

ABSTRACT
Primate lentiviruses, including the human and simian immunodeficiency viruses (HIV and SIV), produce infections marked by persistent, ongoing viral replication. This occurs despite the presence of virus-specific adaptive immune responses, including antibodies targeting the viral envelope glycoprotein (Env), and evolution of antibody-escape variants is a well-documented feature of lentiviral infection. Here, we examined the evolutionary dynamics of the SIV env gene during early infection (≤29 weeks postinfection) in a cohort of four SIVmac251-infected rhesus macaques. We tracked env evolution during acute and early infection using frequent sampling and ultradeep sequencing of viral populations, capturing a transmission bottleneck and the subsequent reestablishment of Env diversity. A majority of changes in the gp120 subunit mapped to two short clusters, one in the first variable region (V1) and one in V4, while most changes in the gp41 subunit appeared in the cytoplasmic domain. Variation in V1 was dominated by short duplications and deletions of repetitive sequence, while variation in V4 was marked by short in-frame deletions and closely overlapping substitutions. The most common substitutions in both patches did not alter viral replicative fitness when tested using a highly sensitive, deep-sequencing-based competition assay. Our results, together with the observation that very similar or identical patterns of sequence evolution also occur in different macaque species infected with related but divergent strains of SIV, suggest that resistance to early, strain-specific anti-Env antibodies is the result of temporally and mutationally predictable pathways of escape that occur during the early stages of infection.
IMPORTANCE The envelope glycoprotein (Env) of primate lentiviruses mediates entry by binding to host cell receptors followed by fusion of the viral membrane with the cell membrane. The exposure of Env complexes on the surface of the virion results in targeting by antibodies, leading to selection for virus escape mutations. We used the SIV/rhesus macaque model to track in vivo evolution of variation in Env during acute/early infection in animals with and without antibody responses to Env, uncovering remarkable variation in animals with antibody responses within weeks of infection. Using a deep-sequencing-based fitness assay, we found substitutions associated with antibody escape had little to no effect on inherent replicative capacity. The ability to readily propagate advantageous changes that incur little to no replicative fitness costs may be a mechanism to maintain continuous replication under constant immune selection, allowing the virus to persist for months to years in the infected host.
KEYWORDS: HIV, SIV, simian immunodeficiency virus, env, viral fitness
INTRODUCTION
The ability of primate lentiviruses to replicate continuously in the face of innate and virus-specific adaptive immune responses stems from a complement of different immune evasion strategies (1, 2). Primate lentiviruses generate viral populations of genetically distinct variants through a high mutation rate, rapid cycles of replication (∼1 to 2 days), and high turnover of viral particles and infected CD4+ T cells (3–5). Mutants generated by the low-fidelity reverse transcriptase (RT) may be acted upon by selective pressures, including host immune responses, leading to the rapid selection of escape variants (6–23).
The envelope glycoprotein (Env) of human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV), which is expressed as a trimeric complex on the surfaces of virions and virus-producing cells, serves as the primary target of antibody-based B-cell responses. During synthesis, the Env precursor protein (gp160) trimerizes, is modified with glycans, and is cleaved to produce the surface subunit (gp120) and the transmembrane subunit (gp41), which remain noncovalently associated. Together the gp120:gp41 heterodimers form the trimeric Env complex. Env mediates entry through direct interactions with the CD4 cell surface molecule and coreceptor molecules (multimembrane-spanning chemokine molecules, most notably CCR5 and CXCR4). Receptor and coreceptor interactions lead to conformational changes in gp120 that expose the fusion peptide located at the N terminus of gp41, causing fusion of the viral membrane with the host cell membrane.
In a manner similar to that of HIV-1- and HIV-2-infected patients, rhesus macaques infected with pathogenic SIV strains develop continuous and robust antibody responses to Env (17, 24–26). However, in both hosts, escape from antibody responses often occurs rapidly due to the generation of populations of genetically distinct variants from which escape variants are selected, making early neutralizing antibody (NAb) responses highly strain specific and low in potency (11, 16, 22, 25, 27, 28).
The SIV/macaque model allows for tracking of env sequence variation in animals inoculated with genetically defined SIV strains and tracing of evolution of env from a well-defined source inoculum through transmission and over the course of infection. Analysis of env sequence by bulk PCR and cloning of env sequences isolated from chronically SIV-infected macaques has revealed evidence for positive selection, specifically in the V1 and V4 loops (17, 29, 30). Variation in V1 and V4 was similarly observed in other SIV/macaque models of infection, including SIVmac251 infection of rhesus macaques, SIVmne infection of pig-tailed macaques, and SIVsm infection of rhesus macaques (31–39), as well as in the context of natural infection in sooty mangabeys (40). More recently, an analysis of env sequence variation in an animal with a potent NAb response to the parental challenge strain, SIVmac239, identified individual substitutions within the V1 and V4 loop that, when introduced into SIVmac239, provided resistance to the high-titer neutralizing plasma (17).
We began the current study by surveying the literature on SIV env evolution and antibody escape, noting strikingly similar patterns of amino acid substitutions and insertions/deletions even when comparing studies based on different viral strains and/or using different species of macaques as hosts. However, many of these studies predated high-throughput deep-sequencing technology and involved isolating and sequencing small numbers of clones representing a few well-separated time points. Moreover, most early studies were based on high-titer intravenous (i.v.) inoculation with virus stock, whereas more recent practice is to initiate infection by repeated low-dose mucosal exposure, which is thought to more accurately mimic natural modes of transmission, during which infection by one or a small number of virions results in a severe genetic bottleneck. In this study, we revisit these earlier observations by examining a small cohort of animals experimentally infected by low-dose mucosal exposure with an uncloned viral stock (swarm). Specifically, we tracked evolution of env from an initial bottleneck through 29 weeks of early infection using intensive longitudinal sampling and deep sequencing of the viral population at each time point. More frequent and deeper sampling allowed us to track the kinetics of sequence change with more accuracy. In addition, we tracked the antibody response and noted striking differences in env evolution and pathogenesis across animals with differing antibody responses. We confirmed patterns noted in earlier studies and tested the most common substitutions in a deep-sequencing-based fitness assay, noting that the changes most commonly associated with antibody escape in the literature appear to have little or no significant impact on inherent replicative capacity of the virus. Taken together, prior reports and our results indicate that env evolution and escape from antibody responses follows defined, predictable pathways in SIV/macaque models, and that escape may be effected through the rapid appearance of substitutions in V1 and V4 that are effectively neutral with respect to viral replicative capacity but which permit facile generation of escape mutations from immunodominant antibody responses.
RESULTS
Reproducible appearance and fixation of common amino acid substitutions in SIV Env.
We began with a comparison of previous studies, revealing a highly reproducible pattern of env coding sequence evolution in SIV-infected macaques with respect to amino acid replacements in the V1 and V4 loops of gp120; furthermore, we observed that these different studies describe similar and sometimes identical substitutions regardless of SIV strain or species of macaque (Table 1). Thus, while experimental macaques are genetically diverse, outbred hosts, with respect to env evolution and antibody escape, similar patterns will often emerge in multiple individuals in the same cohort. This contrasts with cytotoxic T-lymphocyte (CTL) escape, which is a function of each individual animal's unique MHC genotype in combination with the primary amino acid sequences of the proteins encoded by the infecting virus strain, such that the pattern of CTL escape will differ from one animal to the next and from one SIV strain to the next.
TABLE 1.
Env substitutions in the gp120 subunit involved in neutralizing antibody escape found in previous reports
| Virus | Subunit | Region | Residue | Reference(s) | Reference for neutralization data |
|
|---|---|---|---|---|---|---|
| Plasma | MAb | |||||
| SIVmac239 | gp120 | V1 | R120 | 30, 41 | ||
| SIVmac239 | gp120 | V1 | S132 | 17, 29 | ||
| SIVmac239, SIVmneCL8 | gp120 | V1 | S135 | 29, 30, 41 | ||
| SIVmac239 | gp120 | V1 | T136 | 17, 29, 41 | ||
| SIVmac239, SIVmneCL8 | gp120 | V1 | A138 | 17, 30 | 17 | |
| SIVmac239 | gp120 | V1 | S139 | 41 | ||
| SIVmac239 | gp120 | V2 | N198 | 29 | ||
| SIVmac239 | gp120 | V2 | D201 | 29, 41 | ||
| SIVmac239, SIVmac251 | gp120 | V2 | N202 | 39 | ||
| SIVmac239, SIVmac251, SIVmneCL8, HIV-27312A | gp120 | V3 | W345 | 17, 29, 30, 37, 39, 41, 42 | 42 | |
| SIVmneCL8 | gp120 | V4 | D415 | 30, 43 | ||
| SIVmac239, SIVmneCL8 | gp120 | V4 | A417 | 17, 29, 30, 41, 43 | 17 | |
| SIVmac239, SIVmac251, SIVmneCL8 | gp120 | V4 | P421 | 17, 29, 30, 38, 39, 43 | 17 | |
| SIVmac239, SIVmac251 | gp120 | V4 | R424 | 17, 37, 39, 41 | 37 | |
Study cohort: viral load and Env-specific antibody responses in four SIVmac251-infected rhesus macaques.
To understand SIV Env sequence evolution in the face of Env-specific antibody responses, we first determined the presence of antibody responses in a cohort of four male rhesus macaques experimentally infected by repeated low-dose mucosal exposure to uncloned SIVmac251 (Table 2). To account for the reported impact of major histocompatibility complex (MHC) class I and TRIM5 on viral replication, all four animals were genotyped for specific TRIM5 and MHC class I alleles (Table 2). SIV viral RNA (vRNA) loads measured from longitudinal plasma samples verified all four animals were successfully infected, with acute peak viral replication levels ranging from 5.5 × 106 to 2.3 × 108 copies/ml (Table 2 and Fig. 1A). One animal, macaque monkey (Mm) 156-10, here identified as rapid progressor 1 (RP1), maintained viral loads above 10 million copies/ml through 15 weeks of infection and had to be euthanized due to the rapid development of AIDS-related signs. The remaining three animals had postacute viral loads within a range that is characteristic of SIVmac-infected macaques (Fig. 1A). Two animals, Mm 10-10 and Mm 198-08, had typical viral load courses; however, Mm 174-08 had a markedly low viral load course, suggestive of control of viral replication.
TABLE 2.
Cohort of four Indian-origin rhesus macaques infected by repeated low-dose exposure to uncloned SIVmac251
| Animal no. | Animal ID | Viral load peak (copies/ml) | Viral set point | MHC-I genotype | TRIM5 genotype | Binding antibodyb | Neutralization of TCLA SIVmac251b | CD20+ B cellsc |
|---|---|---|---|---|---|---|---|---|
| RP1 | Mm 156-10 | 2.8 × 108 | High (>1 × 108) | −a | TFP/CypA | − | − | + |
| SP1 | Mm 174-08 | 5.5 × 106 | Low (<1 × 105) | A*08 | TFP/TFP | + | + | + |
| TP1 | Mm 10-10 | 2.3 × 108 | Typical (1 × 105–3 × 106) | A*08 | TFP/Q | + | + | + |
| TP2 | Mm 198-08 | 1.8 × 108 | Typical (1 × 105–3 × 106) | A*08 | TFP/Q | + | + | + |
−, animal was negative for all nine MHC class I alleles tested.
−, response was low/not detected. +, response was detected.
+, CD20+ B cells were detected.
FIG 1.

Plasma viral RNA load and antibody-based immune responses following repeated low-dose SIVmac251 mucosal challenge. (A) Viral loads of four rhesus macaques infected by repeated low-dose challenge with SIVmac251 were measured 1, 2, 3, 4, 6, 8, 10, 14, 18, 23, 25, and 29 weeks from initial challenge by real-time RT-PCR of plasma viral RNA and reported as SIV RNA copy equivalents/ml of plasma. All four male rhesus macaques were positively infected, showing peak of viral replication 2 weeks after the last SIV-negative assay. Viral loads were synchronized to the last week in which vRNA was not detectable in each infected animal. (B) Env-specific antibody responses were measured by ELISA using recombinant soluble SIVmac239 gp140. The level of binding in plasma from each animal was tested by serial 4-fold dilution in duplicate. Antibody titer was calculated as the inverse of the lowest dilution with a value at least 2-fold above background. (C) Neutralizing antibody titers in plasma were measured against laboratory-adapted SIVmac251. Plasma was tested over eight serial 2-fold dilutions in duplicate to determine the concentration of plasma which reduced infection by 50%, termed the IC50. The IC50 was reported for every time point tested for each animal in the cohort. (D) Two million mononuclear cells were stained with anti-human monoclonal antibodies CD3-V450, CD20-allophycocyanin (APC)-H7, CD4-APC (BD Bioscience, San Jose, CA), and Dead/Live Aqua dead cell stain (Invitrogen, Carlsbad, CA). Data were acquired using an Aria I flow cytometer (BD Bioscience, San Jose, CA) and analyzed using FlowJo software (TreeStar, Cupertino, CA). (E and F) Mononuclear cells from peripheral blood (PBMC), bone marrow (BM), spleen (SP), axillary lymph node (AxLN), and inguinal lymph node (InLN) were stimulated with a pool of overlapping peptides corresponding to the complete amino acid sequence of SIVmac239 Env (E) and Gag (F). The average number of spot-forming cells (SFC) is reported for duplicate wells seeded at 200,000 cells per well. Results correspond to PBMC at 3 weeks p.i. and BM, SP, AxLN, and InLN at necropsy.
