

ABSTRACT
Cysteine-linked antibody-drug conjugates (ADCs) produced from IgG2 monoclonal antibodies (mAbs) are more heterogeneous than ADCs generated from IgG1 mAbs, as IgG2 ADCs are composed of a wider distribution of molecules, typically containing 0 – 12 drug-linkers per antibody. The three disulfide isoforms (A, A/B, and B) of IgG2 antibodies confer differences in solvent accessibilities of the interchain disulfides and contribute to the structural heterogeneity of cysteine-linked ADCs. ADCs derived from either IgG2-A or IgG2-B mAbs were compared to better understand the role of disulfide isoforms on attachment sites and distribution of conjugated species. Our characterization of these ADCs demonstrated that the disulfide configuration affects the kinetics of disulfide bond reduction, but has minimal effect on the primary sites of reduction. The IgG2-A mAbs yielded ADCs with higher drug-to-antibody ratios (DARs) due to the easier reduction of its interchain disulfides. However, hinge-region cysteines were the primary conjugation sites for both IgG2-A and IgG2-B mAbs.
KEYWORDS: antibody-drug conjugate, ADC, IgG2, conjugation positions, disulfide isoforms, drug-to-antibody ratio, DAR, drug loading
Introduction
Antibody-drug conjugates (ADCs) are a class of targeted cancer therapeutics that benefit from the specificity of antibodies to deliver potent cytotoxic drug molecules directly to cancer cells.1,2 ADCs that are conjugated using thiol-maleimide chemistry via partial reduction of the interchain disulfides form a heterogeneous distribution of conjugated molecules with varying numbers of drug-linkers per mAb.3,4 The number of conjugated drug-linkers can range from 0 – 8 for an IgG1 ADC, or 0 – 12 for an IgG2 ADC. Antibody-drug conjugates with an even number of drug-linkers conjugated per antibody are typically observed from both IgG1 and IgG2 mAbs.4–6
IgG1 mAbs have been the preferred antibody framework for the development of ADCs, due at least in part to the decades of development history of IgG1 antibodies as biotherapeutics, which has resulted in a vast collection of biochemical and biophysical information for cysteine-linked IgG1 ADCs.6–15 However, the Fc regions of IgG1 antibodies may stimulate effector functions that can interfere with the mechanism of action of ADCs, thereby increasing the potential for undesired extracellular release of the cytotoxic drug and off-target cytotoxicity.16,17 IgG2 mAbs have similar in serum half-life compared to IgG1 mAbs, but have lower affinity for Fc-γ receptors, and are therefore weaker at eliciting effector responses.16 These features of IgG2 mAbs could make IgG2 ADCs better candidates to fulfill their intended mechanism of action upon internalization of the ADC and intracellular release of the drug. Moreover, a previous study comparing cysteine-linked ADCs derived from IgG1, IgG2, and IgG4 antibodies showed similar tolerability and toxicity profiles between the IgG1 and IgG2 ADCs.18
In-depth characterization of the drug substance and drug product is an important aspect during clinical development of therapeutic candidates, as deeper understanding of molecular attributes enhance the quality and batch consistency of the drug product. Recent analytical improvements have allowed the optimization of tandem and novel mass spectrometry methods developed specifically for detailed structural analysis of ADCs.19–21 Nevertheless, limited structural and biophysical information is available regarding IgG2 ADCs because these molecules have not been a popular choice for clinical development. This gap in the knowledge of the characteristics of an IgG2 ADC poses a challenge to the design of better ADC therapeutics with enhanced drug product homogeneity, stability, efficacy, and improved therapeutic window. The aim of this investigation is to understand how the disulfide isoforms of IgG2 mAbs influence the site(s) of drug attachment in cysteine-linked ADCs, and determine whether the disulfide isoform distribution should be controlled as a critical attribute of the mAb intermediate.
Characterization of IgG2 mAbs and ADCs is more challenging when compared to their IgG1 counterparts. The complexity of IgG2 ADCs results from the multiple configurations of the mAb interchain disulfide bonds (Fig. 1). In addition to a higher possible number of drug-linkers conjugated per mAb compared to an IgG1, the increased number of interchain disulfide bonds in an IgG2 mAb also equates to a larger number of conjugation positional isoforms. Moreover, some IgG2 mAbs potentially contain more trisulfides in the hinge region than IgG1s, which can affect the quality attributes of cysteine-linked ADCs.22–25 To our knowledge, the effect of the mAb disulfide isoforms on the conjugation profile has not been previously evaluated.
Figure 1.
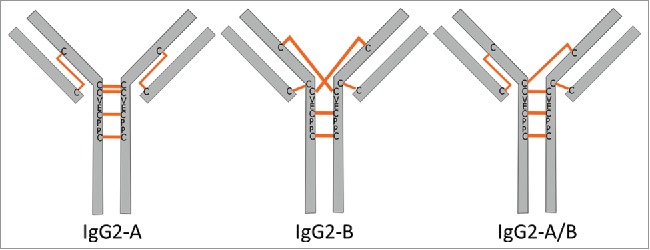
Cartoon representations of the IgG2 interchain disulfide configurations as previously reported.26,27 The A and B isoforms differ by the bond configuration of the LC-HC disulfide. The A/B configuration is a hybrid structure containing both A and B disulfide configurations.
The IgG2 mAb disulfide isoforms, defined as A, A/B, or B isoforms, are distinguished by the arrangement of the light chain-heavy chain (LC-HC) disulfide bonds.26–29 The classical IgG interchain disulfide bond configuration connects the LC and HC at the antigen-binding fragment (Fab) region and the two HCs at the hinge region via parallel disulfides. The IgG2-A contains disulfides configured in the canonical way. The IgG2-B isoform connects the CH1 domain to a hinge cysteine of the opposite HC, resulting in a crossed disulfide configuration. The LC of the IgG2-B is in turn disulfide-bonded to the N-terminal hinge cysteine. The A/B disulfide isoform contains one Fab arm with the canonical disulfide connectivity, and the other Fab arm resembles the B disulfide configuration. Previous studies have shown that IgG2 disulfide isoforms exhibit differences in solvent accessibilities, hydrophobicity, isoelectric points, and antigen binding.30–34 The physicochemical attributes imparted by the higher-order structural differences as a result of disulfide connectivity could influence the conjugation positions in cysteine-linked ADCs. Additionally, the biological activity, safety, and off-target cytotoxicity of the ADCs produced using the IgG2 framework could be affected by the locations of drug attachment.35,36 Since the sites of conjugation are considered critical quality attributes (CQAs) in ADCs, a precise understanding of the distribution and positions of conjugated drug-linkers are important for the development of effective and stable ADCs.
Previous results have shown that the primary conjugation sites of cysteine-linked ADCs are influenced by the IgG subclass. The main locations of drug attachment in IgG1 ADCs are at the Fab region cysteines that form the LC-HC interchain disulfide bond, and correlate with the rank order of reduction susceptibility of the interchain disulfide bonds.4,5,37 In contrast to the IgG1 ADCs, the primary conjugation positions in cysteine-linked IgG2 ADCs derived from a mAb containing a mixture of the A, A/B, and B disulfide isoforms are the cysteines located in the hinge region.5
For this study, we generated and evaluated the ADCs derived from IgG2-A and IgG2-B mAbs produced under the same reaction conditions. The first reaction step involved the release of cysteines from interchain disulfide bonds via partial reduction using low concentrations of tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl), followed by conjugation of the cysteines with molar excess of maleimidocaproyl valine-citrulline para-aminobenzoic acid monomethyl auristatin E (vcMMAE). The partially reduced mAbs and the corresponding ADCs were characterized with respect to distribution and locations of conjugated drug-linkers.
Results
Enrichment and purification of IgG2-A and IgG2-B antibodies
The starting material used for this study was a Chinese hamster ovary cell-derived human IgG2 mAb, containing 22% IgG2-A, 33% IgG2-A/B, and 45% IgG2-B (Table 1). Enrichment for either the IgG2-A or IgG2-B isoform was achieved by first shuffling the disulfide bonds under redox conditions in a buffer containing L-cysteine and cystamine dihydrochloride at pH 8.0, modified from previously published procedures.26,27 Conditions that promote rearrangement of disulfide bonds into the A isoform required the addition of 1.5 M guanidine-HCl to the redox buffer, consistent with previous reports.26,27
Table 1.
