

Abstract
The use of first- and second-generation tyrosine kinase inhibitors (TKIs) significantly improves prognosis for patients with early chronic phase chronic myeloid leukemia (CML) and efficiently counteracts leukemia in most patients with CML bearing a disease characterized by the expression of BCR–ABL1 mutants. However, the so-called ‘tinib’ TKIs (e.g. imatinib, nilotinib, dasatinib, and bosutinib) are both ineffective in patients who undergo blastic transformation and unable to eradicate CML at the stem cell level. This raises a few important questions. Is BCR–ABL1 expression and/or activity essential for blastic transformation? Is blastic transformation the result of genetic or epigenetic events that occur at the stem cell level which only become apparent in the granulocyte-macrophage progenitor (GMP) cell pool, or does it arise directly at the GMP level? As altered mRNA metabolism contributes to the phenotype of blast crisis CML progenitors (decreased translation of tumor suppressor genes and transcription factors essential for terminal differentiation and increased translation of anti-apoptotic genes), one attractive concept is to restore levels of these essential molecules to their normal levels. In this review, we discuss the mechanisms by which mRNA processing, translation, and degradation are deregulated in BCR–ABL1 myeloid blast crisis CML progenitors, and present encouraging results from studies with pharmacologic inhibitors which support their inclusion in the clinic.
Keywords: Myeloid leukemias and dysplasias, RNA binding proteins, blast crisis
Introduction
Chronic myeloid leukemia (CML) is a disease that, without treatment, progresses from the indolent chronic phase (CML-CP), to an accelerated phase (CML-AP), and then a rapidly fatal blast crisis (CML-BC). The chronic phase of the disease is characterized by the accumulation of myeloid precursors in peripheral blood, bone marrow, and extramedullary sites that display enhanced survival and reduced susceptibility to drug-induced apoptosis while retaining the ability to terminally differentiate. CML-BC, which now occurs mostly in patients with CML-CP refractory to tyrosine kinase inhibitor (TKI)-based therapies or with non-manageable TKI resistance, is characterized by a marked increase in myeloid (M-BC) or lymphoid (L-BC) progenitors in the aforementioned tissues that demonstrate enhanced proliferation and self-renewal capability, increased survival and genomic instability, and, unlike CML-CP, differentiation arrest [1,2].
The hallmark of the disease is the presence of the Philadelphia chromosome (Ph1), which results from the reciprocal translocation of the breakpoint cluster region (BCR) gene on chromosome 22 and the c-ABL gene on chromosome 9, t(9;22)(q34:q11). In hematopoietic progenitors, BCR–ABL1 functions as a constitutively active tyrosine kinase that has been shown to activate numerous pathways both in vitro and in vivo, including Ras/mitogen activated protein kinase (MAPK) [3], phosphatidylinositol 3-kinase (PI3K)/Akt [4], and signal transducer and activator of transcription (STAT) [5–7] pathways. In CML-CP, presence of the Ph1 chromosome is, to date, the sole cytogenetic alteration [1], although it is plausible that BCR–ABL1-induced genomic instability is likely to induce a milieu of important mutations also in CML-CP [8,9], which ultimately may determine the time of emergence of a cell clone undergoing progression. Indeed, additional molecular and chromosomal abnormalities are often detected in CML-BC. For example, secondary genetic lesions found in patients in M-BC include mutations leading to inactivation of the p53 tumor suppressor (12–30%) [1,10], and enhanced transcription of target genes resulting from the GATA2 L359V single-nucleotide polymorphism (SNP) or point mutation (10%) [11]. In rare cases of CML-BC, mutation of the Ras oncogene has led to its constitutive activity [1]. In L-BC, mutation of the p16/ARF gene, which helps stabilize p53 by sequestering MDM2, is found in 50–60% of patients [1]. Additional mutations/deletions identified in L-BC include deletion of the tumor suppressor Rb (18%) [1], and amplification of IKZF1 (70%) [12]. Interestingly, the absence of a secondary mutation common to all patients diagnosed with M-BC suggests that there are likely one or more unidentified molecular mechanisms leading to disease progression [8]. One attractive hypothesis is that increased levels of BCR–ABL1 activity are responsible for this transition. This notion is supported by the increased BCR–ABL1 expression/activity in CD34+ common myeloid progenitors (CMPs) and granulocyte-macrophage progenitors (GMPs) from patients in M-BC [13–20]. The increased BCR–ABL1 activity leads to genetic instability [21], which causes secondary genetic abnormalities that, in many cases, inactivate tumor suppressor genes [1]. This hypothesis is supported by the enhanced survival and persistence of Ph+ CD34+ progenitors in TKI-treated patients in M-BC.
The employment of imatinib mesylate (IM) as first-line therapy has greatly improved the prognosis for patients diagnosed in CML-CP; however; its ineffectiveness in patients in CML-BC and those with Ph+ B-cell acute lymphoblastic leukemia (B-ALL) [22–24] has led to the introduction of second-generation tyrosine kinase inhibitors (e.g. dasatinib, nilotinib, and bosutinib) in combination with conventional chemotherapy as first-line treatment for CML-BC. While these compounds have shown promise, they are by no means a curative therapy as not all patients show long-term response, and relapse within 18 months. Thus, additional molecular targets for CML-BC therapy downstream of BCR–ABL1 need to be identified.
As altered mRNA metabolism seems to play a pivotal role in CML-BC, in that the processing, export, translation, and degradation of specific mRNAs regulating proliferation, survival, and differentiation of myeloid progenitors are aberrantly regulated by increased BCR–ABL1 expression and activity, restoring mRNA metabolism to basal levels within these cells is an attractive concept. In this review, we describe the mechanisms by which mRNA metabolism is deregulated in BCR–ABL1 cells, and present results from studies employing pharmacologic agents impairing them, thereby supporting their inclusion in blast crisis CML therapy.
Altered mRNA processing in chronic myeloid leukemia
Several mRNA species, including BCR–ABL1 [25,26], have been shown to be alternatively spliced in Ph+ leukemias. In Ph+ B-ALL, expression of BCR–ABL1 leads to the absence of IKAROS, a transcription factor required for early lymphoid lineage commitment, resulting from the BCR–ABL1-induced dominant-negative splice variant of IKAROS, termed IK6. As a consequence of defective IKAROS signaling, these cells express myeloid lineage-specific molecules [27]. Inhibition of IK6 in BCR–ABL1 cell lines by RNA interference partially restores B-lymphoid lineage commitment [27]. Another species that has been found to be alternatively spliced in CML, in this instance leading to increased levels, is the β1-integrin-responsive non-receptor tyrosine kinase PYK2 [28]. It remains to be determined whether altered splicing of PYK2 contributes to CML leukemogenesis, but it is reasonable to suspect its involvement, as β1-integrin signaling is important for maintenance, proliferation, and differentiation of hematopoietic stem cells and is increased in BCR–ABL1 cells [28–32]. Additionally, glycogen synthase kinase 3β (GSK3β) has also been shown to be alternatively spliced in GMP cells isolated from patients in CML-BC [33]. Sequencing of cDNA from GMPs revealed an in-frame splice deletion of the GSK3β kinase domain. This misspliced gsk3b results in increased β-catenin expression, a protein shown to be essential for the acquisition of self-renewal of CML-BC GMPs [34]. Reintroduction of full-length GSK3β mRNA reduced both in vitro replating and in vivo leukemic engraftment, thereby limiting the self-renewal ability of the CML-BC leukemia-initiating GMP cells [33]. Altogether, these studies indicate that BCR–ABL1 expression affects pre-mRNA splicing, leading to deregulated expression of genes contributing to the CML-BC phenotype, and provide additional mechanisms (e.g. β-catenin inhibitors) for therapeutic intervention.
