

Summary
The Cucurbita genus (squashes, pumpkins and gourds) includes important domesticated species such as C. pepo, C. maxima and C. moschata. In this study, we present a high‐quality draft of the zucchini (C. pepo) genome. The assembly has a size of 263 Mb, a scaffold N50 of 1.8 Mb and 34 240 gene models. It includes 92% of the conserved BUSCO core gene set, and it is estimated to cover 93.0% of the genome. The genome is organized in 20 pseudomolecules that represent 81.4% of the assembly, and it is integrated with a genetic map of 7718 SNPs. Despite the small genome size, three independent lines of evidence support that the C. pepo genome is the result of a whole‐genome duplication: the topology of the gene family phylogenies, the karyotype organization and the distribution of 4DTv distances. Additionally, 40 transcriptomes of 12 species of the genus were assembled and analysed together with all the other published genomes of the Cucurbitaceae family. The duplication was detected in all the Cucurbita species analysed, including C. maxima and C. moschata, but not in the more distant cucurbits belonging to the Cucumis and Citrullus genera, and it is likely to have occurred 30 ± 4 Mya in the ancestral species that gave rise to the genus.
Keywords: genome, crop, zucchini, whole‐genome duplication, Cucurbitaceae, transcriptome
Introduction
Cucurbita pepo L. is the main crop of the Cucurbita genus. At the subspecies level, three taxa are recognized as follows: subsp. pepo, known only in cultivation (zucchini, pumpkins and summer and winter squashes), subsp. ovifera (L.) Decker (=subsp. texana (Scheele) Filov), known in cultivation and in the wild (scallop and acorn squashes, ornamental gourds), and subsp. fraterna (L. H. Bailey) Lira, Andres & Nee (=C. fraterna L. H. Bailey), known only in wild populations (Andres, 1987; Decker‐Walters, 1990; Paris et al., 2015). Subspecies pepo and ovifera include many edible‐fruited cultivar groups, such as pumpkin, vegetable marrow, cocozelle, zucchini, acorn, scallop, straightneck and crookneck. There is evidence of an early domestication of this species (Smith, 1997), with more than one domestication event in Mexico and the United States (Smith, 2006), and it has had two different diversification processes: one in America and one in Europe (Paris et al., 2012), where zucchini and other elongated forms, such as vegetable marrow and cocozelle, were developed.
Cucurbita pepo is an economically important crop. Its production reached 25 million tons in 2014, with nearly two million cultivated hectares (http://www.fao.org/faostat/en). Cultivated varieties display a rich diversity of vine, flowering and fruit traits, and among them, cultivars of the zucchini group rank among the highest‐valued vegetables worldwide (Formisano et al., 2012). The Cucurbita genus and the Cucurbitaceae family contain other important crops, such as other squashes, pumpkins and gourds (Cucurbita maxima Duchesne and Cucurbita moschata (Duchesne ex Lam.) Duchesne ex Poir.), melon (Cucumis melo L.), cucumber (Cucumis sativus L.) and watermelon (Citrullus lanatus (Thunb.) Mansf).
Despite the agronomic importance of the species, prior to the genome assembly presented here, few C. pepo genetic and genomic resources were available: a first generation of genetic maps constructed with AFLP, RAPD and SSR markers (Brown and Myers, 2002; Gong et al., 2008; Lee et al., 1995; Zraidi and Lelley, 2004, 2007) that were later improved with SNPs (Esteras et al., 2012) and several transcriptomes (Blanca et al., 2011; Vitiello et al., 2016; Wyatt et al., 2015; Xanthopoulou et al., 2016). More recently, a high‐density SNP‐based genetic map was developed using a RIL population derived from the cross between two C. pepo subspecies (subsp. pepo Zucchini × subsp. ovifera Scallop) (Montero‐Pau et al., 2017). This map was developed to assist us with the de novo assembly process.
In this study, we present a de novo assembly of the C. pepo genome, a high coverage transcriptome of C. pepo and 40 transcriptomes of 12 species of the Cucurbita genus. Comparative and phylogenetic analyses show that a whole‐genome duplication (WGD) occurred just before the speciation events that created this genus. All these resources and several previous transcriptome and draft genome versions are publicly available at https://bioinf.comav.upv.es/downloads/zucchini.
Results
De novo genome assembly
The complete genome of Cucurbita pepo has been sequenced using a whole‐genome shotgun sequencing approach. The zucchini type (C. pepo subsp. pepo) accession MU‐CU‐16 was selfed four times before sequencing. This accession is characterized by early flowering, a bushy growth habit, high production and uniform, cylindrical, dark‐green fruits. This accession was also used as parental in two previous genetic maps (Esteras et al., 2012; Montero‐Pau et al., 2017). One paired‐end library, with an insert size of 500 bp, and four mate‐pair libraries, with sizes of 3, 7, 10 and 20 Kb, were created and sequenced in five Illumina Hiseq2000 lanes, resulting in a genome coverage of 254× for the pair‐end library and 54, 46, 65 and 62 X for the 3‐, 7‐, 10‐ and 20‐Kb libraries, respectively (Table S2). All reads were quality‐trimmed and filtered. Additionally, approximately 40% of the 3‐ and 7‐Kb mate‐pair reads were found to be chimeric and were filtered out by comparing them against a preliminary assembly (see Table S2, Figure S1). This chimeric filtering doubled the contig N50 and tripled the scaffold N50 of the final assembly. Finally, 503‐M filtered pair‐end reads and 185‐M mate‐pair reads were used in the assembly. The genome was assembled by SOAPdenovo2 (Luo et al., 2012). A k‐mer size of 41 was chosen for the final assembly because it yielded the highest N50 values (Figure S2). The SOAPdenovo2 scaffolds were broken, and the scaffolding was redone with SSPACE (Boetzer et al., 2011) and GapCloser (Luo et al., 2012). The final assembly covered 263 Mb in 26 005 scaffolds and 32 754 contigs with a contig N50 of 110 Kb (L50 = 606 contigs) and a scaffold N50 of 1.8 Mb (L50 = 42 scaffolds) (Table 1). Completeness of the de novo assembly was assessed with BUSCO using a plant‐specific database of 1440 genes. Of this total, 92.1% were found to be complete (73.1% as single genes and 19.0% as duplicated genes) and 2.1% were found to be fragmented. The Illumina RNAseq reads obtained from the MU‐CU‐16 accession were mapped with HISAT2 with this genome as reference with a 91.9% success rate. The pair‐end reads used to build the assembly were mapped against the assembly with a success rate of 99.4%. From the k‐mer distribution, the genome size was estimated to be 283 Mb; thus, 93.0% of the genome would be covered by the assembly. Chloroplastic and mitochondrial scaffolds were detected using Blast: 250 scaffolds were identified as mitochondrial and 13 as chloroplastic (Table S3).
Table 1.
Assembly statistics of C. pepo genome version 4.1
| Parameter | Value |
|---|---|
| GC content (%) | 36.52 |
| No. of contigs (≥0 bp) | 32 754 |
| No. of contigs (≥500 bp) | 13 896 |
| No. of contigs (≥1000 bp) | 8217 |
| Bases in contigs (≥0 bp) | 247 816 249 |
| Bases in contigs (≥1000 bp) | 238 245 128 |
| Largest contigs (bp) | 639 487 |
| N50 contig size (bp) | 110 136 |
| N75 contig size (bp) | 49 377 |
| L50 contig number | 606 |
| L75 contig number | 1407 |
| No. of scaffolds (≥0 bp) | 26 025 |
| No. of scaffolds (≥500 bp) | 7994 |
| No. of scaffolds (≥1000 bp) | 3709 |
| Bases in scaffolds (≥0 bp) | 263 500 453 |
| Bases in scaffolds (≥500 bp) | 258 108 973 |
| Bases in scaffolds (≥1000 bp) | 255 237 628 |
| Largest contig (bp) | 6 123 784 |
| N50 scaffold size (bp) | 1 749 822 |
| N75 scaffold size (bp) | 453 344 |
| L50 scaffold number | 42 |
| L75 scaffold number | 112 |
The genetic map developed with the RIL population of zucchini x scallop (accessions MU‐CU‐16 × UPV‐196) (Montero‐Pau et al., 2017) was used to detect chimeric scaffolds and to anchor and order the scaffolds into pseudomolecules. A total of 7718 SNPs (average of 386 markers/linkage group) were located in the map. Based on the relationship of physical and genetic distances and the presence of the same scaffold in more than one linkage group, 22 of the 26 005 scaffolds were identified as chimeric. Those scaffolds were visually inspected and split. In a first attempt, a total of 181 scaffolds could be anchored to 21 pseudomolecules, which represents the 81.4% of the assembled genome. Finally, after the integration of this genetic map with the genetic maps developed by Esteras et al. (2012), which were based on data from the F2 of the same cross, and the genetic map of Holdsworth et al. (2016), all scaffolds were grouped into 20 pseudomolecules (Table 2 and Table S4). Between 4 and 19 scaffolds were anchored to each pseudochromosome with a length between 8.1 Mb and 21.3 Mb (Table 2). Of the remaining 25 344 scaffolds, 3295 were longer than 1 Kb and 365 were longer than 20 Kb. The average correlation between physical distance and genetic distance was 0.98 (0.94–1.00; Figure S3). This assembly constitutes genome version 4.1. Some other previous versions were made available to the Cucurbita community, but none were published.