Disulfide isoform distribution of the IgG2 mAb preparations
| Enriched for IgG2-A |
Enriched for IgG2-B |
||||
|---|---|---|---|---|---|
| Disulfide Isoforma | Starting Materialb | Redox onlyc | Redox + CEXd | Redox onlyc | Redox + CEXd |
| A + A’ | 22% | 65% | 100% | 16% | 0% |
| A/B | 33% | 26% | 0% | 14% | 0% |
| B | 45% | 9% | 0% | 70% | 100% |
Distribution of disulfide isoforms were determined via nrRP-UHPLC.
Initial IgG2 mAb sample (“starting material”) is comprised of a mixture of A, A/B, and B disulfide isoforms.
Distribution of disulfide isoforms after dialysis in redox buffer for 5 days at 4°C, protected from light.
Distribution of disulfide isoforms of redox-enriched A and B mAb further purified via CEX-HPLC fractionation.
Levels of the desired mAb isoforms after incubation in redox buffer increased to 65 – 70% (Table 1), and cation exchange high-pressure liquid chromatography (CEX-HPLC) was used to fractionate each isoform.27,38 The disulfide isoforms were resolved on a weak cation exchange (WCX) analytical column using a sodium acetate/NaCl mobile phase system optimized for this IgG. Both IgG2-A and IgG2-B mAbs were 100% pure after CEX fractionation as determined by a non-reduced reversed-phase ultra-high pressure liquid chromatography (nrRP-UHPLC) method that resolves disulfide isoforms (Fig. 2). The nrRP-UHPLC method utilizes a trifluoroacetic acid (TFA)/ isopropanol (IPA)/ acetonitrile (CH3CN) gradient and provides better resolution than CEX-HPLC. However, the severe conditions of the nrRP-UHPLC assay irreversibly denatures the mAb, which precluded its use for fractionation purposes. The IgG2-B elutes first by nrRP-UHPLC, followed by the A/B and A isoforms. The IgG2-A isoform is further resolved as two species using this method, and were assigned as the A and A’ species.30 The A’ species has the same disulfide configuration as the A-form, but contains a hinge disulfide that is protected from reduction.39
Figure 2.

nrRP-UHPLC chromatograms of the starting material (A), redox-enriched A and B isoforms (B), and CEX-fractionated IgG2-A and IgG2-B mAbs (C).
Susceptibility of interchain disulfide bond reduction based on antibody isoform
Partial reduction of the mAb is the first step in the preparation of ADCs conjugated at the cysteines involved in interchain disulfide bonding. The starting material, purified IgG2-A, and purified IgG2-B mAbs were partially reduced at pH 5.3 with TCEP-HCl:mAb molar ratios ranging from 4:1 – 46:1. Typical antibody reduction and drug-linker conjugation reactions involved in the production of cysteine-linked ADCs use buffers with pH ≥ 7, and normally generate two moles of reactive thiol per mole of reducing reagent.4 Because IgG2 disulfide bonds can rearrange in alkaline or reducing environments, the partial reduction conditions were optimized at pH 5.3 to control for disulfide scrambling.31,40,41 However, preliminary experiments determined that the reduction kinetics of the interchain disulfide bonds are slower at pH 5.3, thereby requiring higher molar equivalents of TCEP-HCl:mAb to tune the generation of ADCs with higher average DAR values. Control experiments were also performed using typical ADC manufacturing parameters at a higher pH (data not shown).
We monitored the extent of interchain disulfide reduction using via RP-UHPLC analyses of the partially reduced mAbs. The chromatographic conditions for this method were different than those used for the separation of disulfide isoforms. The TFA/CH3CN gradient used in this analysis provides baseline resolution of the peaks corresponding to the intact mAb and LC, but does not resolve the mAb disulfide isoforms (Fig. 3). The HC and other covalently associated antibody fragments partially co-elute with the intact mAb peak, and the increasing amplitude of these peaks correspond to higher amount of interchain disulfide reduction.
Figure 3.
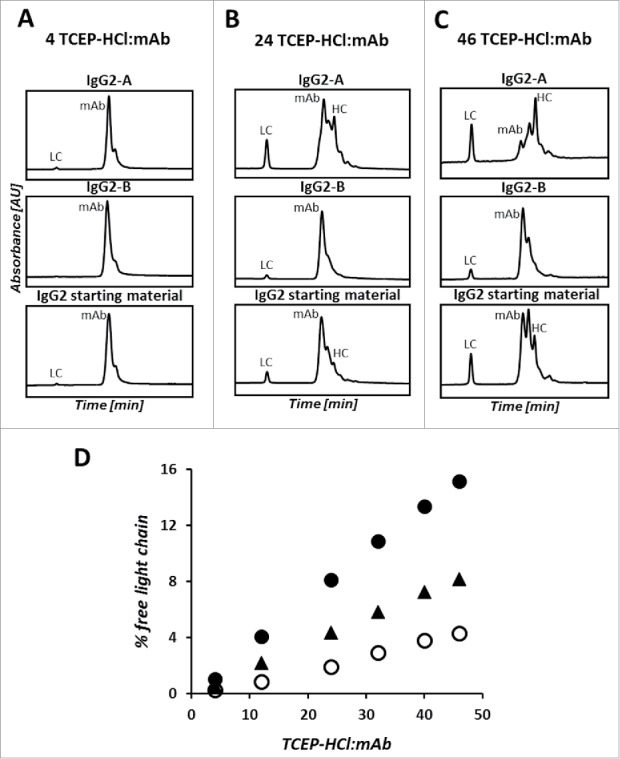
Overlay of the nrRP-UHPLC chromatograms of the partially-reduced IgG2-A (top), IgG2-B (middle), and IgG2 starting material (bottom) at TCEP-HCl:mAb ratios of 4 (A), 24 (B), and 46 (C). Bottom plot shows the percent of LC quantified by nrRP-UHPLC analysis of the partially-reduced mAb as a function of TCEP-HCl:mAb ratio (D). Closed circles (•) are the IgG2-A, open circles (o) are the IgG2-B, and filled triangles (▴) are the non-enriched IgG2 starting material.
Increased levels of antibody fragments and free LC with a concomitant decrease of the intact mAb peak were observed for the IgG2-A when compared to the IgG2-B and the starting material treated with the same amount of reducing reagent. Focusing specifically on the extent of LC-HC bond reduction, a higher percent of free LC was generated after partial reduction of the IgG2-A antibody. The amount of free LC increased linearly with the concentration of TCEP-HCl, topping at 16% for the IgG2-A mAb and 4% for the IgG2-B mAb. With respect to the B isoform, our analysis of the partially reduced IgG2-B mAb showed a higher amount of intact mAb and other covalently associated species in the samples treated with higher TCEP-HCl:mAb ratios.
The control for this experiment was the starting material containing a mixture of A, B, and A/B isoforms. The chromatographic profiles and percent free LC of the starting material align in between the results observed for the purified A and B isoforms. The higher similarity of the starting material to the B isoform reflects the distribution of disulfide isoforms of the starting material, where 39% of Fab arms are in the A configuration and 61% in the B configuration.
Size variant distributions of the partially reduced mAbs were evaluated under non-denaturing conditions and found to be mostly in monomeric form. The combined amounts of high and low molecular weight species (HMWS and LMWS, respectively) were less than 2% across the range of TCEP-HCl:mAb ratios (Figure S1 of the Supplementary Information).
Characterization of conjugate distribution of IgG2-A and IgG2-B ADCs
The ADCs were produced by conjugating vcMMAE to the partially reduced mAbs using the same reagent stoichiometries and reaction conditions for both A and B isoforms. To highlight the influences conferred by the mAb disulfide configuration on the ADCs, we selected TCEP-HCl:mAb molar equivalents of 4, 24, and 46 to represent conditions necessary to target relatively low, intermediate, and high average DARs. We characterized the distribution of conjugated species in the resulting ADC samples under intact, reduced, and denaturing conditions. The ADCs were determined to be >97% monomeric for the TCEP-HCl range evaluated (Figure S1 of the Supplementary Information). While an increase in LMWS was observed for the IgG2-A ADC in comparison to its mAb counterparts, the highest relative percent of LMWS detected was only 1.6% for the ADC.
