

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
With 5-year median follow-up, continuous single-agent ibrutinib therapy was well tolerated with deepening of response.
Previously untreated patients, even those with TP53 aberration, achieved durable responses.
Abstract
The safety and efficacy of ibrutinib (420 mg) in chronic lymphocytic leukemia (CLL) were evaluated in a phase 2 study; 51 patients had TP53 aberration (TP53 cohort) and 35 were enrolled because of age 65 years or older (elderly cohort). Both cohorts included patients with treatment-naive (TN) and relapsed/refractory (RR) CLL. With the median follow-up of 4.8 years, 49 (57.0%) of 86 patients remain on study. Treatment was discontinued for progressive disease in 20 (23.3%) patients and for adverse events in 5 (5.8%). Atrial fibrillation occurred in 18 (20.9%) patients for a rate of 6.4 per 100 patient-years. No serious bleeding occurred. The overall response rate at 6 months, the primary study endpoint, was 95.8% for the TP53 cohort (95% confidence interval, 85.7%-99.5%) and 93.9% for the elderly cohort (95% confidence interval, 79.8%-99.3%). Depth of response improved with time: at best response, 14 (29.2%) of 48 patients in the TP53 cohort and 9 (27.3%) of 33 in the elderly cohort achieved a complete response. Median minimal residual disease (MRD) in peripheral blood was 3.8 × 10−2 at 4 years, with MRD-negative (<10−4) remissions in 5 (10.2%) patients. In the TP53 cohort, the estimated 5-year progression-free survival (PFS) was 74.4% in TN-CLL compared with 19.4% in RR-CLL (P = .0002), and overall survival (OS) was 85.3% vs 53.7%, respectively (P = .023). In the elderly cohort, the estimated 5-year PFS and OS in RR-CLL were 64.8% and 71.6%, respectively, and no event occurred in TN-CLL. Long-term administration of ibrutinib was well tolerated and provided durable disease control for most patients. This trial was registered at www.clinicaltrials.gov as #NCT01500733.
Visual Abstract
Introduction
The combination of chemotherapy with an anti-CD20 monoclonal antibody, referred to as chemoimmunotherapy, has been the mainstay of therapy for chronic lymphocytic leukemia (CLL). Although a subset of patients achieves durable remissions, most relapse within a few years.1,2 Clonal evolution leads to the expansion of genetically altered cells that dominate at the time of relapse, limiting efficacy of repeat chemotherapy.3 In particular, patients with TP53 aberration, either because of deletion of chromosome 17p (del[17p]) or TP53 mutation, relapse early after frontline chemoimmunotherapy, with a median progression-free survival (PFS) of less than 2 years.4,5 Toxicity of chemoimmunotherapy can be limiting, especially for the majority of patients with CLL who are older than 65 years. Although reduced-intensity regimens are less toxic, durability of response is inferior.6-8
Targeted therapy with kinase inhibitors is emerging as an alternative to conventional therapy for CLL.9-13 Activation of B-cell receptor signaling through self-association or interactions with autoantigens is a key driver of CLL pathogenesis.14-17 Ibrutinib covalently binds Bruton’s tyrosine kinase (BTK), leading to sustained inhibition of B-cell receptor and NF-κB signaling and tumor proliferation.18,19 In randomized trials, ibrutinib induced higher overall response rates (ORRs) and extended survival compared with the comparator treatment of both relapsed and/or refractory CLL (RR-CLL) and treatment-naive CLL (TN-CLL).9,10 In contrast to the experience with chemoimmunotherapy, response to ibrutinib appears to be independent of TP53 aberration.11,20,21 However, in RR-CLL with del(17p), 1 study reported 2-year PFS of 63%.11 In contrast, we previously reported 2-year PFS of 82% in 51 patients with TP53 aberration (del[17p] and/or TP53 mutation), the majority of whom received ibrutinib as first-line therapy.21 At the time of progression, mutations in PLCG2 and BTK that reduce the inhibitory effects of ibrutinib are often present.22-27 Computational models that incorporate estimates of mutation frequency and growth kinetics suggest that subclones carrying these mutations likely exist before initiation of ibrutinib therapy.28
Ibrutinib has generally been found to have a favorable safety profile. However, 1 center reported an estimated cumulative incidence of nonrelapse discontinuation of ibrutinib in 15.6% at 18 months.29 Further, the rate of atrial fibrillation is higher in ibrutinib-treated patients compared with control groups in randomized trials.30 Finally, the effect of chronic inhibition of BTK on infection risk remains to be better defined. Although the frequency of infections, in particular respiratory infections, has been reported to decrease on continuous therapy,20 and increases in serum IgA on ibrutinib are associated with a lower risk for infections,31 others report an apparent increase in some opportunistic infections during ibrutinib therapy, including Pneumocystis jirovecii pneumonia and invasive fungal infections.32,33
Additional information on the safety and efficacy of long-term therapy with ibrutinib will help estimate risks and expected benefit for different patient groups. Here we report the results with single-agent ibrutinib in patients with TP53 aberration or age 65 years or older enrolled in an investigator-initiated phase 2 study. Our data suggest that for a large proportion of patients with CLL, ibrutinib monotherapy provides durable disease control.
