

Abstract
The ability to control the motion of single biomolecules is key to improving a wide range of biophysical and diagnostic applications. Solid-state nanopores are a promising tool capable of solving this task. However, molecular control and the possibility of slow readouts of long polymer molecules are still limited due to fast analyte transport and low signal-to-noise ratios. Here, we report on a novel approach of actively controlling analyte transport by using a double-nanopore architecture where two nanopores are separated by only a ∼ 20 nm gap. The nanopores can be addressed individually, allowing for two unique modes of operation: (i) pore-to-pore transfer, which can be controlled at near 100% efficiency, and (ii) DNA molecules bridging between the two nanopores, which enables detection with an enhanced temporal resolution (e.g., an increase of more than 2 orders of magnitude in the dwell time) without compromising the signal quality. The simplicity of fabrication and operation of the double-barrel architecture opens a wide range of applications for high-resolution readout of biological molecules.
Keywords: Single-molecule sensing, double nanopore architecture, biophysics
Nanopores have emerged as versatile biophysical tools capable of probing molecules one at a time.1−3 Nowadays, nanopore applications widely range from DNA/RNA sequencing to unraveling the underlying mechanisms of biological processes.4−6 The appeal of nanopore sensing stems partly from the beautiful simplicity of its operating principle: modulations in ionic current reflect the passage of single biomolecules through a nanometric aperture.7 Despite the significant contributions that nanopore sensing has made to date,8 high-resolution readouts are still limited by fast analyte transport9 and low signal-to-noise ratios (SNR).10 The ability to slow down and control the motion of biomolecules would pave the way for a broader range of biophysical and diagnostic applications including protein sequencing and studying DNA–protein interaction and manipulation.11−14 Since the inception of solid-state nanopores,15,16 researchers have proposed many solutions aimed toward addressing these limitations: tuning the nanopore shape and geometry,17,18 use of two-dimensional materials,19 tuning the physicochemical parameters of the electrolyte solutions (e.g., viscosity, temperature, pH, ionic strength gradients, physical-confinement),20−26 chemically modifying the pore surface,27−29 and more.30−34
Recently, a promising new strategy based upon solid-state multinanopore architectures was introduced to control molecular transport. These architectures bear some similarity to two-pore channels protein complexes found in eukaryotic cells, where a subset of voltage- and ligand-gated cation channels consist of subunits that give rise to multiple adjacent pores.35,36 For instance, Pud et al.37 proposed a double-nanopore system fabricated in a Si/SiN membrane in which a single dsDNA molecule could be engaged in a “tug-of-war” between the nanopores, resolving into a temporary stall of the analyte across the pores. Furthermore, Cadinu et al.38 successfully slowed down both DNA and small proteins using a zeptolitre “nanobridge”. While these new approaches are innovative, their potential is somewhat limited, as they rely on passive mechanisms of slowing down the analyte and the nanopores could not be electrically addressed independently.
Here we report on a new strategy, double barrel nanopores, which enables novel modes of single-molecule manipulation and allows for actively controlling the molecular transport. This device consists of two independently addressable nanopores that are located at the tip of a double barrel quartz nanopipette and separated by a gap of approximately 20 nm. The approach introduces a novel nanopore sensing platform in which the forces applied to the different ends of a single molecule can be tuned in real time. We demonstrate that this double barrel nanopore platform is capable of actively controlling DNA transport and efficiently bridging molecules between two pores (where up to 60% of all translocations can be bridged, compared to 0.8% reported previously).37 DNA molecules suspended between the two apertures can be sensed with an enhanced temporal resolution (viz., an increase of at least 2 orders of magnitude in the dwell time) with the possibility of complete trapping of the molecules without affecting the SNR and capture rate.
