

Abstract
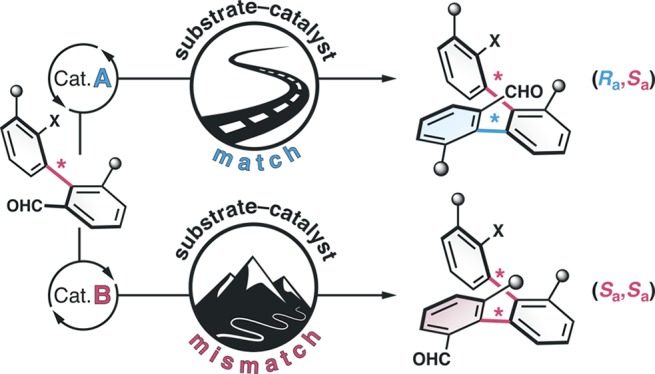
Molecular scaffolds with multiple rotationally restricted bonds allow a precise spatial positioning of functional groups. However, their synthesis requires methods addressing the configuration of each stereogenic axis. We report here a catalyst-stereocontrolled synthesis of atropisomeric multiaxis systems enabling divergence from the prevailing stereochemical reaction path. By using ion-pairing catalysts in arene-forming aldol condensations, a strong substrate-induced stereopreference can be overcome to provide structurally well-defined helical oligo-1,2-naphthylenes. The configuration of up to four stereogenic axes was individually catalyst-controlled, affording quinquenaphthalenes with a unique topology.
Short abstract
The synthesis of structurally well-defined atropisomeric multiaxis systems is enabled by efficient catalytic processes that allow divergence from prevailing stereochemical reaction paths.
A defined relative orientation of groups in space is essential for the rational design of functional molecular systems. As structurally well-defined scaffolds, multiaxis systems offer a particularly broad range of topologies as compared to small, bridged, or linearly arranged compounds. Atropisomers with multiple configurationally stable axes would hence permit geometrical control over an extended and more characteristic molecular arrangement. However, for the configuration of each stereogenic axis to be individually addressed, a stereochemically versatile synthetic strategy is required,1−3 where the choice of specific catalysts allow divergence of the diastereoselective reaction paths.
Seminal studies on diastereodivergent catalyst control over the stereocenter configuration underline the intricacies of the substrate–catalyst mismatch scenario, where selectivity for the unfavored product requires a particularly pronounced lowering of the activation energy (Scheme 1 top, cat. 2B, TSBsubs vs TSBcat).4−12 By implementing this consideration for the preparation of atropodiastereoisomers, stereodivergent synthesis of multiaxis systems under catalyst control would impose selectivity for an array of stereogenic axes, which induce a topologically unique structure (Scheme 1 bottom). We therefore envisaged that a catalyst-accelerated diastereoselective arene-forming aldol condensation13,14 would provide discretely configured oligo-1,2-naphthylenes with multiple configurationally stable stereogenic axes. By catalyst variation, either the selectivity of a substrate-stereocontrolled reaction15−26 could be increased, or more notably, the stereochemical course of the reaction could be inverted to provide otherwise inaccessible atropisomeric multiaxis systems with a characteristic molecular shape (substrate–catalyst mismatch case).
Scheme 1. Catalyst-Controlled Diastereodivergent Synthesis.
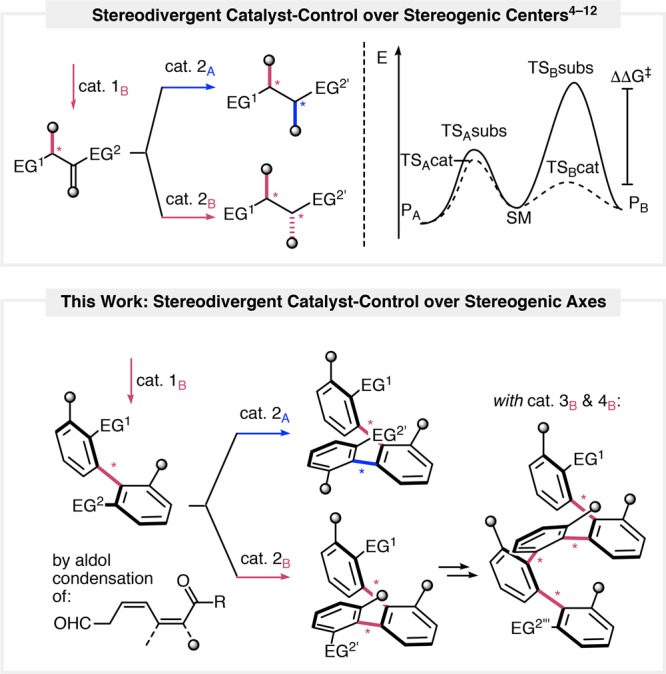
Selectivity for the unfavored product (PB) of a substrate-stereocontrolled reaction (TSAsubs vs TSBsubs) requires a more pronounced lowering of the activation energy (ΔΔG⧧) under catalyst stereocontrol (TSAcat vs TSBcat). EG: end group, SM: starting material.
