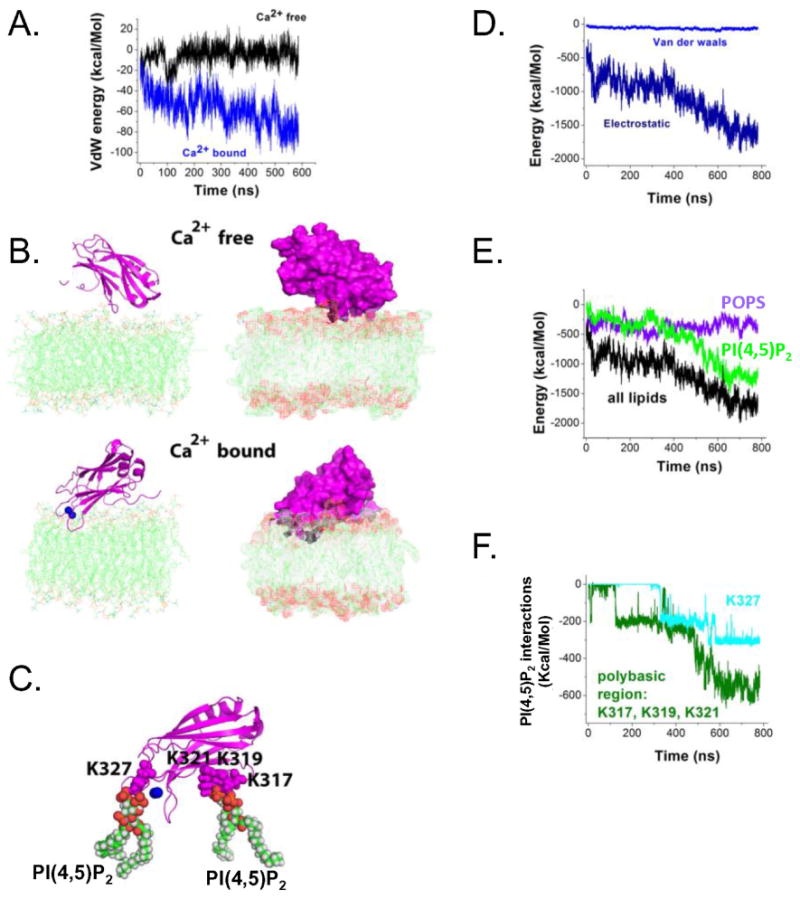
Molecular dynamics simulations show lipid penetration by Ca2+-binding loops of the Ca2+ -bound C2B domain of DOC2B, which is promoted by the interaction of the polybasic region (K317, K319, and K321) with PI(4,5)P2. (A) Van der Waals (VdW) energy profile along the MD trajectories of Ca2+-free and Ca2+-bound forms of DOC2B C2B domain indicate lipid penetration for the Ca2+-bound but not Ca2+-free form. The graph depicts VdW energy of protein–lipid interactions. Note steadily decreasing negative energy for the Ca2+-bound form, indicating penetration (blue line), as well as a constant energy fluctuating around zero for the Ca2+-free form, corresponding to relatively weak interactions (black line). (B) Ca2+-free and Ca2+-bound forms of the DOC2B C2B domain (magenta) at the end of the trajectory. In its Ca2+-free form, the protein remains on the surface of the lipid bilayer. In contrast, the Ca2+-bound form of DOC2B penetrates the lipids. Ca2+ ions are shown as blue spheres. Two different views are shown: line/cartoon representation (left) and surface representation (right). Note deep penetration of the DOC2B C2B domain in the Ca2+-bound form. (C) PI(4,5)P2 interactions of the DOC2B C2B domain in the Ca2+-bound form. Three residues of the polybasic region, K317, K319 and K321, form salt bridges with a PI(4,5)P2 molecule, anchoring the protein to the lipid bilayer. In addition, interaction of another PI(4,5)P2 molecule with the residue K327 positioned at one of the Ca2+-binding loops enhances the penetration. (D) Energy components of protein–lipid interactions show domination of electrostatics. (E) The interactions of the protein with PI(4,5)P2 prevail at the end of the trajectory, although PI(4,5)P2 and POPS lipids make comparable contributions to the overall energy of the protein–lipid interactions at the beginning of the trajectory. (F) The energy of the interactions of PI(4,5)P2 with the polybasic residues K317, K319, and K321, as well as with residue K327 of a Ca2+-binding loop. The energy decrease along the trajectory reflects the formation of multiple salt bridges.
In contrast, the Ca2+-bound form of DOC2B's C2B domain was consistently penetrating into the bilayer (note a steady decrease in the energy of VdW interactions between the protein and lipids; Figure 6A, blue line). At the final point of the trajectory (Figure 6B), the Ca2+-binding loops of the protein deeply penetrated the lipid bilayer. Importantly, this state was stabilized by a salt bridge between lysine 319 and a PI(4,5)P2 molecule (Figure 6C). In addition, salt bridges were formed between the same PI(4,5)P2 molecule and the residues K317 and K321. Finally, a salt bridge was formed between another PI(4,5)P2 molecule and lysine 327 belonging to one of the Ca2+-binding loops. This interaction is likely to promote penetration of the Ca2+-binding loops into the lipid bilayer. Overall, fairly deep penetration of the Ca2+-bound C2B domain was observed (Figure 6C), involving the polybasic region of the domain as well as its Ca2+-bound loops.
The analysis of energy components revealed that the protein–lipid interactions are largely governed by electrostatics (Figure 6D). Interactions of the protein with POPS and PI(4,5)P2 lipids made the major contribution to the overall energy of the protein–lipid interactions (Figure 6E). The energy of the interactions between the protein and PI(4,5)P2 molecules was close to zero at the initial time point, but it decreased along the trajectory (Figure 6E) as the salt bridges between the protein and PI(4,5)P2 molecules formed (Figure 6F). In contrast, the energy of the interactions between the protein and POPS lipids remained at a steady level (Figure 6E). Although at the beginning of the trajectory, the interactions of the protein with POPS and PI(4,5)P2 made roughly equal contributions to the overall energy of the protein–lipid interactions, at the end of the trajectory, interactions with PI(4,5)P2 strongly prevailed (Figure 6E). The residues of the polybasic region K317, K319, and K321 made the major contribution to these interactions, and they were also fortified by the salt bridge between PI(4,5)P2 and K327 (Figure 6C,F).
To validate the simulation, and assess the contribution of the basic regions to translocation and PI(4,5)P2 binding, we performed translocation assays to compare the translocation of C2BWT, C2BK319E and C2BK327E. While C2BWT translocated to the PM following KCl stimulation, translocation of C2BK319E was completely abolished. The translocation of C2BK327E was also significantly impaired, albeit to a lesser extent. This might be due to the fact that the lysine-to-glutamate exchange at position 319 interferes with the charge of neighboring basic residues in the polybasic region, magnifying its effect (Figure 7A,B).
We further tested the effect of the mutations in lysines 237, 319, 327 on the translocation of full-length DOC2B to learn more about the contribution of each C2 domain to PI(4,5)P2 binding and PM association. We found that impairing the C2B–PI(4,5)P2 interaction significantly impairs translocation of the full-length protein. Impairing PI(4,5)P2 binding in C2A by the K237E mutation 1 also reduced the full-length protein's translocation, although this reduction did not reach statistical significance (Figure 7C,D). R-GECO was co-expressed in the cells to verify that the Ca2+ dynamics was similar under all tested conditions (Figure 7B,D, bottom graphs).