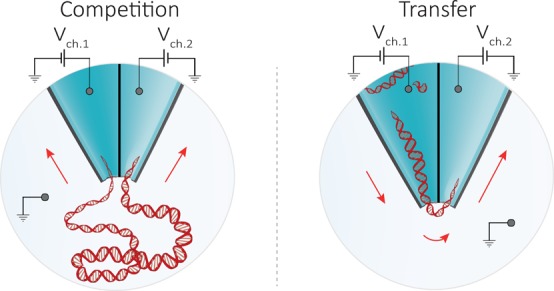
Double barrel nanopores were fabricated by laser-assisted pulling38−40 of double-barrel quartz capillaries, resulting in a nanopipette with two adjacent nanopores at the tip, as shown in Figure 1. Both barrels had comparable internal diameters of 23 ± 9 nm and were separated by a 20 ± 2 nm gap, as measured by scanning electron microscope (SEM) and transmission electron microscope (TEM), Figure 1b,c, SI Figure 1. An electrolyte solution of 2 M LiCl was used to fill both barrels, yielding an average pore conductance of 33 ± 4 nS (n = 20, Figure 1d). In all experiments, each barrel contained an independent working electrode, corresponding to an independent detection channel, and the common ground/reference electrode was introduced in the bath along with DNA. The ability to individually modulate the bias in the two detection channels enhanced the molecular control on DNA translocation, allowing either to capture a single DNA molecule into both pores leading to a “tug-of-war” between the two nanopores or to transport the analyte from one barrel to the other. We define this distinct modus operandi as “competition” and “transfer” mode, respectively (Figure 1a).
Figure 1.

Experimental setup and characterization of the double nanopore platform. (a) Schematic representation of the experimental setup showing a double barrel nanopore. In all experiments, the ground electrode was placed in the bath along with the DNA, while each barrel contained an independent working electrode, allowing each nanopore to be addressed individually. Depending on the polarity of the bias applied to the channels, two modes of operation are possible: competition and transfer mode. In the competition mode, where a positive bias is applied to both barrels, DNA molecules are attracted toward the two pores and can result in a single molecule being trapped between them. In the transfer mode, the pores are biased with voltages of opposite polarity enabling DNA molecules to be ejected from one pore and immediately threaded into the second one. (b,c) TEM and SEM images of the double barrel quartz nanopipette, respectively, showing pore diameters of approximately 23 ± 9 nm (n = 4 devices) separated by ≃20 nm gap and a cone angle of ∼0.11 radians. (d) Current–voltage characteristics of the two barrels measured in 2 M LiCl buffered in TE at pH 8 showing comparable a pore size. Errors denote one standard deviation. Insets show conductance histograms of the two pores.
In competition mode, the same positive bias is applied to both channels (Figure 2a). The negatively charged DNA is therefore attracted from the bath toward the tip of the pipet. Whereas the DNA can independently thread through each of the apertures, a significant fraction (from 6% to 62% depending on DNA size and potentials applied) ends up captured in both pores, leading to a molecule that temporarily gets stretched across the two pores. This results in opposing forces being exerted on the two ends of the molecule, significantly prolonging its residence time in the detection area. Eventually, in most cases the DNA slides out from one of the two pores (iv) and completes the translocation through the second nanopore (v). We refer to these types of translocations as “double pore events” as the same DNA molecule is being detected in the ionic current traces of both nanopores (Figure 2b).
Figure 2.

DNA translocations in competition mode. (a) Schematic of a DNA molecule being trapped between the two pores in competition mode. A positive bias is applied to each of the barrels, causing a DNA molecule to move from the bath toward the tip of the nanopipette (i). Subsequently, the molecule starts threading into one of the pores (ii) inducing a sharp drop in the ionic current of the corresponding channel. Given the proximity of the two apertures, the nontranslocated part of the molecule can get captured by the second pore (iii) resulting in a double pore event. Competing forces are exerted on the DNA by each of the nanopores, leading to a prolonged residence time across the pores. Eventually, the molecule slips out of the channel exerting the weakest force on the DNA sharply ending the current blockade of its recording (iv), and escapes through the other nanopore (v). Notably, when the DNA completes its translocation into the second nanopore, the corresponding ionic current recording returns to baseline following an exponential profile. (b) Current–time traces of the two detection channels acquired for 300 pM 10 kbp DNA molecules in 2 M LiCl at 400 mV bias applied to both barrels. At the bottom of the panel, two examples of double pore event current traces are shown. The exit channel recording (Ch.1 top current trace in both examples) of the double pore events shows the characteristic monoexponential profile with time-constant τ. (c) A histogram of τ revealed a peak maximum of ∼100 μs. (d) Histograms of the time offset at the start (δ1) and end (δ2) of double pore events.
