

Abstract
Extracellular hemoglobin, a byproduct of hemolysis, can release its prosthetic heme groups upon oxidation. This produces metabolically active heme that is exchangeable between acceptor proteins, macromolecules and low molecular weight ligands, termed here labile heme. As it accumulates in plasma labile heme acts in a pro‐oxidant manner and regulates cellular metabolism while exerting pro‐inflammatory and cytotoxic effects that foster the pathogenesis of hemolytic diseases. Here, we developed and characterized a panel of heme‐specific single domain antibodies (sdAbs) that together with a cellular‐based heme reporter assay, allow for quantification and characterization of labile heme in plasma during hemolytic conditions. Using these approaches, we demonstrate that when generated during hemolytic conditions labile heme is bound to plasma molecules with an affinity higher than 10−7 m and that 2–8% (~ 2–5 μm) of the total amount of heme detected in plasma can be internalized by bystander cells, termed here bioavailable heme. Acute, but not chronic, hemolysis is associated with transient reduction of plasma heme‐binding capacity, that is, the ability of plasma molecules to bind labile heme with an affinity higher than 10−7 m. The heme‐specific sdAbs neutralize the pro‐oxidant activity of soluble heme in vitro, suggesting that these maybe used to counter the pathologic effects of labile heme during hemolytic conditions. Finally, we show that heme‐specific sdAbs can be used to visualize cellular heme. In conclusion, we describe a panel of heme‐specific sdAbs that when used with other approaches provide novel insights to the pathophysiology of heme.
Keywords: antibody engineering, heme, hemolysis, labile heme, single‐domain antibody
Abbreviations
- β‐ME
β‐mercaptoethanol
- ATR
attenuated total reflectance
- biotin
N‐[‐5‐(Hydrazinocarboxy) pentyl]‐D‐biotinamide
- BR
bilirubin
- BV
Biliverdin
- CCD
charge‐coupled device
- CD
circular dichroism
- CDR
complementary determining region
- CN
cysteine‐asparagine
- CoPP
cobalt PP
- CS
cysteine‐serine
- Cytc
cytochrome c
- DeuP
deuteroporphyrin IX
- DHB
dihydroxybenzoic acid
- Fe
iron
- FePP
heme
- FTIR
Fourier transform infrared
- GaPP
gallium protoporphyrin IX
- HABA
4′‐hydroxyazobenzene‐2‐carboxylic acid
- HA
hemagglutinin
- HBC
heme‐binding capacity
- Hb
hemoglobin
- HBM
heme‐binding motifs
- HPLC
high performance liquid chromatography
- HPX
hemopexin
- HRP
horseradish peroxidase
- HSA
human serum albumin
- iRBC
infected RBC
- LB
Luria broth
- mAb
monoclonal antibody
- MALDI
matrix‐assisted laser desorption/ionization
- Mb
myoglobin
- MetHb
oxidized Hb
- NO
oxide
- PBS
Dulbecco's phosphate buffered saline
- pNPP
para‐nitrophenylphosphate
- PP
protoporphyrin
- RBC
red blood cell
- RR
Resonance Raman
- SA
succinylacetone
- SdAb
single‐domain antibody
- SDS/PAGE
sodium dodecyl sulfate/polyacrylamide gel electrophoresis
- SnPP
tin PP
- TNF
tumor necrosis factor
- TOFMS
time‐of‐flight mass spectrometry
- ZnPP
zinc protoporphyrin
Introduction
Heme consists of a tetrapyrrole ring bound to iron (Fe) via several nitrogens. Heme exists essentially as a prosthetic group of hemoproteins 1, including hemoglobin (Hb) 2, where Fe binds to gaseous molecules such as oxygen (O2), nitric oxide (NO) 3 or carbon monoxide (CO) 4, via effective back‐donation of Fe dπx electrons to low‐lying π* orbitals of these diatomic molecules. Heme displays subtle chemical modifications, giving rise to the a, b, and c heme variants 5. The most common and abundant heme type in mammals is heme b present in Hb and myoglobin (Mb), among other hemoproteins. Whereas heme c can bind covalently to proteins via two thioester bonds, heme b and a cannot and can be released from hemoproteins, such as observed upon Hb oxidation 6, 7, 8, 9. While often referred as ‘free heme’ 10, 11, when released from hemoproteins heme is most probably never in ‘free form’. Labile heme is the term used to refer to the pool of metabolically active heme, which is loosely bound to acceptor proteins, therefore becoming exchangeable with other macromolecules and low molecular weight ligands 12. Of note, while heme is a stable molecule, the term ‘labile’ is used to emphasis that ‘labile heme’ is more readily prone to alteration of its redox state, as imposed by its immediate environment.
In adult humans, the sheer number of red blood cells (RBC; ~ 2–3 × 1013) coupled to their high Hb (~ 3 ×108 molecules/RBC) and heme (~ 1.2 × 109 molecules per RBC) 13 content makes that clinically silent levels of intravascular hemolysis can be associated with the release of relatively high amounts of Hb into plasma. Upon oxidation, extracellular Hb can release its noncovalently bound heme b groups 6, 7, 8, 9, generating pro‐oxidant labile heme 8, 14, 15 that acts as a alarmin 16, sensed by pattern recognition receptors, such as Toll like receptor 4 17 or NACHT, LRR and PYD domains‐containing protein 3 (NALP3) 18. This endows labile heme with pro‐inflammatory 16, 17, 18, 19, 20, vasoactive 21, and cytotoxic 10, 22, 23, 24, 25 effects, while also regulating metabolism 26 and interfering with coagulation 27. Presumably, these pathophysiologic effects of labile heme contribute critically to the pathogenesis of hemolytic conditions, such as malaria caused upon infection by the protozoan parasites of the genus Plasmodium 6, 28, severe sepsis caused by polymicrobial infections 10 or sickle cell disease caused by mutations in the gene encoding the β chain of Hb 21, 29, 30.
Current limitations in methodologies allowing for accurate quantification of labile heme or providing further information on its biological environment make it particularly challenging to ascertain the contribution of labile heme to the pathogenesis of these hemolytic conditions. Here, we developed a panel of heme‐specific sdAbs that, when used together with other approaches 31, allow to quantify and characterize labile heme in plasma, its rate of cellular internalization and cellular localization. We report that intravascular hemolysis in mice is associated with the accumulation of labile heme in plasma, which is bound to plasma molecules with an affinity higher than 10−7 m. We estimate that about 2–8% (~ 2–5 μm) of the heme released in plasma becomes ‘bioavailable’, being internalized by bystander cells. Heme‐specific sdAb can be used to neutralize the pro‐oxidant effect of labile heme in solution, an effect that could have therapeutic implications in the treatment of infectious as well as noninfectious hemolytic conditions. Finally, we demonstrate that heme‐specific sdAb can be used to monitor intracellular heme, detected primarily in the mitochondria.
Results
Generation of heme‐specific SdAbs
We screened a synthetic phage display sdAb library 32, derived from a highly soluble and stable VL scaffold 33 against heme b, biotinylated in a single carboxylic group, corresponding to a molecular weight of 969.2 Da (Fig. 1A,B). Phages encoding sdAb recognizing biotinylated‐heme were selected using streptavidin‐magnetic beads (Fig. 1C,D), essentially as described 34. An ELISA, using nonbiotinylated‐heme as a solid‐phase antigen, was used to determine the relative heme‐binding capacity of the selected sdAb. Heme recognition was normalized to the relative amount of sdAb, as detected by ELISA, using a monoclonal antibody (mAb) against the human influenza hemagglutinin (HA) epitope expressed in the C‐terminus of these sdAb. Eleven sdAbs were selected through this assay (Fig. 1E), expressed in Escherichia coli BL21 and purified under denaturing conditions, before refolding by step‐wise dialysis 35. Protein yield was typically around 10 mg·L−1 with > 90% purity, as assessed by SDS/PAGE (Fig. 1E). Expression of the ~ 15 kDa protein corresponding to the full‐length sdAb containing the MKKTAIAIAVALAGFATV leader peptide sequence and a ~ 13 kDa protein corresponding to the leaderless sdAb, was confirmed by western blot (Fig. 1E).
Figure 1.
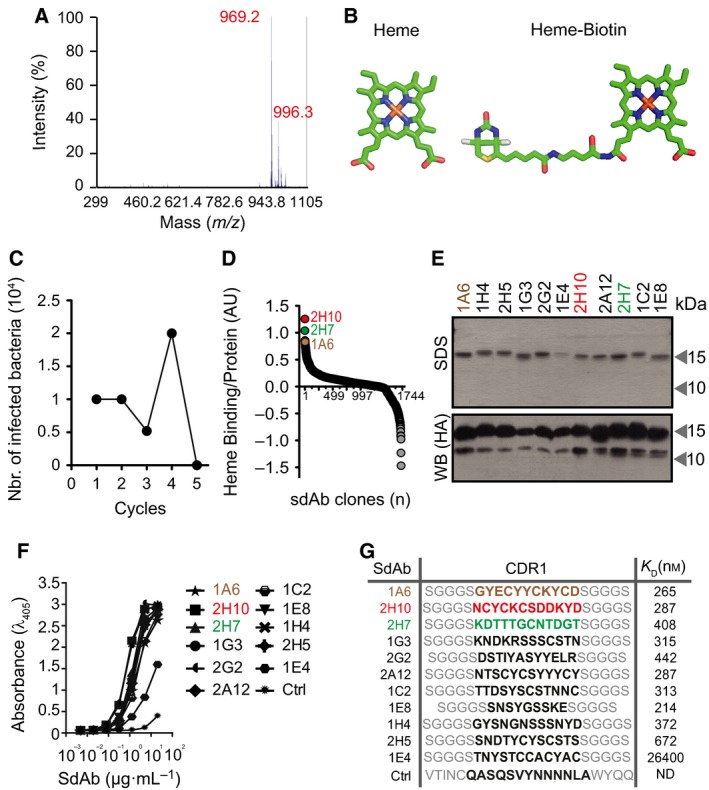
Selection of heme‐binding sdAbs using phage display technology. (A) MALDI‐TOF/TOF analysis of biotinylated‐heme. Peak of mass‐to‐charge (m/z) 969.2 Da with characteristic isotopic cluster pattern, corresponding to biotinylation of a single hemin carboxylic acid residue. (B) Schematic representation (Accelrys draw 4.1 (BIOVIA, San Diego, CA, USA) and 3D representations; pymol software, PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC, New York, NY, USA) of heme and biotinylated‐heme. (C) Elution cycles outputs of bacteria infected with phages displaying sdAbs recognizing heme. (D) Ratio of heme binding to protein expression of 1721 sdAb (circles) screened by ELISA, as described in Experimental procedures. SdAbs 2H10, 2H7, and 1A6, with highest heme binding to protein expression ratio, are highlighted. (E) SDS/PAGE of purified sdAbs stained by coomassie‐based stain or detected by western blot using an anti‐HA mAb. (F) ELISA for recognition of solid‐phase heme by purified sdAb. Ctrl: Control sdAb that does not recognize heme. (G) SdAb CDR1 aminoacid sequences, as determined by DNA sequencing. Binding affinity of SdAbs toward heme, determined by BIAcore surface Plasmon resonance. ND, not detectable.
We compared the relative capacity of the purified sdAbs, when used at the same concentration, to bind a solid‐phase heme in an ELISA (Fig. 1F). The affinity (K D) of these sdAb toward heme was determined by surface plasmon resonance and estimated to be in the order of 10−7 m, with the exception of sdAb 1E4, which has an affinity of 10−5 m toward heme (Fig. 1G). Further in silico analyses revealed that the complementarity‐determining region (CDR) 1 of all the selected sdAbs carries potential heme‐binding motifs 36, including Cysteine‐X‐X‐Cysteine‐Lysine (CXXCK; were X is any amino acid (aa)) characteristic of hemoproteins that bind heme c 37, CK, i.e. sdAb 1A6, CN, i.e. sdAb 2H7 or CS, i.e. sdAb 2H10, 1G3, 2A12, 1C2 and 2H5, characteristic of hemoproteins that bind heme b (Fig. 1G). Based on their heme affinity and the presence of putative heme‐binding motifs, we selected sdAbs 1A6, 2H7 and 2H10 for further analyses. Of note, CDR3 sequences were absent in these sdAbs.
SdAbs specificity
To determine the relative specificity of sdAbs 1A6, 2H7 and 2H10, these were preincubated with a variety of heme‐related molecules and their capacity to bind solid‐phase bound heme was assessed by ELISA. Heme (FePP) was used a positive control, gallium protoporphyrin (PP) IX (GaPP) as a metallated porphyrin containing redox inert gallium instead of Fe and PP IX (PP) as a metal‐free precursor of heme. As expected, preincubation with FePP abolished subsequent recognition of solid‐phase bound heme by all three sdAbs (Fig. 2A). Preincubation with PP inhibited heme recognition by sdAb 1A6 but not by sdAbs 2H7 and 2H10 (Fig. 2A). This suggests that Fe, present in FePP but not in PP, is required for heme recognition by sdAbs 2H7 and 2H10 but not by sdAb 1A6. Preincubation with GaPP abolished heme recognition by all three sdAbs (Fig. 2A). Given that gallium is not readily reduced, mimicking Fe3+, this suggests that all three sdAbs recognize preferentially oxidized Fe3+PP, consistent with their initial selection under aerobic conditions favoring heme oxidation into Fe3+PP. To assess the relative contribution of the porphyrin side chains to heme recognition by the sdAb, these were preincubated with Fe deuteroporphyrin IX (DeutP), a synthetic heme derivative lacking the heme vinyl group or with FePP IX dimethyl ester chloride (FePPCH3) in which the two‐propionate side chains are methylated (Fig. 2B). While DeutP inhibited heme recognition by all three sdAb (Fig. 2B), FePPCH3 inhibited heme recognition by sdAb 1A6 and 2H7 but only to a lesser extent by sdAb 2H10. This suggests that heme recognition by sdAb 2H10 involves a carboxylic acid in the propionate side chains (Fig. 2B).
Figure 2.