Conjugate distributions and average DARs are CQAs for the ADC, and can be characterized by a combination of chromatographic analyses of both the intact and reduced molecules. The distributions of conjugated species were evaluated by hydrophobic interaction chromatography (HIC), where the ADCs elute in order of the increasing number of conjugated drug-linkers per mAb in a gradient of decreasing ammonium sulfate and increasing isopropanol. The relative abundance of ADCs conjugated with two (D2) and four drug-linkers (D4) per antibody at the low TCEP-HCl:mAb level is similar between the two isoforms, although the IgG2-B sample contained a higher amount of unconjugated mAb (Fig. 4). The differences between ADCs produced from A and B isoforms become more apparent at the intermediate TCEP-HCl:mAb condition, and grow further accentuated at the high conjugation level. The multiply conjugated D4, D6, and D8 molecules are the predominant species in the IgG2-A ADC derived from 46 molar equivalents of TCEP-HCl, and contrast with D2 and D4 being the major conjugated species for the B isoform produced under the same conditions. D10 and D12 species were not detected by HIC, which we attributed to a limitation of the sample preparation for the HIC analysis where those species irreversibly precipitate when diluted with sample diluent containing NaCl. Although particulates were not observed upon visual inspection, low protein recovery was a problem during the analyses of the highly conjugated ADCs, particularly for the purified IgG2-A isoform. Nonetheless, the non-covalently associated fragments related to D8, D10, and D12 were detected under denaturing assay conditions of non-reduced ADCs (nrRP-UHPLC).
Figure 4.
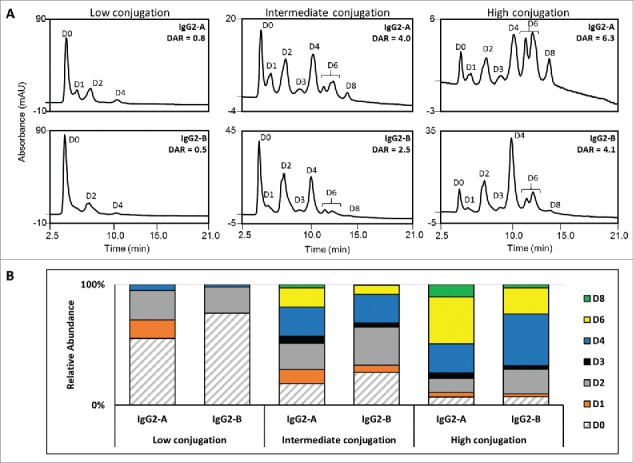
Hydrophobic interaction chromatograms (A) and distribution of the drug-loaded species (B) for the IgG2-A and IgG2-B ADCs at the low, intermediate, and high conjugation levels. Unconjugated mAb (D0) is filled with diagonal lines and the various conjugated species are filled with solid color.
The relative peak areas integrated from the HIC profile are widely used for the calculation of average DAR in IgG1 cysteine-linked ADCs. In the case of the IgG2 ADCs, however, poor resolution of the D8, D10, and D12 species precludes accurate integration and leads to errors in the average DAR calculation. Recent advances in analytical technologies have provided alternate methods for quantifying drug loading based on mass, such as coupling two-dimensional LC with mass spectrometry or via determination of the concentration of drug-linker after enzymatic deconjugation.42–46 Instead, we used the distributions of conjugated light and heavy chains derived from the RP-UHPLC analysis of the reduced ADCs (rRP-UHPLC) to calculate the average DAR values, where species elute according to differences in hydrophobicity between the LC and HC and the number of drug-linkers conjugated. For an IgG2 ADC, the expected species are unconjugated LC (L0), LC conjugated with one drug-linker (L1), unconjugated HC (H0), and HC conjugated with one to five drug-linker molecules (H1 – H5).
Average DAR was plotted as a function of the TCEP-HCl:mAb ratio to compare the conjugation levels of the A and B isoforms (Fig. 5). Both IgG2-A and IgG2-B ADCs had similar average DARs when reduced with low (4) TCEP-HCl equivalents, calculated to be 0.8 and 0.5 average DAR, respectively. The average DAR vs. TCEP-HCl:mAb curves of the A and B isoforms diverged as the concentration of TCEP-HCl increased, with the IgG2-A showing higher drug loading than the IgG2-B ADCs. This corresponded to 1.9 drug-linkers per mAb for every 10 TCEP-HCl molar equivalents for the A isoform, and 1.2 drug-linkers per mAb for the B isoform. The average DARs of the IgG2-A ADCs were calculated as 4.0 and 6.3 at the intermediate and high conjugation conditions, in contrast to 2.5 and 4.2 for the IgG2-B ADCs produced under the same conditions.
Figure 5.
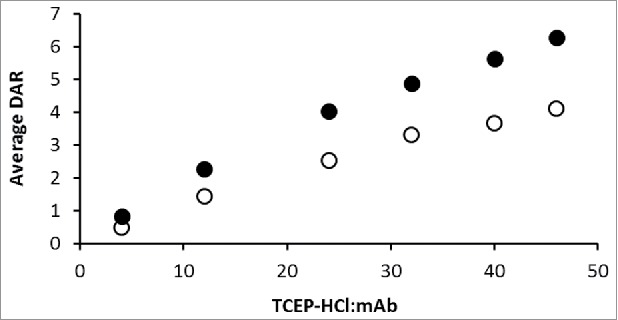
Average DAR as a function of TCEP-HCl:mAb ratio. IgG2-A ADCs are represented by closed circles (•) and IgG2-B ADCs by open circles (o).
The distributions of conjugated HCs were significantly influenced by the mAb disulfide isoform and correlated with the average DAR values (Fig. 6). Higher levels of HC conjugated with three or more drug-linkers (H3, H4, H5) were detected in the IgG2-A ADC at the intermediate and high TCEP-HCl:mAb levels. In the case of the IgG2-B ADCs, HCs conjugated with zero, one, or two drug-linkers (H0, H1, H2) were the predominant species when generated using the same reaction conditions. Interestingly, the highly conjugated IgG2-A included a higher percentage of H0 (17%) compared to the B isoform (12% H0). Notably, a very low amount of conjugation to the LC was detected (0 – 5%), depending on the TCEP-HCl:mAb levels.
Figure 6.
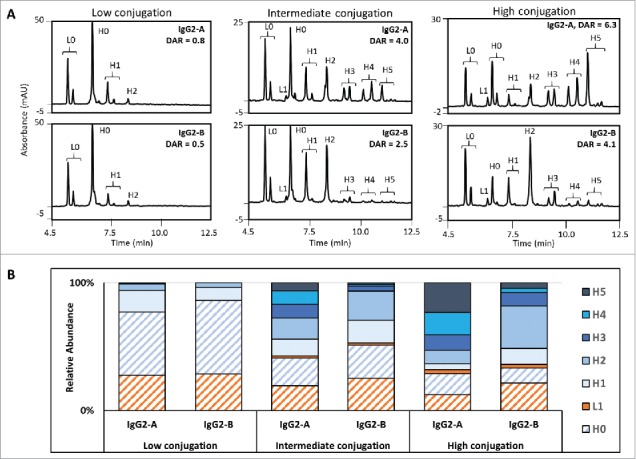
Reduced RP-UHPLC chromatograms (A) and distribution of the reduced species (B) for the IgG2-A and IgG2-B ADCs at the low, intermediate, and high conjugation levels. Light chains are shaded orange and heavy chains are shaded blue. The unconjugated species (L0 and H0) are filled with diagonal lines and conjugated species (L1, H1, H2, H3, H4, and H5) are filled with solid color.
Conjugation positional isomer distributions of IgG2-A and IgG2-B ADCs
The preceding experiments showed prevalent reduction/conjugation at the HC for both disulfide isoforms. We therefore elucidated the main conjugation positional isomers of the D2, D4, D6, and D8 species. The conjugation sites were assigned to cysteines in the Fab or the hinge regions by reconstructing the ADCs based on the species detected under denaturing conditions. Peaks detected via nrRP-UHPLC were identified by matching the observed masses obtained by LC/MS-ESI-TOF to the theoretical masses, as listed in Table S1 of the Supplementary Information.
The collection of possible component species associated with each conjugated variant are cataloged in Fig. 7. For example, a D2 molecule conjugated at the LC-HC interchain cysteines on one Fab arm would result in the detection of a conjugated LC (L1) and a heavy-heavy-light chain fragment with one drug ((HH)1L0). nrRP-UHPLC profiles of IgG2-A and IgG2-B ADCs with average DARs of ∼2.5 and ∼4.0 shown in Fig. 8 represent the differences observed in distribution of conjugated species as a function of average DAR and mAb disulfide configuration. The primary conjugation positional isomers reconstructed from the data are illustrated in Fig. 9.