Patients, materials, and methods
Study design and participants
This phase 2, open-label, single-center study was approved by the Institutional Review Board (clinicaltrials.gov: NCT01500733). All patients provided written informed consent. Eligibility criteria included active CLL or small lymphocytic lymphoma requiring therapy,34 and del(17p) by fluorescence in situ hybridization in 10% or more of nuclei or TP53 mutation for the TP53 cohort, or age 65 years or older for the elderly cohort, Eastern Cooperative Oncology Group performance status of 2 or less, neutrophil count of 0.5 × 109/L or higher, and platelet count of at least 30 × 109/L. Exclusion criteria included any histologic transformation of CLL (Richter’s syndrome or prolymphocytic leukemia), autoimmune cytopenia requiring steroids, impaired organ function (total bilirubin ≥1.5 or aspartate aminotransferase/alanine aminotransferase ≥2.5 × upper limit of normal; creatinine ≥2.0 mg/dL or creatinine clearance of 50 mL/min or less), active hepatitis B infection, HIV infection, concomitant prednisone more than 20 mg/day, and/or anticoagulation with warfarin. A 4-week interval from prior therapy was required. Pretreatment evaluation included history, physical examination, laboratory evaluations, bone marrow aspiration and biopsy, peripheral blood and bone marrow flow cytometry, and computed tomography (CT). Fluorescence in situ hybridization and sequencing of the immunoglobulin heavy chain variable gene (IGHV) were performed as previously described.21
Procedures
Ibrutinib was administered orally at the dose of 420 mg once daily until disease progression or the development of unacceptable toxicity. Clinical safety monitoring was performed every other week for the first month, then every 4 weeks until 24 weeks, and every 3 months thereafter. Ibrutinib was held for grade 4 neutropenia lasting more than 7 days or grade 4 thrombocytopenia. For the first occurrence of grade 3 or higher adverse events (AEs), ibrutinib was held until the toxicity resolved to grade 1 or lower or baseline, and restarted at the original dose of 420 mg. For subsequent occurrences of grade 3 or higher AEs, ibrutinib was dose-reduced by the increment of 140 mg. Patients experiencing a first occurrence of grade 3 diarrhea, constitutional symptoms, or infection were restarted at the same dose. For new-onset or recurrent grade 2 or higher atrial fibrillation, ibrutinib was held and cardiac evaluation was completed before restarting therapy. After a first occurrence of grade 2 or higher atrial fibrillation, ibrutinib could be restarted at the same dose after toxicity resolved to grade 1 or lower or baseline. Subsequent occurrences of atrial fibrillation required dose reduction by 140-mg increments. In March 2015, dose modifications for nonhematologic AEs were harmonized with the ibrutinib prescriber information (USPI, reference ID 3694103). Laboratory testing included complete blood counts; basic metabolic, hepatic, and mineral panels; lactate dehydrogenase; and uric acid. The trial is fully enrolled.
Outcomes
The primary endpoint was response after 6 cycles of therapy. Secondary endpoints included safety, tolerability, overall survival (OS), PFS, and best response. Data cutoff for this report was 31 December 2017. Nonhematologic AEs were graded by using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0. Hematologic AEs and responses were called according to the International Workshop on CLL 2008 criteria, incorporating recent updates (supplemental Table 1, available on the Blood Web site).34 At 2, 6, and 12 months, and yearly thereafter, CT, bone marrow biopsies, and flow cytometry were performed. Spleen volume was calculated from CT, using the Vitrea Core Workstation Server, version 6.6 (Vital Images, Minnetonka, MN). Normal spleen volume was considered less than 315 mL. We previously reported the activity and safety data of the TP53 cohort (51 patients with TP53 aberration) at a median follow-up of 24 months.21
Flow cytometry
Flow cytometry to quantify residual disease used methods recommended by the European Research Initiative in CLL.35 Minimal residual disease (MRD) negativity was defined as fewer than 1 CLL cell in 10 000 (<10−4) leukocytes assessed.
Statistical analysis
We used a Simon’s minimax 2-stage design to test the null hypothesis that the ORR is 15% or less vs the 1-sided alternative. If 3 or more responses were observed among 16 patients of the first stage, an additional 11 would enter the second stage. With 8 or more responses, the null hypothesis would be rejected. For each cohort, 27 patients provide 90% power at the 0.05 significance level when the true response rate is 40%. Eight additional patients could be enrolled to account for non-treatment-related discontinuations before 6 months. In September 2012, the study was amended to allow enrollment of up to 35 patients with TN-CLL in the TP53 cohort, adding to 16 patients with RR-CLL. The total for the TP53 cohort was 51 patients; 35 patients were enrolled in the elderly cohort.