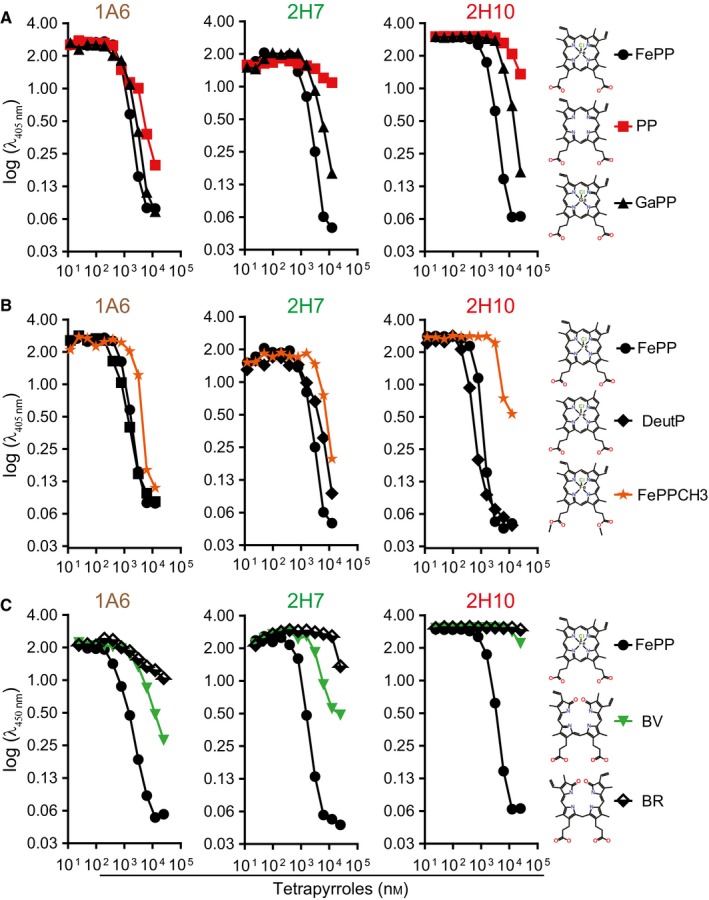
Assessment of sdAbs specificity against heme versus related tetrapyrroles. Inhibitory effect of preincubation of sdAb with increasing concentrations of (A) FePP, PP or GaPP, (B) FePP, DeutP or FePPCH3, (C) biliverdin (BV) or bilirubin (BR) on sdAb binding to solid‐phase bound heme, measured by ELISA. Data from one out of three independent experiments with similar trend are shown.
Preincubation with biliverdin (BV) or bilirubin (BR), a direct and an indirect end‐product of heme catabolism, respectively, partially inhibited heme recognition by sdAb 1A6 but not by sdAb 2H10 (Fig. 2C). Recognition of solid‐phase bound heme by sdAb 2H7 was also partially inhibited by BV but not by BR (Fig. 2C). This suggests that sdAb 2H10 is probably the only, among these three sdAbs, that recognizes specifically heme. This is consistent with the preferred V‐shaped ridge tile conformation of BR 38, being quite distinct from heme and as such unlikely to be recognized by the same linear aa motif used to recognize heme in 2H10 sdAb.
We then asked whether these sdAbs recognize porphyrins containing divalent metals, other than Fe, such as tin PP (SnPP), cobalt PP (CoPP), or Zinc PP (ZnPP). Preincubation of sdAbs 1A6, 2H7, and 2H10 with SnPP or CoPP inhibited their binding to solid‐phase bound heme (Fig. 3A), while preincubation with ZnPP inhibited heme recognition by sdAb 2H7 but not by sdAb 1A6 and 2H10 (Fig. 3A). This suggests that the chemical properties of the metal at the center of the protoporphyrin ring may be important for heme recognition by sdAb 1A6 and 2H10 but not by sdAb 2H7.
Figure 3.
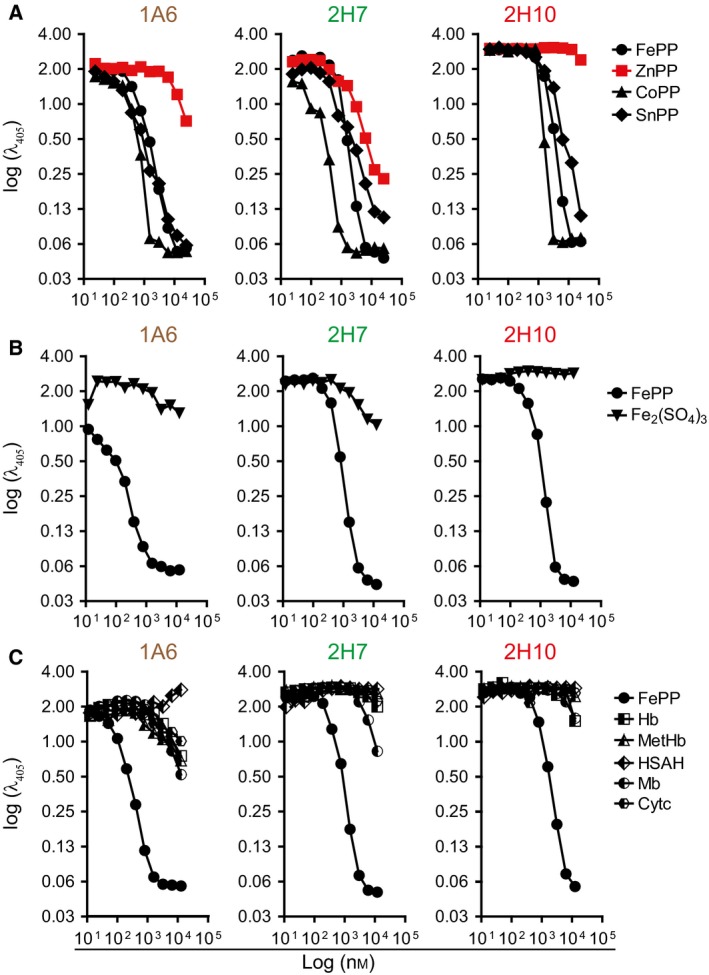
Assessment of sdAbs specificity against heme versus heme analogs or heme contained in hemoproteins. Inhibitory effect of preincubation of sdAb with increasing concentrations of (A) FePP, ZnPP, CoPP or SnPP, (B) FePP or Fe2 (SO 4)3 (C) FePP, Hb, MetHb, Mb, Cytochrome c (Cytc) or human serum albumin bound to heme (HSAH), measured by ELISA. Data from one out of three independent experiments with similar trend are shown.
We also asked whether these sdAbs recognized Fe in the absence of a porphyrin ring. Preincubation with Fe2(SO4)3 failed to inhibit the recognition of solid‐phase‐bound heme by all three sdAbs (Fig. 3B), suggesting that Fe is not sufficient per se for recognition by these sdAb. Finally, we asked whether these sdAbs recognize heme contained inside hemoproteins. Preincubation of the sdAbs with Hb, oxidized Hb (MetHb), Mb, albumin bound to heme b or cytochrome c failed to inhibit the recognition of solid‐phase bound heme by all three sdAbs (Fig. 3C). This suggests that while these sdAbs recognize soluble heme they fail to recognize heme when incorporate in hemoproteins. Table 1 summarizes these results.
Table 1.
Summary of the sdAbs binding specificity against different tetrapyrroles. SdAbs 1A6, 2H7 and 2H10 recognize (+) or not (−) the different tetrapyrroles and some only when at higher concentration (+/−)
| Tetrapyrroles | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| sdAbs | FePP | PP | GaPP | ZnPP | CoPP | SnPP | DeutPP | FePPCH3 | BV | BR |
| 1A6 | + | + | + | − | + | + | + | + | +/− | +/− |
| 2H7 | + | − | + | + | + | + | + | +/− | +/− | − |
| 2H10 | + | − | + | − | + | + | + | − | − | − |
Structural characterization of sdAbs‐heme complexes
We used a heme‐biotin pull‐down assay to compare the relative capacity of sdAbs 1A6, 2H7, and 2H10 to recognize heme in solution. All three sdAbs bound heme‐biotin in solution, with 1A6 and 2H10 showing higher binding capacity, as compared to 2H7 (Fig. 4A), which is consistent with their relative affinity toward heme (Fig. 1G).
Figure 4.
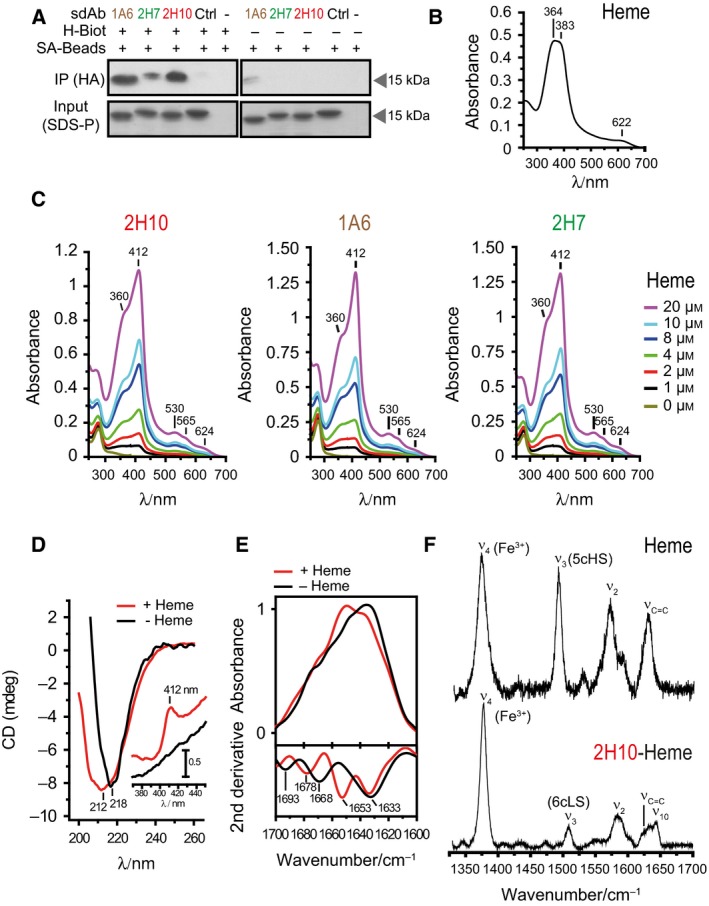
Analysis of heme binding by SdAbs. (A) SdAbs bound to biotinylated‐heme in solution were pooled‐down using streptavidin (SA) beads and detected by western blot using anti‐HA mAb. Input was measured by Coomassie‐based stain. (B) UV‐Visible spectra of hemin. Soret region at approximately 364 and 383 nm and a CT band at 622 nm are shown, representative of three independent experiments. (C) UV‐visible spectra of sdAb 2H10, 1A6 and 2H7 bound to heme at different concentrations. Soret (412 nm), Q1 (530 nm), Q0 (565 nm), and CT (624 nm) bands are highlighted. (D) Far UV CD spectra of sdAb 2H10 in the apo (black) and heme‐bound (red) forms. Shift from 212 to 218 is due to heme‐driven conformational rearrangement of the sdAb secondary structure. The inset shows the Soret region, with the appearance of the 412 nm band, due to heme binding to the sdAb. (E) ATR FTIR absorption spectra (top) and second derivative (bottom) of sdAb 2H10 in the apo (black) and heme‐bound (red) forms in the amide I region (1700–1610 cm−1), showing structural modification upon heme coordination. (F) High frequency Resonance Raman spectra of hemin and sdAb 2H10 bound to hemin, obtained with 413 nm excitation.
We used UV‐visible spectroscopy to monitor heme (Fig. 4B) and heme binding to sdAbs (Fig. 4C). The UV‐visible spectrum of heme (Fig. 4B) shows two overlapping bands, at ~ 363 and 383 nm, in the Soret region and a charge transfer band at 622 nm, indicating high spin Fe3+ state. The UV‐visible spectrum of sdAb bound to heme shows a δ band at 360 nm, an intense Soret band at 412 nm, two weak Q bands at 530 and 565 nm and another 624 nm band in the charge transfer region (Fig. 4C). The Soret band indicates the presence of Fe3+, the 530 and 565 nm bands are consistent with the presence of a six‐coordinated low spin Fe3+ state, the δ band at 360 nm suggests the presence of a thiol axial heme ligation 39 and the 624 nm band is indicative of either Cys or porphyrin to high spin Fe3+ charge transfer (Fig. 4C). With the exception of the charge transfer band, UV‐visible features of the sdAb/heme complex are similar to the spectra of ferric cytochrome P450cam, carrying Cys and H2O axial ligands 40.
Heme binding was associated with alterations in the secondary structure of the sdAb, as assessed by circular dichroism (CD) and Attenuated total reflection Fourier Transform infrared (ATR‐FTIR) spectroscopies for sdAb 2H10. Namely, the far‐UV CD spectrum revealed a negative band centered at 218 nm, indicative of a β‐sheet protein fold (Fig. 4D). Heme binding resulted in broadening of this band, suggesting a conformational rearrangement of secondary structure elements (Fig. 4D). Analysis of the Soret region reflects heme coordination with the appearance of a positive CD band at 412 nm, absent in apo‐2H10 sdAb (Fig. 4D, inset).
The impact of heme coordination on the secondary structure of sdAb 2H10 was further analyzed by ATR‐FTIR, which is sensitive to different types of β‐sheet structures. A comparison of the absorption spectra of sdAb 2H10 in the presence/absence of heme, in the amide I region (1700–1610 cm−1) is indicative of structural alterations upon heme coordination (Fig. 4E, top). Second derivative analysis of the ATR‐FTIR spectra, performed to identify individual components under the intrinsically broad amide I band envelope 41, revealed major contributions typical of β‐sheet structures for apo‐2H10, in agreement with CD data (Fig. 4E, bottom). The presence of 1633 and 1693 cm−1 bands simultaneously is most likely associated with an antiparallel arrangement of β‐strands in sdAb 2H10, which are typically characterized by a strong band at ~ 1630 cm−1 and a weaker one at ~ 1690 cm−1. Upon heme ligation, there was a rearrangement in β structures with the β‐turn band at 1668 cm−1 shifting to 1678 cm−1 and a lack of the contribution at 1693 cm−1, which denotes a loss of the antiparallel β‐sheet fingerprint. Thus, the sdAb 2H10 is likely composed of antiparallel β‐sheets, which undergo conformational changes upon heme binding.
Resonance Raman (RR) spectroscopy was used to further characterize the heme moiety in the sdAb heme‐2H10 complex. Upon excitation into the Soret electronic transition band, RR spectra display in the high frequency region (1300–1700 cm−1) core size marker bands (e.g. ν4, ν3, ν2, and ν10), which are sensitive to coordination, spin, and redox state of the heme‐Fe. A comparison of RR spectra of hemin (heme) versus the sdAb‐heme complex (Fig. 4F) demonstrates that the sdAb 2H10 binds in a specific manner to heme b, revealed by changes in frequency and relative intensity of spin state sensitive ν3, ν2, and ν10 modes. Also, the bandwidth of the well‐defined ν3 mode revealed one spin species (i.e. one type of ligation) in the complex. The ν4 at 1377 and 1373 cm−1 in the heme‐2H10 complex and heme, respectively, indicate in both cases a Fe3+ state of the heme‐Fe. The heme‐2H10 complex revealed the ν3 and ν2 at 1508 and 1585 cm−1 respectively, characteristic for six‐coordinated low spin heme‐Fe (Fig. 4F). These modes were found at 1492 and 1571 cm−1, respectively in the heme spectra, indicating a five‐coordinated high spin (Fig. 4F) state, with most likely a Cl− ion acting as the fifth axial ligand. The most probable candidates for heme axial ligands in the six‐coordinated low spin 2H10‐heme complex are tyrosine (Y) and lysine (K), besides cysteine (C), as established by the UV‐Vis spectra. These aa are present in the CDR1 of sdAb 2H10 (Fig. 1G) as well as axial ligands in several hemoproteins 36. Also, the H2O molecule, which in some heme proteins acts as a strong axial ligand, cannot be excluded.
