

Abstract
Introduction
Scientific interest in the gut microbiota is increasing due to improved understanding of its implications in human health and disease. In patients with kidney disease, gut microbiota-derived uremic toxins directly contribute to altered nonrenal drug clearance. Microbial imbalances, known as dysbiosis, potentially increase formation of microbiota-derived toxins, and diminished renal clearance leads to toxin accumulation. High concentrations of microbiota-derived toxins such as indoxyl sulfate and p-cresol sulfate perpetrate interactions with drug metabolizing enzymes and transporters, which provides a mechanistic link between increases in drug-related adverse events and dysbiosis in kidney disease.
Areas Covered
This review summarizes the effects of microbiota-derived uremic toxins on hepatic phase I and phase II drug metabolizing enzymes and drug transporters. Research articles that tested individual toxins were included. Therapeutic strategies to target microbial toxins are also discussed.
Expert Commentary
Large interindividual variability in toxin concentrations may explain some differences in nonrenal clearance of medications. Advances in human microbiome research provide unique opportunities to systematically evaluate the impact of individual and combined microbial toxins on drug metabolism and transport, and to explore microbiota-derived uremic toxins as potential therapeutic targets.
Keywords: Drug metabolism, drug transporters, nonrenal clearance, human microbiome, kidney disease, microbiota-derived uremic toxins, microbial toxins, uremic toxins
1. Introduction
The gut microbiota is a dynamic community of metabolically active microorganisms that interact bi-directionally with human physiological systems. The average human gut microbiota contains over 38 trillion microorganisms that make important contributions to overall human health [1–3]. Microbial composition is impacted by several factors, including diet, medications, host genetics, environmental interactions, disease specific changes and transient bacterial species. Microbiota-derived toxins, byproducts of the metabolism of dietary nutrients by gut microbiota, are implicated in multiple diseases including kidney disease [4], cardiovascular disease [5–7], cancer [8], neurological diseases [9], and inflammatory states [10]. Several microbiota-derived uremic toxins such as indoxyl sulfate, p-cresol sulfate, hippuric acid, 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid (CMPF) and indole-3-acetic acid have been shown to accumulate extensively in kidney disease patients, likely due to increased toxin producing bacterial species and simultaneously decreased renal clearance (Figure 1) [11]. However, they may represent only the tip of the iceberg as many more microbial toxins likely circulate in humans.
Figure 1.
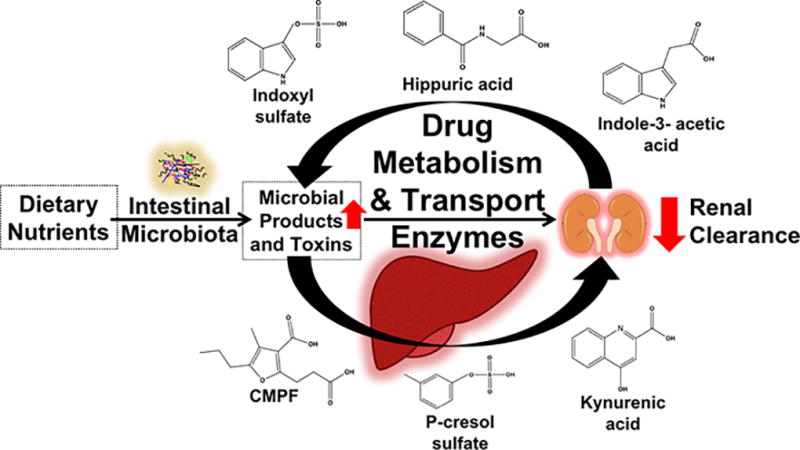
The intestinal microbiota produces several microbial toxins that extensively accumulate in kidney disease likely due to increased toxin producing bacterial species and decreased renal clearance. Microbiota-derived uremic toxins such as p-cresol sulfate, indoxyl sulfate, indole-3-acetic acid, CMPF, kynurenic acid, and hippuric acid perpetrate interactions with phase I/II metabolic enzymes and drug transporters. These interactions occur in the liver and kidneys at the translational and post-translational levels. Patients with kidney disease are at increased risk for adverse drug events, and these accumulated microbiota-derived uremic toxins likely contribute to altered drug disposition.
