

Abstract
Unknown fifteen years ago, proprotein convertase subtilisin/kexin type 9 (PCSK9) is now common parlance amongst scientists and clinicians interested in prevention and treatment of atherosclerotic cardiovascular disease. What makes this story so special is not its recent discovery nor the fact that it uncovered previously unknown biology, but rather that these important scientific insights have been translated into an effective medical therapy in record time. Indeed, the translation of this discovery to novel therapeutic serves as one of the best examples of how genetic insights can be leveraged into intelligent target drug discovery. The PCSK9 saga is unfolding quickly but is far from complete. Here, we review major scientific understandings as they relate to the role of PCSK9 in lipoprotein metabolism and atherosclerotic cardiovascular disease and the impact that therapies designed to inhibit its action are having in the clinical setting.
Keywords: Proprotein convertase subtilisin/kexin type 9 (PCSK9), PCSK9 inhibitors, low-density lipoprotein (LDL), LDL receptor (LDLR), familial hypercholesterolemia (FH), atherosclerotic cardiovascular disease, dyslipidemia, clinical trials, clinical management guidelines
Introduction
It has only been fifteen years since proprotein convertase subtilis/kexin type 9 (PCSK9) was identified as an important regulator of low-density lipoprotein (LDL) metabolism. As its name suggests, PCSK9 is the ninth member of the proprotein convertase family, a group of serine proteases that are characterized by their ability to hydrolyze peptide bonds in their cognate substrates for activation 1. Initial clues were provided by a French family with familial hypercholesterolemia in 2003 2. Abifadel et al. linked gain-of-function (GOF) mutations in PCSK9 with autosomal dominant hypercholesterolemia and ultimately uncovered a key new player in lipid metabolism. This seminal discovery led to a series of investigations that demonstrated that loss-of-function (LOF) mutations in PCSK9 associate with life-long low cholesterol levels and marked reductions in the risk of atherosclerotic cardiovascular disease (ASCVD) 3-6. The very rare individuals with homozygous LOF mutations in PCSK9 (and no circulating protein) demonstrated extremely LDL-cholesterol (LDL-C) [≈15 mg/dL], normal health and reproductive capacity, and no evidence of neurological or cognitive dysfunction 5, 7. This complementary set of observations has been leveraged into the most important therapy for the treatment of hypercholesterolemia and ASCVD since the introduction of the statins over thirty years ago. Indeed, the so-called PCSK9 inhibitors, fully human monoclonal antibodies that bind PCSK9, reduce LDL-C by approximately 60% and risk of myocardial infarction (MI) and stroke by approximately 20% after over two years of treatment 8. Remarkably, these agents antagonizing PCSK9 action were approved by regulatory agencies spanning the globe only a decade after its discovery. While the scientific and medical communities have swiftly uncovered many facets of PCSK9 biology, there is still much to learn. Here we survey the most salient aspects of PCSK9 biology and therapeutic modulation as it relates to lipoprotein metabolism, atherosclerosis, and prevention of atherosclerotic cardiovascular events.
A new player in cholesterol homeostasis
Much of the excitement surrounding the discovery of PCSK9 relates to revelations in lipoprotein metabolism, necessitating a reworking of models previously held for decades. In the pre-PCSK9 era, it was thought that all regulatory systems of cholesterol homeostasis were strictly intracellular 9, with the role played by extracellular proteins limited to modulation of packaging, processing, and clearance of plasma lipoproteins. Remarkably, PCSK9 impacts lipoprotein metabolism both within (prior to secretion) and outside (after secretion into the circulation) of the cell 10. Plasma cholesterol is mostly manufactured, exported, and eventually recaptured by hepatocytes. Cholesterol synthesis is a complex, multistep, and highly regulated pathway, and 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG CoA-R) is its key rate-limiting enzyme. Statins antagonize the activity of HMG CoA-R, reduce hepatic cholesterol synthesis, and up-regulate the transcription of the LDL receptor (LDLR) gene via a sensing mechanism operated by the sterol regulatory element binding protein (SREBP) pathway 11. Each cell in the body must maintain membrane cholesterol at a critical concentration in order to ensure proper membrane function. It is thus evident that the cell uses a series of regulatory and counter- regulatory steps to respond to increases and decreases in membrane cholesterol straying from the critical value range. These include synthetic, assembly, secretory, and re-uptake activities. The lipid cargo, mostly triglycerides and cholesterol, is packaged within apolipoprotein B (apoB)-containing very low-density lipoproteins (VLDL), the intravascular precursors of LDL, which primarily transport triglycerides from the liver to peripheral tissues, with cholesterol packaged to enhance stability.
The mechanism by which LDLR internalize LDL was described by Goldstein and Brown in the early 1970s 12, leading to one of the most exciting series of discoveries in the history of medicine and the cataloguing of a critical aspect of cellular life, the sensing and regulation of membrane cholesterol levels. Receptor mediated endocytosis is facilitated by the binding of apoB on the LDL particle to the LDLR and coordinated by an adaptor protein (LDLRAP) that positions LDLR on the sinusoidal side of the polarized hepatocyte, clustered in coated pits 13. The LDL/LDLR complex then gets internalized within coated vesicles and expands to join the endosomal compartment, where it eventually merges with the lysosome. The pH gradient in the descent toward the lysosome induces dissociation between receptor and cargo. In the lysosome, the LDL particle is digested and the cholesterol and triglycerides are de-esterified for transport into the cytosol, where they can take on myriad fates. On the other hand, the LDLR is recycled back to the hepatocyte surface to participate for many more rounds of LDL binding and endocytosis 14, 15. LDLR recycles every 10 minutes with a life span of 20 hours. This process allows a single LDLR to internalize hundreds of LDL particles during its lifespan. For decades, it was assumed that a generic ubiquitin-related sorting of altered molecules eventually terminated this recycling process. The discovery of PCSK9 heralded a new era of understanding; this low abundance circulating protein binds cell-surface LDLR on coated pits and triggers the internalization of the receptor. The interaction between PCSK9 and LDLR locks the receptor in its “open” conformation in the endosome and routes the ligand-receptor pair to the lysosomal compartment for degradation, thus inhibiting the LDLR recycling that follows internalization of ligands such apoB on LDL and apoE on remnants 16-19. In other words, the normal recycling loop is short-circuited as PCSK9 disables the LDLR from escaping lysosomal digestion, thereby reducing cell surface receptor density and resulting in raised plasma LDL-C.
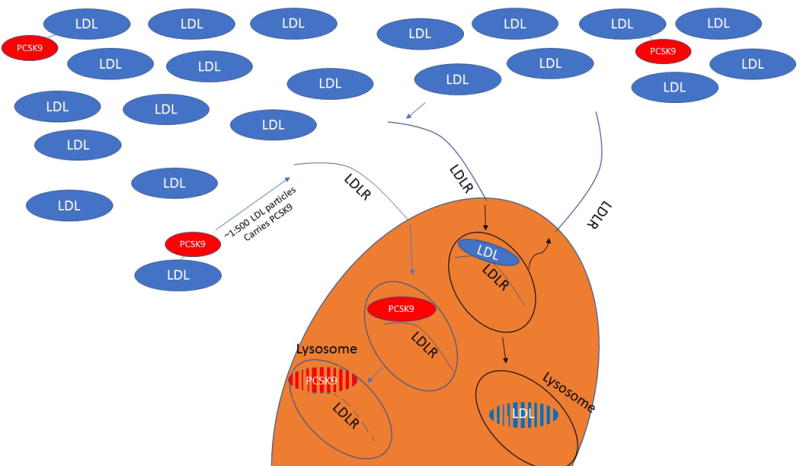
It must be noted that up to half of plasma PCSK9 is associated with the LDL particle, for a frequency of one PCSK9 molecule for every 500–1000 LDL particles 20, 21. This introduces the intriguing possibility that the carefully orchestrated cellular regulation of cholesterol concentration is ultimately under the control of a stochastic extracellular system, where every few hundred encounters with canonical LDL, the LDLR meets its fate by interacting with a PCSK9-carrying LDL that terminates its life cycle (Figure 1).
Figure 1. Stochastic interaction between the LDLR and LDL-bound PCSK9 terminate the receptor life.
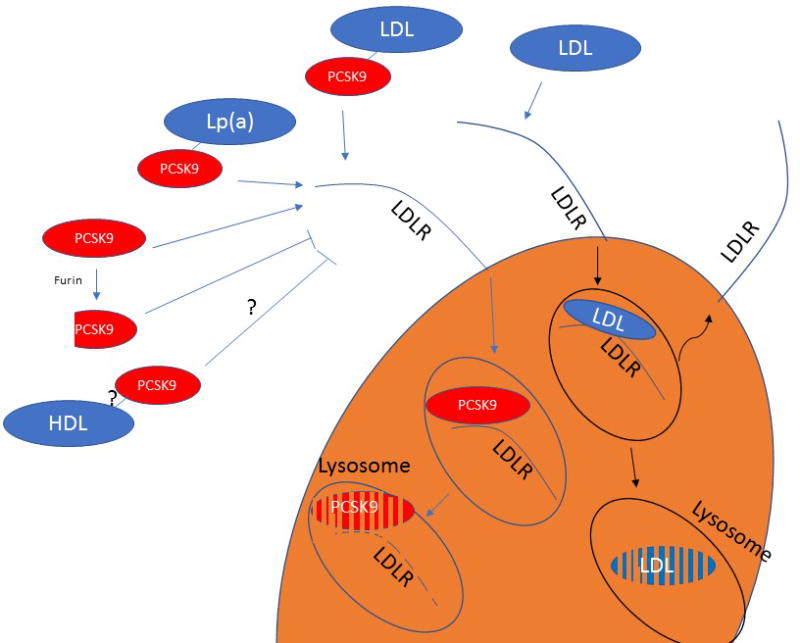
Before the discovery of PCSK9 it was understood that the LDLR can recycle hundreds of time in its ~20-hours life span. Since PCSK9 is found on one in every 500-1000 LDL particle, one can envision a scenario where one in every 500 encounters, an LDLR binds to an LDL particle harboring a molecule of PCSK9. Such a stochastic interaction will then lead to the degradation of the receptor rather to its recycling, explaining, at least in part, why LDLR recycle hundreds of times.
LDLR = low-density lipoprotein receptor; LDL = low-density lipoprotein; PCSK9 = proprotein convertase subtilisin/kexin type 9
PCSK9 biology
Structure
PCSK9 is synthesized predominantly in the liver as a 75 kDa proprotein. Based on its protein structure 22-24, removal of the signal peptide (amino acids 1-30) produces a secreted heterodimer protein with three domains:
A pro-domain (amino acids 31-152), which undergoes autocatalytic cleavage but continues to associate with the rest of the protein.
A catalytic domain (amino acids 153-454), which contains a proteolytic active site (catalytic triad amino acids 186, 226, and 386) inactivated by the associated pro-domain. The proteolytic active site is only required for the autocatalytic cleavage, has no other known targets, and is not related to the LDLR reducing activity of PCSK9 25, 26. The LDLR degradation capacity of PCSK9 is based on a protein-protein interaction between the epidermal growth factor-like A domain of the LDLR and amino acids 367-380 in the catalytic domain region of PCSK9 independent of the proteolytic active site.
A C-terminal domain (residues 455-692), which consists of three similar modules. Most of the N-terminal residues and some of the C-terminal residues are not visible in the proposed crystal structures due to poor electron density.
LDLR degradation and PCSK9 kinetics
The kinetics of PCSK9 binding to cell surface LDLR exhibit Kd values that range from 90 to 840 nM at neutral pH. Its affinity for the LDLR is increased by two orders of magnitude at lower pH with Kd values ranging from 1 to 8 nM 22, 24, 27, 28. The increased affinity at acidic pH facilitates PCSK9 capture of the LDLR in the late endosome and ensures that the PCSK9-LDLR complex will be targeted to the lysosome for degradation 29. PCSK9 binding to LDLR occurs in two phases:
Rapid-phase binding - accounts for one-third of overall equilibrium binding and is characterized by a binding half-time of 5-10 minutes and half-time dissociation of 20 minutes 30.
Slow-phase binding - accounts for two-thirds of overall equilibrium binding and is characterized by a binding half-time of ~1.5 hours and half-time dissociation of ~5 hours 30.
Although PCSK9-LDLR binding, internalization, and lysosomal shuttling occurs within 2-3 hours from initial contact 31, PCSK9-mediated degradation of LDLR in vitro is only evident 12-24 hours after adding PCSK9 to cultured cells 32, 33. In mice, PCSK9 remains intact in the liver for at least 4 hours after its LDLR-mediated internalization 20. Therapeutic PCSK9 inhibition in humans only significantly reduces LDL-C levels after 2-3 days from start of therapy34. Based on these observations, it is clear that there is a delay between the first PCSK9 interaction with the LDLR and the eventual loss of LDLR. In contemplating this apparent paradox, there are several possibilities to consider. First, the PCSK9-LDLR interaction may not lead to immediate shuttling of both proteins to the lysosome for degradation and may require additional steps and interactions. Alternatively, intracellular LDLR concentrations may be in vast excess relative to cell-surface LDLR density 35, 36, so that the initial elimination of cell-surface LDLR by PCSK9 is rapidly replenished until intracellular stores are also depleted.
Upon synthesis, the PCSK9 proprotein is directed to the endoplasmic reticulum by a signal sequence, subsequently removed by a signal peptidase. The proprotein form then undergoes autocatalytic cleavage, forming a heterodimer (62+13 kDa). This form is then transported to the Golgi via a COPII complex involving Sec24a 37, leading to PCSK9 secretion into the blood stream. Even though PCSK9 and LDLR co-exist within the secretory pathway of hepatocytes and their co-localization was suggested to result in reduced LDLR levels, it was later demonstrated that the interaction does lead to LDLR degradation 38,39 40. Binding of PCSK9 to GRP94, an ER-resident protein expressed in hepatocytes, protects LDLR from degradation by preventing early binding of PCSK9 to LDLR within the ER 38. Several LOF mutations in PCSK9 are known to cause impaired processing (e.g., S386A) 41, trafficking (e.g. R46L) 42, or secretion (e.g. S462P) 43 of PCSK9, leading to low plasma PCSK9 levels and consequent hypocholesterolemia. Interestingly, some of the GOF mutations of PCSK9 causing hyperlipidemia are also not secreted (e.g. S127R, D129G) 42. It is possible that these mutations are able to interact with LDLR in the secretory pathway leading the complex to degradation, bypassing GRP94 protection in the ER.
Regulation of PCSK9 levels and function
As for all plasma proteins, PCSK9 levels represent the balance between production and clearance. Human PCSK9 is expressed in multiple tissues, with liver, small intestine, and kidney the major sources of its plasma levels 44. The human protein shares substantial homology with its murine counterpart, with 76.6% identity 45. The gene is found on chromosome 1 in humans and chromosome 4 in mice. PCSK9 is regulated by the SREBP through a sterol-regulatory element (SRE) motif in the promoter region 46, 47. In addition, an Sp1 motif also controls transcriptional regulation through the SREBP pathway 48. SREBP regulation of cholesterol synthesis, LDLR, and PCSK9 results in an apparently paradoxical scenario where depletion of intracellular cholesterol levels, leads to the simultaneous up-regulation of both LDLR and PCSK9 expression 48, 49. This SRE-mediated up-regulation of PCSK9 attenuates the LDL-C lowering effect of medications such as statins and ezetimibe. The PCSK9 promoter also contains a hepatic nucleic factor 1 motif (between the SRE and Sp1 sites), which likely functions as a liver-specific regulatory sequence 50-52. Metabolic studies in humans have shown that the production rate of PCSK9 is ~20 µg/kg/day with a plasma pool size of ~1000 µg 53. Thus, given a blood pool of 5 liters, the average plasma concentration of PCSK9 is ~200 ng/ml in an average adult, and the plasma pool turns over very fast, by over 2 pools a day 53. For comparison, the production rate of VLDL-apoB is 1000-fold higher (~20 mg/kg/day) 53.
At a high level, it would appear that the primary role of PCSK9 is to carry out a suicide mission that ultimately leads to the demise of the LDLR. However, careful study of PCSK9 physiology shows a less threatening picture. PCSK9 is simply another ligand for the LDLR (just like apoB and apoE) and uses the LDLR to exit the plasma compartment. However, if PCSK9 uses LDLR as the main clearance route of elimination and at the same time it causes degradation of LDLR, then a reciprocal regulation between these two proteins controls plasma PCSK9 levels, hepatic LDLR expression, and plasma LDL-C levels 20, 54, rendering the evaluation of these inextricably linked processes at any given time in the steady state extremely difficult, as changes in production or efficiency in any of the two proteins will have consequences on the other and on LDL-C levels. In mice, plasma PCSK9 levels are highly regulated by LDLR expression 20, 55. In mice, complete removal of the LDLR results in a substantial increase in the plasma half-life of PCSK9 56 whereas overexpression of hepatic LDLR results in increased clearance of PCSK9 20. Humans with homozygous or heterozygous familial hypercholesterolemia (FH) manifest higher levels of plasma PCSK9 compared to non-FH controls 54. Strangely, LDLR mutations in humans have a larger effect on LDL-C than on PCSK9 concentrations, while the opposite happens in mice 57. One possible explanation is the human LDLR mutations are classified as either receptor-defective or receptor-negative (<2% of normal LDL internalization ability), with receptor-defective (2-25% of normal LDL internalization ability) mutations being far more common 58, 59. Thus, the most human LDLR mutants have some residual activity toward binding both LDL and PCSK9, while studies mice are done in the context of a complete absence of the LDLR.