We used descriptive statistics to summarize findings. Duration of follow-up was calculated for surviving patients. OS and PFS were estimated by the Kaplan-Meier method and compared between subgroups by the log-rank test. Response rates were estimated by the proportions for all patients and subgroups, and their 95% confidence intervals (CIs) were computed and compared between subgroups by Fisher’s exact test. Wilcoxon signed-rank test was used to assess the change in the quantification of MRD. Spearman’s analysis was used to assess the correlation between blood and bone marrow MRD. Statistical analyses were conducted using R version 3.4.0 (R Foundation for Statistical Computing).
Role of the funding source
The trial was designed by the investigators, and a draft of the protocol was submitted to Pharmacyclics for comments. The study was funded by the Intramural Research Program of the National Heart, Lung, and Blood Institute and National Cancer Institute at the National Institutes of Health (NIH). Pharmacyclics provided the study drug. All data were collected by the investigators and stored at NIH. The investigators analyzed the data and wrote the manuscript. A draft of the manuscript was submitted to Pharmacyclics for comments. The corresponding author had full access to all the data in the study and had final responsibility for the content of the report and the decision to submit for publication.
Results
Patient characteristics and disposition
Between December 2011 and January 2014, 86 patients were enrolled; 51 were eligible because of TP53 aberration (TP53 cohort) and 35 because of age 65 years or older (elderly cohort; supplemental Figure 1; Table 1). Overall, 53 (61.6%) patients had TN-CLL, 58 (67.4%) had advanced Rai stage (III/IV), and 57 (66.3%) had unmutated IGHV. As of December 31, 2017, 49 (57.0%) patients remained on study (median time on study, 4.8 years; range, 4.0-6.0 years). Supplemental Table 2 summarizes the disposition of 37 patients who discontinued the study. Four (4.7%) patients died on study for reasons other than disease progression: 3 from infections and 1 because of sudden, presumably cardiac death. Twenty (23.3%) patients discontinued ibrutinib because of progressive disease, and 5 (5.8%) because of AEs, including newly diagnosed lung and ovarian cancer in 1 patient each. Six (7.0%) patients withdrew consent for various reasons. Two patients who did not meet enrollment criteria were removed and are included in the safety analysis only.
Table 1.
Baseline characteristics
| All (n = 86) | TP53 cohort (n = 51) | Elderly cohort (n = 35) | |
|---|---|---|---|
| Age, median (range), y | 66 (33-85) | 62 (33-82) | 69 (63*-85) |
| ≥65, N (%) | 55 (64.0) | 21 (41.2) | 34 (97.1)* |
| Sex, N (%) | |||
| Female | 36 (41.9) | 20 (39.2) | 16 (45.7) |
| Male | 50 (58.1) | 31 (60.8) | 19 (54.3) |
| Prior treatment status, N (%) | |||
| Treatment-naïve | 53 (61.6) | 35 (68.6) | 18 (51.4) |
| Relapsed/refractory† | 33 (38.4) | 16 (31.4) | 17 (48.6) |
| Rai stage, N (%) | |||
| I/II | 28 (32.6) | 19 (37.3) | 9 (25.7) |
| III/IV | 58 (67.4) | 32 (62.7) | 26 (74.3) |
| Bulky adenopathy (≥5 cm), N (%)‡ | 31 (36.0) | 19 (37.3) | 12 (34.3) |
| Splenomegaly, N (% evaluable)§ | 74 (88.1) | 44 (88.0) | 30 (88.2) |
| IGHV unmutated, N (%)‖ | 57 (66.3) | 34 (66.7) | 23 (65.7) |
| TP53 aberration, N (%) | 54 (62.8) | 51 (100) | 0 (0) |
| Deletion 17p | 50 (58.1) | 47 (92.2) | 3 (8.6)¶ |
| TP53 mutation | 4 (4.7) | 4 (7.8) | 0 (0) |
| β2-microglobulin | |||
| Median (range), mg/dL | 4·0 (1.7-12.9) | 3.9 (1.7-12.3) | 4.4 (1.9-12.9) |
| >4 mg/dL, N (%) | 44 (51.2) | 24 (47.1) | 20 (57.2) |
One patient not meeting age requirement was removed from study.
Median number of prior therapies was 3 (range, 1-7).
Target lymph nodes and spleen were assessed with CT scans.
Two patients had splenectomy. Normal spleen volume is less than 315 mL.
Unmutated IGHV indicates a less than 2% change in IGHV sequence compared with germ line.