Figure 7.
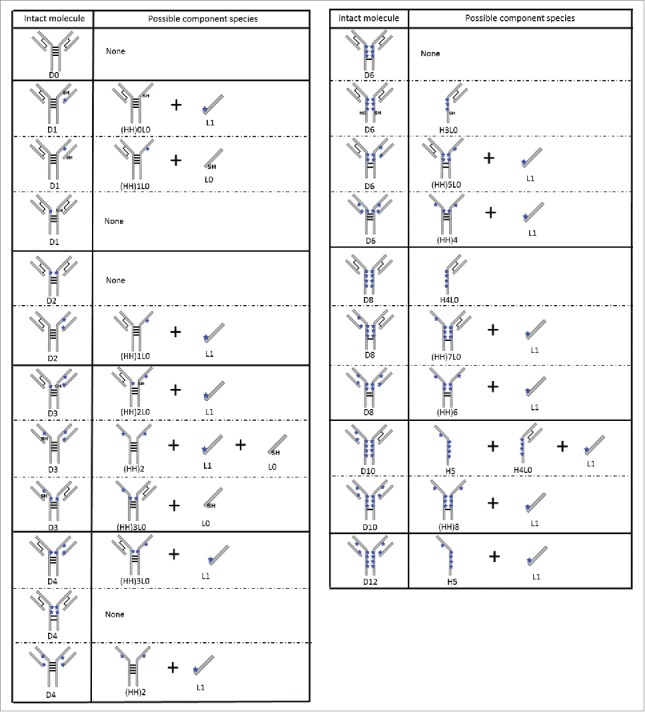
Possible component species for IgG2 ADC conjugation positional isomers analyzed under denaturing conditions. For simplicity, only cartoon representations of the A isoform ADCs are shown. The hinge conjugations are shown for illustrative purposes and do not represent the specific hinge cysteine that was reduced and conjugated. The black lines represent interchain disulfide bonds and the blue starts represent the drug-linker conjugated at an interchain cysteine.
Figure 8.

nrRP-UHPLC chromatograms comparing ADCs with average DAR ∼2.5 (A, B) and average DAR ∼ 4.0 (C, D) conjugated from IgG2-A (A, C) and IgG2-B (B, D). Peak codes listed in the figure were identified by matching observed molecular weights (MW) by LC-MS to the calculated MW of the expected component species.
Figure 9.

Main conjugation positional isomers detected IgG2 ADCs via nrRP-UHPLC analysis. Two possible pathways for the formation of D6 to D10 species are shown. Only the IgG2-A isoform is depicted for clarity, where the black lines indicate disulfide bonds and the blue stars represent drug-linkers attached at cysteines. The “SH” labels indicate reduced interchain cysteines that are not conjugated.
Intact D2 and D4 were detected as the predominant conjugation isomers in both A and B isoforms. However, unique peak shapes and retention times were observed for the conjugates derived from each disulfide isoform. Referring to Fig. 7, detection of intact D2 and D4 species reveal that the drug-linkers are attached at the hinge cysteines, although we were unable to pinpoint the exact conjugated hinge cysteines using this approach. A noteworthy difference between the two isoforms is that the D2 species in the IgG2-B ADCs was observed as a peak doublet, and LC/MS data showed a 2 Dalton (Da) mass difference between the peaks. Fab conjugations were detected at minor levels in component species derived from D2 and D4 of IgG2-A ADCs. The higher peak amplitudes of L1, (HH)0L0, (HH)1L0, and (HH)2L0 species in the IgG2-A ADCs provide further evidence of conjugation at the LC-HC cysteines. Also unique to the A isoform ADCs was the detection of a 23.8 kDa fragment co-eluting with the L1 peak. This fragment was not characterized, but it could be derived from in-source fragmentation of the drug-linker of the singly conjugated LC or from peptide bond hydrolysis of the HC.47,48
Component species that correspond to D6 in both A and B isoforms were detected primarily as (HH)5L0, which is the HHL fragment with 5 drug-linkers distributed on the HCs. This component species was observed as a peak doublet with equivalent masses, and potentially represents positional isomers of (HH)5L0. The A isoform ADCs showed higher abundance of the later eluting (HH)5L0 peak in the doublet pair, whereas the IgG2-B ADCs contained higher abundance of the earlier eluting peak. Reconstruction of the D6 molecule shows that the primary isomer contains four HC conjugations and two Fab conjugations. No intact D6 was detected, but baseline amounts of H3L0 were detected in both isoforms.
The main component species corresponding to D8 was H4L0, indicating that the hinge-conjugated D8 is the prevalent isomer. Constituents reflecting D8 with Fab conjugations (i.e., (HH)7L0 and (HH)6) were detected at baseline levels in the highly conjugated ADCs derived from both isomers (data not shown). Components pertaining to D10 and D12 were identified as L1, H5, (HH)8, and (HH)10. As discussed earlier, D10 and D12 were not detected via HIC due to their higher propensity to precipitate during sample preparation. We were unable to determine the main positional isomer of D10 using the current approach due to the low abundance of its component species.
Curiously, intact D8 and (HH)10 peaks were only detected in the IgG2-B ADCs. These species were not expected under the denaturing assay conditions, as all interchain disulfides have been disrupted. It is possible that either species was formed via conjugation to cysteines involved in both interchain and intrachain disulfide bonding, but no orthogonal evidence supported reduction of intrachain disulfide bonds.
Discussion
Most ADCs in clinical development utilize the IgG1 framework, and previous studies have shown that physicochemical attributes may differ between IgG1 and IgG2 cysteine-linked ADCs.1,5,49 Therefore, characterization knowledge gathered using IgG1 ADCs as the model may not translate to the IgG2 ADCs. One critical difference between the IgG subclasses is the presence of two additional hinge disulfide bonds in the IgG2 antibodies, which allow for a distribution of 0 – 12 drug-linkers. Also unique to the IgG2s is the existence of antibody disulfide isoforms, and their impact on the generation of ADCs was previously unknown. In this study, we characterized the partially reduced mAbs and ADCs derived from pure IgG2-A and IgG2-B isoforms.
Consistent differences in retention times of the partially reduced antibodies and the intact ADCs from the purified isoforms suggest that no disulfide scrambling occurred during the reduction step under the conditions used in this study. Results obtained from the material containing a mixture of disulfide isoforms also supports this inference. Data acquired from the partially reduced mAbs also showed that the reduction kinetics is influenced by the disulfide configuration, with IgG2-A mAbs revealing a higher rate of reduction than the B isoform. This result indicates that IgG2-B hinge disulfides are more solvent-protected, which is in agreement with the closer Fab/CH2 interdomain interactions reported for this isoform as measured by HDX and EM.50,51 Additional evidence was observed as the hinge-conjugated D8 and (HH)10 species in the IgG2-B ADCs analyzed under denaturing conditions. Though all hinge disulfides have been disrupted by conjugation, the HCs likely remained associated under denaturing conditions due to the strong non-covalent interactions between the Fab arms and the CH2 domains in the conformation imparted by the B configuration. In contrast, H4L0 and H5 fragments were detected in the IgG2-A samples, which indicates weaker Fab-CH2 interactions.50 Future improvements to mass spectrometric applications for disulfide mapping in heterogeneous samples should allow for a straightforward examination of disulfide configurations in ADCs.
