

Abstract
Unbiased genetic association studies, including genome-wide association and whole-genome sequencing studies, have uncovered many novel disease-associated variants. Relatively few of these associated regions, however, have led to insights that are biologically mechanistic or clinically actionable due in part to the difficulty in designing appropriate functional validation studies to understand how variants contribute to disease. Asthma is a complex inflammatory lung disease for which many genetic associations have been identified. Using asthma as a disease model, we designed Reducing Associations by Linking Genes And transcriptomic Results (REALGAR), an app that facilitates the design of functional validation studies by integrating cell- and tissue-specific results of diseaserelevant gene expression and other omics studies. Via specific examples, we demonstrate how integrated gene- centric and disease-specific information leads to asthma insights, and more broadly, can help understand complex diseases.
Introduction
The results of genome-wide association studies (GWAS) for various complex diseases have contributed to the growing credibility of using unbiased genetic association approaches for uncovering novel disease variants1. In most cases, however, findings from GWAS have not yet led to significant progress toward understanding how identified genomic variants are functionally and clinically relevant2. Recent exome sequencing and whole-genome sequencing (WGS) studies are finding an even greater number of variants associated with complex diseases, most of which have not yet been related functionally to outcomes3, 4 Among the factors that contribute to the slow translation of genetic association results into functional insights are1, 2: (1) functional studies are time-consuming, as each follow-up experiment has to be tailored to a particular complex disease phenotype and type of polymorphism in a genomic region, (2) in order to test genes and variants for function, complex diseases have to be simplified into assays that may not capture the cell-specific, developmental or environmental context necessary for functional elucidation of the gene, and (3) unlike older candidate gene studies, GWAS and WGS studies have identified loci in gene deserts and in genes with no annotated function, making the design of functional experiments even more difficult. In silico approaches that screen genes and variants for potential function are needed to guide the efficient experimental validation of top hits.
Functional validation studies of gene associations often begin with searches for what is known about a specific locus, including what genes are nearby and whether associated variants are expression quantitative trait loci (eQTL) and/or regulators of transcription of these genes. Although this information serves as a starting point, it does not offer phenotype-specific mechanistic clues about how the genes in question modify relevant biological pathways. To obtain such information, researchers often search public databases to find out the tissue(s) where identified genes are expressed and under what disease and treatment conditions they are differentially expressed. Public gene expression data from sources such as the Gene Expression Omnibus (GEO) are a primary resource for answering these questions, but many wet-lab and clinical researchers do not have the proper expertise or dedicated computational resources necessary to obtain and integrate gene expression microarray, RNA-Seq, and other omics results. Even researchers who do have such resources often repeat similar analytical tasks every time a new association locus is found. Having an integrated resource of tissue-specific results from expression and other omics studies that is guided by disease-specific knowledge would facilitate prioritization and rational design of experiments that can provide clinically actionable insights.
Asthma is an episodic inflammatory lung disease characterized by variable airflow limitation and airway hyperresponsiveness that affects over 25 million Americans5. One of the most common pharmacologic treatments of asthma consists of glucocorticoid medications, given in inhaler form as maintenance therapy or oral form to alleviate exacerbations or severe disease6. Glucocorticoids, which are also used to treat various inflammatory diseases, act by modulating transcription of genes in a tissue-dependent fashion7. The genetics of asthma has been studied for over 20 years, and consortium GWAS carried out in Europeans and diverse North American populations8, 9 have identified robust associations at loci such as the GSDMB-ORMDL3-ZPBP2 17q21 locus, HLA-DQ, IL1RL1, IL18RL1, IL33, TSLP, SLC22A5, SMAD3, and RORA. Most strikingly, the 17q21 locus association has been replicated in over 15 independent diverse populations and has been linked to childhood-onset asthma. How associations in this region are functionally related to asthma remains unclear10. Because asthma features are heterogeneous and genetic variation also contributes to asthma-related phenotypes such as glucocorticoid response11, there is difficulty in determining what specific traits are modulated by measured statistical associations with asthma.