DOC2B translocation to the PM is not affected by other phosphoinositides (PIs)
We next examined whether manipulations of other PIs would have an effect similar to that of PI(4,5)P2 on DOC2B translocation. We manipulated phosphatidylinositol (3,4)-bisphosphate [PI(3,4)P2] and phosphatidylinositol (3,4,5)-trisphosphate [PI(3,4,5)P3] at the PM and examined DOC2B-GFP translocation under these conditions. To substantially reduce PI(3,4)P2 and PI(3,4,5)P3 at the PM, we used the PI3 kinase (PI3K) inhibitor wortmannin 38, and monitored their localization using the PI(3,4)P2 and PI(3,4,5)P3 biosensor PH-Akt-GFP 39-42. Indeed, wortmannin induced loss of PI(3,4)P2 and PI(3,4,5)P3, as indicated by the relocalization of PH-Akt-GFP from the PM to the cytosol (Figure 8A). However, DOC2B translocation persisted. We further quantified DOC2B translocation to find any abnormalities following incubation with wortmannin. In some of our experimental repeats, PI(3,4,5)P3 manipulations affected the Ca2+ influx, and we therefore simultaneously measured translocation and Ca2+ using the Ca2+ indicator Fura4F-AM in epifluorescence. However, no abnormalities in translocation were detected in cells with sufficient Ca2+ elevation (above the EC50 required for DOC2B translocation; Figure 8B,C).
Figure 8.
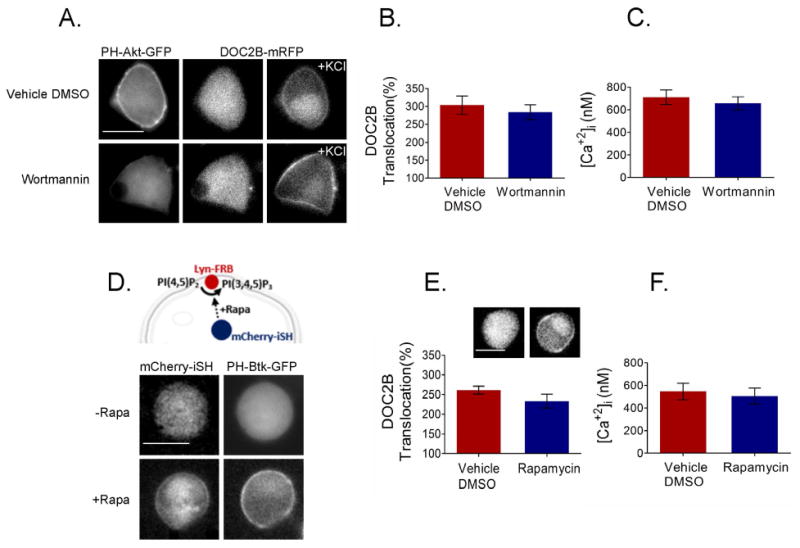
PI(3,4)P2 and PI(3,4,5)P3 are not crucial for DOC2B translocation. (A) DOC2B translocation was not affected by loss of PI(3,4)P2 or PI(3,4,5)P3, induced by wortmannin and reported as detachment of the PI(3,4)P2 and PI(3,4,5)P3 biosensor PH-Akt-GFP from the PM (n > 10). (B) Quantification of DOC2B KCl induced translocation under the wortmannin and control conditions (n = 9 for control and 8 for wortmannin-treated cells; t-test P > 0.05). (D) PI(3,4,5)P3 enhancement using the rapamycin system. (top) schematic representation of the rapamycin (Rapa)-induced PI(3,4,5)P3 synthesis at the PM. Rapamycin induced dimerization of mCherry-iSH and Lyn-FRB resulting in (bottom) robust translocation of the PI(3,4,5)P3-specific biosensor PH-Btk-GFP to the PM, indicating PI(3,4,5)P3 synthesis at the PM (n > 5). (E) DOC2B-GFP remains cytosolic under conditions of increased PI(3,4,5)P3 and translocates normally following KCl stimulation (inset: representative DOC2B-GFP images; n = 9 under both conditions; t-test P > 0.05). (C,F) Quantification of calcium using Fura4F- AM in the cells presented in B and E, respectively (t-test P > 0.05 under both conditions). Data presented as mean ± SEM. Scale bar = 10 μm.
To increase PI(3,4,5)P3 levels at the PM, we used the rapamycin system. Here, rapamycin induced the dimerization of a fluorescently tagged FKBP-iSH and the PM anchor Lyn11-FRB. The iSH unit is an inter-Src homology 2 (iSH2) domain from p85, which complexes in cells with the endogenous PI3K p110 to promote PI(3,4,5)P3 synthesis without affecting PI(4,5)P2 levels 20. PH-Btk-GFP, a specific biosensor for PI(3,4,5)P3, was used to verify the latter's synthesis at the PM 41. Rapamycin induced robust translocation of PH-Btk-GFP to the PM, indicating PI(3,4,5)P3 synthesis (Figure 8D). Nevertheless, DOC2B remained cytosolic under basal [Ca2+]i and its translocation to the PM was similar to that of the control upon KCl-induced depolarization (Figure 8E). Figure 8F shows that the average [Ca2+]i peaks, as measured by Fura4F-AM, were similar under enhanced PI(3,4,5)P3 and control conditions. These results suggest that neither PI(3,4)P2 nor PI(3,4,5)P3 play a significant role in the targeting of DOC2B to the PM, and that the observed requirement for PI(4,5)P2 in DOC2B translocation is specific to this PI.
Ca2+-dependent translocation of DOC2B to the PM is ATP-independent, driven by diffusion
After we established the target of DOC2B at the PM, we examined its mode of translocation. This translocation can be active—ATP-dependent, or passive—driven by diffusion. We thus examined DOC2B translocation under conditions aimed at influencing ATP dependent/enzymatically mediated processes: low temperature and ATP reduction.
Most active biological systems have a Q10 of 2–3, meaning that lowering the temperature by 10°C slows the kinetics of the process by a factor of 2–3 43-45. Thus, in these experiments, we induced a temperature change of at least 10°C to cause significant inhibition of all enzymatic activity. In the second set of experiments, ATP levels were reduced by a short (10 min) incubation with 2-deoxyglucose and sodium azide 44,46-48. As low temperature and low ATP reduce calcium-channel activation and calcium currents, we pre-incubated PC12 cells overexpressing DOC2B-GFP with NP-EGTA-AM. A flash of UV light was applied to uncage Ca2+ from NP-EGTA and elevate [Ca2+]i, bypassing any effect on calcium currents. Translocation was examined by TIRFM and its kinetics and amplitude were measured. We verified that these two manipulations indeed affect the ATP-dependent process of vesicular transport in cells, as evidenced by inhibition of the directed movement of large dense-core vesicles stained with the vesicle marker NPY-mRFP (Figure 9A,E). Under conditions of reduced ATP or low temperature, the kinetics and amplitude of DOC2B translocation remained unaffected (Figure 9B–D, F–H).
Figure 9.