Interestingly, the signal shapes of the ionic current blockades (Figure 2a,b) allow one to distinguish in which pore the DNA molecule enters first and in which nanopore it completes its translocation. When DNA translocates from the outside bath to the inside of the pipette (out-to-in), events are characterized by a sharp onset of the current blockade and a monoexponential return to the baseline at the beginning and the end of the event respectively (SI Figure 2). This shape can be explained by considering the varying electrical resistance along the conical nanopipette tip, as the DNA is quickly transported from the opening of the nanopore, a region of high local resistance, to the inside of the conical nanopipette tip, a region governed by access resistance. Conversely, pipet-to-bath (in-to-out) events follow the opposite trend, where a monoexponential current blockade is followed by a sharp return to baseline (SI Figure 3). These profiles are in good agreement with Bell et al.41 for single pore DNA translocation in 2 M LiCl. For our double pore events, the barrel in which the molecule concludes the translocation can thus be identified from the ionic current trace, as it contains the characteristic exponential profile at the end of the current blockade (Figure 2b). For 10 kbp DNA, the double pore event profiles were fitted using a monoexponential function with a time constant τ, yielding to a τ distribution which peaks at ∼100 μs (Figure 2c) and is consistent with the values measured in the single barrel recordings (SI Figures 2 and 3). It should be noted that τ is instrumentation independent, and it has a value larger than the rise/fall times of the amplifiers used (35 μs at the 10 kHz cutoff frequency used). The signal shapes also allowed to discriminate double pore events from the (rare, <0.2%, SI Figure 4) events in which two different DNA molecules simultaneously thread through each of the pores.
Double pore events start with a single DNA molecule entering one of the nanopores, followed by insertion of the second part of the molecule into the other nanopore after a time offset δ1. Selecting one of the pores as a fixed reference, δ1 can be either positive or negative depending on the nanopore in which the DNA molecule enters first. For equal bias applied, the δ1 distribution spreads symmetrically around 0 (Figure 2d), showing that molecules have no preferential entrance pore. Notably, a preference for the pore of entrance can be tuned by inducing an imbalance between the forces that the nanopores exert on the DNA in the access region. This can be achieved either by using nanopores of different sizes (SI Figure 5) or by biasing them with different voltages (SI Figures 6 and 7). The ending of a double pore event is characterized by δ2, the time offset between the moment that the DNA molecule is released from the first pore and its escape through the second pore. This δ2 represents the (very short) time that the DNA takes to travel the ∼20 nm gap. The measured δ2 distribution exhibited a much narrower range than δ1. This can be understood by considering the different nature of δ1 and δ2 offset times. Right after the start of the event, the second part of the DNA polymer requires time to find the second pore, a time which will depend on the polymer configuration and its position with respect to the pores. On the other hand, at the end of the event, the time of passage between the two channels is very well-defined by the distance between the nanopores and the translocation speed of the linearly stretched DNA.
A significant benefit of operating in competition mode is the long translocation time of double pore events. When both channels were held at an equal potential, 10 kbp DNA translocations events were recorded with dwell times up to 100 ms (Figure 3a,b). This value is more than 2 orders of magnitude higher than the dwell times of single pore events, Figure 3c, and the more conventional single barrel experiments (SI Figure 8). Importantly, the magnitude of the peak currents was comparable for double pore, and single-pore events at all biases applied (Figure 3d, SI Figure 9). With increasing applied voltage, the distributions of δ1 narrowed considerably, indicating that at higher fields, the DNA takes less time to find the second pore (Figure 3e,f).
Figure 3.

Voltage dependence analysis of DNA translocations in competition mode. Equal voltages ranging from 400 to 1000 mV were applied to both nanopores. Detection was carried out using 300 pM 10 kbp DNA in 2 M LiCl. (a) Representative current–time traces measured at the two detection channels at the different voltages. (b) Scatter plots of dwell times of double pore events plotted for channel 2 versus channel 1. Distributions are symmetric with respect to the diagonal of the plot (dotted line) meaning that the recorded dwell times are comparable for both channels. (c,d) Voltage dependence of dwell time and peak current for double pore (dark blue) and single pore (light blue) events. Double pore events experience a significant increase in dwell times, compared to single pore events. Both double pore and single pore events show peak currents increasing and dwell times decreasing with increasing voltages. (e,f) Distribution of start (δ1) and end (δ2) offsets for the different voltages. The width of the distributions narrows as voltages increase, with δ1,δ2 < |1| ms at 400 mV and δ1,δ2 < |0.5| ms at 1000 mV.
The percentage of double pore events was vastly higher than the 0.8% value reported previously.37 For instance, at all voltages applied double pore events constituted 13% of all events recorded for 10 kbp DNA and even 62% for 48.5 kbp DNA (Figure 4). Such a high bridging probability likely originates from the DNA radius of gyration being significantly greater than the tip dimensions. In some cases, the molecule was permanently trapped across the two pores, likely because the net force exerted in the two channels canceled out (Figure 4b). Such permanently trapped molecules could only be released by reversing the voltage applied to one of the channels.