To determine whether each animal mounted an antibody response specific for Env, we screened longitudinal plasma samples by enzyme-linked immunosorbent assay (ELISA) for antibodies that could bind to soluble SIVmac239 Env gp140 (Fig. 1B). We found that animals Mm10-10 (here typical progressor 1 [TP1]), 198-08 (TP2), and 174-08 (here slow progressor 1 [SP1]) had binding antibody responses specific to Env that increased in titer over time; however, we were unable to detect an antibody response to Env in RP1 at any time point tested. We further tested plasma samples for the ability to neutralize a laboratory-adapted, neutralization-sensitive stock of SIVmac251 (Fig. 1C). We found that plasma from all animals except for RP1 could neutralize laboratory-adapted SIVmac251. Despite the lack of an Env-specific antibody response, the overall frequency of CD20+ B cells in RP1 was similar to that of the other three animals (Fig. 1D). Further, we detected CTL responses by gamma interferon (IFN-γ) activity of peripheral blood mononuclear cells (PBMCs) isolated from SP1 directed to Gag and Env by enzyme-linked immunospot (ELISPOT) assay (Fig. 1E and F, respectively). Therefore, our cohort consisted of two animals with typical viral load courses and detectable antibody responses, one animal with a low viral load course and detectable antibody response and one rapid progressor that did not mount a detectable antibody response to Env. We used this small cohort of animals to investigate the early, posttransmission evolution of the SIVmac env gene in an environment of a sustained Env-specific antibody response.
Emergence and distribution of Env amino acid diversity during early SIV infection.
To investigate SIV env sequence evolution during the earliest stages of SIV infection, we used Illumina deep sequencing of reverse transcription-PCR (RT-PCR)-amplified SIV RNA extracted from the SIVmac251 stock and from longitudinal plasma samples from each animal. Sequence data were produced using an overlapping two-amplicon system that targeted the entire env coding region (∼4 kb). The mean read coverage of the env gene across all samples was 13,171 reads/site, ranging from 670 reads/site to 24,647 reads/site (data not shown).
Diversity of the SIVmac251 stock.
We used next-generation sequence analysis to characterize a sample of the SIVmac251 stock used to infect the animals and compared this to longitudinal samples spanning acute and early infection in each animal. Variant calls were made relative to the consensus sequence at each site (codon) in env, and diversity was defined as the frequency of codons present at each site that differed from the majority codon. We further determined the level of nonsynonymous diversity (codons encoding an amino acid differing from the majority codon) and synonymous diversity at each site. For the SIVmac251 stock, the mean diversity across all sites in Env was 0.555 (nonsynonymous diversity) and 0.416 (synonymous diversity) (Fig. 2A). The mean nonsynonymous diversity measurements for gp120 and gp41 were 0.425 and 0.751, respectively, while the synonymous diversity measurements for gp120 and gp41 were 0.353 and 0.511, respectively.
FIG 2.

Nonsynonymous and synonymous diversity in the uncloned SIVmac251 stock followed by genetic bottleneck at transmission. (A) The level of nonsynonymous (blue) and synonymous (red) diversity was determined for every codon in Env in the uncloned SIVmac251 stock inoculum. Nonsynonymous diversity was determined as the frequency of codons with a nonsynonymous change relative to the majority codon of the stock consensus sequence, and synonymous diversity was determined as the frequency of codons with a synonymous change relative to the majority codon of the stock consensus sequence. (B) We split Env into 90-nucleotide windows (30 codons). For each animal and the stock inoculum, we collected NGS reads spanning each window and characterized the number of variants. For the animals, sequences from the first sample time postinfection were considered. Each bar shows particular variants and their frequency (boxed in black). Variants either were found in the inoculum (blue) or not (red). Variants not found in the inoculum, labeled de novo, could either have been generated during infection through mutation or represent low-frequency inoculum variants that were not identified. Missing bars represent windows with poor NGS coverage, with <100 high-quality reads. Bars that do not reach a frequency of 1 reflect variants with a frequency <1%, which are not shown for readability. (C) Divergence was determined as the frequency of a majority codon (frequency of 50% or more) at the sampled time point that differed from the majority codon of the SIVmac251 stock inoculum. Nonsynonymous sites are reported in blue, and synonymous sites are reported in red.
Loss of diversity and divergence from stock consensus after mucosal transmission of SIVmac251.
In each animal, we used sequences collected at the first sampling time, either week 2 or 3 postinfection, to determine the transmitted/founder (T/F) variants. Sequence analysis suggested 2 to 4 T/F variants in all four animals (Fig. 2B). In each animal, there was one major T/F variant, present at a frequency of >0.9. The inoculum was much varied; for example, the inoculum possessed at least eight different V1 variants at a range of frequencies, and in many cases minor inoculum variants became the major T/F variant, as discussed below. We examined sequence divergence from the stock consensus sequence at the earliest sampled time points and calculated divergence at each site in Env as a change in the majority codon for a given time point relative to the majority codon of the stock inoculum. We identified several sites of divergence at the earliest sampled time point in each animal clustering in the V1 loop of gp120 and the C-terminal region of the cytoplasmic tail of gp41 (Fig. 2C). The gp41 cytoplasmic tail (CT) exhibited a high level of genetic variation in the stock inoculum that clustered between amino acid positions 750 and 800, and during transmission the gp41 CT underwent a transmission bottleneck similar to that observed for gp120.
Reestablishment of sequence diversity during acute infection.
We next compared the nonsynonymous and synonymous diversity at acute infection (2 to 3 weeks postinfection [p.i.]) to that at postacute (13 to 15 weeks p.i.) and early chronic (29 weeks p.i.) stages of infection (Fig. 3). As expected, we observed a severe loss of diversity in samples taken at or near the peak of viral load in each animal (either 2 or 3 weeks postinfection) relative to the diversity observed in the stock inoculum, indicative of transmission bottleneck events in all four animals (Fig. 3). We observed recovering diversity in animals TP1 and TP2 by 14 weeks p.i. and found that the total (nonsynonymous and synonymous) mean Env diversity reached the same level of diversity found in the stock inoculum by 23 weeks p.i. (0.97 for stock, 1.08 for TP1, and 1.10 for TP2). Although variation was present throughout Env, variation was highest in the V1 and V4 variable loop domains of gp120 and the cytoplasmic tail of gp41. Comparing the mean diversity of each gp120 variable region to the total mean diversity of Env, we observed the highest diversity emerge in V1 and V4 in animals TP1 and TP2 (the two animals with typical acute and postacute viral loads) (data not shown). In contrast, Env diversity in RP1 and in the animal with low viral loads (SP1) never recovered to the level found in the stock inoculum or even to the level achieved in the other two animals (Fig. 3).
FIG 3.

Nonsynonymous and synonymous diversity at multiple time points spanning early SIV infection. The level of nonsynonymous (A) and synonymous (B) diversity was determined for every codon in Env at 2 to 3 weeks p.i. (green bars), 13 to 15 weeks p.i. (red bars), and 29 weeks p.i. (blue bars). Nonsynonymous diversity was determined as the frequency of codons with a nonsynonymous change relative to the majority codon of the sample time point consensus sequence, and synonymous diversity was determined as the frequency of codons with a synonymous change relative to the majority codon of the sample time point consensus sequence.
Evolution of gp120 coding sequence dominated by two short clusters of variation in V1 and V4.
We next undertook a closer examination of the evolution of the V1 and V4 loop regions in the viral population throughout early infection using a haplotype-based analysis, where we analyzed sequence variation in windows of 90 nucleotides using reads assembled to the env gene of SIVmac239, allowing us to analyze variation within the entire length of the V1 and V4 loops (44). Haplotypes found in the inoculum were labeled I haplotypes, whereas novel haplotypes found in animal samples were labeled H haplotypes. In the following subsections we describe patterns of sequence evolution unique to each of the two domains (V1 and V4), including insertions, deletions, and amino acid replacements.
Evolution of V1 includes expansion and contraction of three-nucleotide repeats and a tightly clustered patch of amino acid replacements.
The haplotype analysis revealed a remarkable level of in-frame insertions and deletions in the V1 loop, similar to reports of other SIVmac-infected animals (30, 31, 43). In TP2, insertions were found at four different loci within a short span of 13 residues (positions 126 to 139), rich in serine and threonine residues coded for by TCA and ACA codons (Fig. 4). The inoculum contained variants with a 3-amino-acid insertion, relative to the consensus sequence of the stock, at the first locus (position 132), making up 23.3% of the viral population. Among the four animals in the cohort, a haplotype containing a longer V1 loop was only transmitted in SP1. TP2 had no haplotypes with insertions in V1 at transmission or over the first 18 weeks of infection, but at 23 weeks p.i., several haplotypes emerged in TP2 with insertions ranging from a single amino acid up to five amino acids. The most common insertion, consisting of a 5-amino-acid duplication, TTTAS, resulted from a direct duplication of codons 128 to 132. Insertions that emerged at 25 weeks p.i. ranged from a single amino acid up to 5 amino acids, with all insertions consisting of GCA, TCA, ACA, and CCA codons. Therefore, the highly repetitive nature of this stretch of V1 led to the emergence of several haplotypes that contained multiple amino acid insertions and deletions, leading to the rapid length fluctuation within a short span of 6 weeks of viral replication.
FIG 4.

Nucleotide insertions and deletions in the V1 loop arise after 18 weeks in TP2. The frequency of haplotypes was determined from deep sequence reads of each sample spanning Env amino acid residues 115 to 144 and assembled to the SIVmac239 reference sequence. Inoculum haplotypes were observed in the source inoculum and/or during infection, in contrast to haplotypes only observed during infection of individual animals.
Evolution of V4 during early infection dominated by small in-frame deletions.
We applied the haplotype analysis to reads spanning the V4 loop region and detected the presence of deletions and substitutions in both TP1 and TP2 (Fig. 5). In both animals, haplotypes containing substitutions emerged prior to haplotypes that contained multiple in-frame deletions. Strikingly, in-frame deletions that flanked or spanned a proline at position 421 eventually dominated the viral populations in both animals. V4 loop deletions in TP1 were present by 18 weeks p.i. and increased to 43.5% by 23 weeks p.i. (Fig. 5A and B). By 25 weeks p.i., two additional deletion variants were detectable: Δ423ERHR426 and Δ421PKERHRR427. At 29 weeks p.i., the frequencies of Δ423ERHR426 and Δ415DLTTQR420 were reduced to below 5%, Δ421PKERHRR427 remained at a similar frequency, but Δ418TQRPKERH425 rose to a frequency of 53.8%. A deletion spanning the same positions (residues 418 to 425) was found at chronic time points in two additional rhesus macaques from an unrelated SIVmac239-infected cohort (Fig. 5E). Interestingly, all three deletion variants either flanked or spanned the P421 site in V4, providing strong evidence of selection continuously targeting the V4 loop at or around the proline at position 421, resulting in the dominance of V4 loop deletions in the viral population (Fig. 5B and D). By 23 weeks p.i. in TP1, greater than 40% of the viral population was composed of V4 loop deletion variants, increasing to 74% by 29 weeks p.i. In TP2, deletion variants emerged to greater than 40% by 14 weeks p.i. and eventually dominated the viral population from 23 to 29 weeks p.i. at a total frequency of 95% (C and D).
FIG 5.

Evolution of in-frame deletions in SIV Env V4 loop observed in TP1 and TP2. (A to D) Deletions in V4 and frequencies for animal TP1 (A and B) and animal TP2 (C and D). (A and C) Deletions are depicted relative to the V4 amino acid sequence of SIVmac239 and the consensus of the SIVmac251 stock inoculum. In-frame multiple-amino-acid deletions flank or overlap the P421 site (highlighted in red in the 239 and 251 V4 reference sequences). (B and D) The relative frequency of deletion variants detected in the reads spanning the entire V4 loop are reported at 10, 14, 18, 23, 25, and 29 weeks p.i. Reads lacking in-frame deletions were reported as having no deletions (in gray). (E) Viral RNA from MM001 and MM002 were deep sequenced as part of a separate study (A. K. Hill, S. Ita, R. Newman, and W. E. Johnson, unpublished data). The relative frequency of deletion variants detected in the reads spanning the V4 loop from amino acid positions 410 to 439 at 2 and 40 weeks p.i. are reported. Reads that did not possess any in-frame deletions were reported as no deletion (in gray).
We observed a unique pattern of deletions in the V4 loop in animal TP2; however, the emergence of deletions occurred much earlier, with deletion variants rising to a high frequency by 14 weeks p.i. (Fig. 5). Three deletion variants were detectable at 14 weeks: Δ422KERH425, Δ415DLTTQR420, and Δ423ERHR426. While Δ423ERHR426 was also observed in TP1, this deletion rose to dominate the viral population only in TP2, staying above a level of 75% from 18 weeks p.i. to 29 weeks p.i. and rising as high as 95.8% at 18 weeks p.i. It has been reported that deletions spanning P421 in V4 can result in loss of recognition by V4-specific antibodies (31). Thus, the emergence of Δ423ERHR426 in two animals in our cohort, the rise to high frequency early in infection, and the persistence of this deletion variant over several weeks of replication (to high levels within TP2) together suggest that early Env-specific antibody responses target either the V4 loop or a conformational epitope spanning the V4 loop region. Notably, TP2 maintained viral loads above 105 copies/ml during this period of early infection.
Patterns of substitution in V1.
In the V1 loop of the stock inoculum we found eight haplotypes, with one haplotype (I1) present at just over 50% (Fig. 6). I1 was found at transmission in every animal except SP1 but was not the dominant haplotype at acute infection in TP1 or TP2. TP1 haplotype I5, which was present at just 2.2% in the stock inoculum, was the dominant haplotype at 2 weeks p.i., at 90.2% (Fig. 6). In TP2, we found inoculum haplotype I3 present at 83.5% at week 2 p.i., and it rose to 99.3% by 3 weeks p.i. In both animals, all changes among haplotypes were tightly clustered within a patch of 13 codons of highly repetitive GCA, CCA, ACA, and TCA codons spanning positions 128 to 141 (commonly described as the hypervariable region of V1). Interestingly, at 6 weeks p.i., we observed a small, transient increase in V1 codon diversity at several sites due to the rise of single haplotypes that were present in the stock inoculum. In the case of TP2, haplotype I1 rose to a frequency of 39%, replacing I3 as the dominant haplotype at 8 weeks p.i. and maintaining dominance through 14 weeks p.i. before being replaced by novel haplotypes not found in the inoculum at 18 weeks p.i. In TP1, haplotypes I1 and I2 both increased in frequency at 6 weeks p.i., resulting in a small, transient increase in variation at multiple codons, including 127, 132, 139, and 142.