The prevalence of the mAb peak and other covalently associated fragments (e.g., HHL and HH) point towards the hinge disulfides being the main targets for reduction in IgG2 antibodies. As disulfide isoforms differ in the bonding patterns to the hinge cysteines, each of the four hinge disulfides may exhibit differences in the rank order of vulnerability to reduction based on their connectivity. The influence of the disulfide bonding pattern on the sequence of hinge disulfide bond reduction is currently being investigated. We considered the possibility that the preferential reduction and conjugation sites for the A isoform could be the cysteines involved in the LC-HC interchain disulfide bond, given that the IgG2-A configuration is similar to an IgG1.6,37 Comparison of the starting material and purified IgG2-A and IgG2-B mAbs showed that higher amounts of free LC were generated after partial reduction of the IgG2-A antibody. However, the absolute amount of free LC is relatively low in the A isoform, implying that the LC-HC disulfide is not the main target of reduction. One explanation could be attributed to the position of the CH1 cysteine involved in interchain disulfide bonding, which resides in the middle of the Fab rather than proximal to the hinge region as is the case for the IgG1 isotype.31,52
Our characterization of the ADCs showed that a higher rate of disulfide bond reduction correlates with a higher average DAR. Analysis of the intact and reduced ADCs via HIC and RP-UHPLC shows that highly conjugated species were more prevalent in the A isoform. Hence, unconjugated mAb and lesser-conjugated species were the major components in the IgG2-B ADCs. However, an interesting result was observed in the highly conjugated ADCs, where the A isoform contained more unconjugated HC (H0) than the B isoform. While we did not further characterize the H0 sub-population in the IgG2-A, we speculated that perhaps the presence of the A’ isoform with a protected hinge disulfide could play a role in this instance.39,53
The preference for hinge conjugation is consistent with the observations from the partially reduced mAb analysis. We can therefore infer that the release of reduction of disulfide bonds determines the locations of drug-linker conjugation. Examination of the major conjugation isomers of each variably conjugated ADC allowed us to discern the regional progression of disulfide reduction. Our approach directly analyzed the heterogeneous DAR mixture under denaturing conditions and used the detected components to reconstruct the original conjugation isomers. Identical primary conjugation isomers were detected for the A and B isoforms, suggesting that the disulfide configuration does not play a significant role in directing the reduction/conjugation locations. Hinge-conjugated isomers were the primary species detected for the D2, D4, and D8 conjugates. The low abundance of D10 in our samples precluded the identification of its primary conjugation isomer, but all isomers shown in Fig. 9 were detected in both the A and B isoforms.
Formation of D6 deviated from the hinge conjugation trend, as its main isomer contains a pair of Fab conjugations. A low level of hinge-conjugated D6 was identified as the H3L0 species, indicating that all four hinge disulfides were reduced but only three cysteines were conjugated. Therefore, the reduction of three hinge disulfide bonds likely progresses promptly towards the reduction of the fourth disulfide. This hypothesis also rationalizes the predominance of the hinge-conjugated D8, as this molecule probably progressed from further hinge reduction of the mAb that generated D4. Under native conditions, the hinge-conjugated D6 likely remains associated in LHHL form via non-covalent interactions.
The hypothesis of rapid reduction of two hinge disulfides has been previously suggested for the IgG1 ADC, where the main D6 conjugation positional isomer is derived from a minor conjugation isomer of D4 that likely results from the consequent reduction of both hinge disulfides.6 The IgG2 antibody possibly undergoes global conformational changes after the reduction of two hinge disulfides, thereby increasing solvent accessibility at the hinge region. Other mechanisms such as thiol-disulfide exchange aided by a nearby reduced cysteine, or conversion of interchain to intrachain disulfides similar to the putative mechanism suggested for the IgG4 could also facilitate the rapid reduction of the remaining two hinge disulfides in the IgG2 mAb.54–56
Peak profile differences observed during the denatured analysis of ADCs suggested that the disulfide isoforms potentially drive the reduction/conjugation to different hinge region cysteines. The most remarkable differences between isoforms are the D2 and (HH)5L0 species. The intact D2 peak appeared as a single peak in the A isoforms, whereas a peak doublet was detected in the B isoforms. The 2 Da mass difference between the two peaks suggests the possibility that one isomer contains a pair of reduced, but unconjugated, hinge cysteines. However, the possibility that the two peaks pertain to different hinge conjugation sites should also be considered. Similarly for the (HH)5L0 species, the distribution of the doublet pair is specific to each disulfide isoform.
In this study, we analyzed the effect of the IgG2 disulfide isoforms on the formation of cysteine-linked ADCs. The mAb disulfide configurations influence the solvent accessibilities of the interchain disulfide bonds, but do not affect the primary sites of reduction and conjugation. The impact of expected process variabilities on conjugation sites, potency, and cytotoxicity remains to be addressed. Future explorations will focus on characterizing the hinge conjugation positions, and the correlations between biological activity and the ADCs derived from different disulfide isoforms.
Greater availability of data derived from IgG2 ADCs will contribute to the development of ADCs with improved quality profiles and clinical benefit. While IgG2 ADCs may be more structurally heterogeneous compared to IgG1 or IgG4 ADCs, batch consistency with respect to disulfide isoform distribution, average DAR, and composition of drug-loaded species is presently attainable in large-scale processes. Moreover, cysteine-linked IgG2 ADCs are composed of higher amounts of the proper D4 species where the intended DAR target is 4 drugs/mAb, compared to the binomial distribution typically reported for cysteine-linked IgG1 ADCs.4,5,57 Improved understanding of process parameters on the CQAs of IgG2 mAbs and ADCs will allow fine-tuning of the product quality. From a biological perspective, the reduced ability of IgG2s to stimulate effector functions could be a key benefit that can improve internalization into the target cells and minimize the incidence of off-target toxicity. Recent efforts for improving drug product consistency have popularized the development of site-specific ADCs with homogeneous DAR,58,59 which makes this an attractive modality with regard to the chemistry, manufacturing, and controls requirements. It remains unclear, however, whether DAR homogeneity correlates to clinical benefit for all molecules, as other factors such as off-target binding and drug-linker chemistry may also contribute to the reported dose-limiting toxicities.60–62 Continuing efforts evaluating the associations between product quality and clinical activity will help elevate the impact of ADCs as therapeutic options for oncology and other indications.
Materials and methods
Materials and equipment
Human IgG2 monoclonal antibody was obtained from the Process Sciences and Manufacturing group at Agensys, Inc., an affiliate of Astellas. The IgG2 mAb starting material containing a mixture of A, A/B, and B disulfide isoforms was produced using upstream and downstream protocols established in-house. The antibody was stored as a 25 mg/mL formulated bulk in acetate/sucrose buffer at pH 5.0. High-purity maleimidocaproyl valine-citrulline para-aminobenzoic acid monomethyl auristatin E (mc-vc-PABA-MMAE, or vcMMAE) was supplied by the ADC Chemistry group as a 10 mM solution in dimethyl sulfoxide (DMSO). L-cysteine, N-acetyl-L-cysteine, and DMSO were purchased from Sigma Aldrich (Cat #s 168149, A7250, and D2650). Cystamine dihydrochloride (AAB2287314), TRIS base (T395-500), 5 M NaCl solution (BMA51202), glacial acetic acid (A38-212), TCEP-HCl (PI20491), sucrose (S6-12), LC/MS-grade TFA (A116-10 × 1AMP), 0.5 M sodium phosphate monobasic solution (50-843-058), 0.5 M sodium phosphate dibasic solution (50-843-060), ammonium sulfate (A702-500), No-Weigh® dithiothreitol (DTT, PI20291), and LC/MS-grade formic acid (A11710 × 1-AMP) were all purchased from Fisher Scientific. Guanidine HCl (IC048205-40), disodium EDTA salt (BDH4616-500G), HPLC-grade IPA (BDH20880.400) and LC-MS grade CH3CN (BDH83640.400) were purchased from VWR. EMD Millipore Amicon Ultra centrifugal devices with 10 kDa MWCO Ultracel membranes were used for buffer exchange, unless otherwise noted. All chromatographic separations were carried out on a Thermo Scientific Ultimate 3000RS UHPLC equipped with a 100 µL split-loop injector, pre-column heater, post-column cooler, and a diode-array detector. Chromatography data analyses were performed using Thermo Scientific Chromeleon version 6.8 software.
Enrichment of IgG2-A and IgG2-B isoforms by reduction-oxidation
Enrichment for either the IgG2-A or IgG2-B disulfide isoform was done by modifying conditions previously reported26 using the IgG2 starting material containing a mixture of A, A/B, and B disulfide isoforms. For enrichment of the A and B isoforms, the starting material was diluted to 5 mg/mL in enrichment buffer and transferred into Thermo Scientific Slide-A-Lyzer™ 10K MWCO dialysis cassettes (PI66453). The mAb was incubated in redox enrichment buffer for 5 days at 4°C, protected from light. The enrichment buffer to promote conversion to the B isoform was composed of 0.3 M TRIS-HCl, 1.5 mM cystamine dihydrochloride, 9 mM L-cysteine, pH 8.0. The same buffer composition with an additional 1.5 M guanidine-HCl was used as the enrichment buffer for the conversion to the A isoform. The enriched samples were buffer exchanged using Amicon Ultra-15 (UFC901024) into 20 mM sodium acetate, pH 5.0, in preparation for loading onto the CEX column.