Microbial imbalance (i.e., disturbed microbial composition) or maladaptation of microbial communities to the diseased environment, also known as dysbiosis, is partially mediated by microbial toxins and influences gut microbiota-host interactions [12,13]. Microbiota-derived uremic toxins interact with several nonrenal clearance pathways, thereby potentially modifying drug dosing-exposure-response relationships in patients with kidney disease [14–17]. In fact, alterations in the functional expression of drug metabolizing enzymes and transporters in kidney disease are well documented [18–21], and several microbial toxins are potential mediators [22,23]. The uremic milieu is complicated, containing thousands of compounds including numerous endogenous solutes (e.g., urate, parathyroid hormone, inflammatory cytokines) that are not derived from the microbiota, yet may elicit independent pathophysiological effects. Therefore, this review highlights the effects of microbiota-derived uremic toxins on drug metabolizing enzymes and transporters, focusing on individual toxins, rather than uremia in general.
We present a comprehensive review of the effects of microbiota-derived uremic toxins on hepatic phase I and phase II drug metabolizing enzymes and drug transporters. The MEDLINE bibliographic database was searched through PubMed using relevant search terms, including uremic toxins, uremic retention solutes, uremia, drug metabolism, drug transport, kidney disease, renal disease, nonrenal clearance, and human microbiome. Other articles were identified from reference lists within the retrieved manuscripts. A total of 36 articles that explicitly tested individual microbial toxin effects on drug metabolizing enzymes or transporters. All other citations are included as supporting evidence that the microbiome and microbiota-derived toxins impact nonrenal clearance.
2. Intestinal Microbiota-Host Liver Interactions
The Human Microbiome Project and the Metagenomics of the Human Intestinal Tract initiative revealed important information about the vast diversity and complexity of the human microbiome [24–26]. The diversity and function of the intestinal microbiota directly influences bacterial metabolism, which in turn impacts the systemic exposure of orally administered nutrients and xenobiotics and the formation of microbial toxins that elicit pathophysiological effects [27]. Some of the nutrients and xenobiotics transformed by gut microbiota reach the liver via portal circulation where they interact with drug metabolizing enzymes and transporters.
Hepatic drug metabolizing enzymes interact with the microbiota through biotransformation of select microbial toxins. For example, dietary tryptophan is metabolized by microbial tryptophanase to indole, which is subsequently hydroxylated by CYP2E1 to indoxyl and finally sulfonated by sulfotransferases (SULT1A1) to indoxyl sulfate. The biotransformation of indoxyl sulfate requires CYP2E1 and SULT1A1 hepatic enzymes and demonstrates the importance of these pathways in microbiota-derived uremic toxin formation (Figure 2). Formation of p-cresol sulfate and indoxyl sulfate is decreased in chronic kidney disease patients and animal models with advanced liver cirrhosis (and thus impaired hepatic metabolism) [28]. Cirrhotic patients also exhibit higher serum concentrations of metabolic precursors like phenol and indole, suggesting decreased metabolic conjugation [29,30]. The role of hepatic metabolism in biotransformation of some microbiota-derived uremic toxins indicates a potential for competitive interactions with both Phase I and II metabolic pathways.
Figure 2.