Three patients had 7%-9% of nuclei with deletion 17p by fluorescence in situ hybridization; inclusion criteria for the TP53 cohort was more than 10% of nuclei with deletion 17p.
Safety
Treatment-related AEs leading to treatment discontinuation were, in 1 patient each, asymptomatic interstitial pulmonary infiltrates, progressive multifocal leukoencephalopathy on concurrent mycophenolate mofetil, and persistent grade 3 diarrhea with biopsy-proven microscopic colitis. Two (2.3%) patients discontinued because of second malignancies requiring systemic therapy. Nine (10.5%) patients required a dose reduction: 6 for atrial fibrillation, 1 for grade 3 rash, and 1 for grade 3 diarrhea, and 1 patient requested a reduction for grade 2 arthralgia. Treatment-emergent grade 3 or 4 hematologic AEs were neutropenia in 33 (38.4%), thrombocytopenia in 13 (15.1%), and anemia in 6 (7.0%) patients, primarily during the first few months receiving ibrutinib (supplemental Table 3). Most hematologic AEs were attributable to disease and improved with ibrutinib. Most nonhematologic AEs were grade 1 or 2 and were consistent with prior experience and USPI. Grade 3 or 4 nonhematologic AEs reported in 2 or more patients were infection (9.3%), atrial fibrillation (5.8%), diarrhea (3.5%), rash (2.3%), and arthritis (2.3%). Infection of any grade occurred in 24 (27.9%) patients. Notably, the overall frequency of infections, irrespective of attribution, decreased with time receiving therapy, suggesting improvements in immune function.31 Atrial fibrillation occurred in 18 (20.9%) patients, 13 grade 1 to 2 events, and 5 grade 3 events (supplemental Table 4). The rate of atrial fibrillation was 6.4 per 100 patient-years. Three patients underwent electrical cardioversion or ablation. All patients with atrial fibrillation restarted ibrutinib; 5 patients had dose reduction to 280 mg/day, and 1 patient to 140 mg/day. One patient with recurrent atrial fibrillation chose to stop ibrutinib after 2 years on study. Seven patients received apixaban, and 6 patients received aspirin. No grade 3 or higher bleeding event occurred on study.
Response
Eighty-one patients were evaluable for response at 6 months, the protocol-defined primary endpoint (Table 2). Three patients were not evaluable: 2 had died in the first 2 months, and 1 patient developed lung cancer. ORR for all patients was 95.1% (95% CI, 87.8%-98.6%; 95.8% for the TP53 cohort [95% CI, 85.7%-99.5%] and 93.9% for the elderly cohort [95% CI, 79.8%-99.3%]) and was not significantly different between subgroups stratified by treatment history, age, bulky lymphadenopathy, splenomegaly, IGHV mutation status, and baseline β2-microglobulin (all P > .05).
Table 2.
Response to treatment
| All (n = 81)* | TP53 cohort (n = 48)* | Elderly cohort (n = 33)* | |
|---|---|---|---|
| Response after 6 mo, N (%) | |||
| CR | 0 (0) | 0 (0) | 0 (0) |
| PR | 51 (63.0) | 27 (56.3) | 24 (72.7) |
| PR-L | 26 (32.1) | 19 (39.6) | 7 (21.2) |
| SD | 1 (1.2) | 0 (0) | 1 (3.0) |
| Progression | 3 (3.7)† | 2 (4.2) | 1 (3.0) |
| Best response, N (%) | |||
| CR | 23 (28.4) | 14 (29.2) | 9 (27.3) |
| PR | 54 (66.7) | 32 (66.7) | 22 (66.7) |
| PR-L | 2 (2.5) | 1 (2.1) | 1 (3.0) |
| SD | 0 (0) | 0 (0) | 0 (0) |
| Progression | 2 (2.5) | 1 (2.1) | 1 (3.0) |
CR, complete response; PR, partial response; PR-L, partial response with lymphocytosis; SD, stable disease.
Of 86 enrolled, 5 patients were not evaluable for response. Two patients did not meet inclusion criteria and were removed from study, 2 patients died within the first 2 months, and 1 patient could not be evaluated because of newly diagnosed lung cancer. Supplemental Table 2 summarizes patient disposition.
Two patients never responded; 1 patient achieved a partial response at 2 mo and progressed (compared with 2-mo nadir) at 6 mo.