Here, we present REducing Associations by Linking Genes And transcriptomic Results (REALGAR), an app that integrates results from gene expression, RNA-Seq and other omics studies to help guide validation experiments for gene associations. We use asthma as a disease model to show how gene-centric information that is gathered in a disease-specific fashion can be useful to better understand associations.
Methods
Data Sources
Gene Expression. A search for all human gene expression microarray datasets matching search terms related to “asthma” and “glucocorticoid” was performed in GEO12. Raw signal intensity and phenotype files from relevant datasets were downloaded. The R package arrayQualityMetrics was used to perform quality control analysis on each dataset13. Samples that did not pass quality control measures within datasets were excluded from subsequent analyses, and any provided covariates that were deemed to influence outcomes and/or produce batch effects via cluster analyses were noted. Pre-processing of raw signal intensities was performed with Robust Multi-Array Average (RMA).14 Three asthma-related RNA-Seq datasets15–17 were obtained from GEO.
Gene Association. Publicly available single nucleotide polymorphism (SNP) association results from asthma GWAS studies were obtained from three sources: (1) the Genome-Wide Repository of Associations Between SNPs and Phenotypes (GRASP), a catalog hosted by the National Heart, Lung and Blood Institute that contains all publicly available SNPs with reported p-values <0.0518, (2) the GABRIEL Consortium8, and (3) the EVE Consortium9. GABRIEL results were filtered to select SNPs with p-values <0.05, and the EVE results, which contain distinct p- values for different racial/ethnic groups, were filtered to select SNPs that had p-value <0.05 in at least one group. The UCSC liftOver executable and chain files19 were used to convert the GABRIEL genome coordinates from the hg17 genome build into the hg19 genome build, which is the version used by GRASP and EVE. SNP locations were mapped to genes (within 10kb on either end) using BEDTools20 according to the hg19 reference genome.
Glucocorticoid Receptor DNA Binding Sites. ChIP-Seq results for a variety of tissues and transcription factors have identified protein-DNA binding events that may modulate transcription21. Of relevance to asthma, one study identified regions of the genome where glucocorticoid receptors bind after 1 hour of treatment with a glucocorticoid (100nM dexamethasone) in A549 cells, an adenocarcinomic human alveolar basal epithelial cell line22. ChIP-Seq results from this dataset were obtained via the UCSC genome browser, where they are reported accorded to the hg19 genome build, and mapped to genes (within 10kb on either end) using BEDTools20.
Gene Expression Analysis
For microarray data, differential expression was quantified using the R package limma23. For datasets with noticeable batch effects, the R package SVA was used to perform differential expression analyses while adjusting for batch effect24. Fold changes were computed from the log2 fold changes output by limma or SVA, and standard deviations were computed as log2 fold change divided by the moderated t-statistic output by limma or SVA. Results from 27 asthma and glucocorticoid response datasets were saved for inclusion in the app.
RNA-Seq data was analyzed with Kallisto25 to quantify transcript abundance according to reads mapped to the hg38 reference genome and Sleuth26 to compute transcript-level differential expression results. Gene-level results displayed in REALGAR correspond to those of the transcript with the lowest differential expression p-value, in cases where genes had more than one transcript with reported results. The fold change and standard deviation reported in the app correspond to the beta and standard error of beta measures that are output by Sleuth.
For both microarray and RNA-Seq, differential expression results were computed for the test condition of interest relative to within-study controls (i.e. asthma vs. non-asthma samples, or glucocorticoid treatment vs. vehicle). Results of analyzed datasets were stored for use with the app.
To obtain an illustrative hypothesis related to our datasets, we conducted a meta-analysis of all available glucocorticoid response microarray datasets (Table 1) using the R package GeneMeta27. Microarray probes were matched to official gene symbols using Bioconductor database packages for corresponding microarray platforms, and gene-level intensities were computed by obtaining the mean of probe intensities for all probes matching to an official gene symbol. Matrix files for overlapping genes of each study were used by GeneMeta to compute combined two-sided z-scores.
Table 1.