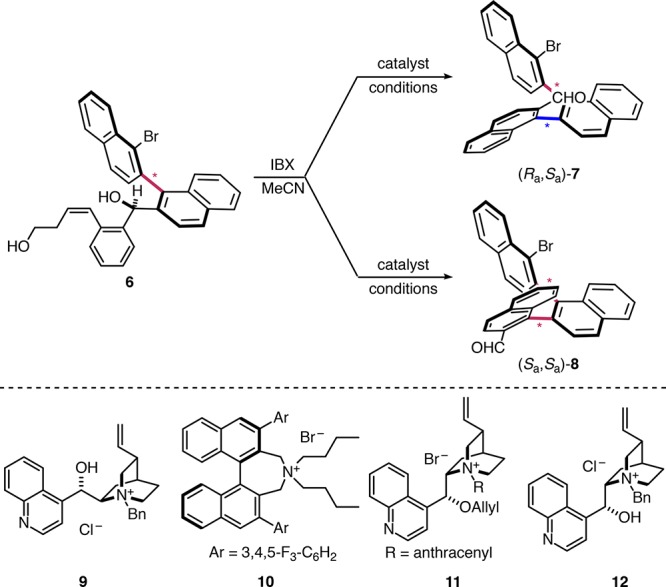
To evaluate the feasibility of the catalyst-controlled stereodivergent synthesis of multiaxis atropisomers, we prepared a substrate with a previously defined stereogenic axis using our established procedure.27 Addition of diaryl magnesium alkoxide reagent 1 to 1-bromonaphthalene-2-carbaldehyde (2) was followed by in situ double oxidation of diol (±)-3 and an enantioselective arene-forming aldol condensation with (S)-pyrrolidinyl tetrazole catalyst 4 (99:1 er, Scheme 2). Building block 1 was subsequently added to the configurationally stable binaphthalene (Sa)-528,29 to give prerequisite precursor diol 6.
Scheme 2. Substrate Synthesis.

Upon double-oxidation of 6 using IBX, the keto-aldehyde substrate was first converted to the ternaphthalene with two stereogenic axes under substrate stereocontrol (Table 1). Treatment with aqueous KOH thus revealed a 4:1 preference for (Ra,Sa)-7 over (Sa,Sa)-8 (entry 1).30 We next investigated the stereodivergent synthesis of the atropisomeric two-axis system by evaluating the level of catalyst control of selected amine and ion-pairing catalysts under multiple reaction conditions.31
Table 1. In Situ Double Oxidationa and Optimization of the Catalyst-Controlled Stereodivergent Synthesis of an Atropisomeric Two-Axis Systemb.
| entry | catalyst (mol %) | solvent/additive | drc (7:8) | yieldd |
|---|---|---|---|---|
| 1 | CHCl3/aq KOH | 4:1 | 65% | |
| 2 | 4 (40) | CHCl3 | 1:12 | 13% |
| 3 | 4 (40) | DMF/H2O | 1:32 | 55% |
| 4e | 4 (40) | CHCl3/DMF/H2O | 1:24 | 54% |
| 5e,f | 4 (40) | CHCl3/DMF/aq bufferg | 1:32 | 75% |
| 6e | ent-4 (40) | CHCl3/DMF/aq bufferg | 24:1 | 64% |
| 7 | 9 (10) | CHCl3/aq KOH | 8:1 | 54% |
| 8 | 10 (10) | CHCl3/aq KOH | 3.5:1 | 44% |
| 9 | 11 (10) | CHCl3/aq KOH | 3.5:1 | 18% |
| 10 | 12 (10) | CHCl3/aq KOH | 16:1 | 72% |
| 11 | 12 (5) | CHCl3/aq KOH | 16:1 | 70% |
| 12 | 12 (1) | CHCl3/aq KOH | 16:1 | 69% |
In situ double oxidation with 100 μmol substrate precursor 6 in MeCN and 300 μmol IBX at 60 °C for 4 h.