Plasma PCSK9 can be found in two main forms, an intact heterodimer (62+13 kDa), which is often considered the (more) active form (stronger binding to and degradation of LDLR), and a furin-cleaved heterodimer (55+13 kDa) 60, which binds the LDLR less avidly (two-fold reduced affinity) 61, and is thus considered the less active form61-63. In contrast, intracellular PCSK9 is only found in its proprotein form (75 kDa) or as an intact heterodimer ready to be secreted 64. These findings suggest that the cleavage of PCSK9 by furin occurs outside the cell, by interaction of PCSK9 with either membrane-bound or circulating furin. Although PCSK9 and furin co-exist in the Golgi, there is no clear evidence that furin is able to cleave PCSK9 intracellularly 60. Another direct regulator of PCSK9 function in plasma is the LDL particle itself 65, 66 It has been shown that PCSK9 associates with LDL with a Kd in the range of 160 to 320 nM 67, 68. PCSK9 association with LDL is thought to occur via apoB and requires the presence of the PCSK9 pro-domain 67. We also found that plasma PCSK9 associates with Lp(a), although it does not bind to other apoB-containing lipoproteins such as VLDL or chylomicrons 69. The in vivo relevance of PCSK9 association with LDL and Lp(a) was first shown in patients undergoing lipoprotein apheresis, where together with a ~70% reduction in LDL levels, plasma PCSK9 levels were also reduced by over 50% 70. Nevertheless, the physiologic role of PCSK9 binding to lipoproteins is not clear. In vivo data suggest that the PCSK9 species associated with LDL is primarily the intact heterodimer form, whereas the non-LDL-bound (free) PCSK9 is mainly found in the furin-cleaved conformation 70. Our in vivo and in vitro data further suggest that LDL “protects” PCSK9 from furin cleavage and that LDL-bound PCSK9 has a two-fold stronger binding affinity for the LDLR compared with non-LDL-bound PCSK9 71. These observations support the notion that LDL-bound PCSK9 is the more functional form of this protein. Furthermore, the LDL-PCSK9 interaction has therapeutic potential as its modulation may lead to increased proteolytic cleavage and reduced plasma PCSK9 activity. Compartmentalization of PCSK9 and its effects on PCSK9 activity are summarized in Figure 2.
Figure 2. PCSK9 compartmentalization and function in plasma.
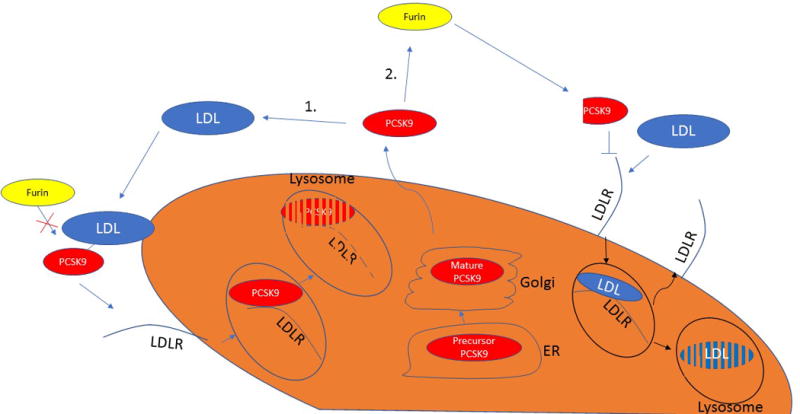
(A) PCSK9 is found in plasma in primarily two monomeric forms; an active form representing the full-length plasma protein and an inactive/less-active shorter fragment, which is a cleavage product of the full length protein by the protease furin. The active PCSK9 is found predominantly on LDL and Lp(a) particles, but not on VLDL or chylomicron remnants. In contrast, the furin-cleaved PCSK9 is not found in association with these apoB-lipoproteins. While it is not clear whether PCSK9 (active or furin cleaved) is found in association with HDL, it was suggested the HDL can inhibit PCSK9 function. (B) PCSK9 is secreted as an active form representing the full-length plasma protein. Upon secretion, PCSK9 can take on one of two fates, which ultimately determines its function. (i) PCSK9 can interact with an LDL particle, which protects PCSK9 from being cleaved by furin and leaves the protein bound to the particle in its active form, or alternatively, (ii) PCSK9 can interact with furin, which leads to the formation of a shorter fragment of PCSK9 that exhibits at least two-fold lower affinity to LDLR with limited ability/inability to degrade it.
PCSK9 = proprotein convertase subtilisin/kexin type 9; LDL = low-density lipoprotein; Lp(a) = lipoprotein(a); VLDL = very low-density lipoprotein; apoB = apolipoprotein B; HDL = high-density lipoprotein; LDLR = low-density lipoprotein receptor
On the other hand, it was shown that adding LDL to recombinant PCSK9 in vitro leads to reduced affinity of PCSK9 for the LDLR, likely due to competition of LDL and PCSK9 for the LDLR 67, 71. Thus, it is possible that LDL-bound PCSK9 is a physiologically less functional fraction of plasma PCSK9 72. The importance of defining and elucidating the factors that govern the partitioning of plasma PCSK9 and understanding the physiological role of the LDL-bound PCSK9 cannot be overstated. Experimental and translational studies will ultimately determine whether this partitioning in general, and the LDL-PCSK9 interaction in particular, can be exploited for therapeutic gains.
Receptors and other partner proteins
Immediately following the discovery of LDLR-mediated degradation by PCSK9, it was found that this protein interacts with other proteins in both hepatic and extra-hepatic tissues. Similar to its interaction with the LDLR, PCSK9 may engage other members of the LDLR family, such as the VLDL receptor 10, apoE receptor 2 (apoER2) 10, and LDLR-related protein 1 (LRP1) 73. However, it is possible that PCSK9 interaction with these receptors does not lead to their degradation, or at least not in all tissues 74, 75, and that, perhaps, its interaction with LRP1 and apoER2 leads to downstream signaling instead.
The surface scavenger receptor CD36 76 and the tetraspanin receptor CD81 77 were also proposed as possible targets of PCSK9. The nature of these interactions remains unknown, as these proteins do not share homology with the LDLR. CD36 is a scavenger receptor that also plays a role in muscle lipid utilization, adipose energy storage, and hepatic triglyceride storage and secretion. Thus, systemic modulation of its levels by PCSK9 may have implications on basic human physiology. CD81 plays a role in the regulation of cell development, activation, growth and motility and also serves as an important receptor for hepatitis C virus entry to the cell.
PCSK9 has also been reported to interact with proteins other than receptors, as it reduces levels of beta-secretase 1 78, an aspartic-acid protease responsible for the proteolytic processing of the amyloid precursor protein, and endothelial sodium channel 79, a membrane-bound ion channel that plays a major role in determining extracellular fluid osmolality. Amyloid-like protein 2 80 and Annexin A2 81 may affect the formation and processing of the PCSK9-LDLR complex though interaction with the C-terminal domain of PCSK9. In addition, it was shown that the adrenal cells are insensitive to LDLR-mediated degradation by PCSK9 independently of Annexin A2 levels 20.
Heparan sulfate proteoglycans (HSPG) play an important role in hepatic metabolism through several mechanisms including endocytosis of lipoproteins 82. It was recently suggested that HSPG lining the hepatocyte surface capture PCSK9 bound to the LDLR and heparin mimetics appear to have PCSK9 inhibitor activity 82. In order to understand the potential role of HSPG on PCSK9, it remains to be determined whether HSPG knock down in vivo (e.g through hepatic Ndst1 knockout 83) affects PCSK9-mediated LDLR degradation, PCSK9 turnover, and/or PCSK9 levels.
PCSK9 as a biomarker to predict ASCVD risk
PCSK9 and Atherosclerosis Imaging
Since PCSK9 is a circulating protein, it could have direct effects on the plaque beyond its ability to regulate hepatic LDLR levels. Several lines of investigation have also explored the relationship between plasma PCSK9 and subclinical atherosclerosis. Chan et al. performed carotid intima-media wall thickness (CIMT) measurements in 295 asymptomatic subjects and found a significant and direct relationship between PCSK9 levels and carotid thickness 84. These findings corroborated earlier studies on the correlation between PCSK9 and CIMT 85-87. However, a sub-analysis of the FATE (Firefighters and Their Endothelium) study found no relationship between PCSK9 levels and measures of subclinical atherosclerosis (CIMT and flow-mediated dilation) in 1,527 middle-aged men free of vascular disease 88. The utility of serial CIMT as a means to evaluate the association of serum PCSK9 with progression of carotid plaque is limited. However, Xie et al. performed serial studies in 643 Chinese healthy participants from the general population as part of the Chinese Multi-provincial Cohort Study 89 and found a statistically significant relationship between plasma PCSK9 concentration and progression of atherosclerosis as reflected by total plaque area, independent of plasma LDL-C concentration.
The coronary artery calcium (CAC) score has also been utilized as a means to investigate the relationship of PCSK9 to coronary atherosclerosis. Alonso et al. evaluated 161 genetically confirmed FH patients who underwent CAC scoring 90 and found that serum PCSK9 concentrations independently predicted the extent of CAC. Importantly, after full adjustment, only PCSK9 and apolipoprotein(a) [apo(a)] remained predictive of CAC in this cohort of asymptomatic FH patients. Similarly, Zhao et al. evaluated the association of plasma PCSK9 concentrations and CAC in 403 untreated patients presenting with chest pain and found that PCSK9 was independently associated with CAC 91.
The relationship of circulating PCSK9 concentration to atherosclerosis has also been explored in the context of invasive diagnostic imaging as well. Cheng et al. utilized intravascular ultrasound (IVUS) virtual histology to evaluate the association of serum PCSK9 levels and necrotic core within coronary atherosclerotic lesions in subjects with known CAD confirmed at the time of angiography 92. The results demonstrated a direct linear relationship between plasma PCSK9 and the necrotic core fraction in coronary plaque. Importantly, this endpoint remained significant in all subgroups, independently of LDL-C levels and use of statins. These observational studies cannot provide insight into the directionality of the association; thus, many questions remain. Can plasma PCSK9 influence atherogenesis through direct effect on endothelial cells or by direct transit into the subendothelial space? Does PCSK9 production within the plaque ultimately influence plasma levels? Or, is the association of these two factors mediated through other pathways entirely?
Epidemiologic studies
Beyond evaluations of the relationship between PCSK9 and subclinical atherosclerosis, a number of clinical investigations have explored PCSK9 as a biomarker of atherosclerotic risk in both primary and secondary prevention. Four major studies in primary prevention examined this issue. In the FATE study, 1,527 middle aged firefighters who were free of vascular disease at baseline were followed longitudinally for a mean of 7.2 ± 1.7 years 88. While plasma PCSK9 concentration correlated with LDL-C, insulin, and triglyceride levels, it did not correlate with cardiovascular events. As a sub-analysis of the Women’s Health Study, Ridker et al. performed a nested case–control evaluation from a prospective cohort of 28,000 initially healthy American women over the age of 45 years old and not on statin therapy 93. Plasma PCSK9 was measured at baseline among 358 cases (MI, ischemic stroke, CV death) and 358 controls matched for age, smoking, and hormone replacement therapy (women who remained free of cardiovascular disease during 17 years of follow-up). While there was a modest, positive association between PCSK9 level and apoB and triglycerides, no difference was seen in median PCSK9 concentrations in cases vs. controls (304.4 ng/mL vs. 299.7 ng/mL). Moreover, baseline apoB levels predicted incident cardiovascular events but baseline PCSK9 levels did not. Leander et al. prospectively evaluated 4,232 apparently healthy 60-year-old men and women living in Stockholm County to investigate the correlation between PCSK9 and future cardiovascular events (composite primary outcome of fatal or non-fatal MI, angina, chronic ischemic heart disease, sudden cardiac death, and fatal or non-fatal ischemic stroke) 94. During the 15 years of follow-up, the cumulative incidence of the primary outcome was 13%. Consistent with other studies, they noted a modest relationship between plasma PCSK9 and LDL-C (r=0.18, p<0.0001) and triglyceride levels (r=0.12, p<0.0001). Unlike the Ridker study, they observed a significant direct relationship between quartiles of PCSK9 concentration and incident cardiovascular events. However, incorporation of plasma PCSK9 levels into their clinical risk prediction model did not lead to significant improvement in discrimination or net reclassification, metrics that relate to clinical utility. A provocative sub-analysis suggested that subjects with discordant PCSK9 and LDL-C levels (i.e., high PCSK9 – low LDL-C) had the highest future hazard of the primary outcome, even compared to those with high PCSK9 – high LDL-C 94. More recently, Laugsand et al. evaluated the utility of PCSK9 as a circulating biomarker for prediction of incident MI in a nested case–control evaluation from a prospective cohort of a general population sample in Norway (1,488 cases vs. 3,1819 controls, 11.1 years of follow-up) 95. Risk of MI was 47% higher in those subjects in the highest quartile of PCSK9 relative to those in the lowest quartile after adjustment for age and sex. However, when adjusting for LDL-C, the relationship no longer remained significant.
Results from secondary prevention studies are equally non-definitive. Werner et al. prospectively tested whether fasting serum PCSK9 concentration predict cardiovascular events in 504 patients with stable coronary artery disease on background statin therapy with well-controlled LDL-C levels 96. Although serum PCSK9 levels predicted atherosclerotic events the association was lost when adjusting for fasting triglyceride levels. Similarly, Li et al. followed 616 Chinese subjects with stable coronary artery disease for 17 months to assess the relationship between PCSK9 levels and atherosclerotic cardiovascular disease 97. An association was found between PCSK9 concentrations and the severity of coronary artery disease by SYNTAX score. Additionally, at 17 months, cardiovascular event rates were higher in those with higher PCSK9 levels, though this relationship was noted only amongst those subjects who did not receive coronary revascularization. Finally, Gencer et al. evaluated this issue in an acute coronary syndrome (ACS) cohort. They assayed plasma PCSK9 in 2,030 individuals presenting with ACS and undergoing coronary angiography 98. Plasma PCSK9 levels correlated with measures of inflammation, lipid-lowering therapy, and the clinical onset of ACS, but did not predict mortality at one year.
Based on the above observational trials in primary and secondary prevention, no firm conclusions regarding plasma measures of PCSK9 as a predictor of future cardiovascular events can be drawn. On that basis, a number of systematic reviews and meta-analysis have been performed in the hopes of shedding further light on this relationship 99-101. In three meta-analyses, when PCSK9 concentration was considered as a categorical value, the highest category of PCSK9 was associated with cardiovascular outcomes. However, when treated as a continuous variable, disparate results emerge. Part of the challenge in evaluating this issue has to do with differences in study design, clinical outcomes, and measurement methods for PCSK9. Based on the available data, clinical measurement of plasma PCSK9 for CVD risk prediction or for prognostic assessment is not recommended.
As discussed earlier in this review, experimental evidence demonstrates that PCSK9 is compartmentalized within the plasma, with approximately 40% of PCSK9 bound to LDL and lipoprotein(a) [Lp(a)] particles, and the remainder not associated with apoB-lipoproteins 20, 21, 70, 102. Given the stoichiometry between LDL and PCSK9 in plasma, approximately only 1 LDL particle in every 500-1000 carries at least one molecule of PCSK9. If, as we speculate, LDL-bound PCSK9 is the biologically more active from, future analyses should evaluate the separate association of LDL-bound and free PCSK9 with atherosclerotic events. We are currently developing a practical and reproducible assay to quantify PCSK9 in its two major compartments, which may elevate the status of plasma PCSK9 as a viable clinical biomarker.
Why would PCSK9 predict CVD independently of LDL?
While experimental work supports a critical role of PCSK9 in atherosclerosis, the value of circulating PCSK9 as a biomarker for ASCVD risk assessment in patients remains unclear. Since there is an inextricable link between PCSK9 and LDL-C, one would not necessarily expect plasma PCSK9 concentrations to predict risk above and beyond LDL-C unless PCSK9 has pleiotropic vascular effects.
Mendelian randomization analyses have demonstrated a common theme – genetic variants that are associated with lower LDL-C (including variants in PCSK9) also associate with a lower risk of ASCVD 103, 104. Furthermore, the magnitude of genetically mediated LDL-C reduction relates linearly to the magnitude of ASCVD risk reduction, irrespective of the gene under study 103, 105, 106. The results of these Mendelian randomization analyses are strikingly similar to those seen in randomized controlled clinical trials testing statins, ezetimibe, and, more recently, PCSK9 inhibitors 107. The major difference in genetically vs. pharmacologically mediated LDL-C lowering relates to a larger magnitude of risk reduction observed in the genetic analyses, given the lifetime exposure of lower LDL-C. If PCSK9, and its pharmacologic inhibition, manifest clinically important pleiotropic effects on atherosclerotic events beyond LDL-C lowering, there should be differences in the magnitude of CVD risk reduction in analyses of PCSK9 variants and in the randomized controlled trials testing PCSK9 inhibitors compared with other gene variants and drugs that target LDL-C lowering. However, the perfectly consistent linear relationship between magnitude of LDL-C reduction and magnitude of ASCVD risk reduction 108, irrespective of gene or drug, implies the theoretical pleiotropic effects of PCSK9 are likely not clinically significant.