Glucocorticoid response datasets used for meta-analysis
| GEO Accession Number | Microarray Platform | Study Description* |
|---|---|---|
| GSE55876 | Affymetrix Human Gene 1.0 ST Array | Lymphoblastic leukemia cells; (N=6) |
| 3 dexamethasone (100nM; 6h), 3 vehicle | ||
| GSE55877 | Affymetrix Human Gene 1.0 ST Array | Lymphoblastic leukemia cells; (N=6) |
| 3 dexamethasone (100nM; 6h), 3 vehicle | ||
| GSE4917 | Affymetrix Human Genome U133A Array | MCF10A-Myc cells; (N=6) |
| 3 dexamethasone (1uM; 24h), 3 vehicle | ||
| GSE22152 | Affymetrix Human Genome U133 Plus 2.0 Array | Lymphoblastic leukemia cells; (N=12) |
| 6 dexamethasone (100nM; 6h), 6 vehicle | ||
| GSE22779 | Affymetrix Human Genome U133 Plus 2.0 Array | Peripheral blood mononuclear cells; (N=6) |
| 3 dexamethasone (10uM; 6h), 3 vehicle | ||
| GSE61880 | Affymetrix HT HG-U133+ PM Array Plate | Monocyte derived macrophages; (N=6) |
| 3 dexamethasone (100nM; 10h), 3 vehicle |
*Each entry contains: cell type; total sample size; sample size and treatment condition for cases, sample size of vehicle controls
REALGAR Design and Features
The RStudio package Shiny, which enables creation of web applications that users can easily engage without needing a background in programming28, was used to create REALGAR, as its end users include wet-lab researchers with little computing experience. The app code, results of analyzed datasets and GENCODE hg19 reference genome files were saved on a DigitalOcean droplet containing a Shiny server that includes various R packages to retrieve and display results (i.e., forestplot, lattice, and Gviz). The app is displayed at http://himeslab.org/realgar, and the full code is available at https://github.com/HimesGroup/realgar.
To use the app, an official gene symbol or SNP ID is provided by a user (Figure 1). Users can select (1) tissue and (2) asthma types to be included by clicking on corresponding checkboxes, (3) what asthma-related drug treatment gene expression results to display (e.g., glucocorticoid, beta-agonist or vitamin D treatment), and (4) which of the GWAS results (GRASP, EVE and/or GABRIEL) to include. If EVE is selected, an additional option box allows the user to choose results from specific populations (i.e., African American, Latino, European American, or all subjects). As selections are made, the analyzed GEO datasets that match the selected criteria appear in a table in a separate “Datasets loaded” tab that provides: (1) GEO accession numbers that link directly to GEO entries, (2) PMIDs for papers, when available, that link directly to PubMed entries, and (3) a brief description of each dataset.
Study-specific results are visualized as a forest plot in which the selected gene’s fold changes for all selected tissue and asthma type comparisons are shown as computed relative to within-study controls (Figure 2A). If a SNP ID, rather than a gene symbol, is entered, results for the gene closest to that SNP are displayed. If one or more of the treatment options are selected, a second forest plot displays the fold changes of this gene under the treatment(s) selected, relative to within-study controls, for all selected tissue types with available data. Forest plot rows are colored according to the negative log10 of the corresponding false-discovery rate adjusted p-values (i.e., q-values), and an adjoining table displays study information, including tissue type and endotype or treatment, as applicable.
Figure 2.
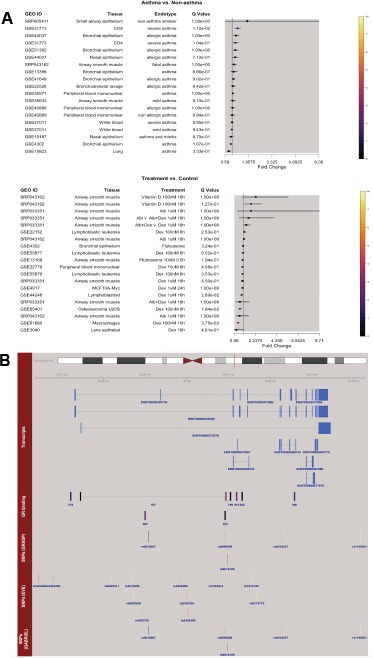
REALGAR output. A) Forest plots show gene fold-changes of differential expression, with colors corresponding to q-values. Example shown for SRC indicates that its mRNA is increased in CD8+ cells of severe asthma subjects and decreased with glucocorticoid treatment in macrophages. B) Genome tracks show gene transcripts, GR binding sites and SNPs with GWAS results. Example shown for SRC reveals that asthma- associated SNPs are within/near GR binding sites.
