

Abstract
Thailanstatin A has been isolated recently from the fermentation broth of B. thailandensis MSMB43. We describe here an enantioselective convergent synthesis of thailanstatin A methyl ester and evaluation of its splicing activity. Synthesis of both highly functionalized tetrahydropyran rings were carried out from commercially available tri-O-acetyl-D-glucal as the key starting material. Our convergent synthesis involved the synthesis of both tetrahydropyran fragments in a highly stereoselective manner. The fragments were then coupled using cross-metathesis as the key step. The synthesis of the diene subunit included a highly stereoselective Claisen rearrangement, a Cu(I)-mediated conjugate addition of MeLi to set the C-14 methyl stereochemistry, a reductive amination reaction to install the C16-amine functionality and a Wittig olefination reaction to incorporate the diene unit. The epoxy alcohol subunit was synthesized by a highly selective anomeric allylation, a Peterson olefination and a vanadium catalyzed epoxidation that installed the epoxide stereoselectively. Cross-metathesis of the olefins provided the methyl ester derivative of thailanstatin A. We have carried out in vitro splicing studies of the methyl ester derivative, which proved to be a potent inhibitor of the spliceosome.
Graphical abstract

Introduction
Pre-mRNA splicing is a fundamental event in gene expression in eukaryotes.1,2 Splicing is catalyzed by a complex ribonucleoprotein machinery called the spliceosome, which is formed from uridine-rich small nuclear RNAs and dozens of proteins.3,4 During splicing, the spliceosome removes intervening sequences (introns) while joining coding sequences (exons) to form mature mRNA, which is exported to the cytoplasm for translation into proteins.5,6 Alternative splicing provides cell transcriptome diversity; recent studies have shown that splicing is pathologically altered to promote the initiation and maintenance of cancer.7,8,9 Splicing requires numerous protein-protein and protein-RNA interactions.10,11 These interactions and regulatory steps provide a wide range of opportunities to manipulate the splicing cascade for therapeutic purposes. Currently, a series of natural products and their derivatives, in particular, pladienolide B (1), spliceostatin A (2), and herboxidiene (3) are known to inhibit spliceosome function by binding to the SF3B subunit of U2 snRNP.12,14,15 The precise biochemical interactions of these natural products with these proteins are not clear. Interestingly, the effect of these natural products on cell cytotoxicity is completely abrogated by mutation of a single residue of SF3B1 (SF3B1; R1074H), which suggests that these compounds function through specific interactions with SF3B1.16,17 Despite potent splicing inhibitory activity by these natural products, clinical use has been limited due to inadequate physicochemical properties.18,19 Thus far, a semisynthetic derivative E707,6, which showed improved pharmacological profiles, underwent clinical trials.20,21 Also, several spliceostatin derivatives have been recently modified for specific delivery of spliceostatin payloads using antibody-drug conjugates.22,23,24 Liu and co-workers in 2013, reported a new class of spliceostatins, one of which is thailanstatin A (1, Figure 1), isolated from the fermentation broth of B. thailandensis MSMB43.25 The structure of thailanstatin A was elucidated with a combination of high resolution mass spectrometry and extensive NMR studies. He and co-workers in 2014, isolated spliceostatins as well as thailanstatins from a culture broth of Burkholderia sp. FERM BP-3421.26 Thailanstatin A showed potent antiproliferative activity in human cancer cell lines with IC50 values ranging from 59–320 nM in various cancer cell lines.25 The methyl ester derivative of thailanstatin A, 2, exhibited remarkably potent in vitro potency in multiple cancer cell lines with IC50 values ranging from 0.25–0.78 nM.26 Because of its potential for development of cancer drugs, thailanstatin A has been produced in quantities via an optimized fermentation process.27,28 Over the years, total synthesis and further design of structural derivatives of spliceostatins such as FR901464 (3), spliceostatin A (4) and pladienolides, in particular pladienolide B (5), have been pursued to improve stability and drug properties.29–35 Recently, Nicolaou and co-workers reported the first synthesis of thailanstatin A, 1 and its methyl ester derivative, 2.36 Our interest in thailanstatin A arose from its potent antitumor activity, and its structural features for incorporating linkers for the preparation of antibody drug conjugates. Herein, we report our convergent synthesis of thailanstatin A methyl ester and our preliminary studies of in vitro splicing activity of the methyl ester derivative. The synthesis provides rapid access to structural variants of thailanstatins and spliceostatins.
Figure 1.
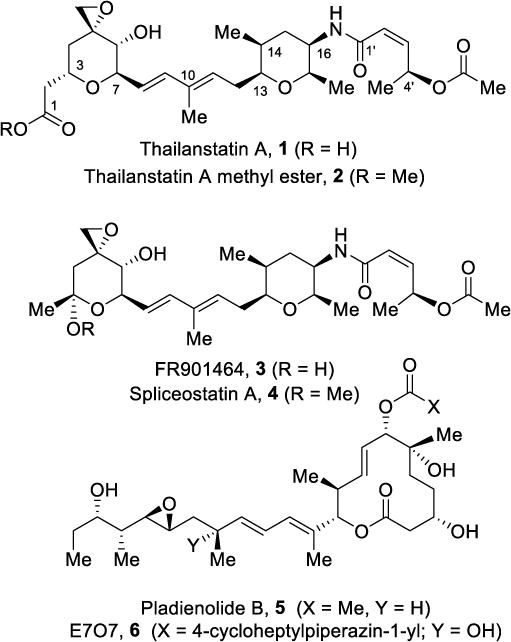
Structures of splicing inhibitors 1–6.
Results and Discussion
The thailanstatin class of natural products consists of complex and sensitive functionalities. Thailanstatin A contains nine stereogenic centers, and two highly functionalized tetrahydropyran ring systems attached via a conjugated diene functionality.25,26 Furthermore, the molecule contains a sensitive epoxide and an allylic acetate group. Some of these functional groups not only created a synthetic challenge, but also pose significant obstacles for advancing these agents to cancer therapy. Specifically, both the epoxide and the diene functionalities are very sensitive to acid and the epoxide ring has proven to be subjected to nucleophilic attack readily. Our retrosynthetic analysis of thailanstatin A is shown in Scheme 1. Our strategic disconnections of the diene (C8-C9) provided subunits 7 and 8, respectively. Our plan was to utilize cross-metathesis of these subunits at a late stage to construct the molecular architecture of thailanstatin A. We planned to assemble the diene subunit by amide coupling of amine 9 and carboxylic acid 10. The functionalized tetrahydropyran ring 9 would be constructed stereoselectively from the aldehyde intermediate 11. Synthesis of this aldehyde derivative can be carried out by utilizing a Claisen rearrangement of a vinyl ether derivative which would be obtained from the commercially available tri-O-acetyl-D-glucal (13). The synthesis of the epoxy alcohol subunit 8 would also be carried out from tri-O-acetyl-D-glucal by a stereoselective allylation reaction to obtain intermediate 12 followed by functional group manipulations. The overall chiron approach from tri-O-acetyl-D-glucal would construct eight of the nine stereogenic centers of thailanstatin A.
Scheme 1.
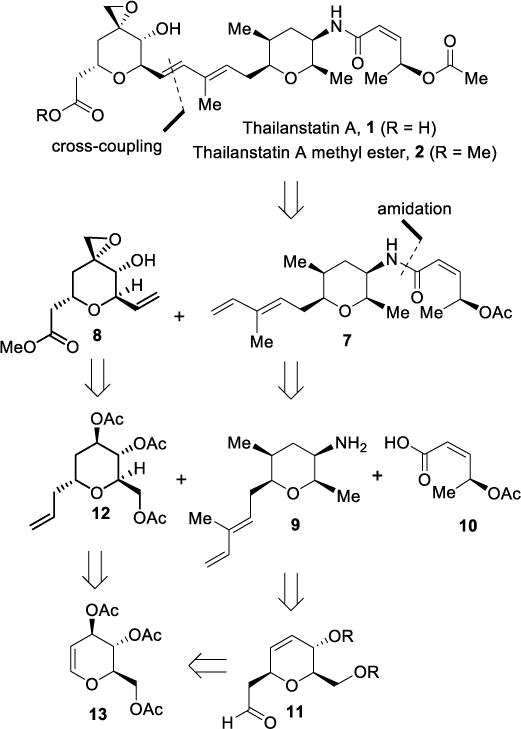
Retrosynthetic analysis of thailanstatin A.
Our synthesis of functionalized amine 9 from tri-O-acetyl-D-glucal is shown in Scheme 2. The preparation of vinyl ether derivative 14 was accomplished in multigram scale from 13 as reported in the literature.37,38 The three-step reaction sequence involved (1) saponification with K2CO3 in MeOH; (2) reaction of the resulting triol with di-t-butylsilyl triflate and pyridine to provide the corresponding silyl acetal and (3) reaction of the resulting allylic alcohol with an excess of ethyl vinyl ether in the presence of Hg(OAc)2 to provide 13 in 54% yield over 3-steps. Vinyl ether 14 was dissolved in toluene and the resulting solution was heated in a sealed tube at 190 °C for 12 h to furnish aldehyde 15 in 95% yield. Wittig olefination of aldehyde 15 with 2-(triphenylphosphoranylidene)propanal in toluene at 110 °C for 12 h furnished the corresponding α,β-unsaturated aldehyde. Subsequent Wittig olefination of this aldehyde with methylenetriphenylphosphorane, generated from the reaction of Ph3PCH3Br and tBuOK in THF at 0 °C, afforded diene 16 in 76% yield over 2-steps. To install the C-17 methyl group, compound 16 was subjected to tetrabutylammonium fluoride (TBAF) in THF at 0 °C to 23 °C for 12 h to provide the corresponding diol. Reaction of this diol with 2,4,6-triisopropylbenzenesulfonyl chloride (TPSCl) in pyridine at 0 °C to 23 °C for 24 h furnished sulfonate derivative 17 in 88% yield over 2-steps.39 Sulfonate 17 was reduced with LAH in THF at 0 °C to 65 °C for 1.5 h to furnish the reduced product 18 in 95% yield.
Scheme 2.
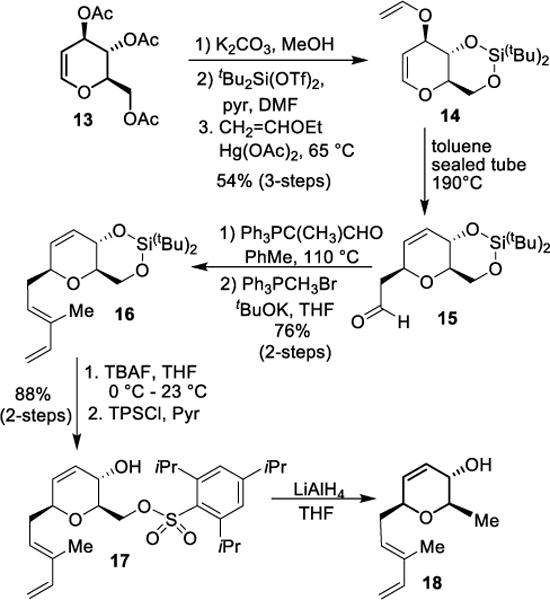
Stereoselective synthesis of allylic alcohol 18.
The synthesis of diene subunit 7 is shown in Scheme 3. For the introduction of the C14 methyl group, we planned to utilize a Cu(I)-mediated 1,4-addition developed by us previously.31 Accordingly, alcohol 18 was oxidized with Dess-Martin periodinane in the presence of NaHCO3 in CH2Cl2 at 0 °C to 23 °C for 1.5 h to provide the corresponding enone. Reaction of this enone with the CuBr•SMe2 complex at −78 °C for 2 h resulted in pyranone 19 in 64% yield over 2-steps. Pyranone 19 was obtained as a single diastereomer by 1H-NMR and 13C-NMR analysis. The high degree of diastereoselectivity can be rationalized based upon the conformation of the enone in transition state model 20. Here, axial attack by the cuprate is favored due to coordination of the Cu-complex to the endocyclic pyranone oxygen on the top face. Thus, the equatorial attack is clearly less favored due to developing non-bonded interactions with the nucleophile as shown. To install the C16 amine functionality, we planned a substrate-controlled stereoselective reduction that we previously carried out on a similar system.31 Thus, reduction of ketone 19 with NaBH(OAc)3 and ammonium acetate in the presence of trifluoroacetic acid (TFA) at 23 °C for 2 h afforded amine 9 as a mixture of diastereomers (8.5:1 by 1H-NMR analysis).40 The amine mixture was reacted with known31 optically active acid 10 using HATU in the presence of diisopropylethylamine (DIPEA) to provide amide derivative 7 in 85% yield.
Scheme 3.