Stereodivergent aldol condensation with the specified catalyst and additive at RT for 24 h.
Ratio of (Ra,Sa)-7:(Sa,Sa)-8 as determined by 1H NMR of the crude reaction mixture.
Yield of isolated product over two steps.
Reaction time of 48 h.
Using 4.00 mmol substrate.
Sodium citrate buffer, pH 5.
Intriguingly, (S)-pyrrolidinyl tetrazole catalyst 4 led to an inverted selectivity of 1:12 in favor of (Sa,Sa)-8, however, in low yield (entry 2, substrate-catalyst mismatched case). Upon optimization of the reaction conditions, chloroform, DMF, and aqueous citrate buffer as medium were found to provide (Sa,Sa)-8 with increased selectivity and a significantly improved yield (75% over two steps, 32:1 dr, entries 3–5). We next corroborated that catalysts govern the stereochemical course of the reaction by using (R)-pyrrolidinyl tetrazole (ent-4) under identical conditions, affording the opposite diastereomer (Ra,Sa)-7 with an atropodiastereoselectivity of 24:1 (entry 6, substrate–catalyst matched case). Moreover, the catalyst loading could be dramatically reduced by the use of ion-pairing catalysts (aq KOH, entries 7–10). Whereas N-benzylcinchoninium chloride (9),32−34 Maruoka catalyst 10,35−37 and Corey catalyst 11(38) showed variable activity and selectivity, the cinchonidine-derived Lygo catalyst 12(39,40) induced a high dr combined with a particularly high catalytic activity even if reduced to 1.0 mol % (entries 10–12).31
Having confirmed that the atropodivergent synthesis of a two-axis system enables the preparation of both atropodiastereoisomers under catalyst control, we became intrigued by the prospect of governing the configuration of an atropisomeric stereotriad. Building block addition to (Sa,Sa)-8 (75%) and subsequent in situ oxidation of the corresponding diol expeditiously provided the corresponding keto-aldehyde, the substrate for the stereodivergent arene-forming aldol condensation in question. Interestingly, the level of substrate stereocontrol increased to 7:1 in favor of the homologous diastereomer (Ra,Sa,Sa)-13 (Scheme 3). For this more pronounced substrate bias for the terminally (Ra)-configured atropisomer to be overcome, several catalysts and conditions were thoroughly examined.31 Whereas primary and secondary amines proved inefficient, catalyst 12 was capable of forming helical (Sa,Sa,Sa)-14 with a selectivity of 6:1, which was further improved to 8:1 at 0 °C (1.0 mol % 12, aq KOH). The inversion of this considerable substrate preference with 1.0 mol % of 12 under otherwise identical conditions highlights the high efficiency of ion-pairing catalysts and indicates their particular utility for the substrate–catalyst mismatch scenario in the stereodivergent synthesis of multiaxis systems.
Scheme 3. Stereodivergent Synthesis of an Atropisomeric Three-Axis System.