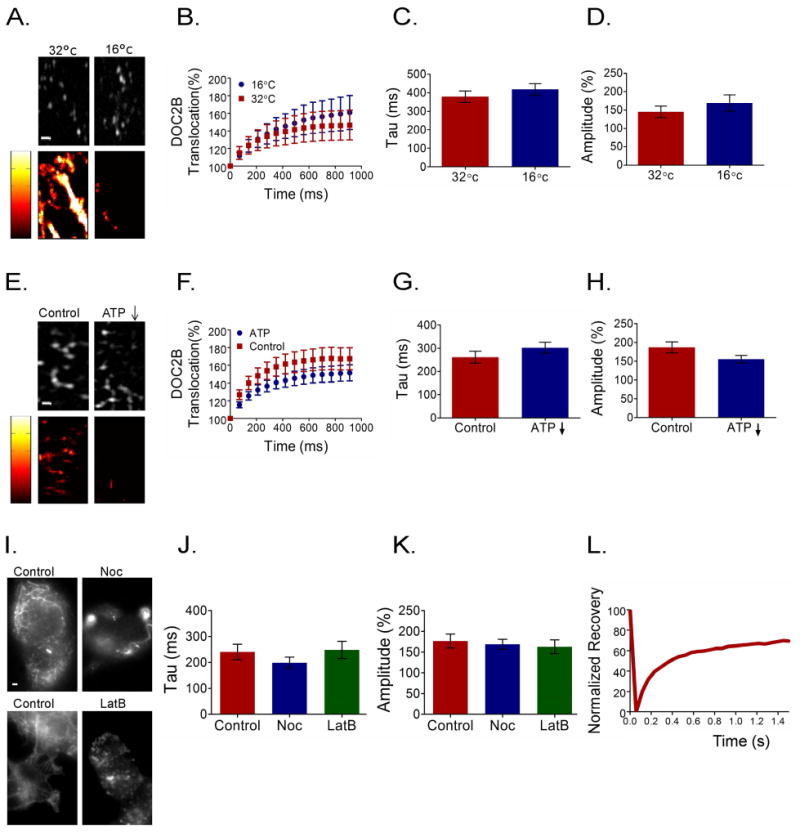
DOC2B translocation is ATP-independent with diffusion-like kinetics. (A,E) Vesicle trafficking in BHK-21 cells as a control for an ATP-dependent process that is severely affected by temperature change and ATP reduction. NPY-mRFP tagged vesicles (top panels) and their mobility maps (bottom panels) within a BHK-21 cell under elevated and decreased temperature conditions (A) or under reduced ATP and control conditions (E). High mobility and low mobility are represented by bright and dark colors, respectively (mobility scores were: 30.4 ± 3.8 and 7.7 ± 2.7 for the high- and low-temperature conditions, respectively, and 16.3 ± 2.4 and 1.77 ± 0.9 for the control and ATP-reduced conditions, respectively; n = 5 in each group, Mann–Whitney P<0.005). (B,F) Averaged TIRFM traces of DOC2B-GFP translocation under the temperature-change and ATP-reduction conditions, respectively. (C,D,G,H) Quantification of the TIRFM DOC2B-GFP translocation parameters: amplitude and kinetics in the temperature (n = 10 and 11 for 16°C and 32°C, respectively; t-test P > 0.05 for both tau and amplitude) and ATP-reduction experiments (n = 10 and 11 for control and ATP-depletion conditions, respectively; t-test P > 0.05 for both tau and amplitude). (I) Control PC12 staining for microtubules (top panels) and actin filaments (bottom panels) before and after incubation with nocodazole (Noc) or latrunculin B (LatB), respectively (n > 5 in each group). (J,K) TIRFM quantification of DOC2B-GFP translocation in the presence of nocodazole or latrunculin B in terms of amplitude and kinetics (n = 10,11 and 10 for control, nocodazole and latrunculin B respectively; for both tau and amplitude, when each condition was compared separately to the control: t-test P > 0.05). Cells were pre-incubated with NP-EGTA-AM and translocation was induced by a flash of UV light. (L) Representative FRAP trace of DOC2B-GFP. Data presented in bar graphs as mean ± SEM. Scale bar = 1 μm.
We further examined whether impairing the cytoskeleton would affect DOC2B translocation, as most active transport of proteins and vesicles in the cytoplasm is based on elements of the cytoskeleton network. We incubated PC12 cells overexpressing DOC2B-GFP with latrunculin B or nocodazole to depolymerize actin or microtubules, respectively. As can be seen in the control images of actin and microtubule staining, these networks were completely disassembled under our experimental conditions (Figure 9I). Nevertheless, both the kinetics and amplitude of flash-induced DOC2B-GFP translocation remained unchanged (Figure 9J,K).
If translocation of DOC2B to the PM is ATP-independent, it might occur through simple diffusion. We thus measured DOC2B cytoplasmic diffusion rate by fluorescence recovery after photobleaching (FRAP). In this experiment, DOC2B-GFP-expressing PC12 cells were photobleached by a laser beam in a small round area of the cytoplasm (radius ≈ 2 μm), and recovery of fluorescence in this area was measured (Figure 9L). The recovery time constant (τ) value of DOC2B-GFP was 82 ± 4 ms (n = 10), yielding an estimated diffusion coefficient (D) of 8 μm2/s 49, which is in accordance with diffusion coefficients reported in the literature for small proteins in the cytoplasm 50,51, and allows a translocation time constant of 200–400 ms for DOC2B-GFP in a typical PC12 cell with a radius of ∼5 μm (Figure 9C,G,J). We thus concluded that DOC2B translocation is ATP-independent, and driven by diffusion.
Discussion
In this study, we elucidated the Ca2+-dependent translocation mechanism and targeting of DOC2B to the PM. We demonstrated that DOC2B is targeted to the PM by PI(4,5)P2 following elevation of [Ca2+]i, and that its arrival to the membrane is ATP-independent with diffusion-like kinetics.
To elucidate the molecular component that directs DOC2B to the PM under elevated [Ca2+]i, we screened the expression patterns of the known DOC2B-interacting partners in the PC12 platform: PS, PI(4,5)P2 and the t-SNARE proteins syx1A and SNAP25 1,3,4. We assumed that the targeting element of DOC2B to the PM should also be predominantly localized to the PM. As was demonstrated in this work and by others before e.g., 16,17,19, only PI(4,5)P2 and to some extent SNAP25 are restricted to the PM in a manner similar to DOC2B•Ca2+. In contrast, syx1A and PS are distributed in the PM but also in intracellular membranes where DOC2B•Ca2+ is absent. However, there is some evidence suggesting that DOC2B interacts with binary SNAP•syx complexes, which can be found predominantly in the PM of PC12 cells 4,17. This suggested that t-SNAREs, PI(4,5)P2 or both, target DOC2B to the PM. The fact that the full-length DOC2B mutant, DOC2BK237,319E, which is deficient in t-SNARE and PI(4,5)P2 binding, failed to translocate to the PM in the presence of elevated [Ca2+]i strengthened this notion. To understand which of the two—t-SNARE or PI(4,5)P2 binding—is responsible for this effect, we manipulated each component separately at the PM and examined DOC2BWT translocation. This led to the conclusion that PI(4,5)P2 is crucial for DOC2B translocation, and thus targets DOC2B to the PM. The facts that the calcium dynamics remained similar among our experimental conditions and that DOC2BD218,220N, which is found constitutively at the membrane under basal [Ca2+]i 4,36, detached from the PM following PI(4,5)P2 hydrolysis, suggested that our observation regarding the requirement of PI(4,5)P2 for DOC2B translocation indeed stems from loss of PI(4,5)P2 and not from aberrations in Ca2+ dynamics. Furthermore, our ability to control and redirect DOC2B translocation to new destinations by synthesizing PI(4,5)P2 in intracellular membranes further strengthened our hypothesis that DOC2B is targeted to PI(4,5)P2-containing membranes. The latter constitutes a novel approach to examining protein targeting in living cells by demonstrating the spatial control of PI(4,5)P2 on DOC2B localization.
Enhancing PI(4,5)P2 levels at the PM did not result in enhanced DOC2B translocation. This is consistent with previous studies which also found that the PI(4,5)P2 biosensor itself, PLCδ-PH-GFP, is not affected by this manipulation 20. The synthesis of PI(4,5)P2 in intracellular membranes demonstrated that the CF-PIPK construct induces PI(4,5)P2 synthesis but there are additional possible explanations for this result: (1) basal PI(4,5)P2 levels are sufficient to induce maximal DOC2B translocation; (2) this method may only expand existing PI(4,5)P2 clusters without generating new ones, making it challenging to monitor changes in the recruitment of proteins; (3) intracellular regulation of PI(4,5)P2 levels prevents its excessive synthesis at the PM.
The necessity of PI(4,5)P2 for Ca2+-dependent translocation of DOC2B to the PM in living cells was unique to PI(4,5)P2, as depletion or synthesis of PI(3,4)P2 and PI(3,4,5)P3 did not affect DOC2B translocation. In vitro experiments with syt1 also showed a preference for PI(4,5)P2-containing liposomes over PI(3,4,5)P3 [but not PI(3,4)P2] under elevated [Ca2+]i 52. In addition, specific interactions between PI(4,5)P2 and DOC2B C2 domains, as well as other homologous C2 domains, have been described 1,26.