Table 2.
Pseudochromosome summary. Number of scaffolds anchored to each pseudochromosome, total length, length without the 1000 N spacers and number of genes
| Molecule | #Scaffolds | Length (bp) | Length without N spacers (bp) | Number of genes |
|---|---|---|---|---|
| Cp4.1LG01 | 19 | 21 320 769 | 21 302 769 | 3258 |
| Cp4.1LG02 | 16 | 14 376 414 | 14 361 414 | 1981 |
| Cp4.1LG03 | 12 | 13 772 414 | 13 761 414 | 2178 |
| Cp4.1LG04 | 5 | 12 709 140 | 12 705 140 | 1870 |
| Cp4.1LG05 | 8 | 10 865 678 | 10 858 678 | 1753 |
| Cp4.10LG06 | 11 | 10 677 745 | 10 667 745 | 1407 |
| Cp4.1LG07 | 14 | 10 147 556 | 10 134 556 | 1404 |
| Cp4.1LG08 | 4 | 10 059 303 | 10 056 303 | 1503 |
| Cp4.1LG09 | 10 | 9 920 322 | 9 911 322 | 1608 |
| Cp4.1LG10 | 8 | 9 835 092 | 9 828 092 | 1432 |
| Cp4.1LG11 | 11 | 9 833 969 | 9 823 969 | 1319 |
| Cp4.1LG12 | 5 | 9 824 194 | 9 820 194 | 1388 |
| Cp4.1LG13 | 8 | 9 354 089 | 9 347 089 | 1400 |
| Cp4.1LG14 | 5 | 8 955 933 | 8 951 933 | 1263 |
| Cp4.1LG15 | 4 | 8 816 444 | 8 813 444 | 1136 |
| Cp4.1LG16 | 10 | 8 691 934 | 8 682 934 | 1114 |
| Cp4.1LG17 | 9 | 8 680 504 | 8 672 504 | 1281 |
| Cp4.1LG18 | 7 | 8 333 454 | 8 327 454 | 1186 |
| Cp4.1LG19 | 8 | 8 246 682 | 8 239 682 | 1386 |
| Cp4.1LG20 | 7 | 8 120 804 | 8 114 804 | 1000 |
Transcriptome and genome annotation
Two cDNA libraries were created for the parent accessions of the RIL population using pooled RNA from different vegetative and reproductive tissues. More than 228 million reads were added to the previously available 454‐based transcriptome (Blanca et al., 2011). They were used to create a new transcriptome assembly (version 3.0, available at https://bioinf.comav.upv.es/downloads/zucchini) and to annotate the genome. The transcriptome assembly identified 108 062 transcripts, 65 990 of which included an ORF. GO terms could be assigned to 71.5% of the coding transcripts. Completeness of the de novo assembly assessed with BUSCO showed that 91.0% of the 1440 BUSCO groups searched were found to be complete and 4.0% were found to be fragmented.
The genome annotation resulted in 34 240 predicted gene models, of which 27 870 were protein‐coding genes (Table 3). These results are similar to those found in melon and cucumber (Garcia‐Mas et al., 2012; Huang et al., 2009). The average gene size was 3450 bp with an average of 5.4 exons (Figure S4). The gene models covered 118 Mb, and their coding regions 35 Mb, which represent 45.3% and 13.7% of the assembled genome, respectively, and indicate a high degree of genome compaction (Figure 1a). GO terms could be assigned to 19 784 protein‐coding genes out of 27 870 (71.0%; Figures S5 and S6). Functional descriptions were added to 76.6% of transcripts using AHRD, and 79.2% were tagged with an InterPro protein domain.
Table 3.
Genome annotation summary
| Number of genes | 34 240 |
| Number of protein‐coding genes | 27 870 |
| Number of exons | 184 243 |
| Number of CDSs | 166 271 |
| Number of introns | 150 003 |
| Number of 5′ UTRs | 21 701 |
| Number of 3′ UTRs | 22 296 |
| Number of tRNAs | 6370 |
| % of gene with introns | 70.8 |
| Mean number of exons per gene | 5.4 |
| Mean gene length (bp) | 34503.4 |
| Mean exon length (bp) | 274.9 |
| Mean intron length (bp) | 450.0 |
Figure 1.

Genome organization. (a) Circos plot showing paralogous gene pairs in Cucurbita pepo (red lines). Outer plots represent the proportion of repetitive (blue) or gene‐encoding (green) DNA by 200‐Kb windows. (b) Genomic synteny between Cucurbita pepo and Cucumis melo, Cucumis sativus and Citrullus lanatus. Lines join single‐copy orthologs. (c) Summary diagram with the species phylogeny showing the WGD event.
Repetitive elements
We identified that 93 Mb (37.8% of the assembly) consisted of repetitive elements (REs) (Figure S7 and Table S5). Long terminal repeats (LTR) represented 50.7% of the identified REs. Gypsy and Copia were the most abundant LTR superfamilies (24.2% and 19.8% of identified REs and 3.3% and 2.7% of the total genome). The Gypsy LTR abundance is similar to that found in C. melo, C. lanatus and C. sativus, which ranged from 19.5% to 34.4%, whereas the Copia family was less represented than in other Cucurbitaceae genomes (30.9%–34.4%). Other two LTR superfamilies were more abundant in the C. pepo genome compared to the other cucurbits: Cassandra (3% of identified RE vs. 0.1%–0.8%) and Caulimovirus (2.1% vs. 0.26%–0.9%). Satellites and simple repeats constituted 25.2% of all identified REs, which is a larger fraction than in related Cucurbitaceae species (4.4%–12.4%). Copia and Gypsy REs were assigned to their distinct families by building two phylogenetic trees (one for Copia and one for Gypsy; Figure S7). All Copia and Gypsy families previously identified in C. melo, C. lanatus and C. sativus were also present in C. pepo, except for Copia/Bianca and Gypsy/Ogre families. In these trees, the Gypsy/Galadriel and Copia/Tork4 families were overrepresented in C. pepo, so they seem to have undergone a diversification process in this species. Finally, approximately, 24% of REs were not assigned to any class of repetitive or transposable elements (TEs).
Comparative genomics
Genes, represented by the longest protein of the four cucurbit crops (Cucurbita pepo, Cucumis melo, Citrullus lanatus and two Cucumis sativus cultivars var. sativus, Chinese long; and var. hardiwickii, PI 183967), were grouped into gene families using OrthoMCL. The percentage of genes that could be assigned to a gene family in these species ranged from 91.2% to 72.8% (Table S6). In C. pepo, the number of gene families with two or more paralogs was higher than in the other crops (Figure 2a). Most C. pepo gene families were also present in the other cucurbits (Figure 2b); however, many of them had more than one gene in C. pepo (Figure 2c). Most of the zucchini paralogs were organized in large syntenic regions that covered most of the genome (Figure 1). Synteny with the other cucurbit species showed that, despite some conserved synteny, an extensive chromosomal rearrangement has occurred (Figure 1). The high number of paralogous genes detected and their synteny suggests that C. pepo could have undergone a WGD (Tables S7, S8 and S9).
Figure 2.

Genome duplication. (a) Distribution of the number of gene families based on the number of gene copies for Cucurbita pepo, Cucumis melo, Cucumis sativus and Citrullus lanatus; (b) Venn diagram showing the number of gene families, and (c) the number of duplicated gene families shared among the cucurbit genomes; (d) distribution of the rate of transversions on fourfold degenerate synonymous sites (4DTv) among paralogs for the five studied genomes. The inset shows the boxplots for the 4DTv distribution between ortholog copies of C. pepo and the rest of cucurbit species. The red dashed line shows the duplication event in C. pepo.