FIG 6.

Nucleotide repeats are associated with variation in V1 loop of TP1 during early infection. The frequency of haplotypes based on 90-nucleotide windows were determined from deep sequence reads of each sample that spanned the V1 loop region from position 115 to 144 and assembled to the SIVmac239 reference sequence. Haplotypes contained unique amino acid sequences, with inoculum haplotypes being found in the source inoculum and novel haplotypes being found in the viral population of posttransmission longitudinal samples. Haplotypes that contained novel N-linked glycosylation motifs are labeled with a green tree icon.
Over the course of early infection, we identified the emergence of novel haplotypes with a predominance of variation at serine, threonine, and alanine sites, specifically S127, S132, T135, T136, A138, and S139, in addition to one site outside this hypervariable region, K120 (Fig. 7; positions are identified as the consensus residue in the stock inoculum but based on SIVmac239 numbering). In TP1, substitutions at these sites followed a similar pattern of change, with all sites having a single dominant residue present at 100% at acute infection until the emergence of variants beginning at either 18 or 23 weeks p.i. Variation at positions 120, 132, 135, 136, and 138 resulted in a newly emerged codon replacing the ancestral codon present at peak viral load and becoming dominant in the viral population.
FIG 7.

Evolution of amino acid substitutions in SIV Env V1 loop in TP1 and TP2. The frequency of codons was determined from deep sequence reads of each sample assembled to the de novo consensus sequence of the same sample using the V-Profiler software program (Broad Research Institute). The six sites within the hypervariable region of V1 are plotted over the first 29 weeks of in vivo viral replication and are representative of sites with the highest codon variation for TP1 (A) and TP2 (B).
The dynamics of V1 variation in TP2 followed a pattern similar to that seen in animal TP1, with substitutions clustering within the same hypervariable region and at many of the same sites, including positions 120, 127, 132, 135, and 139 (Fig. 7). In fact, in contrast to the dynamics observed in TP1, the level of diversity in the hypervariable region of V1 in TP2 continued to increase from 14 weeks until 29 weeks p.i., the end of the sampled time period (data not shown).
Interestingly, threonine at position 136 in the V1 loop is coded for by ACG, whereas all other threonine codons in V1 are coded for by ACA. This ACG codon was present in the SIVmac251 stock inoculum and was maintained in three of four animals in our cohort (ACG was replaced by AAT by 29 weeks p.i. in TP1) and is observed in other SIVmac variants, including SIVmac239. Strikingly, during the large fluctuations in V1 length observed in TP2, this ACG codon was never lost or replaced, and no length variation in V1 was observed beyond this ACG codon in any animal in our cohort.
In contrast to TP1 and TP2, animal SP1 did not display a similar level of diversity through 13 weeks p.i. in Env (Fig. 3) or in the V1 and V4 loops of gp120 (data not shown). Interestingly, SP1 did mount a detectable antibody response similar to that of TP1 and TP2 (Fig. 1), thus the selective pressure of an Env-specific antibody response was present. An important difference in the viral infection of animal SP1 was a pronounced decrease in viral load over the course of infection. Similar to SP1, we did not observe the same level of Env diversity in RP1 at any time point up to 15 weeks in V1, yet this animal had no detectable antibody titers and maintained viral loads above 6 million copies/ml through 15 weeks of infection.
Patterns of substitution in V4.
As with V1, evolution of the SIVmac251 Env V4 loop during early infection included the emergence of substitutions at the same or similar sites in multiple animals and in analyses reported by others (Table 1), indicating that there is a reproducible and predictable pattern of evolution in response to selective pressures on Env. The mean diversity of V4 began to rise at 14 weeks p.i. in TP1 and as early as 10 weeks p.i. in TP2, but we found no increase in diversity in animals SP1 and RP1 during the same time period (data not shown).
Substitutions in V4 were tightly clustered within a region spanning nine amino acids from positions 415 to 424; notably, this is the same stretch of residues comprising the short deletions described in the previous section on indels in V4. Substitutions predominated at three sites in the V4 loops of TP1 and TP2: D415, P421, and R424 (Fig. 8). D415N introduces a novel N-linked glycosylation motif in the V4 loop of both TP1 and TP2.
FIG 8.

Evolution of sequence diversity in SIV Env V4 loop. The frequency of codons was determined from deep sequence reads of each sample assembled to the de novo consensus sequence of the same sample using the V-Profiler software program (Broad Research Institute). (A to D) Three sites within V4 are plotted over the first 29 weeks of in vivo viral replication of TP1 (A) and TP2 (B), over the first 15 weeks of in vivo viral replication in RP1 (C), and over the first 13 weeks of in vivo viral replication of SP1 (D) and are representative of sites with the highest codon variation in the V4 loop.
Position P421 experienced some of the highest variation within a single given site in gp120 (Fig. 8). Variation at P421 first emerged with a P421S substitution by 14 weeks in TP1 and 10 weeks in TP2. Strikingly, in TP1, we observed the emergence of four variants at position 421 in parallel by 18 weeks p.i. (P421S, P421T, P421L, and P421Q), with P421T becoming the dominant residue by the last sampled time point. P421S and P421L emerged in TP2, albeit to a lesser extent, likely due to the low frequency of variants without deletions in the V4 loop (Fig. 5). P421Q was previously defined as an antibody escape change (17); therefore, we hypothesized the degree of variation at position 421 was due to antibody targeting at or near the apex of the V4 loop region. We tested many of the adaptations observed at P421 for their effect on binding to Env-specific rhesus monoclonal antibodies (Rh MAbs; including P421A, P421L, P421Q, and P421S) and found they abrogated binding to several V4 loop-specific Rh MAbs (Fig. 9A). Further, we found that P421Q, in the context of fully replicating SIV, could increase neutralization resistance in a neutralization-sensitive background (Fig. 9B), supporting an earlier report that introducing P421Q into SIVmac239 conferred neutralization resistance to high-titer neutralizing plasma (17).
FIG 9.

Substitutions at V4 loop P421 site lead to a loss of recognition of V4 loop-specific MAbs, and P421Q confers neutralization resistance to V4 loop-specific MAbs in neutralization-sensitive background. (A) We tested the effect of SIVmac239 adaptations at position 421 on antibody recognition by a panel of Env-specific MAbs with specificity to the V1, V3, and V4 loops. Fifty nanograms of recombinant SIVmac239 gp140 WT or mutant protein were added to 96-well plates and incubated overnight at 4°C. Binding was measured over seven serial 6-fold dilutions of MAb in triplicate at a starting concentration of 10 μg/ml against soluble gp140 WT or mutant proteins. An HRP-conjugated anti-IgG antibody was used with TMB substrate for detection of binding. Absorbance values were read at 450 nm and experiments were performed in triplicate. (B) The level of virus neutralization of Env-specific MAbs was measured over 10 serial 2-fold dilutions of MAb against SIVmac316 WT or with SIVmac316 P421Q in triplicate. Ten nanograms of p27 of WT and mutant virus stock was used to infect 5 × 103 C8166-SEAP cells/well in a 96-well plate. MAb 3.11H targets a V3 linear epitope and was used as a control. Infectivity is reported as the level of SEAP activity above background from uninfected C8166-SEAP cells.
SIVmac251 possesses three closely spaced arginine residues, at positions 424, 426, and 428 in V4. Variation was observed at each of these positions, but only 424 had variation that rose above 5%. R424Q was observed in both TP2 and TP1 (Fig. 8).
Variation in V2 and at the base of the V3 loop.
Outside of the V1 and V4 loops, which exhibited the highest levels of change, other minor sites of notable variation included the V2 loop and just outside the base of the V3 loop. The emergence of adaptations in the V2 loop involved N-linked glycosylation sites (NLGS) in both animals TP1 and TP2 (Fig. 10A and B). Although variations differed between TP1 and TP2, we found the evolution of changes resulting in a loss of a NLGS in the V2 loop. For TP1 we observed a decrease in N202 from weeks 18 to weeks 25 p.i., with the concurrent emergence of a lysine and later a serine at this position (Fig. 10A). However, the frequency of N202 increased to 88.88% by 29 weeks p.i. Asparagine at position 202 was never lost from the population, therefore the NLGS at position 202 remained in the virus population over the course of the infection; however, a reduction of asparagine at this position could be interpreted as a loss in the frequency of variants in the population that contained an N-linked glycan at position 202. In TP2, we found a similar pattern of change, with asparagine at position 198 (N198), which contains an NLGS. Similar to N202 in TP1, we found a decrease in the frequency of N198 from 100% to 64.01% beginning at week 18 to week 25, which was then followed by an increase in N198 to 84.52% by week 29 p.i. (Fig. 10B). Interestingly, in TP2 we observed the emergence of a D201E substitution that became dominant by 29 weeks p.i. Again, similar to the V1 and V4 loops, we observed no changes at any of these positions in RP1 or SP1 over the course of the infection that we sampled (Fig. 10C and D).
FIG 10.

Evolution of substitutions in Env V2 and V3 loop of SIVmac251. The frequency of codons was determined from deep sequence reads of each sample assembled to the de novo consensus sequence of the same sample using the V-Profiler software program (Broad Research Institute). (A to D) Sites within V2 and V3 site W345 are plotted over the first 29 weeks of in vivo viral replication in TP1 (A) and TP2 (B), over the first 15 weeks of in vivo viral replication in RP1 (C), and over the first 13 weeks of in vivo viral replication in SP1 (D) and are representative of sites with the highest codon variation in V2 and V3 loops.
Although we did not detect variation in the V3 loop to the level found in the V1/V2 and V4 loops, we did find a single site just outside the base of the V3 loop, position 345, where a W345R substitution emerged in both TP1 and TP2 (Fig. 10). The same W345R substitution was also detected in patients infected with HIV-2 and was found to disrupt binding and increase neutralization resistance to Env-specific MAbs (42). In TP1, we found that two different codons for arginine emerged between 10 and 14 weeks p.i., suggesting strong selection for arginine at position 345. In TP2 the emergence of arginine (coded for by AGG) continued steadily to become the dominant residue by 29 weeks p.i. As in TP1, Arg (CGG) emerged but not to the level of Arg (AGG). The W345R substitution was not detected in RP1, which was sacrificed at week 15, or in SP1, the animal with low-set-point viral loads (Fig. 10).
A majority of amino acid substitutions that emerge in V1 and V4 do not impact viral replicative fitness.
When we tested individual V1 and V4 adaptations that arose in vivo in standard infectivity assays, we noted little or no impact on viral titer (data not shown). Similarly, Sato et al. tested a subset of these same changes (Env A138T, A417T, and P421Q), also in the context of SIVmac239, and found little or no impact on infectivity (17). This raises the possibility that these frequently occurring adaptations incur little or no fitness trade-offs, which could explain the rapidity with which they expand in the population in vivo. This is in contrast to many CTL escape mutations (45–51) and HIV drug resistance mutations (52–55), which often incur large trade-offs in viral replicative fitness. Adaptations that have little to no impact on viral replication are effectively neutral in the absence of antibody and therefore can rapidly contribute to immune escape and viral persistence (56, 57). However, standard infectivity assays usually are not sensitive enough to detect small (but significant) differences in replicative fitness and often do not measure the impact of mutations on complete viral replication cycles. In order to ask whether frequently selected adaptations we observed in vivo incur fitness costs and to quantify the impact of these changes on viral replication, we developed a highly sensitive viral fitness assay based on the use of deep-sequencing technology. We refer to this approach as FITseq.
We pooled single-point mutant virus stocks of V1 or V4 loop SIVmac239 mutants and infected two independently derived rhesus macaque T-cell lines with the pooled inocula at a low multiplicity of infection (MOI). Mutants were selected based on in vivo mutations observed in our cohort and others (Table 1), with the exception of V142I and A417T. In the case of V1421, valine is the wild-type (WT) residue in SIVmac239, but it was an isoleucine in the SIVmac251 swarm stock. We observed the I142V substitution in TP1 at transmission before reverting back to isoleucine at 29 weeks p.i. Therefore, we tested the V142I mutation in SIVmac239. In the case of A417T, this mutation results in the introduction of an NLGS motif at position 415 in SIVmac239, similar to the D415N mutation observed in SIVmac251. We next measured relative replicative capacity (RRC) by FITseq and found that nearly all single-point mutants tested had little to no effect on viral replicative fitness relative to the parental virus (Fig. 11). Specifically, none of the V1 substitutions resulted in RRC values (slopes) that differed significantly from 0 (Table 3). Of the four V4 substitutions tested, two did not differ significantly from 0. The remaining substitutions, P421S and P421Q, had very slight deviations from 0, and P421Q only differed in the Rh 221 cell line. We additionally cloned two multiple-amino-acid in-frame V4 loop deletions into SIVmac239 that emerged in two animals in our cohort and one which also emerged in two of three SIVmac239-infected macaques from another cohort (Fig. 5). Of note, no other deletions in the V4 loop were found in the viral populations of these two animals. When we pooled the two V4 loop deletion mutants, we found that deletion mutant 1 (ΔV4 1; Δ423EQHK426) had no effect on viral replicative fitness (for Rh 221, slope of −0.046, P = 0.719, R2 = 0.014; for Rh 444, slope of 0.050, P = 0.692, R2 = 0.016), while deletion mutant 2 (ΔV4 2; Δ418NQKPKEQH425) had a negative impact on viral replicative fitness (Rh 221, slope of −0.292, P = 0.104, R2 = 0.242; Rh 444, slope of −0.463, P = 0.020, R2 = 0.434) (Fig. 11 and Table 3).