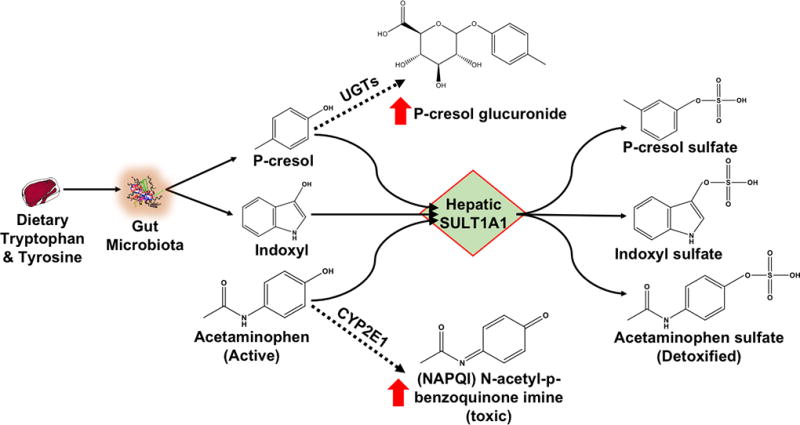
P-cresol, indoxyl and acetaminophen are substrates for hepatic SULT1A1. Uremic toxins perpetrate interactions with SULT1A1, such that metabolism of substrates can be shunted to alternate metabolic pathways. For example, acetaminophen metabolism may be shunted (dotted line) towards CYP2E1, forming the toxic metabolite NAPQI, and p-cresol metabolism may be shunted (dotted line) towards UGT, forming p-cresol glucuronide.
The gut microbiota regulates hepatic phase I and II metabolizing enzyme and transporter expression [31,32], therefore alterations of gut microbiota constituents might also affect these processes [31,33]. Experimental models investigating the effects of the gut microbiota on drug metabolism and transport include un-colonized or germ-free animals. Germ-free and normally colonized mice differentially express important drug metabolism and transporter genes [33–36]. For example, germ-free mice exhibit increased expression of Cyp1a2 (+51%) and Cyp4a14 (+202%) mRNA, but decreased expression of Cyp3a11 (−87%) and Cyp2b10 (−57%) mRNA. Germ-free mice more efficiently metabolize intravenous pentobarbital, measured by total anesthesia time, suggesting enzymatic activity increases with gene expression [33]. One potential mechanism may be the continuous passing of microbial metabolites from the intestine to the liver via the portal circulation. Since these metabolites may be ligands for nuclear receptors, this could lead to altered hepatic gene expression [37]. Collectively, the evidence suggests that the gut microbiota interacts with hepatic enzymes, which might link dysbiosis in kidney disease to altered nonrenal clearance.
3. Dysbiosis in Kidney Disease
Dysbiosis in kidney disease influences microbial toxin production and absorption. Alterations to the gut microbiota in patients with kidney disease are due to a combination of factors, including concomitant use of numerous medications, frequent antibiotic use, changes in intestinal pH, and intestinal barrier dysfunction [13,27,39]. These factors may lead to an increase in intestinal bacteria that produce uremic toxins [12,40]. For example, bacteria that produce precursors to indoxyl sulfate and p-cresol sulfate are increased in patients with advanced kidney disease [40] and animal models of kidney disease [41]. Uremic toxins in the gastrointestinal tract may engender inflammation and promote a “leaky gut” [42]. Dysbiosis also contributes to impairment of the gut epithelial barrier, which facilitates greater absorption of microbial toxins, increased serum endotoxin concentrations and bacterial translocation [11,43]. In fact, increased microbial toxin production and absorption is associated with nearly half of the most highly accumulated uremic toxins [44]. Constipation may also worsen uremia and increase the risk of adverse renal outcomes [45]. Overall, the altered gut microbiota observed in kidney disease produces more microbial toxins that accumulate, become constituents of the uremic milieu, and are readily available at high concentrations to interact with drug metabolizing enzymes and transporters.