Figure 4.

DNA size dependence in competition mode. (a) Ionic current traces recorded in competition mode for 50 pM 48.5 kbp DNA (red), 150 pM 20 kbp DNA (yellow), and 300 pM 10 kbp DNA (green) when 400 mV is applied to both nanopores (the color code for different DNA length is maintained throughout the whole figure). (b) Representative examples of 48.5 kbp DNA molecule trapped between the pores when 200 mV was applied to both nanopores. The molecule could only be released after reversing the potential in one of the channels. (c) Double pore event rate versus voltage for 10, 20, and 48.5 kbp DNA (2 M LiCl in TE buffered at pH 8.0). The rates show a strong dependence on the DNA size and a moderate dependence on the voltage applied. (d) Comparison between the probability density function of dwell times for double pore and single pore events recorded at 400 mV. (e,f) Scatter plots of the dwell times and peak current of double pore events. (g) Start (δ1) and end (δ2) offset distributions. For all DNA lengths, double pore events show longer start offsets compared to end offsets with the majority of the double pore translocations of 48.5 kbp molecules having δ1 < |5 ms| and δ2 < |1 ms|. The width of both distributions decreases with decreasing DNA length: the time constant of the exponential fittings for the start offset was calculated to be 0.93 ± 0.14, 0.43 ± 0.07, and 0.32 ± 0.01 ms for 48.5, 20, and 10 kbp DNA respectively.
The double pore event rate was only moderately influenced by the applied voltages (Figure 4c), but quite strongly dependent on DNA size. Longer molecules experienced longer dwell times in the nanopore, thus providing more time for the second end of the DNA to find the second pore and hence increasing the likelihood of a double pore event. The behavior of the δ1 offset distribution for different DNA sizes also confirms this, as δ1 is distributed over a broader range for longer DNA molecules (e.g., up to 3 ms for λ-DNA, Figure 4g). The longer DNA molecules also showed longer double pore translocation times (Figure 4d,e). For 48.5 kbp and 10 kb DNA, the maximum dwell times for double pore events were measured to be as long as 1.8 s and 55 ms at 400 mV, respectively.
An alternative method to control the DNA transport was also explored, which we dub transfer mode; in this mode of operation, a single molecule can be efficiently transferred from one barrel to the other (Figure 5). In these experiments, molecules are initially loaded electrokinetically into the pipette from the bath. The transfer is realized by holding one of the barrels at negative bias while keeping the other at positive bias with respect to the grounded bath. DNA molecules are thus ejected from the channel held at negative bias (panel (i) in Figure 5a), then attracted to (ii) and threaded into the other channel (iii,iv). Because of the symmetrical geometry of the double-barrel nanopipettes, the delivery and recipient nanopore can be chosen at will by the experimenter. As for competition mode, the shape of the ionic current blockade is different for the delivery and the recipient nanopore (see Figure 5b) thus enabling to discriminate between single pore events and double pore events. Much like in competition mode, the event profile was fit using a monoexponential function with time constant τ (Figure 5c) with values again being consistent with single barrel recordings (SI Figures 2 and 3). By tuning the voltages of the two channels, we found that it is possible to drive all molecules from the delivery to the receiving nanopore with near 100% efficiency (Figure 5e, SI, Figures 10–12).
Figure 5.

DNA translocations in transfer mode. (a) Schematic of a pore-to-pore translocation in transfer mode. Voltages of opposite polarity are applied to both detection channels. The exit of a DNA molecule from the nanopore held at negative bias can be observed as an exponential rise being recorded on the current time trace of Channel 1 which is defined as the delivery detection channel (dark blue) (i). Before being fully released into the bath, the molecule is attracted toward the second nanopore (recipient detection channel), inducing a sharp blockade onset in the current trace (light blue) (ii). The DNA then exits the delivery nanopore (iii) and translocates through the recipient nanopore (iv), resulting in an gradual rise in the ionic current of the recipient channel. (b) Current–time traces of the two channels acquired for 300 pM 10 kbp DNA in 2 M LiCl when a −400 mV (Ch. 1) and 400 mV (Ch. 2) are applied. At the bottom of the panel examples of double pore transfer events are shown. representative profiles of individual translocation event measured in the delivery (dark blue) and the recipient (blue) channel. Monoexponential fits are highlighted with red and black dashed fitting lines, respectively. (c) Histograms of exponential decay fittings of double pore transfer events recorded for both delivery (τ1) and transfer (τ2). (d) Distributions of start (δ1) and end (δ2) offsets of the transfer events. Transfer events show faster end offsets than beginning offsets, with δ1 < |1.5| ms and δ2 < |0.5| ms.(e) Transfer efficiency as a function of the bias applied to the recipient nanopore for voltages of −200, −400, and −600 mV, applied at the delivery channel.
