

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by a selective loss of upper and lower motor neurons. Recent studies have shown that mutations in SQSTM1 are linked to ALS. SQSTM1 encodes SQSTM1/p62 that regulates not only autophagy via the association with MAP1LC3/LC3 and ubiquitinated proteins but also the KEAP1-NFE2L2/Nrf2 anti-oxidative stress pathway by interacting with KEAP1. Previously, we have demonstrated that loss of SQSTM1 exacerbates disease phenotypes in a SOD1H46R-expressing ALS mouse model. To clarify the effects of SQSTM1 overexpression in this model, we generated SQSTM1 and SOD1H46R double-transgenic (SQSTM1;SOD1H46R) mice. SQSTM1;SOD1H46R mice exhibited earlier disease onset and shorter lifespan than did SOD1H46R mice. Conversely, disease progression after the onset rather slightly but significantly slowed in SQSTM1;SOD1H46R mice. However, there were observable differences neither in the number of Nissl positive neurons nor in the distribution of ubiquitin-positive and/or SQSTM1-positive aggregates between SOD1H46R and SQSTM1;SOD1H46R mice. It was noted that these protein aggregates were mainly observed in neuropil, and partly localized to astrocytes and/or microglia, but not to MAP2-positive neuronal cell bodies and dendrites at the end-stage of disease. Nonetheless, the biochemically-detectable insoluble SQSTM1 and poly-ubiquitinated proteins were significantly and progressively increased in the spinal cord of SQSTM1;SOD1H46R mice compared to SOD1H46R mice. These results suggest that overexpression of SQSTM1 in SOD1H46R mice accelerates disease onset by compromising the protein degradation pathways.
Electronic supplementary material
The online version of this article (10.1186/s13041-018-0373-8) contains supplementary material, which is available to authorized users.
Keywords: Amyotrophic lateral sclerosis, SOD1, SQSTM1/p62, Ubiquitin-positive aggregates
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by a selective loss of upper and lower motor neurons. ALS patients exhibit enhanced tendon reflex caused by loss of upper motor neurons and muscle atrophy, impaired motor control caused by loss of lower motor neurons without sensory disturbance, and ultimately die within 3 to 5 years due to respiratory failure [1]. An approximately 10% of ALS cases is familial and the remaining 90% are sporadic [2]. Although more than 50 different genes that are linked to ALS have been identified, molecular mechanisms by which motor neurons are selectively and progressively degenerated are still unknown [3].
Recently, mutations in the SQSTM1 gene have been identified in patients with ALS and ALS/frontotemporal dementia (FTD) [4–8]. SQSTM1 mutations have originally been identified in Paget disease of bone [9]. The SQSTM1 gene product, sequestosome1 (SQSTM1/p62), is a multi-functional adapter protein. SQSTM1 regulates not only autophagy via the association with microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3) and poly-ubiquitinated proteins [10, 11] but also the Kelch-like ECH-associated protein 1 (KEAP1)-nuclear factor related erythroid 2-related factor 2 (NFE2L2/Nrf2) anti-oxidative stress pathway by interacting with KEAP1 [12]. Indeed, ALS-linked SQSTM1 missense mutant (Leu341Val) [7] showed a decreased affinity to LC3, resulting in impaired autophagy [13]. It has also been shown that SQSTM1 is multiply phosphorylated. Especially, the phosphorylation of SQSTM1 at serine (Ser) 403 (corresponding to mouse Ser405) increases the binding affinity to poly-ubiquitin molecules and enhances the autophagic degradation of poly-ubiquitinated proteins [14, 15]. In addition, it has been reported that the phosphorylation at Ser349 (corresponding to mouse Ser351) enhances the interaction between SQSTM1 and KEAP1, thereby activating the KEAP1-NFE2L2/Nrf2 pathway [16]. With respect to the relationship between SQSTM1 and neurodegenerative diseases, it has been demonstrated that SQSTM1-positive aggregates are accumulated in the spinal cord and brain of ALS and ALS/FTD patients [17–19].
On the other hand, the SOD1 gene has been identified as a causative gene for an autosomal dominant form of familial ALS in 1993. Thus far, more than 180 different SOD1 mutations have been identified (http://alsod.iop.kcl.ac.uk/als/). Several transgenic (tg) ALS mouse models expressing different SOD1 mutants have been generated [20]. Among them, SOD1H46R mutation has originally been identified in Japanese kindred, showing a slow progression of symptoms [21]. SOD1H46R-tg mouse recapitulates many neurological and pathological features observed not in only familial but also in sporadic ALS patients, which include progressive motor neuron degeneration, impaired motor control, and accumulation of SQSTM1 and ubiquitin-positive aggregates in the spinal cord [19, 20, 22, 23].
It has been shown that, in other motor neuron disease (MND) mouse model, i.e., spinal and bulbar muscular atrophy (SBMA) mice, loss of SQSTM1 causes earlier disease onset, whereas overexpression of it ameliorates disease symptoms [24]. We have previously demonstrated that loss of SQSTM1 exacerbates disease phenotypes, including a shorter lifespan, accelerated body weight decline and motor dysfunction, in a mutant SOD1H46R-tg ALS mouse model [25]. These findings suggest a neuroprotective role of SQSTM1 in vivo. However, the effect of SQSTM1 overexpression on the disease onset and progression in mutant SOD1-expressing mice remains unknown.
In this study, to clarify the effect of systemic overexpression of SQSTM1 on the disease onset and progression as well as the accumulation of aggregated proteins in SOD1H46R mice, we generated SQSTM1-overexpressing SOD1H46R double-tg (SQSTM1;SOD1H46R) mice, and analyzed lifespan, body weight, the number of motor neurons, and the distribution of ubiquitin-positive aggregates in the spinal cord. In addition, we conducted a series of western blot analysis to quantify the amount of expressed protein levels including phosphorylated forms of SQSTM1. We here revealed that overexpression of SQSTM1 accelerated disease onset by compromising the protein degradation pathways in a SOD1H46R-expressing ALS mouse model.
Methods
Animals
SOD1H46R-tg mice express human SOD1 carrying the H46R mutation under control of the human SOD1 promotor [22]. CAG-SQSTM1-HA (SQSTM1)-tg mice express human SQSTM1 with hemagglutinin (HA) tag at the C-terminus under control of the CAG [(C) the cytomegalovirus (CMV) early enhancer element, (A) the promoter, the first exon and the first intron of chicken β-actin gene, and (G) the splice acceptor of the rabbit β-globin gene] promoter [24]. Both SOD1H46R-tg and SQSTM1-tg mice were backcrossed to 57BL/6 N (B6) mice more than 10 generations, and were maintained as B6 congenic lines. We generated SQSTM1-overexpressing SOD1H46R mice (SQSTM1;SOD1H46R) by crossing male SOD1H46R mice with female SQSTM1 mice. The offsprings were genotyped by PCR using genomic DNA extracted from ear tissues. Primers used were as follows; SOD1H46R: hSOD1_ex2: 5′-TCAGAAACTCTCTCCAACTTTGC-3′ and 5′-CAAGTATGGGTCACCAGCAC-3′, hSOD1_ex4: 5′-GGCATCAGCCCTAATCCATC-3′ and 5′-CCGCGACTAACAATCAAAGTG-3′, SQSTM1-HA: 5′-AGCTGCCTTGTACCCACATC-3′ and 5′-AGCGTAATCTGGAACATCGT-3′. Mice were housed at 22 °C with a 12 h light-dark cycle. Food and water were fed ad libitum.
Antibodies
Primary antibodies used for immunohistochemistry were listed as follows; anti-human Ubiquitin (MBL MK-11-3; mouse monoclonal, 1:300), anti-human Ubiquitin (Dako Z0458; rabbit polyclonal, 1:500), anti-human SQSTM1 (Thermo PA5–20839; rabbit polyclonal, 1:300), anti-human p62-C (PROGEN GP62-C; guinea-pig polyclonal, 1:500), anti-rat MAP2 (SIGMA M9942; mouse monoclonal, 1:500), anti-GFAP (NICHIREI 422251; rabbit polyclonal, 1:2), anti-mouse Iba1 (Abcam ab178896; rabbit monoclonal, 1:500), and anti-HA-tag (Cell Signaling 3724; rabbit monoclonal, 1:500) antibodies. Secondary antibodies included Alexa 594 conjugated anti-mouse IgG (Molecular Probes, 1:500), Alexa 594 conjugated anti-Rabbit IgG (Molecular Probes, 1:500), Alexa 594 conjugated anti-guinea-pig IgG (Molecular Probes, 1:500), Alexa 488 conjugated anti-mouse IgG (Molecular Probes, 1:500), and Alexa 488 conjugated anti-Rabbit IgG (Molecular Probes, 1:500) antibodies.
Primary antibodies used for western blotting included anti-human SOD1 (SANTA CRUZ cs-11,407; rabbit polyclonal, 1:15000), anti-human misfolded SOD1 (C4F6; mouse monoclonal, 1:3000) [26], anti-bovine ubiquitin (SANTA CRUZ cs-8017; mouse monoclonal, 1:3000), anti-human SQSTM1 (MBL PM045; rabbit polyclonal, 1:2000), anti-human phosphorylated SQSTM1 (S403) (MBL D343–3; rat monoclonal, 1:3000), anti-mouse phosphorylated SQSTM1 (S351) (MBL PM074; rabbit polyclonal, 1:2000), anti-human LC3 (SIGMA L8918; rabbit polyclonal, 1:5000), anti-human NQO1 (Abcam ab34173; rabbit polyclonal, 1:3000), anti-rabbit GAPDH (MBL M171–3; mouse monoclonal, 1:5000), anti-Actin (SIGMA A5060; rabbit polyclonal, 1:1000), and anti-HA-tag (SANTA CRUZ sc-805; rabbit polyclonal, 1:3000) antibodies. Secondary antibodies included horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Jackson, 1:5000), HRP-conjugated anti-rabbit IgG (GE Healthcare NA934, 1:5000), and HRP-conjugated anti-rat IgG (SANTA CRUZ sc-2006, 1:5000) antibodies.
Lifespan analysis
Lifespan (endpoint) of tg mice with each genotype (SOD1H46R and SQSTM1;SOD1H46R) were determined by the observations that mice were unable to crawl by their forelimb and to eat/drink by themselves.
Growth curve analysis
Body weight of mice with each genotype [wild-type (WT), SQSTM1, SOD1H46R, and SQSTM1;SOD1H46R] was weekly monitored from 5 weeks to a maximum of 32 weeks of age.