4. Considerations for Interpreting Microbiota-Derived Uremic Toxin Studies
Caution is warranted when interpreting studies involving microbiota-derived uremic toxins due to frequent discrepancies between concentrations and conformations of toxins observed clinically and those used experimentally. The highest clinically observed total concentrations of microbial toxins in kidney disease patients range from 3.7-fold (indole-3-acetic acid) to 83-fold (indoxyl sulfate) higher than in patients with normal kidney function (Table 3). Moreover, many microbial toxins are extensively protein bound, and since only unbound toxins elicit biological activity and/or undergo transport into cells, interpretation of data based on unbound versus total (free and bound) concentrations may lead to different conclusions. For example, some studies acknowledge protein binding [46], while others use total concentrations [47] to interpret clinical relevance of results. The unbound concentrations at the site of interactions are likely the most relevant but are difficult to estimate [48]. Only considering plasma concentrations of microbial toxins may not accurately reflect tissue distribution. For instance, some microbial toxins accumulate to higher concentrations in liver and kidney tissues than in plasma [49].
Table 3.
Clinically Observed Microbiota-Derived Uremic Toxin Concentrations
| Microbial Toxin | Normal Total Concentration (SD) [µM] |
Mean Uremic Free Concentration (SD) [µM] |
Mean Uremic Total Concentration (SD) [µM] |
Highest Uremic Total Concentration (SD or Range) [µM] |
|---|---|---|---|---|
| CMPF | 15 (1) | - | 25 (10) | 135 (46) [109] |
| Hippuric Acid | 17 (11) | 231 (80) | 398 (77) | 487 (345) |
| Indole-3-acetic acid | 3 (1.7) | 2.1 (0.6) | 12 (2) | 14 (13) |
| Indoxyl sulfate | 2.5 (1.4) | 15 (5.7) | 109 (61) | 210 (72) |
| Kynurenic acid | .03 (.007) | - | - | 0.8 (0.4) |
| P-cresol sulfate | 10 (7) | 9.3 (6.4) | 111 (65) | 218 (71) |
CMPF, 3-Carboxy-4-methyl-5-propyl-2-furan-propanoic acid.
Adapted from Duranton et al., J Am Soc Nephrol. 2012 Jul;23(7): 1258–1270 [44]
Another important consideration when interpreting studies involving microbiota-derived uremic toxins is the moiety studied. For instance, p-cresol is frequently employed in experimental models, but the sulfate and glucuronide conjugates are the only measurable forms in human plasma [50]. This raises concern over the clinical relevance of results derived in experiments using unconjugated p-cresol. Nevertheless, p-cresol is likely present in portal circulation, and must also be present at some lower concentrations in hepatocytes and kidney tissue [46]. Clinically observed total microbial toxin concentrations are included in Table 3 and used as a benchmark to evaluate clinical relevance of individual studies [44].
Lastly, it is important to consider limitations of experimental methods employed in individual studies. For example, experiments assessing the effect of individual microbial toxins on metabolism or transport pathways often use in vitro techniques (e.g. microsomes, isolated hepatocytes, functional assays) that may not completely translate to in vivo human processes. The strengths and weaknesses inherent to experimental techniques used should be considered in the interpretation of results.
5. Phase I Metabolic Pathways
Phase I oxidative enzymes, including cytochrome P450 (CYPs) and flavin-containing monooxygenases (FMOs), metabolize approximately 80% of commonly used medications [51]. Patients with kidney disease are often prescribed 10–12 different medications [52] and many are substrates for Phase I drug metabolizing enzymes. Therefore, disruption in the expression and/or function of these pathways by uremic toxins may alter drug exposure and thereby lead to suboptimal response or increased drug-related adverse events. Indeed, several microbiota-derived uremic toxins accumulate in patients with kidney disease and interfere with hepatic metabolism (Tables 1–3). For example, the combined effects of hippuric acid, indoxyl sulfate and p-cresol result in a >50% decrease in CYP3A4, CYP1A2, CYP2C9, and CYP2E1 human microsomal activity [46]. The combination of microbial toxins may be either additive or synergistic. Determination of individual and combined effects of microbiota-derived toxins on hepatic enzymes responsible for xenobiotic and endobiotic metabolism is therefore critical.