Altered mRNA translation in chronic myeloid leukemia
Increased mammalian target of rapamycin signaling in CML
The study of Ras and PI3K/Akt pathways, two pathways that have been shown to be essential for transformation in CML [35–37], revealed that BCR–ABL1 expression, through PI3K/Akt, results in activation of the mammalian target of rampamycin (mTOR) [38–40]. In addition, independent of PI3K/Akt, a pathway involving phospholipase Cγ1 was also shown to activate mTOR in BCR–ABL1-expressing cell lines and CML mononuclear cells (MNCs) [41]. mTOR is a serine–threonine kinase that functions as two complexes, TORC1 (target of rapamycin complex-1) and TORC2 (target of rapamycin complex-2) [4], which regulate cell growth and proliferation in response to growth factors [43]. Targets of mTOR include the 40S ribosomal protein p70S6-kinase, which promotes translation of specific mRNA through its phosphor-ylation of S6, and the eukaryotic initiation factor 4E-binding protein-1 (4E-BP1), a negative repressor of mRNA translation [43–45]. Phosphorylation of S6 leads to its activation, while phosphorylation of 4E-BP1 results in its inactivation, as it no longer binds eukaryotic initiation factor 4E (eIF4E), allowing formation of the eIF4F complex which performs 5′-cap-dependent translation [46]. In fact, over-expression and phosphorylation of eIF4E is essential for β-catenin activation in CML-BC [47]. Imatinib treatment of BCR–ABL1-expressing cell lines and primary cells from patients with CML (CP and BC) indicates that S6 and 4E-BP1 are phosphorylated, in part, in a BCR–ABL1 kinase-dependent manner [38,41,48]. Treatment of IM-sensitive and -resistant CML cell lines, as well as primary CML (CP and BC) progenitors, with mTOR antagonists suggests that reinstating normal levels of mTOR signaling may be effective in inhibiting BCR–ABL1-driven leukemogenesis [38,41,48,49]. Interestingly, rapamycin, an mTOR antagonist, acts synergistically with IM, and co-treatment of peripheral blood from patients with CML (CP and BC) leads to greatly reduced numbers of colony-forming cells [48]. Recently, the application of PP242, a more potent mTOR antagonist than rapamycin, reduced the viability of Ph+ B-ALL or CML-BC cell lines, and the percentage of blasts in the bone marrow and peripheral blood in a Ph B-ALL mouse model [49]. Moreover, CGP57380,+a compound that regulates translation initiation and polysomal assembly independently of mTOR, synergistically acts with IM to impair polysomal mRNA loading in BCR–ABL1-expressing cells, which, in turn, results in inhibited growth and survival of Ba/F3-BCR–ABL1 and K562 cells via impaired cell cycle entry and increased apoptosis [50]. The importance of increased mTOR signaling in BCR–ABL1 cells was further illustrated upon treatment of leukemic cells with arsenic trioxide [51]. Treatment of various acute leukemia cell lines with arsenic trioxide resulted in the phosphorylation of 4E-BP1, leading to its above-mentioned inactivation, suggestive of increased mTOR signaling, as part of a negative feedback loop that counteracts signals triggered by this treatment [51]. Two mRNAs have been reported to be more efficiently translated within BCR–ABL1-expressing cells as a result of increased mTOR activity. First, treatment of the Ba/F3-BCR–ABL1 cell line and primary cells from a patient in blast crisis with IM revealed a decrease in protein levels of the cell cycle progression-promoting cyclin D3, while its mRNA levels remained the same [48]. Similar results were observed upon treatment of these cells with rapamycin, an inhibitor of mTOR signaling [48]. Interestingly, IM and rapamycin acted synergistically against primary CML progenitor cells resulting in significantly decreased levels of cyclin D3, thereby causing cell cycle arrest [48]. A second mRNA whose translation is increased due to increased mTOR activity in BCR–ABL1-expressing cell lines is hypoxia inducible factor-1α (HIF-1α) [52], a transcription factor for vascular endothelia growth factor (VEGF), leading to increased levels of VEGF, a protein implicated in leukemia-associated angio-genesis [53]. These studies indicate that BCR–ABL1 kinase-deregulated mTOR signaling results in altered mRNA metabolism of genes involved in pathways that contribute to the phenotype of these cells. Taken together, these studies indicate the potential efficacy of mTOR and mRNA translation inhibitors in IM-sensitive and, more important, IM-resistant CML and warrant further investigation. In fact, clinical trials with a combination of IM and mTOR antagonists are currently being performed in patients with CML who were unable to achieve a complete cytogenetic response with IM; however, whether this combination therapy will prolong survival of patients with CML-BC needs yet to be determined.
Autophagy in CML cells and altered mRNA translation
Autophagy is a cellular process in eukaryotic cells that results from the destruction of intracellular materials (e.g. long-lived proteins, organelles such as mitochondria and ribosomes [54]) within lysosomes in the cytoplasm under normal conditions and in response to stress stimuli [55,56]. Originally described as an alternative cell death mechanism [57,58], it has become more evident that this process can serve as a cell survival mechanism, as autophagy occurs in cells deprived of growth factors [59,60], allowing them to evade apoptosis. Treatment of the K562 cell line with IM induces autophagy as indicated by the expression of autophagy-related genes and increased presence of translucent vacuoles within these cells [61]. More important, the addition of the pharmacologic autophagy inhibitor chloroquine to IM treatment potentiated IM-induced cell death in CML cell lines and primary CML cells, including those carrying partially IM-resistant BCR–ABL1 mutations, and reduced the colony formation of CD34+ CD38− CML cells and long-term culture-initiating cells (LTC-ICs) [61]. Based on these results, it is reasonable to speculate that during IM-induced autophagy the number of ribosomes within the cytoplasm is reduced, leading to decreased translation of tumor suppressor genes, thereby providing a novel mechanism by which CML cells evade apoptosis and supporting the inclusion of autophagy inhibitors (e.g. chloroquine) in the treatment of both IM-resistant CML and CML-BC.
Increased RNA binding protein expression in blast crisis chronic myeloid leukemia
RNA binding proteins play an essential role in RNA metabolism as they regulate mRNA fate from the active site of transcription to that of translation [62]. Throughout this journey the mRNA is not unaccompanied, as different RNA binding proteins participate in binding to regulatory elements or are involved in maintaining specific mRNA secondary structures [62]. Thus, while some RNA binding proteins are general regulators of mRNA transcription, processing, nuclear export, stability, and translation, others bind mRNA in a sequence-specific manner and determine the fate of a specific mRNA [63,64]. Therefore, it is plausible that BCR–ABL1-induced alteration in the expression or function of these RNA binding proteins has a profound effect on Ph+ progenitor cell proliferation, survival, and differentiation (Figure 1). Indeed, enhanced expression of various RNA binding proteins that bind mRNA in a sequence-specific manner is among the changes in gene expression found in primary mono-nuclear and CD34+ cells from patients with CML-BC and in BCR–ABL1-transformed murine myeloid progenitors. Enhanced expression of specific RNA binding proteins correlates with the levels of BCR–ABL1 and is sensitive to treatment with IM [65,66]. Mechanisms by which an increase in these proteins is observed include enhanced gene transcription (e.g. heterogeneous nuclear ribonucleoprotein K; hnRNP K [67]) or increased protein stability (e.g. TLS/FUS [68], hnRNP A1 [69], hnRNP E2 [18], and La/SSB [20]). Increased expression of these RNA binding proteins correlates with enhanced activity, which is positively regulated by BCR–ABL1-activated pathways (e.g. PI3K/Akt, MAPK/extracellular signal-regulated kinase 1/2 [MAPKERK1/2], protein kinase C). By contrast, expression of the RNA binding protein CUGBP1 inversely correlates with BCR–ABL1 activity and diminishes in CML-BC compared with CML-CP and normal bone marrow progenitors [70].
Figure 1.
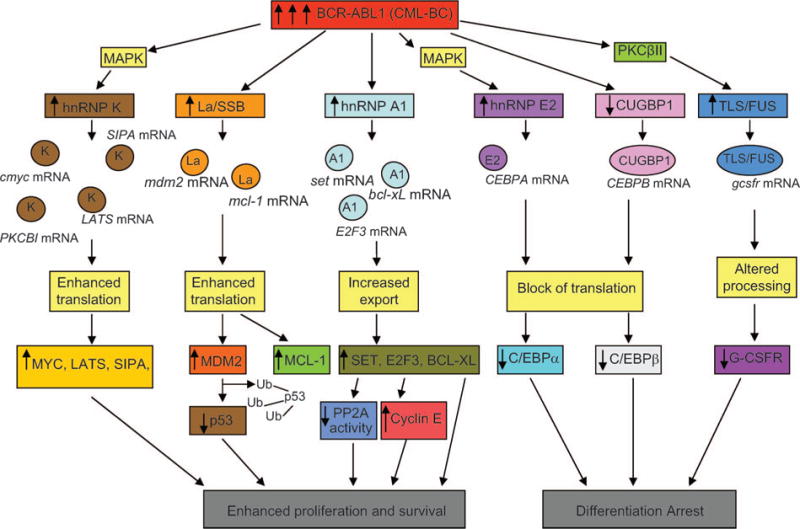
Altered mRNA metabolism in CML-BC resulting from deregulated expression of RNA binding proteins. The differential expression of RNA binding proteins in CML-BC, when compared with CML-CP, results from increased BCR–ABL expression and activity levels, and leads to the altered mRNA metabolism of tumor suppressor genes, genes involved in regulating proliferation, and those responsible for differentiation. These altered pathways subsequently contribute to the malignant phenotype of this disease.