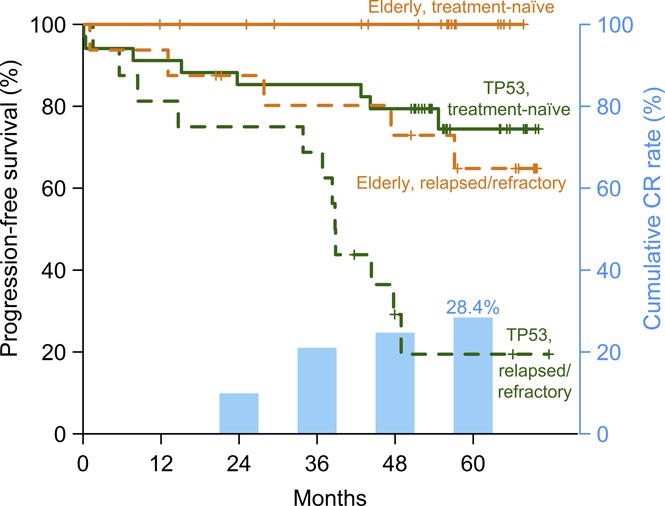
Disease burden progressively decreased in all anatomic compartments (supplemental Figure 2) with time receiving ibrutinib. At 3 years, the median reduction was 97% in absolute lymphocyte count, 94% in bone marrow, 89% in sum of the product of target lymph nodes, and 89% in spleen volume. The cumulative CR rate was 9.9% at 2 years, 21.0% at 3 years, and 28.4% at 5 years (Figure 1A). No clinical or laboratory parameter was significantly associated with different CR rates (Figure 1B). Next, we scored each of the International Workshop on CLL response criteria in each patient to investigate which criteria were not consistent with CR (Figure 1C). In order of decreasing frequency, the 3 most common were persistent splenomegaly (volume >315 mL) in 33 (40.7%) of 81 patients, residual lymph nodes (diameter ≥1.5cm) in 27 (33.3%), and persistent lymphocytosis (absolute lymphocyte count ≥4 × 109/L) in 23 (28.4%). In 36 (44.4%) patients, more than 1 requirement for CR was not met.
Figure 1.

Complete response on ibrutinib. (A) Cumulative best response in 81 evaluable patients. (B) Forest plot showing CR rates at best response in subgroups divided by clinical and laboratory criteria; the dashed line marks the CR rate in all patients (23.5%). Whiskers indicate 95% CI. *Trend in CR rate difference between low and advanced Rai stages (P = .054). **Spleen size was not evaluated in 2 patients with history of splenectomy. (C) Status of CR criteria in 79 patients; spleen (volume assessed by CT), target lymph nodes (LN), absolute lymphocyte count (ALC), bone marrow infiltration and cellularity (Marrow), and hematologic recovery (Heme). Twenty-three (28%) patients met all CR criteria (blue blocks); red blocks mark CR criteria not met. B2M, β2-microglobulin; IGHV-M, IGHV mutated; IGHV-U, IGHV unmutated.
Minimal residual disease
CLL burden, quantified by flow cytometry at 6 and 12 months and at annual points thereafter, significantly decreased in both blood and bone marrow (Figure 2A-B). Past the first year, the disease burden decreased by 33% in blood and 25% in marrow with each additional year of treatment (supplemental Figure 3). The median MRD at 3 years was 5.3 × 10−2 CLL cells/leukocyte in blood and 7.3 × 10−2 in bone marrow. The median MRD at 4 years was 3.8 × 10−2 CLL cells/leukocyte in blood and 6.2 × 10−2 in bone marrow. Measurements in the 2 compartments were highly correlated (Spearman’s ρ = 0.70 at 3 years and 0.89 at 4 years; both P < .0001; Figure 2C-F).
Figure 2.

MRD assessment in blood and bone marrow. (A-B) MRD was measured by flow cytometry in peripheral blood (PB) and bone marrow (BM). One patient who had undetectable PB MRD at 4 years was plotted at the lowest margin of detection (10−6). Patients were stratified by IGHV subgroups: M for mutated, U for unmutated. Red lines indicate median, boxes indicate interquartile ranges. *P < .05; **P < .01; n.s., not significant. (C-E) PB and BM MRD in matched samples at 24, 36, and 48 months. (F) Correlation between PB and BM MRD measurements.
Residual disease burden was similar in patients stratified by cohort or prior treatment status. However, patients with mutated IGHV had significantly more residual disease in the blood than patients with unmutated IGHV (Figure 2A). MRD levels in bone marrow were not significantly different between 2 IGHV subgroups (Figure 2B). MRD negativity (<10−4) was achieved in 1 of 64 patients in peripheral blood, in 2 of 51 in bone marrow aspirate at 3 years, in 5 of 49 patients in peripheral blood, and in 2 of 25 patients in bone marrow aspirate at 4 years. Seven patients in CR achieved MRD negativity in either blood or bone marrow. One patient with MRD negativity in blood at 3 and 4 years was not in CR because of persistent splenomegaly (residual spleen volume, 442 mL at 4 years).