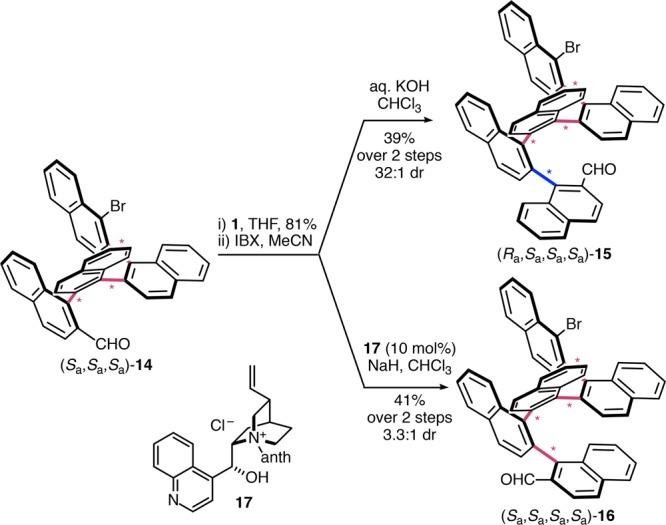
Encouraged by these results, we set out to investigate systems with four stereogenic axes individually addressed by catalyst stereocontrol. The required keto-aldehyde substrate poised for the arene-forming aldol condensation was accessible after a third building block addition to (Sa,Sa,Sa)-14 (Scheme 4, 81%) and a subsequent in situ double oxidation. Under substrate stereocontrol, the additional repeating unit had a dramatic effect on the preference for quinquenaphthalene (Ra,Sa,Sa,Sa)-15, which was formed with a selectivity of 32:1. This increasingly prevalent bias however hampers the synthesis of (Sa,Sa,Sa,Sa)-16, as it requires diverging from a remarkably dominant reaction path. With N-benzylcinchonidinium chloride catalyst 12, this preference was diminished but could not be overcome, and (Ra,Sa,Sa,Sa)-15 remained the major product (1:3 dr, substrate–catalyst mismatch case). After meticulous experimentation using various prototypical catalysts and conditions,31 anthracene-bearing catalyst 17 with an increased steric demand was identified to provide a 1:1 diastereoisomeric mixture. This suggests that activation energy parity was reached by compensation of the substrate bias for stereoisomer (Ra,Sa,Sa,Sa)-15. Gratifyingly, subsequent optimization revealed that a combination with sodium hydride allows the inversion of selectivity to give the desired (Sa,Sa,Sa,Sa)-16 as a major product with a dr of 3.3:1.31 An efficient catalytic system hence prevails even over a marked substrate bias (32:1 dr), enabling divergence of the atropodiastereoselectivity for the synthesis of a previously elusive stereotetrad.
Scheme 4. Stereodivergent Synthesis of an Atropisomeric Four-Axis System.
Having both quinquenaphthalene diastereoisomers in hand, we next studied their characteristic structures by NMR spectroscopy. Large upfield shifts of the aldehyde protons (>1.0 ppm, 8.86 ppm) of the terminally (Ra)-configured atropisomer 15 indicate a strong influence of the ring current from the second naphthalene unit to the aldehyde in proximity. Furthermore, substantial shifts of the aromatic protons (5.6 ppm) and clear NOE correlations between every third naphthalene unit of (Sa,Sa,Sa,Sa)-configured 16 were consistent with a compact helical secondary structure of the oligomers. These findings were further supported by the crystal structure of (Ra,Sa,Sa,Sa)-15 (Figure 1)41 with distances between the naphthalene moieties as small as 3.4 Å. The P-helical structure, particularly along the uniformly configured (Sa,Sa,Sa)-motif, is evident by the one and a half helix turns (a third turn per unit) and an average dihedral angle of 37° from alternating minimally distorted arene (5 × 4° in average) and their interstitial binaphthyl bonds (4 × 79° in average). These coil the configurationally stable ortho-arylene into a compact and distinctive geometry. We assume that only two conformers, resulting from the rotation about the arene-carbaldehyde bond, are accessible, whereas the oligo-1,2-naphthylene scaffold provides a configurationally entirely stable structure.
Figure 1.