FIG 11.

Viral replicative fitness of V1 substitutions, V4 substitutions, and V4 deletions relative to SIVmac239. Rhesus 221 and 444 immortalized T-lymphocyte cell lines were infected with a pooled mixture of SIVmac239 and Env single-point mutant virus stocks (A and B) or with a pooled mixture of SIVmac239 and Env V4 loop deletion mutant virus stocks (C) at a low MOI (≤0.01). Viral RNA extracted from mixed-infection supernatants was used as the template in RT-PCR using target region-specific primers. Amplicon products were barcoded to produce libraries that were deep sequenced by Illumina MiSeq. Reads were directly assembled to the SIVmac239 reference sequence. The frequency of reads with the introduced point mutation relative to the total number of reads was plotted over time for V1 loop mutants (A), V4 loop mutants (B), and V4 loop deletion mutants (C) in Rh 221 cells and Rh 444 cells. The results are reported as the means ± standard deviations (SD) from four replicates performed in parallel. Linear regression analysis was performed to determine the linear slope of the frequency of the mutant virus over time (reported as the relative replication capacity of the mutant virus), and the R2 value of each line and the P value of an F-test, testing whether the slope of the line is significantly different from zero, was determined.
TABLE 3.
Relative replication capacity of SIVmac239 single-point substitutions and in-frame deletionsa
| Mutation | Region | Rhesus 221 cells |
Rhesus 444 cells |
||||
|---|---|---|---|---|---|---|---|
| RRC | R2 | P value | RRC | R2 | P value | ||
| S132P | V1 loop | −0.131 | 0.373 | 0.061 | −0.040 | 0.063 | 0.432 |
| T136N | V1 loop | −0.206 | 0.020 | 0.664 | −0.083 | 0.269 | 0.084 |
| A138T | V1 loop | −0.173 | 0.073 | 0.396 | −0.036 | 0.051 | 0.482 |
| S139P | V1 loop | −0.087 | 0.027 | 0.609 | 0.056 | 0.086 | 0.380 |
| V142I | V1 loop | −0.224 | 0.015 | 0.705 | −0.011 | 0.005 | 0.830 |
| A417T | V4 loop | 0.040 | 0.028 | 0.604 | −0.023 | 0.015 | 0.704 |
| P421Q | V4 loop | 0.152 | 0.670 | 0.001 | 0.078 | 0.064 | 0.427 |
| P421S | V4 loop | −0.172 | 0.352 | 0.042 | −0.173 | 0.349 | 0.043 |
| P421T | V4 loop | 0.008 | 0.004 | 0.842 | 0.045 | 0.014 | 0.711 |
| ΔV4 1 | V4 loop | −0.046 | 0.014 | 0.719 | 0.050 | 0.016 | 0.692 |
| ΔV4 2 | V4 loop | −0.292 | 0.242 | 0.104 | −0.463 | 0.434 | 0.020 |
The RRC of single-point and multiple-amino-acid deletion mutants was calculated by linear regression analysis of mean frequency values. The R2 value of each line and the P value of an F-test, testing whether the slope of the line is significantly different than zero, is reported. Significant P values (<0.05) are reported in boldface.
Although no structural information exists for the SIV V1/V2 domain, the V4 loop is present in a crystal structure of the gp120 subunit (58). The structure places D415 near the base of the V4 loop and P421 at the middle of the V4 loop; both are exposed on the higher-order Env trimer (Fig. 12) (58). The D415N adaptation introduces an N-linked glycosylation site, which was not present in the stock population of the SIVmac251 swarm.
FIG 12.
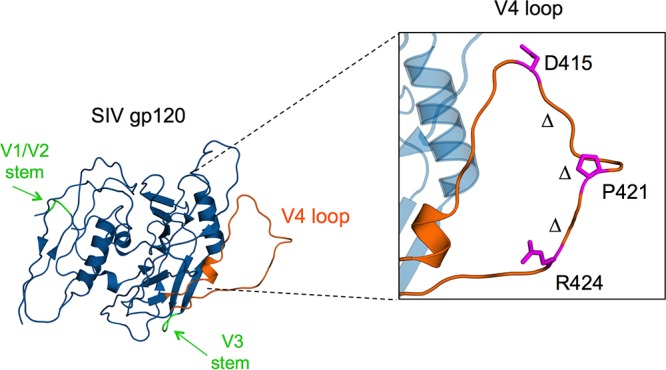
SIVmac gp120 V4 loop is the target of Env-specific antibodies, leading to substitutions and in-frame deletions. A crystal structure of the SIVmac gp120 subunit (58) showing the presence of the V4 loop (orange) on the external surface of the gp120 monomer where substitutions and in-frame deletions emerge rapidly during early SIV infection in macaque hosts. The D415 adaptation leads to a novel N-linked glycosylation site in the V4 region and is found near the apex of the V4 loop. P421 is a hot spot of adaptation, with several substitutions emerging at this site. The presence of a bend near the Pro site suggests adaptation leads to a loss of rigidity within the loop that is maintained by the Pro. An R424Q change was observed in two of four animals in our cohort and occurs near the bottom of the V4 loop. The Δ symbols denote the span of in-frame multiple-amino-acid deletions that emerged to become dominant in two animals (TP1 and TP2) by 18 to 23 weeks p.i. The image was produced with PDB entry 3FUS.
DISCUSSION
We took advantage of the resolution of Illumina ultradeep sequencing to examine the evolution of SIV env during acute and early infection and to track the initial emergence of changes in regions of Env targeted by antibody. Specifically, we tracked the in vivo sequence evolution of the SIV env gene in four rhesus macaques infected with an uncloned SIVmac251 stock using high-resolution Illumina deep sequencing of the SIVmac251 stock inoculum and longitudinal samples ranging from 2 to 29 weeks p.i. We captured the population bottleneck at the point of transmission from the inoculum into each animal and the subsequent emergence of Env diversity, defining the time frame of the earliest Env sequence variation in SIV populations. Furthermore, we tracked the appearance and spread of specific substitutions and in-frame insertions/deletions in the viral populations in each animal and found that a majority of changes were highly concentrated in two short stretches of sequence, one each in the surface-exposed V1 and V4 loops (which are known targets of Env-specific antibodies in SIV-infected macaques) (17, 31, 34, 37, 59). We also implemented a novel high-resolution virus competition fitness assay, FITseq, to ask whether any of these common changes associated with escape come at a cost to viral replicative fitness. Interestingly, we found that nearly all of the most common amino acid substitutions were neutral (although one high-frequency eight-amino-acid deletion in V4 did incur a replicative fitness cost when introduced into SIVmac239).
Variation in env manifested in a time frame that correlated with the appearance of host Env-specific antibody responses, evidence that the initial antibody response poses an early selective barrier to the SIV population. The repeated emergence of similar or identical adaptations in our cohort and in cohorts described in previous reports using older methodology reflects a pattern of variation that likely provides a mechanism of viral evasion from a targeted and constant antibody response. The observation that the same patterns of early adaptation occur in different animals (and in different studies) raises the interesting possibility that future vaccine strategies could be devised to redirect the early, strain-specific antibody response in order to block the most common pathways of escape, perhaps driving selection for lower-fitness viral variants.
We tracked the earliest SIV env sequence changes starting from an uncloned SIVmac251 stock inoculum in a cohort of four rhesus macaques, and in two of the four animals we tracked env evolution through week 29 postinfection. Env diversity of the SIVmac251 stock inoculum was present in both the gp120 and gp41 subunits of Env, with the level of diversity being similar to those of other uncloned stocks of SIVmac251 as measured by single-genome amplification (SGA) (60). In all animals, the transmission bottleneck led to a major T/F variant, found at frequencies of >0.9, and several minor T/F variants, collectively found at frequencies of <0.1. For many loci, the major and minor T/F variant haplotypes were at low frequencies in the stock inoculum. In particular, for two of four animals, the dominant V1 loop haplotypes found at acute infection were minor variants in the stock inoculum. Although the transmission of low-frequency inoculum variants was not statistically significant, our results demonstrate the importance of low-frequency inoculum variants in shaping early infection. Similar observations have recently been made for HIV-1 infections (61).
Previous analysis of HIV-1 transmission found variants with shorter V1/V2 loops (62, 63), and a recent study investigating signatures of HIV/SIV transmission in SGA sequences of several SIV and SHIV cohorts found shorter V1 and V4 loop regions in some, but not all, SIV cohorts (64). Here, we also observed shorter V1 and V4 loop lengths at transmission. In three of the four animals, the T/F variants contained inoculum haplotypes with short V1 loops, and all four animals contained inoculum haplotypes with short V4 loops, although the transmission of short V1 and V4 loops was not statistically significant. Future studies focusing on the T/F variants identified in our study could help to determine what fitness advantages, if any, these rare variants possess at transmission.
We identified the emergence of sequence diversity as early as 6 weeks p.i. in the V1 loop of the two typical progressor animals in the cohort, TP1 and TP2, due to the rise of inoculum haplotypes initially present at low frequency during acute infection. This indicates that diversity during early infection arose from fluctuation in variants already present in the viral population, as opposed to de novo variation. This transient increase in diversity was followed by the emergence of novel haplotypes (comprising substitutions, insertions, and deletions) beginning around 10 to 14 weeks p.i., when we detected the emergence of changes in several regions of gp120, including the V1/V2 loops, the base of the V3 loop, and the V4 loop.
Despite the length of the V1 and V4 loops, the highest levels of diversity within V1 and V4 were surprisingly limited to short patches (10 to 12 residues) of surface-exposed sequence. We captured the rise of variation within the Ser/Thr-rich stretch of V1 that was consistent with the selection of O-linked glycosylation attachment sites predicted to exist in the V1 loop of SIVmac (and SIVsm strains) (43, 65). As mentioned in the results, Thr at position 136 is encoded by codon ACG, in contrast to all other Thr residues in the V1 loop, which are coded by ACA. We speculate that the ACA/TCA/GCA/CCA codon-rich region in V1 could be more prone to slippage of the polymerase (RT) during reverse transcription, generating short in-frame insertions and deletions and providing a mechanism for rapid generation of variants and adaptation. If true, perhaps the rare ACG codon at position 136 delimits the region of elevated insertion/deletions; however, this mechanism would need to be verified through further investigation. Several substitutions also emerged in V1 at low frequencies (>5%) and then increased to replace the ancestral residue, becoming the majority variant, most notably between V1 residues 132 and 139. When we tested several V1 substitutions for their effect on viral replicative fitness by FITseq, we found these mutations incurred little to no viral fitness cost, including A138T, a known NAb escape adaptation (17) that also emerged independently in two animals in our cohort. A structure of the HIV-1 V1/V2 domain in complex with neutralizing antibody PG9 revealed that much of the V1/V2 domain is arranged in secondary beta sheets, such that only a limited number of residues actually reside in unstructured loops (giving rise to the hypervariation observed in variable loop regions, as we observed during early infection).
Adaptations at sites of either NAb escape or novel N-linked glycosylation sites in V4 occurred as early as 10 to 14 weeks p.i. in TP1 and TP2 but were not observed to the same degree in RP1 through 15 weeks p.i. The highest degree of variation among any single site in our study occurred at NAb escape site P421, with several adaptations emerging in parallel in TP1 and three of these same adaptations (P421L, P421S, and P421T) similarly emerging in TP2 (Fig. 8). Introduction of these changes individually into recombinant SIVmac239 gp140 abrogated binding by several V4 loop-specific Rh MAbs (Fig. 9A). Interestingly, several P421 adaptations, with the exception of P421S, resulted in little to no viral fitness cost, similar to substitutions in V1. These results are evidence that commonly occurring changes within the variable loop regions of SIV Env incur no cost in terms of inherent replication capacity, including several adaptations previously shown to confer escape from Env-specific antibodies (17).
Single-site substitutions were followed by the emergence of multiple-amino-acid in-frame deletions that spanned the apex of the V4 loop and lead to a loss of binding to Env-specific MAbs (31). Several V4 deletion variants increased in frequency to become the dominant variants in the viral populations of both TP2 and TP1 by 18 to 23 weeks p.i. Variants with the 8-amino-acid deletion Δ418TQKPKERH425 were found in TP1, and a deletion spanning the same residues (418 to 425) was found at chronic time points in two animals from an unrelated SIVmac239-infected cohort (Fig. 5E). Unlike single substitutions, deletion Δ418TQKPKERH425 had a viral replicative fitness cost (Fig. 11 and Table 3), supporting previous evidence that deletions of the entire V1, V2, V3, or V4 loop significantly reduce replication and infectivity in vitro (66–70) and result in increased sensitivity to antibody in vivo (69).
The rapid time frame in which these V4 loop deletions emerge and rise to dominance is striking evidence that deletion variants are continuously selected over multiple weeks, despite in some instances incurring a replicative fitness cost. Taken together with the highly exposed nature of the V4 loop (Fig. 12 and reference 58), adaptations within this short span of the loop may represent a key strategy toward maintaining replicative fitness while balancing the need to adapt to continuous antibody selection. The short stretches of sequence within V1 and V4 experienced an overwhelming majority of variation over the course of early infection in two of our animals, suggesting these two patches of SIV gp120 have arisen as an evolutionary answer to deal with constant targeting by antibody, unlike adaptations in functionally critical regions, such as the CD4 binding site, which do incur replicative fitness costs to escape from broadly neutralizing antibodies that target the CD4 binding site (71).
We did not observe substantial variation in the V3 loop itself, consistent with previous reports of variation in SIV Env (17, 29, 30), but did observe the emergence of substitution W345R at the base of the V3 loop. The W345R substitution was also reported in HIV-2 Env sequences, and the alanine substitution mutant, W345A, conferred neutralization resistance to several distinct groups of MAbs targeting the base of the V3 loop, suggesting that primate lentiviruses evolve along shared sequence pathways that lead to antibody escape and viral persistence (42). Included in the epitope of several broadly neutralizing antibodies from the PGT class is an N-linked glycan at the base of the V3 loop in HIV-1, providing further evidence that this region of Env is a common target among primate lentiviruses (72–74).