Disease onset and progression analyses
Onset of the disease of mice with each genotype (SOD1H46R and SQSTM1;SOD1H46R) was defined as the point (date) at which body weight was successively decreased from its peak-value in each animal without any increases thereafter. Post-onset survival was defined as the duration from the onset of disease to the endpoint.
Preparation of paraffin sections
We obtained tissue samples of the spinal cord from SOD1H46R and SQSTM1;SOD1H46R mice at 16 and 22 weeks of age, and at end-stage. We also obtained the samples from WT and SQSTM1 mice at 16, 22, and 28 weeks of age. Samples from WT and SQSTM1 mice at 28 weeks of age were used to compare to those from tg mice at their end-stage. Mice were anesthetized with 4% isoflurane by inhalation and perfused with physiological saline containing 100 U/ml heparin, followed by 4% paraformaldehyde (PFA)/0.1 M phosphate buffer (PB) (pH 7.2). Spinal cord together with the spinal column was removed and fixed with 4% PFA/PB overnight at 4 °C. Lumbar cord (L4-L5) was removed from the spinal column, cut by an approximately 2 mm length (rostral to caudal direction), and post-fixed with 4% PFA/PB overnight at 4 °C. After washing with PB overnight twice, lumbar cord was immersed in 100% methanol for 1 h followed by soaking to fresh methanol overnight. Lumbar cord was further immersed in methanol:chloroform = 1:1 solution for 30 min, and in 100% chloroform for 2 h followed by the treatment with fresh chloroform overnight. The resulting lumbar cord samples were embedded in paraffin. Paraffin-embedded transverse sections at a thickness of 6 μm were prepared by microtome (LEICA RM2165) for Nissl staining and immunohistochemistry.
Nissl staining
Lumbar cord paraffin sections from mice with four different genotypes at 16 and 22 weeks of age (WT, SQSTM1, SOD1H46R, and SQSTM1;SOD1H46R), and at end-stage (SOD1H46R and SQSTM1;SOD1H46R) or 28 weeks of age (WT and SQSTM1), were treated with xylene (5 min × 3 times) and ethanol (5 min × 3 times), and hydrated. Slices were permeabilized in 1% Triton X-100/phosphate buffered saline (PBS) for 15 min and stained with 50 μl of NeuroTrace green fluorescent Nissl stains (ThermoFisher N21480) (1:500)/PBS for 25 min. Slices were washed with PBS for 2 h and mounted by VECTASHIELD Mounting Medium with DAPI for nuclear staining. A total of 5 representative Nissl staining images of every tenth serial sections from each sample was captured by fluorescence microscope (KEYENCE BZ-9000).
Counting the number of large Nissl-positive neurons
Large Nissl-positive neurons, representing motor neurons, were identified by using Dynamic cell count (KEYENCE). In brief, tones of the captured images were binarized, and the area covering 600 pixels (px) × 400 px corresponding to anterior horn of the spinal cord was selected. Automatic cell separation procedures (setting resolution; 14, threshold; 45 with excluding cells whose cross-sectional areas were less than 240 px) were executed. Neurons identified were defined as large Nissl-positive neurons and counted. The numbers of large Nissl-positive neurons from 5 representative images from each sample were summed.
Immunohistochemistry
Spinal cord sections were deparafinized in xylene (5 min × 3 times) and ethanol (5 min × 3 times), and were hydrated. Sections were heated by microwave oven in 300 ml of 0.1 M citric acid (pH 5.0) for 15 min. After cooling at room temperature (RT), sections were washed with PBS and blocked with 50 μl of 0.1% Triton X-100/5% normal goat serum (NGS)/PBS for 30 min. Sections were incubated with primary antibody in 50 μl of the antibody diluted solution (0.05% Triton X-100, 2% NGS/PBS) overnight at 4 °C. Sections were washed with PBS for 2 h and incubated with secondary antibody in 50 μl of the antibody diluted solution for 2 h. Sections were washed with PBS for 1 h and mounted using VECTASHIELD Mounting Medium with DAPI. Signals were analyzed by fluorescence microscope (KEYENCE BZ-710).
Preparation of tissue samples for western blot analysis
To analyze the expression and distribution of transgene product; i.e., SQSTM1-HA, we obtained tissue samples from WT and SQSTM1 mice at 16 weeks of age. Tissues including the liver, skeletal muscle, olfactory bulb, cerebral cortex, hippocampus, cerebellum, and whole spinal cord were removed, immediately frozen on dry ice, and stored at − 80 °C until use. To analyze the expression of particular proteins of interest in the spinal cord, we obtained the samples from mice with four different genotypes (WT, SQSTM1, SOD1H46R, and SQSTM1;SOD1H46R) at 16 and 22 weeks of age, and at end-stage (SOD1H46R and SQSTM1;SOD1H46R) or 28 weeks of age (WT and SQSTM1). The whole spinal cord was removed from each mouse, immediately frozen on dry ice, and stored at − 80 °C until use.
Preparation of protein samples
Spinal cord tissues were weighted and homogenized in 2 weight-volume (mg/μl) of PBS by sonication (Output 2, 10–20 times, BRANSON Sonifier 450). One-hundred microliters of homogenates and 700 μl of Lysis buffer A’ [25 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1% Triton X-100, Complete Protease Inhibitor Cocktail (Roche), Phosphatase inhibiter Cocktail I (SIGMA)] were mixed, followed by centrifugation at 23,000×g for 20 min at 4 °C. Supernatant was collected as 1% Triton X-100 soluble fraction. The insoluble pellet was washed by mixing with 600 μl of Lysis buffer A’ and centrifuged again, and resulting pellet was suspended in 400 μl of Lysis buffer B′ [25 mM Tris-HCl (pH 7.5), 50 mM NaCl, 5% sodium dodecyl sulfate (SDS)], sonicated (Output 2, 5–10 times), and used as 1% Triton X-100 insoluble fraction. Protein concentration was determined by Micro BCA (Thermo, MicroBCA Protein Assay kit #23235). Each protein fraction was diluted by the corresponding lysis buffer and adjusted its concentration at 0.75 μg/μl. One-hundred microliters of diluted samples were mixed with 50 μl of 3xSDS buffer [187 mM Tris-HCl (pH 6.8), 6% SDS, 30% glycerol, 0.015% bromophenol blue (BPB), 15% 2-melcaptoethanol] and heated for 5 min at 95 °C.
Western blot analysis
Equal amount of proteins was subjected to SDS-polyacrylamide gel electrophoresis (PAGE) (Wako Supersep™Ace, 5–20% 17well) in SDS-PAGE buffer [25 mM Tris-HCl, 192 mM glycine, 0.1% (w/v) SDS]. Precision Plus Protein standards dual color standard (BIO-RAD 1610374) was used as molecular weight markers. Proteins were electro-transferred onto polyvinylidene fluoride (PVDF) membrane (Merckmillipore Immobilon-P) in the transfer buffer [25 mM Tris-HCl, 192 mM glycine, 20% (w/v) methanol]. The membranes were blocked with 0.5% skimmed milk/TBST [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% Tween20] for 1 h at RT. When using anti-phosphorylation SQSTM1 antibody, membranes were blocked with 50% blocking one (Wako)/TBST for 30 min at RT. Membranes were incubated with the primary antibody in TBST overnight at 4 °C. After washing with TBST (10 min, 6 times), membranes were incubated with HRP-conjugated secondary antibody in TBST for 2 h at RT. After washing with TBST, signals were visualized by Immobilon (Merck Millipore) and detected by Ez-Capture MG (ATTO).
Quantification of the signal intensities on western blotting
We conducted a quantitative analysis of the signal intensities on western blotting to determine the expression levels of proteins of interest in 1% Triton X-100 soluble and insoluble factions from mice with four different genotypes (WT, SQSTM1, SOD1H46R, and SQSTM1;SOD1H46R) at 16 and 22 weeks of age, and at end-stage (SOD1H46R and SQSTM1;SOD1H46R) or 28 weeks of age (WT and SQSTM1). The signal intensities were quantified by using CS Analyzer Ver3.0 (ATTO) and were normalized by the levels of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for soluble fractions or β-actin for insoluble fractions.
Statistical analysis
All statistical analyses were conducted using Prism 5 (Graph pad). Data for lifespan, onset of disease, and post-onset survival between SOD1H46R and SQSTM1;SOD1H46R mice were compared using Kaplan-Meier survival analysis with Log-rank test. Statistical significances for body weight, the number of spinal motor neurons, and quantitative western blotting data between groups were evaluated by One-way or Two-way ANOVA with Bonferroni post hoc tests. A p-value < 0.05 was considered as reaching statistical significance.
Results
Expression and tissue distribution of SQSTM1-HA in SQSTM1 transgenic mice
SQSTM1-tg mouse carries the expression-cassette that can express the C-terminally HA-tagged human SQSTM1 (SQSTM1-HA) protein under control of the CAG promoter (Additional file 1: Figure S1a) [24]. To determine the distribution and expression levels of SQSTM1-HA in SQSTM1 mice, we prepared protein samples from the liver, skeletal muscle, olfactory bulb, cerebral cortex, hippocampus, cerebellum, and spinal cord from WT and SQSTM1 mice, and performed western blotting using anti-SQSTM1 and anti-HA antibodies. Mouse endogenous SQSTM1 was detected in all tissues examined with highest in the cerebral cortex and hippocampus (Additional file 1: Figure S1b, see WT). The CAG promoter-driven human SQSTM1-HA was highly expressed in skeletal muscle, and also detected in the central nervous system including the spinal cord, while it was undetectable in the liver (Additional file 1: Figure S1b).
Overexpression of SQSTM1 accelerates body weight declines in SOD1H46R mice
We performed growth curve analysis of WT, SQSTM1, SOD1H46R, and SQSTM1;SOD1H46R mice. Both WT and SQSTM1 mice were viable for at least 32 weeks of age. Throughout the experimental periods, body weights of SQSTM1 mice tended to be lower than those of WT mice in female (Fig. 1a). Body weights of female SQSTM1;SOD1H46R mice were significantly lower than those of SOD1H46R mice at 5, 8–13, 16–25, and 28 weeks of age (Fig. 1a). In male, body weights of SQSTM1;SOD1H46R mice were significantly lower than those of SOD1H46R mice at 25 and 26 weeks of age (Fig. 1b). These results indicate that overexpression of SQSTM1 in SOD1H46R mice accelerates weight loss.
Fig. 1.