hnRNP A1 and SET/protein phosphatase 2A interplay
hnRNP A1 is a nucleocytoplasmic shuttling protein that binds the nascent pre-mRNA in a sequence-specific manner and regulates splice site selection, exon skipping or inclusion, nuclear export of mature mRNA, mRNA turnover, and translation [71]. In addition to its interaction with mRNA species, hnRNP A1 has been shown to be required for the processing of miR-18a [72], a miRNA that is part of the miR-17–92 cluster which has been reported to be differentially expressed in CD34+ progenitors from patients with CML-CP and CML-BC [73]. As a consequence of enhanced protein stability, hnRNP A1 expression is induced by BCR–ABL1 in a dose-and kinase-dependent manner [17,69]. In fact, hnRNP A1 levels are higher in CML-BC than in CML-CP progenitors [17,69]. The use of a nucleus-localized dominant-negative hnRNP A1 mutant deficient in shuttling activity indicates that the hnRNP A1 mRNA export activity is required for cytokine-independent proliferation, survival, and tumorigenesis of acute phase CML blasts and BCR–ABL1-expressing myeloid progenitor cell lines [69], indicating in turn that hnRNP A1 controls the nuclear export of mRNAs encoding factors that are important for the leukemic phenotype of CML-BC progenitors. For example, the anti-apoptotic factor BCL-XL and the pro-oncogene SET, the physiologic inhibitor of protein phosphatase 2A (PP2A), are among some of the cytoplasmic mRNAs whose export and translation depend on hnRNP A1 shuttling activity [17,69,74]. SET expression is induced by high levels of BCR–ABL1 activity, similar to those detected only in CML-BC GMPs, and increases during CML disease progression [17]. Interestingly, enhanced SET levels lead to inactivation of the tumor suppressor PP2A in CML-BC and Ph+ B-ALL progenitors [17]. In these cells, PP2A loss-of-function accounts for increased and sustained BCR–ABL1 activity in CML-BC progenitors [17]. In fact, molecular or pharmacologic reactivation of PP2A phosphatase activity inhibits BCR–ABL1 activity and triggers BCR–ABL1 proteosomal degradation through a mechanism that requires activity of the SHP-1 tyrosine-phosphatase [17]. As a result of the rescue of PP2A activity, IM-sensitive and -resistant (T315I included) BCR–ABL1+ cell lines and CML-BC patient-cells cease to proliferate and undergo apoptosis [17]. Furthermore, impaired clonogenicity of CML-BC patient-cells and decreased in vivo BCR–ABL1-driven leukemogenesis are observed on treatment with PP2A-activating drugs (e.g. forskolin, 1,9-dideoxy forskolin, and FTY720) used at pharmacologic doses that do not exert toxic in vivo effects in rodents and/or in ex vivo human normal hematopoietic cells [75]. Interestingly, SET-dependent inactivation of PP2A may also occur through the activity of another ribonucleoprotein, hnRNP A2. In this regard, it has been reported that hnRNP A2 cooperates with SET in the inhibition of PP2A [76]. In agreement, hnRNP A2/B1 is overexpressed in human CD34+ progenitors ectopically expressing p210 BCR–ABL1, suggesting the possible involvement of hnRNP A2/B1 in blastic transformation of CML [77]. Another mRNA that has been shown to interact with hnRNP A1 in a BCR–ABL1 kinase-dependent manner is the transcription factor E2F3 [74]. The mRNA encoding E2F3 associates to hnRNP A1 through a conserved binding site located in the E2F3 3′-untranslated region (UTR). E2F3 levels were up-regulated in CML-BCCD34+ in a BCR–ABL1 kinase- and hnRNP A1 shuttling-dependent manner [74]. Furthermore, shRNA-mediated E2F3 knock-down and BCR–ABL1-transduced lineage-negative bone marrow cells from E2F3+/+ and E2F3−/− mice demonstrated that E2F3 is important for BCR–ABL1 clonogenic activity in vitro and in vivo and leukemogenic potential [74].
hnRNP K and MYC overexpression in CML-BC
In BCR–ABL1-expressing myeloid and lymphoid cell lines and in primary CML-BC but not CML-CP progenitor cells, hnRNP K transcription and mRNA stability are enhanced by MAPKERK1/2 in a BCR–ABL1 dose- and kinase-dependent manner [67]. Expression of hnRNP K is essential for BCR–ABL1 leukemogenesis as its down-modulation impairs cytokine-independent clonogenicity and leukemogenesis of BCR–ABL1+ cells [67]. These effects mostly depend on decreased levels of MYC, an oncogene essential for BCR–ABL1 leukemogenesis [78] and whose expression is transcriptionally and translationally controlled by hnRNP K [79,80]. While hnRNP K transcriptional regulation is dispensable for BCR–ABL1 oncogenic potential, its MAPK-dependent translational-regulatory activity is required both in vitro and in vivo [67]. In fact, hnRNP K binds to the internal ribosome entry site element of MYC mRNA and enhances its translation in a BCR–ABL1-MAPK-dependent manner [67]. These data are consistent with MYC protein but not mRNA overexpression in most CML-BC progenitors [67,81]. Several studies suggest that targeting molecules responsible for deregulated expression of the RNA binding protein hnRNP K in CML-BC may be effective in impairing BCR–ABL1-driven leukemogenesis. First, treatment of the 32D-BCR–ABL1 and K562 cell lines with the MAPK inhibitors PD098059, U0126, and the clinically relevant CI-1040 resulted in substantially decreased levels of hnRNP K protein [67]. Second, the introduction of hnRNP K shRNA in BCR–ABL1+ cells reduced MYC levels and resulted in decreased viability and clonogenic potential of these cells [67]. Third, 32D-BCR–ABL1 cells transduced with a MAPK-resistant hnRNP K cDNA demonstrated impaired cytokine-independent growth and colony formation. Consistent with these effects in vitro, transplantation of these cells into SCID (severe combined immunodeficiency) mice resulted in a longer latency in tumor formation and reduced tumor burden in these recipient mice when compared with controls. These studies suggest that treatment of CML-BC with the MAPK inhibitor PD098059 may be of therapeutic benefit. In addition to MYC, other mRNAs found associated with and translationally regulated by hnRNP K in Ph1 cell lines in a BCR–ABL1 kinase-dependent manner include LATS1, SIPA1, and protein kinase-Cβ1 (PKCβI) [74]. LATS1 promotes proliferation by binding to cyclin dependent kinase 1 (Cdk1), thereby freeing cyclins whose transcriptional functions are necessary for cell cycle progression, while SIPA1 levels have been found to correlate with metastatic capability both in vitro and in vivo [82]. PKCβ1 is a member of the protein kinase C family of proteins that play a key role in the regulation of cell growth and differentiation. Other mRNAs found to interact with hnRNP K in BCR–ABL1-expressing cells are those of reticulocyte 15-lipoxygenase (r15-lox) and c-Src [83]. r15-lox is important for erythroid precursor maturation and is regulated by hnRNP K at the translational level. Ectopic expression of human r15-lox in the K562 cell line and in primary human CD34+ induced terminal erythroid differentiation [83]. In addition, hnRNP K was also found to bind to the 3′-UTR of c-Src and inhibit its translation by blocking 80S ribosome formation in K562 [83]. Taken together, these studies indicate that hnRNP K contributes to leukemogenesis by BCR–ABL1 by enhancing or repressing the translation of mRNA species responsible for the enhanced survival, increased proliferation, and differentiation arrest that are characteristic of CML-BC progenitors.
La/SSB and MDM2: implication for p53 functional inactivation in CML-BC
The RNA binding protein La/SSB controls RNA metabolism at different levels: it binds and protects newly RNA polymerase III-transcribed RNAs, regulates the processing of 5′ and 3′ ends of pre-tRNA precursors, functions as an RNA chaperone, and controls translation of specific mRNAs [84]. Expression of La is markedly increased by BCR–ABL1 and correlates with that of MDM2 [20]. La is more abundant in CML-BC than CML-CP patient-cells, and its levels appear to correlate with BCR–ABL1 levels and activity [20]. Interestingly, La is a positive regulator of mdm2 translation because: (1) it recognizes a specific conserved sequence in the intercistronic region of mdm2 mRNA that is required for efficient MDM2 expression; (2) a dominant-negative La inhibits mdm2 mRNA translation in BCR–ABL1+ cells; (3) La down-regulation leads to a marked decrease in MDM2 levels; and (4) over-expression of wild-type La increases MDM2 expression [20]. That La-mediated effect on MDM2 expression is functionally relevant for BCR–ABL1 leukemogenesis is indicated by the changes in susceptibility of BCR–ABL1-expressing cells to p53-dependent drug-induced apoptosis [20]. Inactivating mutations of the p53 gene are rarely found in CML-CP but are common in M-BC [10], suggesting that loss of function of p53 contributes to disease progression. Indeed, loss of wild-type p53 potentiates the leukemic-inducing effects of BCR–ABL1 [85,86]. Because genetic inactivation of p53 is in only ~30% of patients with CML-BC [10], the La-dependent MDM2 overexpression may represent a mechanism whereby BCR–ABL1 functionally inactivates p53 in patients with CML-BC with a wild-type p53 gene. Thus, the La-dependent stimulation of MDM2 translation is not only relevant for survival of CML-BC progenitors but may also contribute to disease progression through functional inactivation of the p53 tumor suppressor. In addition to MDM2, other mRNA species have been found to be associated with La in BCR–ABL1+ cells in a BCR–ABL1 kinase-dependent manner. A few examples include the previously mentioned LATS1 [74], and the potent suppressor of apoptosis Mcl-1 [74], which has been implicated in CML [87–89].