Below the expression results, an ideogram of the selected gene’s chromosome with its location indicated by a red line is shown, along with a genome track displaying the gene’s transcripts (Figure 2B). If there are SNPs with GRASP, EVE or GABRIEL asthma association results and/or glucocorticoid receptor (GR) binding sites within10kb of the gene’s boundaries, then association and/or GR binding site tracks will also be present. GR binding sites are colored according to the ENCODE-provided binding scores, with higher scores corresponding to brighter colors. SNP association results are colored according to negative log10 of p-value, so that lower p-values correspond to brighter colors. Individual plots can be downloaded in.png format, and tables of expression results and SNPs can be downloaded in.csv format. GWAS results displayed can be downloaded, allowing users to obtain quantitative results of the associations across populations.
Results
Generating Hypotheses That Link Gene Associations to Function with REALGAR. The tissue-specific results available in REALGAR offer insights about the role of prominent asthma-associated genes. Looking up the 17q21 loci genes GSDMB, ORMDL3 and ZPBP2 in the app reveals that glucocorticoid treatment does not alter the expression of these genes, nor are they differentially expressed among subjects with asthma in any of the available tissue types. Thus, these genes are not involved in glucocorticoid or immune responses, nor are they useful as asthma biomarkers, according to the evidence provided. In contrast, REALGAR shows that IL1R1 is highly glucocorticoid responsive in macrophages, where dexamethasone treatment induces a 3.41 (95% CI: 2.65-4.39) increased fold-change vs. control (q-value 3.13x10-06). Additionally, IL1R1 mRNA has decreased expression in nasal epithelium of asthma subjects with allergic rhinitis vs. non-asthma controls, exhibiting a fold-change of 0.60 (95% CI: 0.49-0.73; q-value 0.020). These tissue-specific results help suggest experiments that may clarify the role of IL1R1 in asthma.
Using REALGAR to Re-Prioritize Disease-Associated Genes for Functional Validation Studies. By combining GR- binding site results with SNP results from GRASP, EVE and GABRIEL, 511 unique asthma-associated SNPs that were within GR-binding sites were identified. Among these was rs6090585, within an intron of the SRC ProtoOncogene, Non-Receptor Tyrosine Kinase (SRC) gene, which had a nominally significant association with asthma in the GABRIEL GWAS consortium study (p-value 0.010; OR 1.13 [1.03-1.24])8. Although this SNP would not be a preferred candidate for further study based on the GWAS results alone, the integrated data in REALGAR provides further evidence that the locus may be relevant to asthma: (1) it is located in a putative GR binding site, (2) SRC mRNA expression is increased in CD8+ cells in severe asthma (fold change 1.33 [1.17-1.5] vs. non-asthma controls; q-value 0.012), and (3) SRC mRNA expression is decreased in macrophages treated with the glucocorticoid dexamethasone (fold change 0.47 [0.35, 0.64] vs. control; q-value 0.0038) (Figure 2). Another asthma-associated SNP within a GR-binding site was rs3759324, a SNP located within introns of two genes: Lymphotoxin Beta Receptor (LTBR) and Sodium Channel Epithelial 1 Alpha Subunit (SCNN1A). This SNP was nominally associated with asthma in the GRASP study (p-value 0.0044; OR 1.07 [1.02-1.12]), and thus would not be prioritized for further study based on GWAS data. Considering other data in REALGAR, however, increases support for the hypothesis that this locus is related to asthma: (1) rs3759324 is located in a putative GR-binding site with the highest possible ENCODE binding score (1000), (2) the GR-binding site in question occurs fewer than 30kb upstream of several SCNN1A transcripts, suggesting mRNA levels of this gene are modulated directly by glucocorticoids, (3) SCNN1A mRNA expression is increased in CD8+ cells in severe asthma (fold change 1.53 [1.22-1.92] vs. nonasthma controls; q-value 0.027), and (4) SCNN1A mRNA expression is increased in lens epithelial cells treated with dexamethasone (fold change 14.98 [9.34, 24.01] vs. control; q-value 0.023). In contrast, no significant changes were observed for LTBR mRNA levels in any disease state or tissue type tested, although the GR-binding site containing the SNP of interest lies upstream of several LTBR transcripts. According to these results, SCNN1A should be prioritized for study as an asthma gene, and specifically as a modulator of glucocorticoid and/or immune response. These examples highlight how REALGAR’s integrated data can aid in re-prioritizing associations for functional validation studies.