Analysis of lifespan, disease onset, and progression in SQSTM1;SOD1H46R mice. a Growth curve in female mice with four different genotypes [wild-type (WT; n = 8–12), SQSTM1 (SQSTM1; n = 9–12), SOD1H46R (H46R; n = 2–12), and SQSTM1;SOD1H46R (SQSTM1;H46R; n = 1–12)]. b Growth curve in male mice with four different genotypes [WT; n = 4–12, SQSTM1; n = 11–12, H46R; n = 2–12, and SQSTM1;H46R; n = 4–12]. a, b Body weight of SQSTM1;SOD1H46R mice was significant lower than that of SOD1H46R mice (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). There are no significantly differences in body weight between WT and SQSTM1 mice. Values were mean ± s.d.. Statistical significance was evaluated by one-way ANOVA with Bonferroni’s post hoc test. c Lifespan analysis of SOD1H46R mice (H46R; n = 31) and SQSTM1;SOD1H46R mice (SQSTM1;H46R; n = 28). Kaplan-Meier analysis demonstrated that lifespan in SQSTM1;SOD1H46R mice (mean ± s.d.; 179.3 ± 2.6 days) was significantly shorter than that in SOD1H46R mice (189.3 ± 2.2 days) (Log-rank test; p = 0.0013). d Onset of the disease in SOD1H46R (H46R; n = 24) and SQSTM1;SOD1H46R (SQSTM1;H46R; n = 24) mice. Onset of disease is defined as the turning-point of body weight successive reduction. Onset in SQSTM1;SOD1H46R mice (mean ± s.d.; 21.5 ± 2.1 weeks) was significantly earlier than that in SOD1H46R mice (23.8 ± 2.1 weeks) (Log-rank test; p = 0.0007). e Survival after the onset of disease (post-onset survival interval) in SOD1H46R (H46R; n = 24) and SQSTM1;SOD1H46R (SQSTM1;H46R; n = 24) mice. Survival after the onset is defined as the period between the onset of disease and end-point. Post-onset survival interval in SQSTM1;SOD1H46R mice (mean ± s.d.; 3.9 ± 1.3 weeks) was significantly longer than that in SOD1H46R mice (3.0 ± 1.1 weeks) (Log-rank test; p = 0.0314)
Overexpression of SQSTM1 shortens lifespans in SOD1H46R mice
In the previous study, we have shown that loss of SQSTM1 significantly shortens lifespans in SOD1H46R mice [25]. Further, it has been reported that loss of SQSTM1 shortens lifespan while overexpression of it prolongs lifespan in SBMA mice [24]. To clarify the effect of overexpression of SQSTM1 on the onset and progression in an ALS mouse model (SOD1H46R mice), we performed lifespan analysis using SOD1H46R and SQSTM1;SOD1H46R mice. Since there were no gender differences of lifespans in B6 congenic SOD1H46R mice [27], we used the gender-combined data. Kaplan-Meier survival analysis revealed that the lifespan of SQSTM1;SOD1H46R mice (179.3 ± 2.6 days) was significantly shorter than that of SOD1H46R mice (189.3 ± 2.2 days) (Fig. 1c; Log-rank test; p = 0.0013).
Overexpression of SQSTM1 accelerates onset of disease but slightly extends post-onset survival in SOD1H46R mice
Previously, it has been reported that onset of disease can be estimated by the turning-point of the body weight from gain to loss in mutant SOD1-tg mice [27–29]. Growth curve analysis revealed that onset of disease in SQSTM1;SOD1H46R mice (21.5 ± 2.1 weeks) was significantly earlier than those of SOD1H46R mice (23.8 ± 2.1 weeks) (Fig. 1d and Table 1; Log-rank test; p = 0.0007). To investigate the difference in disease progression between SOD1H46R and SQSTM1;SOD1H46R mice, we analyzed the survival interval after disease onset (post-onset survival). Contrary to the disease onset, survival interval after the onset of disease in SQSTM1;SOD1H46R mice (3.9 ± 1.3 weeks) was slightly but significantly extended when compared to SOD1H46R mice (3.0 ± 1.1 weeks) (Fig. 1e and Table 1; Log-rank test; p = 0.0314).
Table 1.
Summary of survival and onset point analysis in SOD1H46R and SQSTM1;SOD1H46R mice
| Genotype | Number | End point (weeks) | s.d. | Onset (weeks) | s.d. | Post-onset survival interval (weeks) | s.d. |
|---|---|---|---|---|---|---|---|
| SOD1H46R | 24 | 26.7 | 1.5 | 23.8 | 2.1 | 3.0 | 1.1 |
| SQSTM1;SOD1H46R | 24 | 25.4 | 1.5 | 21.5 | 2.1 | 3.9 | 1.3 |
Number represents the number of mice used in survival and onset analyses. Values represent mean ± s.d. (weeks)
Progressive decreases in the number of motor neurons in the lumbar spinal cord of SOD1H46R-expressing mice
To investigate whether earlier disease onset observed in SQSTM1;SOD1H46R mice was associated with motor neuron loss in the spinal cord, we performed Nissl staining and counted the number of large neurons in the anterior horn of lumbar spinal cord. In this analysis, mice with four different genotypes; WT, SQSTM1, SOD1H46R, and SQSTM1;SOD1H46R mice at 16 and 22 weeks of age, and at end-stage (SOD1H46R and SQSTM1;SOD1H46R) or 28 weeks of age (WT and SQSTM1), were used. The numbers of large Nissl-positive neurons were progressively decreased both in SOD1H46R and SQSTM1;SOD1H46R mice compared to WT and SQSTM1 mice (Fig. 2 and Additional file 2: Fig. S2a). However, there were no significant differences in the numbers of Nissl positive large neurons between groups both at 22 weeks of age and the end-stage, suggesting that decrease in the number of motor neurons did not explain the reasons why overexpression of SQSTM1 not only accelerates the onset of disease but also slows survival after the onset in SOD1H46R mice.
Fig. 2.
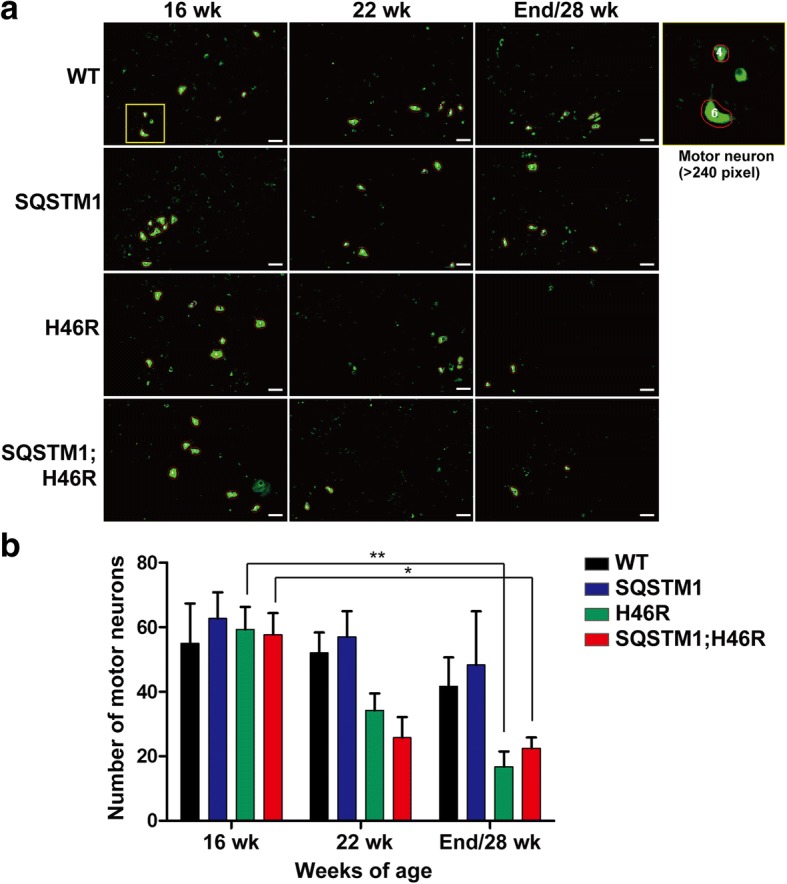
Quantitative analysis of motor neurons in the anterior horn of lumbar spinal cord. a Representative images for fluorescence Nissl staining in the anterior horn. Nissl positive neurons were detected in the anterior horn of lumbar spinal cord (L4–5) from the mice with four different genotypes; wild-type (WT), SQSTM1, SOD1H46R (H46R), and SQSTM1;SOD1H46R (SQSTM1;H46R) at 16 and 22 weeks of age (wk), and at end-stage (H46R and SQSTM1;H46R) or at 28 wk. (WT and SQSTM1). Cells covering the area of over 240 square pixels (px), corresponding to large motor neurons, are surrounded by red circles, and counted. Scale bars = 50 μm. b Number of large Nissl-positive neurons in the anterior horn of lumbar spinal cord. Vertical axis represents the cumulative number of motor neurons identified in 5 representative Nissl staining images of every tenth serial sections from each sample (n = 3–4, SOD1H46R and SQSTM1;SOD1H46R mice at 22 wk. and end-stage; n = 4, others; n = 3). Values are mean ± s.e.m.. Statistical significance was evaluated by two-way ANOVA with Bonferroni’s post hoc test (*p < 0.05, **p < 0.01)
Accumulation of ubiquitin/SQSTM1-positive aggregates and the activation of glial cells in SOD1H46R-expressing mice
SQSTM1 acts as an adaptor protein that directly binds to poly-ubiquitinated proteins destined for degradation in autophagy [10]. It has been reported that accumulation of ubiquitin- and SQSTM1-positive aggregates is observed not only in ALS patients but also in a SOD1H46R ALS mouse model [22], and that proliferation of astrocytes and the activation of microglia also occur in SOD1H46R mice as disease progresses. To clarify the effect of SQSTM1 overexpression on the localization of ubiquitin-/SQSTM1-positive aggregates and the activation of glial cells, we conducted immunohistochemical analysis of ubiquitin and SQSTM1 in the lumbar cord (L4–5) from WT, SQSTM1, SOD1H46R, and SQSTM1;SOD1H46R mice at 16 and 22 weeks of age, and at end-stage (SOD1H46R and SQSTM1;SOD1H46R) or 28 weeks of age (WT and SQSTM1) (Fig. 3 and Additional file 2: Figure S2b). We also conducted immunohistochemistry for microtubule-associated protein 2 (MAP2), glial fibrillary acidic protein (GFAP), and ionized calcium binding adaptor molecule 1 (Iba1) using mice with four different genotypes at end-stage (SOD1H46R and SQSTM1;SOD1H46R) or 28 weeks of age (WT and SQSTM1). MAP2, GFAP, and Iba1 were used as cellular markers for neuronal soma and dendrite, astrocytes, and microglia, respectively (Figs. 4 and 5, and Additional file 2: Figure S2c).