Purification of disulfide isoforms by cation exchange high-pressure liquid chromatography
The enriched IgG2-A and IgG2-B antibodies in acetate buffer were fractionated using CEX-HPLC on an Ultimate 3000RS UHPLC equipped with an AFC-3000 fraction collector. The redox-enriched samples were loaded onto a Sepax Technologies Antibodix WCX-NP5 column (602NP5-4625), 10 mm ID x 250 mm L, that had been equilibrated with the mobile phase A (MPA) composed of 20 mM sodium acetate, pH 5.0. The column was set to 40°C and gradient elution occurred at a flow rate of 0.5 mL/min, from 35% B to 65% B over 14 minutes. The mobile phase B (MPB) was composed of 20 mM sodium acetate, 450 mM sodium chloride, pH 5.0 + 5% isopropanol. Collected fractions were concentrated and buffer exchanged into the acetate/sucrose formulation buffer at pH 5.0 using Amicon Ultra-15 centrifugal devices. Fractionated samples were stored frozen at –80°C until conjugation or analysis. The final concentrations were measured using a Thermo Scientific Nanodrop 2000c, determined as 48 mg/mL for the IgG2-A and 23 mg/mL for the IgG2-B antibodies.
Antibody partial reduction and drug-linker conjugation
The IgG2-A, IgG2-B, and starting IgG2 material were diluted to 5 mg/mL in the reduction/conjugation buffer composed of 20 mM sodium acetate, 150 mM sodium chloride, 1 mM EDTA, pH 5.3. Partial reduction of the antibody was carried out in an Eppendorf Thermomixer set at 37°C and 300 RPM via incubation in nanomolar concentrations of TCEP-HCl prepared in the reduction/conjugation buffer. After the 3-hour partial reduction period, TCEP-HCl was removed using Amicon Ultra-0.5 (UFC5010BK). The partially reduced samples were adjusted to a minimum concentration of 8% DMSO (v/v) prior to addition of the drug-linker. vcMMAE was added at a theoretical 5-fold molar excess to the amount of reduced cysteines, and allowed to react for 1 hour at 25°C and 300 RPM. The unreacted vcMMAE was quenched using excess N-acetyl-L-cysteine, and incubated for 30 minutes at room temperature. The antibody-drug conjugate solution was purified of excess reagents via buffer exchange into the acetate/sucrose buffer at pH 5.0.
Analysis of disulfide isoform distribution via non-reduced reversed-phase ultra-high pressure liquid chromatography
The disulfide isoform distributions of the purified fractions were analyzed by nrRP-UHPLC as follows. Five micrograms of sample diluted in formulation buffer to a target between 2.5 and 5.0 mg/mL were injected onto an Agilent Technologies Zorbax RRHD 300SB-C8 (857750-906), 2.1 mm ID x 50 mm L column with a 1.8 µm particle size. The column had been previously equilibrated at 82°C in 75% MPA (0.1% TFA, 2% IPA, 97.9% water) and 25% MPB (0.1% TFA, 24.9% CH3CN, 75% IPA). Separation of the antibody disulfide isoforms was achieved at 0.4 mL/min using a multi-step gradient ramping from 25.0% B to 27.5% B over 6 minutes, 27.5% B to 28.0% B over 1 minute, and 28.0% B to 30.0% B over 1 minute. A post-column cooler set to 30°C was used to cool the solution prior to UV detection at λ = 280 nm.
Hydrophobic interaction chromatography
HIC analysis utilized a Tosoh Bioscience TSKgel Butyl-NPR column (4.6 mm ID x 35 mm L, cat # 14947). The column was equilibrated with 100% MPA, composed of 25 mM sodium phosphate, 1.1 M ammonium sulfate, pH 6.7. Fifty micrograms of sample containing 2.5 M NaCl were injected onto a 35°C column, and eluted using gradient from 0 – 100% MPB (25 mM sodium phosphate, pH 6.7 + 25% IPA) at 0.8 mL/min over 18 minutes. Absorbance was detected at λ1 = 214 nm and λ2 = 280 nm.
RP-UHPLC chromatography of mAb and ADC
Analysis of the partially reduced mAb intermediate and the ADC in both reduced and non-reduced forms utilized the same Agilent Zorbax RRHD 300SB-C8 column described previously. Gradient elution was carried out with MPA composed of 0.1% TFA in water and MPB composed of 0.08% TFA in 90% CH3CN, 10% water. UV absorbances at λ1 = 280 nm and λ2 = 248 nm were used for peak integration.
The partially reduced mAb intermediate and the non-reduced ADCs (nrRP-UHPLC) were diluted with equal volumes of 2% formic acid, 50% CH3CN, 48% water. Five micrograms of protein were loaded onto a 75°C column. Separation of the component species was carried out at a flow rate of 0.5 mL/min by ramping up from 30% to 60% B over 14 minutes. Peak identification by matching theoretical to observed masses via LC-MS is described in the Supplementary Information (Table S1).
Analyses of the reduced ADC (rRP-UHPLC) were carried out by reducing the remaining interchain disulfide bonds via incubation in 20 mM DTT for 15 minutes at 37°C to generate variably conjugated light and heavy chain fragments. The ADC samples were adjusted to pH ∼8 using 1 M TRIS-HCl, pH 9 prior to reduction with DTT. Excess reducing reagent was quenched by addition of equal volume of 2% formic acid, 50% CH3CN, 48% water. Seven and a half micrograms of protein were loaded to the column set at 70°C, and elution used a multi-step gradient at 0.8 mL/min. Initially, 34.5% B was ramped up to 38.0% B over 3 minutes, then to 38.5% B over 2.5 minutes, and finally to 55.0% B over 20 minutes.
Average DAR was calculated by weighting the relative peak areas of each light and heavy chain based on the number of drug linkers attached (n), as described in the following equation:
Supplementary Material
Funding Statement
Agensys, Inc., an affiliate of Astellas.
Disclosure statement
All work in this article was financially supported by Agensys, Inc., an affiliate of Astellas. LL and GR are current employees of Agensys, Inc. AF was employed by Agensys, Inc. at the time of this research. The drug-linker technology used in this research is licensed from Seattle Genetics (Bothell, WA).
Acknowledgments
We would like to thank the Agensys Process Sciences and Manufacturing group for the production of the IgG2 mAb used in this study, the Agensys ADC Chemistry group for providing the vcMMAE reagent, and Jihea Park for the critical review of this manuscript. We appreciate the support expressed by the late Dr. Wolfgang Noe, former VP of Technical Operations at Agensys, and by Gary Welch, VP of Technical Operations at Agensys for the execution of this study.