Insights Provided By Meta-Analysis of Single-Modality Data in REALGAR. In addition to using REALGAR to understand genetic associations, integration of data types in the app provides novel insights about the role of genes in disease. For example, a meta-analysis of six of the glucocorticoid-response expression microarray datasets in REALGAR found that Suppressor of Cytokine Signaling 1 (SOCS1) gene ranked as the “top” gene according to magnitude of two-sided z-score (6.2; two-sided z-score false discovery rate <10-5) (Figure 3). While SOCS1 was highly responsive to glucocorticoids in most tissues, structural cells were an exception: SOCS1 was not responsive in epithelium, and results in airway smooth muscle were mixed, with a nominally significant decrease observed in one study (fold change 0.74 [0.65, 0.85]; p-value 6.1x10-3; q-value 0.089) and an increase in another (fold change
Figure 3.
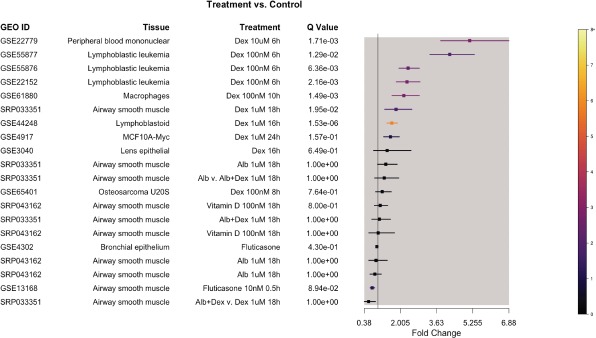
SOCS1 was the most significant differentially expressed gene in response to glucocorticoids across all tissues. Its levels increased highly in immune cells and were not changed in structural cells, with the exception of airway smooth muscle, which had mixed results between datasets.
1. 81 [1.30, 2.52]; p-value 3.58 x10-3; q-value 0.020). Previous studies have identified SOCS1 as a target via which glucocorticoids regulate inflammation and a mediator of rhinovirus-induced asthma exacerbations29, 30. REALGAR results provide complementary information: (1) a GR-binding site near the gene suggests that SOCS1 is directly activated by glucocorticoids, and (2) the results of 17 microarray studies and RNA-Seq show that SOCS1 mRNA levels are not significantly different between asthma and non-asthma subjects, except for in bronchial epithelial cells, where they were decreased in asthma subjects (fold change 0.83 [0.76, 0.91]; q-value 0.021). These observations suggest that glucocorticoids may not be able to rectify differences in SOCS1 levels that are present in bronchial epithelial cells of asthma subjects. Thus, aggregating single-modality data allows for detection of patternsnot apparent in single datasets, while complementary information available in REALGAR allows for quick insights into disease-specific gene function.