To summarize, we presented two novel double-nanopore sensing configurations for active control of single-molecule transport in solid-state nanopores. By using a double-barrel nanopipette, it is possible to obtain a three-terminal architecture where the electric field in each of the nanopores can be adjusted individually. We showed that DNA molecules could be efficiently confined and trapped between the two pores leading to a molecular tug-of-war which slows down the translocation process by several orders of magnitude. The yield of these double pore events ranged between 13% and 60% depending on the DNA length and only moderately on the applied voltages. Furthermore, we demonstrated that DNA molecules could be loaded from the bath to the barrel and then either released back or transferred to the other barrel. By tuning the voltages applied, we could obtain near 100% pore-to-pore transfer efficiency.
The strength and reliability of the platform open a plethora of possible applications. For example, implementation of a feedback control mechanism on the system would increase the degrees of freedom in manipulating the DNA. Such a mechanism could be used to perform multiple subsequent readings of the same DNA molecule, yielding more accurate information about proteins bound along the DNA strand or alternatively to study changes in DNA conformations induced by small molecules by performing multiple readings of the same DNA molecule.
Materials and Methods
Double Barrel Nanopore Fabrication
Nanopipettes were fabricated using a P-2000 laser puller (Sutter Instrument Co, U.S.A.) from quartz theta capillaries (QF120-90-7.5; Sutter Instrument Co, U.S.A.) with an outer diameter of 1.2 mm and length of 7.5 cm. Prior laser-pulling, nanopipettes were oxygen plasma cleaned for 15 min to remove organic contaminants. Nanopipettes were fabricated according to the following settings: (1) HEAT, 850; FIL, 4; VEL, 30; DEL, 160; PUL, 100 followed by (2) HEAT, 860; FIL, 3; VEL, 20; DEL, 140; PUL, 160. The final nanopipettes had pore diameters of 23 ± 9 nm (calculated as the average between the major and minor elliptical axes) and cone semiangles of about 0.11 radians, as measured by scanning electron microscope (SEM) and transmission electron microscope (TEM), (Figure 1b,c). Pulling resulted in two symmetrical pores with a conductance of 35 ± 4 nS for the larger aperture and 32 ± 4 nS for the smaller one (n = 20, measured in 2 M LiCl). It is noteworthy that the pulling parameters are instrument specific and will vary from a puller to puller. For the fabrication of symmetric nanopores, we found out that the septum separating the two barrels has to be aligned parallel to the incident laser. Under these conditions, ∼75% of all pulled pippetes resulted in symmetric (conductance variation within 10%), and functional nanopores, if the pipettes are filled with the solution immediately after pulling. After the solution is filled, the pipettes were subjected to negative (back) pressure to ensure that there are no air bubbles left in the solution or the pipette tips. The nanopipettes underwent a silanization process to avoid crosstalk and current leakages between barrels. The back end of the nanopipette was exposed to vapors of Trichloro(1H,1H,2H,2H-perfluorooctyl)silane (product number 448931-10G, Sigma-Aldrich) for no more than 10 s. If after pulling the pipettes are stored in air for long periods of time (i.e., >1 day), we found that these devices are difficult to fill with solution and require oxygen plasma cleaning.
Ionic Current Recordings
The ionic current recordings were performed with a Multiclamp 700B low-noise current amplifier (Molecular Devices, U.S.A.) in voltage clamp mode. The recorded analog signal was low-pass filtered using a built-in four-pole Bessel filter with a cutoff frequency of 10 kHz. All signals were acquired at 100 kHz using Digidata 1550B data-acquisition module (Molecular Devices, U.S.A.). The two nanopore channels were voltage-addressed with two separate headstages which were connected to Ag/AgCl electrodes. The two patch electrodes were connected in the barrels of the nanopipette while the ground electrode was placed in the bath. In all experiments, the DNA sample was first introduced in the bath. All data was postprocessed using custom-written Matlab scripts.