Therapeutic approaches to inhibit PCSK9 and lower plasma lipids
LDL causes atherosclerosis
The importance of atherogenic lipoproteins as the central actors in the development of ASCVD is now readily accepted 109. The development and clinical testing of the statins with corresponding significant reductions in atherosclerotic events has been one of the great triumphs of medicine in the 20th century and has provided the key line of evidence in support of the “cholesterol hypothesis.” The Cholesterol Treatment Trialists’ Collaborators (CTTC) produced an authoritative meta-analysis that included >170,000 participants in 26 randomized controlled trials testing statins. The bottom line: reducing LDL-C by 39 mg/dL yielded a 22% reduction in the risk of major vascular events and 10% reduction in all-cause mortality over 5 years and independently of baseline LDL-C 108. Despite the outstanding efficacy of the statins, we must recognize that two thirds of the expected ASCVD events in statin-treated patients cannot be prevented. In addition, additional LDL-C cholesterol lowering interventions are needed for patients who cannot tolerate statin therapy or fail to attain adequate LDL-C lowering.
The IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial) study demonstrated that the addition of ezetimibe, a cholesterol absorption inhibitor, on top of simvastatin to patients right after an acute coronary event provided a statistically significant, though clinically modest additional 2% absolute risk reduction in major adverse cardiovascular events, without a change in mortality 110. Interestingly, this incremental event reduction is precisely what the CTTC meta-analysis regression line predicts, e.g., the magnitude of LDL-C reduction is directly and linearly related to the magnitude of event reduction. The results of IMPROVE-IT suggest that statins are not unique in their ability to reduce ASCVD events, and that LDL-C lowering is the reason for improved outcomes. The results of this trial also undid the notion of statin exceptionalism, provided the basis to pursue development of additional LDL-C lowering agents, and ushered in the era of PCSK9 inhibitors 111.
The action of PCSK9 on LDLR can be antagonized in different ways, to include monoclonal antibodies (mAbs), small interfering RNAs (siRNA), antisense oligonucleotides (ASO), adnectins, mimetic peptides, and vaccination strategies. While small molecule inhibitors typically are the favored first approach, their development has been challenging given the flat–surface interaction between PCSK9 and LDLR 112. Thus far, targeting plasma (extracellular) PCSK9 with mAbs is the farthest along with two different fully human mAbs approved for clinical use by regulatory agencies around the world.
Therapeutic monoclonal antibodies
Since 2012, many clinical trials have been performed to evaluate the LDL-C lowering efficacy of anti-PCSK9 monoclonal antibodies in subjects with different levels of CVD risk, alone or combination therapy (statin or ezetimibe), in statin-intolerant patients, and in both heterozygous (HeFH) and homozygous (HoFH) familial hypercholesterolemia. The mAbs have consistently demonstrated remarkable efficacy in reducing LDL-C (≈50% as monotherapy and ≈70% reduction in combination with a statin) with a good short-term safety and tolerability profile 113. Thus far, three mAbs to PCSK9 (PCSK9 inhibitors) have been tested, with two of them (alirocumab and evolocumab) approved by the U.S. FDA for the management of patients with either FH or ASCVD who require additional LDL-C lowering as an adjunct to diet and maximally tolerated statin therapy. Alirocumab and evolocumab are fully human antibodies and have been in the US market now for nearly 3 years, whereas the third (bococizumab, now abandoned)) was a humanized antibody which retains ~3% of murine protein sequence and for this reason induced immune responses limiting its effectiveness (neutralizing antibodies, discussed below) 114. It is interesting to note that the FDA approved the two fully human mAbs (alirocumab and evolocumab) on the basis of their LDL-C lowering efficacy, prior to the results of randomized controlled cardiovascular outcome trials.
As discussed earlier, the measurement of total plasma PCSK9 concentration is not likely to be useful in risk prediction models. However, we recently proposed that measurement of total plasma PCSK9 levels in patients on PCSK9 inhibitor therapy may become useful as a diagnostic tool 115. We demonstrated that patients treated with PCSK9 mAb exhibit an ~7-fold increase in total plasma PCSK9 levels relative to pretreatment levels. The change in total plasma PCSK9 levels is likely due to delayed clearance of the antibody-PCSK9 complex from the circulation and/or due to an increase in hepatic PCSK9 production, though it is not clear at present which of these mechanisms is quantitatively more important. From a clinical perspective, the change in plasma PCSK9 levels can be used to confirm adherence to therapy and/or optimal injection technique in patients that do not show the expected LDL-C lowering response to PCSK9 inhibitor therapy.
Silencing RNA
Whereas mAbs targeting PCSK9 only antagonize plasma PCSK9, siRNA interferes with its intracellular production, e.g., the translation of PCSK9 mRNA to protein. The siRNA selectively and catalytically silences the translation of their complementary target mRNA in a sequence-specific manner through the formation of effector RNA induced silencing complexes 116, 117. A phase I trial of the siRNA against PCSK9 named inclisiran, revealed similar LDL-C lowering efficacy as the PCSK9 mAbs 118. Interestingly, while the mAbs are dosed every two weeks (or once a month at a larger dose), inclisiran has a more durable effect with sustained lowering of LDL-C by an average of 53% and up to 81% at day 180, with a time-adjusted mean of greater than 50% through day 270 after a single injection. The phase II trial of inclisiran, ORION-1 (Trial to Evaluate the Effect of ALN-PCSSC Treatment on Low Density Lipoprotein Cholesterol) enrolled 501 subjects at high ASCVD risk with hypercholesterolemia despite maximally tolerated statin therapy 119. Subjects randomized to inclisiran sustained dose-dependent reductions in PCSK9 and LDL-C levels. LDL-C was reduced to the greatest extent (53%) in those who received the highest dose (two 300-mg injections) regimen of inclisiran. Changes in other plasma lipids and lipoproteins (including Lp(a) levels) with inclisiran were also similar to those seen with the anti-PCSK9 monoclonal antibodies.
Inclisiran distinguishes itself from mAbs to PCSK9 in several major ways. First, its extended duration of action may hold significant advantages. Should this sustained LDL-C lowering efficacy be realized, leveraging this therapeutic approach to PCSK9 inhibition may also help overcome issues related to medication adherence. Second, whereas the mAbs block only plasma PCSK9, the siRNA approach also reduces hepatocellular levels of PCSK9. Whether this intracellular approach has additional effects remains to be determined. Third, the lowering of plasma PCSK9 with siRNA truly reflects reduced levels of the circulating protein, which is similar to that seem in some PCSK9 LOF, whereas mAbs cause a significant accumulation of PCSK9 bound to the antibody 115. Fourth, the siRNA approach does not affect extrahepatic production of PCSK9 120, and thus the concentration of PCSK9 in the atheroma will be higher than for subjects treated with mAbs. Phase III studies with inclisiran are planned.
Vaccination
Vaccination represents an orthogonal approach to PCSK9 inhibition. A vaccine, AT04A, is in development as an agent to induce an antibody response to PCSK9 121. In one study, APOE*3Leiden/CETP mice vaccinated with AT04A developed elevated and persistent levels of antibody against PCSK9, with marked reductions in plasma total (-53%, p<0.001) and LDL-C levels compared with controls 121. Additionally, biochemical measures of inflammation were significantly reduced in vaccinated animals. Total atherosclerotic lesion area was reduced by 64% (p=0.004). Interestingly, antibody concentrations remained high at the end of the study, which may translate to continued reductions in atherogenic lipoprotein concentrations for some time afterwards, resulting in a long-lasting effect. A phase I study with AT04A is ongoing (NCT02508896).
More recently, another group has developed a virus like particle – PCSK9 (PCSK9Qβ-003) vaccine and tested it in both Balb/c mice and LDLR+/− mice 122. Vaccination resulted in significant reductions in total cholesterol and plasma PCSK9 expression. Additionally, the injected animals were found to have significant up-regulation of hepatic LDLR, SREBP-2, hepatocyte nuclear factor 1α, and HMG CoA-R. Positive developments in this area may lead to a viable approach to immunize humans against PCSK9, hindering the development of hypercholesterolemia and atherosclerosis. If the vaccination approach pans out, it could prove more affordable than mAbs, especially because the vaccine would be given once per year, versus once or twice a month. In addition, the vaccination strategy would have the advantage of use in younger patients, and the possibility of truly preventing atheroma formation rather than stabilizing preexisting plaques. The value of life-long exposure to low cholesterol has been proven over and over in natural randomization studies 103, 104.
Beyond these approaches to therapeutic antagonism of PCSK9, other novel methods are being pursued. Recently, a novel targetable pocket in the catalytic subunit of PCSK9 was identified. This structural identification may allow development of oral small molecule inhibitors to antagonize the action of PCSK9 123, 124. There is also interest in pursuing in vivo based editing of PCSK9 using CRISPR/Cas9 125, another approach that could have an application in younger subjects for true primary prevention purposes.
PCSK9 inhibition: Effects on other lipid parameters
Lipoprotein(a)
Lp(a) is an atherogenic LDL-like particle with its apoB covalently bound to apo(a) by a disulfide bond. Its plasma levels are largely genetically determined 126. Observational and genetic epidemiology data provide compelling evidence that Lp(a) has a causal role in atherosclerosis 127. The regulation of its production and clearance is poorly understood, and no effective targeted therapies exist. Interestingly, PCSK9 inhibitors have demonstrated unexpected reductions in Lp(a), on the order of 25-30% 128, 129. While the potent reduction in LDL-C achieved by PCSK9 inhibition is mediated through its profound effect on LDLR preservation, the mechanism by which they lower Lp(a) is unknown. Some suggest that the Lp(a) reduction achieved with PCSK9 inhibition is also secondary to the profound increase in LDLR expression, though that notion poses substantial challenges, as: 1) Lp(a) is not an avid ligand for the LDLR 130; 2) Lp(a) metabolism in FH subjects is similar to that non-FH subjects 131; 3) Lp(a) levels do not change with statin treatment, which also upregulates LDLR 132; 4) PCSK9 inhibition in two subjects with homozygous LDLR-null FH lowered Lp(a) but failed to reduce LDL-C levels 133; 5) loss-of-function PCSK9 mutation carriers do not demonstrate significantly different Lp(a) levels compared to controls 3, 134, 135; and 6) epidemiological studies do not consistently demonstrate a correlation between plasma PCSK9 and Lp(a) concentrations 84, 90, 93, 94, 136. On average, the lowering effect of PCSK9 inhibitors on Lp(a) is about half of that on LDL-C. The modest correlation between LDL-C and Lp(a) lowering with PCSK9 inhibition is tempered by significant discordance in the reduction of these two lipid fractions in approximately 40% of treated individuals, who show a robust LDL-C with minimal Lp(a) response 137. This observation suggests that PCSK9 inhibition activates alternative mechanisms beyond the LDLR and that additional factors ultimately determine the degree to which Lp(a) levels are reduced. It is likely that Lp(a) binds to and is cleared by the LDLR, at least to some extent, as its lowering by PCSK9 inhibitors is inversely related to plasma LDL-C levels 130. Additionally, Lp(a) clearance may also be determined by the length of the apo(a) isoform. It is possible that some Lp(a) isoforms may be cleared through LRP1 138 or CD36 139, two receptors that are also influenced by PCSK9 inhibition 73, 76. Other data suggest that PCSK9 does not affect Lp(a) catabolism, but rather enhances apo(a) secretion and Lp(a) assembly through unknown mechanisms 140. Potential mechanisms underlying the impact of PCSK9 inhibition on plasma Lp(a) concentration are summarized in Figure 3.
Figure 3. Possible effects of PCSK9 inhibition on Lp(a) metabolism.
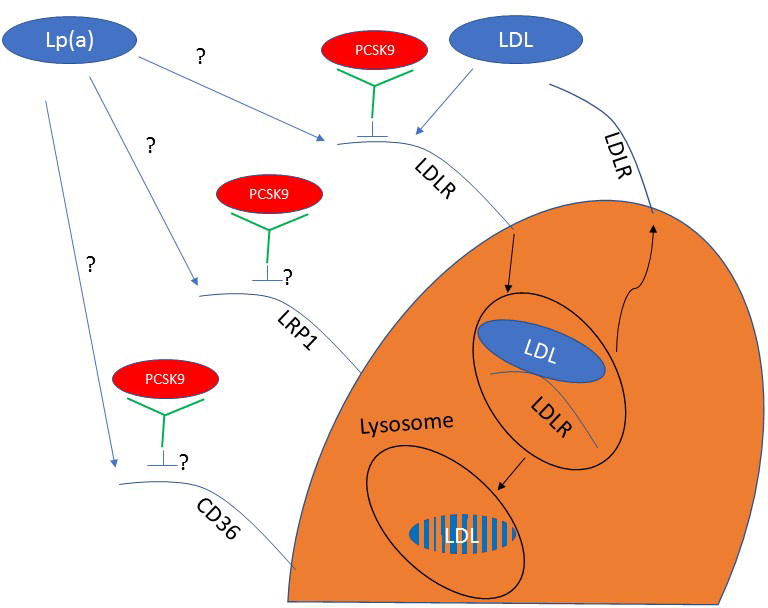
(i) Therapeutic PCSK9 inhibition prevents PCSK9 interaction with the LDLR, therefore facilitating continued recycling of the receptor and efficient clearance of Lp(a) particles; (ii and iii) PCSK9 inhibition affects LRP1 and CD36 levels or function, which results in increased Lp(a) clearance through these receptors; (iv) PCSK9 directly regulates apo(a) secretion, and the inhibition of PCSK9 prevents this process.
PCSK9 = proprotein convertase subtilisin/kexin type 9; Lp(a) = lipoprotein(a); LRP1 = low- density lipoprotein receptor-related protein 1; CD36 = cluster of differentiation 36; apo(a) = apolipoprotein(a)
Triglyceride-Rich Lipoproteins
Studies suggest a role for PCSK9 in triglyceride-rich lipoprotein (TRL) metabolism through LDLR-mediated clearance and possibly through an effect on hepatic and intestinal apoB-lipoprotein production 66, 141, 142. Animal models have shown both LDLR-dependent and independent roles for PCSK9 on TRL metabolism in the liver 143, 144 and small intestine 145, 146, while individuals with the GOF PCSK9 mutation S127R have a three-fold elevation in apoB100 production rates compared with noncarriers 147. However, the effect of PCSK9 inhibition on plasma triglyceride levels is not clear, as most studies only show a modest reduction that does not always reach statistical significance 148-152. Although the absolute changes in triglyceride levels are similar to what is seen with statin therapy (average reduction of ~15%), the effect of PCSK9 inhibition on triglyceride levels is dwarfed by its impact on LDL-C reduction. Metabolic studies in humans have failed to demonstrate an effect of PCSK9 inhibition on VLDL-apoB and VLDL-triglyceride production rate but show an increased fractional catabolic rate for both VLDL-apoB and VLDL-triglycerides, suggesting that PCSK9 does not impact TRL production but only its clearance 153. PCSK9 inhibition also did not influence post-prandial triglyceride or apoB48 levels, consistent with the notion that PCSK9 is not directly involved in TRL production in the small intestine in humans 153.
High-Density Lipoprotein
Studies in mice support a direct relationship between high-density lipoprotein (HDL)-cholesterol (HDL-C) levels, as deletion of the PCSK9 gene 154, injection of PCSK9 blocking antibodies 155, or administration of antisense oligonucleotides against PCSK9 156 all led to reductions in HDL-C levels by 30%-50%. In contrast, PCSK9 transgenic animals exhibit a mild increase in HDL-C levels 20. In contrast, clinical trials with PCSK9 inhibitors demonstrate a modest (<10%) increase in HDL-C and apoAI levels 157. The absolutes changes in HDL-C levels are similar to what is observed after treatment with statins 158. A critical difference from humans is that HDL in mice is rich in apoE, which makes it a good target for LDLR-mediated clearance. Thus, the effect of PCSK9 on HDL-C metabolism in mice is directly linked to the effect of PCSK9 on the LDLR. To date, there is no clear evidence that PCSK9 directly effects HDL production or clearance in humans. One possibility is that the rapid clearance of LDL due to PCSK9 impairs the CETP-mediated cholesterol exchange between HDL and LDL, leading to modestly increased HDL-C levels.
PCSK9 inhibition: Clinical Outcome studies
Cardiovascular outcomes
Prior to the results of the first dedicated randomized controlled cardiovascular outcome trial with PCSK9 inhibition, there were post-hoc analyses of the alirocumab and evolocumab clinical trial programs that suggested that these drugs may be of significant benefit for cardiovascular event reduction. The OSLER (Open-Label Study of Long-Term Evaluation Against LDL Cholesterol) trial examined the long-term effects of evolocumab as an extension of the open-label, randomized controlled OSLER 1 and 2 trials and included 4,465 patients 159. The majority of patients (~80%) had other cardiovascular risk factors including hypertension, diabetes, metabolic syndrome, current cigarette use, or family history of premature coronary artery disease or of inherited hypercholesterolemia. The baseline LDL-C of 120 mg/dL was reduced by 61% to a mean of 48 mg/dL. In this post-hoc analysis, the incidence of cardiovascular events (death, MI, unstable angina requiring hospitalization, coronary revascularization, stroke, transient ischemic attack, and heart failure requiring hospitalization) occurred in 1% of the evolocumab group versus 2% of the standard therapy group (HR 0.47, p = 0.0003).