The rate of transversions on fourfold degenerate synonymous sites (4DTv) is a neutral genetic distance that can be used to estimate relative timing of evolutionary events. The distribution of 4DTvs among paralog pairs for all species other than C. pepo showed a wide peak that ranged from 0.4 to 1.1 with a maximum approximately 0.6 (Figure 2d), whereas for C. pepo, a more recent and narrower peak centred approximately 0.12 was found. The relative date of speciation can also be predicted by computing the 4DTvs between orthologous genes of any pair of species. These analyses showed that the speciation event that gave rise to the Cucurbita genus, represented by the pairwise 4DTv distributions of C. pepo against C. lanatus, C. sativus and C. melo, occurred almost simultaneously with the duplication event found in C. pepo (Figure 2d).
A total of 40 transcriptomes were assembled by Trinity from Illumina reads for 12 species, resulting in 18 446–67 366 genes and 18 902–92 522 transcripts (Table S1). The species and gene family trees were reconstructed using Phyldog, including both the genomes and these 40 transcriptomes. Phyldog identified duplication events in the gene family trees and calculated the number of duplications per branch in the species tree. According to Phyldog, most gene families underwent a duplication event (90%) in the branch that originated the Cucurbita genus (Figure 3). Additionally, a maximum‐likelihood phylogeny was reconstructed using a concatenated alignment in IQ‐TREE. The topologies recovered by both methods are highly congruent. The species trees based on genomic data showed that xerophytic perennial species (C. cordata, C. pedatifolia and C. foetidissima) were in a basal position, whereas mesophytic annuals and short‐lived perennial species of the genus were derived from them and formed a monophyletic taxon. The only remarkable difference between both methods was the position of C. ficifolia: IQ‐TREE grouped it with the mesophytic species, whereas Phyldog grouped it with the xerophytic species. There are some other minor differences related with the position of some accessions within a particular species between both trees. Some of these differences are related to suspected hybrid accessions like PI540737 (between C. pedatifolia and C. foetidissima) or PI532392 (between C. scabridifolia and C. foetidissima). In general, all nodes are supported by bootstrap values close to 100 except those related with hybrids.
Figure 3.
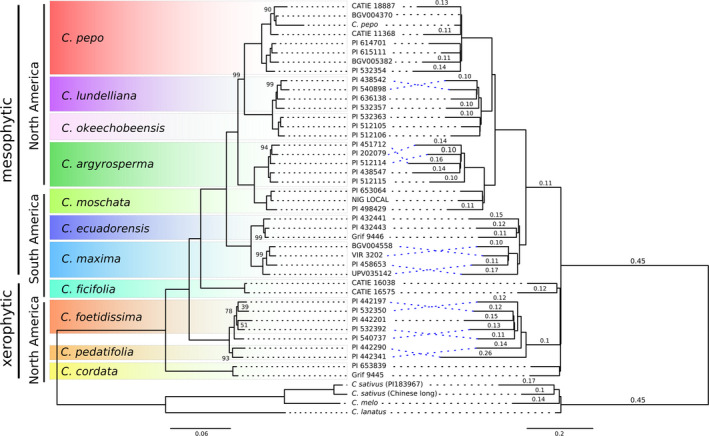
Phylogeny of the Cucurbita genus based on a concatenated method (left tree) or a joint estimation of gene and species trees (right tree). Left tree: branch lengths represent genetic distance and only bootstrap values lower than 100 are shown. Right tree: branch length represents proportion of duplicated genes per branch, values shown those proportions of duplicated genes higher than 0.1
A GO enrichment analysis was carried out on three sets of genes: (i) single‐copy C. pepo genes, (ii) all duplicated C. pepo genes and (iii) duplicated C. pepo genes found to be single copy in the rest of cucurbits (i.e. melon, watermelon and cucumber). Single‐copy genes were enriched in nucleic acid metabolic processes, DNA repair, DNA replication, DNA recombination, rRNA and tRNA processing, lipid metabolism and embryo development (Table S10 and Figure S8). In the gene set found to be duplicated in all species, the most significantly enriched GO terms were transcription and translation regulation, protein metabolism, transmembrane transport, ribosome biogenesis and signal transduction. The terms enriched in the C. pepo exclusive duplication were NAD biosynthesis, regulation of signal transduction, mitochondrial respiratory chain, regulation of cell cycle and cell structure, intracellular protein transport, pollen and vegetative development, photosynthesis light harvesting and photoperiodism of flowering. Interestingly, other genes related to flower development were also found among the exclusively duplicated genes in C. pepo such as EARLY FLOWERING 4, zinc finger CONSTANS‐LIKE 3, flowering locus T, RTF1, CDF73, KNUCKLES, CTR9, flowering locus K, GID1b, FLC EXPRESSOR, FRIGIDA and FPA. Additionally, seven of 34 genes annotated as ‘similar to CONSTANS‐LIKE protein’ were exclusively duplicated in C. pepo, as well as five of nine genes annotated as ‘similar to FRIGIDA’.
Genome assembly and transcriptomic and genomic raw sequences are deposited in NCBI under BioProject PRJNA386743. Genome v. 4.1, genome annotation and transcriptome v. 3.0 are also available at http://bioinf.comav.upv.es.
Discussion
In this study, we present the first description of the C. pepo genome. This new assembly is organized into 20 pseudomolecules, has a scaffold N50 of 1.8 Mb and is integrated with a high‐density genetic map. According to the coverage (92.1%) of the BUSCO conserved gene core set and the percentage of the RNAseq reads (91.1%) and genomic reads (99.4%) mapped against it, the current assembly covers most of the zucchini genome. The genome size inferred by k‐mer analysis was 283 Mb, so this assembly would constitute 93.0% of the genome. Thus, this assembly is an almost complete representation of the C. pepo genome.
Our results show that the C. pepo genome has undergone a WGD that occurred in the origin of the Cucurbita genus. Three independent lines of evidence support this: the topology of the gene family phylogenies, the karyotype organization and the distribution of 4DTv distances. Phyldog reconstructed the phylogeny of every gene family, and by comparing it with the species tree topology, we inferred where the duplication event likely occurred in each family. According to this analysis, most duplications occurred in the branch that separated the Cucurbita genus from the rest of species in the Cucurbitaceae family. The genome structure, shown by the physical location of the pairs of Zucchini paralog genes, was characterized by large syntenic regions within this species. These syntenic regions cover most of the genome and are likely to have been generated by a pseudodiplodization of an ancestral tetraploid followed by various chromosomal rearrangements. Interestingly, all species of the Cucurbita genus (tribe Cucurbiteae) present n = 20 chromosomes (Kocyan et al., 2007; Šiško et al., 2003/9), whereas species of Benincaseae tribe, which include the Cucumis and Citrullus genera, have a different chromosomal organization with n = 12 (melon), n = 11 (watermelon) or n = 7 (cucumber) (Kocyan et al., 2007). A possible polyploidy in the origin of Cucurbita was previously proposed based on the chromosome number and the number of isoenzyme copies (Kirkpatrick et al., 1985; Weeden, 1984). Despite the WGD, the size of the zucchini genome is similar to that of the other sequenced cucurbits, and the number of genes is not much higher. This suggests that most genes were deleted after the WGD event. It might well be the case that there has been a selective pressure to keep the genome size of these species within a certain range and that the maintained genes were specifically selected.
The 4DTv distribution found in C. melo, C. sativus and C. lanatus showed no evidence of a recent WGD (Garcia‐Mas et al., 2012; Huang et al., 2009). These three species present a peak in the 4DTv distribution around (0.6) that corresponds to the ancestral paleohexaploidy (γ) event that occurred in the divergence of monocotyledons and dicotyledons (~300 Mya) (Bowers et al., 2003). However, the 4DTv distances between paralog genes within zucchini showed shorter distances, characterized by a mode of 0.12. Thus, most paralogs seem to have been created by a recent duplication. Additionally, the 4DTv peaks found in the distribution calculated for the orthologous genes between C. pepo and melon, cucumber and watermelon can be used to date the Zucchini duplication. These peaks are all very close to the zucchini duplication peak. Therefore, both the 4DTv and gene family phylogenies are consistent with a duplication that occurred in the ancestral species that gave rise to the Cucurbita genus shortly after its split from the ancestor of C. melo, C. sativus and C. lanatus approximately 30 ± 4 Mya (Schaefer et al., 2009). The evolutionary rate derived from this time estimation is consistent with that found in other plants with a recent WGD like Nelumbo nucifera (4DTv = 0.17, 18 Mya) (Wang et al., 2013), Glycine max (0.057, 13 Mya) (Schmutz et al., 2010), Zizania latifolia (0.07, 13 Mya) (Guo et al., 2015) and Setaria italica (0.38, 70 Mya) (Zhang et al., 2012). Populus trichocarpa would be an exception with a much lower evolution rate (0.1, 60–65 Mya) (Tuskan et al., 2006), but this discrepancy could be due to the longer generation time of this plant (Smith and Donoghue, 2008). This WGD might have provided the Cucurbita species with a way to generate new gene functions to adapt to new habitats. In fact, this genus includes both xerophytic species, perennials adapted to dry climates and species adapted to moister or mesophytic environments, either annuals or short‐lived perennials, and extends from tropical to temperate regions of America. For instance, the duplication of the photosynthesis and flower development regulation genes found in the GO enrichment analysis could have provided mechanisms to adapt flowering to variations in temperature and the duration of days found from the southern USA to southern South America. Genes in these pathways have been implicated in the adaptation of several crops to different photoperiods and geographical adaptations (Blümel et al., 2015). FLOWERING LOCUS T has been described as a possible long‐distance florigenic signal in the cucurbits (Lin et al., 2007). The genus also includes an amazing degree of variation in morphological traits related to vine, fruit and seeds.