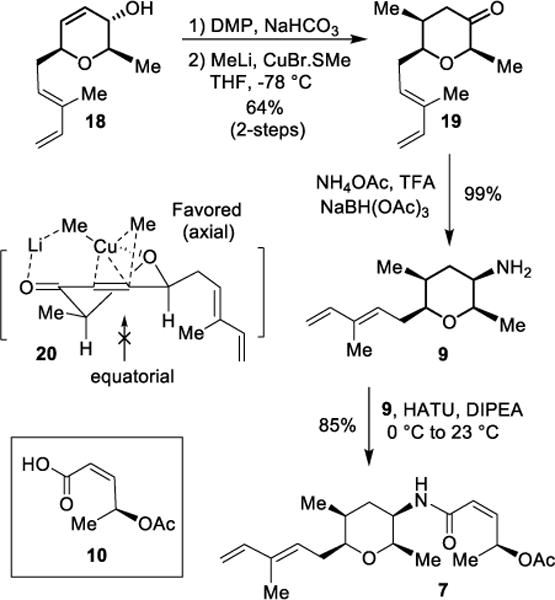
Stereoselective 1,4-addition and synthesis of amide 7.
Our next synthetic plan was to develop an enantioselective route to the other functionalized tetrahydropyran derivative 8. Our initial attempt for the synthesis of this epoxyalcohol subunit 8 is shown in Scheme 4. Tri-O-acetyl-D-glucal 13 was converted to the corresponding methyl acetal by reaction with CeCl3•7H2O and NaI in MeOH as described by Yadav and co-workers.41 Reaction of the resulting acetal with allyltrimethylsilane in the presence of TMSOTf in acetonitrile at 23 °C provided allyl derivative 22 diastereoselectively (10:1 mixture).42 The mixture was separated by silica gel chromatography. Treatment of triacetate 22 with K2CO3 in MeOH at 23 °C for 4 h afforded the corresponding triol. Reaction of the triol with p-anisaldehyde dimethyl acetal in the presence of a catalytic amount of CSA in DMF at 23 °C for 2 h afforded benzylidene acetal 23 selectively in 85% yield over 2-steps. DIBAL-H reduction of 24 in CH2Cl2 at −15 °C for 12 h provided the corresponding PMB-protected alcohol. Selective protection of the primary alcohol as a TBS-ether with TBSCl and imidazole in the presence of DMAP in CH2Cl2 at 23 °C furnished protected allyl derivative 24 in 86% yield over 2-steps. Allyl derivative 24 was converted to methyl ester derivative 25 by exposure to ozonolytic conditions in a mixture of 2.5 M NaOH solution in MeOH and CH2Cl2 at −78 °C.43 This methyl ester 25 was isolated in 36% yield. The alcohol functionality was oxidized with Dess-Martin periodinane and the resulting ketone was reacted with methylenetriphenylphosphorane at 0 °C in THF, to provide olefin 26 in 72% yield over 2-steps.
Scheme 4.
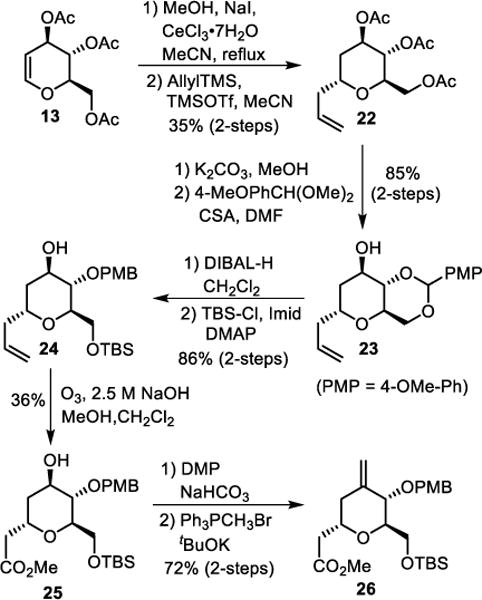
Synthesis of methyl ester derivative 26
Conversion of olefin 26 to epoxy alcohol derivative 8 is shown in Scheme 5. TBS-ether 26 was exposed to TBAF in THF at 23 °C to provide the corresponding alcohol. Oxidation of this primary alcohol to the corresponding aldehyde was accomplished by reaction with Dess-Martin periodinane. Wittig-olefination of the resulting aldehyde with methylenetriphenylphosphorane at 0 °C provided diene derivative 27 in 33% yield over 3-steps. For the directed epoxidation, the PMB-group was removed using DDQ in a phosphate buffer (pH 7) at 0 °C for 3 h. The resulting allylic alcohol was reacted with a catalytic amount (20 mol %) of VO(acac)2 in the presence of t-butyl hydroperoxide in CH2Cl2 at 0 °C to 23 °C for 2 h to provide epoxide 7 as a single diastereomer by 1H-NMR analysis.44
Scheme 5.
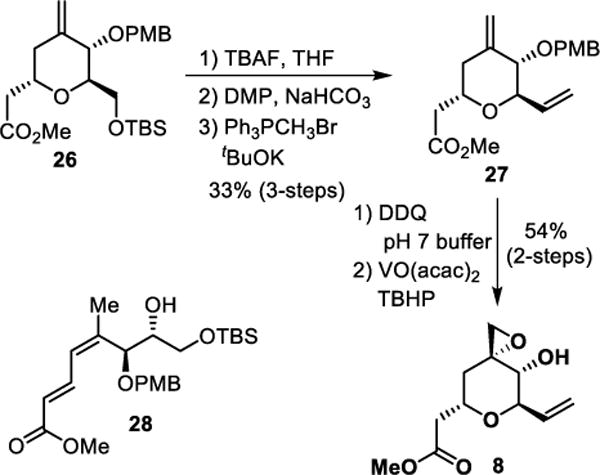
Stereoselective synthesis of epoxy alcohol 8.
Overall, the route provided access to the epoxy alcohol subunit 8, however, there were major drawbacks for this route with one being the conversion of olefin 24 to ester 25 which provided only 36% yield under optimized conditions. Additionally, the Wittig olefination step for the conversion of 25 to 26 provided very low yields and other side products. When the reaction was scaled up, one of the major side products being the formation of conjugated ester 28. Thus, we turned to explore an alternate route utilizing homoallyl derivative 23 and also planned to install the carboxylic acid functionality at a later stage.
Our optimized route to epoxy alcohol 8 is shown in Scheme 6. Ozonolytic cleavage of olefin 23 followed by reduction of the resulting aldehyde with NaBH4 provided the corresponding alcohol. The alcohol was protected as a TBDPS-ether by reaction with TBDPS-Cl and imidazole in CH2Cl2 at 23 °C for 1 h, providing silyl ether derivative 29 in 73% yield over 2-steps. Oxidation of the alcohol 29 with a catalytic amount of TPAP (10 mol %) in the presence of NMO and 4Å MS in CH2Cl2 at 23 °C for 30 min provided the corresponding ketone. Peterson olefination of this ketone was carried out by addition of TMSCH2MgCl in ether at 0 °C for 30 min followed by addition of KHMDS in THF at 0 °C to 23 °C for 1.5 h. Olefin 30 was obtained in 68% yield over 3-steps.46,47 The Peterson olefination step provided convenient access to olefin 30 in a reproducible yield during scale-up of these reactions. DIBAL-H reduction of 30 in PhCH3 at −78 °C to 0 °C for 1 h afforded the PMB-ether derivative 31 regioselectively in 69% yield. Oxidation of the primary alcohol using TPAP in the presence of NMO as described above, provided the corresponding aldehyde. Wittig olefination of the aldehyde furnished diene derivative 32 in 31% yield over 2-steps. Diene 32 was converted to methyl ester derivative 27 in a three-step sequence involving, (i) removal of the TBDPS-ether by exposure to TBAF in THF at 23 °C for 2 h; (ii) oxidation of the resulting alcohol with TEMPO in the presence of BAIB in a mixture (1:1) of CH2Cl2 and H2O and (iii) esterification of the resulting acid with TMSCHN2 in a mixture (3:2) of toluene and MeOH at 23 °C for 2 h. Methyl ester 27 was obtained in 84% yield over 3-steps. It was converted to epoxy alcohol 8 as described in Scheme 4.
Scheme 6.

An alternative synthesis of epoxy alcohol.
With the stereoselective syntheses of functionalized diene subunit 6 and epoxy alcohol subunit 8, we explored cross metathesis as shown in Scheme 7.48,49 The reaction was carried out under optimized conditions as follows. A solution of epoxy alcohol 8 and Grubbs II catalyst was prepared in CH2Cl2 under an argon atmosphere. In a separate flask, to a stirred solution of diene 7 in CH2Cl2, a one-third portion of epoxy alcohol 8 and the catalyst mixture was added and the resulting mixture was heated to 55 °C for 1.5 h. Two other portions of epoxy alcohol and catalyst were successively added to the reaction mixture at 55 °C in 1.5 h intervals. The reaction was carried out for a total of 6 h at 55 °C. After this period, the reaction was cooled down to 23 °C and concentrated. The resulting residue was purified by silica gel chromatography over silica gel to provide the methyl ester derivative of thailanstatin A, (2) in 46% yield. The 1H and 13C-NMR of our synthetic thailanstatin A methyl ester are in full agreement with the reported spectra of synthetic thailanstatin A methyl ester by Nicolaou and co-workers.36 To complete the synthesis of thailanstatin A, we were required to hydrolyze the methyl ester functionality. Our subsequent attempts to hydrolyze the methyl ester under various conditions including trimethyltin hydroxide however, resulted in decomposition to a mixture of unidentified products.50 Since thailanstatin A methyl ester has been reported to have significantly better cytotoxic properties than the acid, we sought to test its activity as a splicing inhibitor in vitro.
Scheme 7.
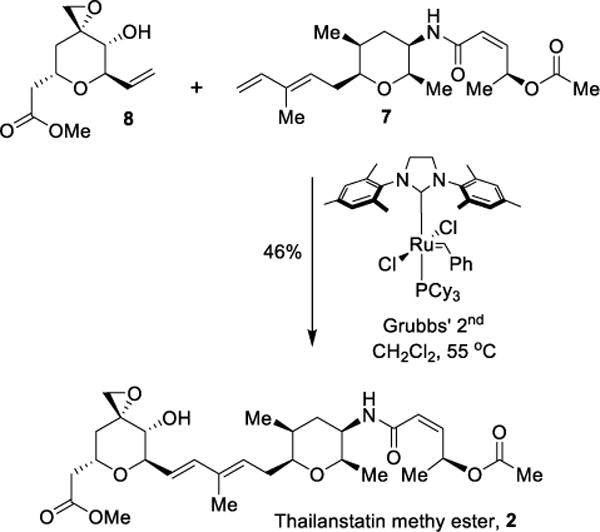
Synthesis of thailanstatin methyl ester.
(III) Biological Activity Studies
The activity of thailanstatin methyl ester (2) was measured in an in vitro splicing assay in which a synthetic pre-mRNA substrate is incubated in nuclear extract from HeLa cells with increasing concentrations of the compound. We determined splicing efficiency by separating the RNA substrate and products by denaturing PAGE and quantifying percent of pre-mRNA converted to mRNA relative to a DMSO control, which does not affect splicing. Compound (2) inhibits in vitro splicing in a dose dependent manner, with an IC50 of approximately 0.4 μM (Figure 2a and 2c). We also used native gel analysis of the same in vitro splicing reactions to examine the effect of (2) on spliceosome assembly. Spliceosomes assemble on pre-mRNA substrates via an ordered series of complexes with differing mobility in native gel electrophoresis. H/E and A complexes represent early intermediates that convert to B and then to C complex, in which splicing reaction is catalyzed. All of the spliceosome complexes appear in splicing reactions with DMSO, but spliceosome assembly is halted at an A-like complex in the presence of increasing amounts of (2) (Figure 2b).The block in complex assembly correlates with los of splicing catalysis, and is consistent with the activity other compounds that target SF3B1, such as spliceostatin A.
Figure 2.
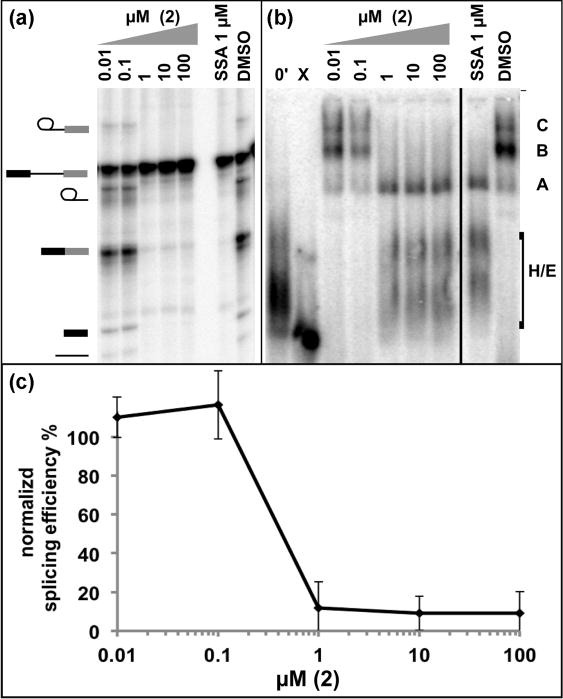
In vitro splicing analysis of thailanstatin methyl ester (2) (a) Denaturing gel analysis of radiolabeled RNA isolated after a 30 minute incubation in splicing reactions including compounds and the indicated concentration. DMSO (1%) and spliceostatin A (SSA) were included as controls. Identities of bands are schematized to the left as (from top to bottom) lariat intermediate, pre-mRNA, lariat intron product, mRNA product, 5′ exon intermediate, and linearized intron product. (b) Native gel analysis of aliquots of the splicing reactions shown in (a) separated under native conditions. The identity of splicing complexes is denoted with assembly occurring in the following order: H/E → A → B → C. (c) Splicing efficiency normalized to DMSO control vs. inhibitor concentration for the splicing reactions shown in (a).