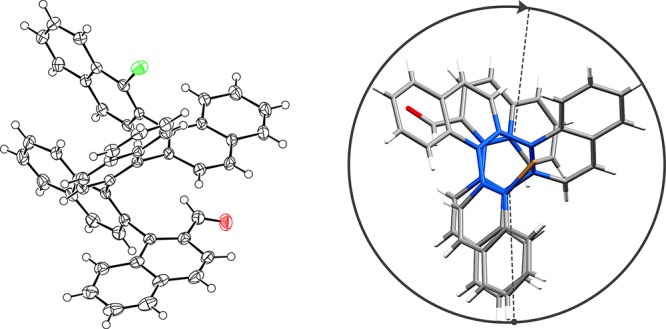
X-ray crystal structure of (Ra,Sa,Sa,Sa)-15. Thermal ellipsoids are drawn at the 50% probability level (left). View along the helix axis (right) with 1.53 turns (0.30 per unit) and an average torsional angle of 37° (average of 79° for four biaryls; average of 4° for five arenes).15
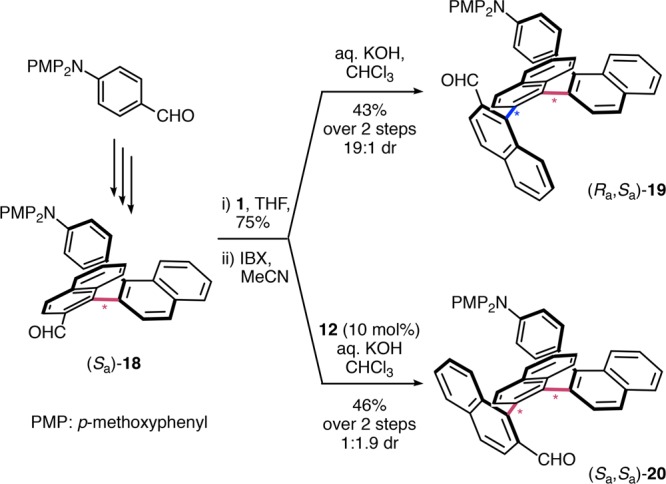
To evaluate the higher substrate bias with increasing oligomer length, we examined differences in their structures in solution. Interestingly, a comparison of the ring current effects of (Sa,Sa,Sa,Sa)-16 with the shorter quaternaphthalene diastereoisomer (Sa,Sa,Sa)-14 indicates that, with length, the helical arrangement is increasingly compact (ΔδH(34–144′′′) = 1.56 ppm vs ΔδH(34–164′′′′) = 1.91 ppm), supporting the notion of Hartley that, in solution, the termini of ortho-arylenes are partially disordered.42,43 Moreover, the lower substrate control observed in aromatic solvents is in agreement with partial disintegration of otherwise densely packed helices (2.7:1 dr (toluene) vs 7:1 dr (CHCl3) for 13:14).31 To further evaluate the interplay of repulsive and attractive effects that influences the level of substrate stereocontrol, we investigated an arene substituent that diminishes aromatic interactions ((Sa)-18, triaryl amine end group, Scheme 5). Remarkably, with an electron-rich end group, the substrate bias is dramatically increased (19:1 dr for 19 vs 7:1 dr for 13), whereas solvent effects remain minimal.31 Although the addition of catalyst 12 has a comparable impact on the diastereoselectivity towards (Sa,Sa)-20 (1:1.9 dr), attractive aromatic interactions likely facilitate the formation of the helical oligomers 14 and 16. In combination, these results are in accordance with prevailing steric effects of the nucleophilic side chain with compact helices (for Ra) and opposing attractive interactions of the aromatic moieties (for Sa, helical).
Scheme 5. Effect of Substrate Substitution.
In conclusion, the configuration of multiaxis systems was individually addressed by catalyst-controlled atropodiadivergent synthesis. Up to four stereogenic axes were governed by amine and ion-pairing catalysts in arene-forming aldol condensations. Even in cases of a marked substrate–catalyst mismatch, otherwise inaccessible multiaxis atropisomers were efficiently prepared. To the best of our knowledge, this is the first example of a catalyst-controlled, stereodivergent synthesis of compounds with multiple stereogenic axes. The configuration of the rotationally restricted oligomers imposes a characteristic helical shape with a defined spatial arrangement. The stereodynamic behavior of the extended quinquenaphthalenes is reduced to a rotation about the terminal arene-carbaldehyde bond. The remarkable differences in substrate bias were attributed to opposing steric and aromatic interactions. Our current studies focus on the spatial positioning of pertinent groups by using structurally well-defined diastereoisomeric scaffolds.