A similar investigation of the long-term safety, tolerability, and efficacy of alirocumab vs. placebo was conducted in patients at high cardiovascular risk from the ODYSSEY LONG TERM Study of 2,341 subjects. LDL-C levels were reduced by 61% in the alirocumab group to a mean of 48 mg/dL compared to an increase of 0.8% in the placebo group. The post-hoc analysis of cardiovascular events (composite of death from coronary heart disease, nonfatal MI, fatal or nonfatal ischemic stroke, or unstable angina requiring hospitalization) again demonstrated much lower rates with alirocumab than with placebo (1.7% vs. 3.3%, HR 0.52, p = 0.02) 160. It is important to realize that these studies contained few events and were not adequately powered to address cardiovascular outcomes. Regardless, many predicted that the PCSK9 mAbs would yield extraordinary outcomes in the dedicated randomized controlled trials given the magnitude and consistency of suggested cardiovascular benefit from these two post-hoc analyses.
In the meantime, Ference et al. performed a Mendelian randomization analysis in an attempt to predict the results of the randomized controlled cardiovascular outcomes trials 104. The investigators used data from 112,772 individuals from 14 studies, with 14,120 cardiovascular events and 10,635 cases of diabetes, to categorize individuals based on inheritance of the number of LDL–lowering alleles, either variants in the genes encoding PCSK9 and/or HMG CoA reductase (HMGCR), the target of statins. Long-term exposure to either PCSK9 variants or HMGCR variants was associated with a remarkably similar reduction in risk of cardiovascular events per unit reduction in LDL cholesterol (by 19% per 10 mg/dl decrease in LDL-C). In combination, the effects of these variants were additive.
Moreover, a detailed atherosclerosis imaging study reported on the impact of evolocumab on coronary plaque utilizing IVUS. Prior IVUS trials demonstrated statistically significant reductions in percent atheroma volume in individuals who were started on high-intensity statin therapy and achieved LDL-C levels <70 mg/dL 161. It was not clear if further lowering of LDL-C with PCSK9 inhibition might have more profound effects on plaque regression. The GLAGOV (Global Assessment of Plaque Regression With a PCSK9 Antibody as Measured by Intravascular Ultrasound) trial evaluated changes in coronary atherosclerosis using serial IVUS evaluations in subjects taking statins or statins plus evolocumab 152. Patients with angiographic coronary artery disease on baseline statin therapy were randomized to monthly evolocumab (n=484) or placebo (n=484). A greater decrease in percent atheroma volume (1% difference) at 76 weeks was seen among those who received evolocumab. Furthermore, more patients on evolocumab than on statin experienced plaque regression (64.3% vs. 47.3%; p<0.001). Several questions remained after GLAGOV; (1) will incremental plaque regression be associated with incremental cardiovascular event reduction as the prior statin monotherapy trials demonstrated, (2) if lowering LDL-C to unprecedented levels only regresses atheroma volume by 1%, have we reached the limits of what can be achieved by dramatic LDL-C lowering, and more fundamentally, (3) if attaining extreme hypocholesterolemia does not dramatically alter plaque, what other orthogonal approaches to ASCVD risk reduction need to be considered?
The FOURIER trial was the first of the randomized controlled cardiovascular outcomes trials with a PCSK9 inhibitor, evolocumab 107. The investigators randomized 27,564 patients with established ASCVD on optimized statin therapy to either evolocumab or placebo and monitored the rate of major cardiovascular events (cardiovascular death, MI, stroke, hospitalization for unstable angina, or coronary revascularization in the primary outcome measure). At 48 weeks, evolocumab therapy was associated with a 59% reduction in LDL-C from a median baseline of 92 mg/dL to 30 mg/dL. At a median follow-up of 26 months, evolocumab was associated with an absolute 1.5% reduction in the primary outcome, driven primarily by reductions in nonfatal MI, stroke, and revascularization. Effects of evolocumab were consistent regardless of baseline LDL-C level or intensity of background statin use. Evolocumab therapy, with its associated intense LDL-C reduction, did not decrease mortality, though the event curves between the evolocumab and placebo groups were still diverging at the time of trial termination. While there was no overall or cardiovascular-specific mortality benefit with evolocumab, death from cardiovascular disease was remarkably low (< 2%) in both groups. Interestingly, the 7-year IMPROVE-IT trial of ezetimibe added to statin therapy also did not show a reduction in mortality 110. Other than a modest 2% incidence in injection-site reactions (which led to no excess drug discontinuations vs. placebo), there was no increase in key adverse events including new-onset diabetes or neurocognitive effects in patients receiving evolocumab despite the dramatic LDL-C reduction.
Did FOURIER underperform or were the post-hoc analyses that preceded it simply way off? The landmark analysis presented in FOURIER demonstrated event reductions similar to those reported in years 0–2 of prior statin trials 162. An enthusiastic interpretation suggests that longer duration of therapy should be associated with greater reduction in major adverse cardiovascular events and perhaps a corresponding mortality reduction. Given the significant cost of PCSK9 inhibitors and lack of established mortality benefit, their routine addition to standard-of-care statin therapy in patients with established ASCVD has thus far been reserved for those perceived to be at particularly high risk for cardiovascular events.
Subsequent analyses from FOURIER have been encouraging. One investigation assessed the efficacy and safety of evolocumab according to degree of LDL-C reduction at one month 163. The primary composite outcome declined steadily as LDL-C levels decreased, with no association between LDL-C level and adverse events. A similar reduction was observed in the key secondary endpoint, with 2,669 subjects in the lowest LDL-C category (<20 mg/dL) at 4 weeks experiencing the lowest rate for cardiovascular death, or MI (adjusted hazard ratio 0.69, 95% CI 0.56-0.85, P=0.0001) compared to the group with highest LDL-C (>100 mg/dL). Exploratory analyses in a subgroup of 504 patients with an LDL-C <10 mg/dL showed even further reduction in cardiovascular events with no increase in safety events. While it remains to be seen if prolonged treatment with a PCSK9 inhibitor with profound LDL-C lowering results in mortality benefit, at the very least this therapeutic approach and iatrogenic extreme hypocholesterolemia appear to be safe. In another FOURIER analysis that evaluated 22,351 subjects with a history of prior MI, those with recent MI, multiple prior MIs, and residual multi-vessel coronary artery disease had 34-90% greater risk for vascular events and enjoyed the greatest benefit from PCSK9 inhibition (absolute risk reduction of 2.6-3.4% over three years). 164. Given expense and barriers to access, this data may help to define appropriate allocation of PCSK9 inhibitors to those with established, high-risk ASCVD. Lastly, the FOURIER investigators evaluated the impact of evolocumab in patients with peripheral arterial disease (PAD) 165. This analysis of 3,642 patients with PAD demonstrated that evolocumab significantly reduced the primary composite endpoint. Evolocumab reduced the primary endpoint consistently in patients both with and without PAD, but the drop was numerically greater in the PAD patients. In patients with PAD the absolute risk reduction on evolocumab versus placebo was 3.5% (vs. 1.5% in the main FOURIER analysis) yielding an attractive number-needed-to-treat (NNT=29) over 2.5 years. In the no-PAD group, the absolute risk reduction was only 1.4%, for an NNT of 72. Interestingly, evolocumab also reduced the risk of major adverse limb events in all patients compared with placebo (HR 0.58; 95% CI 0.38-0.88), with consistent effects for those with and without PAD.
Bococizumab is the other PCSK9 mAb that has been evaluated in a randomized controlled cardiovascular outcome trials. In the SPIRE (Studies of PCSK9 Inhibition and the Reduction of vascular Events) trials, participants were randomly assigned to receive either bococizumab 150 mg subcutaneously every 2 weeks or placebo. The SPIRE program included six parallel, multinational studies designed to assess the variability and durability of the LDL-C-lowering efficacy of bococizumab and two dedicated cardiovascular outcome trials 114, 166. The lipid-lowering trials included 4,300 patients on background statin therapy who were followed up for 1 year. At 12 weeks, bococizumab treatment was associated with a reduction in LDL-C levels of 55% relative to placebo. However, treatment with bococizumab often led to the development of antidrug antibodies and specific neutralizing antibodies, which attenuated the LDL-C-lowering response (48% and 29% of patients at 1 year, respectively) 166. In those who developed higher neutralizing antibody titers (upper tertile and top decile, respectively) in response to bococizumab, LDL-C concentration decreased by only 31% and 12% from baseline. Not surprisingly, given its immunogenicity, bococizumab was also associated with significantly higher rates of injection-site reactions than placebo (10.4% versus 1.3%; P < 0.001).
The two dedicated CVOT in the SPIRE program included a total of 27,438 patients at high risk of ASCVD (93% receiving background statin therapy) and who had either a previous cardiovascular event or history of diabetes mellitus, chronic kidney disease, PAD, or familial hypercholesterolemia (high-risk, primary prevention cohort) 167. The primary end point included nonfatal MI, nonfatal stroke, hospitalization for unstable angina requiring urgent revascularization, and cardiovascular death. The studies had a very short duration due the decision of the manufacturer to abandon the development of the drug. In the combined trials, those receiving bococizumab achieved the anticipated 56% mean reduction in LDL-C levels 114. In the lower-risk subject SPIRE-1 trial (baseline LDL-C ≥70 mg/dl; median follow-up 7 months), bococizumab did not reduce the incidence of the primary composite end point compared with placebo. In contrast, in the higher-risk subject SPIRE-2 trial (baseline LDL-C ≥100 mg/dl; median follow-up 12 months), the rate of major cardiovascular events was significantly lowered by bococizumab (HR 0.79, 95% CI 0.65–0.97, P = 0.02). When the SPIRE-1 and SPIRE-2 data were pooled, the incidence of the composite primary end point was not significantly different between groups (median follow-up 10 months), although patients with the largest percent reduction in LDL-C levels had a 25% decrease in events (HR 0.75, 95% CI 0.61–0.92, P = 0.006), and those who had longer treatment duration (mean 13.6 months) had a 17% reduction in events (HR 0.83, 95% CI 0.70–0.98, P = 0.03).
Most recently, the ODYSSEY OUTCOMES trial was presented at the American College of Cardiology Meeting (Presented by Dr. Philippe Steg at the American College of Cardiology Annual Scientific Session (ACC 2018), Orlando, FL, March 10, 2018.). ODYSSEY OUTCOMES enrolled 18,924 subjects within 1-12 months after an index ACS after a run-in phase of 2-16 weeks on high intensity statin therapy. Individuals who demonstrated LDL-C ≥70 mg/dL (or non-HDL-C ≥100 mg/dL or ApoB ≥80 mg/dL) despite high-intensity therapy were randomized to alirocumab every 2 weeks or placebo. Alirocumab was titrated between 75 and 50 mg to target an LDL-C between 25-50 mg/dL, but above 15 mg/dL. The primary outcome was a composite endpoint including coronary heart disease death, MI, ischemic stroke, or unstable angina. Treatment with alirocumab (on-treatment analysis) was associated with a 54.7% reduction in LDL-C and an absolute risk reduction in the primary endpoint of 1.6%. Of note, alirocumab was discontinued in almost 8% of the treatment group due to two consecutive LDL-C measurements below the prespecified threshold of 15 mg/dL. Several of the secondary outcomes favored treatment with alirocumab, though total mortality is to be considered only nominally significant given that the two proximal endpoints in the hierarchical analysis did not reach statistical significance. Nevertheless, the signal suggesting a reduction in total mortality is reassuring given that the opposite was noted in the FOURIER trial.
The three dedicated cardiovascular outcomes trials with therapeutic monoclonal antibodies targeting PCSK9 have taught us much about what can be expected from the provision of therapeutic monoclonal antibodies to high risk patients. First, the type of antibody used to inhibit PCSK9 matters greatly. Humanized antibodies against PCSK9 are immunogenic and are associated with injection-site reactions, the development of neutralizing antibodies, and an attenuated LDL-C lowering effect. Second, consistent with the ‘lower is better for longer’ hypothesis, clinical benefits with bococizumab were greater and significant only for those patients who achieved and sustained large reductions in LDL-C. Third, while FOURIER pushed this hypothesis to ‘lowest-is-best’, the overall cardiovascular risk reduction afforded by the 60% reduction in LDL-C with evolocumab was modest. Fourth, and not surprisingly, the magnitude of absolute benefit is greatest amongst the highest risk patients and maximized with longer exposure to drug.
Even when considering the PCSK9 monoclonal antibodies in the most positive light, questions remain with regards to the cost-effectiveness of this therapeutic approach. Extrapolation of the clinical trial results reveals a NNT of 74 for 2 years of treatment in FOURIER and NNT of 64 over the duration of ODYSSEY Outcomes. Is this tenable for a drug that is priced at ~$14,000 per year, or almost $1,000,000 to prevent one event? This question is at the center of a debate amongst all stakeholders and remains unresolved.
Safety outcomes
In early 2014, the FDA directed developers of PCSK9 inhibitors to monitor neurocognitive adverse effects given concerns over putative cognitive impairment due to extreme LDL-C lowering. EBBINGHAUS (Evaluating PCSK9 Binding Antibody influence on Cognitive Health in High Cardiovascular Risk Subjects), a substudy of FOURIER, evaluated longitudinal neurocognitive changes in patients receiving a combination of evolocumab plus statin vs. statin alone 168. Before that, a Mendelian randomization study evaluated the association between low LDL-C and risk of dementia using genetic variation in HMGCR and PCSK9 as instrumental variables 127, and found no increased risk of dementia, Parkinson’s disease, or epilepsy. More recently an analysis from REGARDS (The Reasons for Geographic and Racial Differences in Stroke) study recapitulated these findings. The investigators evaluated the association between PCSK9 LOF variants and neurocognitive impairment and decline among participants in REGARDS in study participants with (n=241) and without (n=10,454) C697X or Y142X LOF variants of PCSK9, using a comprehensive battery of neurocognitive tests. They found no differences in neurocognitive decline between the two groups, again suggesting that life-long exposure to low LDL-C is not associated with cognitive dysfunction 169.
In EBBINGHAUS, a total of 1,204 patients with both baseline and follow-up cognitive testing were evaluated 159. Patient baseline characteristics were consistent with those enrolled in FOURIER, with a mean age of 63 years, 72% male and 20% with a prior stroke. Cognitive function was assessed using a standardized, well-validated computer tablet-based testing platform (Cambridge Neuropsychological Test Automated Battery, or CANTAB), which evaluates spatial working memory strategy index of executive function (primary endpoint), as well as other memory assessments, including survey of everyday cognition and investigator-initiated reports of neurocognitive adverse effects. Over a median follow-up of 19.8 months, there was no difference between patients in the evolocumab or placebo treatment groups, with respect to either primary or secondary endpoints. There was also no evidence to suggest differences in cognitive tests in patients attaining very low LDL cholesterol levels, including those with levels <25 mg/dL.
More recently, a group of investigators assessed the incidence of adverse neurocognitive adverse events amongst participants from fourteen phase II and III trials testing alirocumab 170. Neurocognitive events were reported by 22 (0.9%) alirocumab-treated patients vs. 9 (0.7%) with placebo in placebo-controlled trials [hazard ratio (HR) 1.24, 95% confidence interval (CI) 0.57–2.68] and 10 (1.2%) with alirocumab vs. 8 (1.3%) with ezetimibe in ezetimibe-controlled trials (HR 0.81, 95% CI 0.32–2.08). Rates of neurocognitive events were similar in patients with LDL-C levels <25 mg/dL (n = 5/839; 0.6%; 0.5/100 patient-years) vs. ≥25 mg/dL (n = 26/2501; 1.0%; 0.8/100 patient-years).
Of course, the last word has not been spoken on this issue. What are the consequences of longer-term therapeutic reductions in LDL-C? Besides the informative genetic analyses, it is important to bear in mind that these PCSK9 mAbs do not cross the blood brain barrier and thus are unlikely to be associated with direct adverse effects on the central nervous system, whose lipid homeostasis is under separate regulation from the systemic one.
Another theoretical complication of PCSK9 inhibitor induced hypocholesterolemia is diabetes. The concern of drug induced diabetes stems from multiple lines of evidence that demonstrate that statins are associated with a modest risk of new-onset hyperglycemia, especially in patients susceptible to develop diabetes by standard markers 171-173. While this observation has been fairly consistent across trials, the mechanisms at play have not been elucidated. Clinical trials of PCSK9 inhibitors did not show a signal for new-onset diabetes, though these studies are of short duration 174, 175. Two Mendelian randomization analyses provide insight on this issue as well. In one, LOF variants of PCSK9 and HMGCR were associated with increased risk of diabetes (11% and 13%, respectively) 104. In another, using data from more than 550,000 individuals and 51,623 type 2 diabetics, long-term exposure to LOF variants of PCSK9 associated with lower LDL-C and higher fasting glucose concentration and waist-to-hip ratio, and increased risk of type 2 diabetes (odds ratio 1.29, 95% CI 1.11 to 1.50) 176. Both of these Mendelian randomization studies therefore suggest that, like statins, long-term treatment with a PCSK9 inhibitor may predispose vulnerable patients to increased risk of type 2 diabetes. Again, the mechanism that underpins these observations is unknown, though if this relationship is real, it may be related to upregulation of the LDLR with increased lipid uptake by the pancreatic beta-cell. Although the FOURIER trial did not demonstrate an excess of diabetes in those treated with evolocumab 107, the median duration of therapy was just over two years. It remains to be seen whether the Mendelian randomization analyses or the clinical trials with PCSK9 inhibitors correctly forecast the association between PCSK9 efficiency and diabetes.