hnRNP E2 and CCAAT/enhancer binding protein α: differentiation arrest in M-BC
The main feature of CML-BC is the inability of myeloid progenitors to undergo terminal differentiation. This is primarily dependent on inhibition of CCAAT/enhancer binding protein α (C/EBPα) [18], a transcription factor essential for granulocytic differentiation [90,91]. In M-BC CD34+ progenitors, loss of C/EBPα depends on BCR–ABL1-induced activity of the RNA binding protein hnRNP E2, a poly(rC)-binding protein (also referred to as PCBP2) that, like hnRNP K, controls the translation of specific mRNAs [65,92]. In fact, hnRNP E2, upon interaction with the 5′-UTR of C/EBPA mRNA, inhibits C/EBPA translation [18]. As a result, C/EBPα protein but not mRNA expression is down-modulated in primary bone marrow cells from patients with CML-BC and inversely correlated with BCR–ABL1 levels [18]. Accordingly, hnRNP E2 expression was inversely correlated with that of C/EBPα [17] because hnRNP E2 levels were abundant in CML-BC but undetectable in CML-CP mononuclear marrow cells. In fact, hnRNP E2 expression is induced by BCR–ABL1 in a dose- and kinase-dependent manner through constitutive activation of MAPKERK1/2 that, in turn, post-translationally increases hnRNP E2 protein stability [93]. The importance of loss of C/EBPα as a central mechanism leading to differentiation arrest of CML myeloid blasts is supported by the finding that: (1) ectopic C/EBPα expression induces maturation of differentiation-arrested BCR–ABL1-expressing myeloid precursors [18]; (2) a blast crisis-like process emerges in mice transplanted with BCR–ABL1-transduced c/ebpα-null but not heterozygous or wild-type fetal liver cells [94]; and (3) genetic or functional inactivation of C/EBPα is a common event in differentiation-arrested acute myeloid leukemia [95]. In addition to translational repression of C/EBPα by hnRNP E2, hnRNP E2 has been shown to affect levels of the transcription factor FOXO1A in a BCR–ABL1 kinase-dependent manner [74]. While the major consequence of increased hnRNP E2 expression in CML-BC progenitors is their aforementioned arrest in differentiation, the introduction of hnRNP E1 and hnRNP E2 shRNA together in the K562 cell line results in decreased proliferation as the cells become arrested in the G1 phase of the cell cycle [96]. Analysis of mRNP complexes in K562 revealed the association of hnRNP E1 and hnRNP E2 with the cyclin dependent kinase inhibitor 1A, also known as p21WAF, thereby inhibiting its translation [96]. These studies illustrate how BCR–ABL1 kinase-dependent increased levels of hnRNP E2 contribute to the block in differentiation observed in CML-BC myeloid progenitors and how hnRNP E1 and E2 promote proliferation of these cells.
Redundancy of the C/EBP network: CUGBP1 and C/EBPβ
C/EBPβ is another transcription regulator that controls myeloid maturation, and is a functional equivalent of C/EBPα based on its ability to restore granulocytic differentiation in C/EBPα-null mice [97]. In BCR–ABL1-expressing cells, IM treatment restores loading of C/EBPB mRNA onto polysomes. The IM effect results from restored activity of the RNA binding protein CUGBP1 that binds a CUG-repeat region located between the first and the third AUG of C/EBPB mRNA and enhances its translation [70,98]. Like C/EBPα, expression of C/EBPβ is repressed in primary CML-BC progenitors [70], suggesting that loss of C/EBPα and C/EBPβ activity contributes to differentiation arrest of CML-BC cells. Accordingly, levels of CUGBP1 are higher in normal and CML-CP CD34+ cells than in CD34+ CML-BC progenitors [70]. Ectopic expression or+ inducible activation of C/EBPβ inhibits proliferation and promotes granulocytic maturation of differentiation-arrested BCR–ABL1+ cells through a mechanism that depends on C/EBPβ transcriptional activity [70]. Thus, complete loss of C/EBP activity might be necessary to disrupt the differentiation potential of CML-BC progenitors. In addition to hnRNP K, treatment of BCR–ABL1-expressing cell lines with PD098059 resulted in substantially lower levels of hnRNP E2 [93]. As a consequence, C/EBPα expression is restored both in vitro and in vivo and these cells regain the ability to terminally differentiate as a result of granulocyte-colony stimulating factor (G-CSF) stimulation [93]. Taken together, these data support the inclusion of clinically relevant MAPK inhibitors in the therapy of CML-BC.
FUS and the G-CSFreceptor
FUS, also known as TLS, is a nucleocytoplasmic shuttling hnRNP protein that contributes to the block in differentiation of BCR–ABL1+ myeloid progenitors by altering the expression of cytokine receptors [68]. Expressed at high levels in hematopoietic and non-hematopoietic tissues, FUS is involved in pre-mRNA processing and nucleocytoplasmic shuttling, as well as in the regulation of basal transcription [99]. FUS expression and its DNA binding activity are induced in a BCR–ABL1 kinase-dependent manner in hematopoietic cells [68] through a protein kinase CβII-mediated mechanism that increases FUS protein stability by preventing its proteasome degradation [100]. Knockdown of FUS in differentiation-arrested BCR–ABL1+ myeloid cells rescues, in part, granulocytic differentiation by restoring expression of the C/EBPα-regulated G-CSF receptor (G-CSFR) and modestly impairs tumorigenesis [100]. Alternatively, ectopic FUS expression delays G-CSFR up-regulation in response to treatment with G-CSF [100]. The observation that FUS binds in vitro to a segment of G-CSFR mRNA [100] suggests that FUS binds to the G-CSFR pre-mRNA in the nucleus, thereby interfering with its processing or export to the cytoplasm. These studies implicate that the activity of another deregulated RNA binding protein contributes to the differentiation arrest observed in myeloid CML-BC progenitors.
MicroRNA conventional and decoy activities in chronic myeloid leukemia
MicroRNAs (miRNAs) regulate many cellular functions including cell proliferation, differentiation, and apoptosis. These non-coding RNAs silence specific target genes through translational repression or direct mRNA degradation [101]. Recently it has been shown that deregulated expression of specific miR-NAs regulating the expression of oncogenes, including BCR–ABL1 [102], tumor suppressors (e.g. PTEN [103]), and cell cycle regulators (e.g. E2F1 [104] and transforming growth factor β [TGFβ] [105]), is associated with the development of malignancies. Furthermore, specific miRNA expression signatures can be used to effectively classify human tumors [106]. Analysis of Ph1 B-ALL and CML primary cells, along with CML cell lines, revealed that the upstream region of the chromosome containing the human miR-203 miRNA is heavily methylated in BCR–ABL1-expressing cells. This methylation correlates with decreased miR-203 expression and suggests a specific pressure to downregulate miR-203 in Ph+ cells [102]. It was predicted that miR-203 binds to the 3′-UTR of c-ABL and BCR–ABL1 mRNA, thereby inhibiting their translation. Indeed, ectopic expression of miR-203 in the K562 and KCL-22 Ph1 cell lines resulted in substantially lower levels of BCR–ABL1 protein, leading to decreased proliferation, and increased levels of apoptosis [102].
Initial studies indicated that miRNA expression is higher in normal tissues compared to tumors, and led to the hypothesis that global miRNA expression reflects the state of cellular differentiation [106]. MicroRNA profiling of the myeloid leukemia cell line HL-60 following treatment with all-trans retinoic acid, a potent inducer of neutrophilic differentiation [107], revealed that the induction of many miRNAs coincided with differentiation. Similar results have been found with primary human hematopoietic progenitors undergoing erythroid differentiation in vitro. These studies suggest that the miRNA profile within these cells is a consequence of the stage at which they arrested [106]. However, there is substantial evidence indicating that the kinase activity of BCR–ABL1 is responsible for the deregulation of miRNAs in CML (Table I). This includes the observations that: (1) treatment of K562 with imatinib and BCR–ABL1 shRNA resulted in decreased expression of the miR-17–92 polycistron [73], and levels of miR-328 increased following the treatment of K562 and 32Dcl3-BCR–ABL1 cell lines with imatinib [108]; (2) in vivo miR-328 levels were much lower in lineage-negative cells from SCLtTA-BCR–ABL1 [109] mice and CML-BC CD34+ bone marrow, when compared with wild-type controls and CML-CP progenitors, respectively [108]; (3) bone marrow MNCs from patients unresponsive to IM and lacking common point mutations in the kinase domain of BCR–ABL1 (e.g. T315I, Y253H, Y253F, E225K, and E255V) demonstrated lower levels of miR-7, miR-23a, miR-26a, miR-29a, miR-29c, miR-30b, miR-30c, miR-100, miR-126#, miR-134, miR-141, miR-183, miR-196b, miR-199a, miR-224, miR-326, miR-422b, and miR-520a, while miR-191 was up-regulated when compared with patients with CML sensitive to IM [110]; (4) miR-96 was up-regulated while the down-regulation of miR-150 and miR-151 was observed in both BCR-ABL1+ CD34+ cells and MNCs when compared with cells from healthy donors [111]; (5) increased expression of the RNA binding proteins Lin28 and Lin28b, which block let-7 precursors from being processed to mature miRNAs [112–115], was detected in the peripheral blood of patients in CML-BC (42.8%) more frequently than in individuals with CML-CP (9.1%) [116]; and (6) miRNA profiles of CD34+ cells from patients with childhood ALL resulting from several different genetic abnormalities revealed an expression profile unique to Ph1-positive patients [117]. These studies indicate that levels of miRNAs are deregulated by BCR–ABL1 in a dose- and kinase-dependent manner, rather than the miRNA profile of the stage in differentiation at which these cells are arrested.
Table I.