We divided patients into 2 subgroups on the basis of 3-year peripheral blood MRD levels, using the cutoff of 10−2, as previously described by others: MRD-low (<10−2), and MRD-high (≥10−2).36 Among 64 patients receiving ibrutinib at 3 years, 16 (25%) patients were MRD-low, 47 (75%) were MRD-high, and 1 patient missed MRD assessment. The CR rate was 37.5% in the MRD-low group and 21.3% in the MRD-high group (P = .3; supplemental Figure 4). Nine (19.1%) of 47 patients in the MRD-high group and 1 (6.3%) of 16 patients in the MRD-low group subsequently progressed. PFS was not statistically different between the 2 groups (P = .5).
Disease progression and survival
With the median follow-up of 57 months, the estimated 5-year PFS was 58.2% for the TP53 cohort (95% CI, 44.5%-74.5%) and 81.2% for the elderly cohort (95% CI, 67.1%-98.3%; P = .026), and the median OS was 75.7% (95% CI, 64.7%-88.7%) and 83.8% (95% CI, 70%-100%), respectively (P = .10). In both cohorts, PFS was more favorable for patients receiving ibrutinib as a first-line therapy (Figure 3). In the TP53 cohort, the estimated 5-year PFS was 74.4% (95% CI, 60.2%-92.1%) for patients with TN-CLL compared with 19.4% (95% CI, 6.3%-60%) for those with RR-CLL (P = .0002), and OS was 85.3% (95% CI, 74.2%-98.1%) vs 53.7% (95% CI, 33.4%-86.4%), respectively (P = .023). In the elderly cohort, no progression or death occurred in TN-CLL, and in RR-CLL, the estimated 5-year PFS was 64.8% (95% CI, 43.9%-95.7%), and OS was 71.6% (95% CI, 51.2%-100%). There was no statistically significant difference in PFS and OS between subgroups divided by IGHV mutation status (all P > .1; supplemental Figure 5).
Figure 3.

PFS and OS. Kaplan-Meier estimates of PFS and OS of all patients on study (A-B), and by cohort and treatment status (C-D and E-F, respectively); (C-D) for the TP53 cohort and (E-F) for the elderly cohort B. RR, relapsed and/or refractory CLL; TN, treatment-naïve CLL.
Overall, 20 (23.8%) of 84 evaluable patients progressed. The median time to progression was 37.6 months (range, 0.4-54.7 months). Four (4.8%) patients had Richter’s transformation, and 2 (2.4%) had prolymphocytic transformation.21,25 All transformation events occurred within the first 15 months on study. Two patients with Richter’s transformation never achieved a response; all others initially responded, with 3 having achieved CR before progressing (supplemental Figure 6). Fourteen patients progressed with CLL; 12 (85.7%) of these had BTK and/or PLCG2 mutations at progression.25 Of 9 patients maintained on dose-reduced ibrutinib, only 1 has progressed to date. Thirteen patients with progressive CLL received salvage therapy with venetoclax or idelalisib plus rituximab. One patient died before initiation of subsequent therapy. The estimated median survival for patients with progressive CLL was 20.5 months from the time of progression, with 7 (35.0%) of 20 patients alive at data cutoff.
Discussion
In most cancers, with the notable exception of chronic myeloid leukemia, single-agent therapy is limited by the rapid emergence of drug resistance. Our data with extended follow-up of patients on ibrutinib suggests that a large proportion of patients with CLL achieve durable disease control while receiving single-agent ibrutinib, with excellent tolerability. The study population is representative of the clinical spectrum of CLL, comprising previously untreated patients as well as patients relapsing after up to 7 prior regimens, with a majority of patients older than 65 years. However, because of the preferential inclusion of patients with TP53 aberration, the genetic risk profile is skewed toward high-risk disease.
We previously reported 2-year PFS of 82% for the TP53 cohort.21 With the median follow-up of 57 months, the estimated 5-year PFS for these patients decreased to 58.2% (95% CI, 44.5%-74.5%). The majority of patients progressing had both TP53 aberration and RR-CLL. Notably, the estimated 5-year PFS in the current study was 74.4% (95% CI, 60.2%-92.1%) for TN-CLL patients with TP53 aberration compared with 19.4% (95% CI, 6.3%-60%) for those with RR-CLL (P = .0002). These results compare favorably to those achieved with chemoimmunotherapy. In the CLL8 trial, 68% of TN-CLL patients with del(17p) achieved a response to fludarabine, cyclophosphamide, and rituximab, with a median PFS of 11.3 months.4 In the MD Anderson series, 23% of patients with del(17p) were refractory to first-line therapy; the median PFS was 14 months, and 23% developed Richter’s transformation.37 In the current study report, none of the previously untreated patients without TP53 aberration progressed while receiving ibrutinib. Also, patients with RR-CLL without TP53 aberration did well, consistent with data from company-sponsored trials with 3 years of follow-up.38 Thus, TP53 aberration and prior treatment history are important determinants of PFS on ibrutinib. These observations raise the question of whether use of chemoimmunotherapy in first-line could compromise success of subsequent therapy with ibrutinib, or conceivably other targeted agents.39
Onset of response was quick, providing symptomatic relief within days, but long-term therapy was required for CRs. The most common reason for not achieving CR was residual lymphadenopathy or splenomegaly on CT scans. We routinely included bone marrow biopsies, which allowed continuous assessment of disease status in this compartment. Only in 3 patients were bone marrow findings responsible for not meeting CR criteria. MRD-negative remissions, defined as fewer than 10−4 CLL cells per leukocyte, remained uncommon. MRD-low remissions (<10−2) in the blood were observed in 25% of patients at 3 years. MRD measurements in blood and bone marrow were highly correlated. Thus, bone marrow examination seems to add little to response assessments in patients receiving ibrutinib.