Conclusion
We have accomplished an enantioselective synthesis of thailanstatin A methyl ester and carried out an evaluation of its in vitro splicing activity. The convergent synthesis of thailanstatin A methyl ester involved construction of both highly functionalized tetrahydropyran rings utilizing commercially available inexpensive tri-O-acetyl-D-glucal as the key starting material. The synthesis of tetrahydropyran segment 9 was carried out using a highly stereoselective Claisen rearrangement to set the C13 stereocenter. Installation of the C14 methyl center was accomplished by a Cu(I)-mediated conjugate addition of MeLi. Selective formation of a triisopropylbenzene sulfonate derivative of the C17 primary alcohol, followed by LAH reduction installed the C17 methyl center. Stereoselective construction of epoxy ester segment 8 was carried out by a highly stereoselective anomeric C-allylation of tri-O-acetyl-D-glucal derived methyl glycoside, a Peterson olefination to install the C5 methylene group and a vanadium(II)-catalyzed highly selective allylic epoxidation to install the C5 oxirane ring. A cross metathesis of two segments using Grubbs’ 2nd generation catalyst afforded thailanstatin A methyl ester in good yield. Our attempted hydrolysis of the methyl ester under selective hydrolytic conditions resulted in the decomposition of the product.
Since the methyl ester was reported to have significantly potent cytotoxic activity26, we carried out in vitro splicing studies of the synthetic thailanstatin A methyl ester. The methyl ester derivative showed and IC50 value of ~0.4 μM, which is close to the value reported for natural product thalanstatin A (~0.65 μM).25 Thailanstatin A methyl ester blocks spliceosome assembly at an A-like complex like other compounds that target SF3B1, suggesting that it targets the spliceosome by a similar mechanism.33 Further studies of thailanstatin A derivatives are in progress in our laboratory.
Experimental Section
General Experimental Details
Those reactions which required anhydrous conditions were carried out under an argon atmosphere using oven-dried glassware (120 °C). All chemicals and reagents were purchased from commercial suppliers and used without further purification. Anhydrous solvents were obtained as follows: anhydrous tetrahydrofuran and diethyl ether were distilled from sodium metal under argon, anhydrous dichloromethane was dried via distillation from CaH2 immediately prior to use under argon, and anhydrous methanol and ethanol were distilled from activated magnesium under argon. All other solvents were reagent grade. TLC analysis was conducted using glass-backed Thin-Layer Silica Gel Chromatography Plates (60 Å, 250 μm thickness, F-254 indicator). Flash chromatography was performed using 230–400 mesh, 60 Å pore diameter silica gel. 1H NMR spectra were recorded at 400, 500, or 800 MHz. 13C NMR spectra were recorded at 100 or 150 MHz. Chemical shifts are reported in parts per million and are referenced to the deuterated residual solvent peak. NMR data is reported as: δ value (chemical shift, J-value (Hz), integration, where s = singlet, d = doublet, t = triplet, q = quartet, brs = broad singlet). IR spectra were recorded on a Varian 2000 Infrared spectrophotometer and are reported as cm−1. LRMS and HRMS spectra were recorded at the Purdue University Department of Chemistry Mass Spectrometry Center. Melting points were measured on a melting point apparatus and was uncorrected.
(4aR,8R,8aS)-2,2-Di-tert-butyl-8-(vinyloxy)-4,4a,8,8a-tetrahydropyrano[3,2-d][1,3,2]diox asiline (14)
To a solution of glucal 13 (10 g, 36.73 mmol, 1 equiv) in MeOH (40 mL) was added K2CO3 (508 mg, 3.67 mmol, 0.1 equiv) at room temperature and the reaction was set to stir. After 12 h, the reaction was concentrated and the crude residue was purified by column chromatography (5% MeOH in EtOAc) to give 5.20 g (97% yield) of the corresponding triol derivative as a white solid. 1H NMR (300 MHz, CD3OD-d) δ (ppm): 6.34 (dd, J = 6.1, 1.7 Hz, 1H), 4.67 (dd, J = 6.0, 2.3 Hz, 1H), 4.10 (dt, J = 7.1, 2.0 Hz, 1H), 3.93 – 3.64 (m, 2H), 3.55 (dd, J = 9.6, 7.1 Hz, 1H), 3.33 – 3.28 (m, 1H).
To a solution of the above triol derivative (5.20 g, 35.60 mmol, 1 equiv) dissolved in dry DMF (15 mL) was added pyridine (14.4 mL, 178.00 mmol, 5 equiv). The reaction was cooled to −30 °C and tBu2Si(OTf)2 (12.70 mL, 39.14 mmol, 1.1 equiv) was added dropwise. The reaction was slowly warmed to room temperature. After 1.5 h, the reaction was diluted with EtOAc and the organic layer was washed with a 10% solution of CuSO4 (×2), water (×3) and brine (×3). The combined organic layer was dried over anhydrous Na2SO4 and the crude product was purified by column chromatography (10% EtOAc/Hexanes) to give 8.67 g (85% yield) of the resulting dioxasiline derivative as a white solid. Experimental data are consistent with the data reported in literature1. 1H NMR (300 MHz, CDCl3) δ (ppm): 6.27 (ddd, J = 6.1, 1.9, 0.5 Hz, 1H), 4.76 (dd, J = 6.1, 1.9 Hz, 1H), 4.36 – 4.23 (m, 1H), 4.23 – 4.05 (m, 1H), 4.01 – 3.77 (m, 3H), 2.35 (br, 1H), 1.07 (s, 9H), 1.00 (s, 9H).
To a solution of the alcohol (1.66 g, 5.79 mmol, 1 equiv) in ethyl vinyl ether (7.5 mL), in an oven dried sealed tube was added Hg(OAc)2 (551 mg, 1.73 mmol, 0.3 equiv). The mixture was heated to 65 °C, and Hg(OAc)2 (551 mg, 1.73 mmol, 0.3 equiv) was added every 24 h for an additional four times. After five days, the reaction was diluted with EtOAc and the organic layer was washed with water (×2), brine and then dried over anhydrous Na2SO4. Purification by column chromatography (2% EtOAc/Hexanes) gave 1.18 g (65% yield) of vinyl ether 14 as a white solid. Experimental data are consistent with the data reported in literature1. 1H NMR (300 MHz, CDCl3) δ (ppm): 6.53 (dd, J = 14.0, 6.5 Hz, 1H), 6.38 – 6.24 (m, 1H), 4.78 (dd, J = 6.1, 2.0 Hz, 1H), 4.46 – 4.34 (m, 2H), 4.25 – 4.07 (m, 2H), 4.07 – 3.93 (m, 2H), 3.87 (dt, J = 10.2, 5.1 Hz, 1H), 1.06 (s, 9H), 1.00 (s, 9H).
2-((4aR,6S,8aS)-2,2-Di-tert-butyl-4,4a,6,8a-tetrahydropyrano[3,2-d][1,3,2]dioxasilin-6-yl)acetaldehyde (15)
A solution of vinyl ether 14 (4.20 g, 13.46 mmol, 1 equiv) in toluene (40 mL) was heated at 190 °C in a sealed tube. After 12 h, the reaction was concentrated and the resulting residue was purified by column chromatography (10% EtOAc/Hexanes) to give 4.00 g (95% yield) of aldehyde 15 as a white solid. Experimental data are consistent with the data reported in literature1. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.75 (t, J = 2.1 Hz, 1H), 5.93 (d, J = 10.4 Hz, 1H), 5.64 (dt, J = 10.4, 1.8 Hz, 1H), 4.72 (m, 1H), 4.39 (m, 1H), 4.16 (dd, J = 10.0, 5.1 Hz, 1H), 3.84 (t, J = 10.2 Hz, 1H), 3.54 (ddd, J = 10.2, 8.5, 5.0 Hz, 1H), 2.58 (d, J = 2.1 Hz, 1H), 2.56 (d, J = 2.1 Hz, 1H), 1.05 (s, 9H), 0.99 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm): 200.6, 131.4, 128.0, 74.8, 71.1, 70.1, 67.2, 48.4, 27.6, 27.2, 22.8, 20.2.
(4aR,6S,8aS)-2,2-di-tert-Butyl-6-((E)-3-methylpenta-2,4-dien-1-yl)-4,4a,6,8a-tetrahydrop yrano[3,2-d][1,3,2]dioxasiline (16)
To a solution of aldehyde 15 (3.35 g, 10.70 mmol, 1 equiv) in dry toluene (70 mL) was added 2-(triphenylphosphoranylidene)propanal (4.08 g, 12.80 mmol, 1.2 equiv) and the reaction was set to stir at 110 °C. After 12 h, the reaction was concentrated and the crude residue was purified by column chromatography (4.5% EtOAc/Hexanes) to give 3.40 g (90% yield) of the resulting α,β-unsaturated aldehyde as a colorless oil. [α]D20 = −58.6 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3) δ (ppm): 9.41 (s, 1H), 6.52 (t, J = 7.0 Hz, 1H), 5.93 (d, J = 10.0 Hz, 1H), 5.59 (d, J = 10.3 Hz, 1H), 4.49 – 4.32 (m, 2H), 4.17 (dd, J = 9.9, 5.0 Hz, 1H), 3.84 (t, J = 10.1 Hz, 1H), 3.52 (ddd, J = 10.5, 8.3, 5.0 Hz, 1H), 2.68 – 2.44 (m, 2H), 1.74 (s, 3H), 1.05 (s, 9H), 0.99 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm): 195.0, 149.1, 141.2, 131.4, 128.4, 74.8, 74.1, 70.2, 67.2, 34.7, 27.6, 27.2, 22.8, 20.2, 9.6; LRMS-ESI (+) m/z 375.2 [M+Na]+.
To a suspension of methyl triphenylphosphonium bromide (5.65 g, 15.81 mmol, 2.5 equiv) in dry THF (100 mL) at 0 °C was added tBuOK (15.81 mL, 15.81 mmol, 2.5 equiv, 1 M in THF). The mixture was stirred at 0 °C for 30 min and then a solution of the above aldehyde (2.23 g, 6.33 mmol, 1 equiv) in dry THF (50 mL) was added via cannula and the reaction was slowly warmed to room temperature. After 2 h, the reaction was quenched with H2O, extracted with EA (×3), washed with brine and dried over Na2SO4. Purification by column chromatography (4.5% EtOAc/Hexanes) gave 1.86 g (84% yield) of diene 16 as a pale-yellow oil. [α]D20 = + 66.1 (c 9.0, CHCl3); 1H NMR (300 MHz, CDCl3) δ (ppm): 6.37 (dd, J = 17.4, 10.7 Hz, 1H), 5.86 (d, J = 10.4 Hz, 1H), 5.61 (dt, J = 10.4, 2.0 Hz, 1H), 5.49 (t, J = 6.8 Hz, 1H), 5.11 (d, J = 17.4 Hz, 1H), 4.96 (d, J = 10.8 Hz, 1H), 4.40 (m, 1H), 4.26 (m, 1H), 4.18 (dd, J = 10.0, 5.1 Hz, 1H), 3.87 (t, J = 10.2 Hz, 1H), 3.50 (ddd, J = 10.5, 8.4, 5.0 Hz, 1H), 2.37 (t, J = 6.9 Hz, 2H), 1.73 (s, 3H), 1.06 (s, 9H), 0.99 (s, 9H); 13C NMR (75 MHz, CDCl3) δ (ppm): 141.2, 136.3, 130.2, 129.2, 127.3, 111.3, 75.3, 74.8, 70.4, 67.4, 34.4, 27.7, 27.3, 22.9, 20.3, 12.2; LRMS-ESI (+) m/z 351.2 [M+H]+.
((2R,3S,6S)-3-Hydroxy-6-((E)-3-methylpenta-2,4-dien-1-yl)-3,6-dihydro-2H-pyran-2-yl) methyl2,4,6-triisopropylbenzenesulfonate (17)
To a solution of diene 16 (1.86 g, 5.31 mmol, 1 equiv) dissolved in dry THF (40 mL) at 0 °C was added TBAF (10.60 mL, 10.60 mmol, 2 equiv, 1 M in THF) and the reaction was warmed to room temperature. After 12 h, the reaction was diluted with H2O, extracted with EtOAc (×3), washed with brine and dried over Na2SO4. Purification by column chromatography (50% EtOAc/Hexanes) gave 1.08 g (97% yield) of the corresponding diol as a colorless oil. [α]D20 = + 99.8 (c 6.9, CHCl3); 1H NMR (300 MHz, CDCl3) δ (ppm): 6.35 (dd, J = 17.4, 10.7 Hz, 1H), 5.84 – 5.63 (m, 2H), 5.48 (t, J = 6.9 Hz, 1H), 5.10 (d, J = 17.4 Hz, 1H), 4.95 (d, J = 10.6 Hz, 1H), 4.26 – 4.06 (m, 2H), 3.94 – 3.69 (m, 2H), 3.40 – 3.30 (m, 1H), 3.25 (br, 1H), 2.90 (br, 1H), 2.45 – 2.27 (m, 2H), 1.72 (s, 3H); 13C NMR (75 MHz, CDCl3) δ (ppm): 141.0, 136.2, 130.6, 129.3, 127.1, 111.3, 78.9, 74.4, 64.1, 63.1, 34.3, 12.1; HRMS-ESI (+) m/z calc’d for C12H19O3 [M + H]+: 211.1334, found 211.1328.