Deregulated microRNA (miRNA) expression in chronic myeloid leukemia.
| miRNAs | Cell type | Mechanism | mRNA targets | Notes | BCR–ABL1 kinase-dependent? |
|---|---|---|---|---|---|
| miR-203 is down-regulated in Ph1 cells [102] | Primary Ph1 B-ALL and CML cells, K562, KCL-22 | Promoter methylation | c-ABL, BCR–ABL1 | Loss of miR-203 leads to increased levels of BCR–ABL1 | Heavy methylation in BCR–ABL1-expressing cells |
| miR-7, miR-23a, miR-26a, miR-29a, miR-29c, miR-30b, miR-30c, miR-100, miR-126#, miR-134, miR-141, miR-183, miR-196b, miR-199a, miR-224, miR-326, miR-422b, and miR-520a are down-regulated in IM-resistant patients; miR-191 is up-regulated [110] | BM MNCs from IM-sensitive and -insensitive patients; none presented common mutations associated with resistance to IM (e.g. T315I, Y253H, Y253F, E225K, and E255V) | Unidentified | Predicted ABCC5 (miR-199a), ABCA1 (miR-183), and ABCB6 (miR-29c), members of the ATP binding cassette (ABC) family of transmembrane transporters that have been implicated in resistance to chemotherapy | Study implicates these miRNAs in resistance to IM | Without the presence of common mutations associated with resistance to IM, it is likely that these patients have higher levels of BCR–ABL1 kinase activity; therefore, the deregulation of these miRNAs between IM-sensitive and -insensitive patients is likely BCR–ABL1-dependent |
| let-7 family members are down-regulated in CML-BC [116] | Peripheral blood MNCs from paired patients with CML-CP and CML-BC, K562, LAMA-84 | Increased expression of Lin28 | c-MYC, K-Ras | Increased proliferation resulting from higher levels of c-MYC and K-Ras | BCR–ABL1 dependence suggested by increased levels of Lin28 being present in CML-BC, presumably where BCR–ABL1 activity is higher |
| miR-328 is down-regulated in CML-BC [108] | BM CD34+ CML-CP and CMP-BC, Lin− WT and SCLtTA-BCR–ABL1 mice, 32D, 32D-BCR–ABL1, K562 ± IM | BCR–ABL1-MAPK-dependent inhibition of C/EBPA translation | PIM1 | Enhanced survival and arrest in differentiation (miR-328 promotes C/EBPA translation by binding to hnRNP E2) | IM treatment of K562 and Lin− from transgenic mice restores levels of miR-328 |
| miR-17–92 cluster is up-regulated in CML-CP compared with CML-BC [73] | CD34+ from normal, CML-CP, and CML-BC, K562 ± IM, LAMA-84, EM-2 | Unidentified | E2F1, PTEN, and TGFβ (previously described by others, references in text) | Increased proliferation and sensitivity to IM | Deregulated miR-17–92 cluster identified following treatment of K562 with IM and shRNA |
| miR-150 and miR-151 are down-regulated in Ph1 MNCs and CD34+ while miR-96 is up-regulated; up-regulation of miR-17–92 cluster not observed [111] | BM MNCs and CD34+ from normal patients and patients with CML | Unidentified | Unidentified | Deregulated expression of these miRNAs in both CD34+ and MNCs strongly implicates them in pathogenesis by BCR–ABL1 | Study demonstrates altered expression of these miRNAs is BCR–ABL1-dependent and not cell-type specific |
| miR-196b is down-regulated and miR-708, miR-181a, b, c, d are up-regulated [117] | Ph1 B-ALL and normal CD34+ from patients with childhood ALL | Unidentified | Unidentified | Expression of these miRNAs are dependent on leukemic subtypes | Expression profiles specific to oncogene responsible for disease |
| Increased expression of miR-150 and miR-146a, reduced expression of miR-142–3p and miR-199b-5p following IM treatment [122] | PB MNCs from newly diagnosed patients with CML-CP, IM-treated CML-CP, and CML-BC | Unidentified | c-MYB, a previously described target of miR-150 | Following IM treatment, miRNA expression profiles in PB MNCs returned to that of normal donors | Deregulated expression of these miRNAs dependent on BCR–ABL1 kinase activity as its impairment restored normal miRNA levels |
Ph1, Philadelphia chromosome; IM, imatinib; CML-BC, chronic myeloid leukemia-blast crisis; CML-CP, chronic myeloid leukemia-chronic phase; MNC, mononuclear cell; B-ALL, B-cell acute lymphoblastic leukemia; BM, bone marrow; MAPK, mitogen activated protein kinase; TGFβ, transforming growth factor β; hnRNP, heterogeneous nuclear ribonucleoprotein.
The mRNA targets of many of these miRNAs have yet to be identified; however, as miRNAs regulate gene expression by impairing translation or initiating degradation of mRNAs [101], and when considering the phenotype of CML-BC progenitors, it is reasonable to speculate that some of these deregulated miRNAs normally affect traslation of genes either regulating or promoting proliferation and survival (e.g., Figure 2), and differentiation (e.g., Figure 3), thereby contributing to the phenotype of CML-BC. The knockdown of Lin28b in CML-BC cells restored levels of let-7 family members, reinstating their previously described inhibition of c-Myc [118] and k-Ras [119] processing, leading to substantially reduced protein levels of these oncogenes [116] (Figure 2). The treatment of the CML cell lines K562 and KCL-22 with a combination of 5′-azacytidine and 4-phenylbutyrate demonstrated efficient demethylation of the miR-203 upstream region, resulting in substantially increased levels of miR-203, and miR-203-dependent decreased levels of BCR–ABL1 [102]. As the upstream region of miR-203 is heavily methylated in primary cells from patients with CML and Ph1 B-ALL, this study suggests that these epigenetic drugs may have therapeutic benefits in CML. Furthermore, enhanced survival of CML-BC CD34+ progenitors may result from the absence of miR-328, as ectopic expression of miR-328 greatly reduced PIM1, which is known to promote enhanced survival of these cells [108].
Figure 2.
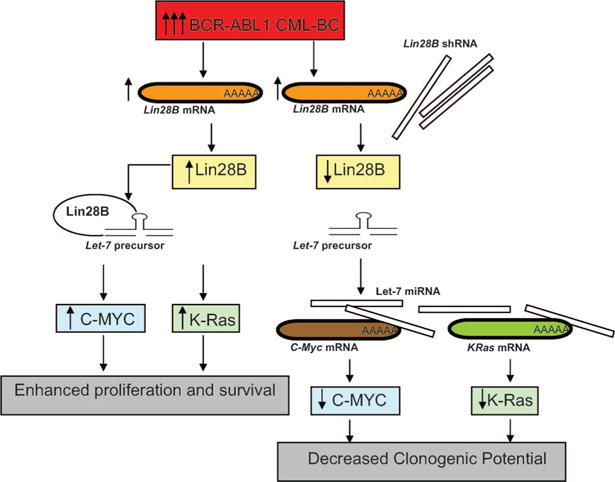
Increased levels of BCR–ABL in CML-BC results in increased levels of the oncogenes c-MYC and K-Ras through inhibition of let-7 family-member processing. In CML-BC, increased levels of BCR–ABL lead to higher levels of the RNA binding protein Lin28B. Lin28B binds let-7 family-member precursors, preventing their processing, and thereby relieving their normal inhibition of c-MYC and K-Ras translation. Introduction of Lin28B shRNA in cell lines and primary cells restores processing of let-7 family members and, subsequently, decreased levels of C-MYC and K-Ras protein are observed.
Figure 3.
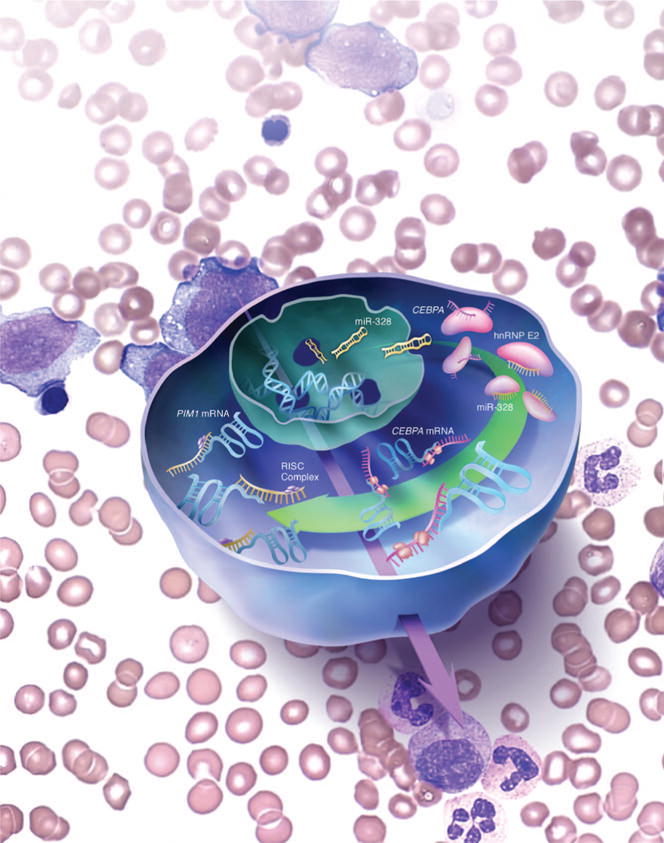
Schematic representation of the dual miR-328 activity in CML-BC. miR-328 decoy activity is essential for the regulation of neutrophil differentiation and its loss determines the blocks of C/EBPα expression and, therefore, the differentiation arrest of myeloid CML-BC blasts. The canonical miR-328 activity controls, at least in part, survival of CML-BC progenitors by regulating the expression of PIM1 serine–threonine kinase.