Consistent with previous Cucurbita phylogenetic studies (Jobst et al., 1998; Kates et al., 2017; Kistler et al., 2015; Sanjur et al., 2002; Wilson et al., 1992; Zheng et al., 2013), the xerophytic perennial species (C. cordata, C. pedatifolia and C. foetidissima) were basal to the Cucurbita genus. The current analysis supports the relationship among mesophytic species found by Kates et al. (2017), and additionally, it clarifies the clustering of the sister species C. foetidissima and C. pedatifolia, and C. lundelliana and C. okeechobeensis that were not previously resolved (Kates et al., 2017). The position of C. ficifolia remains controversial. The concatenated method clusters it as a basal species to the annual mesophytic taxa, showing a paraphyletic relationship with respect to the perennial taxa, in agreement with Wilson et al. (1992) and Kates et al. (2017). However, based on Phyldog analysis, C. ficifolia appears as a sister species of C. pedatifolia and C. foetidissima, in agreement with Zheng et al. (2013). Cucurbita ficifolia is a mesophytic species, but shares some morphological features with the xerophytic species. More data are needed to establish the relationship of C. ficifolia to the mesophytic/xerophytic species of the genus. This incongruence between trees may also be due to hybridization, as some partially fertile hybrids have been obtained between C. ficifolia and C. lundelliana, C. foetidissima and C. pedatifolia (Lira‐Saade, 1995), or it might be the result of very close speciation events.
This genome assembly constitutes a key resource for the study and breeding of the economically important C. pepo. Previous unpublished drafts, made available by us, of this genome have already been used in several publications related to the detection of resistance genes, the study of fruit development and the generation of molecular marker sets (Holdsworth et al., 2016; Martínez et al., 2013; Xanthopoulou et al., 2016). Additionally, we have assembled 40 transcriptomes for 11 species of the Cucurbita genus, which can be a valuable source of molecular markers as well as the foundation of comparative genomic studies.
Experimental procedures
Plant material, genetic material isolation and NGS sequencing
Genomic DNA was isolated from nuclei of the Cucurbita pepo subsp. pepo cultivar‐group zucchini, accession BGV004370 (also referred to as MU‐CU‐16 and held at the COMAV‐UPV GenBank, https://www.comav.upv.es). Leaves were frozen in liquid nitrogen, crushed in a mortar and put in a solution of 0.4 mm sucrose, 10 mm Tris–HCL pH 8.0, 10 mm MgCl2 and 5 mm β‐mercaptoethanol (20 mL per g of leaf tissue). This mixture was incubated on ice for 5 min. To eliminate debris and cellular fragments, samples were successively filtered through two filters (140 and 70 μm) and then centrifuged at 3000 g for 20 min at 4 °C. The pellet was resuspended in a solution of 0.25 mm sucrose, 10 mm Tris–HCl pH 8.0, 10 mm MgCl2, 1% Triton X‐100 and 5 mm β‐mercaptoethanol (1 mL per g of leaf tissue) and centrifuged again at 12 000 g for 10 min at 4 °C. Finally, the pellet was resuspended in 0.5 mL of 1.7 mm sucrose, 10 mm Tris–HCl ph 8.0, 2 mm MgCl2, 0.15% Triton X‐100 and 5 mm β‐mercaptoethanol and then centrifuged at 18 000 g during 1 h at 4 °C. The precipitated nuclei were resuspended in CTAB buffer, and the DNA was extracted using the CTAB protocol (Doyle and Doyle, 1990). Five genomic libraries were prepared: a 500‐bp pair‐end library and four mate‐pair libraries of 3‐, 7‐, 10‐ and 20‐Kb insert size. The first three libraries were prepared and sequenced by Macrogen (Seoul, Republic of Korea) using two Illumina Hiseq2000 lanes, one for the pair‐end library and another for the 3‐ and 7‐Kb mate‐pair libraries. The 10‐ and 20‐Kb libraries were prepared by the Boyce Thompson Institute (Ithaca, New York) using the Nextera protocol and sequenced in a single Illumina Hiseq 2000 lane.
Two different sets of transcriptomes were obtained: (i) a multitissue transcriptome from two cultivars, representing the two main C. pepo subspecies to assist the genome annotation and (ii) a group of 40 transcriptomes from 12 different wild and cultivated species of the Cucurbita genus for the phylogenetic and comparative analyses (see Table S1). In all cases, RNA was isolated using TRI reagent (Sigma), treated with DNAse and purified with a chloroform and ethanol precipitation. For the C. pepo transcriptome, two cultivars with contrasting phenotypes were used (BGV004370 or MU‐CU‐16, subsp. pepo cultivar‐group zucchini; and BGV005382 or UPV‐196, subsp. ovifera cultivar‐group scallop). RNA was extracted from different tissues: roots, leaves, apical shoots from plants in the male and female phase of development. Flower buds were collected at two early stages of flower development: mature flowers, preharvest fruits at various days after pollination, and postharvest fruits subjected to various postharvest treatments (ethylene, methylcyclopropene and cold). Equivalent amounts of RNA from each tissue were mixed into two pools, one per cultivar, and two independent cDNA libraries were prepared and sequenced in an Illumina Hiseq2000 lane by Macrogen (Seoul, Republic of Korea).
In the case of the 40 transcriptomes, in addition to the two C. pepo cultivars used in the multitissue transcriptome (zucchini and scallop), the analysed species included five additional genotypes of C. pepo (one subsp. ovifera (Acorn), two subsp. pepo (pumpkin) and two subsp. fraterna). Additionally, four additional domesticated taxa within the species were represented: C. moschata (three transcriptomes), C. maxima (three) and its wild ancestor C. maxima subsp. andreana Naudin (South America and Africa; one), C. argyrosperma Huber (southern United States and Central America; five) and C. ficifolia Bouché (Guatemala; two), as well as six wild species occurring in Mexico and Central and South America: the mesophytic annuals C. ecuadorensis Cutler & Whitaker (three), C. okeechobeensis (small) L.H Bailey subsp. martinezii (L.H. Bailey) T.C. Andres & G.P. Nabhan ex T.W. Walte (three), and C. lundelliana L.H Bailey (four) and the xerophytic perennials C. foetidissima Kunth (four), C. cordata S. Watson (two) and C. pedatifolia L.H. Bailey (three). RNA was extracted exclusively from young leaves, and the cDNA libraries were prepared and sequenced in a Hiseq2000 lane at the Boyce Thompson Institute (Ithaca, New York).