References
- 1.Sau S, Alsaab HO, Kashaw SK, Tatiparti K, Iyer AK. Advances in antibody-drug conjugates: A new era of targeted cancer therapy. Drug Discov Today. 2017;22(10):1547–56. doi: 10.1016/j.drudis.2017.05.011. PMID:28627385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parslow A, Parakh S, Lee F-T, Gan H, Scott A. Antibody–drug conjugates for cancer therapy. Biomedicines. 2016;4:14. doi: 10.3390/biomedicines4030014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al.. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70. doi: 10.1158/1078-0432.CCR-04-0789. PMID:15501986. [DOI] [PubMed] [Google Scholar]
- 4.Sun MMC, Beam KS, Cerveny CG, Hamblett KJ, Blackmore RS, Torgov MY, Handley FGM, Ihle NC, Senter PD, Alley SC.. Reduction−alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjugate Chem. 2005;16:1282–90. doi: 10.1021/bc050201y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiggins B, Liu-Shin L, Yamaguchi H, Ratnaswamy G. Characterization of cysteine-linked conjugation profiles of immunoglobulin G1 and immunoglobulin G2 antibody–drug conjugates. J Pharmaceutical Sci. 2015;104:1362–72. doi: 10.1002/jps.24338. [DOI] [PubMed] [Google Scholar]
- 6.Le LN, Moore JMR, Ouyang J, Chen X, Nguyen MDH, Galush WJ. Profiling antibody drug conjugate positional isomers: A system-of-equations approach. Anal Chem. 2012;84:7479–86. doi: 10.1021/ac301568f. PMID:22913809. [DOI] [PubMed] [Google Scholar]
- 7.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–33. doi: 10.1111/j.1476-5381.2009.00190.x. PMID:19459844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen T, Chen Y, Stella C, Medley CD, Gruenhagen JA, Zhang K. Antibody-drug conjugate characterization by chromatographic and electrophoretic techniques. J Chromatography B. 2016;1032:39–50. doi: 10.1016/j.jchromb.2016.07.023. [DOI] [PubMed] [Google Scholar]
- 9.Ross PL, Wolfe JL. Physical and chemical stability of antibody drug conjugates: Current status. J Pharm Sci. 2016;105:391–7. doi: 10.1016/j.xphs.2015.11.037. PMID:26869406. [DOI] [PubMed] [Google Scholar]
- 10.Singh SK, Luisi DL, Pak RH. Antibody-drug conjugates: Design, formulation and physicochemical stability. Pharm Res. 2015;32:3541–71. doi: 10.1007/s11095-015-1704-4. PMID:25986175. [DOI] [PubMed] [Google Scholar]
- 11.Lambert JM. Typical Antibody–Drug Conjugates In: Olivier KJJ, Hurvitz SA, eds. Antibody-Drug Conjugates. Hoboken, New Jersey: John Wiley & Sons, Inc, 2016:1–32. [Google Scholar]
- 12.Beckley NS, Lazzareschi KP, Chih HW, Sharma VK, Flores HL. Investigation into temperature-induced aggregation of an antibody drug conjugate. Bioconjug Chem. 2013;24:1674–83. doi: 10.1021/bc400182x. PMID:24070051. [DOI] [PubMed] [Google Scholar]
- 13.Guo J, Kumar S, Chipley MT, Marcq O, Gupta D, Jin Z, Tomar DS, Swabowski C, Smith J, Starkey JA, et al.. Characterization and higher-order structure assessment of an interchain Cysteine-Based ADC: Impact of drug loading and distribution on the mechanism of aggregation. Bioconjug Chem. 2016;27:604–15. doi: 10.1021/acs.bioconjchem.5b00603. PMID:26829368. [DOI] [PubMed] [Google Scholar]
- 14.Guo J, Kumar S, Prashad A, Starkey J, Singh SK. Assessment of physical stability of an antibody drug conjugate by higher order structure analysis: impact of thiol- maleimide chemistry. Pharm Res. 2014;31:1710–23. doi: 10.1007/s11095-013-1274-2. PMID:24464270. [DOI] [PubMed] [Google Scholar]
- 15.Wakankar A, Chen Y, Gokarn Y, Jacobson FS. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs. 2011;3:161–72. doi: 10.4161/mabs.3.2.14960. PMID:21441786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Frontiers Immunol. 2014;5:520. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol. 2007;25:1369–72. doi: 10.1038/nbt1207-1369. PMID:18066027. [DOI] [PubMed] [Google Scholar]
- 18.McDonagh CF, Kim KM, Turcott E, Brown LL, Westendorf L, Feist T, Sussman D, Stone I, Anderson M, Miyamoto J, et al.. Engineered anti-CD70 antibody-drug conjugate with increased therapeutic index. Mol Cancer Therapeutics. 2008;7:2913–23. doi: 10.1158/1535-7163.MCT-08-0295. [DOI] [PubMed] [Google Scholar]
- 19.Botzanowski T, Erb S, Hernandez-Alba O, Ehkirch A, Colas O, Wagner-Rousset E, Rabuka D, Beck A, Drake PM, Cianférani S, et al.. Insights from native mass spectrometry approaches for top- and middle- level characterization of site-specific antibody-drug conjugates. MAbs. 2017;9:801–11. doi: 10.1080/19420862.2017.1316914. PMID:28406343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Said N, Gahoual R, Kuhn L, Beck A, Francois YN, Leize-Wagner E. Structural characterization of antibody drug conjugate by a combination of intact, middle-up and bottom-up techniques using sheathless capillary electrophoresis – Tandem mass spectrometry as nanoESI infusion platform and separation method. Anal Chim Acta. 2016;918:50–9. doi: 10.1016/j.aca.2016.03.006. PMID:27046210. [DOI] [PubMed] [Google Scholar]
- 21.Huang RY, Chen G. Characterization of antibody-drug conjugates by mass spectrometry: advances and future trends. Drug Discov Today. 2016;21:850–5. doi: 10.1016/j.drudis.2016.04.004. PMID:27080148. [DOI] [PubMed] [Google Scholar]
- 22.Pristatsky P, Cohen SL, Krantz D, Acevedo J, Ionescu R, Vlasak J. Evidence for Trisulfide bonds in a recombinant variant of a human IgG2 monoclonal antibody. Anal Chem. 2009;81:6148–55. doi: 10.1021/ac9006254. PMID:19591437. [DOI] [PubMed] [Google Scholar]
- 23.Kita A, Ponniah G, Nowak C, Liu H. Characterization of cysteinylation and Trisulfide bonds in a recombinant monoclonal antibody. Anal Chem. 2016;88:5430–7. doi: 10.1021/acs.analchem.6b00822. PMID:27115984. [DOI] [PubMed] [Google Scholar]
- 24.Gu S, Wen D, Weinreb PH, Sun Y, Zhang L, Foley SF, Kshirsagar R, Evans D, Mi S, Meier W, et al.. Characterization of trisulfide modification in antibodies. Anal Biochem. 2010;400:89–98. doi: 10.1016/j.ab.2010.01.019. PMID:20085742. [DOI] [PubMed] [Google Scholar]
- 25.Liu R, Chen X, Dushime J, Bogalhas M, Lazar AC, Ryll T, Wang L. The impact of trisulfide modification of antibodies on the properties of antibody-drug conjugates manufactured using thiol chemistry. MAbs. 2017;9:490–7. doi: 10.1080/19420862.2017.1285478. PMID:28136017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, Plant M, Henkle B, Li Y, Deechongkit S, et al.. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem. 2008;283:16206–15. doi: 10.1074/jbc.M709988200. PMID:18339626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, Fodor S, Kelner DN, Flynn GC, Liu YD, et al.. Human IgG2 Antibodies Display Disulfide-mediated Structural Isoforms. J Biol Chem. 2008;283:16194–205. doi: 10.1074/jbc.M709987200. PMID:18339624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Bailey R, Nightlinger N, Gillespie A, Balland A, Rogers R. Characterization of cysteine related variants in an IgG2 antibody by LC–MS with an automated data analysis approach. J Chromatography B. 2015;997:30–7. doi: 10.1016/j.jchromb.2015.04.035. [DOI] [PubMed] [Google Scholar]
- 29.Martinez T, Guo A, Allen MJ, Han M, Pace D, Jones J, Gillespie R, Ketchem RR, Zhang Y, Balland A. Disulfide Connectivity of Human Immunoglobulin G2 Structural Isoforms. Biochemistry. 2008;47:7496–508. doi: 10.1021/bi800576c. PMID:18549248. [DOI] [PubMed] [Google Scholar]
- 30.Liu YD, Chen X, JZ-v Enk, Plant M, Dillon TM, Flynn GC. Human IgG2 Antibody Disulfide Rearrangement in Vivo. J Biol Chem. 2008;283:29266–72. doi: 10.1074/jbc.M804787200. PMID:18713741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs. 2012;4:17–23. doi: 10.4161/mabs.4.1.18347. PMID:22327427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moritz B, Stracke JO. Assessment of disulfide and hinge modifications in monoclonal antibodies. Electrophoresis. 2017;38:769–85. doi: 10.1002/elps.201600425. PMID:27982442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He Y, Lacher NA, Hou W, Wang Q, Isele C, Starkey J, Ruesch M. Analysis of identity, charge variants, and disulfide isomers of monoclonal antibodies with capillary zone electrophoresis in an uncoated capillary column. Anal Chem. 2010;82:3222–30. doi: 10.1021/ac9028856. [DOI] [PubMed] [Google Scholar]