We then analyzed the effect of pH on sdAb binding to heme. Heme binding by sdAb 2H10 was inhibited at pH 4.0–4.5 (Fig. 5A). Restoring pH to 7.0 reestablished heme recognition by sdAb 2H10, despite some level of protein precipitation (Fig. 5B). We also addressed whether Ca2+ or Mg2+ interfere with heme binding by this sdAb. We found that heme binding to sdAb 2H10 was impaired by Ca2+ but not by Mg2+ (Fig. 5C,D). This suggest that while divalent metals, such as Fe2+ or Mg2+, do not appear to interfere with sdAb binding to heme, Ca2+ can modulate heme binding to the sdAb 2H10.
Figure 5.
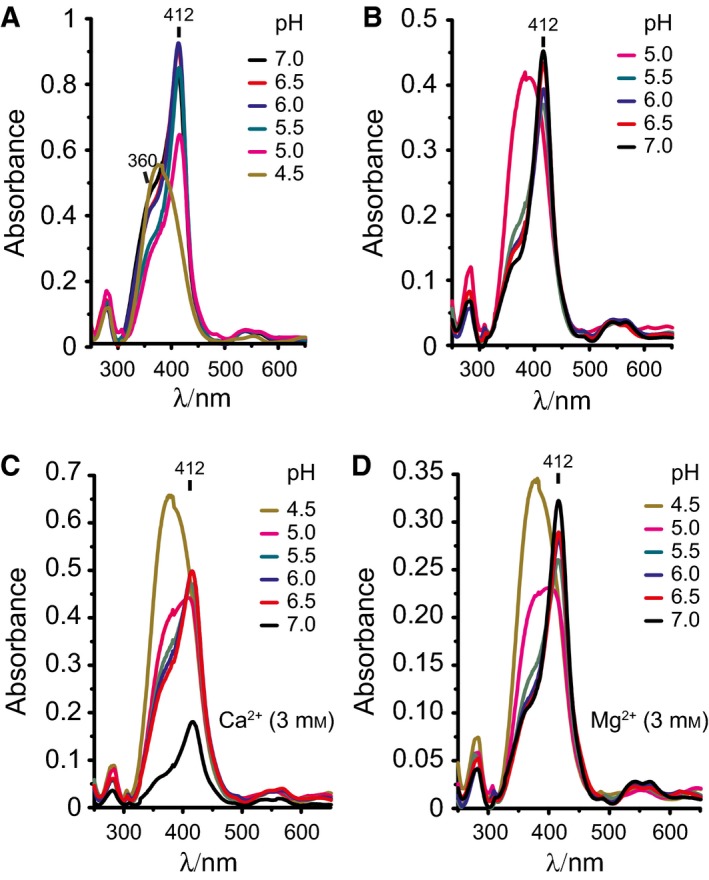
Effect of pH and divalent cations on heme binding by sdAb. (A) UV‐visible spectra of heme binding by sdAb 2H10 bound under gradual pH decrease by the addition of HCl. (B) UV‐visible spectra of sdAb 2H10 bound to heme under gradual pH neutralization by the addition of KOH. (C) UV‐visible spectra of sdAb 2H10 bound to heme under gradual pH neutralization by the addition of KOH in the presence of calcium (Ca2+; 3 mm) or (D) magnesium (Mg2+; 3 mm). The divalent cations Ca2+ impair the binding of the sdAb to heme contrarily to the magnesium (Mg2+). Data are representative of three independent experiments with similar trend.
Characterization of labile heme in plasma during hemolytic conditions
Induction of acute hemolysis in C57BL/6 mice, by the administration of phenylhydrazine, was associated with a marked reduction of circulating RBC numbers (Fig. 6A) and increased heme concentration in plasma (Fig. 6B), i.e. from 23.4 ± 7.1 μm at steady state 29 to 120.8 ± 26.6 μm, 24 h after phenylhydrazine administration (Fig. 6B). There was a negative correlation between the number of circulating RBC and the concentration of heme in plasma, that is, lower numbers of circulating RBC were associated with higher concentration of heme in plasma (Fig. 6C).
Figure 6.
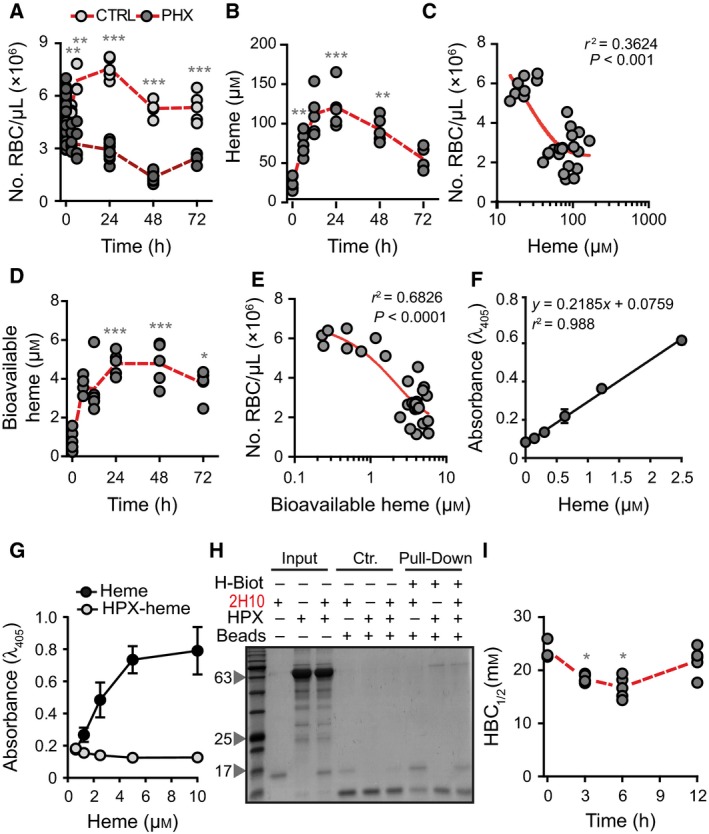
Characterization of labile heme in plasma following acute hemolysis. (A) Number of RBC in C57BL/6 mice receiving Phenylhydrazine (PHX) and control (CTRL) mice receiving PBS. (B) Heme concentration in the plasma of C57BL/6 mice receiving phenylhydrazine. (C) Correlation between circulating RBC numbers (data from A) and heme concentration in plasma (data from B). (D) Concentration of bioavailable heme in plasma of C57BL/6 mice receiving phenylhydrazine, quantified by a heme reporter assay 31. (E) Correlation between circulating RBC numbers (data from A) and concentration of bioavailable heme in plasma (data from D). (F) Soluble hemin quantified by a sandwich ELISA in which the sdAbs 1A6 and 2H7 are used to capture and reveal heme, respectively. (G) Detection of soluble heme versus heme bound to HPX using the same sdAb‐based ELISA as in (F). Note that heme bound to HPX is not detected by ELISA. (H) A pull‐down assay using streptavidin‐beads to capture heme‐biotin. The sdAb 2H10 bound to heme‐biotin was added to HPX at 1/6 SdAb/HPX molar ratio. Streptavidin‐beads pulled down the sdAb 2H10 as well as HPX bound to heme‐biotin, demonstrating that HPX can bind heme‐bound to sdAb 2H10. This is consistent with the higher affinity of HPX toward heme as compared to the sdAb 2H10. Coomassie‐based stain of 15% SDS/PAGE gel loaded with streptavidin‐beads used to pull‐down heme‐biotin from different reaction mixtures. Grey arrowheads indicate the molecular weight of the protein ladder (NZYColour Protein Marker II, Nzytech®) in kDa loaded in the first lane of the gel. Gel is representative of two independent experiments with similar trend. (I) Plasma HBC 1/2 in C57BL/6 mice receiving phenylhydrazine. Circles in A, B, C, D, E, and I correspond to individual mice. Red dash line represents mean ± STD. *P < 0.05, **P < 0.01, ***P < 0.001 versus time 0, calculated by ANOVA and Dunns post‐test.
Heme accumulation in plasma, 24 h after phenylhydrazine administration, was associated with heme uptake by bystander cells, i.e. 4.8 ± 0.6 μm (Fig. 6D), as assessed in vitro using a cellular heme reporter assay 12, 31. Briefly, HEK293 cells transiently transfected with a horseradish peroxidase (HRP) based reporter were exposed to mouse plasma, containing an unknown amount of heme that can be internalized, revealed by intracellular HRP activity. By comparison with a standard curve of soluble heme, it is possible to infer that the amount of bioavailable heme corresponds to about 4% of the total amount of heme in plasma (Fig. 6D). There was a negative correlation between the number of circulating RBC and the concentration of bioavailable heme in plasma, that is, the lower the number of circulating RBC the higher the concentration of bioavailable heme (Fig. 6E).
We developed a sandwich ELISA to quantify heme in solution, using solid‐phase bound sdAb 1A6 to capture soluble heme and biotinylated 2H7 sdAb to detect heme bound to the sdAb 1A6. The assay quantifies heme in solution, in a linear range from 0.15 to 2.5 μm (Fig. 6F). Using this sdAb‐based ELISA, we did not detect heme in the plasma of C57BL/6 mice subjected phenylhydrazine administration (data not shown). Considering that the heme‐specific sdAbs have an affinity of 10−7 m toward heme (Fig. 1G), the ELISA should detect only heme not bound to other molecule with an affinity higher than 10−7 m. This suggests that the bioavailable heme detected in the plasma of C57BL/6 mice receiving phenylhydrazine is bound to plasma molecules with an affinity higher than 10−7 m. In keeping with this notion, the same ELISA did not recognize heme bound to HPX (Fig. 6G).
We then asked whether plasma proteins with high affinity toward heme, such as HPX, can uptake heme bound to the heme‐specific sdAb. We confirmed that HPX can capture heme from the sdAb 2H10, using an in vitro pull‐down assay with streptavidin‐beads capturing biotinylated‐heme bound to sdAb 2H10, in the presence or absence of HPX (Fig. 6H). Namely when apo‐HPX was added to the sdAb 2H10 bound to biotinylated‐heme, the streptavidin‐beads pulled‐down both the sdAb 2H10 and the HPX, revealing that HPX removed the biotinylated‐heme from the sdAb 2H10 (Fig. 6H).
We adapted the heme‐specific ELISA to quantify the relative capacity of plasma macromolecules or other low molecular ligands to bind heme with an affinity higher than 10−7 m, that is, the plasma heme‐binding capacity (HBC1/2). Briefly, plasma is incubated in vitro with increasing concentrations of heme, until saturation of heme‐binding molecules with an affinity toward heme higher than 10−7 m. The heme‐specific sdAb‐based ELISA is used, as described above, to define the concentration of heme required to reduce by 50% the plasma HBC, termed here HBC1/2. Acute hemolysis reduced the HBC1/2 of C57BL/6 mice, from 23.8 ± 1.9 mm at steady state to 16.7 ± 2.1 mm within 6 h of phenylhydrazine administration, returning to steady state levels by 12 h (Fig. 6I). This suggests that the majority of labile heme generated during acute hemolysis is readily bound by plasma molecules with an affinity > 10−7 m and cleared thereafter.
We then asked whether chronic hemolysis induced by Plasmodium infection is associated with the detection of labile heme in plasma. Infection of C57BL/6 mice with Plasmodium chabaudi chabaudi (Pcc) was associated with a progressive increase in the number of infected RBC until day 7, decreasing thereafter until day 9 postinfection (Fig. 7A). The peak of infection was associated with heme accumulation in plasma, i.e. 237.8 ± 28.2 μm, decreasing thereafter to return to basal levels 9 days postinfection (Fig. 7B). There was a positive correlation between the percentage of infected RBC and the concentration of heme in plasma, with higher percentage of infected RBC being associated with higher concentration of heme in plasma (Fig. 7C). Plasmodium infection was not associated with a reduction of plasma HBC1/2 (Fig. 7D) and the levels of labile heme detected by the sdAb‐based ELISA remained above 0.15 μm (data not shown). Nevertheless, 1–2% of the heme detected in plasma at day 7 postinfection, i.e. 3.9 ± 0.3 μm, was bioavailable, as assessed in vitro using a cellular heme reporter assay (Fig. 7E). There was a positive correlation between the percentage of infected RBC and the concentration of bioavailable heme in plasma (Fig. 7F). While this confirms that the blood stage of Plasmodium infection is associated with the accumulation high levels of heme in plasma 6, 10, 11, 22, 28, 42, labile heme is bound to plasma molecules with an affinity higher than 10−7 m, and therefore cannot be detected by the sdAb‐based ELISA. A proportion of the labile heme is bioavailable and therefore likely to participate in the pathogenesis of severe forms of malaria 6, 10, 11, 22, 28, 42.
Figure 7.
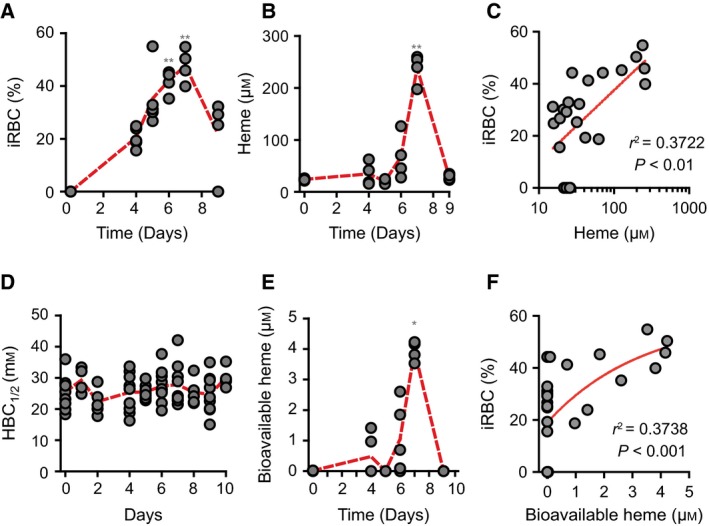
Characterization of labile heme in plasma following Plasmodium infection. (A) Percentage of infected RBC (iRBC) and (B) heme concentration in plasma of C57BL/6 mice (n = 4–5) infected with Pcc. (C) Correlation between percentage of iRBC (data from A) and heme concentration in plasma (data from B). (D) HBC 1/2 in Pcc infected C57BL/6 mice. (E) Bioavailable heme in Pcc infected C57BL/6 mice. (F) Correlation between the percentage of iRBC (data from A) and bioavailable heme (data from E). *P < 0.05 and **P < 0.01 versus time 0, established by ANOVA and Dunns post‐test. Circles correspond to individual mice. Red dash line represents mean from mice pooled from two to three independent experiments with similar trend.