Solution and Reagents
The 48.5 kbp DNA and 10 kbp DNA were purchased from New England Biolabs, U.K. (stock concentration of 500 μg/mL). Twenty kilo-base pair NoLimits DNA fragment with a stock concentration of 500 μg/mL was purchased from ThermoFisher Scientific. All DNA solutions were prepared in 2 M LiCl, 10 mM Tris, 1 mM EDTA at pH 8.0 (product number T9285) using ultrapure DI water (18 MΩ) via serial dilutions. Before use, 48.5 kbp DNA was treated at 65 °C for 4 min to linearize it and then slowly cooled down to room temperature.
TEM and SEM Imaging
The pipette tips were imaged using a JEOL JEM-1400 transmission electron microscope with an acceleration voltage of 120 keV. The pipette tips were glued onto TEM-grid-sized copper windows using epoxy resin and then cut off from the pipette body. Prior to imaging, 5 nm of carbon was sputtered onto the pipette tips in a 208C High Vacuum Turbo Carbon Coater (Cressington Scientific Instruments, U.K.). The tip of the pipette was perpendicular to the direction of sputtering. SEM imaging of the pipette tips was carried out with a FEI NovaNano SEM. Prior to imaging, 7 nm of Pt was sputtered onto the pipette to prevent drifts caused by charging. During sputtering the pipette was positioned with the tip facing the sputtering source in Leica ACE200.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.8b00860.
TEM images of the nanopores, experimental data on transfer and completion modes, control experiments using a conventional nanopore configuration (PDF)
Author Contributions
§ P.C., G.C., and S.P. contributed equally.
J.B.E. has been funded in part by an ERC starting (NanoP), proof of concept (NanoPP), and consolidator (NanoPD) grants. C.D. was funded in part by an ERC advanced grant SynDiv (nr 669598) and The Netherlands Organization for Scientific Research (NWO/OCW) as part of the Frontiers of Nanoscience program. A.P.I. and J.B.E. acknowledge support from EPSRC Grant EP/P011985/1. A.P.I. acknowledges the support of the IC Research Fellowship, G.C. acknowledges the support of the ICB Studentship.
The authors declare no competing financial interest.
Supplementary Material
References
- Dekker C. Solid-State Nanopores. Nat. Nanotechnol. 2007, 2, 209–215. 10.1038/nnano.2007.27. [DOI] [PubMed] [Google Scholar]
- Miles B. N.; Ivanov A. P.; Wilson K.; Doğan F.; Japrung D.; Edel J. B. Single Molecule Sensing with Solid-State Nanopores: Novel Materials, Methods, and Applications. Chem. Soc. Rev. 2013, 42, 15–28. 10.1039/C2CS35286A. [DOI] [PubMed] [Google Scholar]
- Siwy Z. S.; Howorka S. Engineered Voltage-Responsive Nanopores. Chem. Soc. Rev. 2010, 39, 1115–1132. 10.1039/B909105J. [DOI] [PubMed] [Google Scholar]
- Clarke J.; Wu H.; Jayasinghe L.; Patel A.; Reid S.; Bayley H. Continuous Base Identification for Single-Molecule Nanopore DNA Sequencing. Nat. Nanotechnol. 2009, 4, 265–270. 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- Greninger A. L.; Naccache S. N.; Federman S.; Yu G.; Mbala P.; Bres V.; Stryke D.; Bouquet J.; Somasekar S.; Linnen J. M. Rapid Metagenomic Identification of Viral Pathogens in Clinical Samples by Real-Time Nanopore Sequencing Analysis. Genome Med. 2015, 7, 99. 10.1186/s13073-015-0220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zheng D.; Tan Q.; Wang M. X.; Gu L.-Q. Nanopore-Based Detection of Circulating microRNAs in Lung Cancer Patients. Nat. Nanotechnol. 2011, 6, 668–674. 10.1038/nnano.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasianowicz J. J.; Brandin E.; Branton D.; Deamer D. W. Characterization of Individual Polynucleotide Molecules Using a Membrane Channel. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 13770–13773. 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W.; Friedman A. K.; Baker L. A. Nanopore Sensing. Anal. Chem. 2017, 89, 157–188. 10.1021/acs.analchem.6b04260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesa C.; van Loo N.; Ketterer P.; Dietz H.; Dekker C. Velocity of DNA during Translocation through a Solid-State Nanopore. Nano Lett. 2015, 15, 732–737. 10.1021/nl504375c. [DOI] [PubMed] [Google Scholar]