Discussion
Integration of omics data is challenging, as it requires a thorough understanding of heterogeneous data types, as well as computational resources and expertise to store and analyze large datasets31. Various public resources host omics data, but much work remains in maximizing this data’s ability to inform human pathobiology by presenting results derived from omics data integration to wet-lab researchers, who can validate hypotheses. The development of novel computational tools that integrate and visualize omics datasets is vital in bridging the gap between streams of raw data and clinically relevant findings. Several publicly available apps that are helpful in understanding GWAS data exist. For example, HaploReg is a widely used tool that allows users to look up information about a SNP, including others in linkage disequilibrium to it, chromatin state and protein binding annotation from ENCODE, sequence conservation, its effect on regulatory motifs, and associated eQTLs32. A visualization-centered app that provides similar information is LocusExplorer, which is built using the shiny R framework to provide high resolution graphics of omics data for a genomic region33. Despite their utility, these tools’ insights are largely not tissue- specific and hence are limited in their ability to provide insights into disease and treatment mechanisms, which are tissue specific. Apps that focus on tissue-specific analysis include NetWAS, an efficient and visually appealing app that integrates GWAS results with tissue-specific networks derived from gene expression data to re-prioritize disease-gene associations34. While NetWAS includes many tissue-specific results, it does not use disease-specific knowledge to guide integration and presentation of results to wet-lab researchers, who answer specific, hypothesis- driven questions. We built REALGAR as a tissue-specific and disease-focused app to ensure its results are immediately helpful to wet-lab biologists, as well as to researchers who seek to carry out unbiased data analyses.
A well-known challenge in understanding complex diseases such as asthma is that disease-associated polymorphisms vary by race and ethnicity. This factor will be important to address in bridging the gap from omics results to clinically relevant insights. REALGAR allows users to view GWAS results at a population-specific level, and functional validation studies that harness the granular nature of these population-level results can lead to improved understanding of the clinical significance of disease-specific loci. A detailed understanding of thecorrespondence between genetic variants and clinical outcomes will be an important stepping stone towards more personalized medical care.
In ongoing work, we are improving REALGAR by including datasets for more phenotypes, and while we continue to be focused on pulmonary disease phenotypes, future versions will include other complex diseases. In addition to incorporating a broader set of phenotypes, we are adding additional data types, namely eQTL and additional RNA- Seq results. To complement the population-level GWAS results currently included in the app, we will integrate population-specific information for the gene expression results, as this information is available for some of the datasets. Rather than duplicate the compilation of SNP data that exists in other tools, we seek to focus on providing novel integration of disease-focused omics datasets. While scoring metrics across integrated datasets remain highly biased and arbitrary from a biological perspective, future versions of REALGAR will include a scoring metric that weights user-selected datasets to provide an overall ranking that is based on ranks within datasets. As with all in silico tools, the goal of such scores will be to provide evidence-based hypotheses for experimental studies.
Conclusion
We created REALGAR, an app that integrates gene- and disease-specific data to present insights that are helpful for designing functional validation studies in support of genetic associations. REALGAR has an intuitive, user-friendly interface and, while it harnesses the computational power of R, requires no programming background for its use. We provide several specific hypotheses about asthma-related genes based on REALGAR results that may lead to biological insights about asthma and its treatment.
References
- 1.Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118(5):1590–605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363(2):166–76. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 3.Massey J, Eyre S. Rare variants and autoimmune disease. Brief Funct Genomics. 2014;13(5):392–7. doi: 10.1093/bfgp/elu011. [DOI] [PubMed] [Google Scholar]
- 4.Marei HE, Althani A, Suhonen J, El Zowalaty ME, Albanna MA, Cenciarelli C, et al. Common and Rare Genetic Variants Associated With Alzheimer’s Disease. J Cell Physiol. 2016;231(7):1432–7. doi: 10.1002/jcp.25225. [DOI] [PubMed] [Google Scholar]
- 5.Akinbami L, Moorman J, Bailey C, Zahran H, King M, Johnson C, et al. Trends in asthma prevalence, health care use, and mortality in the United States, 2001-2010. Hyattsville, MD: National Center for Health Statistics. 2012 [PubMed] [Google Scholar]
- 6.Fanta CH. Asthma. N Engl J Med. 2009;360(10):1002–14. doi: 10.1056/NEJMra0804579. [DOI] [PubMed] [Google Scholar]
- 7.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N Engl J Med. 2005;353(16):1711–23. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 8.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211–21. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. 2011;43(9):887–92. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ono JG, Worgall TS, Worgall S. 17q21 locus and ORMDL3: an increased risk for childhood asthma. Pediatr Res. 2014;75(1-2):165–70. doi: 10.1038/pr.2013.186. [DOI] [PubMed] [Google Scholar]
- 11.Tantisira KG, Lasky-Su J, Harada M, Murphy A, Litonjua AA, Himes BE, et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N Engl J Med. 2011;365(13):1173–83. doi: 10.1056/NEJMoa0911353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. 2016.