De novo genome assembly
The pair‐end and mate‐pair reads were cleaned using the ngs_crumbs software (code available at https://github.com/JoseBlanca/) to eliminate adapters, low‐quality bases (Phred quality <25 in a 5‐bp window), reads shorter than 50 bp and duplicated sequences. The Nextera mate‐pair reads (10‐Kb and 20‐Kb libraries) were classified by NextClip v0.8 (Leggett et al., 2014) according to the presence of the junction adaptor. Only the mate‐pairs for which NextClip could detect and trim the adaptor were used for the assembly. For the pre‐Nextera mate‐pair libraries, the detection and filtering of possible chimeric pairs were performed by mapping the reads against a first assembly of the genome, and only the pairs with the expected orientation and at the expected distance were kept. The implementation of this process can be found in the classify_chimeras and trim_mp_chimeras binaries of the ngs_crumbs software. The mitochondrial and chloroplastic reads were detected by blasting (Altschul et al., 1990) them against the C. melo organelle genomes (JF412791.1 and NC014050.1). Mitochondrial and chloroplastic reads were also included in the assembly to avoid the elimination of NUPTs (nuclear plastid DNA) and NUMTs (nuclear mitochondrial DNA segments) that could result in an artifactual fragmentation of the assembly, but only a sufficient number of randomly selected reads to reach 150X coverage. Assemblies with k‐mer lengths from 31 to 61 with a step‐size of 4 were made. The final assembly was carried out by SOAPdenovo2 v2.04 (Luo et al., 2012) using k‐mer size of 41. The resulting scaffolds were broken with BreakScaffolds (https://github.com/aubombarely/GenoToolBox) and reassembled with SSPACE (Boetzer et al., 2011). The new scaffolds were improved using SOAPdenovo2's GapCloser (Luo et al., 2012). Gene completeness of the assembly was assessed using BUSCO v.2 (Simão et al., 2015). Mitochondrial and chloroplastic scaffolds were identified using BLAST (Altschul et al., 1990) against the chloroplast and mitochondrial genomes of C. melo. Genome size was estimated from the k‐mer depth distribution as ∑(d · k d )/D, where d is the k‐mer depth, k d is the number of k‐mers for the given depth, and D is the maximum k‐mer depth of the distribution. The leftmost part of the distribution was discarded, as it includes mostly k‐mers due to sequencing errors. The k‐mer distribution was calculated by Jellyfish (Marçais and Kingsford, 2011) using a k‐mer size of 31.
To detect assembly artefacts and to group scaffolds into pseudomolecules, a genetic map was built. A group of 120 individuals of an F8 recombinant inbreed line (RIL) (Montero‐Pau et al., 2017), developed through single seed descent from a previous zucchini (BGV004370) × scallop (BGV005382) F2 (Esteras et al., 2012), were genotyped by genotyping by sequencing (GBS) (Elshire et al., 2011). SNP calling was performed using Freebayes (Garrison and Marth, 2012), and a genetic map was constructed using the R packages R/qtl (Broman et al., 2003) and ASMap (Taylor and Butler, 2014) (see details in Montero‐Pau et al. (2017)). Scaffolds that were present in more than one linkage group in the genetic map were visually explored with Hawkeye (Schatz et al., 2007) and manually split. Scaffolds were ordered and oriented into pseudomolecules according to the genetic map.
De novo transcriptome assembly
Raw reads were processed using ngs_crumbs software to eliminate adapter sequences, low‐quality bases (Phred quality < 25 in a 5‐bp window) and sequences shorter than 40 bp. The transcriptome was assembled with the Trinity assembler v2.0.6 (Grabherr et al., 2011) with default parameters. In the case of the C. pepo transcriptome, reads of both cultivars were merged to obtain a more comprehensive representation of the transcriptome. Additionally, reads from a previous 454‐based transcriptome (Blanca et al., 2011) were also included. The resulting contigs were reassembled with CAP3 (Huang and Madan, 1999) to eliminate redundancies. Low‐complexity transcripts were filtered out using ngs_crumbs. Trinity subcomponents were clustered using BLAST into unigene clusters using transitive clustering. Any two transcripts that shared an overlap longer than 100 bp and a similarity higher than 97% were considered to belong to the same unigene cluster. Finally, transcripts expressed less than 1% of the most highly expressed transcript in each Trinity subcomponent were filtered out using RSEM (http://deweylab.biostat.wisc.edu/rsem/). Gene completeness of the assembly was assessed using BUSCO v.2 (Simão et al., 2015).
Genome annotation
Genome structural annotation was performed using Maker‐P (Campbell et al., 2014) (version 2.31.6) with the default parameters. The C. pepo transcriptome was used to train Augustus (Hoff and Stanke, 2013) (version 3.0.2) with the default parameters. SNAP (Korf, 2004) (version 2006‐07‐28) was also trained with the same data set following the instructions from the Maker‐P manual. Repetitive sequences were extracted from the genome reference using RepeatModeler (Tempel, 2012) (version 1.0.8). The C. pepo transcriptome, repetitive sequences and training ab initio gene predictor files were used for the annotation with Maker‐P. Functional annotation was performed by sequence homology search using BlastP (minimum E‐value of 10−10) with GenBank, TAIR10 and SwissProt protein data sets (downloaded 2014‐12‐21). Additionally, InterProScan (Mulder and Apweiler, 2007) was used to annotate protein domains, extending the annotation to Gene Ontology terms associated with these protein domains. Blast2GO (Conesa and Götz, 2008) was used to annotate based on a Blast search against NCBI's nr database. Functional descriptions were processed using AHRD (https://github.com/groupschoof/AHRD) with weights of 100, 50 and 30 for SwissProt, TAIR and GenBank annotation, respectively.
A structural and homology‐based approach, as described in Campbell et al. (2014), was used to annotate the repetitive DNA. Briefly, miniature inverted‐repeat transposable elements (MITE) and long terminal repeat (LTR) retrotransposons were collected using MITE‐Hunter (Han and Wessler, 2010), LTR‐harvest and LTR‐digest (Ellinghaus et al., 2008; Steinbiss et al., 2009). A MITE and LTR library was built after excluding false positives, and selecting representative sequences (Schnable et al., 2009). This library was used to mask genome sequences with RepeatMasker (Tempel, 2012), and the resulting sequences were then processed by RepeatModeler to identify other repetitive sequences.
Reference sequences of Copia and Gypsy LTR superfamilies of the retrotranscriptase gene were obtained from GyDB (Llorens et al., 2011). Sequences were manually aligned, and the best‐fitting nucleotide substitution model (based on Bayesian information criterion) and maximum‐likelihood tree for each superfamily were obtained using IQ‐TREE (Chernomor et al., 2016; Nguyen et al., 2015). Branch support was computed using the bootstrap ultrafast method.
Transcriptome annotation
Transcripts were blasted against SwissProt, UniRef90 and the Arabidopsis proteins. Orthologues with cucumber and Arabidopsis were detected using a bidirectional BLAST search. The unigenes were associated with GO terms using Blast2GO software (Conesa and Götz, 2008). ORFs were predicted in the unigenes with the aid of the ESTScan software (Iseli et al., 1999).
Comparative genomics
Four complete genomes of three related species belonging to the Cucurbitaceae family were included in the study for comparative genomic analyses: Citrullus lanatus (genome v. 1) (Guo et al., 2012), Cucumis sativus var. sativus (Chinese long; v. 2) (Huang et al., 2009), C. sativus var. hardiwickii (Royle) Gabaer (PI 183967; v. 1) and Cucumis melo (v. 3.5) (Garcia‐Mas et al., 2012). The first three are accessible at www.icugi.org and the latter at http://melonomics.net. To be able to compare among genomes, the repetitive DNA characterization described above was performed in these four genomes.
Detection of gene duplications in the gene families was carried out using OrthoMCL (Fischer et al., 2011; Li et al., 2003) and OrthoMCL DB version 5 on the predicted proteomes of the five cucurbit genomes. In those cases in which more than one transcriptional variant was found for the same gene, only the longest variant was used. Differences in the functional role of the duplicated genes were assessed through GO enrichment tests using R package topGO (Alexa and Rahnenfuhrer, 2010), and REVIGO (Supek et al., 2011) was used to visualize the results. Transversion rates on fourfold degenerate synonymous sites (4DTv) were calculated between pairs of orthologs and paralogs using an in‐house Python script. Values were corrected for multiple substitutions (Tang et al., 2008).
The phylogeny (40 transcriptomes and five genomes) was reconstructed using a concatenated method and through a joint estimation of both species and gene trees carried out by Phyldog (Boussau et al., 2013). For the first approach, single‐copy genes that were detected using OrthoMCL and present in all cucurbit genomes were selected, and then, the corresponding C. pepo transcript was blasted against the 40 Cucurbita spp. transcriptomes. Only the blast hits with an E‐value higher than 10−60 and a match longer than 200 bp were retained. For each gene family, sequence alignments were built using an iterative refinement method implemented in MAFFT (Katoh et al., 2002). Alignments with less than 30 species were excluded. All resulting gene families were concatenated, and the maximum‐likelihood tree was inferred using IQ‐TREE (Nguyen et al., 2015) using a nucleotide substitution model for each gene (Chernomor et al., 2016). For each partition, the best model was selected based on the Bayesian information criterion (BIC). Branch support was obtained by bootstrap using an ultrafast method (Minh et al., 2013).