We then asked whether the development of chronic hemolysis caused by the sickle Hb mutation, is associated with the accumulation of labile heme in plasma. We tested this hypothesis in knock‐in C57BL/6; 129 hα/hα::βS/βS mice, a well‐established experimental model of sickle cell disease 43. Heme concentration in the plasma of C57BL/6;129 hα/hα::βS/βS knock‐in mice was 27.0 ± 3.9 μm, as compared to 18.5 ± 3.9 and 19.9 ± 3.4 μm in control C57BL/6;129 hα/hα::βA/βS and hα/hα::βA/βA mice that do not develop sickle cell disease, respectively (Fig. 8A). There was no apparent reduction of plasma HBC1/2 in hα/hα::βS/βS mice, as compared to control hα/hα::βA/βS or hα/hα::βA/βA mice (Fig. 8B). Detection of labile heme, using the sdAb‐based ELISA, remained above 0.15 μm (data not shown). Nevertheless, about 8% of the heme detected in the plasma of hα/hα::βS/βS mice was bioavailable, i.e. 2.1 ± 0.9 μm, as assessed in vitro using a cellular heme reporter assay (Fig. 8C). This was not the case in control hα/hα::βA/βA or hα/hα::βA/βS mice (Fig. 8C). This confirms that sickle cell disease is associated with the accumulation of labile heme in plasma 21, 30, 44, 45 and supports the notion that labile heme promotes the pathogenesis of this hemolytic condition 21, 30, 44, 45.
Figure 8.
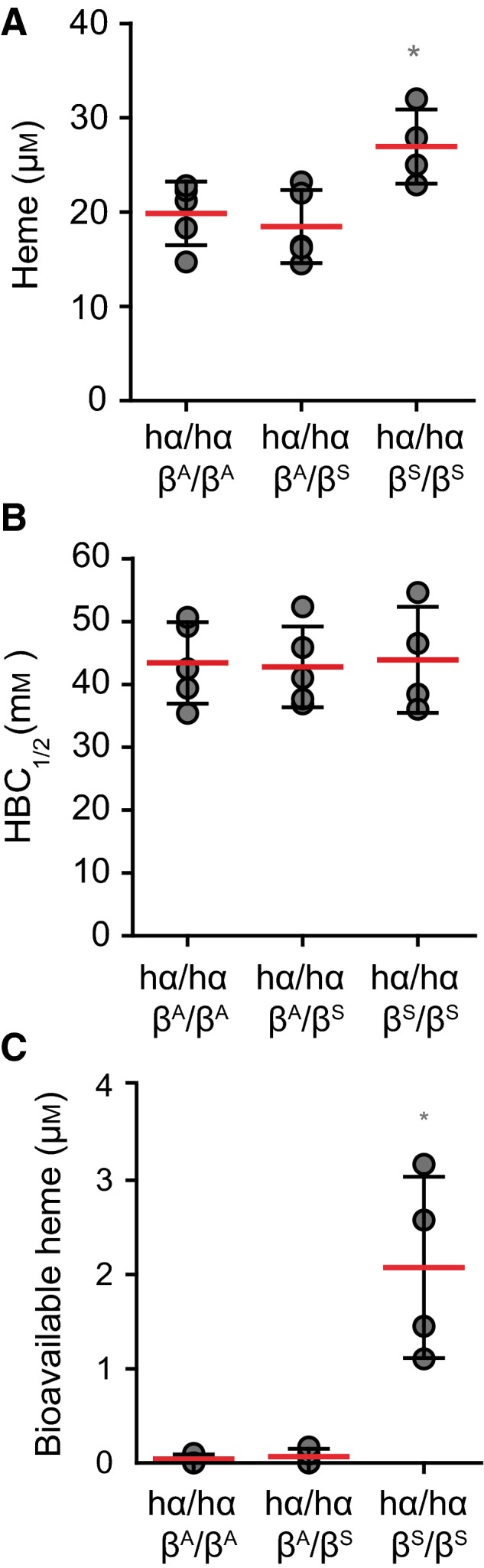
Heme accumulation in plasma in sickle cell disease. (A) Total heme, (B) HBC 1/2 and (C) bioavailable heme in sickle hα/hα::βS/βS versus control hα/hα::βA/βA and hα/hα::βA/βS mice. *P < 0.05 established by ANOVA and Dunns post‐test. Each circle corresponds to an individual mouse. Red dash line represents mean ± STD.
Heme‐specific sdAb modulates the redox activity of labile heme
We determined whether heme binding by sdAb 2H10 interferes with the redox activity of labile heme, as assessed by electron exchange between heme and ascorbic acid in the presence of H2O2. Heme catalyzed ascorbic acid oxidation in a dose‐dependent manner, that is, higher heme concentrations (0–5 μm) depleted ascorbate, as detected by HPLC (Fig. 9A,B). This pro‐oxidant effect of heme was attenuated by sdAb 2H10, as compared to equimolar concentrations of control sdAb or human serum albumin (HSA) (Fig. 9A). Importantly, sdAb 2H10 also inhibited the pro‐oxidant activity of heme/H2O2 in a concentration‐dependent manner, that is, higher sdAb concentrations reduced ascorbate depletion (Fig. 9B). At each concentration tested, the sdAb 2H10 was more effective than the control sdAb or HSA in terms of preventing heme‐driven ascorbate depletion (Fig. 9B).
Figure 9.
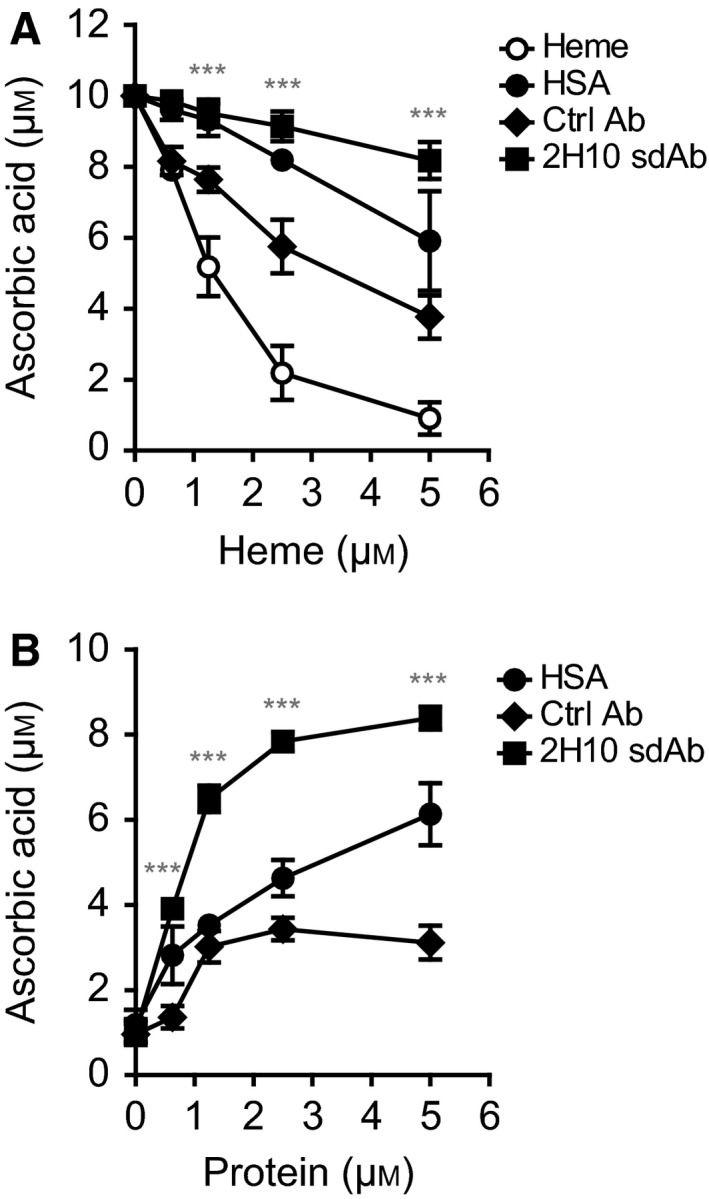
Targeting heme with sdAbs. Ascorbate oxidation by heme in the presence or absence of sdAb 2H10, control (Ctrl) sdAb or human serum albumin (HSA) under (A) increasing heme or (B) protein concentrations. Results shown are the mean ± SEM from four (A) and three (B) independent experiments. ***Mean value of sdAb 2H10 differs from the corresponding mean point value of Ctrl sdAb at the P < 0.001.
Heme‐specific sdAbs detect cellular heme
We asked whether heme‐specific sdAbs can be used to detect intracellular heme by immunofluorescence and/or by flow cytometry. Intracellular heme was detected by immunofluorescence, in the perinuclear area of HeLa cells, using sdAb 1A6, 2H7, and 2H10, as compared to a control sdAb (Fig. 10A). Intracellular heme was also detected by flow cytometry using the same sdAbs (Fig. 10B). Detection of intracellular heme by the sdAb 2H10 was suppressed when the sdAb was preincubated with increasing heme concentrations, as assessed by flow cytometry (Fig. 10C) or by immunofluorescence (Fig. 10D). Preincubation of sdAb 2H10 with PP failed to inhibit intracellular heme detection by flow cytometry (Fig. 10E).
Figure 10.
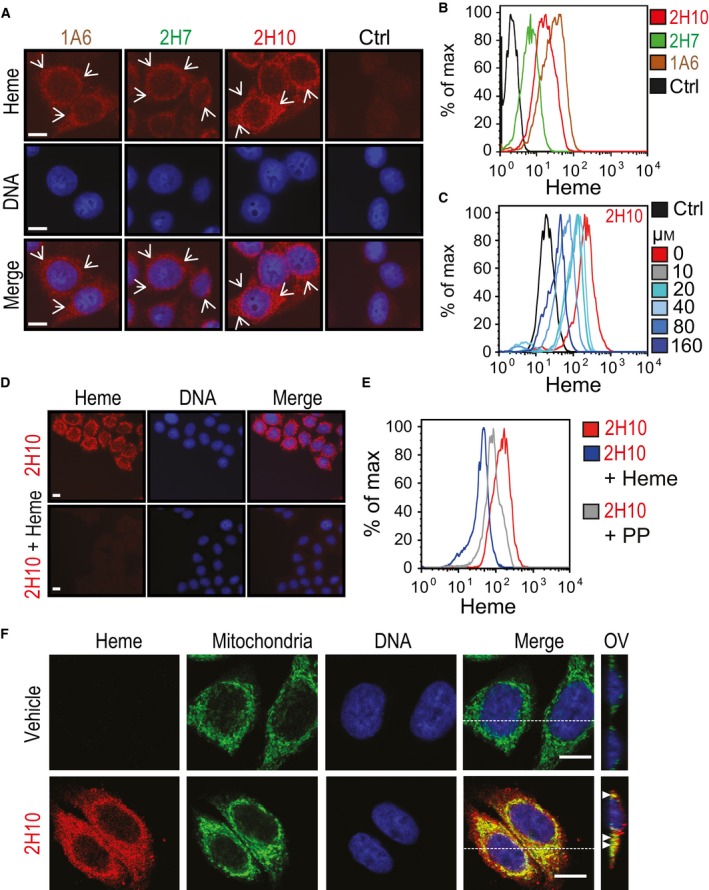
Detection of intracellular heme using sdAbs. (A) Intracellular heme (red) and DNA (blue) in HeLa cells, detected by immunofluorescence using different heme‐specific sdAbs and DAPI, respectively. Staining with a control sdAb (Ctrl) that does not recognize heme, is shown. Scale bar, 10 μm. Arrows highlight heme staining. (B) Detection of intracellular heme by flow cytometry in HeLa cells, using the same sdAbs as in (A). (C) Same as (B), with preincubation of sdAb 2H10 with increasing concentrations (0–160 μm) of hemin in solution. (D) Same as (A), using sdAb 2H10 preincubated or not with hemin (160 μm) in solution. (E) Same as (C), with sdAb 2H10 preincubated with heme (160 μm) or PP (160 μm). (F) Same as (A), in HeLa cells costained with MitoTracker® for mitochondria localization (Green). Heme was detected using the sdAb 2H10 (red) and DNA with DAPI (blue). Merged images show colocalization of heme with mitochondria (yellow). For each condition, Z‐stacks were acquired and orthogonal view (OV) of the dashed line is shown. Arrows highlight colocalization. All images with a scale bar, 10 μm. Data in A–D are representative of three independent experiments with similar trend.
Having established that the heme‐specific sdAbs can be used to detect intracellular heme, we addressed in more detail the intracellular localization of heme. Detection of intracellular heme by sdAb 2H10 overlapped with mitochondria staining by MitoTracker (Fig. 10F) and to a minor extent with endoplasmic reticulum (ER) staining by an anti‐Calnexin monoclonal Ab (mAb) (Fig. 11A) or with early endosome staining by an antiearly endosome antigen 1 (EEA‐1) staining (Fig. 11B), as detected by confocal microscopy. There was no overlap between heme and actin staining (Fig. 11C) and some minimal level of heme colocalization with heme oxygenase‐1 (HO‐1) (Fig. 11D) and BR (Fig. 11E).
Figure 11.
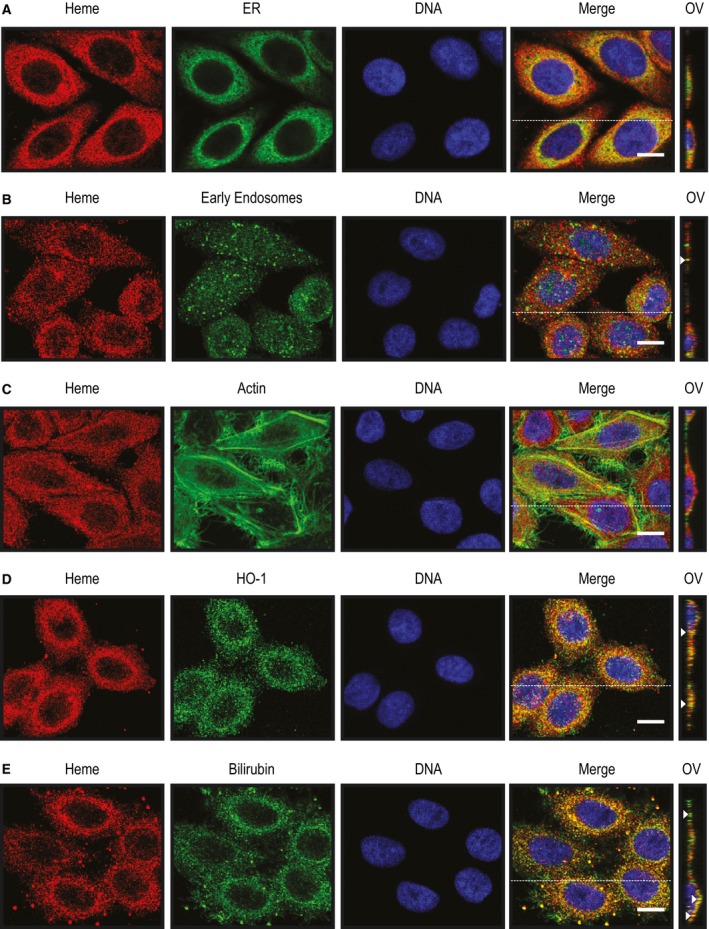
Detection of intracellular heme using sdAbs. Heme (red) and DNA (blue) detected by immunofluorescence in HeLa cells using the sdAb 2H10 and DAPI, respectively. Cells were costained (green) with (A) anti‐Calnexin for endoplasmatic reticulum (ER) (B) anti‐EEA‐1 for endosomes, (C) a phalloidin–tetramethylrhodamine B isothiocyanate for F‐actin, (D) anti‐HO‐1 polyclonal Ab and (E) anti‐bilirubin mAb. Merged figures show colocalizations (yellow). For each condition, Z‐stacks were acquired and orthogonal view (OV) of the dashed line is shown. Arrows highlight colocalization. Data shown are representative of three independent experiments with similar results. All images with a scale bar, 10 μm.