SIVmac251 infection of macaques parallels many pathogenic features of HIV-1 infection and thus is a relevant model for HIV-1 pathogenesis. Although HIV-1 and SIV Env share only 30% homology at the amino acid level, they are structurally and functionally highly conserved. Further, changes within SIV and HIV-1 Env, due to antibody selection, are found within similar regions. Our study highlights the kinetics of Env changes that occurred during sustained antibody responses in these animals. Neutralization titers were measured against laboratory-adapted SIVmac251, but we did not detect neutralization of the more neutralization-resistant (so-called tier III) SIVmac239 (data not shown); thus, variation observed here during early infection may reflect selection from nonneutralizing and strain-specific neutralizing responses. This observation is of note, given the identification that nonneutralizing antibodies, specific to the V2 loop, correlated to partial protection in the RV144 trial and was recapitulated in the SIV/macaque model (75–79).
Reports of host antibody and HIV-1 Env coevolution indicate that Env is highly adaptable in response to neutralizing antibodies, specifically those targeting the V1/V2 domain (71, 80, 81). However, very few studies have directly measured fitness costs of these adaptations in HIV-1, and none have done so for SIV. Early reports of HIV-1 variants both resistant and sensitive to broadly neutralizing antibodies reported no differences in replicative capacity (56, 57). However, a focused study of HIV-1 variants isolated during early infection (<6 months) in three individuals with NAb responses found fitness differences ranging from no cost to detectable reduction in replicative fitness, similar to our results (7). Bar et al. found adaptations mapped to V1, V2, and the base of V3, suggesting that primate lentiviruses use similar pathways of escape to antibody during early infection. Further, escape from broadly neutralizing antibodies that target highly conserved regions, such as the CD4 binding site, do incur fitness costs (71, 82, 83). Interestingly, Lynch et al. identified compensatory changes that restored fitness deficits incurred from escape to broadly neutralizing antibody VRC01 (71). Although our analysis of viral fitness costs was limited to the V1 and V4 regions of SIV Env, our results are in line with a recent exploration of the landscape of fitness costs in HIV-1 within Env (84).
One animal, SP1, had antibody responses similar to those of animals TP1 and TP2, yet we failed to detect any env variation in SP1 up to 13 weeks p.i. Again, we detected CTL responses in SP1 directed to Gag and Env by ELISPOT assay (Fig. 1); however, TP1 and TP2 did not elicit similar responses. Thus, the presence of low viral loads after peak of viral replication during acute infection and the detection of both cellular and antibody-based humoral adaptive immune responses suggests SP1 was able to mount an adaptive immune response leading to control of viral replication, resulting in low Env diversity. The transmitter/founder strain in SP1 differed from the other animals in the cohort in specific regions of Env, including the V1 loop. Unlike the other animals in the cohort, the dominant haplotype in the V1 region at acute infection contained a Pro-Thr-Ala amino acid insertion between positions 132 and 133 in the V1 loop. If the state of the viral population during acute infection represented a decreased viral fitness as a result of these changes, then one possibility is that the virus population was not able to mount the necessary adaptations from this low fitness state to mediate escape prior to adaptive immune responses. Accurately assessing the impact of early variants on subsequent escape will require a broader analysis of substantially larger cohorts and/or experiments based on genetically defined viral strains or mixtures of defined viral strains.
A limitation of our study was linkage among mutations could only be assessed within windows of 90 nucleotides due to short read lengths. Other approaches, such as limiting-dilution single-genome amplification, could link mutations across env with the caveat of lower coverage. Future studies of SIV env evolution could use long read sequencing technologies, such as the Pacific Bioscience single-molecule real-time platform, to overcome these limitations and assess the coevolution of mutations across env identified here.
Strain-specific antibodies evolve early during SIV and HIV infection, selecting for sequence variants that evade antibody through modification of immunodominant epitopes. Our analysis of viral populations during the acute and early stages of SIV infection revealed a characteristic pattern of sequence evolution at common sites within gp120, predominately V1 and V4, with additional variation in V2, V3, and the CT of gp41. Overall, substitutions in the V1 and V4 loop incurred little or no replicative fitness cost. Thus, our results and those reported in other SIV models (Table 1) suggest that there is a temporally and mutationally consistent pattern of early Env evolution involving a finite number of sites (predominately clustered in small stretches of V1 and V4) that can respond rapidly to the early, immunodominant antibody response. Due to their low impact on viral fitness, substitutions at these sites are common in the viral population, and their presence may allow rapid escape as early, strain-specific antibody responses arise. This also raises the intriguing possibility that such patterns ultimately could be predictable, perhaps leading to vaccine strategies that harness the narrow, strain-specific antibody response to block or suppress viral replication before emergence of diversity and establishment of persistent reservoirs.
MATERIALS AND METHODS
Viruses and plasma.
All plasma samples described in this study were obtained from archived material at the New England Regional Primate Research Center. Rhesus macaque monkeys (Mm) 10-10, 198-08, 156-10, and 174-08 were unvaccinated control animals challenged with the uncloned SIVmac251 (swarm) as part of an unrelated vaccine study. All four animals had been challenged with six doses of the SIVmac251 swarm at 300 50% tissue culture infective doses (TCID50) by rectal challenge. The SIVmac251 stock was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH.
Ethics statement.
The animals from which the archived material was originally obtained as part of a previous study were housed at the New England Primate Research Center (NEPRC) of Harvard Medical School and given care in accordance with standards of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and the Harvard Medical School Animal Care and Use Committee. The study was approved by the Harvard Medical Area Standing Committee on Animals, within the Office for Research Subject Protection at Harvard Medical School, and conducted according to the principles described in the Guide for the Care and Use of Laboratory Animals (85). The study was assigned IACUC protocol number 04660.
The Harvard Medical School animal management program is accredited by the AAALAC and meets National Institutes of Health standards as set forth in the Guide for the Care and Use of Laboratory Animals (85). The institution also accepts as mandatory the PHS Policy on Humane Care and use of Laboratory Animals by awardee institutions (86) and the NIH Principles for the Utilization and Care of Vertebrate Animals used in Testing, Research and Training (87). An approved assurance of compliance is on file with the Office of Protection from Research Risks.
Animals in the NEPRC receive a daily health check by both animal care technicians and veterinary professional staff. All animals receive a complete physical examination an average of once every 4 to 6 weeks. In the event that clinical disease is recognized, the attending veterinarian and principal investigator are notified. The animal is examined, the medical record reviewed, and laboratory work repeated if needed. A plan of action is agreed upon between the veterinarian and principal investigator that may involve continued observation, treatment, or euthanasia of the animal. All animals housed in the biocontainment facility have strict criteria for euthanasia, which are determined prior to initiation of experiments through consultation between the attending veterinarians, principal investigators, and members of the Harvard Medical School IACUC.
The NERPRC developed a comprehensive environmental enrichment and psychological well-being plan for primates, which is available for inspection by the United States Animal and Plant Health Inspection Service (APHIS) and by officials of any pertinent funding agency. The Harvard Medical School Standing Committee on Animals (IACUC) must approve an investigator's request for exemption of animals from these plans. In addition, the attending veterinarian may exempt individual animals for health-related reasons. The IACUC documents NERPRC compliance with the plan during semiannual facility inspections.
If warranted, animals may receive standard clinical care, including but not limited to analgesics (buprenex at 0.005 mg/kg of body weight intramuscularly twice a day), intravenous fluids, antibiotics, and other supportive therapy. If animals develop AIDS, euthanasia is performed based on the following criteria: (i) weight loss of >15% in 2 weeks or >30% in 2 months; (ii) documented opportunistic infection; (iii) persistent anorexia for >3 days without explicable cause; (iv) severe, intractable diarrhea; (v) progressive neurologic signs; (vi) significant cardiac and/or pulmonary signs; and (vii) any other serious illness.
The final decision to perform euthanasia is at the discretion of the attending veterinarian and is made following consultation with the principal investigator, review of pertinent laboratory and virological data tests, and physical examination of the animals. Euthanasia is performed following induction of anesthesia with 15 mg/kg ketamine HCl. Sodium pentobarbital (≫50 mg/kg) is administered intravenously. These methods are consistent with the recommendations of the panel on euthanasia of the American Veterinary Medical Association.
Cell lines.
The human embryonic kidney (HEK) cell line 293T/17 was used for transient transfection of full-length SIV plasmids for production of virus stocks of SIVmac239 and protein expression plasmids for recombinant SIVmac239 gp140 protein. HEK293T/17 cells (ATCC ACS-4500, RRID CVCL_4V93) were obtained from the American Type Culture Collection (ATCC; Manassas, VA) and maintained in D10 cell culture medium consisting of Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (FBS) at 10% of the total volume. Human CD4+ C8166 cells engineered to express secreted alkaline phosphatase (C8166-SEAP), as previously described (88), were used for titration of virus infectivity and for virus neutralization assays. C8166-SEAP cells were maintained in R10 medium consisting of Roswell Park Memorial Institute (RPMI) medium supplemented with FBS at 10% of the total volume. TZM-bl cells were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from John C. Kappes, Xiaoyun Wu, and Tranzyme, Inc. (89–92), and were used for titration of viral stocks. The 221 and 444 rhesus immortalized T-lymphocyte cell lines were derived from peripheral blood T cells from rhesus monkeys infected with SIV strains as previously described (93, 94). These cells were maintained in R20 supplemented with interleukin-2.
Cloning of SIVmac239 Env mutants using full-length SIVmac239 viral expression plasmids.
The full-length SIVmac239 expression plasmid p239flSpX and the subclone p239-3′half have been described previously (17, 95). Single point mutations were introduced by site-directed mutagenesis of the p239-3′half plasmid in the env open reading frame and sequenced to verify the desired mutation and the absence of unintended mutations. Full-length single-substitution plasmids were produced by restriction digest of p239-3′half mutant clones with SphI and PmlI and subsequent purification and ligation of the 1.5-kb fragment into the respective sites in p239flSpX. All full-length plasmids were sequenced to verify the absence of any unintended mutations.
Transfection and production of virus stocks.
Virus stocks were produced by transient transfection of 5.5 × 106 HEK293T/17 cells with 10 μg of full-length virus plasmid DNA in D3 medium. Four hours posttransfection, the D3 medium was replaced with fresh D10 medium. Three days posttransfection, the cell supernatant was harvested and centrifuged at 3,000 rpm for 5 min to remove cellular debris. Virus-containing supernatants were aliquoted at 1 ml or 500 μl and immediately stored at −80°C. The virus content of the supernatant stocks was determined by an SIV p27 antigen capture assay kit (Advanced Bioscience Laboratories, Rockville, MD) by following the manufacturer's protocol.
vRNA isolation.
SIV viral RNA (vRNA) was extracted from plasma of SIVmac251-infected rhesus macaques and the uncloned SIVmac251 stock inoculum using a high pure viral nucleic acid kit (Roche) per the manufacturer's protocol. Each sample was processed individually and in chronological order, starting with the SIVmac251 inoculum, in order to prevent cross-contamination. vRNA was eluted in 50 μl of elution buffer, aliquoted, and immediately stored at −80°C.
RT-PCR amplification and library preparation for Illumina deep sequencing.
A unidirectional flow of sample processing was followed for DNase treatment and RT-PCR amplification. RT-PCR, DNase, and Taq control reactions were prepared in a reagent-only hood. DNase treatment and RT-PCR amplification were then carried out in a separate RNA/DNA template-only hood. vRNA samples were thawed from −80°C, and immediately prior to RT and PCR amplification, 8 μl of vRNA was DNase treated using amplification-grade DNase I, followed by a 10-min incubation with EDTA (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Two primer pairs (F1, 5′-GGCCTTCGAATGGCTAAACAG-3′; R1, 5′-CCTGCCTTAACTTAGCTAGC-3′; F2, 5′-TGCACAGGCTTGGAACAAGA-3′; R2, 5′-ACATCCCCTTGTGGAAAG-3′) were synthesized with a 5′ amino modifier C6 (Integrated DNA Technologies) and used in RT-PCRs to produce two overlapping 2.8-kb amplicons that spanned the entire SIV env open reading frame. F1 and R1 were used to produce amplicon 1, and F2 and R2 were used to produce amplicon 2. F1 and R1 were initially reported by Bixby et al. (96) as 412Eyu and 413EYd, respectively, and were designed to amplify SIVmac251 viral RNA. Four to 5 μl of DNase-treated vRNA was reverse transcribed and PCR amplified using the Qiagen OneStep RT-PCR kit (Qiagen). Thermal cycling conditions were 45°C for 120 min and then 95°C for 15 min, followed by 40 cycles of 94°C for 15 s, 50°C for 30 s, and 68°C for 6 min, with a final extension step of 68°C for 6 min. In parallel, 1 μl of DNase-treated vRNA was amplified using the Premix Taq DNA polymerase (Ex Taq version 2.0) (TaKaRa) as a DNA contamination control using the same cycling conditions. An RT-PCR with water instead of template was also performed with each primer pair as a contamination control in parallel with each RT-PCR. For each sample, 5 μl of the RT-PCR and Taq reactions was analyzed on a 1% agarose 1× Tris-acetate-EDTA (TAE) gel to verify the presence of product in RT-PCRs and the absence of any product in the Taq and water control reactions.
Samples were then purified using the QIAquick PCR purification kit (Qiagen, Germantown, MD) per the manufacturer's protocol. Samples were analyzed for DNA concentration using a NanoDrop Lite spectrophotometer (Thermo Scientific, Carlsbad, CA), and approximately 1 ng of each reaction was pooled for shearing, tagmentation, and library preparation using the Nextera XT DNA library preparation kit (Illumina, San Diego, CA). Each sample was uniquely indexed using the Nextera index primer kit (Illumina, San Diego, CA), and the pooled samples were sequenced in a single run on an Illumina MiSeq using a 300-cycle MiSeq v2 kit (Illumina, San Diego, CA).