For the Phyldog approach, sequences were clustered in ortholog groups by blasting all C. pepo genes against all 40 Cucurbita spp. transcriptomes and the four cucurbit genomes. Blast hits with an identity lower than 70% and alignment length shorter than 200 residues were ignored. For each group, three multiple sequence alignments were obtained using Kalign (Lassmann et al., 2009), MUSCLE (Edgar, 2004) and MAFFT (Katoh et al., 2002). Alignment results were combined and evaluated with T‐Coffee (Chang et al., 2014; Wallace et al., 2006), and only alignments with an alignment score higher than 900 were kept. For each alignment, a starting tree for Phyldog was inferred using PhyML (Guindon et al., 2009) assuming the best nucleotide substitution model obtained by jModeltest (Posada, 2008). Phyldog (Boussau et al., 2013) was then used to simultaneously infer species and gene trees and to detect gene duplication events.
Supporting information
Figure S1 Distribution of sequences of k‐mer size 41 for different levels of coverage.
{kind=link}
Figure S2 Distribution of N50 for contigs (A) and scaffolds (B) for different k‐mer size values.
{kind=link}
Figure S3 Correlation between genetic and physical distances for each pseudochromosome. Color scale represents fraction of repetitive DNA.
{kind=link}
Figure S4 Summary of the structural annotation of C. pepo genome.
{kind=link}
Figure S5 Transcriptome GO annotation statistics A) by levels and B) at level 6.
{kind=link}
Figure S6 Genome GO annotation statistics (A) by levels and (B) at level 6.
{kind=link}
Figure S7 Repetitive elements. Fraction of the genome covered by different types of repetitive elements in C. pepo and four Cucurbita genomes (A). Maximum likelihood phylogenetic trees of C. pepo elements of Copia (B) and Gypsy (C) LTR superfamilies based on a fragment of the reverse transcriptase.
{kind=link}
Figure S8 Results of the GO enrichment test. Treemaps for the results of the GO enrichment tests on single‐copy genes in Cucurbita pepo, all duplicated genes in C. pepo and genes that are duplicated in C. pepo but not in other cucurbit genomes. The area of the rectangles represents the negative logarithm of the enrichment test FDR.
{kind=link}
Table S1 Accessions of domesticated and wild Cucurbita spp. used for transcriptomic and phylogenetic analyses. The number of reads used for assembly the transcriptomes, and number of genes and transcripts obtained are also shown.
Table S2 NGS library statistics. Numbers of raw reads, percentage of nucleotides over 30 quality, coverage, % of reads filtered out during the cleaning process, % of reads without adaptor, % of chimeric reads, number of cleaned reads, coverage of cleaned reads, and percentage of nucleotides over 30 quality in the clean reads.
Table S3 Scaffolds of genome assembly v.3.2. containing chloroplastic and mitochondrial regions. The pseudochromosomes were built from the version 3.2 scaffolds.
Table S4 Genome v.4.1 pseudochromosome configuration. The order, orientation and size of genome v. 3.2 scaffolds grouped in each pseudochromosome is shown. Equivalence of pseudochromosomes and linkage groups of Montero‐Pau et al. (2017) genetic map is also shown.
Table S5 Summary of repetitive elements found in Cucurbita pepo, Cucumis melo, Cucumis sativus and Citrullus lanatus. All results are expressed in bp.
Table S6 Gene family (orthogroups and paralogs in OrthoMCL) identification.
Table S7 List of genes that are single copy in Cucurbita pepo. Predicted functions are also shown.
Table S8 List of genes that are duplicated in Cucurbita pepo. Predicted functions are also shown.
Table S9 List of genes that are duplicated in Cucurbita pepo but not in Cucumis melo, Cucumis sativus or Citrullus lanatus. Predicted functions are also shown.
Table S10 GO term enrichment tests. The results are shown for single‐copy genes in Cucurbita pepo, duplicated genes, and genes that are exclusively duplicated in C. pepo when compared with other cucurbit genomes.
Acknowledgements
The authors want to thank the USDA, CATIE, VIR and COMAV‐UPV GenBanks for providing some of the accessions used in this article. Authors also thank Cristina Roig, Gorka Perpiña and Eva Maria Martínez for their technical assistance.
This work was partially funded by the INIA project RTA2011‐00044‐C02‐2 with contributions of E‐RTA2013‐00020‐C04‐03 of the Spanish Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA) cofunded with FEDER funds (EU) and AGL2014‐54598‐C2‐1‐R of the Spanish Ministry of Economy and Competitivity. The authors declare no conflict of interest.
NCBI accession number: PRJNA386743.
Contributor Information
Belén Picó, Email: mpicosi@btc.upv.es.
Joaquín Cañizares, Email: jcanizares@upv.es.
References
- Alexa, A. and Rahnenfuhrer, J. (2010) topGO: Enrichment Analysis for Gene Ontology. R package Version 2.18.0.
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andres, T.C. (1987) Cucurbita fraterna, the closest wild relative and progenitor of C. pepo . Report/Cucurbit Genet. Cooperat. 10, 69–71. [Google Scholar]
- Blanca, J. , Cañizares, J. , Roig, C. , Ziarsolo, P. , Nuez, F. and Picó, B. (2011) Transcriptome characterization and high throughput SSRs and SNPs discovery in Cucurbita pepo (Cucurbitaceae). BMC Genom. 12, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blümel, M. , Dally, N. and Jung, C. (2015) Flowering time regulation in crops—what did we learn from Arabidopsis? Curr. Opin. Biotechnol. 32, 121–129. [DOI] [PubMed] [Google Scholar]
- Boetzer, M. , Henkel, C.V. , Jansen, H.J. , Butler, D. and Pirovano, W. (2011) Scaffolding pre‐assembled contigs using SSPACE. Bioinformatics, 27, 578–579. [DOI] [PubMed] [Google Scholar]
- Boussau, B. , Szöllosi, G.J. , Duret, L. , Gouy, M. , Tannier, E. and Daubin, V. (2013) Genome‐scale coestimation of species and gene trees. Genome Res. 23, 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers, J.E. , Chapman, B.A. , Rong, J. and Paterson, A.H. (2003) Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature, 422, 433–438. [DOI] [PubMed] [Google Scholar]
- Broman, K.W. , Wu, H. , Sen, S. and Churchill, G.A. (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics, 19, 889–890. [DOI] [PubMed] [Google Scholar]
- Brown, R.N. and Myers, J.R. (2002) A genetic map of squash (Cucurbita sp.) with randomly amplified polymorphic DNA markers and morphological markers. J. Am. Soc. Hortic. Sci. 127, 568–575. [Google Scholar]
- Campbell, M.S. , Law, M. , Holt, C. , Stein, J.C. , Moghe, G.D. , Hufnagel, D.E. , Lei, J. et al. (2014) MAKER‐P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 164, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, J.‐M. , Di Tommaso, P. and Notredame, C. (2014) TCS: a new multiple sequence alignment reliability measure to estimate alignment accuracy and improve phylogenetic tree reconstruction. Mol. Biol. Evol. 31, 1625–1637. [DOI] [PubMed] [Google Scholar]
- Chernomor, O. , von Haeseler, A. and Minh, B.Q. (2016) Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 65, 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa, A. and Götz, S. (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker‐Walters, D.S. (1990) Evidence for Multiple Domestication of Cucurbita pepo. Biology and utilization of the Cucurbitaceae, pp. 96–101. Ithaca NY: Cornell University Press. [Google Scholar]
- Doyle, J.J. and Doyle, J.L. (1990) Isolation of plant DNA from fresh tissue. Focus, 12, 13–15. [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus, D. , Kurtz, S. and Willhoeft, U. (2008) LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshire, R.J. , Glaubitz, J.C. , Sun, Q. , Poland, J.A. , Kawamoto, K. , Buckler, E.S. and Mitchell, S.E. (2011) A robust, simple genotyping‐by‐sequencing (GBS) approach for high diversity species. PLoS ONE, 6, e19379 Public Library of Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteras, C. , Gómez, P. , Monforte, A.J. , Blanca, J. , Vicente‐Dólera, N. , Roig, C. , Nuez, F. et al. (2012) High‐throughput SNP genotyping in Cucurbita pepo for map construction and quantitative trait loci mapping. BMC Genom. 13, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, S. , Brunk, B.P. , Chen, F. , Gao, X. , Harb, O.S. , Iodice, J.B. , Shanmugam, D. et al. (2011) Using OrthoMCL to assign proteins to OrthoMCL‐DB groups or to cluster proteomes into new ortholog groups. Current Protocols Bioinform. 35, 6.12.1–6.12.19 Hoboken, NJ, USA: John Wiley & Sons, Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formisano, G. , Roig, C. , Esteras, C. , Ercolano, M.R. , Nuez, F. , Monforte, A.J. and Picó, M.B. (2012) Genetic diversity of Spanish Cucurbita pepo landraces: an unexploited resource for summer squash breeding. Genet. Resour. Crop Evol. 59, 1169–1184 Springer Netherlands. [Google Scholar]
- Garcia‐Mas, J. , Benjak, A. , Sanseverino, W. , Bourgeois, M. , Mir, G. , Gonzalez, V.M. , Henaff, E. et al. (2012) The genome of melon (Cucumis melo L.). Proc. Natl Acad. Sci. 109, 11872–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison, E. and Marth, G. (2012) Haplotype‐based variant detection from short‐read sequencing. arXiv [q‐bio.GN]. http://arxiv.org/abs/1207.3907.