Discussion
About 75–80% of bioavailable Fe in mammals is contained within the prosthetic heme groups of hemoproteins, with Hb containing the major pool of heme 25. Extracellular Hb and labile heme are scavenged in plasma by haptoglobin and HPX, respectively 46. Presumably accumulation of labile heme in plasma only takes place once the scavenging capacity of haptoglobin and HPX are saturated 10, 11, 16, 47, 48. When this occurs, labile heme acts in a pro‐oxidant 49, 50, proinflammatory 17, 51, vasoactive 21, and cytotoxic 10, 22, 23 manner that contributes to the pathogenesis of hemolytic conditions 11, 52, such as illustrated for malaria 6, 28, severe sepsis 10, or sickle cell disease 21, 29, 30. However, the relative lack of analytical methods providing an accurate characterization of labile heme in plasma makes it difficult to prove unequivocally its direct involvement in the pathogenesis of these diseases. Here, we generated and characterized a panel of heme‐specific sdAbs (Figs 1, 2, 3, 4, 5) providing a more accurate characterization of labile heme.
We reasoned that to detect specifically labile heme in plasma, heme‐specific sdAbs should recognize heme not bound to Hb, HPX, high‐ or low‐density lipoprotein or albumin. Therefore, the affinity of the sdAbs toward heme should be lower to the one displayed by Hb (10−14 m) 53, HPX (10−12–10−13 m) 54, 55, high‐ or low‐density lipoproteins (10−11–10−12 m) 56, or even albumin (10−8 m) 5, 57. We selected three heme‐specific sdAbs, i.e. 1A6, 2H7, and 2H10 (Figs 1, 2, 3), with an affinity toward soluble heme in the rage of 10−7 m (Fig. 1G). Analyses of the structural properties of the sdAb 2H10 using Spectrophotometry, Resonance Raman, CD and FITR, suggest that this sdAb interacts with heme (Fig. 4C–F), presumably via a cysteine at the fifth ligand and possibly a lysine or a tyrosine as the sixth axial ligand (Fig. 4F) 36. Conformational changes in the antiparallel β‐sheets of this sdAb are probably required to accommodate heme (Fig. 4D–E).
We found that heme recognition by heme‐specific sdAbs is impaired as pH drops to 4–4.5 (Fig. 5A,B) being also affected by Ca2+ but not by the Mg2+ or Fe2+ (Figs 3B and 5C,D), which is consistent with similar observations for heme binding by the plasma heme scavenger hemopexin (HPX) 42, 58. Of note, however, while heme is bound to HPX by two histidine residues that are protonated at low pH, there are no apparent histidine residues involved in heme binding by the SdAb tested and as such the mechanism via which lowering pH disables heme binding to the sdAb is not clear.
We confirmed that heme accumulates in plasma during acute sterile hemolysis (Fig. 6B) and chronic hemolysis caused by Plasmodium infection (Fig. 7B) as well during the development of sickle cell anemia (Fig. 8A). Approximately 2–8% of the heme detected in plasma during these hemolytic conditions (~ 2–5 μm) is bioavailable (Figs 6D, 7E and 8C). Of note, soluble heme is cytotoxic to parenchyma cells in this concentration range 10, 22, 23, 24, 25, 59 arguing further for the participation of labile (bioavailable) heme in the pathogenesis of these hemolytic conditions. Heme was not detected in plasma using the sdAb‐based ELISA, suggesting that bioavailable heme is contained in a pool of labile heme bound to plasma molecules with an affinity > 10−7 m.
While our data are consistent with the notion that hemolytic conditions are associated with the generation of labile heme bound to plasma molecules with higher affinity than 10−7 m, the identity of these heme‐binding molecules is not clear. Presumably, a significant proportion of the labile heme is captured by HPX (0.6–1.2 mg·mL−1) and by lipoproteins, which bind heme with 10−12 m and 10−10–10−12 m, respectively 23 but also by albumin (43 mg·mL−1) that has a lower affinity (10−8 m) toward heme. Given their range of affinities toward heme (10−8–10−12 m), these plasma proteins can capture heme from the heme‐specific sdAb, as illustrated for HPX (Fig. 6G,H). This also explains why the heme‐specific sdAb‐based ELISA does not detect labile heme in plasma during the different hemolytic conditions tested in this study.
Considering that adult mice have ~ 2 mL of blood, ~ 10 × 109 RBC per mL and ~ 3 × 108 molecules of Hb per RBC, we estimated that 50–75% RBC lysis (Fig. 6A) should release ~ 10–15 mm of heme. This is consistent with the 30% decrease in HBC1/2 following acute hemolysis (Fig. 6I), likely attributed to the saturation of plasma HPX (~ 10–20 μm), high (~ 5–11 μm) and low‐density (~ 0.04 μm) lipoproteins and albumin (~ 450–600 μm) 23, 54, 59, 60 by labile heme. In contrast to acute hemolysis, chronic hemolysis during the development of malaria (Fig. 7) or sickle cell anemia (Fig. 8) was not associated with a reduction of plasma HBC1/2 (Figs 7D and 8B). Our interpretation is that a decrease in plasma HBC1/2 is only detectable in the event of rapid release of large amounts of labile heme, overcoming the HBC1/2 provided by plasma heme scavengers. Chronic hemolysis on the other hand is associated with a more gradual accumulation of labile heme in plasma, presumably allowing for regulatory mechanisms, involving the expression of heme scavengers and the induction of heme catabolism by HO‐1, to prevent plasma HBC1/2 from reaching saturation. This argues for a tight regulatory mechanisms restoring systemic heme homeostasis upon hemolysis. Nevertheless, malaria (Fig. 7) and sickle cell anemia (Fig. 8) are associated with the generation of bioavailable heme that can be readily internalized by bystander cells (Figs 7E and 8C). Our data are consistent with the notion that at the levels detected in plasma, labile heme is likely to participate in the pathogenesis of hemolytic diseases.
There are other methods that allow for the detection and characterization of labile heme in plasma, including a recently described reversed‐high‐performance liquid chromatography (HPLC)‐based approach 61. However, these approaches are less sensitive, as compared to the ones we describe herein, and moreover do not allow discriminating what proportion of the labile heme becomes bioavailable, which is made possible in our study based on the use of a cellular‐based heme reporter assay 31, 62.
Our study shows that heme‐specific sdAbs can also be used to detect intracellular heme compartmentalization (Figs 10 and 11). In human cells the majority of intracellular heme probably localizes in the mitochondria, which is consistent with the last steps of heme synthesis taking place in this organelle. A minor proportion of intracellular heme is also detected in the endoplasmic reticulum of human cells (Fig. 11A) 31, 62. This is consistent with previous studies showing that cellular labile heme concentration range at steady state from 20 to 40 nm and 2.5 nm in the cytoplasm and mitochondria of Saccharomyces cerevisiae, respectively 62. The heme‐specific sdAb also detected cellular heme in close vicinity of HO‐1 (Fig. 11D) and BR (Fig. 11E), a downstream end‐product of heme catabolism. While consistent with the notion that the heme‐specific sdAb recognize intracellular heme, we cannot prove that these react specifically with cellular labile heme. We are also aware that cellular fixation and permeabilization procedures do create changes in protein conformation and some level of protein unfolding, perhaps favoring the detection of loosely bound heme but also heme‐bound proteins with higher affinity.
Ab recognizing heme or related metalloporphyrins have long been appreciated with several of these targeting ferric mesoporphyrin 63 and increasing 64 or reducing 65 its peroxidase activity. Consistent with these findings, binding of the sdAb 2H10 to soluble heme inhibits its peroxidase activity (Fig. 9A,B), which may provide a unique opportunity to prevent the pathogenic effects of labile heme during hemolytic conditions. Given that the affinity of HPX for heme (10−12 m) is about 4–5 logs higher that that of the sdAb (10−7 m) or albumin (10−8 m), it is reasonable to assume that HPX should inhibit ascorbate oxidation with higher efficiency, as compared to the sdAb or to albumin. Because the sdAb 2H10 does not target related metalloporphyrins nor does it bind to heme when contained in Hb or other hemoproteins (Figs 2 and 3), this should make its therapeutic use less toxic, as compared to approaches that target heme within hemoproteins, e.g. carbon monoxide 6, 23, 66.
Experimental procedures
Note: Except when indicated reagents were provide by Sigma (Sintra, Portugal).
Tetrapyrrole preparation
The concentration of different tetrapyrroles (Frontier Scientific, Inc, Logan, UT, USA) was determined spectrophotometrically using appropriated solvents. Briefly, heme (i.e. hemin) was prepared in 0.1 m NaOH and buffered to pH 7.4 using 0.1 m HCl. Alternatively, hemin and other tetrapyrroles were dissolved in dimethyl sulfoxide (DMSO) and concentration calculated using different wave lengths and extinction coefficients (EmM), i.e. hemin (λ405 nm; EmM = 85.82), deuteroporphyrin IX (DeutP; λ392 nm; EmM = 170), gallium protoporphyrin (GaPP; λ413 nm, EmM = 249) 67, protoporphyrin (PP; λ408 nm; EmM = 297; in 1.5 m HCl) 67, Zinc protoporphyrin (ZnPP; λ415 nm; EmM = 150, in ethanol) 67; BV (λ377 nm; EmM = 51.5, in methanol) 68 and BR (λ451 nm, EmM = 60, in chloroform) 69. Cobalt PP, tin PP (SnPP) and FePP IX dimethyl ester chloride (FePPCH3) concentrations in DMSO were calculated gravimetrically according to their molecular weight (MW) = 654.6, 750.26 and 679.99, respectively.
Heme biotinylation
Heme biotinylation was carried out essentially as described 70. Briefly, hemin in dimethylformamide (4.4 mg·mL−1) was incubated with 2‐(1H‐7‐azabenzotriazol‐1‐yl)‐1,1,3,3‐tetramethyluroniumhexafluorophosphate methanaminium and N,N‐diisopropylethylamine (30–60 min; RT). N‐[‐5‐(Hydrazinocarboxy) pentyl]‐D‐biotinamide (biotin) in DMSO was added (60 min; RT) and the reaction was applied onto a C18 reverse‐phase analytic HPLC column (COSMOSIL 5C18‐ARII, Nacalai Tesque, Nijo Karasuma, Japan) to separate biotinylated from nonbiotinylated‐heme and biotin. Fractions were dried (SpeedVac Plus SC110A; Vaccum System Plus UV400A) at < 15 °C.
Matrix‐assisted laser desorption/ionization (MALDI)‐Time‐of‐flight mass spectrometry (TOFMS)
Hemin and biotin in 0.2 m NaOH were spotted onto the MALDI target plate and mixed 1 : 1 with 2,5‐dihydroxybenzoic acid (10 mg·mL−1) in 50% v/v acetonitrile, 5% v/v formic acid and air‐dried. Heme‐biotin in 50% (v/v) acetonitrile was spotted onto a MALDI plate using the Maldi matrix as described above. Data were acquired in positive reflector MS mode in a mass spectrometer (4800 plus MALDI‐TOF/TOF; Ab Sciex, Singapore) and collected (4000 Series Explorer Software v.3.5.3; Applied Biosystems, Foster City, CA, USA). Mass spectrometer calibration was performed using angiotensin II (1046.542 Da), angiotensin I (1296.685 Da), Neurotensin (1672.918 Da), Adrenocorticotropic hormone (ACTH) (1–17) (2093.087 Da), and ACTH (18–39) (2465.199 Da) (Peptide Calibration Mixture 1; LaserBio Labs, Sophia‐Antipolis, France). Each reflector MS spectrum was collected using 500 laser shots per spectra and a fixed laser intensity of 3300 V.
Phage library and selection
A synthetic VL sdAb phage display library previously used against several targets was used in our study 32, 71. The library repertoire (8 × 109 independent clones) was generated on a highly soluble and stable VL scaffold by randomization and accommodating a maximum of 26 aa in CDR1 and 22 aa in CDR3 32. The selection process was divided into four main steps: (a) incubation of phage‐sdAb repertoire with heme‐biotin; (b) streptavidin capture and washing to remove nonspecific phages‐sdAb; (c) heme competition for phage‐sdAb elution and (d) amplification of antigen‐specific phages‐sdAb. Five rounds of binding, elution and amplification were performed to select sdAbs with high binding activities and specificity. Briefly, heme‐biotin (1 μg·μL−1) was preincubated (2 h; RT) with the sdAb VL phage library (~ 1010 mL−1). The selection procedure consisted in using streptavidin coated magnetic beads (Dynabeads; M280; Invitrogen by Thermo Fischer Scientific) to capture phages bound to heme‐biotin, under a magnetic field. Unbound or low affinity bound‐phages were washed out [Dulbecco's Phosphate Buffered Saline (PBS) 0.5% tween 20, 5 ×]. Bound‐phages were released from the heme‐biotin with a competition step by addition of excess hemin (100 μm in PBS; 1 h; RT; 2×). The recovered phages were used to infect E. coli ER2738 (phage display optimized strain; λ600 nm = 0.6; Biocat; 4 mL; 15–30 min 37 °C). Phage‐infected ER2738 bacteria were grown in Super Broth (10 μg·mL−1 tetracycline, 100 μg·mL−1 ampicillin) and plated in Luria Broth (LB) agar (100 μg·mL−1 Amp) at 37 °C. Single clones were randomly selected and screened by PCR to insure the presence of VL sequences. Bacteria growing in liquid medium were then infected with VCSM13 helper phages (1012–1013 pfu in 4 mL; 15–30 min; 37 °C), reselected for kanamycin (100 μg·mL−1) resistance and left ON at 37 °C for phage production. After 16 h, the selected phages were precipitated using PEG‐8000 (4% w/v), NaCl (3% w/v) (1 h on ice), centrifuged (8000 g; 30 min, 4 °C), resuspended (PBS, 15% Glycerol, 1 h in ice), centrifuged (16 000 g, 10–20 min, 4 °C), and filtered (0.22 μm). The resulting ‘purified phages’ were used for one additional panning with 10 washing cycles (PBS, 0.5% Tween 20) and three more additional panning with 15 washing cycles (PBS 0.5% Tween 20). E. coli (TOP10F' strain; λ600 nm = 0.6) were infected with phages selected from the fourth panning (4 mL; 15–30 min 37 °C), grown (Super Broth, 10 μg·mL−1 tetracycline and 100 μg·mL−1 Amp), plated (LB agar, 100 μg·mL−1 Amp), and VL sequences confirmed by PCR. Phage‐infected bacteria were grown (Super Broth, 10 μg·mL−1 tetracycline and 100 μg·mL−1 Amp, ON), the pComb3X phagemid DNA was extracted (Midi prep. Quiagen, Hilden, Germany), isolated and digested with SfiI (Fermentas, Portugal) for the extraction of DNA encoding sdAb fragments. These were purified from agarose gel electrophoresis, cloned into a modified pPT7‐FLAG vector (Technophage SA, Lisboa, Portugal) and used to transform BL21 (DE3) E. coli (Promega, Madison, WI, USA). Bacterial clones were grown in auto induction medium (Novagen, WI, USA), lyzed (PBS, 20% Bugbuster, Novagen, ON, 4 °C) and protein extracts were obtained (16 000 g, 15 min, 4 °C) for selection of sdAb according to their heme‐binding capacity.