To a solution of the above diol (1.14 g, 5.4 mmol, 1 equiv) in dry DCM (10 mL) and dry pyridine (6 mL) at 0 °C was added 2,4,6-triisopropylbenzenesulfonyl chloride (2.13 g, 7.03 mmol, 1.3 equiv). The reaction was heated at 45 °C for 4 h, then cooled to room temperature and another portion of 2,4,6-triisopropylbenzenesulfonyl chloride (0.49 g, 1.62 mmol, 0.3 equiv) was added. After stirring at 45 °C for another 12 h, the reaction was diluted with H2O and DCM, extracted with DCM (×3), washed with 1 N HCl (×2), satd. NaHCO3 (×2) and brine (×3) and dried over Na2SO4. Purification by column chromatography (15 % EtOAc/Hexanes) provided 1.80 g (97% yield) of sulfonate 17 as a white amorphous solid. [α]D20 = + 44.0 (c 4.4, CHCl3); 1H NMR (300 MHz, CDCl3) δ (ppm): 7.18 (s, 2H), 6.34 (dd, J = 17.4, 10.7 Hz, 1H), 5.74 (q, J = 10.2 Hz, 2H), 5.44 (t, J = 7.3 Hz, 1H), 5.10 (d, J = 17.5 Hz, 1H), 4.95 (d, J = 10.7 Hz, 1H), 4.35–4.22 (m, 2H), 4.20 – 4.08 (m, 5H), 3.52 (ddd, J = 8.5, 5.3, 2.5 Hz, 1H), 2.91 (quint, J = 6.9 Hz, 1H), 2.34 – 2.29 (m, 2H), 1.71 (s, 3H), 1.28 – 1.23 (m, 18H); 13C NMR (75 MHz, CDCl3) δ (ppm): 153.7, 150.8, 141.2, 136.3, 131.1, 129.3, 128.7, 127.1, 123.8, 111.4, 74.7, 69.0, 63.8, 60.6, 34.5, 34.2, 29.9, 25.0, 23.8, 14.4, 12.2; HRMS-ESI (+) m/z calc’d for C27H41O5S [M+H]+: 477.2675, found 477.2671.
(2R,3S,6S)-2-Methyl-6-((E)-3-methylpenta-2,4-dien-1-yl)-3,6-dihydro-2H-pyran-3-ol (18)
To a suspension of LiAlH4 (716 mg, 18.90 mmol, 5 equiv) in dry THF (70 mL) at 0 °C was slowly added a solution of sulfonate 17 (1.80 g, 3.80 mmol, 1 equiv) in dry THF (20 mL) via cannula. The reaction was heated at 65 °C for 1 h, then cooled to 0 °C and quenched with EtOAc. A 1 N NaOH solution was carefully added to the mixture and the crude product was filtered through celite. The filtrate was dried over Na2SO4 and purification by column chromatography (30% EtOAc/Hexanes) furnished 700 mg (95% yield) of dihydropyran 18 as a colorless oil. [α]D20 = + 115.7 (c 8.4, CHCl3); 1H NMR (300 MHz, CDCl3) δ (ppm): 6.37 (dd, J = 17.2, 10.8 Hz, 1H), 5.77 – 5.68 (m, 2H), 5.51 (t, J = 7.3 Hz, 1H), 5.10 (d, J = 17.3 Hz, 1H), 4.95 (d, J = 10.7 Hz, 1H), 4.13 (m, 1H), 3.85 (m, 1H), 3.33 (m, 1H), 2.45 – 2.29 (m, 2H), 1.98 (br, 1H), 1.73 (s, 3H), 1.32 (d, J = 6.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ (ppm): 141.2, 136.2, 131.2, 129.3, 127.4, 111.3, 75.7, 74.4, 69.7, 34.5, 18.7, 12.2; HRMS-ESI (+) m/z calc’d for C12H19O2 [M + H]+: 195.1385, found 195.1381.
(2R,5S,6S)-2,5-Dimethyl-6-((E)-3-methylpenta-2,4-dien-1-yl)dihydro-2H-pyran-3(4H)-one (19)
To a solution of 18 (350 mg, 1.80 mmol, 1 equiv) in dry DCM (20 mL) at 0 °C was added DMP (1.15 g, 2.70 mmol, 1.5 equiv) and the reaction was slowly warmed to room temperature. After stirring at room temperature for 1.5 h, the reaction was quenched with a 1:1 mixture of satd. NaHCO3 and satd. Na2S2O3, extracted with DCM (×3), washed with brine and dried over Na2SO4. Purification by column chromatography (10% EtOAc/Hexanes) gave 260 mg (75% yield) of the corresponding enone as a colorless oil. [α]D20 = + 88.1 (c 8.3, CHCl3); 1H NMR (300 MHz, CDCl3) δ (ppm): 6.87 (dd, J = 10.3, 1.5 Hz, 1H), 6.35 (dd, J = 17.4, 10.7 Hz, 1H), 6.05 (dd, J = 10.3, 2.5 Hz, 1H), 5.51 (t, J = 7.3 Hz, 1H), 5.11 (d, J = 17.4 Hz, 1H), 4.96 (d, J = 10.7 Hz, 1H), 4.43 – 4.31 (m, 1H), 4.09 (dd, J = 12.8, 6.4 Hz, 1H), 2.59 – 2.41 (m, 2H), 1.74 (s, 3H), 1.35 (d, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ (ppm): 196.7, 150.6, 140.7, 136.9, 126.7, 126.0, 111.8, 76.9, 73.9, 33.6, 15.5, 12.1; HRMS-ESI (+) m/z calc’d for C12H17O2 [M + H]+: 193.1229, found 193.1225.
To a suspension of CuBr·SMe2 (835 mg, 4.06 mmol, 3 equiv) in dry Et2O (12 mL) at −78 °C, was added MeLi (2.62 mL, 8.31 mmol, 6.2 equiv, 3.1 M solution) dropwise. The reaction was stirred at this temperature for 1 h and then a solution of the above enone (260 mg, 1.35 mmol, 1 equiv) in Et2O (4 mL) was added dropwise. The reaction then continued stirring at −78 °C. After 2 h, the reaction was quenched with H2O, extracted with EtOAc (×3), washed with brine and dried over Na2SO4. Purification by column chromatography (10% EtOAc/Hexanes) gave 240 mg (85% yield) of ketone 19 as a colorless oil in a 10:1 diastereomeric ratio. [α]D20 = −33.4 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ (ppm): 6.39 (dd, J = 17.2, 10.4 Hz, 1H), 5.50 (t, J = 7.2 Hz, 1H), 5.13 (d, J = 14.8 Hz, 1H), 4.98 (d, J = 10.8 Hz, 1H), 4.00 − 3.86 (m, 2H), 2.64 (dd, J = 15.2, 6.0 Hz, 1H), 2.48 (m, 1H), 2.37 − 2.27 (m, 3H), 1.78 (s, 3H), 1.28 (d, J = 6.4 Hz, 3H), 0.98 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 208.7, 141.2, 135.9, 127.9, 111.4, 79.5, 78.8, 46.7, 34.9, 31.4, 15.0, 13.1, 11.9; HRMS-ESI (+) m/z calc’d for C13H20O2Na [M+Na]+: 231.1361, found 231.1355.
(2R,3R,5S,6S)-2,5-Dimethyl-6-((E)-3-methylpenta-2,4-dien-1-yl)tetrahydro-2H-pyran-3-amine (9)
To a solution of ketone 19 (120 mg, 0.58 mmol, 1 equiv) in THF (12 mL) was added NH4OAc (888 mg, 11.52 mmol, 20 equiv), NaBH(OAc)3 (610 mg, 2.88 mmol, 5 equiv) and TFA (44 μL, 0.58 mmol, 1 equiv). The reaction was set to stir at room temperature. After 12 h, the reaction was quenched with satd. NaHCO3, extracted with DCM (×3), washed with brine and dried over Na2SO4. Purification by column chromatography (5% MeOH/DCM) afforded 77 mg (63% yield) of amine 9 as a colorless oil in an 8.5:1 diastereomeric ratio. [α]D20 = −7.0 (c 7.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 6.33 (dd, J = 17.4, 10.6 Hz, 1H), 5.43 (t, J = 7.2 Hz, 1H), 5.06 (d, J = 17.3 Hz, 1H), 4.91 (d, J = 10.7 Hz, 1H), 4.26 (brs, 2H), 3.56 (m, 1H), 3.45 (td, J = 7.1, 2.4 Hz, 1H), 3.39 (s, 1H), 2.83 (brs, 1H), 2.36 (m, 1H), 2.22 (m, 1H), 1.93 (m, 2H), 1.72 (s, 3H), 1.18 (d, J = 6.5 Hz, 3H), 1.07 (d, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ (ppm): 141.4, 135.6, 128.5, 111.0, 80.8, 76.0, 49.3, 37.1, 31.9, 28.9, 17.8, 14.9, 12.0; LRMS-ESI (+) m/z 210.2 [M + H]+.
(S,Z)-5-(((2R,3R,5S,6S)-2,5-Dimethyl-6-((E)-3-methylpenta-2,4-dien-1-yl)tetrahydro-2H-pyran-3-yl)amino)-5-oxopent-3-en-2-yl acetate (7)
To a stirred solution of (S,Z)-4-acetoxypent-2-enoic acid 10 (62 mg, 0.39 mmol, 1.2 equiv) in anhydrous acetonitrile (2 mL) at 0 °C was added HATU (150 mg, 0.39 mmol, 1.2 equiv) and DIPEA (230 μL, 1.32 mmol, 4 equiv). The resulting mixture was then transferred via cannula to a stirred solution of crude amine 9 (68 mg, 0.33 mmol, 1 equiv) in acetonitrile (2 mL) at 0 °C, rinsing with additional acetonitrile (1 mL). After stirring for 24 h at room temperature, the reaction was quenched by the addition of satd. NH4Cl and then diluted with EtOAc. The aqueous phase was extracted with EtOAc (×3), washed with brine and dried over Na2SO4. Purification by silica gel chromatography (25 % EtOAc/Hexanes) afforded 90 mg (78% yield) of amide derivative 7 as a colorless oil. [α]D20 = −58.6 (c 1.05, DCM); 1H NMR (400 MHz, CDCl3) δ (ppm): 6.36 (dd, J = 17.6, 10.8 Hz, 1H), 6.26 (dt, J = 13.6, 6.8 Hz, 1H), 6.00 (d, J = 8.8 Hz, 1H), 5.89 (dd, J = 11.6, 8.0 Hz, 1H), 5.70 (d, J = 11.6 Hz, 1H), 5.46 (t, J = 6.8 Hz, 1H), 5.10 (d, J = 17.6 Hz, 1H), 4.95 (d, J = 10.8 Hz, 1H), 3.94 (t, J = 3.2 Hz, 1H), 3.67 (dd, J = 6.4, 2.0 Hz, 1H), 3.54 (dt, J = 7.6, 2.8 Hz, 1H), 2.39 (m, 1H), 2.24 (m, 1H), 2.04 (s, 3H), 2.00 − 1.86 (m, 2H), 1.80 (m, 1H), 1.75 (s, 3H), 1.39 (d, J = 6.4 Hz, 3H), 1.15 (d, J = 6.4 Hz, 3H), 1.02 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm): 170.4, 164.8, 143.6, 141.3, 135.7, 128.1, 122.5, 111.1, 80.8, 76.0, 68.9, 47.1, 35.8, 31.9, 28.9, 21.2, 20.0, 17.8, 15.0, 11.9; FT-IR (neat) νmax = 3358, 2977, 2934, 1739, 1668, 1634, 1520, 1369, 1243, 1049, 1011 cm−1; HRMS-ESI (+) m/z calc’d for C20H31NO4Na [M +Na]+: 372.2151, found 372.2152.
(2R,3S,4R,6R)-2-(acetoxymethyl)-6-allyltetrahydro-2H-pyran-3,4-diyldiacetate (22)
To glucal 13 (10.0 g, 36.73 mmol, 1, equiv) in acetonitrile (100 mL) was sequentially added MeOH (7.4 mL, 183.65 mmol, 5 equiv), NaI (8.26 g, 55.1 mmol, 1.5 equiv) and CeCl3·7H2O (20.53 g, 55.1 mmol, 1.5 equiv). The reaction was set to reflux overnight at 100 °C. After 12 h, the brown solution was diluted with EtOAc and water. The crude product was extracted with EA (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (20% to 50% EtOAc/Hexanes) gave 6.06 g (54% yield) of methyl acetal as a dark oil.