- Gong, L. , Stift, G. , Kofler, R. , Pachner, M. and Lelley, T. (2008) Microsatellites for the genus Cucurbita and an SSR‐based genetic linkage map of Cucurbita pepo L.. Theoret. Appl. Genet. 117, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr, M.G. , Haas, B.J. , Yassour, M. , Levin, J.Z. , Thompson, D.A. , Amit, I. , Adiconis, X. et al. (2011) Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon, S. , Delsuc, F. , Dufayard, J.‐F. and Gascuel, O. (2009) Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 537, 113–137. [DOI] [PubMed] [Google Scholar]
- Guo, S. , Zhang, J. , Sun, H. , Salse, J. , Lucas, W.J. , Zhang, H. , Zheng, Y. et al. (2012) The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45, 51–58. [DOI] [PubMed] [Google Scholar]
- Guo, L. , Qiu, J. , Han, Z. , Ye, Z. , Chen, C. , Liu, C. , Xin, X. et al. (2015) A host plant genome (Zizania latifolia) after a century‐long endophyte infection. Plant J.: Cell Molecul. Biol. 83, 600–609. [DOI] [PubMed] [Google Scholar]
- Han, Y. and Wessler, S.R. (2010) MITE‐Hunter: a program for discovering miniature inverted‐repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff, K.J. and Stanke, M. (2013) WebAUGUSTUS–a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 41, W123–W128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth, W.L. , LaPlant, K.E. , Bell, D.C. , Jahn, M.M. and Mazourek, M. (2016) Cultivar‐based introgression mapping reveals wild species‐derived Pm‐0, the major powdery mildew resistance locus in squash. PLoS ONE, 11, e0167715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. and Madan, A. (1999) CAP3: A DNA sequence assembly program. Genome Res. 9, 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S. , Li, R. , Zhang, Z. , Li, L. , Gu, X. , Fan, W. , Lucas, W.J. et al. (2009) The genome of the cucumber, Cucumis sativus L. Nat. Genet. 41, 1275–1281 Nature Publishing Group. [DOI] [PubMed] [Google Scholar]
- Iseli, C. , Jongeneel, C.V. and Bucher, P. (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 138–148. [PubMed] [Google Scholar]
- Jobst, J. , King, K. and Hemleben, V. (1998) Molecular evolution of the internal transcribed spacers (ITS1 and ITS2) and phylogenetic relationships among species of the family Cucurbitaceae. Mol. Phylogenet. Evol. 9, 204–219. [DOI] [PubMed] [Google Scholar]
- Kates, H.R. , Soltis, P.S. and Soltis, D.E. (2017) Evolutionary and domestication history of Cucurbita (pumpkin and squash) species inferred from 44 nuclear loci. Mol. Phylogenet. Evol. 111, 98–109 Elsevier. [DOI] [PubMed] [Google Scholar]
- Katoh, K. , Misawa, K. , Kuma, K.‐I. and Miyata, T. (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucl. Acids Res. 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, K.J. , Decker, D.S. and Wilson, H.D. (1985) Allozyme differentiation in the Cucurbita pepo complex: C. pepo var. medullosa vs. C. texana . Econ. Bot. 39, 289–299 Springer. [Google Scholar]
- Kistler, L. , Newsom, L.A. , Ryan, T.M. , Clarke, A.C. , Smith, B.D. and Perry, G.H. (2015) Gourds and squashes (Cucurbita spp.) adapted to megafaunal extinction and ecological anachronism through domestication. Proc. Natl Acad. Sci. USA, 112, 15107–15112 National Acad Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocyan, A. , Zhang, L.‐B. , Schaefer, H. and Renner, S.S. (2007) A multi‐locus chloroplast phylogeny for the Cucurbitaceae and its implications for character evolution and classification. Mol. Phylogenet. Evol. 44, 553–577 Elsevier. [DOI] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinform. 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann, T. , Frings, O. and Sonnhammer, E.L.L. (2009) Kalign2: high‐performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res. 37, 858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y.H. , Jeon, H.J. , Kim, B.D. and Hong, K.H. (1995) Use of random amplified polymorphic DNAs for linkage group analysis in interspecific hybrid F2 generation of Cucurbita . J. Korean Soc. Horticul. Sci. (Korea Republic) 36, 323–330. [Google Scholar]
- Leggett, R.M. , Clavijo, B.J. , Clissold, L. , Clark, M.D. and Caccamo, M. (2014) NextClip: an analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics, 30, 566–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C.J. and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, M.‐K. , Belanger, H. , Lee, Y.‐J. , Varkonyi‐Gasic, E. , Taoka, K.‐I. , Miura, E. , Xoconostle‐Cázares, B. et al. (2007) FLOWERING LOCUS T protein may act as the long‐distance florigenic signal in the cucurbits. Plant Cell, 19, 1488–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira‐Saade, R. (1995) Estudios taxonómicos y ecogeográficos de las Cucurbitaceae Latinoamericanas de importancia económica. Systematic and Ecogeographic Studies on Crop Genepools, No. 9. Rome: International Plant Genetic Resources Institute. [Google Scholar]
- Llorens, C. , Futami, R. , Covelli, L. , Domínguez‐Escribá, L. , Viu, J.M. , Tamarit, D. , Aguilar‐Rodríguez, J. et al. (2011) The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res. 39, D70–D74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, R. , Liu, B. , Xie, Y. , Li, Z. , Huang, W. , Yuan, J. , He, G. et al. (2012) SOAPdenovo2: an empirically improved memory‐efficient short‐read de novo assembler. GigaScience, 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais, G. and Kingsford, C. (2011) A fast, lock‐free approach for efficient parallel counting of occurrences of k‐mers. Bioinformatics, 27, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez, C. , Manzano, S. , Megías, Z. , Garrido, D. , Picó, B. and Jamilena, M. (2013) Involvement of ethylene biosynthesis and signalling in fruit set and early fruit development in zucchini squash (Cucurbita pepo L.). BMC Plant Biol. 13, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh, B.Q. , Nguyen, M.A.T. and von Haeseler, A. (2013) Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero‐Pau, J. , Blanca, J. , Esteras, C. , Martínez‐Pérez, E.M. , Gómez, P. , Monforte, A.J. , Cañizares, J. et al. (2017) An SNP‐based saturated genetic map and QTL analysis of fruit‐related traits in Zucchini using Genotyping‐by‐sequencing. BMC Genom. 18, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder, N. and Apweiler, R. (2007) InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol. Biol. 396, 59–70. [DOI] [PubMed] [Google Scholar]
- Nguyen, L.‐T. , Schmidt, H.A. , von Haeseler, A. and Minh, B.Q. (2015) IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris, H.S. , Lebeda, A. , Křistkova, E. , Andres, T.C. and Nee, M.H. (2012) Parallel evolution under domestication and phenotypic differentiation of the cultivated subspecies of Cucurbita pepo (Cucurbitaceae). Econ. Bot. 66, 71–90 Springer‐Verlag. [Google Scholar]
- Paris, H.S. , Doron‐Faigenboim, A. , Reddy, U.K. , Donahoo, R. and Levi, A. (2015) Genetic relationships in Cucurbita pepo (pumpkin, squash, gourd) as viewed with high frequency oligonucleotide–targeting active gene (HFO–TAG) markers. Genet. Resour. Crop Evol. 62, 1095–1111 Springer Netherlands. [Google Scholar]
- Posada, D. (2008) jModeltest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. [DOI] [PubMed] [Google Scholar]
- Sanjur, O.I. , Piperno, D.R. , Andres, T.C. and Wessel‐Beaver, L. (2002) Phylogenetic relationships among domesticated and wild species of Cucurbita (Cucurbitaceae) inferred from a mitochondrial gene: Implications for crop plant evolution and areas of origin. Proc. Natl Acad. Sci. 99, 535–540 National Acad Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, H. , Heibl, C. and Renner, S.S. (2009) Gourds afloat: a dated phylogeny reveals an Asian origin of the gourd family (Cucurbitaceae) and numerous oversea dispersal events. Proc. Biol. Sci. R. Soc. 276, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz, M.C. , Phillippy, A.M. , Shneiderman, B. and Salzberg, S.L. (2007) Hawkeye: an interactive visual analytics tool for genome assemblies. Genome Biol. 8, R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz, J. , Cannon, S.B. , Schlueter, J. , Ma, J. , Mitros, T. , Nelson, W. , Hyten, D.L. et al. (2010) Genome sequence of the palaeopolyploid soybean. Nature, 463, 178–183. [DOI] [PubMed] [Google Scholar]
- Schnable, P.S. , Ware, D. , Fulton, R.S. , Stein, J.C. , Wei, F. , Pasternak, S. , Liang, C. et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science, 326, 1112–1115. [DOI] [PubMed] [Google Scholar]
- Simão, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Šiško, M. , Ivančič, A. and Bohanec, B. (2003/9) Genome size analysis in the genus Cucurbita and its use for determination of interspecific hybrids obtained using the embryo‐rescue technique. Plant Sci.: Int. J. Experiment. Plant Biol. 165, 663–669 Elsevier. [Google Scholar]
- Smith, B.D. (1997) The initial domestication of Cucurbita pepo in the Americas 10,000 years ago. Science, 276, 932–934 American Association for the Advancement of Science. [Google Scholar]
- Smith, B.D. (2006) Eastern North America as an independent center of plant domestication. Proc. Natl. Acad. Sci. USA, 103, 12223–12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S.A. and Donoghue, M.J. (2008) Rates of molecular evolution are linked to life history in flowering plants. Science, 322, 86–89 science.sciencemag.org. [DOI] [PubMed] [Google Scholar]
- Steinbiss, S. , Willhoeft, U. , Gremme, G. and Kurtz, S. (2009) Fine‐grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 37, 7002–7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek, F. , Bošnjak, M. , Škunca, N. and Šmuc, T. (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE, 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H. , Wang, X. , Bowers, J.E. , Ming, R. , Alam, M. and Paterson, A.H. (2008) Unraveling ancient hexaploidy through multiply‐aligned angiosperm gene maps. Genome Res. 18, 1944–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, J.D. and Butler, D. (2014) ASMap: Linkage map construction using the MSTmap algorithm. R package version 0. 3–3.