The existence of specific interactions between DOC2B and PI(4,5)P2 gained further support from the MD simulations of the Ca2+-bound and unbound forms of DOC2B C2B in the presence of a PI(4,5)P2-containing membrane. The simulations revealed salt bridges between PI(4,5)P2 molecules and several DOC2B residues, including K319 in the polybasic region of C2B. The MD simulation also demonstrated that the interactions between the polybasic residues in C2B and PI(4,5)P2 promote deep penetration of the polybasic residues themselves, as well as of the domain's Ca2+-binding loops, including residue K327, and that this anchors the protein to the membrane. Given this information, it is not surprising that mutating K327 or K319 to glutamate in isolated C2B impaired its translocation. The impairment of translocation was more severe for C2BK319E than C2BK327E. This may be explained by the fact that unlike lysine 327, lysine 319 is surrounded by other basic residues and its replacement by glutamate may interfere with their PI(4,5)P2 interaction as well, thus reducing the overall interaction with the PM. Mutating these residues in the full-length DOC2B also dramatically decreased translocation. The contribution of C2A to PI(4,5)P2 binding was also examined by mutating K237 to glutamate in the full-length DOC2B. DOC2BK237E translocation was also reduced compared to DOC2BWT, however not significantly so, and the reduction was far less prominent than the effect of the mutations in C2B.
Although in vitro studies, as well as our MD simulations, demonstrated that DOC2B interact with PS in the presence of Ca2+ 1,5,6, it seems that in living cells, PS is not sufficient to induce DOC2B translocation. This is supported by previous experiments 5,8,36, that did not detect DOC2B•Ca2+ translocating to intracellular organelles enriched with PS under normal conditions. For instance, PS constitutes 12% of the synaptic vesicle (SV) membrane 15, a concentration that should allow DOC2B translocation to these vesicles as it does in vitro, with liposomes containing similar concentrations of PS 3. In addition, if PS was sufficient to induce DOC2B translocation, our PI(4,5)P2 hydrolysis experiment would not have had such drastic results. Along the same lines, the C2A of DOC2B cannot translocate to the PM following KCl-induced depolarization in PC12 cells. However, it is found bound to PS-containing liposomes in the presence of submicromolar [Ca2+] in vitro 4-6. These differences might stem from cellular factors that regulate C2 domain–PM binding and lead to the difference between in vitro and in vivo results. Hence, a direct interpretation of in vitro binding data to in vivo conditions should be made cautiously. The MD simulation performed here provides some possible explanations and suggests that interactions with PI(4,5)P2 prevail over those with PS, and anchor the protein to the membrane within less than a millisecond. It is possible that intracellular conditions such as ionic strength or intracellular factors regulate the interaction and allow translocation only when strong interactions with PI(4,5)P2 occur. One of these factors may be Munc13-1 53-55 that has been shown to interact with DOC2 proteins via their Munc13 interacting domains (MID) and can affect their translocation and interaction with the PM. Further studies are required to elucidate the significance of PS and other factors to DOC2B localization and function in a physiological environment.
Finally, we tested the ATP dependence of DOC2B's arrival to the PM. We performed several manipulations that severely affected ATP-dependent, enzyme-mediated processes in the context of intracellular active transport events, including low temperature, ATP reduction and disruption of cytoskeleton integrity. DOC2B translocation to the PM was unaffected by these manipulations, suggesting that the translocation is ATP-independent. Furthermore, we propose that DOC2B's arrival to the PM is driven by diffusion. This is supported by a similarity in the timescale of DOC2B translocation and the diffusion coefficient extracted from the FRAP measurements. Accordingly, we suggest the following scenario for the translocation: upon calcium influx, DOC2B•Ca2+ molecules that are close to the PM bind to it immediately. The accumulation of DOC2B at the PM causes the area near the PM to be devoid of DOC2B, which further drives diffusion of DOC2B•Ca2+ from inner parts of the cell toward the PM. The final stage of the translocation mechanism likely includes an electrostatic attraction between DOC2B•Ca2+ and the PLs near the PM 56,57. This promotes specific PI(4,5)P2 interactions and penetration of adjacent hydrophobic residues, some of which are exposed as a result of Ca2+ activation, into the lipid bilayer 1,58. The MD simulation suggests that charge neutralization in the Ca2+-binding site of the C2B domain by Ca2+ reduces the electrostatic repulsion of the domain from the PM and promotes association with PI(4,5)P2, as recently described for syt1 27 and the constitutive PM localization of DOC2BD218,220N. We thus conclude that both Ca2+ and PI(4,5)P2 are required for DOC2B translocation in living cells.
The present findings suggest that the molecular basis for the DOC2B–PM interaction is very similar to that for syt1. These similarities raise questions as to why syt1 affects synchronous release whereas DOC2B affects spontaneous and asynchronous release. This can be explained by differences in the proteins' primary localization and Ca2+ sensitivity. Syt1 is localized to SVs at high concentrations, about 15 copies per SV 15, near Ca2+ channels 59-61, whereas DOC2B is located further from the Ca2+ source and has to translocate from the cytosol to the PM. The Ca2+ sensitivities of syt1 and DOC2B are perfectly adapted to their basal localization: syt1 binds calcium at high, micromolar concentrations 62, suitable for a reaction to intense Ca2+ entry from the Ca2+ channels following depolarization, whereas DOC2B possesses a higher Ca2+ affinity, within the submicromolar range 6,8, optimized to sense even the slightest increase in [Ca2+]i diffusing from the PM to the cytosol following the depolarization. DOC2B then translocates to the PM within a few hundred milliseconds to promote additional release, observed as delayed, asynchronous release. These properties make DOC2B an ideal protein to support asynchronous release: its accumulation at the PM near fusion sites occurs after the synchronous release is over, but its high calcium sensitivity allows it to drive asynchronous release for an additional 20–100 ms. It is reasonable to assume that the interplay between synchronous and asynchronous release is set by local levels of syt1 and DOC2B, by calcium levels, PI(4,5)P2 and additional proteins, lipids and other factors. It is even possible that attachment of DOC2B C2 domains to resting SVs will cause their spontaneous release due to some small local Ca2+ fluctuation. These hypotheses await future experiments. In summary, our findings suggest that following submicromolar [Ca2+] elevation in the cytosol, DOC2B binds Ca2+ and PI(4,5)P2-containing membranes. This drives its translocation, which is characterized by diffusion-like kinetics.
Materials and Methods
Cell cultures and transfections
PC12 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% horse serum, 5% fetal bovine serum (FBS), and 1% penicillin/streptomycin. COS7 and BHK-21 cells were cultured in high-glucose DMEM-based medium containing 10% FBS, 1% penicillin/streptomycin, and 1% glutamine. All cells were incubated at 37°C and 5% CO2. For experiments, cells were plated on 18- or 25-mm glass coverslips, and for PC12 cells, coverslips were coated with 0.1 mg/ml PDL. All cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions, and used 24–48 h later.
DNA constructs
Mutations were introduced into DOC2B-GFP N2 7, C2AB-GFP N2 or C2B-GFP N2 6, using site-directed mutagenesis PCR. Botulinum toxins BoNT/C and BoNT/E were a generous gift from the laboratory of Prof. Ilana Lotan (Tel-Aviv University). mRFP-syx1A and SNAP25-YFP were a kind gift from Prof. Edward Stuenkel (University of Michigan). R-GECO was a generous gift from the laboratory of Prof. Robert Campbell 31. Lact-C2-GFP was a gift from Prof. Sergio Grinstein (addgene #22852). For PI manipulations using the rapamycin dimerization system, the PH domain of PLCδ1, PLCδ-PH-GFP (addgene #21179), CF-Inp (addgene #20155), CF-InpD281A (addgene #20156) and Lyn11-FRB (addgene #20147) were a gift from Prof. Tobias Meyer. CF-PIPK, CF-PIPKi, mcherry-iSH were a generous gift from the laboratory of Prof. Takanari Inoue (Johns Hopkins University). PLCδ-PH-mKate was a kind gift from Prof. Thomas Martin (University of Wisconsin-Madison). Rab7-FRB (addgene #51613), PH-Akt-GFP (addgene #51465), and PH-Btk-GFP (addgene #51463) were a gift from Prof. Tamas Balla. NPY-mRFP was a kind gift from Prof. Matthijs Verhage (Vrije University).