SdAb expression and purification
Twenty His‐HA‐tagged sdAbs were selected according to heme‐binding capacity and 11 of those were expressed and purified, essentially as described 32. Briefly, the sdAbs cloned in a pPT7 expression vector were transformed in E. coli BL21 (DE3) cells and sequences were confirmed (Macrogen DNA Sequencing service, Amsterdam, the Netherlands). For protein expression, 1 L of LB, (50 μg·mL−1 Carbenicillin) was inoculated with ON culture of bacterial cells (10 mL), transformed with pPT7‐VL′s plasmids and grown (37 °C) to exponential phase (λ600 nm = 0.6–0.9). SdAb expression was induced by isopropyl β‐1‐thiogalactopyranoside (1 mm; 6 h, 37 °C). Bacteria were harvested by centrifugation (4000 g, 15 min, 4 °C) and resuspended in equilibration buffer (50 mm HEPES, 1 m NaCl, 5 mm CaCl2, 10 mm imidazole, pH 7.8; 50 mL), supplemented with protease inhibitors (Roche, Amadora, Portugal). Bacteria were lysed by sonication (20 min, ice) and inclusion bodies containing the sdAbs were recovered by centrifugation (12 096 g, 30 min, 4 °C). Pellets were washed [50 mm HEPES, 1 m NaCl, 10 mm imidazole, 5 mm CaCl2, 2 m urea, 1 mm β‐mercaptoethanol (β‐ME); pH 7.8], sonicated (20 min, ice) and collected by centrifugation (12 096 g, 30 min, 4 °C). Inclusion bodies were resuspended in a 6 m Urea buffer (50 mm HEPES, 1M NaCl, 10 mm Imidazole, 5 mm CaCl2, 6 m urea, 1 mm β‐ME, pH 8.0) containing benzonase (> 250 U; Sigma) and incubated ON (4 °C, under agitation) to allow for protein denaturation. SdAbs were purified under denaturing conditions using a His Trap Fast Flow (FF) column (GE Healthcare, Carnaxide, Portugal) and washed twice (50 mm HEPES, 1 m NaCl, 6 m Urea, 1 mm β‐ME, 5 mm CaCl2, and 20 mm or 30 mm Imidazole; pH 7.8) to remove nonspecific protein contaminants. SdAbs were eluted in high concentrated imidazole buffer (50 mm HEPES, 1 m NaCl, 6 m urea, 1 mm β‐ME, 5 mm CaCl2, 500 mm Imidazole, pH 7.8). Refolding was achieved by step‐wise dialysis, essentially as described 35. Briefly, purified sdAb were dialyzed (24 h, 4 °C) to Tris buffer (50 mm Tris‐HCl, 1 mm EDTA; 200 mm NaCl, 6 m Urea, 10 mm mm β‐ME) and β‐ME was removed by dialysis (48 h, 4 °C) to the same buffer without β‐ME, allowing thiol group oxidation and consequently disulfide bond formation. SdAbs were refolded by sequential dialysis (24 h, 4 °C) to Tris buffer (50 mm Tris‐HCl, 1 mm EDTA; 200 mm NaCl; 6 m Urea), reducing Urea concentration step wise from 3, 2, 1, 0.5, and 0 m. Dialyses to buffers containing 1 and 0.5 m Urea were supplemented with 0.4 mm oxidized glutathione and 4 mm reduced glutathione plus 400 mm l‐arginine to promote protein refolding (30). SdAbs were centrifuged (4000 g, 4 °C, 10 min), to remove protein precipitates, aliquoted, and stored (−80 °C) until used. SdAbs purity was assessed by sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS/PAGE) gel with 15% acrylamide under denaturing conditions followed by based Coomassie‐based stain (Instant Blue, 30 min, RT; Gentaur, Kampenhout, Belgium) and by western blotting.
SdAb biotinylation
Briefly, sdAbs were incubated with 20‐fold molar excess of biotin (2 h, ice) in PBS using EZ‐Link NHS‐PEG4‐Biotinylation kit (Pierce by Thermo Fischer Scientific, Waltham, MA, USA). Free biotin and free sdAb were removed using a desalting column (Thermo Zeba Spin Desalting column; Pierce) (1000 g, 2 min), preequilibrated in PBS. SdAb biotinylation was confirmed using 4′‐hydroxyazobenzene‐2‐carboxylic acid (HABA) assay. Briefly, biotinylated‐sdAb was mixed (1/10) with a HABA/Avidin (0.5 mg·mL−1 avidin, 0.3 mm HABA, PBS) in a 96 well plate and λ500 nm measured in a microplate reader (Victor3 Multilabel Readers; Perkin Elmer, Waltham, MA, USA). Biotinylated‐sdAbs were stored at −80 °C until used.
Western Blotting
Proteins were subjected to 15% SDS/PAGE gel electrophoresis (15 V, 60 min), transferred into methanol activated polyvinyldifluoridine membrane (2 min), washed (3× in H2O, 3× T‐TBS; 20 mm Tris pH 7.5, 150 mm, 0.05% Tween‐20) and blocked (5%‐skim milk in T‐TBS; 1 h, ON; RT). SdAbs were detected using a HRP‐conjugated rat anti‐HA monoclonal Ab (mAb) (1/2000; Roche®). Peroxidase activity was revealed using SuperSignal West Pico Chemiluminescent substrate (Pierce) in a photoradiograph (Kodak Biomax Light Film; Eastman Kodak, New York, NY, USA). ExPASy – ProtParam tool was used to estimate the MW and isoelectronic point of sdAbs.
ELISA
Hemin (100 μm) in carbonate buffer (0.1 m NaHCO3, pH 8.6) was used bound to 96 well plates (Costar 3690) (1 h, RT or ON at 4 °C), washed (3×, PBS, 0.1% Tween 20), blocked in protein‐free blocking buffer (Pierce; 1 h, RT), and washed (4×, PBS, 0.1% Tween 20). Bacterial supernatants or purified sdAbs were added (1 h 30 min, RT, diluted in PBS), plates were washed (5× PBS, 0.1% Tween 20) and heme‐bound sdAbs were detected using a rat anti‐HA mAb (3F10; Roche; 0.1 μg·mL−1) in protein‐free blocking buffer. Plates were washed (5× PBS, 0.1% Tween 20) and the rat anti‐HA mAb was detected using an alkaline phosphatase labeled rabbit antiwhole rat IgG (Sigma) polyclonal Ab (1 h, RT, 1/2000) in protein‐free blocking buffer. Alkaline phosphatase was revealed with para‐Nitrophenylphosphate (pNPP, 1 mg·mL−1; Sigma). Absorbance was measured in a microplate reader at λ450 nm (Victor3 Multilabel Readers; Perkin Elmer).
To measure the total amount of protein in sdAb supernatants, 96 well plates (Costar 3690; Sigma) were coated with the sdAb supernatant in 50 mm carbonate/bicarbonate buffer, pH 9.6 (1 h, RT or 16 h, 4 °C). The rest of the procedure was performed as described above. Absorbance was measured in a microplate reader at λ450 nm.
To measure labile heme, 96 well plates were coated with SdAb 1A6 (0.3–5 μg·mL−1) in 50 mm carbonate/bicarbonate buffer, pH 9.6 (16 h, 4 °C), washed (5×, PBS 0.1% Tween 20) and blocked (2 h, RT) with protein‐free blocking buffer (Pierce from Thermo Fischer Scientific). Plates were incubated with hemin (0.15–5 μm in PBS), used as standard or with plasma (1 h 30 min, RT). Plates were washed (5×, PBS, 0.1% Tween 20) and heme was detected using biotinylated 2H7 sdAb (2.5–5 ng·μL−1) in PBS. Plates were washed (5×, PBS, 0.1% Tween 20) and biotinylated‐sdAb was detected using Alkaline phosphatase conjugated with ExtrAvidin (1/2500; Sigma) in protein‐free blocking buffer. Plates were washed (5×, PBS 0.1% Tween 20) and Alkaline phosphatase was revealed using pNPP (1 mg·mL−1; Sigma). Absorbance was measured in a microplate reader at λ450 nm.
BIAcore
SdAbs affinity toward heme was determined using surface plasmon resonance (SPR) (BIAcore 2000; BIAcore Inc. by GE Healthcare). Briefly, sdAbs were captured on a CM5 chip using amine coupling at ~ 800 resonance units (RU). Heme (0–3000 nm) was injected for 4 min and sdAb‐bound heme was allowed to dissociate (10 min) before matrix regeneration (10 mm Glycine, pH 2.5). The signal was subtracted from that of an immobilized cell to generate sensorgrams of the amount of bound heme as a function of time. The running buffer, HBS‐P (0.01 m HEPES pH 7.4, 0.15 m NaCl, 0.005% v/v Surfactant P20; BIAcore) was used for all sample dilutions. biacore kinetic evaluation software by BIAcore Inc. version 3.1 (GE Healthcare, Carnaxide, Portugal) was used to determine dissociation constant (K D) from the association and dissociation rates using a one‐to‐one binding model. An irrelevant sdAb was used as negative control.
SdAb pull‐down assay
SdAb binding to soluble hemin was assessed by a pull‐down assay. Briefly, sdAbs were incubated (5 h, 4 °C, agitation) with heme‐biotin and half of the reaction mixture was incubated (2 h, RT, agitation) with of Streptavidin‐Dynabeads (M‐280 Streptavidin; Invitrogen by Thermo Fischer Scientific), previously blocked (5 h, RT) with protein‐free blocking buffer (Pierce). Mixture was washed (3×; 5 min; PBS, 0.05% Tween 20) and magnetic beads were captured (5 min magnetic field) according to the manufacture's instructions. SdAbs‐heme‐biotin complexes were collected (1 min, magnetic field) and incubated (20 min, 100 °C, agitation) in loading buffer (50 mm Tris‐HCl pH 6.8, 2% SDS, 10% Glycerol, 1% β‐ME, 12.5 mm EDTA, 0.02% Bromophenol blue) (pull‐down). The other half of the reaction mixture was denatured (100 °C, 10 min) in loading buffer. Proteins were applied into a 15% SDS/PAGE gel under denaturing conditions and stained with a Coomassie‐based stain (Instant Blue, Gentaur, 30 min, RT). Alternatively, sdAbs were detected by Western blotting using a rat anti‐HA HRP conjugated mAb (1/5000; Roche®) (1 h, RT).
Hemopexin (HPX)
Apo‐HPX was isolated from rabbit serum, as described 10, 72 (kind gift from Ann Smith, University of Missouri‐K.C., Kansas City, MO, USA). Binding of HPX to heme was confirmed by UV‐Vis spectrophotometry of the apo‐protein or heme‐HPX complexes; the concentration of the protein and equimolar heme binding were quantified, as described 73.
Heme/sdAb complexes
SdAbs in PBS were incubated with 2× molar excess hemin in NaOH/HCl pH 7.4 (ON, 4 °C). Free heme and free sdAb were separated using a PD10 column (GE Healthcare) preequilibrated in PBS. Heme binding to the sdAb was confirmed spectrophotometrically. Concentration of sdAb‐heme complex was determined using formic acid assay, as described bellow (total heme). Total amount of protein was determined using Quick Start Bradford Protein Assay (Biorad, Hercules, CA, USA), according to the manufacture's instructions.
Spectroscopy assays
UV‐Visible spectra were recorded (PerkinElmer Lambda 25, UV/Vis spectrometer) using a concentration of 10 μm of sdAb in PBS. Increasing amount of hemin in 0.1 m NaOH (pH 7.4) was added and incubated (5 min) before recording the next spectra. Resonance Raman (RR) spectra were obtained using Raman spectrometer (Join Yvon U1000) coupled to a confocal microscope equipped with 1200 lines·mm−1 grating and a liquid nitrogen‐cooled back‐illuminated charge‐coupled device (CCD) detector. The spectra of 40 μm protein bound to heme and 50 μm heme in PBS buffer were obtained using a rotating cell (Hellma, Müllheim, Germany) to avoid photoreduction and measured with λ413 nm excitation line from a Krypton ion laser (Coherent Innova 302) with 4.5 mW laser power and 60 s accumulation time, at RT; typically 10 spectra were coadded to improve signal to noise ratio. After polynomial background subtraction, spectra were subjected to component analysis to determine the bandwidths and positions, using in‐house created software. Circular Dichroism (CD) Spectroscopy measurements were performed using a Jasco J‐815 spectropolarimeter equipped with a Peltier‐controlled thermostated cell support. CD spectra were recorded with sdAb 2H10 (240 μg·mL−1 in 35% 50 mm TRIS, 200 mm NaCl, 0.1 mm EDTA, 10% glycerol; pH 7.4 in PBS). Spectra were collected (50 nm min−1) and acquired (10×) to improve the signal to noise ratio. ATR‐FTIR spectra were measured on a Bruker IFS 66/S spectrometer equipped with a MCT detector and a thermostatized Harrick BioATR II cell. All measurements were obtained using an ATR cell with sdAb 2H10 (700 μg·mL−1 in 50 mm TRIS, 200 mm NaCl, 0.1 mm EDTA, 10% glycerol at pH 7.4) dissolved in PBS (1×), with or without heme. Each spectrum comprises the mean of 150 scans taken at a resolution of 4 cm−1. Spectral assignments for specific secondary structure elements were made as in Ref. 41.
pH titration
To address the effect of pH on heme binding by sdAb, assays were performed, essentially as described 74. Briefly, sdAbs (10 μm in PBS) were allowed to bind hemin (10 μm; pH 7, 15–20 min, RT) and pH was titrated down to 4.0–4.5 using 0.5 m HCl and restored thereafter using 0.5 m KOH (RT). To evaluate the effect of divalent cations, sdAbs bound to heme were first titrated down using 0.5 m HCl, followed by the addition of CaCl2 or MgCl2 (3 mm) at pH 4.0–4.5 and then the pH was titrated up to neutral using 0.5 m KOH (RT). UV‐visible spectra were recorded (PerkinElmer Lambda 25, UV/Vis spectrometer). Background of the spectra was corrected using the software originpro 8 ® (OriginLab Corporation, Northampton, MA, USA).