Importantly, a non-canonical function of miR-328 was identified, as miR-328 was found to compete with C/EBPA for binding to hnRNP E2 [108]. As the absence of miR-328 in CML-BC progenitors prevents C/EBPA translation, ectopic expression of miR-328 in these cells decreased hnRNP E2-mediated inhibition of C/EBPA translation, and allowed these cells to undergo terminal granulocytic differentiation upon stimulation with G-CSF [108]. Furthermore, ectopic expression of miR-328 in BCR–ABL1+ cell lines, as well as CML-BCCD34+, reduced methylcellulose colony formation, and impaired survival through the miR-328-dependent degradation of PIM1 [108] (Figure 3). These studies, which heavily implicate loss of miR-328 and, generally, altered miRNA expression in determination of the biological characteristics of CML-BC progenitors, highlight the important role of exploiting new alternative avenues envisioning the use of miRNAs as therapeutic drugs.
Concluding remarks
Together, these studies provide a substantial amount of evidence indicating that BCR–ABL1 kinase-dependent deregulation of mRNA metabolism contributes to the progression of CML. Altered mRNA metabolism in CML-BC resulting from differential pre-mRNA splicing, increased mTOR activity, autophagy, altered RNA binding protein expression, and deregulated miRNA expression affects the levels of oncogenes (e.g. BCR–ABL1 [102], PIM1 [108], MYC [67,112], and K-Ras [112]), potent antiapoptotic proteins (e.g. BCL-XL [69] and MCL-1 [74]), a promoter of proliferation (cyclin D3 [48]), a tumor suppressor (p53 [20]), an antagonist of the PP2A known tumor suppressor (SET [17,75]), and transcription factors essential for terminal differentiation (C/EBPα [18] and C/EBPβ [70]) of myeloid progenitors.
The lack of long-term response of patients with CML-BC to tyrosine kinase monotherapy (imatinib, nilotinib, or dasatinib [2,120,121]) highlights the importance of therapies that target multiple oncogenic pathways. As altered mRNA metabolism within CML-BCCD34+ progenitors contributes to the increased proliferation, arrest in differentiation, and enhanced survival observed by these cells, pharmacologic and molecular therapies reinstating normal mRNA metabolism have been successful in impairing leukemogenesis by BCR–ABL1 in cell lines, murine models of CML, and primary cells from patients, providing support for their inclusion in the clinical setting.
Acknowledgments
Declaration of interest: This work was supported in part by grants from the National Cancer Institute CA095512 (D.P.), NIH, Bethesda, MD; and the US Army CML Research Program, W81XWH-07–1–0270 (D.P.). D.P. is a Scholar of The Leukemia and Lymphoma Society.
References
- 1.Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103:4010–4022. doi: 10.1182/blood-2003-12-4111. [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian HM, Keating MJ, Talpaz M, et al. Chronic myelogenous leukemia in blast crisis - analysis of 242 patients. Am J Med. 1987;83:445–454. doi: 10.1016/0002-9343(87)90754-6. [DOI] [PubMed] [Google Scholar]
- 3.Pui L, Liu J, Gish G, et al. Bcr-Abl oncoproteins bind directly to activators of the Ras signaling pathway. EMBO J. 1994;13:764–773. doi: 10.1002/j.1460-2075.1994.tb06319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffin JD. Phosphatidyl inositol signaling by BCR/ABL: opportunities for drug development. Cancer Chemother Pharmacol. 2001;48:S11–S16. doi: 10.1007/s002800100299. [DOI] [PubMed] [Google Scholar]
- 5.Coppo P, Flamant S, De Mas V, et al. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br J Haematol. 2006;134:171–179. doi: 10.1111/j.1365-2141.2006.06161.x. [DOI] [PubMed] [Google Scholar]
- 6.Ilaria RL, VanEtten RA. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem. 1996;271:31704–31710. doi: 10.1074/jbc.271.49.31704. [DOI] [PubMed] [Google Scholar]
- 7.Shuai K, Halpern J, tenHoeve J, Rao XP, Sawyers CL. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996;13:247–254. [PubMed] [Google Scholar]
- 8.Perrotti D, Jamieson CHM, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120:2254–2264. doi: 10.1172/JCI41246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoklosa T, Poplawski T, Koptyra M, et al. BCR/ABL inhibits mismatch repair to protect from apoptosis and induce point mutations. Cancer Res. 2008;68:2576–2580. doi: 10.1158/0008-5472.CAN-07-6858. [DOI] [PubMed] [Google Scholar]
- 10.Feinstein E, Cimino G, Gale RP, et al. p53 in chronic myelogenous leukemia in acute phase. Proc Natl Acad Sci USA. 1991;88:6293–6297. doi: 10.1073/pnas.88.14.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang SJ, Shi JY, Li JY. GATA-2 L359 V mutation is exclusively associated with CML progression but not other hematological malignancies and GATA-2 P250A is a novel single nucleotide polymorphism. Leuk Res. 2009;33:1141–1143. doi: 10.1016/j.leukres.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 12.Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of IKAROS. Nature. 2008;453:110–114. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 13.Cramer K, Nieborowska-Skorska M, Koptyra M, et al. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008;68:6884–6888. doi: 10.1158/0008-5472.CAN-08-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elmaagacli AH, Beelen DW, Opalka B, Seeber S, Schafer UW. The amount of BCR-ABL fusion transcripts detected by the real-time quantitative polymerase chain reaction method in patients with Philadelphia chromosome positive chronic myeloid leukemia correlates with the disease stage. Ann Hematol. 2000;79:424–431. doi: 10.1007/s002770000169. [DOI] [PubMed] [Google Scholar]
- 15.Gaiger A, Henn T, Horth E, et al. Increase of Bcr-Abl chimeric messenger-RNA expression in tumor-cells of patients with chronic myeloid-leukemia precedes disease progression. Blood. 1995;86:2371–2378. [PubMed] [Google Scholar]
- 16.Jamieson CHM, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 17.Neviani P, Santhanam R, Trotta R, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355–368. doi: 10.1016/j.ccr.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 18.Perrotti D, Cesi V, Trotta R, et al. BCR-ABL suppresses C/EBP alpha expression through inhibitory action of hnRNP E2. Nat Genet. 2002;30:48–58. doi: 10.1038/ng791. [DOI] [PubMed] [Google Scholar]
- 19.Schultheis B, Szydlo R, Mahon FX, Apperley JF, Melo JV. Analysis of total phosphotyrosine levels in CD34(+) cells from CML patients to predict the response to imatinib mesylate treatment. Blood. 2005;105:4893–4894. doi: 10.1182/blood-2005-01-0210. [DOI] [PubMed] [Google Scholar]
- 20.Trotta R, Vignudelli T, Candini O, et al. BCR/ABL activates mdm2 rnRNA translation via the La antigen. Cancer Cell. 2003;3:145–160. doi: 10.1016/s1535-6108(03)00020-5. [DOI] [PubMed] [Google Scholar]
- 21.Nieborowska-Skorska M, Koptyra M, Hoser G, et al. Mechanisms generating free radicals in CML stem/progenitor cell populations causing DNA damage and genomic instability. Blood. 2008;112:78–79. [Google Scholar]
- 22.Ottmann OG, Druker BJ, Sawyers CL, et al. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood. 2002;100:1965–1971. doi: 10.1182/blood-2001-12-0181. [DOI] [PubMed] [Google Scholar]
- 23.Sawyers CL, Hochhaus A, Feldman E, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99:3530–3539. doi: 10.1182/blood.v99.10.3530. [DOI] [PubMed] [Google Scholar]
- 24.Van Etten RA. Oncogenic signaling: new insights and controversies from chronic myeloid leukemia. J Exp Med. 2007;204:461–465. doi: 10.1084/jem.20062335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laudadio J, Deininger MWN, Mauro MJ, Druker BJ, Press RD. An intron-derived insertion/truncation mutation in the BCR-ABL kinase domain in chronic myeloid leukemia patients undergoing kinase inhibitor therapy. J Mol Diagn. 2008;10:177–180. doi: 10.2353/jmoldx.2008.070128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nardi V, Azam M, Daley GQ. Mechanisms and implications of imatinib resistance mutations in BCR-ABL. Curr Opin Hematol. 2004;11:35–43. doi: 10.1097/00062752-200401000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Klein F, Feldhahn N, Herzog S, et al. BCR-ABL1 induces aberrant splicing of IKAROS and lineage infidelity in pre-B lymphoblastic leukemia cells. Oncogene. 2006;25:1118–1124. doi: 10.1038/sj.onc.1209133. [DOI] [PubMed] [Google Scholar]
- 28.Salesse S, Dylla SJ, Verfaillie C. p210(BCR/ABL)-induced alteration of pre-mRNA splicing in primary human CD34(+) hematopoietic progenitor cells. Leukemia. 2004;18:727–733. doi: 10.1038/sj.leu.2403310. [DOI] [PubMed] [Google Scholar]
- 29.Bhatia R, Munthe HA, Verfaillie CM. Role of abnormal integrin-cytoskeletal interactions in impaired beta 1 integrin function in chronic myelogenous leukemia hematopoietic progenitors. Exp Hematol. 1999;27:1384–1396. doi: 10.1016/s0301-472x(99)00084-3. [DOI] [PubMed] [Google Scholar]