Fig. 3.
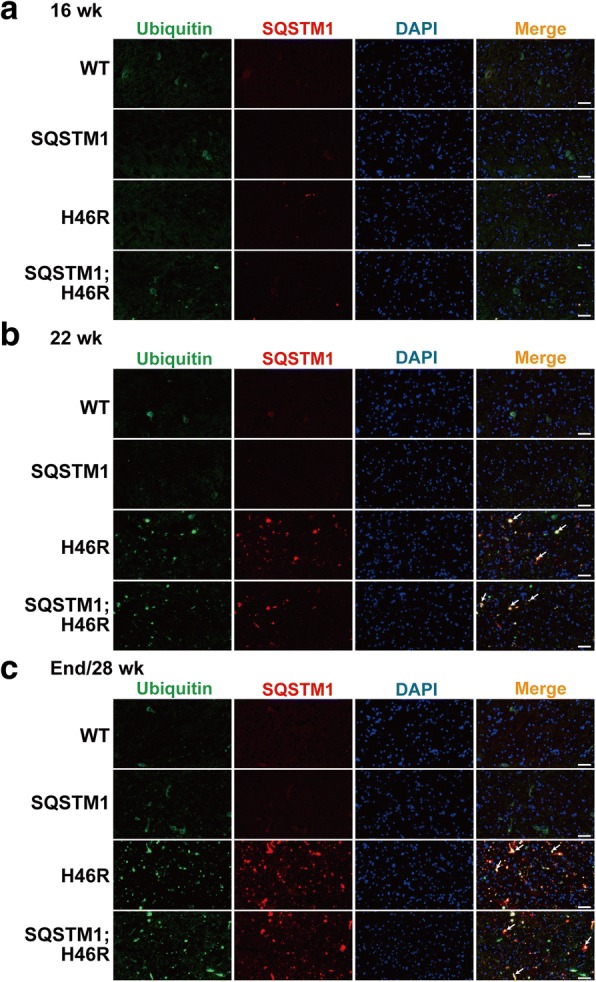
Progressive accumulation of ubiquitin/SQSTM1-positive aggregates in the anterior horn of lumbar spinal cord. a-c Representative images of double immunostaining with Ubiquitin (green) and SQSTM1 (red) in the lumbar spinal cord (L4–5) from wild-type (WT), SQSTM1 (SQSTM1), SOD1H46R (H46R) and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk) (a), 22 wk. (b), and end-stage (H46R and SQSTM1;H46R) or 28 wk. (WT and SQSTM1) (c). The nuclei were counterstained with DAPI (blue). Scale bars = 50 μm. Ubiquitin-positive aggregates and SQSTM1 aggregates were observed in the anterior horn of SOD1H46R (H46R) and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 22 wk. and end-stage. Arrows indicate ubiquitin-positive aggregates that are colocalized with SQSTM1
Fig. 4.
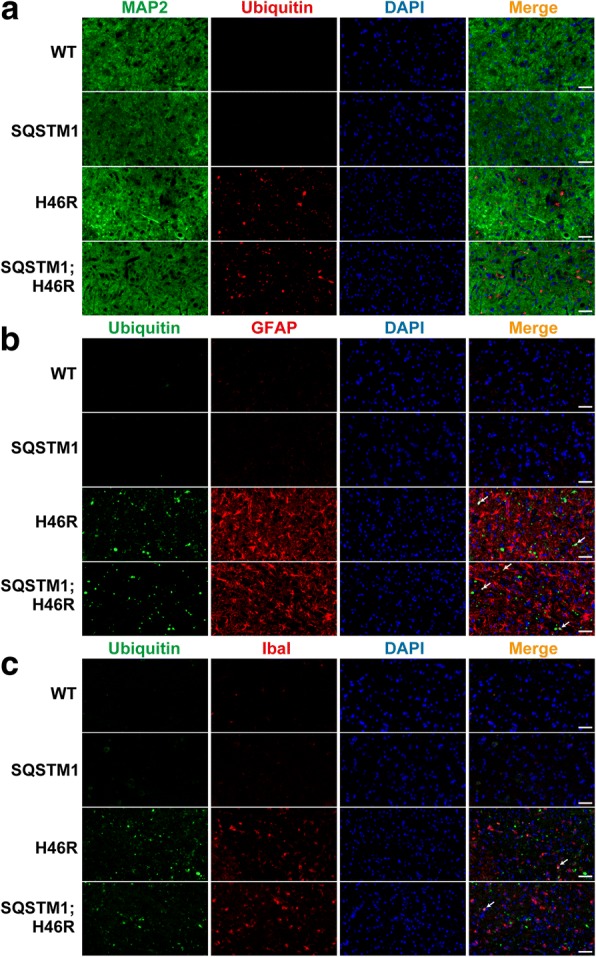
Extensive accumulation of ubiquitin-positive aggregates in the spinal cord from SOD1H46R-expressing mice at end-stage. a-c Representative images of double immunostaining with MAP2 (green; neuron marker) and Ubiquitin (red) (a), Ubiquitin (green) and GFAP (red; astrocyte marker) (b), Ubiquitin (green) and Iba1 (red; microglia marker) (c) in the lumbar spinal cord (L4–5) from wild-type (WT) and SQSTM1 (SQSTM1) at 28 weeks of age, SOD1H46R (H46R) and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at end-stage. The nuclei were counterstained with DAPI (blue). Scale bars = 50 μm. b Arrows indicate colocalization of ubiquitin-positive aggregates with GFAP-positive astrocytes. c Arrows indicate colocalization of ubiquitin-positive aggregates with Iba1-positive microglia
Fig. 5.
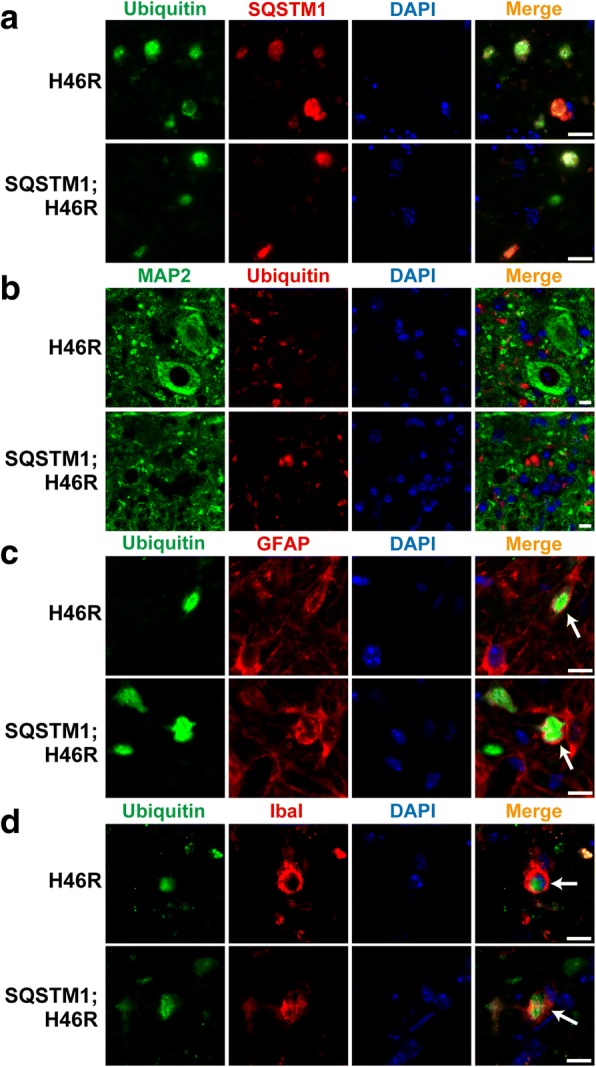
High-magnification images for the distribution of ubiquitin-positive aggregates in the spinal cord from SOD1H46R-expressing mice. a-d Representative images of double immunostaining with Ubiquitin (green) and SQSTM1 (red) (a), MAP2 (green; neuron marker) and Ubiquitin (red) (b), Ubiquitin (green) and GFAP (red; astrocyte marker) (c), Ubiquitin (green) and Iba1 (red; microglia marker) (d) in the lumbar cord (L4–5) from SOD1H46R (H46R) and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at end-stage. The nuclei were counterstained with DAPI (blue). Scale bars = 10 μm. c Arrows indicate that ubiquitin-positive aggregates are present in GFAP-positive astrocytes. d Arrows indicate that ubiquitin-positive aggregates are surrounded by microglia
Double immunostaining of ubiquitin and SQSTM1 revealed that a progressive accumulation of ubiquitin- and SQSTM1-positive aggregates was observed in the spinal cord of SOD1H46R-expressing mice (SOD1H46R and SQSTM1;SOD1H46R) as disease progressed (Fig. 3 and Additional file 2: Figure S2b). It was also noted that a majority of the ubiquitin-positive signals was colocalized with SQSTM1 (Figs. 3 and 5a). Further, SQSTM1-HA aggregates were immunohistochemically detected in SQSTM1;SOD1H46R mice at 22 weeks and end-stage by using the anti-HA-tag antibody (Additional file 3: Figure S3), indicating that overexpressing SQSTM1 was also progressively accumulated in the spinal cord as was endogenous SQSTM1. However, there were no apparent differences in the amount and/or number of ubiquitin-/SQSTM1-positive aggregates between SOD1H46R and SQSTM1;SOD1H46R mice.
Double immunostaining of ubiquitin and MAP2 revealed that ubiquitin-positive aggregates were not present in MAP2-positive neuronal cell bodies or dendrites in the lumbar cord of SOD1H46R-expressing mice (Figs. 4a and 5b and Additional file 2: Figure S2c). On the other hand, double immunostaining of ubiquitin and GFAP demonstrated that the number of GFAP-positive astrocytes was significantly increased in SOD1H46R-expressing mice (Fig. 4b and Additional file 2: Figure S2c), and that small numbers of ubiquitin-positive aggregates were localized to astrocytes (Figs. 4b and 5c; arrows). Activation of Iba1-positive microglia was also observed in SOD1H46R-expressing mice (Fig. 4c and Additional file 2: Figure S2c). Ubiquitin-positive aggregates that were surrounded by microglia were occasionally observed (Fig. 5d; arrows). However, there were no differences in the levels of glial cell activation between SOD1H46R and SQSTM1;SOD1H46R mice.