Translocation experiments
For PC12 cells, unless otherwise stated, recordings were conducted at room temperature in a bath solution (130 mM NaCl, 3 mM KCl, 2 mM MgCl2, 5 mM CaCl2, 10 mM HEPES and 2 g/L glucose), and depolarization was achieved using a high K+ solution (40 mM NaCl, 100 mM KCl, 2 mM MgCl2, 10 mM CaCl2, 10 mM HEPES and 2 g/L glucose), followed by washing in a fresh bath solution. Epifluorescence or TIRF images were acquired as noted. Translocation was quantified in TIRFM as the increase in percent fluorescence relative to the baseline fluorescence prior to depolarization. To verify [Ca2+]i increase in cases of failure to translocate to the PM, we repeated the experiment in the presence of R-GECO which was monitored at λ = 561 nm, 0.2 Hz. For COS7 cells, imaging medium consisted of phenol red-free DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 1% glutamine and 10 mM HEPES. To induce translocation, 10 μM ionomycin and 2 mM Ca2+ were added to the imaging solution.
SNARE cleavage
PC12 cells were transfected with botulinum toxins BoNT/C and BoNT/E together with DOC2B-GFP (4:4:1, respectively) or with DOC2B-GFP only as a control. Translocation experiments were performed 48 h post-transfection.
PI(4,5)P2 manipulations
These were conducted using the rapamycin system as described previously 20 and in the Results section. In these experiments, 5 μM rapamycin was added for 2 min to the bath solution to induce the dimerization of CF-Inp, CF-PIPK or their respective inactive controls CF-InpD281A and CF-PIPKi with the FRB anchor, Lyn11-FRB. Rapamycin was then washed in a fresh bath solution [dimerization of FKBP and FRB is irreversible 20]. PLCδ-PH-GFP to monitor PI(4,5)P2 localization or DOC2B-GFP were co-expressed with the rapamycin system constructs.
For the endosomal synthesis of PI(4,5)P2 in COS7 cells, transfection was optimized to include the following constructs: CF-PIPK (or CF-PIPKi as a control), Rab7-FRB, PLCδ-PH-GFP and DOC2B-mRFP at a ratio of 2:1:1:2, respectively, with a total 0.6 μg DNA. When examining translocation of isolated C2B-GFP and C2BK319E-GFP, PLCδ-PH-mKate was co-expressed together with CF-PIPK and Rab7-FRB in similar amounts. Cells were transferred to the microscope in the imaging solution and 2.5 μM rapamycin was added for up to 20 min as previously suggested 37. This induced the dimerization of CF-PIPK with the Rab7-FRB endosome anchor, used here to initiate PI(4,5)P2 synthesis in intracellular membranes.
Molecular Dynamics
The structure 4ldc obtained by crystallography 6 was used as an initial state for the C2B domain of DOC2B. A Ca2+-free form of DOC2B C2B was obtained by removing Ca2+ ions from the 4ldc structure. The model of the POPC/POPS/PI(4,5)P2 (75:20:5) lipid bilayer was kindly provided by Dr. J. Wereszczynski 63. The molecular system containing the bilayer and the protein in a water box (65 × 65 × 110 Å) was constructed employing VMD software (Visual Molecular Dynamics, Theoretical and Computational Biophysics Group, NIH Center for Macromolecular Modeling and Bioinformatics, University of Illinois at Urbana-Champaign). Potassium and chloride ions were added to neutralize each system and to yield 150 mM KCl. MD simulations were performed with NAMD Scalable Molecular Dynamics software 64 (Theoretical and Computational Biophysics Group, NIH Center for Macromolecular Modeling and Bioinformatics) at the XSEDE (Extreme Science and Engineering Discovery Environment) Stampede cluster (TACC). We used the CHARMM36 65 force field which included parameters for PI(4,5)P2 63,66. All of the simulations were performed with periodic boundary conditions and Ewald electrostatics. The system was minimized for 200 steps, and the heating phase was performed for 1 ps with a 1-fs step. Subsequently, an equilibration phase in the NPT ensemble with a flexible cell and Berendsen barostat was performed for 20 ps. Production runs were performed as described by Bykhovskaia 67, with a 1.5-fs step and employing a Langevin thermostat at 300K. The trajectory analysis was performed with VMD and Vega ZZ (Drug Design Laboratory) software; PyMOL software was used for visualization, molecular graphics, and illustrations.
PI(3,4)P2 and PI(3,4,5)P3 manipulations
PI(3,4)P2 and PI(3,4,5)P3 depletion was achieved using the PIP3K inhibitor wortmannin 38. PH-Akt-GFP, a biosensor for PI(3,4)P2 and PI(3,4,5)P3, was used as an indicator for the reduction of these PIs at the PM 41. Cells were incubated for 1 h prior to recordings with imaging medium containing 400 nM wortmannin and 5 μM Fura4F-AM (Invitrogen), or vehicle dimethyl sulfoxide (DMSO) as a control. PH-Akt-GFP and DOC2B-mRFP images were acquired as detailed in the imaging setup section. PI(3,4,5)P3 synthesis was achieved using the rapamycin system: 5 μM rapamycin induced the dimerization of mcherry-iSH and Lyn11-FRB as described in detail in Results and by Suh et al. 20. PI(3,4,5)P3 synthesis was verified using the PI(3,4,5)P3-specific biosensor PH-Btk-GFP. Cells were incubated in OptiMEM 90 min before the experiments, and 1 h prior to the experiments, 5 μM Fura4F-AM and pluronic acid (1:1, v/v) were added to the OptiMEM. Rapamycin (5 μM) or DMSO vehicle was also added for 2 min prior to the experiment, and then washed away with fresh bath solution. Cells were then taken to a microscope and translocation was induced locally by the high-K+ solution. A combined epifluorescence measurement of translocation and [Ca2+]i was conducted using acquisition of three images at three wavelengths (200 ms exposure each) at 1 Hz: 488 nm was used to view DOC2B-GFP, and 350 nm and 380 nm were used to excite Fura4F-AM. [Ca2+]i was deduced from the Fura4F-AM measurements according to the dual excitation ratio imaging technique and the Grynkiewicz equation 68, and was based on pre-calibration with solutions of known [Ca+2] 8. Translocation was determined as the fluorescence in the membrane divided by the fluorescence at the center of the cell. This ratio was normalized to the baseline ratio for every cell and presented as percentage.
Temperature and ATP-reduction experiments
Prior to these experiments, cells were incubated for 45 min with OptiMEM. Then, 10 μM NP-EGTA-AM (Invitrogen) and pluronic acid (1:1, v/v) were added to the OptiMEM for another 45 min. Following incubation, the cells were transferred to the imaging setting in the bath solution. For the temperature experiments, the temperature of the imaging setup and perfusion solutions was set to either 32°C or 16°C. For ATP-reduction experiments, following the NP-EGTA-AM incubation and immediately prior to imaging, cells were maintained for 15 min in a modified bath solution containing 10 mM 2-deoxyglucose, 10 mM sodium azide, 5% horse serum (HS) and 2.5% fetal calf serum (FCS) with no added glucose (osmolarity of the solution was maintained in the physiological range by NaCl adjustments). The control group for this experiment was examined in a bath solution containing glucose and the serum mix. A flash of UV light (Rapp OptoElectronic; Hamburg, Germany) was applied to release caged Ca2+ and induce translocation. Translocation was measured by TIRFM at 14.3 Hz as described in the imaging setup section. To demonstrate the impact of these manipulations on enzymatic/ATP-dependent cellular processes such as vesicle trafficking, we imaged BHK-21 cells overexpressing the vesicle marker NPY-mRFP 69 at 0.3 Hz as described in the microscopy setup section. A rough estimation of the dynamics of the movie, i.e., the appearance, disappearance, movement and flickering of NPY-mRFP-tagged vesicles, was calculated through a series of simple arithmetic manipulations on the images defining changes in pixel intensity as vesicle dynamics. This was done using an algorithm that was previously developed in our laboratory 70.