- Tempel, S. (2012) Using and understanding RepeatMasker. Methods Mol. Biol. 859, 29–51. [DOI] [PubMed] [Google Scholar]
- Tuskan, G.A. , Difazio, S. , Jansson, S. , Bohlmann, J. , Grigoriev, I. , Hellsten, U. , Putnam, N. et al. (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science, 313, 1596–1604. [DOI] [PubMed] [Google Scholar]
- Vitiello, A. , Scarano, D. , D'Agostino, N. , Digilio, M.C. , Pennacchio, F. , Corrado, G. and Rao, R. (2016) Unraveling zucchini transcriptome response to aphids. PeerJ PrePrints, 4, e1635v1. [Google Scholar]
- Wallace, I.M. , O'Sullivan, O. , Higgins, D.G. and Notredame, C. (2006) M‐Coffee: combining multiple sequence alignment methods with T‐Coffee. Nucleic Acids Res. 34, 1692–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Fan, G. , Liu, Y. , Sun, F. , Shi, C. , Liu, X. , Peng, J. et al. (2013) The sacred lotus genome provides insights into the evolution of flowering plants. Plant J.: Cell Molecul. Biol. 76, 557–567 Wiley Online Library. [DOI] [PubMed] [Google Scholar]
- Weeden, N.F. (1984) Isozyme studies indicate that the genus Cucurbita is an ancient tetraploid. Report/Cucurbit Genet. Cooperat. 7, 84–85 Cucurbit Genetics Cooperative. agris.fao.org. [Google Scholar]
- Wilson, H.D. , Doebley, J. and Duvall, M. (1992) Chloroplast DNA diversity among wild and cultivated members of Cucurbita (Cucurbitaceae). Theoret. Appl. Genet. 84, 859–865. [DOI] [PubMed] [Google Scholar]
- Wyatt, L.E. , Strickler, S.R. , Mueller, L.A. and Mazourek, M. (2015) An acorn squash (Cucurbita pepo ssp. ovifera) fruit and seed transcriptome as a resource for the study of fruit traits in Cucurbita . Horticul. Res. 2, 14070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthopoulou, A. , Psomopoulos, F. , Ganopoulos, I. , Manioudaki, M. , Tsaftaris, A. , Nianiou‐Obeidat, I. and Madesis, P. (2016) De novo transcriptome assembly of two contrasting pumpkin cultivars. Genom. Data, 7, 200–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G. , Liu, X. , Quan, Z. , Cheng, S. , Xu, X. , Pan, S. , Xie, M. et al. (2012) Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 30, 549–554. [DOI] [PubMed] [Google Scholar]
- Zheng, Y.‐H. , Alverson, A.J. , Wang, Q.‐F. and Palmer, J.D. (2013) Chloroplast phylogeny of Cucurbita: Evolution of the domesticated and wild species. J. Syst. Evol. 51, 326–334. [Google Scholar]
- Zraidi, A. and Lelley, T. (2004) Genetic map for pumpkin Cucurbita pepo using random amplified polymorphic DNA markers. In: Progress in cucurbit genetics and breeding research. Proceedings of Cucurbitaceae 2004. ( Lebeda, A. and Paris, H.S. eds), pp. 507–514. Palacký University. [Google Scholar]
- Zraidi, A. , Stift, G. , Pachner, M. , Shojaeiyan, A. , Gong, L. and Lelley, T. (2007) A consensus map for Cucurbita pepo . Molecul. Breed.: New Strateg. Plant Improve. 20, 375–388 Springer Netherlands. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Distribution of sequences of k‐mer size 41 for different levels of coverage.
Figure S2 Distribution of N50 for contigs (A) and scaffolds (B) for different k‐mer size values.
Figure S3 Correlation between genetic and physical distances for each pseudochromosome. Color scale represents fraction of repetitive DNA.
Figure S4 Summary of the structural annotation of C. pepo genome.
Figure S5 Transcriptome GO annotation statistics A) by levels and B) at level 6.
Figure S6 Genome GO annotation statistics (A) by levels and (B) at level 6.
Figure S7 Repetitive elements. Fraction of the genome covered by different types of repetitive elements in C. pepo and four Cucurbita genomes (A). Maximum likelihood phylogenetic trees of C. pepo elements of Copia (B) and Gypsy (C) LTR superfamilies based on a fragment of the reverse transcriptase.
Figure S8 Results of the GO enrichment test. Treemaps for the results of the GO enrichment tests on single‐copy genes in Cucurbita pepo, all duplicated genes in C. pepo and genes that are duplicated in C. pepo but not in other cucurbit genomes. The area of the rectangles represents the negative logarithm of the enrichment test FDR.
Table S1 Accessions of domesticated and wild Cucurbita spp. used for transcriptomic and phylogenetic analyses. The number of reads used for assembly the transcriptomes, and number of genes and transcripts obtained are also shown.
Table S2 NGS library statistics. Numbers of raw reads, percentage of nucleotides over 30 quality, coverage, % of reads filtered out during the cleaning process, % of reads without adaptor, % of chimeric reads, number of cleaned reads, coverage of cleaned reads, and percentage of nucleotides over 30 quality in the clean reads.
Table S3 Scaffolds of genome assembly v.3.2. containing chloroplastic and mitochondrial regions. The pseudochromosomes were built from the version 3.2 scaffolds.
Table S4 Genome v.4.1 pseudochromosome configuration. The order, orientation and size of genome v. 3.2 scaffolds grouped in each pseudochromosome is shown. Equivalence of pseudochromosomes and linkage groups of Montero‐Pau et al. (2017) genetic map is also shown.
Table S5 Summary of repetitive elements found in Cucurbita pepo, Cucumis melo, Cucumis sativus and Citrullus lanatus. All results are expressed in bp.
Table S6 Gene family (orthogroups and paralogs in OrthoMCL) identification.
Table S7 List of genes that are single copy in Cucurbita pepo. Predicted functions are also shown.
Table S8 List of genes that are duplicated in Cucurbita pepo. Predicted functions are also shown.
Table S9 List of genes that are duplicated in Cucurbita pepo but not in Cucumis melo, Cucumis sativus or Citrullus lanatus. Predicted functions are also shown.
Table S10 GO term enrichment tests. The results are shown for single‐copy genes in Cucurbita pepo, duplicated genes, and genes that are exclusively duplicated in C. pepo when compared with other cucurbit genomes.