Acknowledgments
We gratefully acknowledge the Swiss National Science Foundation (BSSGI0-155902/1), the University of Basel, and the NCCR Molecular Systems Engineering for financial support, PD Dr. D. Häussinger and T. Müntener for NMR assistance, Prof. M. Waser for generously providing their ion-pairing catalysts, Prof. O. Wenger and Prof. M. Mayor for helpful discussions, and P. Welsch for skillful technical support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00204.
Experimental details and analytical data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bringmann G.; Price Mortimer A. J.; Keller P. A.; Gresser M. J.; Garner J.; Breuning M. Atroposelective synthesis of axially chiral biaryl compounds. Angew. Chem., Int. Ed. 2005, 44, 5384–5427. 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- Wencel-Delord J.; Panossian A.; Leroux F. R.; Colobert F. Recent advances and new concepts for the synthesis of axially stereoenriched biaryls. Chem. Soc. Rev. 2015, 44, 3418–3430. 10.1039/C5CS00012B. [DOI] [PubMed] [Google Scholar]
- Kumarasamy E.; Raghunathan R.; Sibi M. P.; Sivaguru J. Nonbiaryl and heterobiaryl atropisomers: Molecular templates with promise for atropselective chemical transformations. Chem. Rev. 2015, 115, 11239–11300. 10.1021/acs.chemrev.5b00136. [DOI] [PubMed] [Google Scholar]
- Ko S. Y.; Lee A. W. M.; Masamune S.; Reed L. A.; Sharpless K. B.; Walker F. J. Total synthesis of the l-hexoses. Science 1983, 220, 949–951. 10.1126/science.220.4600.949. [DOI] [PubMed] [Google Scholar]
- Wang A.; Wüstenberg B.; Pfaltz A. Enantio- and diastereoselective hydrogenation of farnesol and O-protected derivatives: Stereocontrol by changing the C = C bond configuration. Angew. Chem., Int. Ed. 2008, 47, 2298–2300. 10.1002/anie.200705521. [DOI] [PubMed] [Google Scholar]
- Kumar R. R.; Kagan H. B. Regioselective reactions on a chiral substrate controlled by the configuration of a chiral catalyst. Adv. Synth. Catal. 2010, 352, 231–242. 10.1002/adsc.200900822. [DOI] [Google Scholar]
- Miller L. C.; Sarpong R. Divergent reactions on racemic mixtures. Chem. Soc. Rev. 2011, 40, 4550–4562. 10.1039/c1cs15069c. [DOI] [PubMed] [Google Scholar]
- Krautwald S.; Sarlah D.; Schafroth M. A.; Carreira E. M. Enantio- and diastereodivergent dual catalysis: α-allylation of branched aldehydes. Science 2013, 340, 1065–1068. 10.1126/science.1237068. [DOI] [PubMed] [Google Scholar]
- Simmons B.; Walji A. M.; MacMillan D. W. C. Cycle-specific organocascade catalysis: Application to olefin hydroamination, hydro-oxidation, and amino-oxidation, and to natural product synthesis. Angew. Chem., Int. Ed. 2009, 48, 4349–4353. 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S.-L.; Wong Z. L.; Buchwald S. L. Copper-catalysed enantioselective stereodivergent synthesis of amino alcohols. Nature 2016, 532, 353–356. 10.1038/nature17191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L.; Feng X. Catalytic strategies for diastereodivergent synthesis. Chem. - Eur. J. 2017, 23, 6464–6482. 10.1002/chem.201604617. [DOI] [PubMed] [Google Scholar]
- Bihani M.; Zhao J. C.-G. Advances in asymmetric diastereodivergent catalysis. Adv. Synth. Catal. 2017, 359, 534–575. 10.1002/adsc.201601188. [DOI] [Google Scholar]
- Link A.; Sparr C. Organocatalytic atroposelective aldol condensation: Synthesis of axially chiral biaryls by arene formation. Angew. Chem., Int. Ed. 2014, 53, 5458–5461. 10.1002/anie.201402441. [DOI] [PubMed] [Google Scholar]
- Witzig R. M.; Lotter D.; Fäseke V. C.; Sparr C. Stereoselective arene-forming aldol condensation: Catalyst-controlled synthesis of axially chiral compounds. Chem. - Eur. J. 2017, 23, 12960–12966. 10.1002/chem.201702471. [DOI] [PubMed] [Google Scholar]