- Smeets R. M. M.; Keyser U. F.; Dekker N. H.; Dekker C. Noise in Solid-State Nanopores. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 417–421. 10.1073/pnas.0705349105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze J. Y. Y.; Ivanov A. P.; Cass A. E. G.; Edel J. B. Single Molecule Multiplexed Nanopore Protein Screening in Human Serum Using Aptamer Modified DNA Carriers. Nat. Commun. 2017, 8, 1552. 10.1038/s41467-017-01584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell N. A. W.; Keyser U. F. Specific Protein Detection Using Designed DNA Carriers and Nanopores. J. Am. Chem. Soc. 2015, 137, 2035–2041. 10.1021/ja512521w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A. P.; Actis P.; Jönsson P.; Klenerman D.; Korchev Y.; Edel J. B. On-Demand Delivery of Single DNA Molecules Using Nanopipets. ACS Nano 2015, 9, 3587–3594. 10.1021/acsnano.5b00911. [DOI] [PubMed] [Google Scholar]
- Yusko E. C.; Bruhn B. R.; Eggenberger O. M.; Houghtaling J.; Rollings R. C.; Walsh N. C.; Nandivada S.; Pindrus M.; Hall A. R.; Sept D. Real-Time Shape Approximation and Fingerprinting of Single Proteins Using a Nanopore. Nat. Nanotechnol. 2017, 12, 360–367. 10.1038/nnano.2016.267. [DOI] [PubMed] [Google Scholar]
- Li J.; Stein D.; McMullan C.; Branton D.; Aziz M. J.; Golovchenko J. a. Ion-Beam Sculpting at Nanometre Length Scales. Nature 2001, 412, 166–169. 10.1038/35084037. [DOI] [PubMed] [Google Scholar]
- Storm A. J.; Chen J. H.; Ling X. S.; Zandbergen H. W.; Dekker C. Fabrication of Solid-State Nanopores with Single-Nanometre Precision. Nat. Mater. 2003, 2, 537–540. 10.1038/nmat941. [DOI] [PubMed] [Google Scholar]
- Wanunu M.; Sutin J.; McNally B.; Chow A.; Meller A. DNA Translocation Governed by Interactions with Solid-State Nanopores. Biophys. J. 2008, 95, 4716–4725. 10.1529/biophysj.108.140475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S.; Wilson J.; Shim J.; Shankla M.; Corbin E. A.; Aksimentiev A.; Bashir R. Slowing DNA Transport Using Graphene-DNA Interactions. Adv. Funct. Mater. 2015, 25, 936–946. 10.1002/adfm.201403719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjmandi-Tash H.; Belyaeva L. A.; Schneider G. F. Single Molecule Detection with Graphene and Other Two-Dimensional Materials: Nanopores and beyond. Chem. Soc. Rev. 2016, 45, 476–493. 10.1039/C5CS00512D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk S. W.; Wells D. B.; Aksimentiev A.; Dekker C. Slowing down DNA Translocation through a Nanopore in Lithium Chloride. Nano Lett. 2012, 12, 1038–1044. 10.1021/nl204273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanunu M.; Morrison W.; Rabin Y.; Grosberg A. Y.; Meller A. Electrostatic Focusing of Unlabelled DNA into Nanoscale Pores Using a Salt Gradient. Nat. Nanotechnol. 2010, 5, 160–165. 10.1038/nnano.2009.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J.; Liu K.; Bulushev R. D.; Khlybov S.; Dumcenco D.; Kis A.; Radenovic A. Identification of Single Nucleotides in MoS2 Nanopores. Nat. Nanotechnol. 2015, 10, 1070–1076. 10.1038/nnano.2015.219. [DOI] [PubMed] [Google Scholar]
- Lu B.; Hoogerheide D. P.; Zhao Q.; Zhang H.; Tang Z.; Yu D.; Golovchenko J. A. Pressure-Controlled Motion of Single Polymers through Solid- State Nanopores. Nano Lett. 2013, 13, 3048–3052. 10.1021/nl402052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedone D.; Langecker M.; Abstreiter G.; Rant U. A Pore-Cavity-Pore Device to Trap and Investigate Single Nanoparticles and DNA Molecules in a Femtoliter Compartment: Confined Diffusion and Narrow Escape. Nano Lett. 2011, 11, 1561–1567. 10.1021/nl104359c. [DOI] [PubMed] [Google Scholar]