The safety profile of continuous therapy with ibrutinib for more than 5 years was similar to what has been reported with shorter treatment duration.9,10,21 No new safety signals emerged, and most AEs were grade 1 or 2 and transient. The most common treatment-related grade 3 or higher AEs were infection (9.3%) and atrial fibrillation (5.8%). With our extended follow-up, the cumulative incidence of any grade atrial fibrillation at 19.8% is the highest reported.30 However, the rate of 6.4 per 100 patient-years in our study is consistent with the incidence of 6 per 100 patient-years reported from the pooled analysis of 4 clinical trials using ibrutinib in CLL and MCL.30 One patient with atrial fibrillation chose to discontinue ibrutinib because of recurrent grade 2 events; all other patients remained on study in consultation with cardiologists. Overall, 5 (5.8%) patients had to discontinue therapy because of treatment-emergent AEs. The low rate of study discontinuation is notable and, in addition to the good tolerability of the drug, might reflect the high proportion of patients with TP53 aberration who were determined to continue ibrutinib for as long as possible.
In summary, long-term administration of ibrutinib was well tolerated with deepening of responses over time. Most previously untreated patients, even those with TP53 aberration, achieved durable responses, making intensification of therapy less urgent and avoidance of unnecessary toxicity more important. Future research should aim to identify patients at risk for early treatment failure who would benefit most from combination therapy.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the patients who participated in this trial and their families, as well as Adriana Byrnes for protocol support, Francine Thomas for imaging analysis, and Tatyana Sarkisova and Manuk Manukyan for data management.
This research was supported by the Intramural Research Program of the NIH, the National Heart, Lung, and Blood Institute, and the National Cancer Institute. C.U.N. was supported by the Danish Cancer Society. Pharmacyclics provided study drug and research support for the study.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.Z.H.F. and A.W. designed the trial; I.E.A., M.Z.H.F., and A.W. conducted the clinical trial, analyzed data, and wrote the report; I.E.A., M.Z.H.F., J.V., C.S., J.L., G.E.M., N.S.S., C.U.N., and G.A. evaluated patients and collected data; S.S. and P.N. provided research nurse support; S.H. provided regulatory support; M.S.-S. and C.M.Y. analyzed flow cytometry; I.M. and K.R.C. evaluated bone marrow biopsies; T.E.H. provided pharmacy support; C.U.N., L.B.P., and C.H.G. performed and analyzed IGHV sequencing; S.P. reviewed lymph node biopsies; S.E.M.H. coordinated laboratory analysis; X.T. performed statistical analysis; R.C. and G.A. provided administrative support; and all authors reviewed and approved the report.
Conflict-of-interest disclosure: M.Z.H.F. is employed by Merck, owns stocks, and has received travel support from Merck. A.W. received research support from Pharmacyclics. N.S.S. received research support from Pharmacyclics. C.U.N. received consultancy fees and/or travel grants outside the current study from Janssen, AbbVie, Novartis, Gilead, and Roche. C.H.G. received consultancy fees from Janssen and Celgene and research support from Novo Nordisk. The remaining authors declare no competing financial interests.
The current affiliation of M.Z.H.F. is Merck & Co., Inc., Kenilworth, NJ.
Correspondence: Adrian Wiestner, Hematology Branch, NHLBI, NIH, Building 10, CRC 3-5140, 10 Center Dr, Bethesda, MD 20892-1202; e-mail: wiestnera@mail.nih.gov.