Mice
Mice were maintained under SPF conditions and experiments approved by the Ethics Committee of the Instituto Gulbenkian de Ciência, Direção Geral de Alimentação e Veterinária (Decreto‐Lei n° 113/2013) and European legislation (Directive 2010/63/EU). C57BL/6J mice were obtained from the Instituto Gulbenkian de Ciência's Animal Facility. C57BL/6xSv129 hα/hα::βA/βA, hα/hα::βA/βS and hα/hα::βS/βS mice 43 were obtained from Jackson Laboratories (Bar Harbor, ME, USA) (Reference 013071).
Phenylhydrazine
Adult C57BL/6 mice were injected (s.c.) with freshly prepared phenylhydrazine (Sigma) (1× PBS, pH7.4; 90 mg·kg−1; body weight).
Plasmodium infection
Adult C57BL/6 mice were infected intraperitoneally (i.p.) with 2 × 106 Pcc AS strain per mouse and parasitemias were determined by Giemsa‐stained blood smears and monitored daily, as described 22, 24.
Mouse plasma
Obtained by cardiac puncture (heparin or EDTA), centrifuged (2 ×; 1600 g, 5 min, 4 °C) and stored (−80 °C).
Total heme
Heme concentration was measured essentially as described 75. Briefly, samples were diluted in H2O in 96 well plates, formic acid (150 μL per well; 98–100%; Merck Portugal, Algés, Portugal) was added and absorbance was measured at λ405 nm using a microplate reader (Victor3 Multilabel Readers; Perkin Elmer). Heme concentration was determined by comparison to a hemin standard curve (0.5–16 μm in H2O).
Bioavailable heme
Bioavailable heme was measured using a previously described cellular heme reporter assay based on heme‐dependent HRP activity 12, 31. Briefly, HEK293 cells (ATCC; 5× 104 cells per well in a 24 well plate) were grown (ON; DMEM, 10% FBS, 1% penicillin 10 000 U·mL−1, streptomycin 10 000 μg·mL−1) and transiently transfected (4–6 h, Lipofectamin 2000; Invitrogen) with an expression vector encoding the HRP gene under the control of the EF‐1α promoter (pEF5/FRT/V5‐DEST‐Golgi‐HRP) in opti‐MEM reduced serum (Gibco by Thermo Fischer Scientific) 12. Expression of HRP was confined to the Golgi with a targeting sequence from galactosyltransferase 12, 31. Control HEK293 cells were transiently transfected with the pEF5/FRT/V5‐DEST vector 12, 31. Transfected cells were cultured (24 h) in DMEM (10% heme‐depleted FBS) with or without succinylacetone (SA; 0.5 mm) to block endogenous heme synthesis. Cells were washed and treated with hemin (ON) in opti‐MEM containing 0.5 mm SA and peroxidase activity was assessed as a red‐out of cellular heme content. Briefly, cells were lysed (20 mm HEPES pH 7.4, 150 mm NaCl, 0.5% Triton X‐100, 2.5× protease inhibitor cocktail set; Calbiochem®, San Diego, CA, USA) (ice, 20 min), centrifuged (16 000 g, 4 °C, 5 min) and peroxidase activity was quantified in a 96 well plate using 3,3′,5,5′‐Tetramethylbenzidine (TMB) substrate reagent set (BD OptEIA by Thermo Fischer Scientific). Reaction was stopped (2 N H2SO4) and absorbance was measured in a microplate reader at λ450 nm (Victor3 Multilabel Readers; Perkin Elmer). Peroxidase activity was determined based on the calibration curve generated with the serial dilution of HRP (type IV; Sigma) and normalized to protein expression (Quick Start Bradford Protein Assay; Biorad). HRP concentration was determined based on Beer‐Lambert law using the extinction coefficient (λ403 nm; EmM = 100).
Heme‐depleted medium
Depletion of heme from culture medium was performed as described 76. Briefly, DMEM (Life Technologies, Thermo Fischer Scientific, Carlsbad, CA, USA) was supplemented with 10% heat inactivated fetal bovine serum (FBS; Life Technologies by Thermo Fischer Scientific) predepleted from heme using ascorbic acid (10 mm; 37 °C, 50 rpm) until λ405 nm = 0.6–0.8 (~ 7 h), dialyzed (3×) in sterile PBS and filtered (0.2 μm). Final culture medium was supplemented with 1% penicillin (10 000 U·mL−1) and streptomycin (10 000 μg·mL−1).
Heme‐biotin pull‐down assay
SdAb 2H10 was incubated (30–60 min, 4 °C, agitation) with heme‐biotin and HPX was added to the mixture at 1/6 SdAb/HPX molar ratio (30–60 min, 4 °C, agitation). To evaluate the ability of HPX to bind heme‐biotin, HPX was incubated (30–60 min, 4 °C, agitation) with heme‐biotin. Half of the reaction mixture was incubated (2 h, RT, agitation) with Streptavidin‐Dynabeads (M‐280 Streptavidin; Invitrogen), previously blocked (5 h, RT) with protein‐free blocking buffer (Pierce). The reaction mixture was washed (3 ×; 5 min; PBS, 0.05% Tween 20), magnetic beads were captured (5 min magnetic field) according to the manufacture's instructions and incubated (20 min, 100 °C, agitation) in loading buffer (50 mm Tris‐HCl pH 6.8, 2% SDS, 10% Glycerol, 1% β‐ME, 12.5 mm EDTA, 0.02% Bromophenol blue). The other half of the reaction mixture was denatured (100 °C, 10 min) in loading buffer. Reaction mixtures in loading buffer were applied into a 15% SDS/PAGE gel under denaturing conditions and stained with a coomassie‐based stain (Instant Blue, Gentaur, 30 min, RT).
Heme‐binding capacity (HBC1/2)
The assay is based on the following principle: Plasma is incubated in vitro with increasing concentrations of heme (1 h30, RT, agitation), until saturation of heme‐binding molecules (with an affinity toward heme higher than 10−7 m). The heme‐specific sdAb‐based ELISA is used, as described above, to detect excess heme. The concentration of heme required to reduce by 50% the plasma heme‐binding capacity is defined as HBC1/2.
Heme competition assays
SdAbs (2.5 μg·mL−1) were preincubated (90 min, RT, mild agitation) with tetrapyrroles or hemoproteins and used to detect solid‐phase bound heme by ELISA, as described above.
Ascorbate oxidation assay
We adapted a previously described assay 77 as a readout for the oxidation activity of heme in the absence or presence of albumin or heme‐specific sdAb. Briefly, the reaction was initiated by the addition of heme (0–5 μm), without or with protein/sdAb (0–5 μm) and hydrogen peroxide (800 μm), to ascorbate (10 μm) in PBS pH 7.4 containing a metal chelator DETAPAC (50 μm) that does not inhibit heme‐mediated oxidation. The reaction was carried out under air at 37 °C and after a certain time, stopped by the addition of one volume of 0.5% metaphosphoric acid to one volume of reaction mix. The final solution was centrifuged and the supernatant collected and analyzed using HPLC with electrochemical detection 78.
Intracellular heme detection by immunofluorescence
HeLa (ATCC) cells were grown (24 h) in DMEM (10% FBS, 1% penicillin 10 000 U·mL−1, streptomycin 10 000 μg·mL−1; Life Technologies), seeded onto coverslips in a 24 well plate until 80% confluence. Cells were washed (1×, PBS), incubated (5 min, RT) in a hypotonic solution (85.5 mm NaCl, 5 mm MgCl2, pH 7.0), fixed (ice cold methanol, 10 min, −20 °C or 4% paraformaldehyde, 10 min at RT), washed (1×, PBS), permeabilized (PBS, 0.1% triton X‐100, 5 min), washed (3×, PBS), and blocked (Protein‐Free Blocking Buffer, Pierce, 1 h, RT). For intracellular heme detection, cells were incubated with heme‐specific sdAbs (2.5 ng·μL−1; 3 h, RT), washed (3×, blocking buffer), and incubated (1 h, RT) with Alexa Fluor ®647 conjugated anti‐HA Ab (clone 6E3; Cell signaling Technology, Danvers, MA, USA). Cells were washed (3×, PBS, 1× milli‐Q Water), DNA was stained with DAPI (Sigma), dried and mounted with coversplips in Mowiol‐Dabco media with DAPI. Images were acquired on a Leica DMRA2 upright microscope, equipped with a CoolSNAP HQ CCD camera, using a 100× 1.4NA Oil immersion objective, DAPI + CY5 fluorescence filtersets, controlled with the metamorph V7.5.1/software (Metamorph from Molecular Devices, Sunnyvale, CA, USA). The analysis was done with ImageJ (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2014). When indicated cells were coimmunostained with an anti‐Calnexin rabbit Ab, an integral endoplasmic reticulum protein (clone C4731; Sigma), antiearly endosome antigen 1 (EEA‐1) rabbit Ab, to localize early endosomes, phalloidin–tetramethylrhodamine B isothiocyanate phalloidin, a toxin that binds F‐actin (P1951; Sigma), anti‐BR mAb (A420; Dojindo) or anti‐HO‐1 polyclonal Ab (SPA‐896; Stressgen). Mitochondria were stained with MitoTracker® Red (Life Technologies). Confocal Z‐series stacks were acquired on a Leica SP5 Live upright microscope, using a 63 × 1.3NA Oil immersion objective, with DAPI + Fluorescein isothiocyanate (FITC) + CY5 or DAPI + Tetramethylrhodamine (TRITC) + CY5 laser lines, and spectral detection adjusted for the emission of the respective fluorochromes, controlled with the Leica software. The analysis was done with imagej (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, MD, USA, http://imagej.nih.gov/ij/, 1997–2014).
Intracellular heme detection by flow cytometry
HeLa cells were tripsinized (0.05% Trypsin‐EDTA; Life Technologies), washed (1×, PBS), fixed (ice cold methanol, 10 min, −20 °C), resuspended, washed (1×, PBS), permeabilized (PBS, 0.1% triton X‐100, 5 min), washed (1×, PBS), and blocked (Protein‐Free Blocking Buffer; Pierce) (1 h, RT). Intracellular heme was detected using antiheme sdAb (2.5 μg·mL−1 in blocking buffer, 2 h 30 min, RT, shaking) followed by Alexa Fluor® 647 conjugated HA‐Tag (6E3) mouse mAb (1/200; Cell Signaling Technology). Cells were washed (1×, PBS), resuspended (PBS) and acquired in a FACScan (Becton Dickinson, Franklin Lakes, NJ, USA) with cellquest software (BD Biosciences). The analysis was done with flowjo (Tree Star, Inc.).
Statistical analysis
When comparing the means of more than two experimental groups, we used analysis of variance (ANOVA) and significance between groups was estimated using Bonferroni post‐test, when Gaussian distribution was confirmed. For samples with non‐Gaussian distribution, comparison between experimental groups was performed using a Kruskal–Wallis test, followed by post‐test Dunns to compare all paired groups. Statistical analyses were performed using the graphpad v.5.0a software (Prism; GraphPad Software, Inc., La Jolla, CA, USA), and P values were represented as *P < 0.05, **P < 0.01, ***P < 0.001.
Author contributions
ZG codesigned, performed or coperformed all the experimental work and analyzed the data with the exception of ascorbate oxidation designed and performed by RS and immunofluorescence and Flow cytometry assays designed and performed by ARC. SR contributed in the malaria experiments. JG and FAS conceived and designed the synthetic phage display library and contributed critically to screening of sdAbs by Phage display technology. CMG and SSL contributed with the analysis by Circular Dichroism and ATR‐FTIR spectroscopy. ST and ZG performed Resonance Raman analysis. OI and ZG performed heme biotinylation. IH and XY developed the Golgi‐HRP assay and assisted ZG; MPS conceived, designed, supervised the study and wrote the manuscript with ZG. All the authors read, corrected, comment and approved the manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
Dr Ana Maria Varela Coelho and Renata Soares at Mass spectrometry and analytic services units of the Instituto de Tecnologia Química e Biológica (ITQB) as well as the Microscopy and Flow Cytometry Unit of the Instituto Gulbenkian de Ciência (IGC). Pierre Crozet and Jorge Carneiro for help in analysis of HBC1/2. Dr C Suarna for help with the ascorbate oxidation studies. The inflammation group at Instituto Gulbenkian de Ciência for critical discussion and comments. This work was supported by Fundação para a Ciência e Tecnologia (FCT), Portugal, (RECI‐IMI‐IMU‐0038‐2012; PTDC/SAU‐TOX/116627/2010; HMSP‐ICT/0018/2011 to MPS, SFRH/BD/44828/2008 to ZG, SFRH/BPD/101608/2014 to ARC, SFRH/BPD/47477/2008 to SSL, PTDC/SAU‐FAR/119173/2010 to JG, IF/01010/2013/CP1183/CT0001 to FAS), ERC‐2011‐AdG 294709‐DAMAGECONTROL to MPS, and NHMRC Senior Principal Research Fellowship 1003484 to RS.