TABLE 1.
Effect of Microbiota-Derived Uremic Toxins on Phase I Drug Metabolizing Enzymes
| Enzyme | Indoxyl Sulfate | P-cresol OR P-cresol Sulfate |
CMPF | Hippuric Acid | Indole-3-Acetic Acid |
Combined Microbial Toxins |
|---|---|---|---|---|---|---|
| CYP3A | ||||||
| CYP1A1 | ||||||
| CYP1A2 | ||||||
| CYP2E1 | ||||||
| CYP2C9 | ||||||
| CYP2B6 | ||||||
| CYP2D6 |
Denotes within clinically relevant concentrations.
Denotes concentration greater than clinically relevant (Table 3).
CMPF, 3-Carboxy-4-methyl-5-propyl-2-furan-propanoic acid. HA, hippuric acid. IS, Indoxyl sulfate. I3A, Indole-3-acetic acid. PC, P-cresol.
Weak inhibitor <29%, Inhibitor 30–69%, Strong inhibitor >70%
5.1. CYP3A4/5
Microbiota-derived uremic toxins may affect the activity and expression of CYP3A4, a critically important isozyme that contributes to the metabolism of approximately 40% of marketed drugs [51]. P-cresol is a potent competitive inhibitor of CYP3A4 and perpetrates a >60% decrease in human microsomal activity [46]. Hippuric acid, indoxyl sulfate, CMPF and p-cresol all individually inhibit CYP3A4 mediated metabolism of testosterone in human microsomes [47]. Additionally, a combination of hippuric acid, CMPF, indoxyl sulfate and indole-3-acetic acid also perpetrate a significant decrease of CYP3A4 mRNA expression [53]. Indoxyl sulfate also inhibits CYP3A4 activity in human hepatocytes and microsomes, animal models and potentially in kidney disease patients [54–56]. Indoxyl sulfate plasma concentrations are inversely related to CYP3A4 activity measured by endogenous 4β-hydroxycholesterol formation in stable renal transplant patients [57]. Conversely, some reports suggest that indoxyl sulfate or other microbial toxins have no interactions with CYP3A4 activity or expression [53,58]. The reason for discordant findings between studies is unclear, but might be due to differences in experimental procedures. Nevertheless, the evidence to date suggests that microbiota-derived uremic toxins and especially indoxyl sulfate affect CYP3A4 activity and potentially expression.
5.2. CYP1A2
Microbiota-derived uremic toxins also affect CYP1A activity and expression [46,55,58–60], with idole-3-acetic acid and indoxyl sulfate being the most potent. Indole-3-acetic acid decreases CYP1A2 activity by 50% in human microsomes [60]. A combination of hippuric acid, indoxyl sulfate and p-cresol also inhibit CYP1A2 activity in human microsomes [46]. However, contradictory findings have been reported, as indole-3-acetic acid and indoxyl sulfate individually have been shown to increase mRNA expression and enzyme activity of CYP1A2 [58,59]. Although reasons for the disparate findings are unclear, it likely involves the AhR nuclear receptor -- a master regulator that activates CYP1A gene expression. Indoxyl sulfate is both a potent activating ligand of AhR that leads to increased expression of CYP1A2, and a direct inhibitor of CYP1A2 activity [58,59]. Because of this, investigators should consider incubations that are long enough in duration to induce expression when assessing the effect of indoxyl sulfate on CYP1A2. The sum of data suggest that microbiota-derived toxins inhibit CYP1A2 enzymatic activity and that indoxyl sulfate induces CYP1A2 functional expression.