Cost, cost-effectiveness, and barriers to access
The issue that has been at the center of the PCSK9 inhibitor debate relates to their cost and cost-effectiveness. With a wholesale acquisition cost of ~$14,000 and only modest reductions in non-fatal atherosclerotic events, stakeholders have wrestled with appropriate allocation and regulations to restrict and target access to these therapies. From the time that PCSK9 mAbs received regulatory approval, payers have placed hurdles in front of patients and providers. A recent analysis identified five notable features of the prior authorization (PA) requirements for PCSK9 inhibitors, including: 1) a requirement to submit medical records along with PA forms; 2) requirements for data that may be challenging for providers to access (eg, adherence measures typically calculated from pharmacy claims, off-treatment LDL-C levels that may not be available); 3) restriction of approval to specialty prescribers (cardiologists, endocrinologists, lipidologists); 4) requirement for genetic testing even though the same insurers will not cover the cost of testing; and 5) requirement to try multiple lipid-lowering regimens 177.
Of course, the restricted access to PCSK9 inhibitors is a complicated issue. Several cost-effectiveness analyses with these agents have been performed and demonstrate disparate results based on different modeling assumptions, though they all found that these drugs are not cost-effective at their current price 178-180. Clearly on the basis of these analyses, the economic proposition of the therapeutic mAbs is not viable, unless only the highest risk patients are allocated to therapy. It remains to be seen whether other approaches to PCSK9 inhibition associated with longer duration of effect, such as siRNA and vaccination, may be more cost-effective.
Such barriers to access can be insurmountable for the many providers that do not have the resources to challenge an unjustified denial. In that regard, we recently presented the concept of the “PCSK9 Inhibitor Clinic”, a new model that streamlines and coordinates care delivery in an effort to improve approval/access to therapy 181. Now, with over 300 patients on therapy, our rate of approval for the PCSK9 mAbs exceeds 97%. This success is due to a combination of appropriate patient selection and a comprehensive, efficient, and consistent approach to the PA and appeal processes. While this model is successful, it may not be scalable, especially in community practices. Clearly, larger efforts led by the American Heart Association and the American College of Cardiology are necessary to engender policy change at the national level.
Conclusion
The discovery of PCSK9 has ushered in an exciting new era for cholesterol management and CVD risk reduction. Fundamental biological insights have provided a far clearer understanding of lipoprotein metabolism. Based on these understandings, we are no longer limited to the previously held view that cholesterol homeostasis is an intracellular affair, but rather appreciate a model whereby a secreted plasma protein, whose action can be easily inhibited, renders dominant control over lipoprotein metabolism. These findings have been translated into newly approved therapies, PCSK9 mAbs that dramatically reduce LDL-C and incrementally reduce atherosclerotic cardiovascular events, with additional therapeutic antagonists of PCSK9 likely on the way. The excitement surrounding the science and unprecedented efficiency in drug development is tempered by the practical realities of cost-effectiveness and barriers to access - issues that must be resolved if the PCSK9 story is to reach its full potential. Given the prospect of PCSK9 modulators to dramatically alter approaches to cardiovascular disease prevention, all stakeholders (scientists, clinicians, guideline committees, payers, and patient advocacy groups) must work together to ensure that therapies targeting PCSK9 are appropriately evaluated, endorsed, allocated, and reimbursed.
Acknowledgments
Sources of funding
Dr. Tavori was partially supported by AHA-SDG grant 16SDG27520011 and MRF-NI grant 1011656.
Dr. Fazio was partially supported by NIH-NHLBI grant 5R01HL132985.
Abbreviations
- PCSK9
proprotein convertase subtilisin/kexin type 9
- LDL
low-density lipoprotein
- LDLR
low-density lipoprotein receptor
- FH
familial hypercholesterolemia
- HeFH
heterozygous familial hypercholesterolemia
- HoFH
homozygous familial hypercholesterolemia
- GOF
gain of function
- LOF
loss of function
- ASCVD
atherosclerotic cardiovascular disease
- MI
myocardial infarction
- SRE
sterol-regulatory element
- SREBP
sterol regulatory element binding protein
- apoB
apolipoprotein B
- VLDL
very low-density lipoproteins
- LDLRAP
low-density lipoprotein receptor adaptor protein
- apoER2
apoE receptor 2
- LRP1
LDLR-related protein 1
- HSPG
Heparan sulfate proteoglycans
- CIMT
carotid intima-media wall thickness
- FATE
Firefighters and Their Endothelium
- CAC
coronary artery calcium
- apo(a)
apolipoprotein(a)
- IVUS
intravascular ultrasound
- ACS
acute coronary syndrome
- Lp(a)
lipoprotein(a)
- CTTC
Cholesterol Treatment Trialists’ Collaborators
- IMPROVE-IT
Improved Reduction of Outcomes: Vytorin Efficacy International Trial
- mAbs
monoclonal antibodies
- siRNA
small interfering RNAs
- ASO
antisense oligonucleotides
- ORION-1
Trial to Evaluate the Effect of ALN-PCSSC Treatment on Low Density Lipoprotein Cholesterol
- TRL
triglyceride-rich lipoprotein
- HDL-C
high-density lipoprotein-cholesterol
- OSLER
Open-Label Study of Long-Term Evaluation Against LDL Cholesterol
- HMGCR
HMG CoA reductase
- GLAGOV
Global Assessment of Plaque Regression With a PCSK9 Antibody as Measured by Intravascular Ultrasound
- PAD
peripheral arterial disease
- SPIRE
Studies of PCSK9 Inhibition and the Reduction of vascular Events
- EBBINGHAUS
Evaluating PCSK9 Binding Antibody influence on Cognitive Health in High Cardiovascular Risk Subjects
- REGARDS
The Reasons for Geographic and Racial Differences in Stroke
- PA
prior authorization
Footnotes
Disclosures (last 12 months)
Dr. Shapiro has received compensation for advisory activities from Akcea, Amgen, Kastle, Novartis, and Regeneron.
Dr. Tavori has no disclosures to report.
Dr. Fazio has received compensation for advisory activities from Amgen, Amarin, Akcea, Aegerion, and Kowa
References
- 1.Seidah NG, Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov. 2012;11:367–83. doi: 10.1038/nrd3699. [DOI] [PubMed] [Google Scholar]
- 2.Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derre A, Villeger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 3.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–5. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 4.Cohen JC, Boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 5.Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193:445–8. doi: 10.1016/j.atherosclerosis.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 6.Fasano T, Cefalu AB, Di Leo E, Noto D, Pollaccia D, Bocchi L, Valenti V, Bonardi R, Guardamagna O, Averna M, Tarugi P. A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. 2007;27:677–81. doi: 10.1161/01.ATV.0000255311.26383.2f. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Z, Tuakli-Wosornu Y, Lagace TA, Kinch L, Grishin NV, Horton JD, Cohen JC, Hobbs HH. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79:514–23. doi: 10.1086/507488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR, Committee FS and Investigators Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017 doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 9.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–40. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 10.Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah NG. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem. 2008;283:2363–72. doi: 10.1074/jbc.M708098200. [DOI] [PubMed] [Google Scholar]
- 11.Osborne TF, Gil G, Goldstein JL, Brown MS. Operator constitutive mutation of 3-hydroxy-3-methylglutaryl coenzyme A reductase promoter abolishes protein binding to sterol regulatory element. J Biol Chem. 1988;263:3380–7. [PubMed] [Google Scholar]
- 12.Anderson RG, Goldstein JL, Brown MS. Localization of low density lipoprotein receptors on plasma membrane of normal human fibroblasts and their absence in cells from a familial hypercholesterolemia homozygote. Proc Natl Acad Sci U S A. 1976;73:2434–8. doi: 10.1073/pnas.73.7.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–803. doi: 10.1172/JCI18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson RG, Brown MS, Beisiegel U, Goldstein JL. Surface distribution and recycling of the low density lipoprotein receptor as visualized with antireceptor antibodies. J Cell Biol. 1982;93:523–31. doi: 10.1083/jcb.93.3.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson RG, Brown MS, Goldstein JL. Role of the coated endocytic vesicle in the uptake of receptor-bound low density lipoprotein in human fibroblasts. Cell. 1977;10:351–64. doi: 10.1016/0092-8674(77)90022-8. [DOI] [PubMed] [Google Scholar]
- 16.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 17.Brown MS, Anderson RG, Goldstein JL. Recycling receptors: the round-trip itinerary of migrant membrane proteins. Cell. 1983;32:663–7. doi: 10.1016/0092-8674(83)90052-1. [DOI] [PubMed] [Google Scholar]
- 18.Fazio S, Linton MF, Hasty AH, Swift LL. Recycling of apolipoprotein E in mouse liver. J Biol Chem. 1999;274:8247–53. doi: 10.1074/jbc.274.12.8247. [DOI] [PubMed] [Google Scholar]
- 19.Rensen PC, Jong MC, van Vark LC, van der Boom H, Hendriks WL, van Berkel TJ, Biessen EA, Havekes LM. Apolipoprotein E is resistant to intracellular degradation in vitro and in vivo. Evidence for retroendocytosis. J Biol Chem. 2000;275:8564–71. doi: 10.1074/jbc.275.12.8564. [DOI] [PubMed] [Google Scholar]
- 20.Tavori H, Fan D, Blakemore JL, Yancey PG, Ding L, Linton MF, Fazio S. Serum proprotein convertase subtilisin/kexin type 9 and cell surface low-density lipoprotein receptor: evidence for a reciprocal regulation. Circulation. 2013;127:2403–13. doi: 10.1161/CIRCULATIONAHA.113.001592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norata GD, Tavori H, Pirillo A, Fazio S, Catapano AL. Biology of proprotein convertase subtilisin kexin 9: beyond low-density lipoprotein cholesterol lowering. Cardiovasc Res. 2016;112:429–42. doi: 10.1093/cvr/cvw194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cunningham D, Danley DE, Geoghegan KF, Griffor MC, Hawkins JL, Subashi TA, Varghese AH, Ammirati MJ, Culp JS, Hoth LR, Mansour MN, McGrath KM, Seddon AP, Shenolikar S, Stutzman-Engwall KJ, Warren LC, Xia D, Qiu X. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol. 2007;14:413–9. doi: 10.1038/nsmb1235. [DOI] [PubMed] [Google Scholar]
- 23.Hampton EN, Knuth MW, Li J, Harris JL, Lesley SA, Spraggon G. The self-inhibited structure of full-length PCSK9 at 1.9 A reveals structural homology with resistin within the C-terminal domain. Proc Natl Acad Sci U S A. 2007;104:14604–9. doi: 10.1073/pnas.0703402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piper DE, Jackson S, Liu Q, Romanow WG, Shetterly S, Thibault ST, Shan B, Walker NP. The crystal structure of PCSK9: a regulator of plasma LDL-cholesterol. Structure. 2007;15:545–52. doi: 10.1016/j.str.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 25.McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem. 2007;282:20799–803. doi: 10.1074/jbc.C700095200. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Tumanut C, Gavigan JA, Huang WJ, Hampton EN, Tumanut R, Suen KF, Trauger JW, Spraggon G, Lesley SA, Liau G, Yowe D, Harris JL. Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem J. 2007;406:203–7. doi: 10.1042/BJ20070664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher TS, Lo Surdo P, Pandit S, Mattu M, Santoro JC, Wisniewski D, Cummings RT, Calzetta A, Cubbon RM, Fischer PA, Tarachandani A, De Francesco R, Wright SD, Sparrow CP, Carfi A, Sitlani A. Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation. J Biol Chem. 2007;282:20502–12. doi: 10.1074/jbc.M701634200. [DOI] [PubMed] [Google Scholar]
- 28.Pearlstein RA, Hu QY, Zhou J, Yowe D, Levell J, Dale B, Kaushik VK, Daniels D, Hanrahan S, Sherman W, Abel R. New hypotheses about the structure-function of proprotein convertase subtilisin/kexin type 9: analysis of the epidermal growth factor-like repeat A docking site using WaterMap. Proteins. 2010;78:2571–86. doi: 10.1002/prot.22767. [DOI] [PubMed] [Google Scholar]
- 29.Zhang DW, Garuti R, Fau-Tang W-J, Tang Wj, Fau-Cohen JC, Cohen Jc, Fau-Hobbs HH, Hobbs HH. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc Natl Acad Sci U S A. 2008;105:13045–50. doi: 10.1073/pnas.0806312105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mousavi SA, Berge KE, Berg T, Leren TP. Affinity and kinetics of proprotein convertase subtilisin/kexin type 9 binding to low-density lipoprotein receptors on HepG2 cells. FEBS J. 2011;278:2938–50. doi: 10.1111/j.1742-4658.2011.08219.x. [DOI] [PubMed] [Google Scholar]
- 31.Ai X, Fischer P, Palyha OC, Wisniewski D, Hubbard B, Akinsanya K, Strack AM, Ehrhardt AG. Utilizing HaloTag Technology to Track the Fate of PCSK9 from Intracellular vs. Extracellular Sources. Curr Chem Genomics. 2012;6:38–47. doi: 10.2174/1875397301206010038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qian YW, Schmidt RJ, Zhang Y, Chu S, Lin A, Wang H, Wang X, Beyer TP, Bensch WR, Li W, Ehsani ME, Lu D, Konrad RJ, Eacho PI, Moller DE, Karathanasis SK, Cao G. Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. J Lipid Res. 2007;48:1488–98. doi: 10.1194/jlr.M700071-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A. 2004;101:7100–5. doi: 10.1073/pnas.0402133101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, Lisbon E, Gutierrez M, Webb C, Wu R, Du Y, Kranz T, Gasparino E, Swergold GD. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–18. doi: 10.1056/NEJMoa1105803. [DOI] [PubMed] [Google Scholar]
- 35.Hare JF. Compartmentation and turnover of the low density lipoprotein receptor in skin fibroblasts. J Biol Chem. 1990;265:21758–63. [PubMed] [Google Scholar]
- 36.Michaely P, Li WP, Anderson RG, Cohen JC, Hobbs HH. The modular adaptor protein ARH is required for low density lipoprotein (LDL) binding and internalization but not for LDL receptor clustering in coated pits. J Biol Chem. 2004;279:34023–31. doi: 10.1074/jbc.M405242200. [DOI] [PubMed] [Google Scholar]
- 37.Chen XW, Wang H, Bajaj K, Zhang P, Meng ZX, Ma D, Bai Y, Liu HH, Adams E, Baines A, Yu G, Sartor MA, Zhang B, Yi Z, Lin J, Young SG, Schekman R, Ginsburg D. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. Elife. 2013;2:e00444. doi: 10.7554/eLife.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poirier S, Mamarbachi M, Chen WT, Lee AS, Mayer G. GRP94 Regulates Circulating Cholesterol Levels through Blockade of PCSK9-Induced LDLR Degradation. Cell Rep. 2015;13:2064–71. doi: 10.1016/j.celrep.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 39.Mousavi SA, Berge KE, Leren TP. The unique role of proprotein convertase subtilisin/kexin 9 in cholesterol homeostasis. J Intern Med. 2009;266:507–19. doi: 10.1111/j.1365-2796.2009.02167.x. [DOI] [PubMed] [Google Scholar]