References
- 1.Fischer K, Bahlo J, Fink AM, et al. . Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127(2):208-215. [DOI] [PubMed] [Google Scholar]
- 2.Thompson PA, Tam CS, O’Brien SM, et al. . Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood. 2016;127(3):303-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landau DA, Tausch E, Taylor-Weiner AN, et al. . Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hallek M, Fischer K, Fingerle-Rowson G, et al. ; German Chronic Lymphocytic Leukaemia Study Group. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164-1174. [DOI] [PubMed] [Google Scholar]
- 5.Stilgenbauer S, Schnaiter A, Paschka P, et al. . Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123(21):3247-3254. [DOI] [PubMed] [Google Scholar]
- 6.Eichhorst B, Fink AM, Bahlo J, et al. ; German CLL Study Group (GCLLSG). First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016;17(7):928-942. [DOI] [PubMed] [Google Scholar]
- 7.Goede V, Fischer K, Busch R, et al. . Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370(12):1101-1110. [DOI] [PubMed] [Google Scholar]
- 8.Hillmen P, Robak T, Janssens A, et al. ; COMPLEMENT 1 Study Investigators. Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): a randomised, multicentre, open-label phase 3 trial. Lancet. 2015;385(9980):1873-1883. [DOI] [PubMed] [Google Scholar]
- 9.Byrd JC, Brown JR, O’Brien S, et al. ; RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burger JA, Tedeschi A, Barr PM, et al. ; RESONATE-2 Investigators. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Brien S, Jones JA, Coutre SE, et al. . Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): a phase 2, open-label, multicentre study. Lancet Oncol. 2016;17(10):1409-1418. [DOI] [PubMed] [Google Scholar]
- 12.Furman RR, Sharman JP, Coutre SE, et al. . Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amrein PC, Attar EC, Takvorian T, et al. . Phase II study of dasatinib in relapsed or refractory chronic lymphocytic leukemia. Clin Cancer Res. 2011;17(9):2977-2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herishanu Y, Pérez-Galán P, Liu D, et al. . The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117(2):563-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dühren-von Minden M, Übelhart R, Schneider D, et al. . Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489(7415):309-312. [DOI] [PubMed] [Google Scholar]
- 16.Minici C, Gounari M, Übelhart R, et al. . Distinct homotypic B-cell receptor interactions shape the outcome of chronic lymphocytic leukaemia. Nat Commun. 2017;8:15746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. 2018;18(3):148-167. [DOI] [PubMed] [Google Scholar]
- 18.Herman SE, Mustafa RZ, Gyamfi JA, et al. . Ibrutinib inhibits BCR and NF-κB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123(21):3286-3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng S, Ma J, Guo A, et al. . BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. 2014;28(3):649-657. [DOI] [PubMed] [Google Scholar]
- 20.Byrd JC, Furman RR, Coutre SE, et al. . Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farooqui MZ, Valdez J, Martyr S, et al. . Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16(2):169-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furman RR, Cheng S, Lu P, et al. . Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370(24):2352-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woyach JA, Furman RR, Liu TM, et al. . Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burger JA, Landau DA, Taylor-Weiner A, et al. . Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016;7:11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn IE, Underbayev C, Albitar A, et al. . Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood. 2017;129(11):1469-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woyach JA, Ruppert AS, Guinn D, et al. . BTKC481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. 2017;35(13):1437-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kadri S, Lee J, Fitzpatrick C, et al. . Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017;1(12):715-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Komarova NL, Burger JA, Wodarz D. Evolution of ibrutinib resistance in chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci USA. 2014;111(38):13906-13911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maddocks KJ, Ruppert AS, Lozanski G, et al. . Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015;1(1):80-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown JR, Moslehi J, O’Brien S, et al. . Characterization of atrial fibrillation adverse events reported in ibrutinib randomized controlled registration trials. Haematologica. 2017;102(10):1796-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun C, Tian X, Lee YS, et al. . Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015;126(19):2213-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahn IE, Jerussi T, Farooqui M, Tian X, Wiestner A, Gea-Banacloche J. Atypical Pneumocystis jirovecii pneumonia in previously untreated patients with CLL on single-agent ibrutinib. Blood. 2016;128(15):1940-1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lionakis MS, Dunleavy K, Roschewski M, et al. . Inhibition of B Cell Receptor Signaling by Ibrutinib in Primary CNS Lymphoma. Cancer Cell. 2017;31(6):833-843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hallek M, Cheson BD, Catovsky D, et al. ; International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines [published e-Letter response 4 June 2012]. Blood. 2008;111(12):5446-5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rawstron AC, Böttcher S, Letestu R, et al. ; European Research Initiative in CLL. Improving efficiency and sensitivity: European Research Initiative in CLL (ERIC) update on the international harmonised approach for flow cytometric residual disease monitoring in CLL. Leukemia. 2013;27(1):142-149. [DOI] [PubMed] [Google Scholar]
- 36.Böttcher S, Ritgen M, Fischer K, et al. . Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: a multivariate analysis from the randomized GCLLSG CLL8 trial. J Clin Oncol. 2012;30(9):980-988. [DOI] [PubMed] [Google Scholar]
- 37.Strati P, Keating MJ, O’Brien SM, et al. . Outcomes of first-line treatment for chronic lymphocytic leukemia with 17p deletion. Haematologica. 2014;99(8):1350-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byrd JC, Furman RR, Coutre SE, et al. . Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125(16):2497-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stilgenbauer S, Eichhorst B, Schetelig J, et al. . Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17(6):768-778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.