- Miyano S.; Tobita M.; Hashimoto H. Asymmetric synthesis of axially dissymmetric 1,1′-binaphthyls via an intramolecular Ullmann coupling reaction of (R)- and (S)-2,2′-bis(1-bromo-2-naphthylcarbonyloxy)-1,1′-binaphthyl. Bull. Chem. Soc. Jpn. 1981, 54, 3522–3526. 10.1246/bcsj.54.3522. [DOI] [Google Scholar]
- Shimada T.; Kina A.; Hayashi T. A New synthetic route to enantiomerically pure axially chiral 2,2‘-bipyridine N,N‘-dioxides. Highly efficient catalysts for asymmetric allylation of aldehydes with allyl(trichloro)silanes. J. Org. Chem. 2003, 68, 6329–6337. 10.1021/jo0300800. [DOI] [PubMed] [Google Scholar]
- Sue D.; Takaishi K.; Harada T.; Kuroda R.; Kawabata T.; Tsubaki K. Synthesis of chiral dotriacontanaphthalenes: How many naphthalene units are we able to elaborately connect?. J. Org. Chem. 2009, 74, 3940–3943. 10.1021/jo900463t. [DOI] [PubMed] [Google Scholar]
- Takaishi K.; Kawamoto M.; Tsubaki K. Multibridged chiral naphthalene oligomers with continuous extreme-cisoid conformation. Org. Lett. 2010, 12, 1832–1835. 10.1021/ol100582v. [DOI] [PubMed] [Google Scholar]
- Richieu A.; Peixoto P. A.; Pouységu L.; Deffieux D.; Quideau S. Bioinspired total synthesis of (−)-vescalin: A nonahydroxytriphenoylated C-glucosidic ellagitannin. Angew. Chem., Int. Ed. 2017, 56, 13833–13837. 10.1002/anie.201707613. [DOI] [PubMed] [Google Scholar]
- Dherbassy Q.; Djukic J. P.; Wencel-Delord J.; Colobert F. Two stereoinduction events in one C–H activation step: A route towards terphenyl ligands with two atropisomeric axes. Angew. Chem., Int. Ed. 2018, 57, 4668–4672. 10.1002/anie.201801130. [DOI] [PubMed] [Google Scholar]
- Hayashi T.; Hayashizaki K.; Ito Y. Asymmetric synthesis of axially chiral 1,1′:5′,1″- and 1,1′:4′,1″-ternaphthalenes by asymmetric cross-coupling with a chiral ferrocenylphosphine-nickel catalyst. Tetrahedron Lett. 1989, 30, 215–218. 10.1016/S0040-4039(00)95163-3. [DOI] [Google Scholar]
- Shibata T.; Fujimoto T.; Yokota K.; Takagi K. Iridium complex-catalyzed highly enantio- and diastereoselective [2 + 2+2] cycloaddition for the synthesis of axially chiral teraryl compounds. J. Am. Chem. Soc. 2004, 126, 8382–8383. 10.1021/ja048131d. [DOI] [PubMed] [Google Scholar]
- Oppenheimer J.; Hsung R. P.; Figueroa R.; Johnson W. L. Stereochemical control of both C–C and C–N axial chirality in the synthesis of chiral N,O-biaryls. Org. Lett. 2007, 9, 3969–3972. 10.1021/ol701692m. [DOI] [PubMed] [Google Scholar]
- Ogaki S.; Shibata Y.; Noguchi K.; Tanaka K. Enantioselective synthesis of axially chiral hydroxy carboxylic acid derivatives by rhodium-catalyzed [2 + 2 + 2] cycloaddition. J. Org. Chem. 2011, 76, 1926–1929. 10.1021/jo1024255. [DOI] [PubMed] [Google Scholar]
- Barrett K. T.; Metrano A. J.; Rablen P. R.; Miller S. J. Spontaneous transfer of chirality in an atropisomerically enriched two-axis system. Nature 2014, 509, 71–75. 10.1038/nature13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou K. C.; Li H.; Boddy C. N. C.; Ramanjulu J. M.; Yue T.-Y.; Natarajan S.; Chu X.-J.; Bräse S.; Rübsam F. Total synthesis of cancomycin–Part 1: Design and development of methodology. Chem. - Eur. J. 1999, 5, 2584–2601. . [DOI] [Google Scholar]
- Lotter D.; Neuburger M.; Rickhaus M.; Häussinger D.; Sparr C. Stereoselective arene-forming aldol condensation: Synthesis of configurationally stable oligo-1,2-naphthylenes. Angew. Chem., Int. Ed. 2016, 55, 2920–2923. 10.1002/anie.201510259. [DOI] [PubMed] [Google Scholar]
- Compound (Sa)-5 was found to decompose in a mixture of DMF and H2O. The addition of CHCl3 prevented the aldol dehydration step and hence also the decomposition. Dehydration was subsequently performed with Amberlite IRA-96 (polyamine resin).