- 40.Poirier S, Mayer G, Poupon V, McPherson PS, Desjardins R, Ly K, Asselin MC, Day R, Duclos FJ, Witmer M, Parker R, Prat A, Seidah NG. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem. 2009;284:28856–64. doi: 10.1074/jbc.M109.037085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kourimate S, Chetiveaux M, Jarnoux AL, Lalanne F, Costet P. Cellular and secreted pro-protein convertase subtilisin/kexin type 9 catalytic activity in hepatocytes. Atherosclerosis. 2009;206:134–40. doi: 10.1016/j.atherosclerosis.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 42.Poirier S, Hamouda HA, Villeneuve L, Demers A, Mayer G. Trafficking Dynamics of PCSK9-Induced LDLR Degradation: Focus on Human PCSK9 Mutations and C-Terminal Domain. PloS one. 2016;11:e0157230. doi: 10.1371/journal.pone.0157230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cameron J, Holla OL, Laerdahl JK, Kulseth MA, Berge KE, Leren TP. Mutation S462P in the PCSK9 gene reduces secretion of mutant PCSK9 without affecting the autocatalytic cleavage. Atherosclerosis. 2009;203:161–5. doi: 10.1016/j.atherosclerosis.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 44.Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chretien M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. 2003;100:928–33. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.PCSK9 proprotein convertase subtilisin/kexin type 9[Homo sapiens] Gene ID: 255738, updated on 18-Feb-2018
- 46.Costet P, Cariou B, Lambert G, Lalanne F, Lardeux B, Jarnoux AL, Grefhorst A, Staels B, Krempf M. Hepatic PCSK9 expression is regulated by nutritional status via insulin and sterol regulatory element-binding protein 1c. J Biol Chem. 2006;281:6211–8. doi: 10.1074/jbc.M508582200. [DOI] [PubMed] [Google Scholar]
- 47.Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res. 2008;49:399–409. doi: 10.1194/jlr.M700443-JLR200. [DOI] [PubMed] [Google Scholar]
- 48.Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, Prat A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1454–9. doi: 10.1161/01.ATV.0000134621.14315.43. [DOI] [PubMed] [Google Scholar]
- 49.Welder G, Zineh I, Pacanowski MA, Troutt JS, Cao G, Konrad RJ. High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J Lipid Res. 2010;51:2714–21. doi: 10.1194/jlr.M008144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Dong B, Park SW, Lee HS, Chen W, Liu J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem. 2009;284:28885–95. doi: 10.1074/jbc.M109.052407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, Park SW, Liu J. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. 2010;51:1486–95. doi: 10.1194/jlr.M003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ai D, Chen C, Han S, Ganda A, Murphy AJ, Haeusler R, Thorp E, Accili D, Horton JD, Tall AR. Regulation of hepatic LDL receptors by mTORC1 and PCSK9 in mice. J Clin Invest. 2012;122:1262–70. doi: 10.1172/JCI61919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Millar JS, Reyes-Soffer G, Jumes P, Dunbar RL, deGoma EM, Baer AL, Karmally W, Donovan DS, Rafeek H, Pollan L, Tohyama J, Johnson-Levonas AO, Wagner JA, Holleran S, Obunike J, Liu Y, Ramakrishnan R, Lassman ME, Gutstein DE, Ginsberg HN, Rader DJ. Anacetrapib lowers LDL by increasing ApoB clearance in mildly hypercholesterolemic subjects. J Clin Invest. 2015;125:2510–22. doi: 10.1172/JCI80025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raal F, Panz V, Immelman A, Pilcher G. Elevated PCSK9 levels in untreated patients with heterozygous or homozygous familial hypercholesterolemia and the response to high-dose statin therapy. J Am Heart Assoc. 2013;2:e000028. doi: 10.1161/JAHA.112.000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ason B, van der Hoorn JW, Chan J, Lee E, Pieterman EJ, Nguyen KK, Di M, Shetterly S, Tang J, Yeh WC, Schwarz M, Jukema JW, Scott R, Wasserman SM, Princen HM, Jackson S. PCSK9 inhibition fails to alter hepatic LDLR, circulating cholesterol, and atherosclerosis in the absence of ApoE. J Lipid Res. 2014;55:2370–9. doi: 10.1194/jlr.M053207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grefhorst A, McNutt MC, Lagace TA, Horton JD. Plasma PCSK9 preferentially reduces liver LDL receptors in mice. J Lipid Res. 2008;49:1303–11. doi: 10.1194/jlr.M800027-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stein EA, Raal FJ. Insights Into PCSK9, Low-Density Lipoprotein Receptor, and Low-Density Lipoprotein Cholesterol Metabolism: Of Mice and Man. Circulation. 2013;127:2372–4. doi: 10.1161/CIRCULATIONAHA.113.003360. [DOI] [PubMed] [Google Scholar]
- 58.Goldstein JL, Brown MS. The LDL receptor locus and the genetics of familial hypercholesterolemia. Annu Rev Genet. 1979;13:259–89. doi: 10.1146/annurev.ge.13.120179.001355. [DOI] [PubMed] [Google Scholar]
- 59.Goldstein JL, Dana SE, Brunschede GY, Brown MS. Genetic heterogeneity in familial hypercholesterolemia: evidence for two different mutations affecting functions of low-density lipoprotein receptor. Proc Natl Acad Sci U S A. 1975;72:1092–6. doi: 10.1073/pnas.72.3.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benjannet S, Rhainds D, Hamelin J, Nassoury N, Seidah NG. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J Biol Chem. 2006;281:30561–72. doi: 10.1074/jbc.M606495200. [DOI] [PubMed] [Google Scholar]
- 61.Lipari MT, Li W, Moran P, Kong-Beltran M, Sai T, Lai J, Lin SJ, Kolumam G, Zavala-Solorio J, Izrael-Tomasevic A, Arnott D, Wang J, Peterson AS, Kirchhofer D. Furin-cleaved proprotein convertase subtilisin/kexin type 9 (PCSK9) is active and modulates low density lipoprotein receptor and serum cholesterol levels. J Biol Chem. 2012;287:43482–91. doi: 10.1074/jbc.M112.380618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poirier S, Prat A, Marcinkiewicz E, Paquin J, Chitramuthu BP, Baranowski D, Cadieux B, Bennett HP, Seidah NG. Implication of the proprotein convertase NARC-1/PCSK9 in the development of the nervous system. J Neurochem. 2006;98:838–50. doi: 10.1111/j.1471-4159.2006.03928.x. [DOI] [PubMed] [Google Scholar]
- 63.Essalmani R, Susan-Resiga D, Chamberland A, Abifadel M, Creemers JW, Boileau C, Seidah NG, Prat A. In vivo evidence that furin from hepatocytes inactivates PCSK9. J Biol Chem. 2011;286:4257–63. doi: 10.1074/jbc.M110.192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, Trillard M, Abifadel M, Tebon A, Attie AD, Rader DJ, Boileau C, Brissette L, Chretien M, Prat A, Seidah NG. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279:48865–75. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- 65.Fan D, Yancey PG, Qiu S, Ding L, Weeber EJ, Linton MF, Fazio S. Self-association of human PCSK9 correlates with its LDLR-degrading activity. Biochemistry. 2008;47:1631–9. doi: 10.1021/bi7016359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun H, Samarghandi A, Zhang N, Yao Z, Xiong M, Teng BB. Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler Thromb Vasc Biol. 2012;32:1585–95. doi: 10.1161/ATVBAHA.112.250043. [DOI] [PubMed] [Google Scholar]
- 67.Kosenko T, Golder M, Leblond G, Weng W, Lagace TA. Low-density lipoprotein binds to proprotein convertase subtilisin/kexin type-9 (PCSK9) in human plasma and inhibits PCSK9-mediated LDL receptor degradation. J Biol Chem. 2013;288:8279–88. doi: 10.1074/jbc.M112.421370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Romagnuolo R, Scipione CA, Boffa MB, Marcovina SM, Seidah NG, Koschinsky ML. Lipoprotein(a) Catabolism Is Regulated by Proprotein Convertase Subtilisin/Kexin Type 9 through the Low Density Lipoprotein Receptor. J Biol Chem. 2015;290:11649–62. doi: 10.1074/jbc.M114.611988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tavori H, Christian D, Minnier J, Plubell D, Shapiro MD, Yeang C, Giunzioni I, Croyal M, Duell PB, Lambert G, Tsimikas S, Fazio S. PCSK9 Association With Lipoprotein(a) Circ Res. 2016;119:29–35. doi: 10.1161/CIRCRESAHA.116.308811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tavori H, Giunzioni I, Linton MF, Fazio S. Loss of plasma proprotein convertase subtilisin/kexin 9 (PCSK9) after lipoprotein apheresis. Circ Res. 2013;113:1290–5. doi: 10.1161/CIRCRESAHA.113.302655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fazio S, Minnier J, Shapiro MD, Tsimikas S, Tarugi P, Averna MR, Arca M, Tavori H. Threshold Effects of Circulating Angiopoietin-like 3 Levels on Plasma Lipoproteins. J Clin Endocrinol Metab. 2017 doi: 10.1210/jc.2016-4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lagace TA. PCSK9 and LDLR degradation: regulatory mechanisms in circulation and in cells. Curr Opin Lipidol. 2014;25:387–93. doi: 10.1097/MOL.0000000000000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Canuel M, Sun X, Asselin MC, Paramithiotis E, Prat A, Seidah NG. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Can Mediate Degradation of the Low Density Lipoprotein Receptor-Related Protein 1 (LRP-1) PloS one. 2013;8:e64145. doi: 10.1371/journal.pone.0064145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Park SW, Moon YA, Horton JD. Post-transcriptional Regulation of Low Density Lipoprotein Receptor Protein by Proprotein Convertase Subtilisin/Kexin Type 9a in Mouse Liver. JBiolChem. 2004;279:50630–50638. doi: 10.1074/jbc.M410077200. [DOI] [PubMed] [Google Scholar]
- 75.Giunzioni I, Tavori H, Ding L, Zhang Y, Predazzi IM, Linton MF, Fazio S. Mechanisms of Direct PCSK9 Effect on Atherosclerosis. Circulation Reserach. 2014:2–8. 2014 AHA Late-Breaking Basic Science Abstracts. [Google Scholar]
- 76.Levy E, Ben Djoudi Ouadda A, Spahis S, Sane AT, Garofalo C, Grenier E, Emonnot L, Yara S, Couture P, Beaulieu JF, Menard D, Seidah NG, Elchebly M. PCSK9 plays a significant role in cholesterol homeostasis and lipid transport in intestinal epithelial cells. Atherosclerosis. 2013;227:297–306. doi: 10.1016/j.atherosclerosis.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 77.Labonte P, Begley S, Guevin C, Asselin MC, Nassoury N, Mayer G, Prat A, Seidah NG. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology. 2009;50:17–24. doi: 10.1002/hep.22911. [DOI] [PubMed] [Google Scholar]
- 78.Jonas MC, Costantini C, Puglielli L. PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep. 2008;9:916–22. doi: 10.1038/embor.2008.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sharotri V, Collier DM, Olson DR, Zhou R, Snyder PM. Regulation of epithelial sodium channel trafficking by proprotein convertase subtilisin/kexin type 9 (PCSK9) J Biol Chem. 2012;287:19266–74. doi: 10.1074/jbc.M112.363382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DeVay RM, Shelton Dl Fau-Liang H, Liang H. Characterization of proprotein convertase subtilisin/kexin type 9 (PCSK9) trafficking reveals a novel lysosomal targeting mechanism via amyloid precursor-like protein 2 (APLP2) J Biol Chem. 2013;288:10805–18. doi: 10.1074/jbc.M113.453373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Seidah NG, Poirier S, Denis M, Parker R, Miao B, Mapelli C, Prat A, Wassef H, Davignon J, Hajjar KA, Mayer G. Annexin A2 is a natural extrahepatic inhibitor of the PCSK9-induced LDL receptor degradation. PloS one. 2012;7:e41865. doi: 10.1371/journal.pone.0041865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gustafsen C, Olsen D, Vilstrup J, Lund S, Reinhardt A, Wellner N, Larsen T, Andersen CBF, Weyer K, Li JP, Seeberger PH, Thirup S, Madsen P, Glerup S. Heparan sulfate proteoglycans present PCSK9 to the LDL receptor. Nat Commun. 2017;8:503. doi: 10.1038/s41467-017-00568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, Thacker BE, Basu D, Lee RG, Mullick AE, Graham MJ, Goldberg IJ, Crooke RM, Witztum JL, Esko JD. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest. 2016;126:2855–66. doi: 10.1172/JCI86610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chan DC, Pang J, McQuillan BM, Hung J, Beilby JP, Barrett PH, Watts GF. Plasma Proprotein Convertase Subtilisin Kexin Type 9 as a Predictor of Carotid Atherosclerosis in Asymptomatic Adults. Heart Lung Circ. 2016;25:520–5. doi: 10.1016/j.hlc.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 85.Lee CJ, Lee YH, Park SW, Kim KJ, Park S, Youn JC, Lee SH, Kang SM, Jang Y. Association of serum proprotein convertase subtilisin/kexin type 9 with carotid intima media thickness in hypertensive subjects. Metabolism. 2013;62:845–50. doi: 10.1016/j.metabol.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 86.Huijgen R, Fouchier SW, Denoun M, Hutten BA, Vissers MN, Lambert G, Kastelein JJ. Plasma levels of PCSK9 and phenotypic variability in familial hypercholesterolemia. J Lipid Res. 2012;53:979–83. doi: 10.1194/jlr.P023994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Toth S, Fedacko J, Pekarova T, Hertelyova Z, Katz M, Mughees A, Kuzma J, Stefanic P, Kopolovets I, Pella D. Elevated Circulating PCSK9 Concentrations Predict Subclinical Atherosclerotic Changes in Low Risk Obese and Non-Obese Patients. Cardiol Ther. 2017;6:281–289. doi: 10.1007/s40119-017-0092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhu YM, Anderson TJ, Sikdar K, Fung M, McQueen MJ, Lonn EM, Verma S. Association of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) With Cardiovascular Risk in Primary Prevention. Arterioscler Thromb Vasc Biol. 2015;35:2254–9. doi: 10.1161/ATVBAHA.115.306172. [DOI] [PubMed] [Google Scholar]
- 89.Xie W, Liu J, Wang W, Wang M, Qi Y, Zhao F, Sun J, Liu J, Li Y, Zhao D. Association between plasma PCSK9 levels and 10-year progression of carotid atherosclerosis beyond LDL-C: A cohort study. Int J Cardiol. 2016;215:293–8. doi: 10.1016/j.ijcard.2016.04.103. [DOI] [PubMed] [Google Scholar]
- 90.Alonso R, Mata P, Muniz O, Fuentes-Jimenez F, Diaz JL, Zambon D, Tomas M, Martin C, Moyon T, Croyal M, Thedrez A, Lambert G. PCSK9 and lipoprotein (a) levels are two predictors of coronary artery calcification in asymptomatic patients with familial hypercholesterolemia. Atherosclerosis. 2016;254:249–253. doi: 10.1016/j.atherosclerosis.2016.08.038. [DOI] [PubMed] [Google Scholar]
- 91.Zhao X, Zhang HW, Li S, Zhang Y, Xu RX, Zhu CG, Wu NQ, Guo YL, Qing P, Li XL, Liu G, Dong Q, Sun J, Li JJ. Association between plasma proprotein convertase subtisilin/kexin type 9 concentration and coronary artery calcification. Ann Clin Biochem. 2018;55:158–164. doi: 10.1177/0004563217695351. [DOI] [PubMed] [Google Scholar]
- 92.Cheng JM, Oemrawsingh RM, Garcia-Garcia HM, Boersma E, van Geuns RJ, Serruys PW, Kardys I, Akkerhuis KM. PCSK9 in relation to coronary plaque inflammation: Results of the ATHEROREMO-IVUS study. Atherosclerosis. 2016;248:117–22. doi: 10.1016/j.atherosclerosis.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 93.Ridker PM, Rifai N, Bradwin G, Rose L. Plasma proprotein convertase subtilisin/kexin type 9 levels and the risk of first cardiovascular events. Eur Heart J. 2016;37:554–60. doi: 10.1093/eurheartj/ehv568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leander K, Malarstig A, Van’t Hooft FM, Hyde C, Hellenius ML, Troutt JS, Konrad RJ, Ohrvik J, Hamsten A, de Faire U. Circulating Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Predicts Future Risk of Cardiovascular Events Independently of Established Risk Factors. Circulation. 2016;133:1230–9. doi: 10.1161/CIRCULATIONAHA.115.018531. [DOI] [PubMed] [Google Scholar]