- 14.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–15. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 15.Himes BE, Koziol-White C, Johnson M, Nikolos C, Jester W, Klanderman B, et al. Vitamin D Modulates Expression of the Airway Smooth Muscle Transcriptome in Fatal Asthma. PLoS One. 2015;10(7):e0134057. doi: 10.1371/journal.pone.0134057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Himes BE, Jiang X, Wagner P, Hu R, Wang Q, Klanderman B, et al. RNA-Seq transcriptome profiling identifies CRISPLD2 as a glucocorticoid responsive gene that modulates cytokine function in airway smooth muscle cells. PLoS One. 2014;9(6):e99625. doi: 10.1371/journal.pone.0099625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hackett NR, Butler MW, Shaykhiev R, Salit J, Omberg L, Rodriguez-Flores JL, et al. RNA-Seq quantification of the human small airway epithelium transcriptome. BMC Genomics. 2012;13:82. doi: 10.1186/1471-2164-13-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eicher JD, Landowski C, Stackhouse B, Sloan A, Chen W, Jensen N, et al. GRASP v2.0: an update on the Genome-Wide Repository of Associations between SNPs and phenotypes. Nucleic Acids Res. 2015;43(Database issue):D799–804. doi: 10.1093/nar/gku1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, et al. The UCSC Genome Browser database: 2016 update. Nucleic Acids Research. 2016;44(D1):D717–D25. doi: 10.1093/nar/gkv1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quinlan AR. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics. 2014;47:11–2. 1–34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–8. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19(12):2163–71. doi: 10.1101/gr.097022.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 24.JT L, WE J, HS P, EJ F, AE J, JD S. sva: Surrogate Variable Analysis. R package version 3200. 2016 [Google Scholar]
- 25.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34(5):525–7. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 26.Pimentel HJ, Bray N, Puente S, Melsted P, Pachter L. Differential analysis of RNA-Seq incorporating quantification uncertainty. bioRxiv. 2016 doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 27.L L, R G, M R. GeneMeta: MetaAnalysis for High Throughput Experiments. R package version 1440. 2016 [Google Scholar]
- 28.W C, J C, JJ A, Y X, J M. shiny: Web Application Framework for R. R package version 01329005. 2016 [Google Scholar]
- 29.Bhattacharyya S, Zhao Y, Kay TW, Muglia LJ. Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc Natl Acad Sci U S A. 2011;108(23):9554–9. doi: 10.1073/pnas.1017296108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gielen V, Sykes A, Zhu J, Chan B, Macintyre J, Regamey N, et al. Increased nuclear suppressor of cytokine signaling 1 in asthmatic bronchial epithelium suppresses rhinovirus induction of innate interferons. J Allergy Clin Immunol. 2015;136(1):177–88. e11. doi: 10.1016/j.jaci.2014.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez-Cabrero D, Abugessaisa I, Maier D, Teschendorff A, Merkenschlager M, Gisel A, et al. Data integration in the era of omics: current and future challenges. BMC Syst Biol. 2014;8(Suppl 2):I1. doi: 10.1186/1752-0509-8-S2-I1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2011;40(Database issue):D930–4. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dadaev T, Leongamornlert DA, Saunders EJ, Eeles R, Kote-Jarai Z. LocusExplorer: a user-friendly tool for integrated visualization of human genetic association data and biological annotations. Bioinformatics. 2016;32(6):949–51. doi: 10.1093/bioinformatics/btv690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, et al. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet. 2015;47(6):569–76. doi: 10.1038/ng.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]