To the above methyl acetal (6.06 g, 19.92 mmol, 1 equiv) in acetonitrile (60 mL) at 0 °C was added allyltrimethylsilane (9.5 mL, 59.76 mmol, 3 equiv) followed by dropwise addition of TMSOTf (1.8 mL, 9.96 mmol, 0.5 equiv) and the reaction was slowly warmed to room temperature. After 12 h, the black solution was slowly quenched was sat. NaHCO3 until the effervescence ceased. The crude product was extracted with EtOAc (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (20% to 30% EtOAc/Hexanes) provided 4.07 g (65% yield) of glucal 22 as a clear oil. 1H NMR (500 MHz, CDCl3) δ (ppm): 5.76 (m, 1H), 5.08–5.15 (m, 2H), 4.86 (t, J = 7.3 Hz, 1H), 4.37 (dd, J = 12.0, 6.2 Hz, 1H), 4.03–4.09 (m, 2H), 3.88–3.92 (m, 1H), 2.51 (m, 1H), 2.28 (m, 1H), 2.05–2.08 (m, 9H), 1.98 (dt, J = 13.6 4.6 Hz, 1H), 1.85 (ddd, J = 13.7, 8.9, 4.8 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ (ppm): 170.9, 170.2, 169.9, 133.9, 117.8, 70.8, 70.0, 68.9, 68.8, 62.2, 36.8, 32.2, 21.2, 21.0, 20.9; LRMS-ESI (+) m/z [M+Na]+: 337.0.
(4aR,6R,8R,8aS)-6-allyl-2-(4-methoxyphenyl)hexahydropyrano[3,2-d][1,3]dioxin-8-ol (23)
To a pale yellow solution of glucal 22 (2.99 g, 9.52 mmol, 1 equiv) in methanol (10 mL) was added potassium carbonate (132 mg, 0.95 mmol, 0.1 equiv) at room temperature. After 1 h, the orange solution was concentrated and filtered through a silica plug rinsing with 10% MeOH/EtOAc. The crude product was placed under high-vacuum overnight at 40 °C to remove any trace of methanol. The crude product was directly used for the subsequent reaction.
To the above crude triol (9.52 mmol, 1 equiv) and anisaldehyde dimethyl acetal (3.2 mL, 19.0 mmol, 2 equiv) in dry DMF (12 mL) was added CSA (442 mg, 1.90 mmol, 0.2 equiv) at room temperature. After 10 min, the reaction was neutralized with NEt3 (0.74 mL, 5.33 mmol). The crude product was extracted with EtOAc (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (30% to 70% EtOAc/Hexanes) gave 2.17 g (74% yield over 2-steps) of benzylidene acetal 23 as a white amorphous solid. [α]20D = +26.6 (c 0.92, CHCl3); 1H NMR (400 MHz, CDCl3) δ (ppm): 7.42 (d, J = 8.6 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 5.77 (m, 1H), 5.52 (s, 1H), 5.09–5.16 (m, 2H), 4.20 (dd, J = 10.0, 4.6 Hz, 1H), 4.02–4.12 (m, 2H), 3.81 (s, 3H), 3.67 (t, J = 10 Hz, 1H), 3.60 (td, J = 9.8, 4.6 Hz, 1H), 3.43 (t, J = 9.1 Hz, 1H), 2.60 (m, 1H), 2.53 (d, J = 2 Hz, 1H), 2.32 (m, 1H), 2.04 (ddd, J = 13.3, 5.2, 1.0 Hz, 1H), 1.89 (ddd, J = 13.4, 11.4, 6.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm): 160.4, 134.4, 130.0 127.7, 117.6, 113.9, 102.1, 84.7, 73.5, 69.5, 66.3, 64.2, 55.5, 36.2, 35.1; FT-IR (neat) νmax = 3315, 3224, 2940, 2892, 2870, 1615, 1519, 1253, 1102, 1033, 1002, 827 cm−1; HRMS-ESI (+) m/z calc’d for C17H22O5Na [M+Na]+: 329.13649, found 329.13217.
(2R,3S,4R,6R)-6-allyl-2-(((tert-butyldimethylsilyl)oxy)methyl)-3-((4-methoxybenzyl)oxy) tetrahydro-2H-pyran-4-ol (24)
To a solution of benzylidene acetal 23 (500 mg, 1.63 mmol, 1 equiv) in dry DCM (10 mL) at −15 °C was added DIBAL-H (7.30 mL, 7.30 mmol, 4.5 equiv, 1 M solution) over 30 min. After stirring at −15 °C for 12 h, the reaction was quenched with MeOH and satd. sodium potassium tartrate. The mixture was stirred for 2 h, extracted with DCM (×3), washed with brine and dried over Na2SO4. Purification by column chromatography (50% EtOAc/Hexanes) gave 477 mg (95% yield) of the resulting alcohol as a colorless oil. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.29 (d, J = 8.6 Hz, 2H), 6.90 (d, J = 8.6 Hz, 2H), 5.75 (m, 1H), 5.21 – 4.95 (m, 2H), 4.70 (d, J = 11.2 Hz, 1H), 4.61 (d, J = 11.2 Hz, 1H), 4.06 (m, 1H), 3.90 (d, J = 11.4 Hz, 1H), 3.81 (m, 5H), 3.55 – 3.45 (m, 1H), 3.28 (t, J = 8.9 Hz, 1H), 2.51 (m, 1H), 2.23 (m, 1H), 1.95 (m, 2H), 1.74 (m, 1H).
To the above alcohol (500 mg, 1.63 mmol, 1 equiv) in dry DCM (9 mL) at 0 °C was added TBS-Cl (221 mg, 1.47 mmol, 0.9 equiv), imidazole (199 mg, 2.93 mmol, 1.8 equiv) and DMAP (40 mg, 0.33 mmol, 0.2 equiv) and the reaction was slowly warmed to room temperature. After stirring for 5 h, the reaction was diluted with H2O and DCM, extracted with DCM (×3), washed with brine and dried over Na2SO4. Purification by column chromatography (30% EtOAc/Hexanes) gave 620 mg (90% yield) of silyl ether 24 as a colorless oil. [α]D20 +20.3 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.28 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 5.77 (m, 1H), 5.08 – 5.02 (m, 2H), 4.67 (d, J = 11.3 Hz, 1H), 4.59 (d, J = 11.3 Hz, 1H), 4.03 (m, 1H), 3.91 (m, 1H), 3.84 (dd, J = 5.5, 2.6 Hz, 2H), 3.80 (s, 3H), 3.51 (m, 1H), 3.35 (t, J = 7.6 Hz, 1H), 2.55 (d, J = 4.1 Hz, 1H), 2.44 (m, 1H), 2.20 (m, 1H), 1.89 (dt, J = 13.1, 4.3 Hz, 1H), 1.68 (ddd, J = 13.1, 9.8, 5.3 Hz, 1H), 0.92 (s, 9H), 0.09 (d, J = 4.8 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ (ppm): 159.5, 135.0, 130.7, 129.7, 116.9, 114.1, 79.4, 74.0, 73.7, 71.1, 68.4, 63.8, 55.4, 36.9, 34.8, 26.1, 18.5, −5.1, −5.4; LRMS-ESI (+) m/z 445.1.2 [M+Na]+.
Methyl 2-((2S,4R,5S,6R)-6-(((tert-butyldimethylsilyl)oxy)methyl)-4-hydroxy-5-((4-methoxybenz yl)oxy)tetrahydro-2H-pyran-2-yl)acetate (25)
To a solution of 24 (540 mg, 1.19 mmol, 1 equiv) in DCM (9 mL) was added NaOH (2.38 mL, 5.95 mmol, 5 equiv, 2.5 M in MeOH) which was then cooled to −78 °C. Ozone was bubbled through the reaction until the characteristic blue color developed. The reaction was then diluted with H2O and EtOAc, warmed to room temperature, extracted with EtOAc (×3), washed with brine and dried over Na2SO4. Purification by column chromatography (20% EtOAc/Hexanes) provided 209 mg (36% yield) of methyl ester 25 as colorless oil. [α]D20 +25.1 (c 1.0, CDCl3); 1H NMR (400 MHz, CDCl3) δ (ppm): 7.27 (d, J = 7.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 4.67 (d, J = 11.4 Hz, 1H), 4.57 (d, J = 11.4 Hz, 1H), 4.49 (m, 1H), 3.90 – 3.84 (m, 3H), 3.80 (s, 3H), 3.67 (s, 3H), 3.54 (m, 1H), 3.38 (t, J = 7.2 Hz, 1H), 2.72 – 2.66 (m, 2H), 2.46 (dd, J = 14.7, 6.3 Hz, 1H), 1.90 (dt, J = 13.4, 4.5 Hz, 1H), 1.75 (ddd, J = 13.4, 9.3, 5.1 Hz, 1H), 0.91 (s, 9H), 0.09 (d, J = 6.3 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ (ppm): 171.5, 159.6, 130.7, 129.7, 114.2, 78.7, 74.5, 73.5, 68.3, 68.0, 63.8, 55.4, 51.9, 38.2, 35.1, 26.1, 18.5, −5.0, −5.3; LRMS-ESI (+) m/z 477.2 [M+Na]+.
Methyl 2-((2S,5S,6R)-6-(((tert-butyldimethylsilyl)oxy)methyl)-5-((4-methoxybenzyl)oxy)-4-meth ylenetetrahydro-2H-pyran-2-yl)acetate (26)
To a solution of alcohol 25 (152 mg, 0.33 mmol, 1 equiv) in dry DCM (5 mL) at 0 °C, was added NaHCO3 (166 mg, 1.98 mmol, 6 equiv) and DMP (284 mg, 0.66 mmol, 2 equiv) and the reaction was slowly warmed to room temperature. After 1 h, the reaction was quenched with a 1:1 mixture of satd. NaHCO3 and satd. Na2S2O3. After vigorously stirring for 10 min, the mixture was extracted with DCM (×3), washed with brine and dried over Na2SO4. Purification by column chromatography (15% EtOAc/Hexanes) gave 141 mg (95% yield) of the corresponding ketone as a colorless oil. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.28 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 4.85 – 4.76 (m, 1H), 4.72 (d, J = 11.0 Hz, 1H), 4.38 (d, J = 11.0 Hz, 1H), 4.05 (d, J = 6.7 Hz, 1H), 3.89 – 3.83 (m, 1H), 3.83 – 3.80 (m, 2H), 3.78 (s, 3H), 3.66 (s, 3H), 2.78 – 2.58 (m, 2H), 2.58 – 2.38 (m, 2H), 0.88 (s, 9H), 0.05 (d, J = 3.9 Hz, 6H).
To a suspension of methyltriphenylphosphonium bromide (354 mg, 1.00 mmol, 3 equiv) in dry THF (2 mL) at 0 °C was added tBuOK (830 μL, 0.83 mmol, 2.5 equiv, 1 M in THF). After stirring at 0 °C for 1 h, a solution of the ketone (150 mg, 0.33 mmol, 1 equiv) in THF (1 mL) was added via cannula. After 10 min at 0 °C, the reaction was quenched with satd. NH4Cl, extracted with EA (×3), washed with brine and dried over Na2SO4. Purification by column chromatography (15% EtOAc/Hexanes) afforded 113 mg (76% yield) of olefin 26 as a colorless oil. [α]D20 +30.8 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ (ppm): 7.25 (d, J = 8.0 Hz, 2H), 6.85 (d, J = 7.0 Hz, 2H), 5.05 (d, J = 21.8 Hz, 2H), 4.56 (d, J = 11.0 Hz, 1H), 4.31 (d, J = 11.7 Hz, 1H), 4.08 (m, 1H), 3.86 – 3.79 (m, 5H), 3.69 – 3.67 (m, 5H), 2.63 (dd, J = 15.3, 7.1 Hz, 1H), 2.44 (dd, J = 15.3, 4.5 Hz, 1H), 2.38 – 2.24 (m, 2H), 0.82 (s, 9H), 0.00 (d, J = 6.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ (ppm): 171.7, 159.3, 140.7, 130.5, 129.6, 113.9, 113.8, 78.1, 75.9, 70.0, 61.4, 55.4, 51.8, 39.9, 36.6, 25.9, 18.3, −5.3; LRMS-ESI (+) m/z 473.1 [M+Na]+.
(4aR,6R,8R,8aS)-6-(2-((tert-butyldiphenylsilyl)oxy)ethyl)-2-(4-methoxyphenyl)hexahydr opyrano[3,2-d][1,3]dioxin-8-ol (29)
To a stirred solution of olefin 23 (2.74 g, 8.95 mmol, 1 equiv) in CH2Cl2/MeOH (1:1, 40 mL) at −78 °C, ozone was bubbled through until the characteristic blue color developed. The reaction was quenched with PPh3 (3.52 g, 13.43 mmol, 1.5 equiv) and warmed to 0 °C. After 30 min, NaBH4 (677 mg, 17.9 mmol, 2 equiv) was added. After another 30 min, the reaction was slowly quenched with satd. NH4Cl until the effervescence ceased. The crude product was extracted with DCM (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (60% ethyl acetate/hexanes to 100% EtOAc/Hexanes) gave 2.94 g (quantitative) of the corresponding alcohol as an amorphous white solid. [α]20D = +16.0 (c 0.56, CHCl); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.41 (d, J = 8.6 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 5.52 (s, 1H), 4.25–4.31 (m, 1H), 4.19 (dd, J = 9.7, 4.3 Hz, 1H), 4.06 – 3.98 (m, 1H), 3.80 (s, 3H), 3.74 (t, J = 6.0 Hz, 2H), 3.68 (t, J = 9.9 Hz, 1H), 3.61 (td, J = 9.8, 4.1 Hz, 1H), 3.43 (t, J = 9.0 Hz, 1H), 2.64 (brs, 1H), 2.20 (m, 1H), 1.94–1.98 (m, 2H), 1.85 (brs, 1H), 1.62 (m, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm): 160.4, 129.9, 127.7, 113.9, 102.1, 84.6, 72.0, 69.4, 66.4, 64.3, 60.3, 55.5, 36.3, 33.5; FT-IR (neat) νmax = 3415, 2958, 2926, 2852, 1612, 1588, 1517, 1464, 1380, 1302, 1250, 1102, 1035, 1006, 830, 728 cm−1; HRMS-ESI (+) m/z calc’d for C16H22O6Na [M+Na]+: 333.13141, found 333.12733.