5.3. CYP2C9
CYP2C9 metabolism also may be inhibited by uremic toxins. Incubation of human microsomes with a combination of p-cresol and indoxyl sulfate resulted in a 46% decrease in metabolism of the dual CYP2C9 and CYP3A4 substrate losartan [61]. CMPF, indole-3-acetic acid and hippuric acid had no inhibitory effect on losartan metabolism [61]. Since losartan is a dual substrate, any effect ellicited by uremic toxins may be partially attributed to inhibition of CYP3A4 activity. Hippuric acid may inhibit CYP2C9 activity to a greater extent than p-cresol and indoxyl sulfate. Human microsomes were incubated with p-cresol, indoxyl sulfate and hippuric acid alone, and only hippuric acid decreased (by 39%) metabolism of the CYP2C9 substrate phenytoin [46].
5.4. CYP2E1
Microbiota-derived uremic toxins might also interact with CYP2E1. A combination of hippuric acid, indoxyl sulfate and p-cresol decreases CYP2E1 human microsomal activity by >50% [46]. At clinically relevant concentrations, p-cresol individually inhibits CYP2E1 activity by >60% in human microsomes, whereas hippuric acid and indoxyl sulfate had no individual effect [46]. This finding suggests that the microbial toxin precursor p-cresol inhibits CYP2E1 activity and other microbial toxins may only have a combined effect on this enzyme system.
In summary, current evidence suggests that microbiota-derived uremic toxins interact most extensively with CYP3A4 and CYP1A2. Interactions might also occur with CYP2C9 and CYP2E1 enzymes. These metabolic interactions and the wide variation of uremic toxin concentrations (Table 3) may partially explain the interindividual variability of alterations in Phase I drug metabolism in kidney disease patients.
6. Phase II Metabolic Pathways
Microbiota-derived uremic toxins also appear to interact with Phase II drug metabolism pathways (Table 2). Phase II enzymes catalyze conjugation reactions and include sulfotransferases, glutathione S-transferases, uridine diphosphate-glucuronosyltransferases, N-acetyltransferases, and methyltransferases. Like Phase I enzymes, these pathways are essential for both xenobiotic and endobiotic disposition and any disruption in their expression and/or function by uremic toxins may alter drug exposure and response.
TABLE 2.
Effect of Microbiota-Derived Uremic Toxins on Phase II Drug Metabolizing Enzymes
| Enzyme | Indoxyl Sulfate | P-cresol OR P-cresol Sulfate |
CMPF | Indole-3-Acetic Acid |
|---|---|---|---|---|
| UGT1A1 | ||||
| UGT1A4 | ||||
| UGT1A6 | ||||
| UGT1A9 | ||||
| UGT2B4 | ||||
| UGT2B7 | ||||
| GST | ||||
| SULT1A1 | ||||
| UGT1A1 |
Denotes within clinically relevant concentrations.
Denotes concentration greater than clinically relevant (Table 3).
CMPF, 3-Carboxy-4-methyl-5-propyl-2-furan-propanoic acid. IS, Indoxyl sulfate. I3A, Indole-3-acetic acid. PC, P-cresol.
Weak inhibitor <29%, Inhibitor 30–69%, Strong inhibitor >70%
6.1. Sulfotransferases (SULT)
Increased production and systemic exposure of p-cresol, indoxyl and perhaps other gut-derived metabolites in patients with kidney disease can perpetrate metabolic drug interactions, including competitive inhibition of hepatic sulfotransferases, and drug toxicity. The metabolic fate of acetaminophen (paracetamol) provides an important illustration of this phenomenon [62]. P-cresol and indoxyl are conjugated by SULT1A1 to the two well-known uremic toxins p-cresol sulfate and indoxyl sulfate, respectively [63]. Acetaminophen is normally metabolized by SULT1A1 conjugation as a major metabolic detoxification pathway. Interestingly, patients with high systemic concentrations of p-cresol sulfate (and thus likely increased concentrations of the unconjugated precursor) exhibit metabolic shunting of acetaminophen to CYP2E1 (Figure 2). CYP2E1 then generates hepatotoxic N-acetyl-p-benzoquinone imine (NAPQI) metabolites. Therefore, competitive SULT metabolic interactions between p-cresol and acetaminophen likely facilitate acetaminophen shunting towards NAPQI formation. Alterations to SULT function may also have cardiovascular implications, since shifts from p-cresol sulfate to glucuronide conjugation are associated with cardiovascular disease and mortality [64].