Illumina sequence assembly and variant calling.
For animal Mm 198-08, we captured deep sequence data spanning the entire env open reading frame from 11 longitudinal time points (weeks 2, 3, 4, 6, 8, 10, 14, 18, 23, 25, and 29). For animal Mm 10-10, we captured deep sequence data from 10 longitudinal time points (weeks 2, 4, 6, 8, 10, 14, 18, 23, 25, and 29). For animals Mm 156-10 and Mm 174-08, sampling was limited to four longitudinal time points. Animal 156-10, which progressed to disease rapidly, was sacrificed early at 15 weeks postinfection (p.i.); however, we captured deep sequence data at 3, 7, 11, and 15 weeks postinfection. In contrast, Mm 174-08 had continuously decreasing viral loads throughout the course of infection. We captured deep sequence data from 2, 3, 7, and 13 weeks of infection. Paired-end sequence reads (150 bp) for each sample were assembled de novo to produce consensus sequences using the VICUNA software program (97). Variant calling and frequency determination were performed using V-Phaser 2 (98, 99) to compare reads to the consensus sequences generated from the SIVmac251 stock inoculum.
We inferred the number and frequencies of transmitter/founder (T/F) variants through two approaches. First, we considered the number of variants within 90-nucleotide windows spanning Env. For each 90-nucleotide window, we collected spanning reads, called variants, and then counted the number of unique variants. Second, we applied an NGS haplotype reconstruction algorithm, RegressHaplo (44). Both approaches were used to estimate the number of T/F variants.
Virus infectivity assay.
For viral competition experiments, we wanted to carry out viral infections at low multiplicities of infection (MOI). To define the number of infectious virions per milliliter of virus stock, 20 μl of virus-containing supernatant was serially diluted in 180 μl of R10 medium (1:10 dilutional series). One hundred μl of each diluted stock then was added to 100 μl of R10 medium containing 1 × 103 TZM-bl cells, which contain a stably integrated Tat-responsive long terminal repeat (LTR) promoter driving beta-galactosidase and luciferase activity. Infections were performed in 96-well plates. Seventy-two hours postinfection, cellular supernatants were removed and the number of TZM-bl cells expressing beta-galactosidase determined using a beta-galactosidase staining kit (Clonetech Laboratories, Mountain View, CA) by following the manufacturer's protocol. Cells positive for beta-galactosidase were counted over several dilutions to determine the number of infectious virions per milliliter of virus stock.
Deep-sequencing-based viral fitness assay (FITseq).
All SIVmac239 virus stocks were produced and titrated for infectivity as described above. For viral competition experiments, viral stocks of SIVmac239, our WT reference strain, were pooled with a single-substitution SIVmac239 Env mutant (dual infection) or multiple mutants (pooled infection) immediately prior to infection of immortalized rhesus T-cell target cells at an MOI of 0.01 or lower. Infections were performed in 96-well plates with 2 × 105 cells in a final volume of 500 μl of R10 medium. Fifty μl of viral supernatant was harvested every 12 h for 72 h and replaced with fresh R10 medium. Harvested supernatants were stored at −80°C. We extracted vRNA from supernatants using a ZR-96 viral RNA kit (Zymo Research Corp., Irvine, CA) per the manufacturer's protocol. vRNA was eluted in 18 μl of elution buffer and immediately stored at −80°C. Prior to RT-PCR amplification, viral RNA was DNase treated using amplification-grade DNase I, followed by a 10-min incubation with EDTA (Invitrogen, Carlsbad, CA), according to the manufacturer's protocol. Two target region-specific primer pairs were used to amplify ∼150-bp amplicons that spanned the mutation of interest within the V1 (V1 forward, 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCCCATTATGCATTACTATGAGATGCAATAAAAGTGAGACAG-3′; V1 reverse, 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCTTGTTCCAAGCCTGTGCAATTATCCTGGGC-3′) and V4 (V4 forward, 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGGACAAATTGCAGAGGAGAGTTCCTCTACTG-3′; V4 reverse, 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGAGGCAAATAAACATTTTTGCCTACTTTATGCC-3′) regions of SIV Env gp120. The sequence in boldface corresponds to adapter overhang sequences added to the env gene-specific sequences. The adapter sequences were included for subsequent library index PCR amplification. Five μl of DNase-treated vRNA was reverse transcribed and PCR amplified using the Qiagen OneStep RT-PCR kit (Qiagen, Valencia, CA). Thermal cycling conditions were 45°C for 120 min; 95°C for 15 min; 40 cycles of 94°C for 15 s, 50°C for 30 s, and 68°C for 6 min; and a final extension step of 68°C for 6 min. In parallel, 1 μl of DNase-treated vRNA was amplified using the Premix Taq DNA polymerase (Ex Taq version 2.0; TaKaRa, Mountain View, CA) as a DNA contamination control using the same cycling conditions. An RT-PCR with water instead of template was also performed with each primer pair as a contamination control in parallel with each RT-PCR. Five μl of the RT-PCR and Taq reaction mixtures were analyzed for each sample on a 1% agarose 1× TAE gel to verify the presence of product in RT-PCRs and the absence of any product in the Taq and water control reactions. Samples then were purified using Agencourt AMPure XP beads (Beckman Coulter, Danvers, MA) per the manufacturer's protocol.
RT-PCR products from experiments performed in parallel were uniquely barcoded by a second round of PCR using Nextera index primers (Illumina, San Diego, CA) according to the manufacturer's protocol. Thermal cycling conditions were 95°C for 3 min, followed by 8 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, with a final extension step at 72°C for 5 min. Following the index PCR, samples were purified using Agencourt AMPure XP beads (Beckman Coulter, Danvers, MA) per the manufacturer's protocol. Each sample was normalized using the SequalPrep normalization plate kit (Invitrogen, Carlsbad, CA), and ∼1 ng of DNA from each sample was pooled and subjected to deep sequencing using a 300-cycle MiSeq v2 kit (Illumina, San Diego, CA) on an Illumina MiSeq instrument to produce 150-bp paired-end reads. Reads were assembled to the SIVmac239 reference genome (GenBank accession number M33262), and the frequency of reads that mapped to the WT or to the mutant were calculated to determine the WT-to-mutant ratio. The frequency of mutant reads was plotted over time, and linear regression analysis was used to determine the slope. As the frequency of each mutant was relative to that of the WT, the slope of the mutant line was reported as a direct measure of relative fitness (relative replication capacity) (46). An F-test was performed to determine if the slope of the mutant line was significantly different from zero. The P value of the F-test and the R2 value for each line are reported.
Virus neutralization assays.
Neutralization assays were performed to measure the neutralizing activity of Rh MAbs against SIV strains SIVmac239 and SIVmac316 and to measure the effect of substitutions in SIV gp120 on neutralization sensitivity against Rh MAbs in the context of neutralization-sensitive SIVmac316. Neutralization assays were performed using a C8166-derived CD4+ T-cell line transduced with an alkaline phosphatase reporter gene under the transcriptional control of the SIV LTR promoter, as previously described (88). Neutralization was determined as the level of inhibition of SEAP activity over serial 4-fold dilutions of MAb. Different amounts of virus input were used in experiments based on infectivity of SIVmac239 and SIVmac316. Virus equivalent to 1.5 ng p27 was used for experiments with SIVmac239 WT, and 10 ng p27 was used for experiments with SIVmac316 or single-substitution SIVmac316 mutants.
Neutralization assays were performed in 96-well plates using 5,000 C8166-SEAP cells per well. Cells in column 1 were incubated with no virus, providing a background level of SEAP activity. Cells in column 2 received virus but no MAb, providing a maximal level of SEAP activity. First, an appropriate volume of R10 medium was added to all wells. Column 3 next received MAb at a starting concentration of 10 μg/μl, followed by nine 4-fold serial dilutions across the plate to column 12. Virus then was added to columns 2 to 12 in a volume of 50 μl, and the plates were incubated for 1 h at 37°C at 5% CO2 levels. After 1 h, 5,000 SEAP cells were added to every well in a volume of 75 μl, bringing the final volume in each well to 200 μl. The level of SEAP activity in the supernatant was measured after 72 h for wells that received SIVmac239. SEAP activity was measured after 5 days for wells that received SIVmac316 or 316 Env mutant viruses. SEAP activity was measured using the Phospha-Light SEAP detection kit (Applied Biosystems, Foster City, CA) according to the manufacturer's protocol.
Recombinant soluble gp140 protein production.
A codon-optimized expression vector for soluble, recombinant SIVmac239 Env gp140, which contains the gp120 subunit and the N-terminal ectodomain of gp41 but contains a stop codon prior to the TM region, was described previously (100). SIVmac239 gp140 proteins were found to express as stable soluble trimer proteins, similar to membrane-associated Env trimers (100). Soluble recombinant expression-optimized SIVmac239 gp140 was synthesized by transfection of HEK293T/17 cells with SIVmac239 gp140 expression vectors. Cellular supernatants were harvested 2 days posttransfection and 5 days posttransfection. Supernatants were pooled and centrifuged to remove cellular debris, passed through a 0.45-μm filter, and purified using Galanthus nivalis lectin-agarose beads. To confirm expression of SIVmac239 gp140 proteins, we separated the purified nonbiotinylated proteins on a denaturing SDS-PAGE gel under reducing conditions and probed for the presence of soluble gp140 through Coomassie stain and by Western blotting using Rh MAb 3.11H, which recognizes a linear epitope in the V3 loop of SIV gp120 (data not shown).
SIVmac239 gp140 binding by Rh MAbs by ELISA.
Wells of a 96-well plate were coated with 50 ng SIVmac239 WT or mutant gp140 proteins overnight at 4°C. The following morning the plates were washed and blocked with 1× phosphate-buffered saline (PBS) with 5% FBS for 2 h at 37°C at 5% CO2. Plates were washed again before adding a serial 6-fold dilution of the MAb being tested for binding at a starting concentration of 10 μg/μl. The plates then were incubated for 1 h at 37°C at 5% CO2 and washed again, and horseradish peroxidase (HRP)-conjugated goat anti-human IgG was added as a secondary antibody (Santa Cruz Biotechnology, Dallas, TX). The plates were incubated again at 37°C for 1 h. Binding of the MAb to gp140 was measured with the addition of TMB substrate followed by stop solution, and absorbance was read at 450 nm using a Victor X5 plate reader (PerkinElmer, Waltham, MA).
IFN-γ ELISPOT assay.
Measurement of virus-specific T cell responses by enzyme-linked immunospot assays has been previously described (101). Briefly, 200,000 cells corresponding to PBMC, bone marrow (BM), spleen, axillary lymph node (AxLN), and inguinal lymph node (InLN) were seeded in duplicate in an IFN-γ precoated 96-well plate using a monkey IFN-γ ELISpotPRO kit (Mabtech, Mariemont, OH). Cells were stimulated with peptide pools (15-mers overlapping by 11 amino acids, at 2.5 μg/ml for each peptide). RPMI medium was used as a background control, while anti-human CD3 monoclonal antibody was used as a positive control. PBMCs were tested with samples corresponding to day of challenge and 3 weeks postchallenge. Tissue samples (BM, AxLN, spleen, and IgLN) were collected at necropsy, corresponding to 15 weeks postinfection for RP1, 30 weeks postinfection for Mm 174-08, and 29 weeks postinfection for Mm 10-10 and Mm 198-08.
Accession number(s).
All nucleotide sequences generated in this study, including the SIVmac251 stock inoculum and each longitudinal animal sample, were submitted to the Sequence Read Archive under accession numbers SRR4372378 to SRR4372405, SRR4372436, and SRR4372459.
ACKNOWLEDGMENTS
We thank Patrick Autissier for flow data acquisition, Brigitte Lawson for assistance with structural images, and Andrea Kirmaier for critical readings of the manuscript. Viral load data were generated by Jeff Lifson and Michael Piatak, Jr. (NCI Frederick).