To the above primary alcohol (2.78 g, 8.96 mmol, 1 equiv) in CH2Cl2 (45 mL) was added imidazole (1.20 g, 17.92 mmol, 2 equiv) at room temperature. The solution was cooled to 0 °C upon which TBDPS-Cl (2.1 mL, 8.06 mmol, 0.9 equiv) was added. After 30 min, the reaction was diluted with water and the crude product was extracted with CH2Cl2 (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (20% to 60% EtOAc/Hexanes) gave 2.78 g (76% yield) of silyl ether 29 as a white foam. [α]20D = +19.2 (c 2.3, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.69 – 7.66 (m, 4H), 7.44 – 7.37 (m, 8H), 6.90 (d, J = 8.8 Hz, 2H), 5.51 (s, 1H), 4.31 (m, 1H), 4.09 (dd, J = 10.3, 4.3 Hz, 1H), 4.01 – 3.96 (m, 1H), 3.81 (s, 3H), 3.78 – 3.62 (m, 3H), 3.43 – 3.41 (m, 2H), 2.46 (d, J = 2.2 Hz, 1H), 2.08 (m, 1H), 2.01 – 1.90 (m, 2H), 1.75 (m, 1H), 1.06 (s, 9H); 13C NMR (125 MHz, CDCl3) δ (ppm): 160.3, 135.7, 135.7, 133.8, 133.7, 130.0, 129.9, 129.8, 127.8, 127.8, 127.7, 113.9, 102.1, 84.8, 70.8, 69.5, 66.5, 64.1, 60.6, 55.5, 36.0, 34.0, 27.0, 19.3; FT-IR (neat) νmax = 3468, 3070, 2954, 2929, 2855, 1616, 1588, 1521, 1464, 1426, 1380, 1302, 1250, 1172, 1088, 1038, 999, 827, 739, 704, 616, 503 cm−1; HRMS-ESI (+) m/z calc’d for C32H40O6SiNa [M+Na]+: 571.24919, found 571.24933.
Tert-butyl(2-((4aR,6S,8aS)-2-(4-methoxyphenyl)-8-methylenehexahydropyrano[3,2-d][1, 3]dioxin-6-yl)ethoxy)diphenylsilane (30)
To a two-neck flask charged with flame-dried 4 Å MS was added alcohol 29 (1.06 g, 1.93 mmol, 1 equiv) in CH2Cl2 (8 mL) and NMO (452 mg, 3.86 mmol, 2 equiv) in CH2Cl2 (2 mL) via cannula at room temperature. After stirring for 20 min, TPAP (68 mg, 0.193 mmol, 0.1 equiv) was added to the reaction flask. After 30 min, the black mixture was filtered over celite, rinsing with CH2Cl2 and the filtrate was concentrated. Purification by flash chromatography (30% to 40% EtOAc/Hexanes) gave 863 mg (80% yield) of the corresponding ketone as a white foam. [α]20D = +30.3 (c 0.57, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.68 – 7.64 (m, 4H), 7.45 – 7.37 (m, 8H), 6.88 (d, J = 8.8 Hz, 2H), 5.52 (s, 1H), 4.70 (m, 1H), 4.31 (dd, J = 9.3, 1.1 Hz, 1H), 4.18 (dd, J = 9.8, 4.2 Hz, 1H), 3.82 – 3.73 (m, 6H), 3.67 (m, 1H), 2.96 (ddd, J = 14.3, 7.5, 1.4 Hz, 1H), 2.43 (dd, J = 14.3, 1.5 Hz, 1H), 1.98 (m, 1H), 1.76 (m, 1H), 1.05 (s, 9H); 13C NMR (125 MHz, CDCl3) δ (ppm): 200.2, 160.4, 135.7, 135.7, 133.5, 133.5, 130.0, 129.9, 129.3, 127.9, 127.9, 127.8, 113.8, 102.2, 83.8, 73.0, 69.9, 67.6, 59.8, 55.4, 45.4, 34.6, 29.9, 27.0, 19.3.; FT-IR (neat) νmax = 3070, 2959, 2928, 2856, 1736, 1615, 1516, 1464, 1428, 1250, 1129, 1111, 824, 704, 504 cm−1; HRMS-ESI (+) m/z calc’d for C32H38O6SiNa [M+Na]+: 569.23354, found 569.23345.
To the above ketone (2.30 g, 4.28 mmol, 1 equiv) in Et2O (16 mL) at 0 °C was added TMSCH2MgCl (6.0 mL, 6.0 mmol, 1.4 equiv, 1M in Et2O) dropwise. After 10 min, the reaction was quenched with satd. NH4Cl. The crude product was extracted with EtOAc (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (10% to 20% EtOAc/Hexanes) gave 2.40 g (88% yield) of the corresponding quaternary alcohol as a white foam. [α]20D = +24.1 (c 0.72, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.70 – 7.67 (m, 4H), 7.42 – 7.37 (m, 8H), 6.91 (d, J = 8.8 Hz, 2H), 5.55 (s, 1H), 4.21 (m, 1H), 4.13 (dd, J = 10.2, 5.0 Hz, 1H), 3.86 (m, 1H), 3.82 (s, 3H), 3.80 – 3.76 (m, 1H), 3.72 – 3.68 (m, 1H), 3.63 (t, J = 10.3 Hz, 1H), 3.37 (d, J = 9.2 Hz, 1H), 2.49 (m, 1H), 1.99 – 1.86 (m, 4H), 1.25 (d, J = 14.7, 1H), 1.05 (s, 9H), 0.89 (d, J = 14.9 Hz, 1H), 0.06 (s, 9H); 13C NMR (125 MHz, CDCl3) δ (ppm): 160.3, 135.8, 135.7, 134.1, 134.0, 130.3, 129.7, 129.7, 127.8, 127.7, 127.6, 113.8, 101.9, 84.5, 71.8, 69.8, 61.1, 60.9, 55.4, 40.2, 34.9, 30.0, 29.9, 27.0, 19.4, 0.6; FT-IR (neat) νmax = 3482, 2957, 2927, 2855, 1616, 1520, 1466, 1429, 1249, 1104, 836, 703, 501 cm−1; HRMS-ESI (+) m/z calc’d for C36H50O6Si2Na [M+Na]+: 657.30436, found 657.30479.
To the above alcohol (2.39 g, 3.76 mmol, 1 equiv) in freshly distilled THF (19 mL) at 0 °C was added KHMDS (9 mL, 4.52 mmol, 1.2 equiv, 0.5 M in PhMe) dropwise. The yellow solution was warmed to room temperature. After 1.5 h, the reaction was quenched with satd. NH4Cl. The crude product was extracted with EtOAc (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (10% to 20% EtOAc/Hexanes) afforded 1.96 g (96% yield) of olefin 30 as a viscous clear oil. [α]20D = +43.6 (c 0.75, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.69 – 7.66 (m, 4H), 7.46 – 7.37 (m, 8H), 6.90 (d, J = 8.8 Hz, 2H), 5.60 (s, 1H), 5.13 (d, J = 1.8 Hz, 1H), 4.85 (d, J = 1.9 Hz, 1H), 4.32 (m, 1H), 4.07 (dd, J = 10.3, 4.7 Hz, 1H), 3.97 (d, J = 9.4 Hz, 1H), 3.81 (s, 3H), 3.77 – 3.64 (m, 3H), 3.43 (td, J = 9.6, 4.7 Hz, 1H), 2.72 (dd, J = 13.8, 6.4 Hz, 1H), 2.25 (d, J = 13.8 Hz, 1H), 2.06 (m, 1H), 1.72 (m, 1H), 1.06 (s, 9H); 13C NMR (125 MHz, CDCl3) δ (ppm): 160.2, 139.9, 135.8, 135.7, 133.9, 133.8, 130.4, 129.8, 129.8, 127.8, 127.8, 127.7, 113.7, 107.5, 101.6, 80.7, 71.6, 70.0, 67.5, 60.5, 55.4, 38.2, 33.1, 29.9, 27.0, 19.3; FT-IR (neat) νmax = 3073, 2956, 2928, 2856, 1615, 1519, 1461, 1428, 1250, 1099, 1036, 824, 704, 504 cm−1; HRMS-ESI (+) m/z calc’d for C33H40O5SiNa [M+Na]+: 567.25427, found 567.25495.
((2R,3S,6S)-6-(2-((tert-butyldiphenylsilyl)oxy)ethyl)-3-((4-methoxybenzyl)oxy)-4-methyle netetrahydro-2H-pyran-2-yl)methanol (31)
To benzylidene acetal 30 (1.95 g, 3.58 mmol, 1 equiv) in toluene (36 mL) at −78 °C was added DIBAL-H (10.7 mL, 10.74 mmol, 3 equiv, 1 M in Hexanes) dropwise. The reaction was warmed to 0 °C and after 1 h added a few drops of MeOH, diluted with EA and quenched with sodium potassium tartrate. After warming to room temperature and stirring vigorously for 1 h, the crude product was extracted with EtOAc (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (10% to 50% EtOAc/Hexanes) afforded 1.36 g (69% yield) of alcohol 31 as a clear oil. [α]20D = +44.3 (c 0.41, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.67 – 7.64 (m, 4H), 7.44 – 7.36 (m, 6H), 7.26 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 5.06 (s, 1H), 4.99 (s, 1H), 4.60 (d, J = 11.4 Hz, 1H), 4.34 (d, J = 11.5 Hz, 1H), 4.10 (m, 1H), 3.86 – 3.80 (m, 4H), 3.71 – 3.64 (m, 4H), 3.53 (m, 1H), 2.32 (qd, J = 13.2, 4.6 Hz, 2H), 1.93 (dd, J = 7.7, 4.2 Hz, 1H), 1.84 (m, 1H), 1.64 (m, 1H), 1.04 (s, 9H); 13C NMR (125 MHz, CDCl3) δ (ppm): 159.4, 141.7, 135.7, 133.9, 133.8, 130.2, 129.8, 129.7, 127.8, 114.0, 111.6, 76.7, 76.2, 70.9, 69.1, 61.1, 60.3, 55.4, 37.7, 35.4, 27.0, 19.3; FT-IR (neat) νmax = 3449, 3069, 2951, 2930, 2855, 1613, 1514, 1426, 1249, 1110, 1086, 1038, 824, 739, 700, 504 cm−1; HRMS-ESI (+) m/z calc’d for C33H42O5SiNa [M+Na]+: 569.26992, found 569.27013.
Tert-butyl(2-((2S,5S,6R)-5-((4-methoxybenzyl)oxy)-4-methylene-6-vinyltetrahydro-2H-p yran-2-yl)ethoxy)diphenylsilane (32)
To a two-neck flask charged with flame-dried 4 Å MS was added alcohol 31 (756 mg, 1.38 mmol, 1 equiv) in CH2Cl2 (12 mL) and NMO (243 mg, 2.07 mmol, 1.5 equiv) in CH2Cl2 (2mL) via cannula at room temperature. After stirring for 20 min, TPAP (49 mg, 0.14 mmol, 0.1 equiv) was added to the reaction flask. After 15 min, the reaction was quenched with DMS, filtered over a silica plug, rinsing with EtOAc and concentrated. The crude aldehyde was used directly for the next reaction.