- Heating an isolated, analytical sample of (Sa)-5 to 180 °C did not lead to detectable racemization (neither neat nor in DMF).
- Product decomposition by the formation of dichlorocarbene was not observed under these conditions.
- See the Supporting Information for details.
- Dolling U.-H.; Davis P.; Grabowski E. J. J. Efficient catalytic asymmetric alkylations. 1. Enantioselective synthesis of (+)-indacrinone via chiral phase-transfer catalysis. J. Am. Chem. Soc. 1984, 106, 446–447. 10.1021/ja00314a045. [DOI] [Google Scholar]
- O’Donnell M. J.; Bennett W. D.; Wu S. The stereoselective synthesis of α-amino acids by phase-transfer catalysis. J. Am. Chem. Soc. 1989, 111, 2353–2355. 10.1021/ja00188a089. [DOI] [Google Scholar]
- Waser M.; Novacek J.; Gratzer K.. Cooperative Catalysis Involving Chiral Ion Pair Catalysts. Cooperative Catalysis; Peters R., Eds.; Wiley-VCH: Weinheim, Germany, 2015; pp 197–225. [Google Scholar]
- Ooi T.; Kameda M.; Maruoka K. Molecular design of a C2-symmetric chiral phase-transfer catalyst for practical asymmetric synthesis of α-amino acids. J. Am. Chem. Soc. 1999, 121, 6519–6520. 10.1021/ja991062w. [DOI] [PubMed] [Google Scholar]
- Ooi T.; Arimura Y.; Hiraiwa Y.; Yuan L. M.; Kano T.; Inoue T.; Matsumoto J.; Maruoka K. Highly enantioselective monoalkylation of p-chlorobenzaldehyde imine of glycine tert-butyl ester under mild phase-transfer conditions. Tetrahedron: Asymmetry 2006, 17, 603–606. 10.1016/j.tetasy.2006.01.019. [DOI] [Google Scholar]
- Ooi T.; Maruoka K. Recent Advances in asymmetric phase-transfer catalysis. Angew. Chem., Int. Ed. 2007, 46, 4222–4266. 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]
- Corey E. J.; Xu F.; Noe M. C. A rational approach to catalytic enantioselective enolate alkylation using a structurally rigidified and defined chiral quaternary ammonium salt under phase transfer conditions. J. Am. Chem. Soc. 1997, 119, 12414–12415. 10.1021/ja973174y. [DOI] [Google Scholar]
- Lygo B.; Wainwright P. G. A new class of asymmetric phase-transfer catalysts derived from cinchona alkaloids — Application in the enantioselective synthesis of α-amino acids. Tetrahedron Lett. 1997, 38, 8595–8598. 10.1016/S0040-4039(97)10293-3. [DOI] [Google Scholar]
- Formation of product 7 or 8 was not observed without potassium hydroxide (with 10 mol % catalyst 12).
- CCDC 1584599 (for compound (Ra,Sa)-7) and CCDC 1584600 (for compound (Ra,Sa,Sa,Sa)-15) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- Hartley C. S.; He J. Conformational analysis of o-phenylenes: Helical oligomers with frayed ends. J. Org. Chem. 2010, 75, 8627–8636. 10.1021/jo1021025. [DOI] [PubMed] [Google Scholar]
- Wagner J. P.; Schreiner P. R. London dispersion in molecular chemistry— Reconsidering steric effects. Angew. Chem., Int. Ed. 2015, 54, 12274–12296. 10.1002/anie.201503476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.