- 95.Laugsand LE, Asvold BO, Vatten LJ, Janszky I. Circulating PCSK9 and Risk of Myocardial Infarction. JACC: Basic To Translational Science. 2016;1:568–575. doi: 10.1016/j.jacbts.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Werner C, Hoffmann MM, Winkler K, Bohm M, Laufs U. Risk prediction with proprotein convertase subtilisin/kexin type 9 (PCSK9) in patients with stable coronary disease on statin treatment. Vascul Pharmacol. 2014;62:94–102. doi: 10.1016/j.vph.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 97.Li JJ, Li S, Zhang Y, Xu RX, Guo YL, Zhu CG, Wu NQ, Qing P, Gao Y, Sun J, Liu G, Dong Q. Proprotein Convertase Subtilisin/Kexin type 9, C-Reactive Protein, Coronary Severity, and Outcomes in Patients With Stable Coronary Artery Disease: A Prospective Observational Cohort Study. Medicine (Baltimore) 2015;94:e2426. doi: 10.1097/MD.0000000000002426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gencer B, Montecucco F, Nanchen D, Carbone F, Klingenberg R, Vuilleumier N, Aghlmandi S, Heg D, Raber L, Auer R, Juni P, Windecker S, Luscher TF, Matter CM, Rodondi N, Mach F. Prognostic value of PCSK9 levels in patients with acute coronary syndromes. Eur Heart J. 2016;37:546–53. doi: 10.1093/eurheartj/ehv637. [DOI] [PubMed] [Google Scholar]
- 99.Vlachopoulos C, Terentes-Printzios D, Georgiopoulos G, Skoumas I, Koutagiar I, Ioakeimidis N, Stefanadis C, Tousoulis D. Prediction of cardiovascular events with levels of proprotein convertase subtilisin/kexin type 9: A systematic review and meta-analysis. Atherosclerosis. 2016;252:50–60. doi: 10.1016/j.atherosclerosis.2016.07.922. [DOI] [PubMed] [Google Scholar]
- 100.Xiao Y, Peng C, Huang W, Zhang J, Gao Y, Kim JH, Yeoh EK, Su X. Circulating Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Concentration and Risk of Cardiovascular Events- Systematic Review and Meta-Analysis of Prospective Studies. Circ J. 2017;81:1150–1157. doi: 10.1253/circj.CJ-16-1142. [DOI] [PubMed] [Google Scholar]
- 101.Qiu C, Zhou Q, Li X, Zhang Z, Zeng P, Cao Z, Pan B, Li X, Chen AF. High circulating proprotein convertase subtilisin/Kexin type 9 concentration associates with cardiovascular risk: A meta-analysis of cohort studies. Medicine (Baltimore) 2017;96:e8848. doi: 10.1097/MD.0000000000008848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tavori H, Christian D, Minnier J, Plubell D, Shapiro MD, Yeang C, Giunzioni I, Croyal M, Duell PB, Lambert G, Tsimikas S, Fazio S. PCSK9 Association With Lipoprotein(a) Circulation Research. 2016;119:29–35. doi: 10.1161/CIRCRESAHA.116.308811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, Mewada A, Kahn J, Afonso L, Williams KA, Sr, Flack JM. Effect of Long-Term Exposure to Lower Low-Density Lipoprotein Cholesterol Beginning Early in Life on the Risk of Coronary Heart Disease: A Mendelian Randomization Analysis. Journal of the American College of Cardiology. 2012;60:2631–2639. doi: 10.1016/j.jacc.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 104.Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, Voros S, Giugliano RP, Davey Smith G, Fazio S, Sabatine MS. Variation in PCSK9 and HMGCR and Risk of Cardiovascular Disease and Diabetes. N Engl J Med. 2016;375:2144–2153. doi: 10.1056/NEJMoa1604304. [DOI] [PubMed] [Google Scholar]
- 105.Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AF, Barbalic M, Gieger C, Absher D, Aherrahrou Z, Allayee H, Altshuler D, Anand SS, Andersen K, Anderson JL, Ardissino D, Ball SG, Balmforth AJ, Barnes TA, Becker DM, Becker LC, Berger K, Bis JC, Boekholdt SM, Boerwinkle E, Braund PS, Brown MJ, Burnett MS, Buysschaert I, Cardiogenics. Carlquist JF, Chen L, Cichon S, Codd V, Davies RW, Dedoussis G, Dehghan A, Demissie S, Devaney JM, Diemert P, Do R, Doering A, Eifert S, Mokhtari NE, Ellis SG, Elosua R, Engert JC, Epstein SE, de Faire U, Fischer M, Folsom AR, Freyer J, Gigante B, Girelli D, Gretarsdottir S, Gudnason V, Gulcher JR, Halperin E, Hammond N, Hazen SL, Hofman A, Horne BD, Illig T, Iribarren C, Jones GT, Jukema JW, Kaiser MA, Kaplan LM, Kastelein JJ, Khaw KT, Knowles JW, Kolovou G, Kong A, Laaksonen R, Lambrechts D, Leander K, Lettre G, Li M, Lieb W, Loley C, Lotery AJ, Mannucci PM, Maouche S, Martinelli N, McKeown PP, Meisinger C, Meitinger T, Melander O, Merlini PA, Mooser V, Morgan T, Muhleisen TW, Muhlestein JB, Munzel T, Musunuru K, Nahrstaedt J, Nelson CP, Nothen MM, Olivieri O, Patel RS, Patterson CC, Peters A, Peyvandi F, Qu L, Quyyumi AA, Rader DJ, Rallidis LS, Rice C, Rosendaal FR, Rubin D, Salomaa V, Sampietro ML, Sandhu MS, Schadt E, Schafer A, Schillert A, Schreiber S, Schrezenmeir J, Schwartz SM, Siscovick DS, Sivananthan M, Sivapalaratnam S, Smith A, Smith TB, Snoep JD, Soranzo N, Spertus JA, Stark K, Stirrups K, Stoll M, Tang WH, Tennstedt S, Thorgeirsson G, Thorleifsson G, Tomaszewski M, Uitterlinden AG, van Rij AM, Voight BF, Wareham NJ, Wells GA, Wichmann HE, Wild PS, Willenborg C, Witteman JC, Wright BJ, Ye S, Zeller T, Ziegler A, Cambien F, Goodall AH, Cupples LA, Quertermous T, Marz W, Hengstenberg C, Blankenberg S, Ouwehand WH, Hall AS, Deloukas P, Thompson JR, Stefansson K, Roberts R, Thorsteinsdottir U, O’Donnell CJ, McPherson R, Erdmann J, Consortium CA. Samani NJ. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–8. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Consortium CAD. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups K, Konig IR, Cazier JB, Johansson A, Hall AS, Lee JY, Willer CJ, Chambers JC, Esko T, Folkersen L, Goel A, Grundberg E, Havulinna AS, Ho WK, Hopewell JC, Eriksson N, Kleber ME, Kristiansson K, Lundmark P, Lyytikainen LP, Rafelt S, Shungin D, Strawbridge RJ, Thorleifsson G, Tikkanen E, Van Zuydam N, Voight BF, Waite LL, Zhang W, Ziegler A, Absher D, Altshuler D, Balmforth AJ, Barroso I, Braund PS, Burgdorf C, Claudi-Boehm S, Cox D, Dimitriou M, Do R, Consortium D, Consortium C. Doney AS, El Mokhtari N, Eriksson P, Fischer K, Fontanillas P, Franco-Cereceda A, Gigante B, Groop L, Gustafsson S, Hager J, Hallmans G, Han BG, Hunt SE, Kang HM, Illig T, Kessler T, Knowles JW, Kolovou G, Kuusisto J, Langenberg C, Langford C, Leander K, Lokki ML, Lundmark A, McCarthy MI, Meisinger C, Melander O, Mihailov E, Maouche S, Morris AD, Muller-Nurasyid M, Mu TC, Nikus K, Peden JF, Rayner NW, Rasheed A, Rosinger S, Rubin D, Rumpf MP, Schafer A, Sivananthan M, Song C, Stewart AF, Tan ST, Thorgeirsson G, van der Schoot CE, Wagner PJ, Wellcome Trust Case Control C. Wells GA, Wild PS, Yang TP, Amouyel P, Arveiler D, Basart H, Boehnke M, Boerwinkle E, Brambilla P, Cambien F, Cupples AL, de Faire U, Dehghan A, Diemert P, Epstein SE, Evans A, Ferrario MM, Ferrieres J, Gauguier D, Go AS, Goodall AH, Gudnason V, Hazen SL, Holm H, Iribarren C, Jang Y, Kahonen M, Kee F, Kim HS, Klopp N, Koenig W, Kratzer W, Kuulasmaa K, Laakso M, Laaksonen R, Lee JY, Lind L, Ouwehand WH, Parish S, Park JE, Pedersen NL, Peters A, Quertermous T, Rader DJ, Salomaa V, Schadt E, Shah SH, Sinisalo J, Stark K, Stefansson K, Tregouet DA, Virtamo J, Wallentin L, Wareham N, Zimmermann ME, Nieminen MS, Hengstenberg C, Sandhu MS, Pastinen T, Syvanen AC, Hovingh GK, Dedoussis G, Franks PW, Lehtimaki T, Metspalu A, Zalloua PA, Siegbahn A, Schreiber S, Ripatti S, Blankenberg SS, Perola M, Clarke R, Boehm BO, O’Donnell C, Reilly MP, Marz W, Collins R, Kathiresan S, Hamsten A, Kooner JS, Thorsteinsdottir U, Danesh J, Palmer CN, Roberts R, Watkins H, Schunkert H, Samani NJ. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR, Committee FS and Investigators Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 108.Baigent C, Blackwell L, Emberson J, Holland L, Reith C, Bhala N, Peto R, Barnes E, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, Watts GF, Boren J, Fazio S, Horton JD, Masana L, Nicholls SJ, Nordestgaard BG, van de Sluis B, Taskinen MR, Tokgozoglu L, Landmesser U, Laufs U, Wiklund O, Stock JK, Chapman MJ, Catapano AL. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017 doi: 10.1093/eurheartj/ehx144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RM. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. New England Journal of Medicine. 2015;372:2387–2397. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 111.Khera AV, Natarajan P, Kathiresan S. The future of low-density lipoprotein cholesterol lowering therapy: An end to statin exceptionalism? Eur J Prev Cardiol. 2015 doi: 10.1177/2047487315600818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J. Molecular basis for LDL receptor recognition by PCSK9. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1820–1825. doi: 10.1073/pnas.0712064105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shapiro MD, Fazio S, Tavori H. Targeting PCSK9 for therapeutic gains. Curr Atheroscler Rep. 2015;17:499. doi: 10.1007/s11883-015-0499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ridker PM, Revkin J, Amarenco P, Brunell R, Curto M, Civeira F, Flather M, Glynn RJ, Gregoire J, Jukema JW, Karpov Y, Kastelein JJP, Koenig W, Lorenzatti A, Manga P, Masiukiewicz U, Miller M, Mosterd A, Murin J, Nicolau JC, Nissen S, Ponikowski P, Santos RD, Schwartz PF, Soran H, White H, Wright RS, Vrablik M, Yunis C, Shear CL, Tardif JC, Investigators SCO Cardiovascular Efficacy and Safety of Bococizumab in High-Risk Patients. N Engl J Med. 2017;376:1527–1539. doi: 10.1056/NEJMoa1701488. [DOI] [PubMed] [Google Scholar]
- 115.Shapiro MD, Miles J, Tavori H, Fazio S. Diagnosing Resistance to a Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitor. Ann Intern Med. 2017 doi: 10.7326/M17-2485. [DOI] [PubMed] [Google Scholar]
- 116.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–55. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bernards R. Exploring the uses of RNAi–gene knockdown and the Nobel Prize. N Engl J Med. 2006;355:2391–3. doi: 10.1056/NEJMp068242. [DOI] [PubMed] [Google Scholar]
- 118.Fitzgerald K, Simon A, White S, Borodovsky A, Patel N, Bettencourt B, Clausen V, Wijngaard P, Horton J, Kauffman R, Kallend D. A Subcutaneously Administered Investigational RNAi Therapeutic (ALN-PCSsc), Targeting PCSK9 for the Treatment of Hypercholesterolemia: Initial Phase 1 Study Results. European Society of Cardiology. 2015 Abstract presented at. [Google Scholar]
- 119.Ray KK, Landmesser U, Leiter LA, Kallend D, Dufour R, Karakas M, Hall T, Troquay RP, Turner T, Visseren FL, Wijngaard P, Wright RS, Kastelein JJ. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N Engl J Med. 2017;376:1430–1440. doi: 10.1056/NEJMoa1615758. [DOI] [PubMed] [Google Scholar]
- 120.Nair JK, Willoughby JL, Chan A, Charisse K, Alam MR, Wang Q, Hoekstra M, Kandasamy P, Kel’in AV, Milstein S, Taneja N, O’Shea J, Shaikh S, Zhang L, van der Sluis RJ, Jung ME, Akinc A, Hutabarat R, Kuchimanchi S, Fitzgerald K, Zimmermann T, van Berkel TJ, Maier MA, Rajeev KG, Manoharan M. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136:16958–61. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 121.Landlinger C, Pouwer MG, Juno C, van der Hoorn JWA, Pieterman EJ, Jukema JW, Staffler G, Princen HMG, Galabova G. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. European heart journal. 2017;38:2499–2507. doi: 10.1093/eurheartj/ehx260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pan Y, Zhou Y, Wu H, Chen X, Hu X, Zhang H, Zhou Z, Qiu Z, Liao Y. A Therapeutic Peptide Vaccine Against PCSK9. Sci Rep. 2017;7:12534. doi: 10.1038/s41598-017-13069-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang Y, Ultsch M, Skelton NJ, Burdick DJ, Beresini MH, Li W, Kong-Beltran M, Peterson A, Quinn J, Chiu C, Wu Y, Shia S, Moran P, Di Lello P, Eigenbrot C, Kirchhofer D. Discovery of a cryptic peptide-binding site on PCSK9 and design of antagonists. Nat Struct Mol Biol. 2017;24:848–856. doi: 10.1038/nsmb.3453. [DOI] [PubMed] [Google Scholar]
- 124.Seidah NG. Insights into a PCSK9 structural groove: a harbinger of new drugs to reduce LDL-cholesterol. Nat Struct Mol Biol. 2017;24:785–786. doi: 10.1038/nsmb.3471. [DOI] [PubMed] [Google Scholar]
- 125.Chadwick AC, Wang X, Musunuru K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler Thromb Vasc Biol. 2017;37:1741–1747. doi: 10.1161/ATVBAHA.117.309881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg-Hansen A, European Atherosclerosis Society Consensus P Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–53. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Benn M, Nordestgaard BG, Frikke-Schmidt R, Tybjaerg-Hansen A. Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer’s disease and Parkinson’s disease: Mendelian randomisation study. BMJ. 2017;357:j1648. doi: 10.1136/bmj.j1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Raal FJ, Giugliano RP, Sabatine MS, Koren MJ, Langslet G, Bays H, Blom D, Eriksson M, Dent R, Wasserman SM, Huang F, Xue A, Albizem M, Scott R, Stein EA. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol. 2014;63:1278–88. doi: 10.1016/j.jacc.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 129.Gaudet D, Kereiakes DJ, McKenney JM, Roth EM, Hanotin C, Gipe D, Du Y, Ferrand AC, Ginsberg HN, Stein EA. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials) Am J Cardiol. 2014;114:711–5. doi: 10.1016/j.amjcard.2014.05.060. [DOI] [PubMed] [Google Scholar]
- 130.Raal FJ, Giugliano RP, Sabatine MS, Koren MJ, Blom D, Seidah NG, Honarpour N, Lira A, Xue A, Chiruvolu P. PCSK9 inhibition-mediated reduction in Lp (a) with evolocumab: an analysis of 10 clinical trials and the LDL receptor’s role. Journal of lipid research. 2016;57:1086–1096. doi: 10.1194/jlr.P065334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rader DJ, Mann WA, Cain W, Kraft HG, Usher D, Zech LA, Hoeg JM, Davignon J, Lupien P, Grossman M. The low density lipoprotein receptor is not required for normal catabolism of Lp(a) in humans. The Journal of Clinical Investigation. 95:1403–1408. doi: 10.1172/JCI117794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Boffa MB, Koschinsky ML. Update on Lipoprotein(a) as a Cardiovascular Risk Factor and Mediator. Current Atherosclerosis Reports. 2013;15:360. doi: 10.1007/s11883-013-0360-6. [DOI] [PubMed] [Google Scholar]
- 133.Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–20. doi: 10.1161/CIRCULATIONAHA.113.004678. [DOI] [PubMed] [Google Scholar]
- 134.Saavedra YG, Dufour R, Davignon J, Baass A. PCSK9 R46L, lower LDL, and cardiovascular disease risk in familial hypercholesterolemia: a cross-sectional cohort study. Arterioscler Thromb Vasc Biol. 2014;34:2700–5. doi: 10.1161/ATVBAHA.114.304406. [DOI] [PubMed] [Google Scholar]
- 135.Saavedra YGL, Dufour R, Baass A. Familial hypercholesterolemia: PCSK9 InsLEU genetic variant and prediabetes/diabetes risk. J Clin Lipidol. 2015;9:786–793 e1. doi: 10.1016/j.jacl.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 136.Lakoski SG, Lagace TA, Cohen JC, Horton JD, Hobbs HH. Genetic and metabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab. 2009;94:2537–43. doi: 10.1210/jc.2009-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Edmiston JB, Brooks N, Tavori H, Minnier J, Duell B, Purnell JQ, Kaufman T, Wojcik C, Voros S, Fazio S, Shapiro MD. Discordant response of low-density lipoprotein cholesterol and lipoprotein(a) levels to monoclonal antibodies targeting proprotein convertase subtilisin/kexin type 9. J Clin Lipidol. 2017;11:667–673. doi: 10.1016/j.jacl.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 138.Yeang C, Gordts PL, Tsimikas S. Novel Lipoprotein(a) Catabolism Pathway via Apolipoprotein(a) Recycling: Adding the Plasminogen Receptor PlgRKT to the List. Circ Res. 2017;120:1050–1052. doi: 10.1161/CIRCRESAHA.117.310700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Berliner JA, Leitinger N, Tsimikas S. The role of oxidized phospholipids in atherosclerosis. J Lipid Res. 2009;50(Suppl):S207–12. doi: 10.1194/jlr.R800074-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Villard EF, Thedrez A, Blankenstein J, Croyal M, Tran TT, Poirier B, Le Bail JC, Illiano S, Nobecourt E, Krempf M, Blom DJ, Marais AD, Janiak P, Muslin AJ, Guillot E, Lambert G. PCSK9 Modulates the Secretion But Not the Cellular Uptake of Lipoprotein(a) Ex Vivo: An Effect Blunted by Alirocumab. JACC Basic Transl Sci. 2016;1:419–427. doi: 10.1016/j.jacbts.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sun XM, Eden ER, Tosi I, Neuwirth CK, Wile D, Naoumova RP, Soutar AK. Evidence for effect of mutant PCSK9 on apolipoprotein B secretion as the cause of unusually severe dominant hypercholesterolaemia. Hum Mol Genet. 2005;14:1161–9. doi: 10.1093/hmg/ddi128. [DOI] [PubMed] [Google Scholar]