To Ph3PCH3Br (2.46 g, 6.90 mmol, 5 equiv) in freshly distilled THF (7 mL) was added tBuOK (5.5 mL, 5.52 mmol, 4 equiv, 1 M in THF) dropwise at 0 °C. After stirring for 1 h at 0 °C, a portion of the yellow stock solution (3.5 mL, 1.52 mmol, 1.1 equiv, 0.44 M) was added via syringe dropwise to the crude aldehyde (1.38 mmol, 1 equiv) in THF (7 mL) at 0 °C. After 10 min, the reaction was quenched with satd. NH4Cl. The crude product was extracted with EtOAc (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (5% to 20% EtOAc/Hexanes) provided 231 mg (31% yield) olefin 32 as a clear oil. [α]20D = +36.0 (c 0.37, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.67 – 7.65 (m, 4H), 7.43 – 7.35 (m, 6H), 7.26 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.5 Hz, 2H), 5.84 (ddd, J = 17.2, 10.8, 5.7 Hz, 1H), 5.28 (d, J = 17.5 Hz, 1H), 5.21 (d, J = 10.8 Hz, 1H), 5.05 (s, 1H), 4.97 (s, 1H), 4.56 (d, J = 11.8 Hz, 1H), 4.38 (m, 1H), 4.30 (d, J = 11.8 Hz, 1H), 4.04 (m, 1H), 3.87 – 3.80 (m, 4H), 3.70 (m, 1H), 3.64 (d, J = 3.1 Hz, 1H), 2.32 (m, 1H), 2.19 (dd, J = 13.1, 3.2 Hz, 1H), 1.86 (m, 1H), 1.71 (m, 1H), 1.03 (s, 9H); 13C NMR (125 MHz, CDCl3) δ (ppm): 159.3, 141.3, 135.7, 135.7, 135.0, 134.1, 134.0, 130.5, 129.7, 127.7, 118.3, 113.9, 113.3, 79.3, 78.2, 69.6, 68.7, 60.3, 55.4, 38.0, 37.3, 29.9, 27.0, 19.3; FT-IR (neat) νmax = 3072, 2956, 2925, 2856, 1612, 1513, 1464, 1428, 1247, 1111, 1087, 821, 737, 704, 613, 501 cm−1; HRMS-ESI (+) m/z calc’d for C34H42O4SiNa [M+Na]+: 565.27501, found 565.27640.
Methyl-2-((2S,5S,6R)-5-((4-methoxybenzyl)oxy)-4-methylene-6-vinyltetrahydro-2H-pyr an-2-yl)acetate (27)
To silyl ether 32 (398 mg, 0.73 mmol, 1 equiv) in THF (7 mL) at 0 °C was added TBAF (1.5 mL, 1.47 mmol, 2 equiv, 1 M in THF). After 2 h, the orange solution was concentrated and purification by flash chromatography (30% to 70% EtOAc/Hexanes) afforded 213 mg (95% yield) of the corresponding alcohol as a clear oil. [α]20D = +55.8 (c 0.36, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.26 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 5.83 (ddd, J = 17.5, 10.8, 5.7 Hz, 1H), 5.31 – 5.25 (m, 2H), 5.09 (s, 1H), 4.96 (s, 1H), 4.55 (d, J = 11.8 Hz, 1H), 4.48 (m, 1H), 4.28 (d, J = 11.8 Hz, 1H), 3.95 (m, 1H), 3.74–3.80 (m, 5H), 3.66 (d, J = 2.3 Hz, 1H), 2.43 (m, 2H), 2.12 (dd, J = 13.1, 3.0 Hz, 1H), 1.90 (m, 1H), 1.67 (m, 1H); 13C NMR (125 MHz, CDCl3) δ (ppm): 159.3, 140.5, 134.4, 130.3, 129.6, 119.0, 114.2, 113.9, 78.9, 78.5, 71.8, 69.3, 61.3, 55.4, 37.4, 37.0; FT-IR (neat) νmax = 3409, 2924, 2855, 1613, 1514, 1466, 1300, 1249, 1173, 1056, 1035, 908, 821 cm−1; HRMS-ESI (+) m/z calc’d for C18H24O4Na [M+Na]+: 327.15723, found 327.15583.
To the above alcohol (205 mg, 0.67 mmol, 1 equiv) in CH2Cl2/H2O (1:1, 6 mL) was added BAIB (651 mg, 2.02 mmol, 3 equiv) and TEMPO (32 mg, 0.20, 0.3 equiv) at room temperature. An orange color developed. After 4 h, the reaction was quenched with sodium thiosulfate. The crude product was extracted with CH2Cl2 (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (40% EtOAc/Hexanes to 10% MeOH/EtOAc) gave 218 mg (quantitative) of the corresponding acid as a clear oil. [α]20D = +47.9 (c 0.33, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.24 (d, 2H), 6.87 (d, J = 8.6 Hz, 2H), 5.81 (ddd, J = 17.4, 10.8, 5.6 Hz, 1H), 5.33 (dt, J = 17.5, 1.5 Hz, 1H), 5.29 (dt, J = 10.8, 1.5 Hz, 1H), 5.13 (s, 1H), 5.02 (s, 1H), 4.55 (d, J = 11.7 Hz, 1H), 4.50 (m, 1H), 4.28 (d, J = 11.7 Hz, 1H), 4.19 (m, 1H), 3.81 (s, 3H), 3.67 (d, J = 2.6 Hz, 1H), 2.69 (dd, J = 15.9, 7.6 Hz, 1H), 2.55 (dd, J = 15.9, 4.9 Hz, 1H), 2.43 (t, J = 12.9 Hz, 1H), 2.24 (dd, J = 13.1, 3.1 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ (ppm): 174.6, 159.3, 139.6, 133.7, 130.1, 129.7, 119.5, 114.9, 114.0, 78.8, 78.7, 69.5, 68.3, 55.4, 40.1, 36.2, 29.9; FT-IR (neat) νmax = 3075, 2956, 2924, 2854, 1711, 1612, 1514, 1302, 1248, 1173, 1067, 1033, 915, 820 cm−1; HRMS-ESI (–) m/z calc’d for C18H21O5 [M–H]−: 317.13890, found 317.13528.
To the above acid (212 mg, 0.67 mmol, 1 equiv) in PhMe/MeOH (3:2, 5 mL) at 0 °C was slowly added TMSCHN2 (1 mL, 2.00 mmol, 3 equiv, 2 M in Hexanes) and the reaction was warmed to room temperature. After 2 h, the yellow solution was diluted with additional MeOH and concentrated. Purification by flash chromatography (10% to 30% EtOAc/Hexanes) gave 195 mg (88% yield) of methyl ester 27 as a clear oil. [α]20D = +56.4 (c 0.33, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 7.24 (d, 2H), 6.87 (d, J = 8.7 Hz, 2H), 5.80 (ddd, J = 17.5, 10.8, 5.3 Hz, 1H), 5.32 (dt, J = 17.5, 1.6 Hz, 1H), 5.26 (dt, J = 10.8, 1.6 Hz, 1H), 5.09 (s, 1H), 5.00 (s, 1H), 4.55 (d, J = 11.8 Hz, 1H), 4.43 (m, 1H), 4.29 (d, J = 11.8 Hz, 1H), 4.22 (m, 1H), 3.80 (s, 3H), 3.68 (s, 3H), 3.64 (d, J = 3.0 Hz, 1H), 2.70 (dd, J = 15.4, 7.7 Hz, 1H), 2.46 (dd, J = 15.4, 5.7 Hz, 1H), 2.38 (m, 1H), 2.24 (dd, J = 13.0, 3.1 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ (ppm): 171.7, 159.3, 140.3, 134.4, 130.3, 129.6, 118.7, 114.2, 113.9, 78.9, 78.4, 69.6, 68.6, 55.4, 51.8, 40.2, 36.6; FT-IR (neat) νmax = 3074, 2954, 2926, 2855, 1739, 1655, 1612, 1584, 1514, 1461, 1436, 1380, 1302, 1246, 1154, 1066, 1031, 1006, 911, 820 cm−1; HRMS-ESI (+) m/z calc’d for C19H24O5Na [M+Na]+: 355.15214, found 355.15003.
Methyl 2-((3R,5S,7R,8R)-8-hydroxy-7-vinyl-1,6-dioxaspiro[2.5]octan-5-yl)acetate (8)
To PMB ether 27 (189 mg, 0.57 mmol, 1 equiv) in CH2Cl2/pH 7 buffer (10:1, 6 mL) at 0 °C was added DDQ (129 mg, 0.57 mmol, 1 equiv). After stirring 1 h at 0 °C, another portion of DDQ (129 mg, 0.57 mmol, 1 equiv) was added. After stirring for 1 h at 0 °C, the last portion of DDQ (129 mg, 0.57 mmol, 1 equiv) was added. After 1 h, the reaction was quenched with satd. NaHCO3. The crude product was extracted with CH2Cl2 (×3), washed with brine and dried over Na2SO4. Purification by flash chromatography (20% to 60% EtOAc/Hexanes) gave 99 mg (76% yield) of the corresponding alcohol as a clear oil. [α]20D = +40.8 (c 0.13, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 5.86 (ddd, J = 17.1, 10.7, 6.1 Hz, 1H), 5.37 (d, J = 17.4 Hz, 1H), 5.33 (d, J = 10.7 Hz, 1H), 5.12 (s, 1H), 4.96 (s, 1H), 4.34 (m, 1H), 4.12 (t, J = 5.8 Hz, 1H), 3.93 (d, J = 4.9 Hz, 1H), 3.69 (s, 3H), 2.68 (dd, J = 15.2, 7.7 Hz, 1H), 2.50 (dd, J = 15.2, 6.0 Hz, 1H), 2.45 – 2.36 (m, 2H), 2.04 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ (ppm): 171.5, 142.1, 134.9, 119.6, 111.3, 79.0, 72.9, 69.2, 51.9, 38.5, 36.8; FT-IR (neat) νmax = 3440, 2958, 2923, 2853, 1737, 1655, 1461, 1440, 1380, 1320, 1264, 1207, 1161, 1092, 1003, 908 cm−1; HRMS-ESI (+) m/z calc’d for C11H16O4Na [M+Na]+: 235.09463, found 235.09315.
To the above alcohol (96 mg, 0.45 mmol, 1 equiv) in CH2Cl2 (5 mL) was added VO(acac)2 (24 mg, 0.09 mmol, 0.2 equiv) at room temperature. After stirring for 10 min, the reaction flask was cooled to 0 °C and diluted TBHP (0.27 mL, 1.36 mmol, 3 equiv, 5–6 M in decane, 0.27 mL in 3 mL CH2Cl2) was added dropwise and the resulting dark red solution was warmed to RT. After 2 hr, the dark red reaction solution had faded to a light orange and the reaction did not progress past 1:1 starting material:product, therefore, the reaction was quenched with DMS and concentrated. Purification by flash chromatography (30% to 70% EtOAc/Hexanes) gave 56 mg (55% yield) of a single diastereomer of epoxide 8 as a clear oil. [α]20D = +66.0 (c 0.35, CHCl3); 1H NMR (500 MHz, CDCl3) δ (ppm): 5.92 (ddd, J = 17.4, 10.8, 5.4 Hz, 1H), 5.39 (dt, J = 17.4, 1.6 Hz, 1H), 5.30 (dt, J = 10.7, 1.5 Hz, 1H), 4.48 (m, 1H), 4.21 (m, 1H), 3.69 (s, 3H), 3.48 (dd, J = 8.1, 6.6 Hz, 1H), 2.95 (d, J = 4.6 Hz, 1H), 2.89 (dd, J = 15.5, 8.2 Hz, 1H), 2.65 (d, J = 4.6 Hz, 1H), 2.64 (dd, J = 15.4, 6.1 Hz, 1H), 1.98–2.03 (m, 2H), 1.82 (dd, J = 14.0, 5.3 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ (ppm): 171.5, 134.7, 118.5, 76.2, 69.9, 68.7, 57.3, 51.9, 50.3, 38.5, 34.4; FT-IR (neat) νmax = 3458, 2958, 2926, 2855, 1736, 1440, 1327, 1278, 1260, 1204, 1161, 1095, 1073, 1006, 933, 813 cm−1; HRMS-ESI (+) m/z calc’d for C11H16O5Na [M+Na]+: 251.08954, found 251.08462.
Methyl-2-((3R,5S,7R,8R)-7-((1E,3E)-5-((2S,3S,5R,6R)-5-((S,Z)-4-acetoxypent-2-enamido)-3,6-dimethyltetrahydro-2H-pyran-2-yl)-3-methylpenta-1,3-dien-1-yl)-8-hydroxy-1,6-di oxaspiro[2.5]octan-5-yl)acetate (2)
In three separate flasks were added diene 7 (8.0 mg, 0.023 mmol, 1 equiv), epoxide 8 (5.23 mg, 0.023 mmol, 1 equiv) and Grubbs II (3.9 mg, 0.0046, 0.2 equiv) and each were dissolved in 1 mL of CH2Cl2. Approximately 1/3 of the epoxide and Grubbs II solutions (0.33 mL) were sequentially added to the diene flask and set to reflux. After 1.5 h, a second 1/3 portion (0.33 mL) of the epoxide and Grubbs II solutions were added to the reaction flask. After an additional 1.5 h, the last portion of the epoxide and Grubbs II solutions were added, rinsing the flasks with a small amount of CH2Cl2. After 2 h, the reaction was cooled to room temperature and concentrated. Purification by flash chromatography (30% to 80% EtOAc/Hexanes) provided 5.9 mg (46% yield) of thailanstatin A methyl ester 2 as a clear oil. [α]20D = +4.8 (c 0.08, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ (ppm): 6.37 (d, J = 15.8 Hz, 1H), 6.25 (m, 1H), 6.00 (d, J = 9.0 Hz, 1H), 5.89 (dd, J = 11.6, 7.9 Hz, 1H), 5.70 (dd, J = 11.6, 1.3 Hz, 1H), 5.62 (dd, J = 15.7, 6.2 Hz, 1H), 5.51 (t, J = 7.2 Hz, 1H), 4.50 (m, 1H), 4.20 (t, J = 6.9 Hz, 1H), 3.94 (m, 1H), 3.66–3.69 (m, 4H), 3.51–3.54 (m, 2H), 2.99 (d, J = 4.6 Hz, 1H), 2.94 (dd, J = 15.5, 7.9 Hz, 1H), 2.69 (dd, J = 15.5, 6.5 Hz, 1H), 2.64 (d, J = 4.5 Hz, 1H), 2.39 (m, 1H), 2.24 (m, 1H), 2.16 (dd, J = 14.2, 5.3 Hz, 1H), 2.04 (s, 3H), 1.93–1.96 (m, 2H), 1.82 (d, J = 8.7 Hz, 1H), 1.73–1.79 (m, 5H), 1.39 (d, J = 6.5 Hz, 3H), 1.15 (d, J = 6.5 Hz, 3H), 1.01 (d, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ (ppm): 171.6, 170.6, 165.0, 143.8, 138.6, 134.7, 129.6, 123.1, 122.6, 80.9, 76.1, 75.8, 69.8, 69.1, 68.9, 57.3, 52.0, 49.8, 47.2, 38.1, 36.0, 34.6, 32.1, 29.0, 21.4, 20.1, 18.0, 15.2, 12.8; HRMS-ESI (+) m/z calc’d for C29H43NO9Na [M+Na]+: 572.2836, found 572.2831.