Cytoskeleton depolymerization and staining procedures
Depolymerization of actin and microtubules was achieved by incubating PC12 cells with 10 mM latrunculin B for 20 min or 10 mM nocodazole for 90 min, respectively. Translocation experiments were conducted identically to the ATP-reduction and temperature experiments except that the bath solutions also included 5% HS, 2.5% FCS and 10 mM of the depolymerization agent (i.e., latrunculin B or nocodazole). Depolymerization of the cytoskeleton was verified by staining. After incubation in OptiMEM and 10 mM latrunculin B, PC12 cells were fixed in 4% paraformaldehyde and stained with 0.2 μg/mL TRITC-labeled phalloidin (20 min incubation). PC12 cells that were incubated in OptiMEM and 10 mM nocodazole for 90 min were fixed with cold (-20°C) methanol for 5 min. Mouse anti-microtubule antibody (1:200; Sigma) was used and detected by goat anti-mouse Alexa568 (1:1000; Invitrogen).
FRAP
PC12 cells overexpressing DOC2B-GFP or GFP alone were used in these experiments at room temperature. Twelve baseline images were taken prior to the bleaching. The bleach pulse was for 10 ms at maximum laser intensity, aimed toward cytoplasmic areas to induce a round bleaching region with a radius of ≈2 μm; 50 post-bleach images were taken at 17.2 Hz to follow the recovery of fluorescence. Fluorescence before and after bleaching from the bleached area was measured and plotted against time. Time constants, Tau values were then calculated from the plots. The diffusion coefficient (D) was estimated using the equation: D = w2/2nτ, where w is the radius of the bleached area, n is the spatial dimension 49 (n=3 in this experiment due to the use of high magnitude 60× lens, standard microscopy and a large bleaching area) and τ is the time constant of the fluorescent recovery measured in the experiment.
Imaging setups
For the experiments described in this paper, three different microscopes were used, each with its setting optimized for the particular experiment.
Most experiments (DOC2 mutants, SNARE cleavage, FRAP and most PI manipulations, except a few listed in the section below) were conducted on an imaging setup consisting of an iMIC inverted microscope with an oil-immersion Plan-Apochromatic 100× objective (NA = 1.45, Olympus), a polytrope condenser (iMIC Beam-switch for wide field, TIRF, FRAP illumination), and an Andor iXon DU 888D EMCCD camera (Belfast, Northern Ireland). The equipment was controlled by Live Acquisition Software (TILL Photonics, Gräfelfing, Germany). For epifluorescence imaging, a polychrome V system (TILL Photonics) was used for illumination; for TIRF and FRAP imaging, the solid-state laser “Calypso” 491 nm, 100 mW was used; typical exposure time was 200 ms for GFP and mRFP and 500 ms for CFP.
Experiments involving Fura4F-AM measurements and UV flash [PI(3,4)P2 and PI(3,4,5)P3 manipulations; temperature, ATP and cytoskeleton dependency] were conducted on an inverted Olympus IX-70 microscope with an Andor Ixon 887 EMCCD camera controlled by METAFLUOR software (Universal Imaging) for multiple-wavelength measurements and by MetaMorph software (Molecular Devices, Downingtown, PA) for single-wavelength measurements. Epi fluorescence images were taken using polychrome IV illumination (TILL Photonics) and a 40× objective. TIRF images were acquired using a 60× (TIRF) objective (Olympus), a TIRF condenser (TILL Photonics) and a 473-nm solid-state laser (Laser Quantum, Stockport, UK).
Images of cytoskeleton depolymerization staining were taken at an exposure time of 1 s by an inverted Olympus IX-70 equipped with a 100× objective, XM10 camera (Olympus) and X-Cite 120 PC lamp for illumination.
Image processing
Images were processed using ImageJ software 1.37c 71, subtracted for background using simple subtraction and/or brightness/contrast adjustments and then smoothed once (R-GECO images were not smoothed).
Supplementary Material
Supplementary Figure S1 Initial examination of potential DOC2B targeting. Expression patterns of DOC2B and its interacting partners in PC12 cells. Left: DOC2B-GFP before and after Ca2+ elevation induced by KCl depolarization. Right: phosphatidylserine and PI(4,5)P2 distribution, detected by their biosensors, Lact-C2-GFP and PLCδ-PH-GFP, respectively. Expression patterns of the t-SNARE proteins, mRFP-syx1A and SNAP25-YFP were examined by overexpressing them in the cells. Scale bar = 10 μm.
Supplementary Figure S2 Isolated DOC2B C2B follows PLCd-PH to intracellular membranes. (A) Rapamycin (Rapa) induced CF-PIPK translocation to intracellular membranes as a result of the FKBP-FRB dimerization, inducing PI(4,5)P2 synthesis as reported by its PLCδ-PH-mKate biosensor. C2BWT-GFP translocated to PLCδ-PH-mKate-stained organelles following ionomycin application. (B) C2BK319E-GFP, impaired in PI(4,5)P2 binding, did not translocate to the PM or to the intracellular organelles following ionomycin application. Scale bar = 10 μm.
Synopsis.
DOC2B, a Ca2+ sensor, translocates from the cytosol to the plasma membrane (PM) upon calcium elevation and affects vesicle fusion. We show that DOC2B translocation is diffusion-driven and targeted to the PM by specific interactions with PI(4,5)P2, which anchors the protein to the membrane. Molecular dynamics simulations and mutagenesis revealed multiple PI(4,5)P2–DOC2B interactions and suggest that both calcium and PI(4,5)P2 binding are needed for DOC2B translocation. This demonstrates how membrane composition controls vesicle fusion and neuronal communication.
Acknowledgments
We would like to thank Prof. Takanari Inoue (Johns Hopkins University) for providing us with the plasmids required for the lipid manipulations. This work was supported in part by Israel Science Foundation grants 1211/07, 730/11 and 953/16 to U.A. and grant R01 MH099557 from the National Institutes of Health to M.B. The authors declare no competing financial interests.
Abbreviations
- BoNT
botulinum toxin
- [Ca2+]i
intracellular calcium concentration
- DOC2B
double C2 domain protein B
- PI
phosphoinositide
- PI(4,5)P2
phosphatidylinositol (4,5)-bisphosphate
- PM
plasma membrane
- PS
phosphatidylserine
- SV
synaptic vesicle
- syt
synaptotagmin
- syx
syntaxin
- TIRFM
total internal reflection fluorescence microscopy
- t-SNAREs
target SNAREs
Footnotes
Author contributions: L.M. conducted the great majority of the experimental work published here, including the writing of this paper.
I.G. was involved in cloning, writing of the paper and conducted some of the experiments.
M.B. Performed and analyzed the MD simulations of DOC2B C2B domain and contributed to writing the paper.
U.A. supervised the work and was involved in the writing of the paper.