- 142.Lalanne F, Lambert G, Amar MJ, Chetiveaux M, Zair Y, Jarnoux AL, Ouguerram K, Friburg J, Seidah NG, Brewer HB, Jr, Krempf M, Costet P. Wild-type PCSK9 inhibits LDL clearance but does not affect apoB-containing lipoprotein production in mouse and cultured cells. J Lipid Res. 2005;46:1312–9. doi: 10.1194/jlr.M400396-JLR200. [DOI] [PubMed] [Google Scholar]
- 143.Lambert G, Jarnoux AL, Pineau T, Pape O, Chetiveaux M, Laboisse C, Krempf M, Costet P. Fasting induces hyperlipidemia in mice overexpressing proprotein convertase subtilisin kexin type 9: lack of modulation of very-low-density lipoprotein hepatic output by the low-density lipoprotein receptor. Endocrinology. 2006;147:4985–95. doi: 10.1210/en.2006-0098. [DOI] [PubMed] [Google Scholar]
- 144.Tavori H, Giunzioni I, Predazzi IM, Plubell D, Shivinsky A, Miles J, Devay RM, Liang H, Rashid S, Linton MF, Fazio S. Human PCSK9 promotes hepatic lipogenesis and atherosclerosis development via apoE- and LDLR-mediated mechanisms. Cardiovasc Res. 2016;110:268–78. doi: 10.1093/cvr/cvw053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Le May C, Kourimate S, Langhi C, Chetiveaux M, Jarry A, Comera C, Collet X, Kuipers F, Krempf M, Cariou B, Costet P. Proprotein convertase subtilisin kexin type 9 null mice are protected from postprandial triglyceridemia. Arterioscler Thromb Vasc Biol. 2009;29:684–90. doi: 10.1161/ATVBAHA.108.181586. [DOI] [PubMed] [Google Scholar]
- 146.Rashid S, Tavori H, Brown PE, Linton MF, He J, Giunzioni I, Fazio S. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation. 2014;130:431–41. doi: 10.1161/CIRCULATIONAHA.113.006720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ouguerram K, Chetiveaux M, Zair Y, Costet P, Abifadel M, Varret M, Boileau C, Magot T, Krempf M. Apolipoprotein B100 metabolism in autosomal-dominant hypercholesterolemia related to mutations in PCSK9. Arterioscler Thromb Vasc Biol. 2004;24:1448–53. doi: 10.1161/01.ATV.0000133684.77013.88. [DOI] [PubMed] [Google Scholar]
- 148.Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, Wasserman SM, Stein EA, Investigators T Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:341–50. doi: 10.1016/S0140-6736(14)61374-X. [DOI] [PubMed] [Google Scholar]
- 149.Stein EA, Gipe D, Bergeron J, Gaudet D, Weiss R, Dufour R, Wu R, Pordy R. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36. doi: 10.1016/S0140-6736(12)60771-5. [DOI] [PubMed] [Google Scholar]
- 150.Raal F, Scott R, Somaratne R, Bridges I, Li G, Wasserman SM, Stein EA. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation. 2012;126:2408–17. doi: 10.1161/CIRCULATIONAHA.112.144055. [DOI] [PubMed] [Google Scholar]
- 151.Roth EM, Moriarty PM, Bergeron J, Langslet G, Manvelian G, Zhao J, Baccara-Dinet MT, Rader DJ, investigators OCI A phase III randomized trial evaluating alirocumab 300 mg every 4 weeks as monotherapy or add-on to statin: ODYSSEY CHOICE I. Atherosclerosis. 2016;254:254–262. doi: 10.1016/j.atherosclerosis.2016.08.043. [DOI] [PubMed] [Google Scholar]
- 152.Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, Koenig W, Somaratne R, Kassahun H, Yang J, Wasserman SM, Scott R, Ungi I, Podolec J, Ophuis AO, Cornel JH, Borgman M, Brennan DM, Nissen SE. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA. 2016;316:2373–2384. doi: 10.1001/jama.2016.16951. [DOI] [PubMed] [Google Scholar]
- 153.Reyes-Soffer G, Pavlyha M, Ngai C, Thomas T, Holleran S, Ramakrishnan R, Karmally W, Nandakumar R, Fontanez N, Obunike J, Marcovina SM, Lichtenstein AH, Matthan NR, Matta J, Maroccia M, Becue F, Poitiers F, Swanson B, Cowan L, Sasiela WJ, Surks HK, Ginsberg HN. Effects of PCSK9 Inhibition With Alirocumab on Lipoprotein Metabolism in Healthy Humans. Circulation. 2017;135:352–362. doi: 10.1161/CIRCULATIONAHA.116.025253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci U S A. 2005;102:5374–9. doi: 10.1073/pnas.0501652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chan JC, Piper DE, Cao Q, Liu D, King C, Wang W, Tang J, Liu Q, Higbee J, Xia Z, Di Y, Shetterly S, Arimura Z, Salomonis H, Romanow WG, Thibault ST, Zhang R, Cao P, Yang XP, Yu T, Lu M, Retter MW, Kwon G, Henne K, Pan O, Tsai MM, Fuchslocher B, Yang E, Zhou L, Lee KJ, Daris M, Sheng J, Wang Y, Shen WD, Yeh WC, Emery M, Walker NP, Shan B, Schwarz M, Jackson SM. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci U S A. 2009;106:9820–5. doi: 10.1073/pnas.0903849106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Choi S, Korstanje R. Proprotein convertases in high-density lipoprotein metabolism. Biomark Res. 2013;1:27. doi: 10.1186/2050-7771-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Robinson JG, Nedergaard BS, Rogers WJ, Fialkow J, Neutel JM, Ramstad D, Somaratne R, Legg JC, Nelson P, Scott R, Wasserman SM, Weiss R, Investigators L Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA. 2014;311:1870–82. doi: 10.1001/jama.2014.4030. [DOI] [PubMed] [Google Scholar]
- 158.Smilde TJ, van Wissen S, Wollersheim H, Trip MD, Kastelein JJ, Stalenhoef AF. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet. 2001;357:577–81. doi: 10.1016/s0140-6736(00)04053-8. [DOI] [PubMed] [Google Scholar]
- 159.Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, Scott R, Koren MJ, Stein EA. Efficacy and Safety of Evolocumab in Reducing Lipids and Cardiovascular Events. New England Journal of Medicine. 2015;372:1500–1509. doi: 10.1056/NEJMoa1500858. [DOI] [PubMed] [Google Scholar]
- 160.Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, Koren MJ, Lepor NE, Lorenzato C, Pordy R, Chaudhari U, Kastelein JJ, Investigators OLT Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99. doi: 10.1056/NEJMoa1501031. [DOI] [PubMed] [Google Scholar]
- 161.Nicholls SJ, Ballantyne CM, Barter PJ, Chapman MJ, Erbel RM, Libby P, Raichlen JS, Uno K, Borgman M, Wolski K, Nissen SE. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365:2078–87. doi: 10.1056/NEJMoa1110874. [DOI] [PubMed] [Google Scholar]
- 162.Cholesterol Treatment Trialists C. Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–90. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Giugliano RP, Pedersen TR, Park JG, De Ferrari GM, Gaciong ZA, Ceska R, Toth K, Gouni-Berthold I, Lopez-Miranda J, Schiele F, Mach F, Ott BR, Kanevsky E, Pineda AL, Somaratne R, Wasserman SM, Keech AC, Sever PS, Sabatine MS, Investigators F Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the FOURIER trial. Lancet. 2017;390:1962–1971. doi: 10.1016/S0140-6736(17)32290-0. [DOI] [PubMed] [Google Scholar]
- 164.Sabatine MS. Clinical benefit of evolocumab in patients with a history of MI: an analysis from FOURIER; Presented at: American Heart Association 2017 Scientific Sessions; November 13, 2017; Anaheim, CA. [Google Scholar]
- 165.Bonaca MP, Nault P, Giugliano RP, Keech AC, Pineda AL, Kanevsky E, Kuder J, Murphy SA, Jukema JW, Lewis BS, Tokgozoglu L, Somaratne R, Sever PS, Pedersen TR, Sabatine MS. Low-Density Lipoprotein Cholesterol Lowering With Evolocumab and Outcomes in Patients With Peripheral Artery Disease: Insights From the FOURIER Trial (Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk) Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.117.032235. [DOI] [PubMed] [Google Scholar]
- 166.Ridker PM, Tardif JC, Amarenco P, Duggan W, Glynn RJ, Jukema JW, Kastelein JJ, Kim AM, Koenig W, Nissen S, Revkin J, Rose LM, Santos RD, Schwartz PF, Shear CL, Yunis C, Investigators S Lipid-Reduction Variability and Antidrug-Antibody Formation with Bococizumab. N Engl J Med. 2017 doi: 10.1056/NEJMoa1614062. [DOI] [PubMed] [Google Scholar]
- 167.Ridker PM, Revkin J, Amarenco P, Brunell R, Curto M, Civeira F, Flather M, Glynn RJ, Gregoire J, Jukema JW, Karpov Y, Kastelein JJ, Koenig W, Lorenzatti A, Manga P, Masiukiewicz U, Miller M, Mosterd A, Murin J, Nicolau JC, Nissen S, Ponikowski P, Santos RD, Schwartz PF, Soran H, White H, Wright RS, Vrablik M, Yunis C, Shear CL, Tardif JC, Investigators SCO Cardiovascular Efficacy and Safety of Bococizumab in High-Risk Patients. N Engl J Med. 2017 doi: 10.1056/NEJMoa1701488. [DOI] [PubMed] [Google Scholar]
- 168.Giugliano RP, Mach F, Zavitz K, Kurtz C, Im K, Kanevsky E, Schneider J, Wang H, Keech A, Pedersen TR, Sabatine MS, Sever PS, Robinson JG, Honarpour N, Wasserman SM, Ott BR, Investigators E Cognitive Function in a Randomized Trial of Evolocumab. N Engl J Med. 2017;377:633–643. doi: 10.1056/NEJMoa1701131. [DOI] [PubMed] [Google Scholar]
- 169.Mefford MT, Rosenson RS, Cushman M, Farkouh ME, McClure LA, Wadley VG, Irvin MR, Bittner VA, Safford MM, Somaratne R, Monda KL, Muntner P, Levitan EB. PCSK9 Variants, LDL-Cholesterol, and Neurocognitive Impairment: The REasons for Geographic and Racial Differences in Stroke (REGARDS) Study. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.117.029785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Harvey PD, Sabbagh MN, Harrison JE, Ginsberg HN, Chapman MJ, Manvelian G, Moryusef A, Mandel J, Farnier M. No evidence of neurocognitive adverse events associated with alirocumab treatment in 3340 patients from 14 randomized Phase 2 and 3 controlled trials: a meta-analysis of individual patient data. Eur Heart J. 2017 doi: 10.1093/eurheartj/ehx661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Erqou S, Lee CC, Adler AI. Statins and glycaemic control in individuals with diabetes: a systematic review and meta-analysis. Diabetologia. 2014;57:2444–52. doi: 10.1007/s00125-014-3374-x. [DOI] [PubMed] [Google Scholar]
- 172.Maki KC, Ridker PM, Brown WV, Grundy SM, Sattar N, The Diabetes Subpanel of the National Lipid Association Expert P An assessment by the Statin Diabetes Safety Task Force: 2014 update. J Clin Lipidol. 2014;8:S17–29. doi: 10.1016/j.jacl.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 173.Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, Seshasai SR, McMurray JJ, Freeman DJ, Jukema JW, Macfarlane PW, Packard CJ, Stott DJ, Westendorp RG, Shepherd J, Davis BR, Pressel SL, Marchioli R, Marfisi RM, Maggioni AP, Tavazzi L, Tognoni G, Kjekshus J, Pedersen TR, Cook TJ, Gotto AM, Clearfield MB, Downs JR, Nakamura H, Ohashi Y, Mizuno K, Ray KK, Ford I. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–42. doi: 10.1016/S0140-6736(09)61965-6. [DOI] [PubMed] [Google Scholar]
- 174.Colhoun HM, Ginsberg HN, Robinson JG, Leiter LA, Muller-Wieland D, Henry RR, Cariou B, Baccara-Dinet MT, Pordy R, Merlet L, Eckel RH. No effect of PCSK9 inhibitor alirocumab on the incidence of diabetes in a pooled analysis from 10 ODYSSEY Phase 3 studies. Eur Heart J. 2016;37:2981–2989. doi: 10.1093/eurheartj/ehw292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sattar N, Preiss D, Robinson JG, Djedjos CS, Elliott M, Somaratne R, Wasserman SM, Raal FJ. Lipid-lowering efficacy of the PCSK9 inhibitor evolocumab (AMG 145) in patients with type 2 diabetes: a meta-analysis of individual patient data. Lancet Diabetes Endocrinol. 2016;4:403–10. doi: 10.1016/S2213-8587(16)00003-6. [DOI] [PubMed] [Google Scholar]
- 176.Schmidt AF, Swerdlow DI, Holmes MV, Patel RS, Fairhurst-Hunter Z, Lyall DM, Hartwig FP, Horta BL, Hypponen E, Power C, Moldovan M, van Iperen E, Hovingh GK, Demuth I, Norman K, Steinhagen-Thiessen E, Demuth J, Bertram L, Liu T, Coassin S, Willeit J, Kiechl S, Willeit K, Mason D, Wright J, Morris R, Wanamethee G, Whincup P, Ben-Shlomo Y, McLachlan S, Price JF, Kivimaki M, Welch C, Sanchez-Galvez A, Marques-Vidal P, Nicolaides A, Panayiotou AG, Onland-Moret NC, van der Schouw YT, Matullo G, Fiorito G, Guarrera S, Sacerdote C, Wareham NJ, Langenberg C, Scott R, Luan J, Bobak M, Malyutina S, Pajak A, Kubinova R, Tamosiunas A, Pikhart H, Husemoen LL, Grarup N, Pedersen O, Hansen T, Linneberg A, Simonsen KS, Cooper J, Humphries SE, Brilliant M, Kitchner T, Hakonarson H, Carrell DS, McCarty CA, Kirchner HL, Larson EB, Crosslin DR, de Andrade M, Roden DM, Denny JC, Carty C, Hancock S, Attia J, Holliday E, O’Donnell M, Yusuf S, Chong M, Pare G, van der Harst P, Said MA, Eppinga RN, Verweij N, Snieder H, LifeLines Cohort study g. Christen T, Mook-Kanamori DO, Gustafsson S, Lind L, Ingelsson E, Pazoki R, Franco O, Hofman A, Uitterlinden A, Dehghan A, Teumer A, Baumeister S, Dorr M, Lerch MM, Volker U, Volzke H, Ward J, Pell JP, Smith DJ, Meade T, Maitland-van der Zee AH, Baranova EV, Young R, Ford I, Campbell A, Padmanabhan S, Bots ML, Grobbee DE, Froguel P, Thuillier D, Balkau B, Bonnefond A, Cariou B, Smart M, Bao Y, Kumari M, Mahajan A, Ridker PM, Chasman DI, Reiner AP, Lange LA, Ritchie MD, Asselbergs FW, Casas JP, Keating BJ, Preiss D, Hingorani AD, consortium U. Sattar N. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5:97–105. doi: 10.1016/S2213-8587(16)30396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Writing C, Lloyd-Jones DM, Morris PB, Ballantyne CM, Birtcher KK, Daly DD, Jr, DePalma SM, Minissian MB, Orringer CE, Smith SC., Jr 2016 ACC Expert Consensus Decision Pathway on the Role of Non-Statin Therapies for LDL-Cholesterol Lowering in the Management of Atherosclerotic Cardiovascular Disease Risk: A Report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2016;68:92–125. doi: 10.1016/j.jacc.2016.03.519. [DOI] [PubMed] [Google Scholar]
- 178.Kazi DS, Penko J, Coxson PG, Moran AE, Ollendorf DA, Tice JA, Bibbins-Domingo K. Updated Cost-effectiveness Analysis of PCSK9 Inhibitors Based on the Results of the FOURIER Trial. JAMA. 2017;318:748–750. doi: 10.1001/jama.2017.9924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fonarow GC, Keech AC, Pedersen TR, Giugliano RP, Sever PS, Lindgren P, van Hout B, Villa G, Qian Y, Somaratne R, Sabatine MS. Cost-effectiveness of Evolocumab Therapy for Reducing Cardiovascular Events in Patients With Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 2017;2:1069–1078. doi: 10.1001/jamacardio.2017.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Arrieta A, Hong JC, Khera R, Virani SS, Krumholz HM, Nasir K. Updated Cost-effectiveness Assessments of PCSK9 Inhibitors From the Perspectives of the Health System and Private Payers: Insights Derived From the FOURIER Trial. JAMA Cardiol. 2017;2:1369–1374. doi: 10.1001/jamacardio.2017.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kaufman TM, Duell PB, Purnell JQ, Wojcik C, Fazio S, Shapiro MD. Application of PCSK9 Inhibitors in Practice: Challenges and Opportunities. Circ Res. 2017;121:499–501. doi: 10.1161/CIRCRESAHA.117.311532. [DOI] [PubMed] [Google Scholar]