In vitro splicing reactions
In vitro splicing reactions consist of a pre-mRNA substrate incubated in nuclear extract from human cells, which provides the splicing machinery. The template DNA for the pre-mRNA splicing substrate is a derivative of the Adenovirus Major Late gene (REF: O. Gozani, J.G. Patton, R. Reed, EMBO J. 13 (1994) 3356–3367), and capped RNA splicing substrate was generated by standard in vitro T7 RNA polymerase “run-off” transcription including 32P-UTP label and G(5′)ppp(5′)G-cap analog. Nuclear extract was prepared from HeLa cells grown in DMEM/F12 1:1 media supplemented with 5% (v/v) newborn calf serum using the method by Dignam et al (REF: J.D. Dignam, R.M. Lebovitz, R.D. Roeder, Nucleic Acids Res. 11 (1983) 1475–1489). Splicing reactions were carried out by incubating 10 nM pre-mRNA substrate in 60 mM potassium glutamate, 2 mM magnesium acetate, 2 mM ATP, 5 mM creatine phosphate, 0.05 mg ml−1 tRNA, and 50% (v/v) HeLa nuclear extract at 30°C for 30 minutes, and then analyzed by both denaturing polyacrylamide and native agarose gel electrophoresis.
Denaturing gel analysis
RNA was extracted from in vitro splicing reaction and separated on a 15% (v/v) denaturing polyacrylamide gel. 32P-labeled RNA species were visualized by phosphorimaging and quantified with ImageQuant software (Molecular Dynamics). Splicing efficiency is calculated as the amount of mRNA relative to total substrate RNA and normalized to a DMSO control reaction. IC50 values for inhibitors are the concentration of inhibitor that causes 50% decrease of splicing efficiency, which were derived from averaged plots of splicing efficiency vs. compound concentration from three independent assays.
Native gel analysis
Splicing reactions were set up as described, and after 30 minutes 10 μL of splicing reactions were mixed with 10 μL native gel loading buffer (20 mM Trizma base, 20 mM glycine, 25% (v/v) glycerol, 0.1% (w/v) cyan blue, 0.1% (w/v) bromophenol blue, 1 mg ml−1 heparin sulfate) and incubated at room temperature for five minutes before loading onto a 2.1% (w/v) low-melting temperature agarose gel. Gels were run at 72 V for 3.5 hours, dried onto Whatman paper and exposed to phosphorimaging screens, which were digitized with a Typhoon Scanner (Molecular Dynamics).
Supplementary Material
Acknowledgments
Financial support of this work was provided by the National Institutes of Health (GM122279) and Purdue University.
Footnotes
Full spectroscopic data for all compounds are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roybal GA, Jurica MS. Nucleic Acids Res. 2010;38:6664–6672. doi: 10.1093/nar/gkq494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 4.Raymond B. Nat Chem Biol. 2007;3:533–535. doi: 10.1038/nchembio0907-533. [DOI] [PubMed] [Google Scholar]
- 5.Wahl MC, Will CL, Lührmann R. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Nilsen TW, Graveley BR. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper TA, Wan L, Dreyfuss G. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee SC-W, Abdel-Wahab O. Nat Med. 2016;22:976–986. doi: 10.1038/nm.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pal S, Gupta R, Kim H, Wickramasinghe P, Baubet V, Showe LC, Dahmane N, Davuluri RV. Genome Research. 2011;21:1260–1272. doi: 10.1101/gr.120535.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen THD, Galej WP, Bai X-C, Oubridge C, Newman AJ, Schere SHW, Nagai K. Nature. 2016;530:298–302. doi: 10.1038/nature16940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan R, Yan C, Bai R, Wang L, Huang M, Wong CCL, Shi Y. Science. 2016;351:466–475. doi: 10.1126/science.aad6466. [DOI] [PubMed] [Google Scholar]
- 12.Mizui Y, Sakai T, Iwata T, Uenaka T, Okamoto K, Shimizu H, Yamori T, Yoshimatsu K, Asada MJ. Antibiot (Tokyo) 2004;57:188–196. doi: 10.7164/antibiotics.57.188. [DOI] [PubMed] [Google Scholar]
- 13.Nakajima H, Sato B, Fujita T, Takase S, Terano H, Okuhara M. J Antibiot (Tokyo) 1996;49:1196–1203. doi: 10.7164/antibiotics.49.1196. [DOI] [PubMed] [Google Scholar]
- 14.Sakai T, Asai N, Okuda A, Kawamura N, Mizui Y. J Antibiot (Tokyo) 2004;57:180–187. doi: 10.7164/antibiotics.57.180. [DOI] [PubMed] [Google Scholar]
- 15.(a) Lagisetti C, Yermolina MV, Sharma LK, Palacios G, Prigaro BJ, Webb TR. ACS Chem Biol. 2014;9:643–648. doi: 10.1021/cb400695j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao Y, Vogt A, Forsyth CJ, Koide K. ACS Chem Biol. 2012;14:49–52. doi: 10.1002/cbic.201200558. [DOI] [PubMed] [Google Scholar]; (c) Ghosh AK, Lv K, Ma N, Cárdenas EL, Effenberger KA, Jurica MS. Org Biomol Chem. 2016;14:5263–5271. doi: 10.1039/c6ob00725b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yokoi A, Kotake Y, Takahashi K, Kadowaki T, Matsumoto Y, Minoshima Y, Sugi NH, Sagane MH, Iwata M, Mizui Y. FEBS J. 2011;278:4870–4880. doi: 10.1111/j.1742-4658.2011.08387.x. [DOI] [PubMed] [Google Scholar]
- 17.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, Tani T, Horinouchi S, Yoshida M. Nat Chem Biol. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 18.Effenberger KA, Urabe VK, Prichard BE, Ghosh AK, Jurica MS. RNA. 2016;22:350–359. doi: 10.1261/rna.053108.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonnal S, Vigevani L, Valcárcel J. Nat Rev Drug Discov. 2012;11:847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 20.Sato M, Muguruma N, Nakagawa T, Okamoto K, Kimura T, Kitamura S, Yano H, Sannomiya K, Goji T, Miyamoto H, Okahisa T, Mikasa H, Wada S, Iwata M, Takayama T. Cancer Sci. 2014;105:110–116. doi: 10.1111/cas.12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butler MA. In: Natural Product-Derived Compounds in Late State Clinical Development at the End of 2008. Buss A, Butler M, editors. The Royal Chemical Society of Chemistry; Cambridge, UK: 2009. pp. 321–354. [Google Scholar]
- 22.Bonnal S, Vigevani L, Valcárcel J. Nat Rev Drug Discovery. 2012;11:847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 23.Bessire AJ, Ballard TE, Charati M, Cohen J, Green M, Lam M-H, Loganzo F, Nolting B, Pierce B, Puthenveetil S, Roberts L, Schildknegt K, Subramanyam C. Bioconj Chem. 2016;17:1645–1654. doi: 10.1021/acs.bioconjchem.6b00192. [DOI] [PubMed] [Google Scholar]
- 24.Beck A, Goetsch L, Dumontet C, Corvaïa N. Nat Rev Drug Discovery. 2017;16:315–337. doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Biswas S, Berg MG, Antapli CM, Xie F, Wang Q, Tang M-Ch, Tang G-L, Zhang L, Dreyfuss G, Cheng Y-Q. J Nat Prod. 2013;76:685–693. doi: 10.1021/np300913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He H, Ratnayake AS, Janso JE, He M, Yang HY, Loganzo F, Shor B, O’Donnell CJ, Koehn FE. J Nat Prod. 2014;77:1864–1870. doi: 10.1021/np500342m. [DOI] [PubMed] [Google Scholar]
- 27.Eustáquio AS, Chang L-P, Steele GL, O’Donnell CJ, Koehn FE. Metab Eng. 2016;33:67–75. doi: 10.1016/j.ymben.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Dirico KJ, Eustáquio AS, Green ME, He H, He M, Koehn FE, O’Donnell CJ, Puthenveetil S, Ratnayake AS, Subramanyam C, Teske JA, Yang HY. International Patent WO. 2014;068443:A1. [Google Scholar]
- 29.(a) Thompson CF, Jamison TF, Jacobsen EN. J Am Chem Soc. 2000;122:10482–10483. [Google Scholar]; (b) Thompson CF, Jamison TF, Jacobsen EN. J Am Chem Soc. 2001;123:9974–9983. doi: 10.1021/ja016615t. [DOI] [PubMed] [Google Scholar]; (c) Horigome M, Motoyoshi H, Watanabe H, Kitahara T. Tetrahedron Lett. 2001;42:8207–8210. [Google Scholar]; (d) Motoyoshi H, Horigome M, Watanabe H, Kitahara T. Tetrahedron. 2006;62:1378–1389. [Google Scholar]
- 30.(a) Albert BJ, Koide K. Org Lett. 2004;6:3655–3658. doi: 10.1021/ol049160w. [DOI] [PubMed] [Google Scholar]; (b) Albert BJ, Sivaramakrishnan A, Naka T, Koide K. J Am Chem Soc. 2006;128:2792–2793. doi: 10.1021/ja058216u. [DOI] [PubMed] [Google Scholar]; (c) Albert BJ, Sivarama-krishnan A, Naka T, Czaicki NL, Koide K. J Am Chem Soc. 2007;129:2648–2659. doi: 10.1021/ja067870m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Ghosh AK, Chen ZH. Org Lett. 2013;15:5088–5091. doi: 10.1021/ol4024634. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK, Chen ZH, Effenberger KA, Jurica MS. J Org Chem. 2014;79:5697–5709. doi: 10.1021/jo500800k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh AK, Cheng X. Org Lett. 2011;13:4108–4111. doi: 10.1021/ol201626h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Effenberger KA, Anderson DD, Bray WM, Prichard BE, Ma N, Adams MS, Ghosh AK, Jurica MS. J Biol Chem. 2014;289:1938–1947. doi: 10.1074/jbc.M113.515536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh AK, Ma N, Effenberger KA, Jurica MS. Org Lett. 2014;16:3154–3157. doi: 10.1021/ol501345d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. ACS Chem Biol. 2011;6:582–589. doi: 10.1021/cb100356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicolaou KC, Rhoades D, Lamani M, Pattanayak MR, Kumar SM. J Am Chem Soc. 2016;138:7532–7535. doi: 10.1021/jacs.6b04781. [DOI] [PubMed] [Google Scholar]
- 37.Pazos G, Pérez M, Gándara Z, Gómez G, Fall Y. Tetrahedron Lett. 2009;50:5285–5287. [Google Scholar]
- 38.Hoberg JO. Carbohydr Res. 1997;300:365–367. [Google Scholar]
- 39.Ghosh AK, Reddy GC, MacRae AJ, Jurica MS. Org Lett. ASAP; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J Org Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 41.Yadav JS, Reddy BVS, Reddy KB, Satyanarayana M. Tetrahedron Lett. 2002;43:7009–7012. [Google Scholar]
- 42.Fujiwara K, Sato D, Watanabe M, Morishita H, Murai A, Kawai H, Suzuki T. Tetrahedron Lett. 2004;45:5243–5246. [Google Scholar]
- 43.Marshall JA, Garofalo AW. J Org Chem. 1993;58:3675–3680. [Google Scholar]
- 44.Sharpless KB, Michaelson RC. J Am Chem Soc. 1973;95:6136–6137. [Google Scholar]
- 45.Sheng MN, Zajacek JG. J Org Chem. 1970;35:1839–1843. [Google Scholar]
- 46.Peterson DJJ. Org Chem. 1968;33:780–784. [Google Scholar]
- 47.Magnus P. Aldrichimica Acta. 1980;13:43–51. [Google Scholar]
- 48.Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 49.Artman GD, III, Grubss AW, Williams RM. J Am Chem Soc. 2007;129:6336–6342. doi: 10.1021/ja070259i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BB. Ang Chem Int Ed Engl. 2005;44:1378–1382. doi: 10.1002/anie.200462207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.