- 30.Salgia R, Li JL, Ewaniuk DS, et al. BCR/ABL induces multiple abnormalities of cytoskeletal function. J Clin Invest. 1997;100:46–57. doi: 10.1172/JCI119520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhatia R, McCarthy JB, Verfaillie CM. Interferon-alpha restores normal beta 1 integrin-mediated inhibition of hematopoietic progenitor proliferation by the marrow microenvironment in chronic myelogenous leukemia. Blood. 1996;87:3883–3891. [PubMed] [Google Scholar]
- 32.Verfaillie CM, McCarthy JB, McGlave PB. Differentiation of primitive human multipotent hematopoietic progenitors into single lineage clonogenic progenitors is accompanied by alterations in their interaction with fibronectin. J Exp Med. 1991;174:693–703. doi: 10.1084/jem.174.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abrahamsson AE, Geron I, Gotlib J, et al. Glycogen synthase kinase 3 beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci USA. 2009;106:3925–3929. doi: 10.1073/pnas.0900189106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12:528–541. doi: 10.1016/j.ccr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kharas MG, Fruman DA. ABL oncogenes and phosphoinositide 3-kinase: mechanism of activation and downstream effectors. Cancer Res. 2005;65:2047–2053. doi: 10.1158/0008-5472.CAN-04-3888. [DOI] [PubMed] [Google Scholar]
- 36.Kharas MG, Janes MR, Scarfone VM, et al. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL(+) leukemia cells. J Clin Invest. 2008;118:3038–3050. doi: 10.1172/JCI33337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren SY, Xue F, Feng J, Skorski T. Intrinsic regulation of the interactions between the SH3 domain of p85 subunit of phosphatidylinositol-3 kinase and the protein network of BCR/ABL oncogenic tyrosine kinase. Exp Hematol. 2005;33:1222–1228. doi: 10.1016/j.exphem.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 38.Ly C, Arechiga AF, Melo JV, Walsh CM, Ong ST. Bcr-Abl kinase modulates the translation regulators ribosomal protein S6 and 4E-BP1 in chronic myelogenous leukemia cells via the mammalian target of rapamycin. Cancer Res. 2003;63:5716–5722. [PubMed] [Google Scholar]
- 39.Mohi MG, Boulton C, Gu TL, et al. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci USA. 2004;101:3130–3135. doi: 10.1073/pnas.0400063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parmar S, Smith J, Sassano A, et al. Differential regulation of the p70 S6 kinase pathway by interferon alpha (IFN alpha) and imatinib mesylate (STI571) in chronic myelogenous leukemia cells. Blood. 2005;106:2436–2443. doi: 10.1182/blood-2004-10-4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Markova B, Albers C, Breitenbuecher F, et al. Novel pathway in Bcr-Abl signal transduction involves Akt-independent, PLC-gamma 1-driven activation of mTOR/p70S6-kinase pathway. Oncogene. 2010;29:739–751. doi: 10.1038/onc.2009.374. [DOI] [PubMed] [Google Scholar]
- 42.Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 43.Gingras AC, Raught B, Gygi SP, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15:2852–2864. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 45.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 46.Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 47.Lim S, Saw TY, Chuah C, Ong ST. Overexpression and phosphorylation of eIF4E is required for beta-catenin activation in blast crisis chronic myelogenous leukemia. Blood. 2009;114:22. [Google Scholar]
- 48.Prabhu S, Saadat D, Zhang M, Halbur L, Fruehauf JP, Ong ST. A novel mechanism for Bcr-Abl action: Bcr-Abl-mediated induction of the eIF4F translation initiation complex and mRNA translation. Oncogene. 2007;26:1188–1200. doi: 10.1038/sj.onc.1209901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janes MR, Limon JJ, So LM, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang M, Fu WX, Prabhu S, et al. Inhibition of polysome assembly enhances imatinib activity against chronic myelogenous leukemia and overcomes imatinib resistance. Mol Cell Biol. 2008;28:6496–6509. doi: 10.1128/MCB.00477-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altman JK, Yoon P, Katsoulidis E, et al. Regulatory effects of mammalian target of rapamycin-mediated signals in the generation of arsenic trioxide responses. J Biol Chem. 2008;283:1992–2001. doi: 10.1074/jbc.M705227200. [DOI] [PubMed] [Google Scholar]
- 52.Mayerhofer M, Valent P, Sperr WR, Griffin JD, Sillaber C. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1 alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood. 2002;100:3767–3775. doi: 10.1182/blood-2002-01-0109. [DOI] [PubMed] [Google Scholar]
- 53.Bellamy WT, Richter L, Sirjani D, et al. Vascular endothelial cell growth factor is an autocrine promoter of abnormal localized immature myeloid precursors and leukemia progenitor formation in myelodysplastic syndromes. Blood. 2001;97:1427–1434. doi: 10.1182/blood.v97.5.1427. [DOI] [PubMed] [Google Scholar]
- 54.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 56.Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5:886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 57.Baehrecke EH. Autophagy: DUAL roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 58.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 59.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 60.Lum JJ, Bauer DE, Kong M, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 61.Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119:1109–1123. doi: 10.1172/JCI35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 63.Keene JD. Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome. Proc Natl Acad Sci USA. 2001;98:7018–7024. doi: 10.1073/pnas.111145598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keene JD, Tenenbaum SA. Eukaryotic mRNPs may represent posttranscriptional operons. Mol Cell. 2002;9:1161–1167. doi: 10.1016/s1097-2765(02)00559-2. [DOI] [PubMed] [Google Scholar]
- 65.Perrotti D, Calabretta B. Post-transcriptional mechanisms in BCR/ABL leukemogenesis: role of shuttling RNA-binding proteins. Oncogene. 2002;21:8577–8583. doi: 10.1038/sj.onc.1206085. [DOI] [PubMed] [Google Scholar]
- 66.Perrotti D, Turturro F, Neviani P. BCR/ABL, mRNA translation and apoptosis. Cell Death Differ. 2005;12:534–540. doi: 10.1038/sj.cdd.4401606. [DOI] [PubMed] [Google Scholar]
- 67.Notari M, Neviani P, Santhanam R, et al. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood. 2006;107:2507–2516. doi: 10.1182/blood-2005-09-3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perrotti D, Bonatti S, Trotta R, et al. TLS/FUS, a pro-oncogene involved in multiple chromosomal translocations, is a novel regulator of BCR/ABL-mediated leukemogenesis. EMBO J. 1998;17:4442–4455. doi: 10.1093/emboj/17.15.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iervolino A, Santilli G, Trotta R, et al. hnRNP A1 nucleocytoplasmic shuttling activity is required for normal myelopoiesis and BCR/ABL leukemogenesis. Mol Cell Biol. 2002;22:2255–2266. doi: 10.1128/MCB.22.7.2255-2266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guerzoni C, Bardini M, Mariani SA, et al. Inducible activation of CEBPB, a gene negatively regulated by BCR/ABL, inhibits proliferation and promotes differentiation of BICRABL-expressing cells. Blood. 2006;107:4080–4089. doi: 10.1182/blood-2005-08-3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dreyfuss G, Matunis MJ, Pinolroma S, Burd CG. Hnrnp proteins and the biogenesis of messenger-RNA. Annu Rev Biochem. 1993;62:289–321. doi: 10.1146/annurev.bi.62.070193.001445. [DOI] [PubMed] [Google Scholar]
- 72.Guil S, Caceres JF. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat Struct Mol Biol. 2007;14:591–596. doi: 10.1038/nsmb1250. [DOI] [PubMed] [Google Scholar]
- 73.Venturini L, Battmer K, Castoldi M, et al. Expression of the miR-17–92 polycistron in chronic myeloid leukemia (CML) CD34(+) cells. Blood. 2007;109:4399–4405. doi: 10.1182/blood-2006-09-045104. [DOI] [PubMed] [Google Scholar]
- 74.Eiring AM, Neviani P, Santhanam R, et al. Identification of novel posttranscriptional targets of the BCR/ABL oncoprotein by ribonomics: requirement of E2F3 for BCR/ABL leukemogenesis. Blood. 2008;111:816–828. doi: 10.1182/blood-2007-05-090472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neviani P, Santhanam R, Oaks JJ, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest. 2007;117:2408–2421. doi: 10.1172/JCI31095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vera J, Jaumot M, Estanyol JM, Brun S, Agell N, Bachs O. Heterogeneous nuclear ribonucleoprotein A2 is a SET-binding protein and a PP2A inhibitor. Oncogene. 2006;25:260–270. doi: 10.1038/sj.onc.1209050. [DOI] [PubMed] [Google Scholar]
- 77.Perrotti D, Neviani P. From mRNA metabolism to cancer therapy: chronic myelogenous leukemia shows the way. Clin Cancer Res. 2007;13:1638–1642. doi: 10.1158/1078-0432.CCR-06-2320. [DOI] [PubMed] [Google Scholar]