Together, these results indicate that ubiquitin-/SQSTM1-positive aggregates were mainly localized to neuropil and occasionally to astrocytes and/or microglia but not to the cell body of neurons at the end-stage of SOD1H46R-expressing mice. However, there were no discernible differences in the distribution and the degree of colocalization of ubiquitin-positive aggregates to glial cells between SQSTM1;SOD1H46R and SOD1H46R mice.
Overexpression of SQSTM1 increases insoluble poly-ubiquitinated proteins in the spinal cord of SOD1H46R mice
To biochemically investigate the accumulation of SOD1, SQSTM1, and ubiquitin, we extracted Triton X-100 soluble and insoluble proteins from the spinal cord of mice with four different genotypes; WT, SQSTM1, SOD1H46R, and SQSTM1;SOD1H46R mice at 16 and 22 weeks of age, and at end-stage (SOD1H46R and SQSTM1;SOD1H46R) or at 28 weeks of age (WT and SQSTM1) and performed western blot analysis, followed by the quantification of signal intensities.
Although a progressive accumulation of insoluble high-molecular weight SOD1 (SOD1_HMW) was observed both in SOD1H46R and SQSTM1;SOD1H46R mice as disease progressed (Fig. 6a and Additional file 4: Figure S4c), quantitative analyses showed no significant differences of the amount of insoluble SOD1_HMW and monomer SOD1 (SOD1_mono) between SOD1H46R and SQSTM1;SOD1H46R mice (Fig. 6c). Further, we detected misfolded SOD1 molecules using anti-human misfolded SOD1 antibody (C4F6) [26]. Misfolded monomeric SOD1 was observed both in SOD1H46R and SQSTM1;SOD1H46R mice, but not in WT and SQSTM1 mice (Fig. 6b). Quantitative analyses showed that both soluble and insoluble misfolded SOD1 in SQSTM1;SOD1H46R mice at 16 weeks of age were significantly higher than those in SOD1H46R mice, while there were no differences between two groups at the later stages of disease (22 weeks of age and end-stage) (Fig. 6c and Additional file 5: Figure S5).
Fig. 6.

Overexpression of SQSTM1 increases misfolded SOD1 in the spinal cord of SOD1H46R-expressing mice. a Western blot analysis of SOD1. b Western blot analysis of misfolded SOD1. a, b The spinal cord samples were obtained from wild-type (WT), SQSTM1 (SQSTM1), SOD1H46R (H46R), and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk), 22 wk., and end-stage (H46R and SQSTM1;H46R) or 28 wk. (WT and SQSTM1). Two fractions; 1% Triton X-100 soluble and 1% Triton X-100 insoluble/5%SDS soluble fractions were analyzed. SOD1_mono and SOD1_HMW represent monomeric and high molecular-weight forms of SOD1, respectively. SOD1(C4F6) represents misfolded SOD1. GAPDH and β-actin (Actin) were used for a loading control in Triton X-100-soluble and -insoluble fractions, respectively. c Quantification of 1% Triton X-100 soluble SOD1, insoluble SOD1_mono, insoluble SOD1_HMW, soluble misfolded SOD1, and insoluble misfolded SOD1. Values are mean ± s.e.m. (n = 4) in an arbitrary unit relative to H46R mice at 16 weeks of age. Signal intensities were normalized by the levels of GAPDH (soluble fractions) and Actin (insoluble fractions). Statistical significance was evaluated by two-way ANOVA with Bonferroni’s post hoc test (comparisons between different genotypes in the same age; *p < 0.05, **p < 0.01)
Both soluble and insoluble SQSTM1 were significantly and progressively increased in SOD1H46R and SQSTM1;SOD1H46R mice as disease progressed (Fig. 7a, b and Additional file 4: Figure S4d and S4e). Compared to SOD1H46R mice, the accumulated levels of insoluble SQSTM1 in SQSTM1;SOD1H46R mice were much higher at 16 and 22 weeks of age, and at end-stage (Fig. 7b). To distinguish between mouse endogenous and transgene-derived SQSTM1, we performed western blot analysis using anti-HA antibody, and revealed that transgene-encoded SQSTM1-HA was also progressively accumulated as insoluble forms in SQSTM1;SOD1H46R mice (Fig. 7a).
Fig. 7.

Overexpression of SQSTM1 increases the insoluble poly-ubqiuitinated proteins in the spinal cord of SOD1H46R-expressing mice. a Western blot analysis of SQSTM1, SQSTM1-HA (HA) and Ubiquitin (Ub) in the spinal cord from wild-type (WT), SQSTM1 (SQSTM1), SOD1H46R (H46R), and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk), 22 wk., and end-stage (H46R and SQSTM1;H46R) or 28 wk. (WT and SQSTM1). Two fractions; 1% Triton X-100 soluble and 1% Triton X-100 insoluble/5%SDS soluble fractions were analyzed. SOD1_mono and SOD1_HMW represent monomeric and high molecular-weight forms of SOD1, respectively. Ub_mono and Ub_HMW represent monomeric ubiquitin and poly-ubiquitinated proteins, respectively. GAPDH and β-actin (Actin) were used for a loading control in Triton X-100-soluble and -insoluble fractions, respectively. Since the blots used in these western analyses were same as those in Fig. 6a, same images for GAPDH and Actin (asterisks) in Fig. 6a were used again as references. b Quantification of soluble and insoluble SQSTM1. c Quantification of soluble and insoluble poly-ubiquitinated proteins (Ub_HMW). b, c Values are mean ± s.e.m. (n = 4) in an arbitrary unit relative to WT mice at 16 weeks of age. Signal intensities were normalized by the levels of GAPDH (soluble fractions) and Actin (insoluble fractions). Statistical significance was evaluated by two-way ANOVA with Bonferroni’s post hoc test (comparisons between different genotypes in the same age; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
Accumulation of insoluble poly-ubiquitinated proteins that were represented by smear signals was observed in SOD1H46R-expressing mice (Fig. 7a and Additional file 4: Figure S4 g). Although poly-ubiquitinated proteins detected in the spinal cord of SQSTM1;SOD1H46R mice were comparable to those in SOD1H46R mice at 22 weeks of age, those in SQSTM1;SOD1H46R mice at end-stage were significantly higher than that in SOD1H46R mice (Fig. 7a and c).
These results suggest that overexpression of SQSTM1 accelerates the accumulation not only of misfolded SOD1 at the earlier stage, but also of insoluble poly-ubiquitinated proteins at the later stage of disease in the spinal cord of SOD1H46R-expressing mice.
Overexpression of SQSTM1 increases insoluble phosphorylated SQSTM1 in the spinal cord of SOD1H46R mice
Previous studies have shown that phosphorylation of SQSTM1 at Ser403 regulates degradation of poly-ubiquitinated proteins by autophagy [14], and that protein aggregates containing Ser403-phosphorylated SQSTM1 are enriched in the brain of ALS patients [30]. Further, phosphorylation of SQSTM1 at Ser351 increases the binding of KEAP1 to SQSTM1, thereby activating the NFE2L2/Nrf2 mediated anti-oxidative stress pathway [16]. These phosphorylation sites are conserved between human and mouse and can be detected by specific anti-phospho-SQSTM1 antibodies. To elucidate the effect of SQSTM1 overexpression on autophagy and anti-oxidative stress responses in SOD1H46R mice, we performed western blot analysis using antibodies to specifically detect Ser403(human)/Ser405(mouse)-phosphorylated SQSTM1, Ser349(human)/Ser351(mouse)-phosphorylated SQSTM1, MAP1LC3/LC3, and NAD(P)H dehydrogenase [quinone] 1 (NQO1) in the spinal cord of WT, SQSTM1, SOD1H46R, and SQSTM1;SOD1H46R mice at 16 and 22 weeks of age, and at end-stage (SOD1H46R and SQSTM1;SOD1H46R) or at 28 weeks of age (WT and SQSTM1).
The levels of insoluble Ser403/Ser405-phosphorylated SQSTM1 were increased both in SOD1H46R and SQSTM1;SOD1H46R mice at 22 weeks of age and end-stage compared to WT and SQSTM1 mice. However, there were no differences in the amount of Ser403/Ser405-phosphorylated SQSTM1 between SOD1H46R and SQSTM1;SOD1H46R mice, despite that the total amount of SQSTM1 was progressively increased at the end-stage (Figs. 7a, b and 8a, b, and Additional file 6: Figure S6b). Remarkably, Ser349/Ser351-phosphorylated SQSTM1 was specifically detected in insoluble but not in soluble fractions. Further, insoluble Ser349/Ser351-phosphorylated SQSTM1 was significantly increased in SQSTM1;SOD1H46R, but not in SOD1H46R, mice as disease progressed (Fig. 8a, c and Additional file 6: Fig. S6d).
Fig. 8.

Accumulation of phosphorylated SQSTM1 in the spinal cord of SOD1H46R-expressing mice. a Western blot analysis of Ser403(human)/Ser405(mouse)-phosphorylated SQSTM1 (p-S403/405), Ser349(human)/Ser351(mouse)-phosphorylated SQSTM1 (p-S349/351), LC3, and NQO1 in the spinal cord from mice with four different genotypes; wild-type (WT), SQSTM1 (SQSTM1), SOD1H46R (H46R), and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk), 22 wk., and end-stage (H46R and SQSTM1;H46R) or 28 wk. (WT and SQSTM1). Two fractions; 1% Triton X-100 soluble and 1% Triton X-100 insoluble/5%SDS soluble fractions were analyzed. GAPDH and β-actin (Actin) were used for a loading control in Triton X-100-soluble and -insoluble fractions, respectively. To properly compare the intensities of signals between phosphorylated SQSTM1 and total SQSTM1, the results from same blots for SQSTM1 in Fig. 7a, GAPDH, and Actin (asterisks) in Fig. 6a are used again as references. b Quantification of 1% Triton X-100 soluble and insoluble S403/S405-phosphorylated SQSTM1. c Quantification of insoluble S349/S351-phosphorylated SQSTM1. d Quantification of the LC3-II to LC3-I ratio (LC3-II/LC3-I) in insoluble fraction. e Quantification of soluble NQO1. b-e Values are mean ± s.e.m. (n = 4) in an arbitrary unit relative to WT mice at 16 weeks of age. Signal intensities were normalized by the levels of GAPDH (soluble fractions) and Actin (insoluble fractions). Statistical significance was evaluated by two-way ANOVA with Bonferroni’s post hoc test (comparisons between different genotypes in the same age; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
The levels of LC3-II in insoluble fraction were progressively increased in SOD1H46R-expressing mice (Fig. 8a and Additional file 6: Figure S6f). The quantitative analysis of the LC3-II to LC3-I ratio (LC3-II/LC3-I), a marker of the autophagosome formation, demonstrated that increase in the levels of LC3-II/LC-I was more prominent in SOD1H46R mice compared to SQSTM1;SOD1H46R mice (Fig. 8a and d). On the other hand, the levels of NQO1, which was one of the NFE2L2/Nrf2 target proteins, were affected neither by overexpression of mutant SOD1, SQSTM1, nor both of them (Fig. 8a and e, and Additional file 6: Figure S6 g).