References
- 1.Groffen AJ, Martens S, Arazola RD, et al. Doc2b Is a High-Affinity Ca2+ Sensor for Spontaneous Neurotransmitter Release. Science (New York, NY) 2010;327(5973):1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lavi A, Sheinin A, Shapira R, Zelmanoff D, Ashery U. DOC2B and Munc13-1 differentially regulate neuronal network activity. Cerebral cortex (New York, NY : 1991) 2014;24(9):2309–2323. doi: 10.1093/cercor/bht081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yao J, Gaffaney Jon D, Kwon Sung E, Chapman Edwin R. Doc2 Is a Ca2+ Sensor Required for Asynchronous Neurotransmitter Release. Cell. 2011;147(3):666–677. doi: 10.1016/j.cell.2011.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedrich R, Groffen AJ, Connell E, et al. DOC2B Acts as a Calcium Switch and Enhances Vesicle Fusion. Journal of Neuroscience. 2008;28(27):6794–6806. doi: 10.1523/JNEUROSCI.0538-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaffaney JD, Xue R, Chapman ER. Mutations that disrupt Ca2+-binding activity endow Doc2β with novel functional properties during synaptic transmission. Molecular biology of the cell. 2014;25(4):481–494. doi: 10.1091/mbc.E13-10-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giladi M, Michaeli L, Almagor L, et al. The C2B Domain Is the Primary Ca2+ Sensor in DOC2B: A Structural and Functional Analysis. Journal of Molecular Biology. 2013;425(22):4629–4641. doi: 10.1016/j.jmb.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 7.Groffen AJA, Brian EC, Dudok JJ, Kampmeijer J, Toonen RF, Verhage M. Ca2+-induced Recruitment of the Secretory Vesicle Protein DOC2B to the Target Membrane. Journal of Biological Chemistry. 2004;279(22):23740–23747. doi: 10.1074/jbc.M400731200. [DOI] [PubMed] [Google Scholar]
- 8.Groffen AJA, Friedrich R, Brian EC, Ashery U, Verhage M. DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties. Journal of neurochemistry. 2006;97(3):818–833. doi: 10.1111/j.1471-4159.2006.03755.x. [DOI] [PubMed] [Google Scholar]
- 9.Cho W, Stahelin R. Membrane binding and subcellular targeting of C2 domains. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2006;1761(8):838–849. doi: 10.1016/j.bbalip.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 10.Corbalan-Garcia S, Gómez-Fernández JC. Signaling through C2 domains: More than one lipid target. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2014;1838(6):1536–1547. doi: 10.1016/j.bbamem.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Martens S, McMahon HT. C2 domains and membrane fusion. Curr Top Membr. 2011;68:141–159. doi: 10.1016/B978-0-12-385891-7.00006-4. [DOI] [PubMed] [Google Scholar]
- 12.Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 1996;5(12):2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coussens L, Parker PJ, Rhee L, et al. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science. 1986;233(4766):859–866. doi: 10.1126/science.3755548. [DOI] [PubMed] [Google Scholar]
- 14.Knopf JL, Lee MH, Sultzman LA, et al. Cloning and expression of multiple protein kinase C cDNAs. Cell. 1986;46(4):491–502. doi: 10.1016/0092-8674(86)90874-3. [DOI] [PubMed] [Google Scholar]
- 15.Takamori S, Holt M, Stenius K, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127(4):831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 16.Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. Membrane phosphatidylserine regulates surface charge and protein localization. Science. 2008;319(5860):210–213. doi: 10.1126/science.1152066. [DOI] [PubMed] [Google Scholar]
- 17.Bajohrs M, Darios F, Peak-Chew SY, Davletov B. Promiscuous interaction of SNAP-25 with all plasma membrane syntaxins in a neuroendocrine cell. Biochem J. 2005;392(Pt 2):283–289. doi: 10.1042/BJ20050583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gamper N, Shapiro MS. Target-specific PIP2signalling: how might it work? The Journal of physiology. 2007;582(3):967–975. doi: 10.1113/jphysiol.2007.132787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8(6):343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- 20.Suh BC, Inoue T, Meyer T, Hille B. Rapid Chemically Induced Changes of PtdIns(4,5)P2 Gate KCNQ Ion Channels. Science (New York, NY) 2006;314(5804):1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cremona O, De Camilli P. Phosphoinositides in membrane traffic at the synapse. J Cell Sci. 2001;114(Pt 6):1041–1052. doi: 10.1242/jcs.114.6.1041. [DOI] [PubMed] [Google Scholar]
- 22.Kutateladze TG. Translation of the phosphoinositide code by PI effectors. Nat Chem Biol. 2010;6(7):507–513. doi: 10.1038/nchembio.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manna D, Bhardwaj N, Vora MS, Stahelin RV, Lu H, Cho W. Differential Roles of Phosphatidylserine, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 in Plasma Membrane Targeting of C2 Domains: MOLECULAR DYNAMICS SIMULATION, MEMBRANE BINDING, AND CELL TRANSLOCATION STUDIES OF THE PKC C2 Domain. Journal of Biological Chemistry. 2008;283(38):26047–26058. doi: 10.1074/jbc.M802617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marín-Vicente C, Nicolás FE, Gómez-Fernández JC, Corbalán-García S. The PtdIns(4,5)P2 Ligand Itself Influences the Localization of PKCα in the Plasma Membrane of Intact Living Cells. Journal of Molecular Biology. 2008;377(4):1038–1052. doi: 10.1016/j.jmb.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 25.Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol. 2004;11(1):36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- 26.Guillen J, Ferrer-Orta C, Buxaderas M, et al. Structural insights into the Ca2+ and PI(4,5)P2 binding modes of the C2 domains of rabphilin 3A and synaptotagmin 1. Proc Natl Acad Sci U S A. 2013;110(51):20503–20508. doi: 10.1073/pnas.1316179110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pèrez-Lara Á, Thapa A, Nyenhuis SB, et al. PtdInsP2 and PtdSer cooperate to trap synaptotagmin-1 to the plasma membrane in the presence of calcium. Elife. 2016;5 doi: 10.7554/eLife.15886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tucker WC, Edwardson JM, Bai J, Kim HJ, Martin TF, Chapman ER. Identification of synaptotagmin effectors via acute inhibition of secretion from cracked PC12 cells. J Cell Biol. 2003;162(2):199–209. doi: 10.1083/jcb.200302060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato M, Mori Y, Matsui T, et al. Role of the polybasic sequence in the Doc2α C2B domain in dense-core vesicle exocytosis in PC12 cells. Journal of neurochemistry. 2010 doi: 10.1111/j.1471-4159.2010.06739.x. no-no. [DOI] [PubMed] [Google Scholar]
- 30.Giordano F, Saheki Y, Idevall-Hagren O, et al. PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell. 2013;153(7):1494–1509. doi: 10.1016/j.cell.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, Araki S, Wu J, et al. An expanded palette of genetically encoded Ca2+ indicators. Science. 2011;333(6051):1888–1891. doi: 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunger AT, Jin R, Breidenbach MA. Highly specific interactions between botulinum neurotoxins and synaptic vesicle proteins. Cell Mol Life Sci. 2008;65(15):2296–2306. doi: 10.1007/s00018-008-8088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fasshauer D, Margittai M. A transient N-terminal interaction of SNAP-25 and syntaxin nucleates SNARE assembly. J Biol Chem. 2004;279(9):7613–7621. doi: 10.1074/jbc.M312064200. [DOI] [PubMed] [Google Scholar]
- 34.Schiavo G, Shone CC, Bennett MK, Scheller RH, Montecucco C. Botulinum neurotoxin type C cleaves a single Lys-Ala bond within the carboxyl-terminal region of syntaxins. J Biol Chem. 1995;270(18):10566–10570. doi: 10.1074/jbc.270.18.10566. [DOI] [PubMed] [Google Scholar]
- 35.Vaidyanathan VV, Yoshino K, Jahnz M, et al. Proteolysis of SNAP-25 isoforms by botulinum neurotoxin types A, C, and E: domains and amino acid residues controlling the formation of enzyme-substrate complexes and cleavage. J Neurochem. 1999;72(1):327–337. doi: 10.1046/j.1471-4159.1999.0720327.x. [DOI] [PubMed] [Google Scholar]