REFERENCES
- 1.Johnson WE, Desrosiers RC. 2002. Viral persistence: HIV's strategies of immune system evasion. Annu Rev Med 53:499–518. doi: 10.1146/annurev.med.53.082901.104053. [DOI] [PubMed] [Google Scholar]
- 2.Evans DT, Desrosiers RC. 2001. Immune evasion strategies of the primate lentiviruses. Immunol Rev 183:141–158. doi: 10.1034/j.1600-065x.2001.1830112.x. [DOI] [PubMed] [Google Scholar]
- 3.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 4.Mansky LM, Temin HM. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol 69:5087–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH, Saag MS, Shaw GM. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 6.Allen TM, O'Connor DH, Jing P, Dzuris JL, Mothe BR, Vogel TU, Dunphy E, Liebl ME, Emerson C, Wilson N, Kunstman KJ, Wang X, Allison DB, Hughes AL, Desrosiers RC, Altman JD, Wolinsky SM, Sette A, Watkins DI. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407:386–390. doi: 10.1038/35030124. [DOI] [PubMed] [Google Scholar]
- 7.Bar KJ, Tsao CY, Iyer SS, Decker JM, Yang Y, Bonsignori M, Chen X, Hwang KK, Montefiori DC, Liao HX, Hraber P, Fischer W, Li H, Wang S, Sterrett S, Keele BF, Ganusov VV, Perelson AS, Korber BT, Georgiev I, McLellan JS, Pavlicek JW, Gao F, Haynes BF, Hahn BH, Kwong PD, Shaw GM. 2012. Early low-titer neutralizing antibodies impede HIV-1 replication and select for virus escape. PLoS Pathog 8:e1002721. doi: 10.1371/journal.ppat.1002721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MB, Shaw GM. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med 3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 9.Chung AW, Isitman G, Navis M, Kramski M, Center RJ, Kent SJ, Stratov I. 2011. Immune escape from HIV-specific antibody-dependent cellular cytotoxicity (ADCC) pressure. Proc Natl Acad Sci U S A 108:7505–7510. doi: 10.1073/pnas.1016048108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med 3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 11.Moore PL, Ranchobe N, Lambson BE, Gray ES, Cave E, Abrahams MR, Bandawe G, Mlisana K, Abdool Karim SS, Williamson C, Morris L. 2009. Limited neutralizing antibody specificities drive neutralization escape in early HIV-1 subtype C infection. PLoS Pathog 5:e1000598. doi: 10.1371/journal.ppat.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mudd PA, Ericsen AJ, Walsh AD, Leon EJ, Wilson NA, Maness NJ, Friedrich TC, Watkins DI. 2011. CD8+ T cell escape mutations in simian immunodeficiency virus SIVmac239 cause fitness defects in vivo, and many revert after transmission. J Virol 85:12804–12810. doi: 10.1128/JVI.05841-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Connor DH, Allen TM, Vogel TU, Jing P, DeSouza IP, Dodds E, Dunphy EJ, Melsaether C, Mothe B, Yamamoto H, Horton H, Wilson N, Hughes AL, Watkins DI. 2002. Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat Med 8:493–499. doi: 10.1038/nm0502-493. [DOI] [PubMed] [Google Scholar]
- 14.Phillips RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, Ogunlesi AO, Elvin JG, Rothbard JA, Bangham CR, Rizza CR, McMichael AJ. 1991. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- 15.Price DA, Goulder PJ, Klenerman P, Sewell AK, Easterbrook PJ, Troop M, Bangham CR, Phillips RE. 1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc Natl Acad Sci U S A 94:1890–1895. doi: 10.1073/pnas.94.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rong R, Li B, Lynch RM, Haaland RE, Murphy MK, Mulenga J, Allen SA, Pinter A, Shaw GM, Hunter E, Robinson JE, Gnanakaran S, Derdeyn CA. 2009. Escape from autologous neutralizing antibodies in acute/early subtype C HIV-1 infection requires multiple pathways. PLoS Pathog 5:e1000594. doi: 10.1371/journal.ppat.1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato S, Yuste E, Lauer WA, Chang EH, Morgan JS, Bixby JG, Lifson JD, Desrosiers RC, Johnson WE. 2008. Potent antibody-mediated neutralization and evolution of antigenic escape variants of simian immunodeficiency virus strain SIVmac239 in vivo. J Virol 82:9739–9752. doi: 10.1128/JVI.00871-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Gils MJ, Bunnik EM, Burger JA, Jacob Y, Schweighardt B, Wrin T, Schuitemaker H. 2010. Rapid escape from preserved cross-reactive neutralizing humoral immunity without loss of viral fitness in HIV-1-infected progressors and long-term nonprogressors. J Virol 84:3576–3585. doi: 10.1128/JVI.02622-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogel TU, Friedrich TC, O'Connor DH, Rehrauer W, Dodds EJ, Hickman H, Hildebrand W, Sidney J, Sette A, Hughes A, Horton H, Vielhuber K, Rudersdorf R, De Souza IP, Reynolds MR, Allen TM, Wilson N, Watkins DI. 2002. Escape in one of two cytotoxic T-lymphocyte epitopes bound by a high-frequency major histocompatibility complex class I molecule, Mamu-A*02: a paradigm for virus evolution and persistence? J Virol 76:11623–11636. doi: 10.1128/JVI.76.22.11623-11636.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh AD, Bimber BN, Das A, Piaskowski SM, Rakasz EG, Bean AT, Mudd PA, Ericsen AJ, Wilson NA, Hughes AL, O'Connor DH, Maness NJ. 2013. Acute phase CD8+ T lymphocytes against alternate reading frame epitopes select for rapid viral escape during SIV infection. PLoS One 8:e61383. doi: 10.1371/journal.pone.0061383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watkins BA, Reitz MS Jr, Wilson CA, Aldrich K, Davis AE, Robert-Guroff M. 1993. Immune escape by human immunodeficiency virus type 1 from neutralizing antibodies: evidence for multiple pathways. J Virol 67:7493–7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 23.Wu F, Ourmanov I, Kuwata T, Goeken R, Brown CR, Buckler-White A, Iyengar R, Plishka R, Aoki ST, Hirsch VM. 2012. Sequential evolution and escape from neutralization of simian immunodeficiency virus SIVsmE660 clones in rhesus macaques. J Virol 86:8835–8847. doi: 10.1128/JVI.00923-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong R, Li H, Bibollet-Ruche F, Decker JM, Zheng NN, Gottlieb GS, Kiviat NB, Sow PS, Georgiev I, Hahn BH, Kwong PD, Robinson JE, Shaw GM. 2012. Broad and potent neutralizing antibody responses elicited in natural HIV-2 infection. J Virol 86:947–960. doi: 10.1128/JVI.06155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richman DD, Wrin T, Little SJ, Petropoulos CJ. 2003. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc Natl Acad Sci U S A 100:4144–4149. doi: 10.1073/pnas.0630530100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato S, Johnson W. 2007. Antibody-mediated neutralization and simian immunodeficiency virus models of HIV/AIDS. Curr HIV Res 5:594–607. doi: 10.2174/157016207782418515. [DOI] [PubMed] [Google Scholar]
- 27.Gray ES, Moore PL, Choge IA, Decker JM, Bibollet-Ruche F, Li H, Leseka N, Treurnicht F, Mlisana K, Shaw GM, Karim SS, Williamson C, Morris L. 2007. Neutralizing antibody responses in acute human immunodeficiency virus type 1 subtype C infection. J Virol 81:6187–6196. doi: 10.1128/JVI.00239-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li B, Decker JM, Johnson RW, Bibollet-Ruche F, Wei X, Mulenga J, Allen S, Hunter E, Hahn BH, Shaw GM, Blackwell JL, Derdeyn CA. 2006. Evidence for potent autologous neutralizing antibody titers and compact envelopes in early infection with subtype C human immunodeficiency virus type 1. J Virol 80:5211–5218. doi: 10.1128/JVI.00201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burns DP, Desrosiers RC. 1991. Selection of genetic variants of simian immunodeficiency virus in persistently infected rhesus monkeys. J Virol 65:1843–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Overbaugh J, Rudensey LM, Papenhausen MD, Benveniste RE, Morton WR. 1991. Variation in simian immunodeficiency virus env is confined to V1 and V4 during progression to simian AIDS. J Virol 65:7025–7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi WS, Collignon C, Thiriart C, Burns DP, Stott EJ, Kent KA, Desrosiers RC. 1994. Effects of natural sequence variation on recognition by monoclonal antibodies neutralize simian immunodeficiency virus infectivity. J Virol 68:5395–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eastman D, Piantadosi A, Wu X, Forthal DN, Landucci G, Kimata JT, Overbaugh J. 2008. Heavily glycosylated, highly fit SIVMne variants continue to diversify and undergo selection after transmission to a new host and they elicit early antibody dependent cellular responses but delayed neutralizing antibody responses. Virol J 5:90. doi: 10.1186/1743-422X-5-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jurkiewicz E, Hunsmann G, Schaffner J, Nisslein T, Luke W, Petry H. 1997. Identification of the V1 region as a linear neutralizing epitope of the simian immunodeficiency virus SIVmac envelope glycoprotein. J Virol 71:9475–9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kinsey NE, Anderson MG, Unangst TJ, Joag SV, Narayan O, Zink MC, Clements JE. 1996. Antigenic variation of SIV: mutations in V4 alter the neutralization profile. Virology 221:14–21. doi: 10.1006/viro.1996.0348. [DOI] [PubMed] [Google Scholar]
- 35.Rudensey LM, Kimata JT, Long EM, Chackerian B, Overbaugh J. 1998. Changes in the extracellular envelope glycoprotein of variants that evolve during the course of simian immunodeficiency virus SIVMne infection affect neutralizing antibody recognition, syncytium formation, and macrophage tropism but not replication, cytopathicity, or CCR-5 coreceptor recognition. J Virol 72:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rybarczyk BJ, Montefiori D, Johnson PR, West A, Johnston RE, Swanstrom R. 2004. Correlation between env V1/V2 region diversification and neutralizing antibodies during primary infection by simian immunodeficiency virus sm in rhesus macaques. J Virol 78:3561–3571. doi: 10.1128/JVI.78.7.3561-3571.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeh WW, Brassard LM, Miller CA, Basavapathruni A, Zhang J, Rao SS, Nabel GJ, Mascola JR, Letvin NL, Seaman MS. 2012. Envelope variable region 4 is the first target of neutralizing antibodies in early simian immunodeficiency virus mac251 infection of rhesus monkeys. J Virol 86:7052–7059. doi: 10.1128/JVI.00107-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuste E, Bixby J, Lifson J, Sato S, Johnson W, Desrosiers R. 2008. Glycosylation of gp41 of simian immunodeficiency virus shields epitopes that can be targets for neutralizing antibodies. J Virol 82:12472–12486. doi: 10.1128/JVI.01382-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeh WW, Rahman I, Hraber P, Coffey RT, Nevidomskyte D, Giri A, Asmal M, Miljkovic S, Daniels M, Whitney JB, Keele BF, Hahn BH, Korber BT, Shaw GM, Seaman MS, Letvin NL. 2010. Autologous neutralizing antibodies to the transmitted/founder viruses emerge late after simian immunodeficiency virus SIVmac251 infection of rhesus monkeys. J Virol 84:6018–6032. doi: 10.1128/JVI.02741-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Demma LJ, Vanderford TH, Logsdon JM Jr, Feinberg MB, Staprans SI. 2006. Evolution of the uniquely adaptable lentiviral envelope in a natural reservoir host. Retrovirology 3:19. doi: 10.1186/1742-4690-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buckley KA, Li PL, Khimani AH, Hofmann-Lehmann R, Liska V, Anderson DC, McClure HM, Ruprecht RM. 2003. Convergent evolution of SIV env after independent inoculation of rhesus macaques with infectious proviral DNA. Virology 312:470–480. doi: 10.1016/S0042-6822(03)00262-9. [DOI] [PubMed] [Google Scholar]
- 42.Kong R, Li H, Georgiev I, Changela A, Bibollet-Ruche F, Decker JM, Rowland-Jones SL, Jaye A, Guan Y, Lewis GK, Langedijk JP, Hahn BH, Kwong PD, Robinson JE, Shaw GM. 2012. Epitope mapping of broadly neutralizing HIV-2 human monoclonal antibodies. J Virol 86:12115–12128. doi: 10.1128/JVI.01632-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Overbaugh J, Rudensey LM. 1992. Alterations in potential sites for glycosylation predominate during evolution of the simian immunodeficiency virus envelope gene in macaques. J Virol 66:5937–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leviyang S, Griva I, Ita S, Johnson WE. 2017. A penalized regression approach to haplotype reconstruction of viral populations arising in early HIV/SIV infection. Bioinformatics 33:2455–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boutwell CL, Carlson JM, Lin TH, Seese A, Power KA, Peng J, Tang Y, Brumme ZL, Heckerman D, Schneidewind A, Allen TM. 2013. Frequent and variable cytotoxic-T-lymphocyte escape-associated fitness costs in the human immunodeficiency virus type 1 subtype B Gag proteins. J Virol 87:3952–3965. doi: 10.1128/JVI.03233-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boutwell CL, Rowley CF, Essex M. 2009. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J Virol 83:2460–2468. doi: 10.1128/JVI.01970-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandez CS, Stratov I, De Rose R, Walsh K, Dale CJ, Smith MZ, Agy MB, Hu SL, Krebs K, Watkins DI, O'Connor DH, Davenport MP, Kent SJ. 2005. Rapid viral escape at an immunodominant simian-human immunodeficiency virus cytotoxic T-lymphocyte epitope exacts a dramatic fitness cost. J Virol 79:5721–5731. doi: 10.1128/JVI.79.9.5721-5731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins JI, Brander C, Walker BD, Stuart DI, Kiepiela P, Goulder P. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol 80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miura T, Brockman MA, Schneidewind A, Lobritz M, Pereyra F, Rathod A, Block BL, Brumme ZL, Brumme CJ, Baker B, Rothchild AC, Li B, Trocha A, Cutrell E, Frahm N, Brander C, Toth I, Arts EJ, Allen TM, Walker BD. 2009. HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte recognition. J Virol 83:2743–2755. doi: 10.1128/JVI.02265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Troyer RM, McNevin J, Liu Y, Zhang SC, Krizan RW, Abraha A, Tebit DM, Zhao H, Avila S, Lobritz MA, McElrath MJ, Le Gall S, Mullins JI, Arts EJ. 2009. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog 5:e1000365. doi: 10.1371/journal.ppat.1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wright JK, Naidoo VL, Brumme ZL, Prince JL, Claiborne DT, Goulder PJ, Brockman MA, Hunter E, Ndung'u T. 2012. Impact of HLA-B*81-associated mutations in HIV-1 Gag on viral replication capacity. J Virol 86:3193–3199. doi: 10.1128/JVI.06682-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quinones-Mateu ME, Arts EJ. 2002. Fitness of drug resistant HIV-1: methodology and clinical implications. Drug Resist Updat 5:224–233. doi: 10.1016/S1368-7646(02)00123-1. [DOI] [PubMed] [Google Scholar]