These results suggest that overexpression of SQSTM1 affects the autophagy activation and/or the impairment of autophagic degradation of LC3-II positive autophagosomes, whereas it has no impact on the NFE2L2/Nrf2 mediated anti-oxidative stress pathway even in conditions in which the levels of Ser349/Ser351-phosphlylated SQSTM1 is upregulated. It is thus reasonable to assume that phosphorylated SQSTM1 molecules present in the insoluble fraction might be biologically inactive and are present as inert insoluble aggregates in the spinal cord of SOD1H46R mice.
Discussion
We have previously shown that loss of SQSTM1 exacerbates disease phenotypes in SOD1H46R mice [25]. Further, in a SBMA mouse model, loss of SQSTM1 also accelerates the onset of disease, while overexpression of it ameliorates disease phenotypes [24]. These findings suggest a protective role of SQSTM1 in motor neuron degeneration in vivo. In this study, we investigated the effect of systemic overexpression of SQSTM1 on the disease onset and progression in SOD1H46R mice. Contrary to our expectation, we revealed that systemic overexpression of SQSTM1 resulted in earlier onset and shorter lifespan in a SOD1H46R-expressing ALS mouse model.
It has been reported that phosphorylation at Ser349 (corresponding to mouse Ser351) of SQSTM1 enhances the binding of SQSTM1 to KEAP1 which in turn activates the NFE2L2/Nrf2 pathway and NFE2L2/Nrf2 mediated anti-oxidative stress responses in mice [16]. On the other hand, the previous study on NFE2L2/Nrf2 deficient SOD1H46R mice has revealed that loss of NFE2L2/Nrf2 affects neither the onset of disease nor the progression of disease [25]. In this study, despite that the accumulation of Ser349/Ser351-phosphorylated SQSTM1 was increased in SQSTM1;SOD1H46R mice at 22 weeks of age and end-stage (Fig. 8a and c), there were no differences in the expression levels of NQO1, which was one of the NFE2L2/Nrf2 downstream targets, between SOD1H46R and SQSTM1;SOD1H46R mice. Further, immunohistochemical analysis revealed that ubiquitin/SQSTM1-positive aggregates were not localized in the cell body and dendrite of neurons but mainly to neuropil (Figs. 4 and 5), indicative of dysfunctional SQSTM1 molecules in insoluble aggregates. Taken together, we speculate that progressively-increased Ser349/Ser351-phosphorylated SQSTM1 is physiologically inactive, and thus the NFE2L2/Nrf2 anti-oxidative stress pathway plays a minor role both in the disease onset and progression.
We suppose three possible reasons as to why overexpression of SQSTM1 accelerates the onset of disease in SOD1H46R mice, unlike in the case of a SBMA mouse model [24]. One possibility is that protein homeostasis (proteostasis) in the spinal cord of SOD1H46R mice is more severely compromised than those in SBMA mice under conditions in which SQSTM1 is overexpressed. A large number of studies has revealed that the accumulation of aggregated proteins is one of the pathological features of neurodegenerative diseases. Misfolded ubiquitinated proteins are preferentially degraded by the ubiquitin-proteasome system (UPS), but undegradable misfolded proteins by the UPS form aggregates, which in turn are degraded by autophagy [31]. SQSTM1 acts as an autophagy adaptor for the degradation of ubiquitinated proteins [11]. Thus, in addition to the UPS, autophagy is the important intracellular degradation pathway to maintain proteostasis [32], and its impairment results in the accumulation of SQSTM1 and ubiquitinated proteins [10]. Recently, it has been demonstrated that the crossbreeding of conditional Atg7 knockout mice with SOD1G93A mice remarkably extends the lifespan, despite that the onset of hindlimb tremor is accelerated [33]. Further, the pharmacological induction of mTOR-independent autophagy has failed to slow the disease progression of SOD1G93A mice [34]. More importantly, motor neuron-specific disruption of autophagy displays no phenotypic changes in ALS mice, while proteasome inhibition leads to ALS-associated pathology and paralysis [35]. We here showed that misfolded SOD1 in SQSTM1;SOD1H46R mice at 16 weeks of age were significantly higher than those in SOD1H46R mice (Fig. 6c). We also demonstrated that the extensive accumulation of insoluble SQSTM1 occurred in SQSTM1;SOD1H46R mice even prior to the onset of disease; i.e., at 16 weeks of age (Fig. 7a and b). Although phosphorylation of human SQSTM1 at Ser403 was known to induce autophagic degradation of ubiquitinated proteins [14], the levels of Ser403 (corresponding to mouse Ser405)-phosphorylated SQSTM1 between SQSTM1;SOD1H46R and SOD1H46R mice were most comparable (Fig. 8a and b). Further, the ratio of LC3-II/LC3-I in SQSTM1;SOD1H46R mice was lower than that of SOD1H46R mice (Fig. 8a and d) at onset stage; i.e., 22 week of age. These results combined suggest that overexpression of SQSTM1 does not activate autophagy, but rather accelerates the accumulation of SQSTM1 by possibly hindering the UPS and autophagic degradation in SOD1H46R mice. Since the accumulation of autophagosome-like structures and SQSTM1-positive aggregates are among the cardinal pathological features in patients with ALS as well as in murine ALS models [18, 23], it is reasonable to assume that overexpression of SQSTM1 enhances mutant SOD1-associated neurotoxicity by further compromising proteostasis, thereby leading to an earlier onset of disease. On the other hand, SBMA-linked toxic entities; e.g., polyglutamine-mediated transcriptional dysregulation [24], may play an indirect role in the regulation of the UPS and/or autophagy, and thus the SQSTM1-associated beneficial effect becomes more tangible in SBMA mice.
Second possibility is that SQSTM1 expressing in cells and tissues other than neurons affects the onset of disease in SOD1H46R mice. Evidences showing that, in skeletal muscle of a SOD1G93A-expressing ALS mouse model, the UPS is activated prior to the onset of disease, while the autophagic degradation pathway is activated after the onset of disease till at end-stage [36], suggest a possible pathogenic interaction between proteostasis in skeletal muscles and neurodegeneration. Since the expression level of transgene-coded SQSTM1 in skeletal muscle is higher than those in other tissues (Additional file 1: Figure S1b), it is possible that the earlier onset of disease in SQSTM1;SOD1H46R mice is linked to dysregulation of autophagy by SQSTM1 overexpression in skeletal muscles. However, even if that would be the case, the underlying mechanism has yet to be investigated.
Third possibility is that body weight at pre-symptomatic healthy stages affects the onset of disease in SOD1H46R mice. It has been shown that a high fat food feeding in a mutant TAR DNA-binding protein 43-kD (TARDBP)-expressing ALS mouse model extends their lifespans [37], and that the activation of AMP-activated protein kinase (AMPK), which functions as an energy sensor, results in the extension of lifespans in a mutant SOD1-expressing ALS mouse model [38]. These studies on ALS and energy metabolism suggest that nutritional conditions affect the disease phenotypes in ALS animal models. Interestingly, we revealed that body weight of SQSTM1 mice tended to be lower than that of WT mice (Fig. 1a and b), and that body weight of female SQSTM1;SOD1H46R mice was significantly lower than that of SOD1H46R mice throughout their pre-symptomatic period (Fig. 1a). Taken together, it is possible that the reduction of body weight that is associated with SQSTM1 overexpression causes the earlier onset of disease in SOD1H46R mice. Since loss of SQSTM1 in mice causes hyperphagia that results in obese phenotypes [39], a systemic overexpression of SQSTM1 may conversely induce hypophagia thereby decreasing the body weight in mice. In any cases, pathogenic mechanisms by which overexpression of SQSTM1 accelerates the onset in a SOD1H46R-expressing ALS mouse model remain unknown, and thus further studies are required.
One of the interesting findings in this study is that survival after the onset is slightly extended in SQSTM1;SOD1H46R mice, despite that these mice show earlier onset of disease (Fig. 1). However, we are currently considering that, although the extension of post-onset survival reaches to a statistically significant level, its reliability may still not be high enough to conclude that overexpression of SQSTM1 definitely extends survival, because the prediction of disease onset is solely determined by a single measure; i.e., body weight changes, in this study. Previously, it has been shown that mutant SOD1-mediated toxicities in neurons determine the disease onset, whereas those in astrocytes or microglia regulate the disease progression in ALS mouse models [29, 40]. Based on these findings, it could be hypothesized that mutant SOD1-mediated toxicities in glial cells relative to neurons are decreased by systemic overexpression of SQSTM1 in SOD1H46R mice. Although quantitative evaluation of GFAP and Iba1 levels by western blotting remained to be performed, immunohistochemical analysis revealed that the glial cells activation in the spinal cord between SOD1H46R and SQSTM1;SOD1H46R mice were almost comparable (Figs. 4 and 5). Thus, it is fair to assume that such glial contribution may not play a major role in the extension of survival after the onset. On the other hand, we have previously reported that loss of SQSTM1 in SOD1H46R mice decreases the biochemically-detectable insoluble ubiquitinated proteins in the spinal cord in parallel with the accumulation of ubiquitin-positive inclusions in somal area of motor neurons [25]. Since loss of SQSTM1 exacerbates disease symptoms in SOD1H46R mice, we consider that the accumulation of biochemically-detectable insoluble ubiquitinated proteins is beneficial and/or protective rather than harmful to motor neurons, while the intrasomal ubiquitin-positive inclusions are poisonous [25]. Indeed, we showed that biochemically-detectable insoluble ubiquitinated proteins were increased in the spinal cord of SQSTM1;SOD1H46R mice at end-stage (Fig. 7a and c). Further, immunohistochemistry revealed that ubiquitin-positive inclusions were exclusively observed in neuropil but not in somal area of motor neurons of SQSTM1;SOD1H46R mice (Figs. 4 and 5). Thus, the effects of loss and overexpression of SQSTM1 on these factors are obviously opposite to each other. Taken together, it is still possible that the insolubilization of poly-ubiquitinated proteins and the exclusion of ubquitinated protein aggregates from neuronal cell body by SQSTM1 are implicated in some neuroprotective roles, particularly, at the stage of disease progression, thereby slowing disease progression by SQSTM1 overexpression in SOD1H46R mice. However, in order to conclude as such, further studies that includes other methods to determine the onset of disease, such as grip power, the hind-limb reflex, rotarod retention time, and balance-beam test will be required.