References
- 1. Fischer H & Zeile K (1929) Synthese des haematoporphyrins, protoporphyrins und haemins. Justus Liebigs Annalen der Chemie 468, 98–116. [Google Scholar]
- 2. Tsiftsoglou AS, Tsamadou AI & Papadopoulou LC (2006) Heme as key regulator of major mammalian cellular functions: molecular, cellular, and pharmacological aspects. Pharmacol Ther 111, 327–345. [DOI] [PubMed] [Google Scholar]
- 3. Moncada S & Erusalimsky JD (2002) Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat Rev Mol Cell Biol 3, 214–220. [DOI] [PubMed] [Google Scholar]
- 4. Dioum EM, Rutter J, Tuckerman JR, Gonzalez G, Gilles‐Gonzalez MA & McKnight SL (2002) NPAS2: a gas‐responsive transcription factor. Science 298, 2385–2387. [DOI] [PubMed] [Google Scholar]
- 5. Severance S & Hamza I (2009) Trafficking of heme and porphyrins in metazoa. Chem Rev 109, 4596–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha‐Rodrigues M et al (2007) Heme oxygenase‐1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med 13, 703–710. [DOI] [PubMed] [Google Scholar]
- 7. Hebbel RP, Morgan WT, Eaton JW & Hedlund BE (1988) Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc Natl Acad Sci USA 85, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Balla J, Jacob HS, Balla G, Nath K, Eaton JW & Vercellotti GM (1993) Endothelial‐cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci USA 90, 9285–9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bunn HF & Jandl JH (1968) Exchange of heme among hemoglobins and between hemoglobin and albumin. J Biol Chem 243, 465–475. [PubMed] [Google Scholar]
- 10. Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A et al (2010) A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med 2, 51ra71. [DOI] [PubMed] [Google Scholar]
- 11. Ferreira A, Balla J, Jeney V, Balla G & Soares MP (2008) A central role for free heme in the pathogenesis of severe malaria: the missing link? J Mol Med 86, 1097–1111. [DOI] [PubMed] [Google Scholar]
- 12. White C, Yuan X, Schmidt PJ, Bresciani E, Samuel TK, Campagna D, Hall C, Bishop K, Calicchio ML, Lapierre A et al (2013) HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab 17, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith DW (1980) The molecular biology of mammalian hemoglobin synthesis. Ann Clin Lab Sci 10, 116–122. [PubMed] [Google Scholar]
- 14. Balla G, Vercellotti G, Eaton JW & Jacob HS (1990) Heme uptake by endothelium synergizes polymorphonuclear granulocyte‐mediated damage. Trans Assoc Am Physicians 103, 174–179. [PubMed] [Google Scholar]
- 15. Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW & Vercellotti GM (1992) Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem 267, 18148–18153. [PubMed] [Google Scholar]
- 16. Soares MP & Bozza MT (2016) Red alert: labile heme is an alarmin. Curr Opin Immunol 38, 94–100. [DOI] [PubMed] [Google Scholar]
- 17. Figueiredo RT, Fernandez PL, Mourao‐Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca‐Souza AV & Bozza MT (2007) Characterization of heme as activator of Toll‐like receptor 4. J Biol Chem 282, 20221–20229. [DOI] [PubMed] [Google Scholar]
- 18. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, Zamboni DS & Bozza MT (2014) Hemolysis‐induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci USA 111, E4110–E4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Porto BN, Alves LS, Fernandez PL, Dutra TP, Figueiredo RT, Graca‐Souza AV & Bozza MT (2007) Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J Biol Chem 282, 24430–24436. [DOI] [PubMed] [Google Scholar]
- 20. Fernandez PL, Dutra FF, Alves L, Figueiredo RT, Mourao‐Sa D, Fortes GB, Bergstrand S, Lonn D, Cevallos RR, Pereira RM et al (2010) Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)‐dependent reactive oxygen species generation. J Biol Chem 285, 32844–32851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, Smith A, Nath KA, Hebbel RP & Vercellotti GM (2014) Heme triggers TLR4 signaling leading to endothelial cell activation and vaso‐occlusion in murine sickle cell disease. Blood 123, 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, Rebelo S, Penido C, Smith NR, Coutinho A et al (2009) Heme oxygenase‐1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci USA 106, 15837–15842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gozzelino R, Jeney V & Soares MP (2010) Mechanisms of cell protection by heme oxygenase‐1. Annu Rev Pharmacol Toxicol 50, 323–354. [DOI] [PubMed] [Google Scholar]
- 24. Gozzelino R, Andrade BB, Larsen R, Luz NF, Vanoaica L, Seixas E, Coutinho A, Cardoso S, Rebelo S, Poli M et al (2012) Metabolic adaptation to tissue iron overload confers tolerance to malaria. Cell Host Microbe 12, 693–704. [DOI] [PubMed] [Google Scholar]
- 25. Gozzelino R & Soares MP (2014) Coupling heme and iron metabolism via ferritin H chain. Antioxid Redox Signal 20, 1754–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weis S, Carlos AR, Moita MR, Singh S, Blankenhaus B, Cardoso S, Larsen R, Rebelo S, Schäuble S, Del Barrio L et al (2017) Metabolic adaptation establishes disease tolerance to sepsis. Cell 169, 1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roumenina LT, Rayes J, Lacroix‐Desmazes S & Dimitrov JD (2016) Heme: modulator of plasma systems in hemolytic diseases. Trends Mol Med 22, 200–213. [DOI] [PubMed] [Google Scholar]
- 28. Ferreira A, Marguti I, Bechmann I, Jeney V, Chora A, Palha NR, Rebelo S, Henri A, Beuzard Y & Soares MP (2011) Sickle hemoglobin confers tolerance to Plasmodium infection. Cell 145, 398–409. [DOI] [PubMed] [Google Scholar]
- 29. Vinchi F, De Franceschi L, Ghigo A, Townes T, Cimino J, Silengo L, Hirsch E, Altruda F & Tolosano E (2013) Hemopexin therapy improves cardiovascular function by preventing heme‐induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 127, 1317–1329. [DOI] [PubMed] [Google Scholar]
- 30. Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR & Ofori‐Acquah SF (2013) Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest 123, 4809–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yuan X, Rietzschel N, Kwon H, Walter Nuno AB, Hanna DA, Phillips JD, Raven EL, Reddi AR & Hamza I (2016) Regulation of intracellular heme trafficking revealed by subcellular reporters. Proc Natl Acad Sci USA 113, E5144–E5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cunha‐Santos C, Figueira TN, Borrego P, Oliveira SS, Rocha C, Couto A, Cantante C, Santos‐Costa Q, Azevedo‐Pereira JM, Fontes CM et al (2016) Development of synthetic light‐chain antibodies as novel and potent HIV fusion inhibitors. AIDS 30, 1691–1701. [DOI] [PubMed] [Google Scholar]
- 33. Goncalves J, Silva F, Freitas‐Vieira A, Santa‐Marta M, Malho R, Yang X, Gabuzda D & Barbas C 3rd (2002) Functional neutralization of HIV‐1 Vif protein by intracellular immunization inhibits reverse transcription and viral replication. J Biol Chem 277, 32036–32045. [DOI] [PubMed] [Google Scholar]
- 34. Barbas CF, Burton DR, Scott JK & Silverman GJ (2001) Phage Display A Laboratory Manual. Cold Spring Harbor Laboratory, New York, NY. [Google Scholar]
- 35. Umetsu M, Tsumoto K, Hara M, Ashish K, Goda S, Adschiri T & Kumagai I (2003) How additives influence the refolding of immunoglobulin‐folded proteins in a stepwise dialysis system. Spectroscopic evidence for highly efficient refolding of a single‐chain Fv fragment. J Biol Chem 278, 8979–8987. [DOI] [PubMed] [Google Scholar]
- 36. Li T, Bonkovsky HL & Guo JT (2011) Structural analysis of heme proteins: implications for design and prediction. BMC Struct Biol 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pearson AR, Elmore BO, Yang C, Ferrara JD, Hooper AB & Wilmot CM (2007) The crystal structure of cytochrome P460 of Nitrosomonas europaea reveals a novel cytochrome fold and heme‐protein cross‐link. Biochemistry 46, 8340–8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McDonagh AF (2001) Turning green to gold. Nat Struct Biol 8, 198–200. [DOI] [PubMed] [Google Scholar]
- 39. Marvin KA, Kerby RL, Youn H, Roberts GP & Burstyn JN (2008) The transcription regulator RcoM‐2 from Burkholderia xenovorans is a cysteine‐ligated hemoprotein that undergoes a redox‐mediated ligand switch. Biochemistry 47, 9016–9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mouro C, Jung C, Bondon A & Simonneaux G (1997) Comparative Fourier transform infrared studies of the secondary structure and the CO heme ligand environment in cytochrome P‐450cam and cytochrome P‐420cam. Biochemistry 36, 8125–8134. [DOI] [PubMed] [Google Scholar]
- 41. Barth A & Zscherp C (2002) What vibrations tell us about proteins. Q Rev Biophys 35, 369–430. [DOI] [PubMed] [Google Scholar]
- 42. Jeney V, Ramos S, Bergman ML, Bechmann I, Tischer J, Ferreira A, Oliveira‐Marques V, Janse CJ, Rebelo S, Cardoso S et al (2014) Control of disease tolerance to malaria by nitric oxide and carbon monoxide. Cell Rep 8, 126–136. [DOI] [PubMed] [Google Scholar]
- 43. Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J & Townes TM (2006) Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood 108, 1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muller‐Eberhard U, Javid J, Liem HH, Hanstein A & Hanna M (1968) Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood 32, 811–815. [PubMed] [Google Scholar]
- 45. Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, Cerwenka A, Tolosano E & Muckenthaler MU (2015) Hemopexin therapy reverts heme‐induced pro‐inflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 127, 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ascenzi P, Bocedi A, Visca P, Altruda F, Tolosano E, Beringhelli T & Fasano M (2005) Hemoglobin and heme scavenging. IUBMB Life 57, 749–759. [DOI] [PubMed] [Google Scholar]
- 47. Soares MP & Weiss G (2015) The Iron age of host‐microbe interactions. EMBO Rep 16, 1482–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Soares MP & Hamza I (2016) Macrophages and iron metabolism. Immunity 44, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Balla J, Balla G, Jeney V, Kakuk G, Jacob HS & Vercellotti GM (2000) Ferriporphyrins and endothelium: a 2‐edged sword‐promotion of oxidation and induction of cytoprotectants. Blood 95, 3442–3450. [PubMed] [Google Scholar]
- 50. Balla G, Vercellotti GM, Muller‐Eberhard U, Eaton J & Jacob HS (1991) Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab Invest 64, 648–655. [PubMed] [Google Scholar]
- 51. Graca‐Souza AV, Arruda MA, de Freitas MS, Barja‐Fidalgo C & Oliveira PL (2002) Neutrophil activation by heme: implications for inflammatory processes. Blood 99, 4160–4165. [DOI] [PubMed] [Google Scholar]
- 52. Schaer DJ, Buehler PW, Alayash AI, Belcher JD & Vercellotti GM (2013) Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121, 1276–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kassa T, Jana S, Meng F & Alayash AI (2016) Differential heme release from various hemoglobin redox states and the upregulation of cellular heme oxygenase‐1. FEBS Open Bio 6, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Paoli M, Anderson BF, Baker HM, Morgan WT, Smith A & Baker EN (1999) Crystal structure of hemopexin reveals a novel high‐affinity heme site formed between two beta‐propeller domains. Nat Struct Biol 6, 926–931. [DOI] [PubMed] [Google Scholar]
- 55. Hrkal Z, Vodrazka Z & Kalousek I (1974) Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. Eur J Biochem 43, 73–78. [DOI] [PubMed] [Google Scholar]
- 56. Camejo G, Halberg C, Manschik‐Lundin A, Hurt‐Camejo E, Rosengren B, Olsson H, Hansson GI, Forsberg GB & Ylhen B (1998) Hemin binding and oxidation of lipoproteins in serum: mechanisms and effect on the interaction of LDL with human macrophages. J Lipid Res 39, 755–766. [PubMed] [Google Scholar]
- 57. Adams PA & Berman MC (1980) Kinetics and mechanism of the interaction between human serum albumin and monomeric haemin. Biochem J 191, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li RC, Saleem S, Zhen G, Cao W, Zhuang H, Lee J, Smith A, Altruda F, Tolosano E & Dore S (2009) Heme‐hemopexin complex attenuates neuronal cell death and stroke damage. J Cereb Blood Flow Metab 29, 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Larsen R, Gouveia Z, Soares MP & Gozzelino R (2012) Heme cytotoxicity and the pathogenesis of immune‐mediated inflammatory diseases. Front Pharmacol 3, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Smith A & McCulloh RJ (2015) Hemopexin and haptoglobin: allies against heme toxicity from hemoglobin not contenders. Front Physiol 6, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Meng F & Alayash AI (2017) Determination of extinction coefficients of human hemoglobin in various redox states. Anal Biochem 521, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hanna DA, Harvey RM, Martinez‐Guzman O, Yuan X, Chandrasekharan B, Raju G, Outten FW, Hamza I & Reddi AR (2016) Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc Natl Acad Sci USA 113, 7539–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ricoux R, Sauriat‐Dorizon H, Girgenti E, Blanchard D & Mahy JP (2002) Hemoabzymes: towards new biocatalysts for selective oxidations. J Immunol Methods 269, 39–57. [DOI] [PubMed] [Google Scholar]
- 64. Kawamura‐Konishi Y, Asano A, Yamazaki M, Tashiro H & Suzuki H (1998) Peroxidase activity of an antibody–ferric porphyrin complex. J Mol Catal B Enzym 4, 181–190. [Google Scholar]
- 65. Cochran AG & Schultz PG (1990) Peroxidase activity of an antibody‐heme complex. J Am Chem Soc 112, 9414–9415. [Google Scholar]
- 66. Motterlini R & Otterbein LE (2010) The therapeutic potential of carbon monoxide. Nat Rev Drug Discov 9, 728–743. [DOI] [PubMed] [Google Scholar]
- 67. Rish KR, Swartzlander R, Sadikot TN, Berridge MV & Smith A (2007) Interaction of heme and heme‐hemopexin with an extracellular oxidant system used to measure cell growth‐associated plasma membrane electron transport. Biochim Biophys Acta 1767, 1107–1117. [DOI] [PubMed] [Google Scholar]
- 68. Miyamoto Y, Nishimura S, Inoue K, Shimamoto S, Yoshida T, Fukuhara A, Yamada M, Urade Y, Yagi N, Ohkubo T et al (2010) Structural analysis of lipocalin‐type prostaglandin D synthase complexed with biliverdin by small‐angle X‐ray scattering and multi‐dimensional NMR. J Struct Biol 169, 209–218. [DOI] [PubMed] [Google Scholar]
- 69. Clarke JT (1965) Purification and analysis of bilirubin. Clin Chem 11, 681–690. [PubMed] [Google Scholar]
- 70. Ishida M, Dohmae N, Shiro Y & Isogai Y (2003) Synthesis of biotinylated heme and its application to panning heme‐binding proteins. Anal Biochem 321, 138–141. [DOI] [PubMed] [Google Scholar]
- 71. Smith GP (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315–1317. [DOI] [PubMed] [Google Scholar]
- 72. Smith A & Morgan WT (1978) Transport of heme by hemopexin to the liver: evidence for receptor‐mediated uptake. Biochem Biophys Res Commun 84, 151–157. [DOI] [PubMed] [Google Scholar]
- 73. Eskew JD, Vanacore RM, Sung L, Morales PJ & Smith A (1999) Cellular protection mechanisms against extracellular heme. heme‐hemopexin, but not free heme, activates the N‐terminal c‐jun kinase. J Biol Chem 274, 638–648. [DOI] [PubMed] [Google Scholar]
- 74. Smith A, Rish KR, Lovelace R, Hackney JF & Helston RM (2009) Role for copper in the cellular and regulatory effects of heme‐hemopexin. Biometals 22, 421–437. [DOI] [PubMed] [Google Scholar]
- 75. Kuross SA, Rank BH & Hebbel RP (1988) Excess heme in sickle erythrocyte inside‐out membranes: possible role in thiol oxidation. Blood 71, 876–882. [PubMed] [Google Scholar]
- 76. Zhu Y, Sun Y, Jin K & Greenberg DA (2002) Hemin induces neuroglobin expression in neural cells. Blood 100, 2494–2498. [DOI] [PubMed] [Google Scholar]
- 77. Mashima R, Tilley L, Siomos MA, Papalexis V, Raftery MJ & Stocker R (2002) Plasmodium falciparum histidine‐rich protein‐2 (PfHRP2) modulates the redox activity of ferri‐protoporphyrin IX (FePPIX): peroxidase‐like activity of the PfHRP2‐FePPIX complex. J Biol Chem 277, 14514–14520. [DOI] [PubMed] [Google Scholar]
- 78. Behrens WA & Madere R (1987) A highly sensitive high‐performance liquid chromatography method for the estimation of ascorbic and dehydroascorbic acid in tissues, biological fluids, and foods. Anal Biochem 165, 102–107. [DOI] [PubMed] [Google Scholar]