- 34.Wang X, Kumar S, Singh SK. Disulfide scrambling in IgG2 monoclonal antibodies: Insights from molecular dynamics simulations. Pharmaceutical Res. 2011;28:3128–44. doi: 10.1007/s11095-011-0503-9. [DOI] [PubMed] [Google Scholar]
- 35.Strop P, Liu S-H, Dorywalska M, Delaria K, Dushin Russell G, Tran T-T, Ho WH, Farias S, Casas MG, Abdiche Y, et al.. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;20:161–7. doi: 10.1016/j.chembiol.2013.01.010. PMID:23438745. [DOI] [PubMed] [Google Scholar]
- 36.Jackson D, Atkinson J, Guevara CI, Zhang C, Kery V, Moon SJ, Virata C, Yang P, Lowe C, Pinkstaff J, et al.. In vitro and in vivo evaluation of cysteine and site specific conjugated herceptin antibody-drug conjugates. PLoS One. 2014;9:e83865. doi: 10.1371/journal.pone.0083865. PMID:24454709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Chumsae C, Gaza-Bulseco G, Hurkmans K, Radziejewski CH. Ranking the susceptibility of disulfide bonds in human IgG1 antibodies by reduction, differential alkylation, and LC-MS analysis. Anal Chem. 2010;82:5219–26. doi: 10.1021/ac100575n. PMID:20491447. [DOI] [PubMed] [Google Scholar]
- 38.Wang Q, Lacher NA, Muralidhara BK, Schlittler MR, Aykent S, Demarest CW. Rapid and refined separation of human IgG2 disulfide isomers using superficially porous particles. J Separation Sci. 2010;33:2671–80. doi: 10.1002/jssc.201000230. [DOI] [PubMed] [Google Scholar]
- 39.Liu YD, Chou RY, Dillon TM, Poppe L, Spahr C, Shi SD, Flynn GC. Protected hinge in the immunoglobulin G2-A2 disulfide isoform. Protein Sci. 2014;23:1753–64. doi: 10.1002/pro.2557. PMID:25264323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W, Marzilli LA, Rouse JC, Czupryn MJ. Complete disulfide bond assignment of a recombinant immunoglobulin G4 monoclonal antibody. Analytical Biochem. 2002;311:1–9. doi: 10.1016/S0003-2697(02)00394-9. [DOI] [PubMed] [Google Scholar]
- 41.Trivedi MV, Laurence JS, Siahaan TJ. The role of thiols and disulfides in protein chemical and physical stability. Curr Protein Peptide Sci. 2009;10:614–25. doi: 10.2174/138920309789630534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campuzano IDG, Netirojjanakul C, Nshanian M, Lippens JL, Kilgour DPA, Van Orden S, Loo JA. Native-MS analysis of monoclonal antibody conjugates by fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2018;90:745–51. doi: 10.1021/acs.analchem.7b03021. PMID:29193956. [DOI] [PubMed] [Google Scholar]
- 43.Gilroy JJ, Eakin CM. Characterization of drug load variants in a thiol linked antibody-drug conjugate using multidimensional chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1060:182–9. doi: 10.1016/j.jchromb.2017.06.005. PMID:28622622. [DOI] [PubMed] [Google Scholar]
- 44.Beck A, Terral G, Debaene F, Wagner-Rousset E, Marcoux J, Janin-Bussat MC, Colas O, Van Dorsselaer A Cianférani S. Cutting-edge mass spectrometry methods for the multi-level structural characterization of antibody-drug conjugates. Exp Rev Proteomics 2016;13:157–83. doi: 10.1586/14789450.2016.1132167. [DOI] [PubMed] [Google Scholar]
- 45.Sarrut M, Corgier A, Fekete S, Guillarme D, Lascoux D, Janin-Bussat MC, Beck A, Heinisch S. Analysis of antibody-drug conjugates by comprehensive on-line two-dimensional hydrophobic interaction chromatography x reversed phase liquid chromatography hyphenated to high resolution mass spectrometry. I – Optimization of separation conditions. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1032:103–11. doi: 10.1016/j.jchromb.2016.06.048. PMID:27426266. [DOI] [PubMed] [Google Scholar]
- 46.Adamo M, Sun GY, Qiu DF, Valente J, Lan WK, Song HT, Bolgar M, Katiyar A, Krishnamurthy G. Drug-to-antibody determination for an antibody-drug-conjugate utilizing cathepsin B digestion coupled with reversed-phase high-pressure liquid chromatography analysis. J Chromatogr A. 2017;1481:44–52. doi: 10.1016/j.chroma.2016.12.051. PMID:28017567. [DOI] [PubMed] [Google Scholar]
- 47.Cohen SL, Price C, Vlasak J. β-elimination and peptide bond hydrolysis: Two distinct mechanisms of Human IgG1 hinge fragmentation upon storage. J Am Chem Soc. 2007;129:6976–7. doi: 10.1021/ja0705994. PMID:17500521. [DOI] [PubMed] [Google Scholar]
- 48.Vlasak J, Ionescu R. Fragmentation of monoclonal antibodies. mAbs. 2011;3:253–63. doi: 10.4161/mabs.3.3.15608. PMID:21487244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16:315–37. doi: 10.1038/nrd.2016.268. PMID:28303026. [DOI] [PubMed] [Google Scholar]
- 50.Ryazantsev S, Tischenko V, Nguyen C, Abramov V, Zav'yalov V. Three-dimensional structure of the human myeloma IgG2. PLoS One. 2013;8:e64076. doi: 10.1371/journal.pone.0064076. PMID:23762236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang A, Fang J, Chou RYT, Bondarenko PV, Zhang Z. Conformational difference in human IgG2 disulfide isoforms revealed by hydrogen/deuterium exchange mass spectrometry. Biochemistry. 2015;54:1956–62. doi: 10.1021/bi5015216. PMID:25730439. [DOI] [PubMed] [Google Scholar]
- 52.Allen MJ, Guo A, Martinez T, Han M, Flynn GC, Wypych J, Liu YD, Shen WD, Dillon TM, Vezina C, et al.. Interchain disulfide bonding in human IgG2 antibodies probed by site-directed mutagenesis. Biochemistry. 2009;48:3755–66. doi: 10.1021/bi8022174. PMID:19254029. [DOI] [PubMed] [Google Scholar]
- 53.Zhang B, Harder AG, Connelly HM, Maheu LL, Cockrill SL. Determination of Fab−Hinge Disulfide connectivity in structural isoforms of a recombinant human Immunoglobulin G2 antibody. Anal Chem. 2010;82:1090–9. doi: 10.1021/ac902466z. PMID:20039682. [DOI] [PubMed] [Google Scholar]
- 54.Bloom JW, Madanat MS, Marriott D, Wong T, Chan SY. Intrachain disulfide bond in the core hinge region of human IgG4. Protein Sci. 1997;6:407–15. doi: 10.1002/pro.5560060217. PMID:9041643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rispens T, Ooijevaar-de Heer P, Bende O, Aalberse RC. Mechanism of immunoglobulin G4 Fab-arm exchange. J Am Chem Soc. 2011;133:10302–11. doi: 10.1021/ja203638y. PMID:21627172. [DOI] [PubMed] [Google Scholar]
- 56.Michaelsen TE. Alteration of the conformation of human IgG subclasses by reduction of the hinge S-S bonds. Mol Immunol. 1988;25:639–46. doi: 10.1016/0161-5890(88)90099-5. PMID:3419438. [DOI] [PubMed] [Google Scholar]
- 57.Behrens CR, Ha EH, Chinn LL, Bowers S, Probst G, Fitch-Bruhns M, Monteon J, Valdiosera A, Bermudez A, Liao-Chan S, et al.. Antibody–Drug Conjugates (ADCs) Derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. Mol Pharmaceutics. 2015;12:3986–98. doi: 10.1021/acs.molpharmaceut.5b00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Panowksi S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014;6:34–45. doi: 10.4161/mabs.27022. PMID:24423619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou Q, Kim J. Advances in the development of site-specific antibody-drug conjugation. Anticancer Agents Med Chem. 2015;15:828–36. doi: 10.2174/1871520615666150302125448. PMID:25731178. [DOI] [PubMed] [Google Scholar]
- 60.Mantaj J, Jackson PJM, Rahman KM, Thurston DE. From Anthramycin to Pyrrolobenzodiazepine (PBD)‐Containing Antibody–Drug Conjugates (ADCs). Angewandte Chemie (International Ed in English). 2017;56:462–88. doi: 10.1002/anie.201510610. PMID:27862776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Goeij BECG, Lambert JM. New developments for antibody-drug conjugate-based therapeutic approaches. Curr Opin Immunol. 2016;40:14–23. doi: 10.1016/j.coi.2016.02.008. PMID:26963132. [DOI] [PubMed] [Google Scholar]
- 62.Masters JC, Nickens DJ, Xuan D, Shazer RL, Amantea M. Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Invest New Drugs. 2018;36:121–135. doi: 10.1007/s10637-017-0520-6. PMID:29027591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.