- 53.Dykes C, Demeter LM. 2007. Clinical significance of human immunodeficiency virus type 1 replication fitness. Clin Microbiol Rev 20:550–578. doi: 10.1128/CMR.00017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brumme CJ, Huber KD, Dong W, Poon AF, Harrigan PR, Sluis-Cremer N. 2013. Replication fitness of multiple nonnucleoside reverse transcriptase-resistant HIV-1 variants in the presence of etravirine measured by 454 deep sequencing. J Virol 87:8805–8807. doi: 10.1128/JVI.00335-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dapp MJ, Heineman RH, Mansky LM. 2013. Interrelationship between HIV-1 fitness and mutation rate. J Mol Biol 425:41–53. doi: 10.1016/j.jmb.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bunnik EM, Lobbrecht MS, van Nuenen AC, Schuitemaker H. 2010. Escape from autologous humoral immunity of HIV-1 is not associated with a decrease in replicative capacity. Virology 397:224–230. doi: 10.1016/j.virol.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 57.Quakkelaar ED, Bunnik EM, van Alphen FP, Boeser-Nunnink BD, van Nuenen AC, Schuitemaker H. 2007. Escape of human immunodeficiency virus type 1 from broadly neutralizing antibodies is not associated with a reduction of viral replicative capacity in vitro. Virology 363:447–453. doi: 10.1016/j.virol.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 58.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. 2005. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 433:834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- 59.Kuwata T, Takaki K, Yoshimura K, Enomoto I, Wu F, Ourmanov I, Hirsch VM, Yokoyama M, Sato H, Matsushita S. 2013. Conformational epitope consisting of the V3 and V4 loops as a target for potent and broad neutralization of simian immunodeficiency viruses. J Virol 87:5424–5436. doi: 10.1128/JVI.00201-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Del Prete GQ, Scarlotta M, Newman L, Reid C, Parodi LM, Roser JD, Oswald K, Marx PA, Miller CJ, Desrosiers RC, Barouch DH, Pal R, Piatak M Jr, Chertova E, Giavedoni LD, O'Connor DH, Lifson JD, Keele BF. 2013. Comparative characterization of transfection- and infection-derived simian immunodeficiency virus challenge stocks for in vivo nonhuman primate studies. J Virol 87:4584–4595. doi: 10.1128/JVI.03507-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kijak GH, Sanders-Buell E, Chenine AL, Eller MA, Goonetilleke N, Thomas R, Leviyang S, Harbolick EA, Bose M, Pham P, Oropeza C, Poltavee K, O'Sullivan AM, Billings E, Merbah M, Costanzo MC, Warren JA, Slike B, Li H, Peachman KK, Fischer W, Gao F, Cicala C, Arthos J, Eller LA, O'Connell RJ, Sinei S, Maganga L, Kibuuka H, Nitayaphan S, Rao M, Marovich MA, Krebs SJ, Rolland M, Korber BT, Shaw GM, Michael NL, Robb ML, Tovanabutra S, Kim JH. 2017. Correction: rare HIV-1 transmitted/founder lineages identified by deep viral sequencing contribute to rapid shifts in dominant quasispecies during acute and early infection. PLoS Pathog 13:e1006620. doi: 10.1371/journal.ppat.1006620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chohan B, Lang D, Sagar M, Korber B, Lavreys L, Richardson B, Overbaugh J. 2005. Selection for human immunodeficiency virus type 1 envelope glycosylation variants with shorter V1-V2 loop sequences occurs during transmission of certain genetic subtypes and may impact viral RNA levels. J Virol 79:6528–6531. doi: 10.1128/JVI.79.10.6528-6531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frost SD, Liu Y, Pond SL, Chappey C, Wrin T, Petropoulos CJ, Little SJ, Richman DD. 2005. Characterization of human immunodeficiency virus type 1 (HIV-1) envelope variation and neutralizing antibody responses during transmission of HIV-1 subtype B. J Virol 79:6523–6527. doi: 10.1128/JVI.79.10.6523-6527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gonzalez MW, DeVico AL, Lewis GK, Spouge JL. 2015. Conserved molecular signatures in gp120 are associated with the genetic bottleneck during simian immunodeficiency virus (SIV), SIV-human immunodeficiency virus (SHIV), and HIV type 1 (HIV-1) transmission. J Virol 89:3619–3629. doi: 10.1128/JVI.03235-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stansell E, Canis K, Haslam SM, Dell A, Desrosiers RC. 2011. Simian immunodeficiency virus from the sooty mangabey and rhesus macaque is modified with O-linked carbohydrate. J Virol 85:582–595. doi: 10.1128/JVI.01871-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson WE, Morgan J, Reitter J, Puffer BA, Czajak S, Doms RW, Desrosiers RC. 2002. A replication-competent, neutralization-sensitive variant of simian immunodeficiency virus lacking 100 amino acids of envelope. J Virol 76:2075–2086. doi: 10.1128/jvi.76.5.2075-2086.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stamatatos L, Cheng-Mayer C. 1998. An envelope modification that renders a primary, neutralization-resistant clade B human immunodeficiency virus type 1 isolate highly susceptible to neutralization by sera from other clades. J Virol 72:7840–7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cao J, Sullivan N, Desjardin E, Parolin C, Robinson J, Wyatt R, Sodroski J. 1997. Replication and neutralization of human immunodeficiency virus type 1 lacking the V1 and V2 variable loops of the gp120 envelope glycoprotein. J Virol 71:9808–9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Johnson WE, Lifson JD, Lang SM, Johnson RP, Desrosiers RC. 2003. Importance of B-cell responses for immunological control of variant strains of simian immunodeficiency virus. J Virol 77:375–381. doi: 10.1128/JVI.77.1.375-381.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wyatt R, Sullivan N, Thali M, Repke H, Ho D, Robinson J, Posner M, Sodroski J. 1993. Functional and immunologic characterization of human immunodeficiency virus type 1 envelope glycoproteins containing deletions of the major variable regions. J Virol 67:4557–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lynch RM, Wong P, Tran L, O'Dell S, Nason MC, Li Y, Wu X, Mascola JR. 2015. HIV-1 fitness cost associated with escape from the VRC01 class of CD4 binding site neutralizing antibodies. J Virol 89:4201–4213. doi: 10.1128/JVI.03608-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, Kunert R, Zhu P, Wormald MR, Stanfield RL, Roux KH, Kelly JW, Rudd PM, Dwek RA, Katinger H, Burton DR, Wilson IA. 2003. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science 300:2065–2071. doi: 10.1126/science.1083182. [DOI] [PubMed] [Google Scholar]
- 73.Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Koff WC, Wilson IA, Burton DR, Poignard P. 2011. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kwong PD, Mascola JR. 2012. Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity 37:412–425. doi: 10.1016/j.immuni.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gilbert PB, Berger JO, Stablein D, Becker S, Essex M, Hammer SM, Kim JH, Degruttola VG. 2011. Statistical interpretation of the RV144 HIV vaccine efficacy trial in Thailand: a case study for statistical issues in efficacy trials. J Infect Dis 203:969–975. doi: 10.1093/infdis/jiq152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao HX, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH. 2012. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med 366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pegu P, Vaccari M, Gordon S, Keele BF, Doster M, Guan Y, Ferrari G, Pal R, Ferrari MG, Whitney S, Hudacik L, Billings E, Rao M, Montefiori D, Tomaras G, Alam SM, Fenizia C, Lifson JD, Stablein D, Tartaglia J, Michael N, Kim J, Venzon D, Franchini G. 2013. Antibodies with high avidity to the gp120 envelope protein in protection from simian immunodeficiency virus SIV(mac251) acquisition in an immunization regimen that mimics the RV-144 Thai trial. J Virol 87:1708–1719. doi: 10.1128/JVI.02544-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med 361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 79.Rolland M, Edlefsen PT, Larsen BB, Tovanabutra S, Sanders-Buell E, Hertz T, deCamp AC, Carrico C, Menis S, Magaret CA, Ahmed H, Juraska M, Chen L, Konopa P, Nariya S, Stoddard JN, Wong K, Zhao H, Deng W, Maust BS, Bose M, Howell S, Bates A, Lazzaro M, O'Sullivan A, Lei E, Bradfield A, Ibitamuno G, Assawadarachai V, O'Connell RJ, deSouza MS, Nitayaphan S, Rerks-Ngarm S, Robb ML, McLellan JS, Georgiev I, Kwong PD, Carlson JM, Michael NL, Schief WR, Gilbert PB, Mullins JI, Kim JH. 2012. Increased HIV-1 vaccine efficacy against viruses with genetic signatures in Env V2. Nature 490:417–420. doi: 10.1038/nature11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gao F, Bonsignori M, Liao HX, Kumar A, Xia SM, Lu X, Cai F, Hwang KK, Song H, Zhou T, Lynch RM, Alam SM, Moody MA, Ferrari G, Berrong M, Kelsoe G, Shaw GM, Hahn BH, Montefiori DC, Kamanga G, Cohen MS, Hraber P, Kwong PD, Korber BT, Mascola JR, Kepler TB, Haynes BF. 2014. Cooperation of B cell lineages in induction of HIV-1-broadly neutralizing antibodies. Cell 158:481–491. doi: 10.1016/j.cell.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liao HX, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD, Fire AZ, Roskin KM, Schramm CA, Zhang Z, Zhu J, Shapiro L, Mullikin JC, Gnanakaran S, Hraber P, Wiehe K, Kelsoe G, Yang G, Xia SM, Montefiori DC, Parks R, Lloyd KE, Scearce RM, Soderberg KA, Cohen M, Kamanga G, Louder MK, Tran LM, Chen Y, Cai F, Chen S, Moquin S, Du X, Joyce MG, Srivatsan S, Zhang B, Zheng A, Shaw GM, Hahn BH, Kepler TB, Korber BT, Kwong PD, Mascola JR, Haynes BF. 2013. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 496:469–476. doi: 10.1038/nature12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sather DN, Carbonetti S, Kehayia J, Kraft Z, Mikell I, Scheid JF, Klein F, Stamatatos L. 2012. Broadly neutralizing antibodies developed by an HIV-positive elite neutralizer exact a replication fitness cost on the contemporaneous virus. J Virol 86:12676–12685. doi: 10.1128/JVI.01893-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pietzsch J, Scheid JF, Mouquet H, Klein F, Seaman MS, Jankovic M, Corti D, Lanzavecchia A, Nussenzweig MC. 2010. Human anti-HIV-neutralizing antibodies frequently target a conserved epitope essential for viral fitness. J Exp Med 207:1995–2002. doi: 10.1084/jem.20101176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zanini F, Puller V, Brodin J, Albert J, Neher RA. 2017. In vivo mutation rates and the landscape of fitness costs of HIV-1. Virus Evol 3:vex003. doi: 10.1093/ve/vex003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 86.Office of Laboratory Animal Welfare. 2015. Public Health Service policy on humane care and use of laboratory animals. Office of Laboratory Animal Welfare, National Institutes of Health, Washington, DC. [Google Scholar]
- 87.Interagency Research Animal Committee. 1985. Principles for the utilization and care of vertebrate animals used in testing, research and training. National Institutes of Health, Washington, DC. [Google Scholar]
- 88.Means RE, Greenough T, Desrosiers RC. 1997. Neutralization sensitivity of cell culture-passaged simian immunodeficiency virus. J Virol 71:7895–7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. 2000. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol 74:8358–8367. doi: 10.1128/JVI.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Platt EJ, Bilska M, Kozak SL, Kabat D, Montefiori DC. 2009. Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J Virol 83:8289–8292. doi: 10.1128/JVI.00709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol 72:2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Alexander L, Du Z, Rosenzweig M, Jung JU, Desrosiers RC. 1997. A role for natural simian immunodeficiency virus and human immunodeficiency virus type 1 nef alleles in lymphocyte activation. J Virol 71:6094–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goldstein S, Brown CR, Dehghani H, Lifson JD, Hirsch VM. 2000. Intrinsic susceptibility of rhesus macaque peripheral CD4(+) T cells to simian immunodeficiency virus in vitro is predictive of in vivo viral replication. J Virol 74:9388–9395. doi: 10.1128/JVI.74.20.9388-9395.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yuste E, Johnson W, Pavlakis GN, Desrosiers RC. 2005. Virion envelope content, infectivity, and neutralization sensitivity of simian immunodeficiency virus. J Virol 79:12455–12463. doi: 10.1128/JVI.79.19.12455-12463.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bixby JG, Laur O, Johnson WE, Desrosiers RC. 2010. Diversity of envelope genes from an uncloned stock of SIVmac251. AIDS Res Hum Retrovir 26:1115–1131. doi: 10.1089/aid.2010.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang X, Charlebois P, Gnerre S, Coole MG, Lennon NJ, Levin JZ, Qu J, Ryan EM, Zody MC, Henn MR. 2012. De novo assembly of highly diverse viral populations. BMC Genomics 13:475. doi: 10.1186/1471-2164-13-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O, Brander C, Hess C, Gunthard HF, Brumme ZL, Brumme CJ, Bazner S, Rychert J, Tinsley JP, Mayer KH, Rosenberg E, Pereyra F, Levin JZ, Young SK, Jessen H, Altfeld M, Birren BW, Walker BD, Allen TM. 2012. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog 8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Macalalad AR, Zody MC, Charlebois P, Lennon NJ, Newman RM, Malboeuf CM, Ryan EM, Boutwell CL, Power KA, Brackney DE, Pesko KN, Levin JZ, Ebel GD, Allen TM, Birren BW, Henn MR. 2012. Highly sensitive and specific detection of rare variants in mixed viral populations from massively parallel sequence data. PLoS Comput Biol 8:e1002417. doi: 10.1371/journal.pcbi.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fofana IB, Colantonio AD, Reeves RK, Connole MA, Gillis JM, Hall LR, Sato S, Audin CR, Evans DT, Shida H, Johnson RP, Johnson WE. 2011. Flow cytometry based identification of simian immunodeficiency virus Env-specific B lymphocytes. J Immunol Methods 370:75–85. doi: 10.1016/j.jim.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jia B, Ng SK, DeGottardi MQ, Piatak M, Yuste E, Carville A, Mansfield KG, Li W, Richardson BA, Lifson JD, Evans DT. 2009. Immunization with single-cycle SIV significantly reduces viral loads after an intravenous challenge with SIV(mac)239. PLoS Pathog 5:e1000272. doi: 10.1371/journal.ppat.1000272. [DOI] [PMC free article] [PubMed] [Google Scholar]