- 36.Xue R, Gaffaney JD, Chapman ER. Structural elements that underlie Doc2β function during asynchronous synaptic transmission. Proc Natl Acad Sci U S A. 2015;112(31):E4316–4325. doi: 10.1073/pnas.1502288112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammond GRV, Machner MP, Balla T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. The Journal of cell biology. 2014;205(1):113–126. doi: 10.1083/jcb.201312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNamara CR, Degterev A. Small-molecule inhibitors of the PI3K signaling network. Future Medicinal Chemistry. 2011;3(5):549–565. doi: 10.4155/fmc.11.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275(5300):665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 40.James SR, Downes CP, Gigg R, Grove SJ, Holmes AB, Alessi DR. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem J. 1996;315:709–713. doi: 10.1042/bj3150709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Várnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143(2):501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwon Y, Hofmann T, Montell C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol Cell. 2007;25(4):491–503. doi: 10.1016/j.molcel.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Punnonen EL, Ryhanen K, Marjomaki VS. At reduced temperature, endocytic membrane traffic is blocked in multivesicular carrier endosomes in rat cardiac myocytes. Eur J Cell Biol. 1998;75(4):344–352. doi: 10.1016/s0171-9335(98)80067-8. [DOI] [PubMed] [Google Scholar]
- 44.Rotblat B. Ras and Its Signals Diffuse through the Cell on Randomly Moving Nanoparticles. Cancer Research. 2006;66(4):1974–1981. doi: 10.1158/0008-5472.CAN-05-3791. [DOI] [PubMed] [Google Scholar]
- 45.Wada R, Sato D, Nakamura T, Hatori K. Temperature control of the motility of actin filaments interacting with myosin molecules using an electrically conductive glass in the presence of direct current. Arch Biochem Biophys. 2015;586:51–56. doi: 10.1016/j.abb.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 46.Gasparini L, Benussi L, Bianchetti A, et al. Energy metabolism inhibition impairs amyloid precursor protein secretion from Alzheimer's fibroblasts. Neurosci Lett. 1999;263(2-3):197–200. doi: 10.1016/s0304-3940(99)00155-x. [DOI] [PubMed] [Google Scholar]
- 47.He C, Stroink A, Vogel L, Wang CX. Temperature increase exacerbates apoptotic neuronal death in chemically-induced ischemia. PLoS One. 2013;8(7):e68796. doi: 10.1371/journal.pone.0068796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber SC, Spakowitz AJ, Theriot JA. Nonthermal ATP-dependent fluctuations contribute to the in vivo motion of chromosomal loci. Proceedings of the National Academy of Sciences. 2012;109(19):7338–7343. doi: 10.1073/pnas.1119505109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klein C, Waharte F. Analysis of molecular mobility by fluorescence recovery after photobleaching in living cells. In: Mendez-Vilas A, Diaz A, editors. Microscopy: Science, Technology, Applications and Education. Formatex Research Center; 2010. pp. 772–783. [Google Scholar]
- 50.Lippincott-Schwartz J, Snapp E, Kenworthy A. Studying protein dynamics in living cells. Nat Rev Mol Cell Biol. 2001;2(6):444–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- 51.Swaminathan R, Hoang CP, Verkman AS. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys J. 1997;72(4):1900–1907. doi: 10.1016/S0006-3495(97)78835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schiavo G, Gu QM, Prestwich GD, Sollner TH, Rothman JE. Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc Natl Acad Sci U S A. 1996;93(23):13327–13332. doi: 10.1073/pnas.93.23.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fukuda N, Emoto M, Nakamori Y, et al. DOC2B: A Novel Syntaxin-4 Binding Protein Mediating Insulin-Regulated GLUT4 Vesicle Fusion in Adipocytes. Diabetes. 2008;58(2):377–384. doi: 10.2337/db08-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orita S, Naito A, Sakaguchi G, et al. Physical and functional interactions of Doc2 and Munc13 in Ca2+-dependent exocytotic machinery. The Journal of biological chemistry. 1997;272(26):16081–16084. doi: 10.1074/jbc.272.26.16081. [DOI] [PubMed] [Google Scholar]
- 55.Friedrich R, Gottfried I, Ashery U. Munc13-1 Translocates to the Plasma Membrane in a Doc2B- and Calcium-Dependent Manner. Front Endocrinol (Lausanne) 2013;4:119. doi: 10.3389/fendo.2013.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olivotto M, Arcangeli A, Carla M, Wanke E. Electric fields at the plasma membrane level: a neglected element in the mechanisms of cell signalling. Bioessays. 1996;18(6):495–504. doi: 10.1002/bies.950180612. [DOI] [PubMed] [Google Scholar]
- 57.Shao X, Li C, Fernandez I, Zhang X, Sudhof TC, Rizo J. Synaptotagmin-syntaxin interaction: the C2 domain as a Ca2+-dependent electrostatic switch. Neuron. 1997;18(1):133–142. doi: 10.1016/s0896-6273(01)80052-0. [DOI] [PubMed] [Google Scholar]
- 58.Friedrich R, Yeheskel A, Ashery U. DOC2B, C2 Domains, and Calcium: A Tale of Intricate Interactions. Molecular Neurobiology. 2010;41(1):42–51. doi: 10.1007/s12035-009-8094-8. [DOI] [PubMed] [Google Scholar]
- 59.Charvin N, L'Eveque C, Walker D, et al. Direct interaction of the calcium sensor protein synaptotagmin I with a cytoplasmic domain of the alpha1A subunit of the P/Q-type calcium channel. EMBO J. 1997;16(15):4591–4596. doi: 10.1093/emboj/16.15.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sheng ZH, Yokoyama CT, Catterall WA. Interaction of the synprint site of N-type Ca2+ channels with the C2B domain of synaptotagmin I. Proc Natl Acad Sci U S A. 1997;94(10):5405–5410. doi: 10.1073/pnas.94.10.5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wiser O, Tobi D, Trus M, Atlas D. Synaptotagmin restores kinetic properties of a syntaxin-associated N-type voltage sensitive calcium channel. FEBS Lett. 1997;404(2-3):203–207. doi: 10.1016/s0014-5793(97)00130-0. [DOI] [PubMed] [Google Scholar]
- 62.Radhakrishnan A, Stein A, Jahn R, Fasshauer D. The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2009;284(38):25749–25760. doi: 10.1074/jbc.M109.042499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alwarawrah M, Wereszczynski J. Investigation of the Effect of Bilayer Composition on PKCalpha-C2 Domain Docking Using Molecular Dynamics Simulations. J Phys Chem B. 2017;121(1):78–88. doi: 10.1021/acs.jpcb.6b10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. Journal of computational chemistry. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Best RB, Zhu X, Shim J, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J Chem Theory Comput. 2012;8(9):3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lupyan D, Mezei M, Logothetis DE, Osman R. A molecular dynamics investigation of lipid bilayer perturbation by PIP2. Biophys J. 2010;98(2):240–247. doi: 10.1016/j.bpj.2009.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bykhovskaia M. Calcium binding promotes conformational flexibility of the neuronal Ca(2+) sensor synaptotagmin. Biophys J. 2015;108(10):2507–2520. doi: 10.1016/j.bpj.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–3450. [PubMed] [Google Scholar]
- 69.Lang T, Wacker I, Steyer J, et al. Ca2+-triggered peptide secretion in single cells imaged with green fluorescent protein and evanescent-wave microscopy. Neuron. 1997;18(6):857–863. doi: 10.1016/s0896-6273(00)80325-6. [DOI] [PubMed] [Google Scholar]
- 70.Kofer-Geles M, Gottfried I, Haklai R, Elad-Zefadia G, Kloog Y, Ashery U. Rasosomes spread Ras signals from plasma membrane ‘hotspots'. Biochim Biophys Acta. 2009;1793(11):1691–1702. doi: 10.1016/j.bbamcr.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 71.Abrámoff MD, Magalhães PJ, Ram SJ. Image processing with ImageJ. Biophotonics International. 2004;11:36–41. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 Initial examination of potential DOC2B targeting. Expression patterns of DOC2B and its interacting partners in PC12 cells. Left: DOC2B-GFP before and after Ca2+ elevation induced by KCl depolarization. Right: phosphatidylserine and PI(4,5)P2 distribution, detected by their biosensors, Lact-C2-GFP and PLCδ-PH-GFP, respectively. Expression patterns of the t-SNARE proteins, mRFP-syx1A and SNAP25-YFP were examined by overexpressing them in the cells. Scale bar = 10 μm.
Supplementary Figure S2 Isolated DOC2B C2B follows PLCd-PH to intracellular membranes. (A) Rapamycin (Rapa) induced CF-PIPK translocation to intracellular membranes as a result of the FKBP-FRB dimerization, inducing PI(4,5)P2 synthesis as reported by its PLCδ-PH-mKate biosensor. C2BWT-GFP translocated to PLCδ-PH-mKate-stained organelles following ionomycin application. (B) C2BK319E-GFP, impaired in PI(4,5)P2 binding, did not translocate to the PM or to the intracellular organelles following ionomycin application. Scale bar = 10 μm.