Conclusions
In conclusions, we demonstrated that, unlike in SBMA mice, SQSTM1 served as an onset accelerating factor for SOD1-linked toxicities, at least, in a SOD1H46R-expressing ALS mouse model, implying that SQSTM1 upregulation might not always to be beneficial to the treatment of ALS.
Additional files
Figure S1. Schema of the transgene construct for SQSTM1-tg mouse and the distribution of SQSTM1 in tissues. a Schematic diagram of the transgene cassette encoding the C-terminally hemagglutinin (HA)-tagged human SQSTM1 cDNA. Transgene construct consists of cytomegalovirus enhancer (E), chicken β-actin promoter (Pro), rabbit β-globin splice acceptor (S), full length human SQSTM1 cDNA (SQSTM1), hemagglutinin tag (HA), and rabbit β-globin poly A (Poly A). b Expression and distribution of SQSTM1 in the liver, skeletal muscle, olfactory bulb, cerebral cortex, hippocampus, cerebellum, and spinal cord in wild-type (WT) and SQSTM1-tg (SQSTM1) mice. Expression of mouse endogenous SQSTM1 and human SQSTM1 (Endogenous + SQSTM1-HA) was simultaneously detected by western blotting using anti-SQSTM1 antibody. Values shown under each lane were the signal intensities in an arbitrary unit relative to that of the liver in WT mouse. Human SQSTM1 (hSQSTM1-HA) was specifically detected by anti-HA antibody. GAPDH was used as a loading control. (PDF 795 kb)
Figure S2. Whole transverse sectional images of the lumbar spinal cord in Figs. 2, 3 and 4. a Whole transverse sectional images of fluorescence Nissl staining in the lumbar spinal cords in Fig. 2. b Whole transverse sectional images of double immunostaining with Ubiquitin (green) and SQSTM1 (red) in the lumbar spinal cord in Fig. 3. c Whole transverse sectional images of double immunostaining with MAP2 (green) and Ubiquitin (red), Ubiquitin (green) and GFAP (red), and Ubiquitin (green) and Iba1 (red) in the lumbar spinal cord in Fig. 4. a-c Scale bars = 300 μm. b, c The nuclei were counterstained with DAPI (blue). (PDF 7238 kb)
Figure S3. SQSTM1-HA positive aggregates in the anterior horn of lumbar spinal cord. a-c Representative images of double immunostaining with Ubiquitin (green) and HA (SQSTM1-HA; red) in the lumbar spinal cord (L4–5) from wild-type (WT), SQSTM1 (SQSTM1), SOD1H46R (H46R) and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk) (a), 22 wk. (b), and end-stage (H46R and SQSTM1;H46R) or 28 wk. (WT and SQSTM1) (c). The nuclei were counterstained with DAPI (blue). Scale bars = 50 μm. Ubiquitin-positive aggregates and SQSTM1-HA aggregates were observed in the anterior horn of SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 22 wk. and end-stage. Arrows indicate that ubiquitin-positive aggregates colocalizing with SQSTM1-HA. (PDF 1718 kb)
Figure S4. Immunoblot-images used for the quantitative analysis in Figs. 6a and Fig. 7. The immunoblots of (a) soluble SOD1, (b) insoluble SOD1 monomer (Mono), (c) insoluble high-molecular weight (HMW) SOD1, (d) soluble SQSTM1, (e) insoluble SQSTM1, (f) soluble poly-ubiquitinated proteins, (g) insoluble poly-ubiquitinated proteins, (h) soluble GAPDH, and (i) insoluble actin were analyzed for the quantitative analysis (Figs. 6a and 7). The spinal cord from wild-type (WT), SQSTM1 (SQSTM1), SOD1H46R (H46R), and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk), 22 wk., and end-stage (H46R and SQSTM1;H46R) or 28 wk. (WT and SQSTM1) were used. C (control sample) used as internal control indicates soluble and insoluble fractions from 22 week-old SQSTM1;SOD1H46R mouse. Asterisk represents the measured area of poly-ubiquitinated proteins. (PDF 1973 kb)
Figure S5. Immunoblot-images used for the quantitative analysis in Fig. 6b. The immunoblots of (a) soluble misfolded SOD1 and GAPDH, (b) insoluble misfolded SOD1 and actin were analyzed for the quantitative analysis (Fig. 6b). The spinal cord from SOD1H46R (H46R), and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk), 22 wk., and end-stage were used. (PDF 444 kb)
Figure S6. Immunoblot-images used for the quantitative analysis in Fig. 8. The immunoblots of (a) soluble and (b) insoluble Ser403(human)/Ser405(mouse)-phosphorylated SQSTM1 (p-S403/405), (c) soluble and (d) insoluble Ser349(human)/Ser351(mouse)-phosphorylated SQSTM1 (p-S349/351), (e) soluble LC3, (f) insoluble LC3, (g) soluble NQO1, and (h) insoluble NQO1 were analyzed for the quantitative analysis (Fig. 7). The spinal cord from wild-type (WT), SQSTM1 (SQSTM1), SOD1H46R (H46R), and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk), 22 wk., and end-stage (H46R and SQSTM1;H46R) or 28 wk. (WT and SQSTM1) were used. C (control sample) used as internal control indicates soluble and insoluble fractions from 22 week-old SQSTM1;SOD1H46R mouse. (PDF 607 kb)
Acknowledgments
We thank Dr. Makoto Urushitani at Shiga University of Medical Science for the generous gift of anti-misfolded SOD1 antibody (C4F6), and all the members of Support Center for Medical Research and Education at Tokai University for their technical help.
Funding
This study was supported by Japanese Society for Promotion of Science (JSPS) [grant numbers 26290018 and 24650189 to SH], and the National Natural Science Foundation of China (NSFC) and JSPS Bilateral Joint Research Project (to FHS and SH).
Availability of data and materials
The datasets supporting the conclusions of this article is included within the article and its Additional files.
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- AMPK
Adenosine mono-phosphate activated protein kinase
- ANOVA
Analysis of variance
- BPB
Bromophenol blue
- CAG
(C) the cytomegalovirus (CMV) early enhancer element, (A) the promoter, the first exon and the first intron of chicken beta-actin gene, and (G) the splice acceptor of the rabbit beta-globin gene
- DAPI
4′,6-diamidino-2-phenylindole dihydrochloride
- FTD
Frontotemporal dementia
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GFAP
Glial fibrillary acidic protein
- HA
Hemagglutinin
- HRP
Horseradish peroxidase
- Iba1
Ionized calcium binding adaptor molecule 1
- KEAP1
Kelch-like ECH-associated protein 1
- MAP1LC3/LC3
Microtubule-associated protein 1 light chain 3
- MAP2
Microtubule-associated protein 2
- MND
Motor neuron disease
- NFE2L2/Nrf2
Nuclear factor related erythroid 2-related factor 2
- NGS
Normal goat serum
- NQO1
NAD(P)H dehydrogenase [quinone] 1
- PAGE
Polyacrylamide gel electrophoresis
- PB
Phosphate buffer
- PBS
Phosphate-buffered saline
- PFA
Paraformaldehyde
- PVDF
Polyvinylidene difluoride
- RT
Room temperature
- SBMA
Spinal and bulbar muscular atrophy
- SDS
Sodium dodecyl sulfate
- SQSTM1
Sequestosome 1/p62
- TARDBP
TAR DNA-binding protein 43-kD
- tg
Transgenic
- UPS
Ubiquitin-proteasome system
- WT
Wild-type
Authors’ contributions
Conceived and designed the experiments: SM, AO, FHS and SH. Performed the experiments: SM, MN, SO, KS, RS and AO. Analyzed the data: SM. Contributed reagents/materials/analysis tools: HA, MA, GS and SH. Wrote the paper: SM and SH. All authors reviewed and approved the manuscript.
Ethics approval
All animal experimental procedures were carried out in accord with the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and reviewed and approved by The Institutional Animal Care and Use Committee at Tokai University.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13041-018-0373-8) contains supplementary material, which is available to authorized users.
Contributor Information
Shun Mitsui, Email: 7bmud015@mail.u-tokai.ac.jp.
Asako Otomo, Email: asako@tokai-u.jp.
Masahisa Nozaki, Email: matha113@is.icc.u-tokai.ac.jp.
Suzuka Ono, Email: 6bmud003@mail.u-tokai.ac.jp.
Kai Sato, Email: 7bmud007@mail.u-tokai.ac.jp.
Ryohei Shirakawa, Email: heyryo.s@gmail.com.
Hiroaki Adachi, Email: hiadachi@med.uoeh-u.ac.jp.
Masashi Aoki, Email: aokim@med.tohoku.ac.jp.
Gen Sobue, Email: sobueg@med.nagoya-u.ac.jp.
Hui-Fang Shang, Email: hfshang2002@163.com.
Shinji Hadano, Email: shinji@is.icc.u-tokai.ac.jp.