- Liu X.; Mihovilovic Skanata M.; Stein D. Entropic Cages for Trapping DNA near a Nanopore. Nat. Commun. 2015, 6, 6222. 10.1038/ncomms7222. [DOI] [PubMed] [Google Scholar]
- Briggs K.; Madejski G.; Magill M.; Kastritis K.; de Haan H. W.; McGrath J. L.; Tabard-Cossa V. DNA Translocations through Nanopores under Nanoscale Preconfinement. Nano Lett. 2018, 18, 660–668. 10.1021/acs.nanolett.7b03987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X.; Guo W.; Jiang L. Biomimetic Smart Nanopores and Nanochannels. Chem. Soc. Rev. 2011, 40, 2385. 10.1039/c0cs00053a. [DOI] [PubMed] [Google Scholar]
- Ren R.; Zhang Y.; Nadappuram B. P.; Akpinar B.; Klenerman D.; Ivanov A. P.; Edel J. B.; Korchev Y. Nanopore Extended Field-Effect Transistor for Selective Single-Molecule Biosensing. Nat. Commun. 2017, 8, 586. 10.1038/s41467-017-00549-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X.; Ivanov A. P.; Edel J. B. Selective Single Molecule Nanopore Sensing of Proteins Using DNA Aptamer-Functionalised Gold Nanoparticles. Chem. Sci. 2017, 8, 3905–3912. 10.1039/C7SC00415J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H.; Ling X. S. Reverse DNA Translocation through a Solid-State Nanopore by Magnetic Tweezers. Nanotechnology 2009, 20, 185101. 10.1088/0957-4484/20/18/185101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S.; Tsutsui M.; Theodore H.; Yuhui H.; Arima A.; Tsuji T.; Doi K.; Kawano S.; Taniguchi M.; Kawai T. Tailoring Particle Translocation via Dielectrophoresis in Pore Channels. Sci. Rep. 2016, 6, 31670. 10.1038/srep31670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yameen B.; Ali M.; Neumann R.; Ensinger W.; Knoll W.; Azzaroni O. Ionic Transport Through Single Solid-State Nanopores Controlled with Thermally Nanoactuated Macromolecular Gates. Small 2009, 5, 1287–1291. 10.1002/smll.200801318. [DOI] [PubMed] [Google Scholar]
- Luan B.; Stolovitzky G.; Martyna G. Slowing and Controlling the Translocation of DNA in a Solid-State Nanopore. Nanoscale 2012, 4, 1068–1077. 10.1039/C1NR11201E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Yobas L. Slowing DNA Translocation in a Nanofluidic Field-Effect Transistor. ACS Nano 2016, 10, 3985–3994. 10.1021/acsnano.6b00610. [DOI] [PubMed] [Google Scholar]
- Patel S. Function and Dysfunction of Two-Pore Channels. Sci. Signaling 2015, 8, re7. 10.1126/scisignal.aab3314. [DOI] [PubMed] [Google Scholar]
- Kintzer A. F.; Stroud R. M. Structure, Inhibition and Regulation of Two-Pore Channel TPC1 from Arabidopsis Thaliana. Nature 2016, 531, 258–264. 10.1038/nature17194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pud S.; Chao S.-H.; Belkin M.; Verschueren D.; Huijben T.; van Engelenburg C.; Dekker C.; Aksimentiev A. Mechanical Trapping of DNA in a Double-Nanopore System. Nano Lett. 2016, 16, 8021–8028. 10.1021/acs.nanolett.6b04642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadinu P.; Paulose Nadappuram B.; Lee D. J.; Sze J. Y. Y.; Campolo G.; Zhang Y.; Shevchuk A.; Ladame S.; Albrecht T.; Korchev Y. Single Molecule Trapping and Sensing Using Dual Nanopores Separated by a Zeptoliter Nanobridge. Nano Lett. 2017, 17, 6376–6384. 10.1021/acs.nanolett.7b03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodolfa K. T.; Bruckbauer A.; Zhou D.; Korchev Y. E.; Klenerman D. Two-Component Graded Deposition of Biomolecules with a Double-Barreled Nanopipette. Angew. Chem., Int. Ed. 2005, 44, 6854–6859. 10.1002/anie.200502338. [DOI] [PubMed] [Google Scholar]
- Perry D.; Momotenko D.; Lazenby R. A.; Kang M.; Unwin P. R. Characterization of Nanopipettes. Anal. Chem. 2016, 88, 5523–5530. 10.1021/acs.analchem.6b01095. [DOI] [PubMed] [Google Scholar]
- Bell N. A. W.; Chen K.; Ghosal S.; Ricci M.; Keyser U. F. Asymmetric Dynamics of DNA Entering and Exiting a Strongly Confining Nanopore. Nat. Commun. 2017, 8, 1–8. 10.1038/s41467-017-00423-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.