- 78.Sawyers CL, Callahan W, Witte ON. Dominant negative Myc blocks transformation by Abl oncogenes. Cell. 1992;70:901–910. doi: 10.1016/0092-8674(92)90241-4. [DOI] [PubMed] [Google Scholar]
- 79.Evans JR, Mitchell SA, Spriggs KA, et al. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene. 2003;22:8012–8020. doi: 10.1038/sj.onc.1206645. [DOI] [PubMed] [Google Scholar]
- 80.Michelotti EF, Michelotti GA, Aronsohn AI, Levens D. Heterogeneous nuclear ribonucleoprotein K is a transcription factor. Mol Cell Biol. 1996;16:2350–2360. doi: 10.1128/mcb.16.5.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gopal V, Kadam P, Preisler H, et al. Abnormal regulation of the Myc gene in myeloid-leukemia. Med Oncol Tumor Pharmacother. 1992;9:139–147. doi: 10.1007/BF02987745. [DOI] [PubMed] [Google Scholar]
- 82.Park YG, Zhao XH, Lesueur F, et al. Sipa1 is a candidate for underlying the metastasis efficiency modifier locus Mtes1. Nat Genet. 2005;37:1055–1062. doi: 10.1038/ng1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Naarmann IS, Harnisch C, Flach N, et al. mRNA silencing in human erythroid cell maturation - heterogeneous nuclear ribonucleoprotein K controls the expression of its regulator c-Src. J Biol Chem. 2008;283:18461–18472. doi: 10.1074/jbc.M710328200. [DOI] [PubMed] [Google Scholar]
- 84.Wolin SL, Cedervall T. The LA protein. Annu Rev Biochem. 2002;71:375–403. doi: 10.1146/annurev.biochem.71.090501.150003. [DOI] [PubMed] [Google Scholar]
- 85.Honda H, Ushijima T, Wakazono K, et al. Acquired loss of p53 induces blastic transformation in p210(bcr/abl)-expressing hematopoietic cells: a transgenic study for blast crisis of human CML. Blood. 2000;95:1144–1150. [PubMed] [Google Scholar]
- 86.Skorski T, NieborowskaSkorska M, Wlodarski P, et al. Blastic transformation of p53-deficient bone marrow cells by p210(bcr/abl) tyrosine kinase. Proc Natl Acad Sci USA. 1996;93:13137–13142. doi: 10.1073/pnas.93.23.13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aichberger KJ, Mayerhofer M, Florian S, et al. BCR/ABL-dependent expression of Mcl-1 involves the MAP-kinase pathway and contributes to survival of leukemic cells. Blood. 2003;102(Suppl 1) Abstract 4987. [Google Scholar]
- 88.Aichberger KJ, Mayerhofer M, Krauth MT, et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005;105:3303–3311. doi: 10.1182/blood-2004-02-0749. [DOI] [PubMed] [Google Scholar]
- 89.Fukuchi Y, Kizaki H, Yamato K, et al. Mcl-1, an early-induction molecule, modulates activin A-induced apoptosis and differentiation of CML cells. Oncogene. 2001;20:704–713. doi: 10.1038/sj.onc.1204142. [DOI] [PubMed] [Google Scholar]
- 90.Keeshan K, Santilli G, Corradini F, Perrotti D, Calabretta B. Transcription activation function of C/EBP alpha is required for induction of granulocytic differentiation. Blood. 2003;102:1267–1275. doi: 10.1182/blood-2003-02-0477. [DOI] [PubMed] [Google Scholar]
- 91.Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3:89–101. doi: 10.1038/nrc989. [DOI] [PubMed] [Google Scholar]
- 92.Ostareck-Lederer A, Ostareck DH. Control of mRNA translation and stability in haematopoietic cells: The function of hnRNPs K and E1/E2. Biol Cell. 2004;96:407–411. doi: 10.1016/j.biolcel.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 93.Chang JS, Santhanam R, Trotta R, et al. High levels of the BCR/ABL oncoprotein are required for the MAPK-hnRNP-E2 dependent suppression of C/EBPalpha-driven myeloid differentiation. Blood. 2007;110:994–1003. doi: 10.1182/blood-2007-03-078303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wagner K, Zhang P, Rosenbauer F, et al. Absence of the transcription factor CCAAT enhancer binding protein alpha results in loss of myeloid identity in bcr/abl-induced malignancy. Proc Natl Acad Sci USA. 2006;103:6338–6343. doi: 10.1073/pnas.0508143103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perrotti D, Marcucci G, Caligiuri MA. Loss of C/EBP alpha and favorable prognosis of acute myeloid leukemias: a biological paradox. J Clin Oncol. 2004;22:582–584. doi: 10.1200/JCO.2004.12.965. [DOI] [PubMed] [Google Scholar]
- 96.Waggoner SA, Johannes GJ, Liebhaber SA. Depletion of the poly(C)-binding proteins alpha CP1 and alpha CP2 from K562 cells leads to p53-independent induction of cyclin-dependent kinase inhibitor (CDKN1A) and G1 arrest. J Biol Chem. 2009;284:9039–9049. doi: 10.1074/jbc.M806986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jones LC, Lin ML, Chen SS, et al. Expression of C/EBPbeta from the C/ebpalpha gene locus is sufficient for normal hematopoiesis in vivo. Blood. 2002;99:2032–2036. doi: 10.1182/blood.v99.6.2032. [DOI] [PubMed] [Google Scholar]
- 98.Timchenko NA, Welm AL, Lu XH, Timchenko LT. CUG repeat binding protein (CUGBP1) interacts with the 5′ region of C/EBP beta mRNA and regulates translation of C/EBP beta isoforms. Nucleic Acids Res. 1999;27:4517–4525. doi: 10.1093/nar/27.22.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci. 1997;110:1741–1750. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]
- 100.Perrotti D, Iervolino A, Cesi V, et al. BCR-ABL prevents c-Jun-mediated and proteasome-dependent FUS (TLS) proteolysis through a protein kinase C beta II-dependent pathway. Mol Cell Biol. 2000;20:6159–6169. doi: 10.1128/mcb.20.16.6159-6169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 102.Bueno MJ, de Castro IP, de Cedron MG, et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008;13:496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 103.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 104.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 105.Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 107.Stegmaier K, Ross KN, Colavito SA, O’Malley S, Stockwell BR, Golub TR. Gene expression-based high-throughput screening (GE-HTS) and application to leukemia differentiation. Nat Genet. 2004;36:257–263. doi: 10.1038/ng1305. [DOI] [PubMed] [Google Scholar]
- 108.Eiring AM, Harb JG, Neviani P, et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell. 2010;140:652–665. doi: 10.1016/j.cell.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Koschmieder S, Gottgens B, Zhang P, et al. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood. 2005;105:324–334. doi: 10.1182/blood-2003-12-4369. [DOI] [PubMed] [Google Scholar]
- 110.San Jose-Eneriz E, Roman-Gomez J, Jimenez-Velasco A, et al. MicroRNA expression profiling in imatinib-resistant chronic myeloid leukemia patients without clinically significant ABL1-mutations. Mol Cancer. 2009;8:69. doi: 10.1186/1476-4598-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Agirre X, Jimenez-Velasco A, Jose-Eneriz ES, et al. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34(+) cells increases USF2-mediated cell growth. Mol Cancer Res. 2008;6:1830–1840. doi: 10.1158/1541-7786.MCR-08-0167. [DOI] [PubMed] [Google Scholar]
- 112.Heo I, Joo C, Cho J, Ha M, Han JJ, Kim VN. Lin28 mediates the terminal uridylation of Let-7 precursor microRNA. Mol Cell. 2008;32:276–284. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 113.Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA. 2008;14:1539–1549. doi: 10.1261/rna.1155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rybak A, Fuchs H, Smirnova L, et al. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat Cell Biol. 2008;10:987–993. doi: 10.1038/ncb1759. [DOI] [PubMed] [Google Scholar]
- 115.Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of MicroRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Viswanathan SR, Powers JT, Einhorn W, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843–848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schotte D, Chau JCK, Sylvester G, et al. Identification of new microRNA genes and aberrant microRNA profiles in childhood acute lymphoblastic leukemia. Leukemia. 2009;23:313–322. doi: 10.1038/leu.2008.286. [DOI] [PubMed] [Google Scholar]
- 118.Sampson VB, Rong NH, Han J, et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67:9762–9770. doi: 10.1158/0008-5472.CAN-07-2462. [DOI] [PubMed] [Google Scholar]
- 119.Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 120.Cohen MH, Johnson JR, Pazdur R. US food and drug administration drug approval summary: conversion of imatinib mesylate (ST1571; Gleevec) tablets from accelerated approval to full approval. Clin Cancer Res. 2005;11:12–19. [PubMed] [Google Scholar]
- 121.Perrotti D, Neviani P. ReSETting PP2A tumour suppressor activity in blast crisis and imatinib-resistant chronic myelogenous leukaemia. Br J Cancer. 2006;95:775–781. doi: 10.1038/sj.bjc.6603317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Flamant S, Ritchie W, Guilhot J, et al. Micro-RNA response to imatinib mesylate in patients with chronic myeloid leukemia. Haematologica. 2010;95:1325–1333. doi: 10.3324/haematol.2009.020636. [DOI] [PMC free article] [PubMed] [Google Scholar]