References
- 1.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 2.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 3.Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 5.Hirano M, Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Isono C, Miyamoto K, Akamatsu M, Mitsui Y, Kusunoki S. Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology. 2013;80:458–463. doi: 10.1212/WNL.0b013e31827f0fe5. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu H, Toyoshima Y, Shiga A, Yokoseki A, Arakawa K, Sekine Y, Shimohata T, Ikeuchi T, Nishizawa M, Kakita A, et al. Sporadic ALS with compound heterozygous mutations in the SQSTM1 gene. Acta Neuropathol. 2013;126:453–459. doi: 10.1007/s00401-013-1150-5. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Zheng ZZ, Chen X, Huang R, Yang Y, Yuan L, Pan L, Hadano S, Shang HF. SQSTM1 mutations in Han Chinese populations with sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2014;35:726. doi: 10.1016/j.neurobiolaging.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Tang L, Zhang N, Pan L, Hadano S, Fan D. Six SQSTM1 mutations in a Chinese amyotrophic lateral sclerosis cohort. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:378–384. doi: 10.3109/21678421.2015.1009466. [DOI] [PubMed] [Google Scholar]
- 9.Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70:1582–1588. doi: 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 11.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 12.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 13.Goode A, Butler K, Long J, Cavey J, Scott D, Shaw B, Sollenberger J, Gell C, Johansen T, Oldham NJ, et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy. 2016;12:1094–1104. doi: 10.1080/15548627.2016.1170257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–289. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. 2015;24:4429–4442. doi: 10.1093/hmg/ddv179. [DOI] [PubMed] [Google Scholar]
- 16.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Mizuno Y, Amari M, Takatama M, Aizawa H, Mihara B, Okamoto K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J Neurol Sci. 2006;249:13–18. doi: 10.1016/j.jns.2006.05.060. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2011;70:349–359. doi: 10.1097/NEN.0b013e3182160690. [DOI] [PubMed] [Google Scholar]
- 19.Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 20.Kato S. Amyotrophic lateral sclerosis models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:97–114. doi: 10.1007/s00401-007-0308-4. [DOI] [PubMed] [Google Scholar]
- 21.Aoki M, Ogasawara M, Matsubara Y, Narisawa K, Nakamura S, Itoyama Y, Abe K. Mild ALS in Japan associated with novel SOD mutation. Nat Genet. 1993;5:323–324. doi: 10.1038/ng1293-323. [DOI] [PubMed] [Google Scholar]
- 22.Sasaki S, Nagai M, Aoki M, Komori T, Itoyama Y, Iwata M. Motor neuron disease in transgenic mice with an H46R mutant SOD1 gene. J Neuropathol Exp Neurol. 2007;66:517–524. doi: 10.1097/01.jnen.0000263868.84188.3b. [DOI] [PubMed] [Google Scholar]
- 23.Hadano S, Otomo A, Kunita R, Suzuki-Utsunomiya K, Akatsuka A, Koike M, Aoki M, Uchiyama Y, Itoyama Y, Ikeda JE. Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS One. 2010;5:e9805. doi: 10.1371/journal.pone.0009805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doi H, Adachi H, Katsuno M, Minamiyama M, Matsumoto S, Kondo N, Miyazaki Y, Iida M, Tohnai G, Qiang Q, et al. p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J Neurosci. 2013;33:7710–7727. doi: 10.1523/JNEUROSCI.3021-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hadano S, Mitsui S, Pan L, Otomo A, Kubo M, Sato K, Ono S, Onodera W, Abe K, Chen X, et al. Functional links between SQSTM1 and ALS2 in the pathogenesis of ALS: cumulative impact on the protection against mutant SOD1-mediated motor dysfunction in mice. Hum Mol Genet. 2016;25:3321–3340. doi: 10.1093/hmg/ddw180. [DOI] [PubMed] [Google Scholar]
- 26.Urushitani M, Ezzi SA, Julien JP. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104:2495–2500. doi: 10.1073/pnas.0606201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan L, Yoshii Y, Otomo A, Ogawa H, Iwasaki Y, Shang HF, Hadano S. Different human copper-zinc superoxide dismutase mutants, SOD1G93A and SOD1H46R, exert distinct harmful effects on gross phenotype in mice. PLoS One. 2012;7:e33409. doi: 10.1371/journal.pone.0033409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 29.Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurosawa M, Matsumoto G, Sumikura H, Hatsuta H, Murayama S, Sakurai T, Shimogori T, Hattori N, Nukina N. Serine 403-phosphorylated p62/SQSTM1 immunoreactivity in inclusions of neurodegenerative diseases. Neurosci Res. 2016;103:64–70. doi: 10.1016/j.neures.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Lystad AH, Simonsen A. Assays to monitor aggrephagy. Methods. 2015;75:112–119. doi: 10.1016/j.ymeth.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 32.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 33.Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, Piersaint JA, Tapia JC, Rich MM, Maniatis T. Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc Natl Acad Sci U S A. 2017;114:E8294–EE303. doi: 10.1073/pnas.1704294114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perera ND, Sheean RK, Lau CL, Shin YS, Beart PM, Horne MK, Turner BJ. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy. 2018;14:534–551. doi: 10.1080/15548627.2017.1385674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tashiro Y, Urushitani M, Inoue H, Koike M, Uchiyama Y, Komatsu M, Tanaka K, Yamazaki M, Abe M, Misawa H, et al. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem. 2012;287:42984–42994. doi: 10.1074/jbc.M112.417600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olivan S, Calvo AC, Gasco S, Munoz MJ, Zaragoza P, Osta R. Time-point dependent activation of autophagy and the UPS in SOD1G93A mice skeletal muscle. PLoS One. 2015;10:e0134830. doi: 10.1371/journal.pone.0134830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coughlan KS, Halang L, Woods I, Prehn JH. A high-fat jelly diet restores bioenergetic balance and extends lifespan in the presence of motor dysfunction and lumbar spinal cord motor neuron loss in TDP-43A315T mutant C57BL6/J mice. Dis Model Mech. 2016;9:1029–1037. doi: 10.1242/dmm.024786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coughlan KS, Mitchem MR, Hogg MC, Prehn JH. “Preconditioning” with latrepirdine, an adenosine 5′-monophosphate-activated protein kinase activator, delays amyotrophic lateral sclerosis progression in SOD1(G93A) mice. Neurobiol Aging. 2015;36:1140–1150. doi: 10.1016/j.neurobiolaging.2014.09.022. [DOI] [PubMed] [Google Scholar]
- 39.Harada H, Warabi E, Matsuki T, Yanagawa T, Okada K, Uwayama J, Ikeda A, Nakaso K, Kirii K, Noguchi N, et al. Deficiency of p62/Sequestosome 1 causes hyperphagia due to leptin resistance in the brain. J Neurosci. 2013;33:14767–14777. doi: 10.1523/JNEUROSCI.2954-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamanaka K, Boillee S, Roberts EA, Garcia ML, McAlonis-Downes M, Mikse OR, Cleveland DW, Goldstein LS. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc Natl Acad Sci U S A. 2008;105:7594–7599. doi: 10.1073/pnas.0802556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schema of the transgene construct for SQSTM1-tg mouse and the distribution of SQSTM1 in tissues. a Schematic diagram of the transgene cassette encoding the C-terminally hemagglutinin (HA)-tagged human SQSTM1 cDNA. Transgene construct consists of cytomegalovirus enhancer (E), chicken β-actin promoter (Pro), rabbit β-globin splice acceptor (S), full length human SQSTM1 cDNA (SQSTM1), hemagglutinin tag (HA), and rabbit β-globin poly A (Poly A). b Expression and distribution of SQSTM1 in the liver, skeletal muscle, olfactory bulb, cerebral cortex, hippocampus, cerebellum, and spinal cord in wild-type (WT) and SQSTM1-tg (SQSTM1) mice. Expression of mouse endogenous SQSTM1 and human SQSTM1 (Endogenous + SQSTM1-HA) was simultaneously detected by western blotting using anti-SQSTM1 antibody. Values shown under each lane were the signal intensities in an arbitrary unit relative to that of the liver in WT mouse. Human SQSTM1 (hSQSTM1-HA) was specifically detected by anti-HA antibody. GAPDH was used as a loading control. (PDF 795 kb)
Figure S2. Whole transverse sectional images of the lumbar spinal cord in Figs. 2, 3 and 4. a Whole transverse sectional images of fluorescence Nissl staining in the lumbar spinal cords in Fig. 2. b Whole transverse sectional images of double immunostaining with Ubiquitin (green) and SQSTM1 (red) in the lumbar spinal cord in Fig. 3. c Whole transverse sectional images of double immunostaining with MAP2 (green) and Ubiquitin (red), Ubiquitin (green) and GFAP (red), and Ubiquitin (green) and Iba1 (red) in the lumbar spinal cord in Fig. 4. a-c Scale bars = 300 μm. b, c The nuclei were counterstained with DAPI (blue). (PDF 7238 kb)
Figure S3. SQSTM1-HA positive aggregates in the anterior horn of lumbar spinal cord. a-c Representative images of double immunostaining with Ubiquitin (green) and HA (SQSTM1-HA; red) in the lumbar spinal cord (L4–5) from wild-type (WT), SQSTM1 (SQSTM1), SOD1H46R (H46R) and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk) (a), 22 wk. (b), and end-stage (H46R and SQSTM1;H46R) or 28 wk. (WT and SQSTM1) (c). The nuclei were counterstained with DAPI (blue). Scale bars = 50 μm. Ubiquitin-positive aggregates and SQSTM1-HA aggregates were observed in the anterior horn of SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 22 wk. and end-stage. Arrows indicate that ubiquitin-positive aggregates colocalizing with SQSTM1-HA. (PDF 1718 kb)
Figure S4. Immunoblot-images used for the quantitative analysis in Figs. 6a and Fig. 7. The immunoblots of (a) soluble SOD1, (b) insoluble SOD1 monomer (Mono), (c) insoluble high-molecular weight (HMW) SOD1, (d) soluble SQSTM1, (e) insoluble SQSTM1, (f) soluble poly-ubiquitinated proteins, (g) insoluble poly-ubiquitinated proteins, (h) soluble GAPDH, and (i) insoluble actin were analyzed for the quantitative analysis (Figs. 6a and 7). The spinal cord from wild-type (WT), SQSTM1 (SQSTM1), SOD1H46R (H46R), and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk), 22 wk., and end-stage (H46R and SQSTM1;H46R) or 28 wk. (WT and SQSTM1) were used. C (control sample) used as internal control indicates soluble and insoluble fractions from 22 week-old SQSTM1;SOD1H46R mouse. Asterisk represents the measured area of poly-ubiquitinated proteins. (PDF 1973 kb)
Figure S5. Immunoblot-images used for the quantitative analysis in Fig. 6b. The immunoblots of (a) soluble misfolded SOD1 and GAPDH, (b) insoluble misfolded SOD1 and actin were analyzed for the quantitative analysis (Fig. 6b). The spinal cord from SOD1H46R (H46R), and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk), 22 wk., and end-stage were used. (PDF 444 kb)
Figure S6. Immunoblot-images used for the quantitative analysis in Fig. 8. The immunoblots of (a) soluble and (b) insoluble Ser403(human)/Ser405(mouse)-phosphorylated SQSTM1 (p-S403/405), (c) soluble and (d) insoluble Ser349(human)/Ser351(mouse)-phosphorylated SQSTM1 (p-S349/351), (e) soluble LC3, (f) insoluble LC3, (g) soluble NQO1, and (h) insoluble NQO1 were analyzed for the quantitative analysis (Fig. 7). The spinal cord from wild-type (WT), SQSTM1 (SQSTM1), SOD1H46R (H46R), and SQSTM1;SOD1H46R (SQSTM1;H46R) mice at 16 weeks of age (wk), 22 wk., and end-stage (H46R and SQSTM1;H46R) or 28 wk. (WT and SQSTM1) were used. C (control sample) used as internal control indicates soluble and insoluble fractions from 22 week-old SQSTM1;SOD1H46R mouse. (PDF 607 kb)
Data Availability Statement
The datasets supporting the conclusions of this article is included within the article and its Additional files.