6.2. Glutathione S-transferases (GST) and Uridine Diphosphate-glucuronosyltransferases (UGT)
The activity and expression of GST and UGT enzymes also appear to be affected by microbiota-derived toxins. CMPF (by 37% in bovine and 21% in rat enzymes) and indoxyl sulfate (by 27% in bovine enzymes) decrease GST enzyme activity, which suggests that CMPF may be the most potent inhibitor of GST enzymes [65]. The activities of UGT1A1, UGT1A9 and UGT2B7 are decreased by 50% in human microsomes incubated with a combination of hippuric acid, indoxyl sulfate and p-cresol toxins as well as p-cresol alone at clinically observed concentrations, suggesting that p-cresol is mediating the effect [46]. P-cresol, p-cresol sulfate, kynurenic acid, indole-3-acetic acid, indoxyl sulfate and hippuric acid also can significantly decrease UGT activity in human renal proximal tubule cells [66], with p-cresol being the most potent competitive inhibitor. Similar to its effects on CYP1A2, indoxyl sulfate induces mRNA expression of UGT1A1 and UGT1A6 in primary human hepatocytes [59].
In summary, microbiota-derived uremic toxins inhibit SULT, GST and UGT activity, which might contribute to altered drug exposure and response, and high rates of adverse drug events in kidney disease patients. Microbial toxin interactions with phase II enzymes may necessitate careful dosing of drugs that undergo biotransformation through these pathways. The effects of these direct interactions may also extend beyond drug interactions to endogenous substrates such as bilirubin, steroid hormones and bile acids.
7. Effects of Microbial Toxins on Drug Transporters
Transporters are essential transmembrane proteins for hepatic uptake and excretion, as well as renal tubular secretion of drugs. Microbiota-derived uremic toxins have been shown to interact with several transporters located in the kidneys and liver (Table 4).
Table 4.
Effect of Microbiota-Derived Uremic Toxins on Drug Transporters
| Drug Transporter |
Indoxyl Sulfate | p-Cresol OR p-Cresol Sulfate |
Kynurenic Acid |
CMPF | Hippuric Acid | Indole-3-Acetic Acid |
Microbial Toxin Combination |
|---|---|---|---|---|---|---|---|
| OATP1B1 | |||||||
| OATP1B3 | |||||||
| OATP2 | |||||||
| OATP2B1 | |||||||
| OAT1 | |||||||
| OAT3 | |||||||
| MRP2 | |||||||
| MRP4 | |||||||
| BCRP |
Denotes within clinically relevant concentrations,
Higher concentrations than observed in patients.
CMPF, 3-Carboxy-4-methyl-5-propyl-2-furan-propanoic acid. HA, hippuric acid. IS, Indoxyl sulfate. I3A, Indole-3-acetic acid. PC, P-cresol.
7.1. Hepatic Organic Anion Transporting Polypeptides (OATPs)
Hepatic organic anion transporting polypeptides (OATPs) are transporters for large hydrophobic anions (e.g., statins) and other commonly prescribed medications. OATPs transport drugs into hepatocytes, where they may undergo metabolism and biliary excretion. Microbiota-derived uremic toxins affect hepatic OATP1B1, OATP1B3, and OATP2B1 activity and expression [56,67–71]. For example, OATP1B1 and OATP1B3 transport of methotrexate is inhibited by kynurenic acid, indole-3-acetic acid, indoxyl sulfate and p-cresol in HEK293 cells [68]. Kynurenic acid has the strongest inhibitory effect. Interestingly, indoxyl sulfate and kynurenic acid are substrates of OATP1B1 and OATP1B3, and therefore might be competitive inhibitors. Uptake of the OATP1B3 substrate digoxin in rat and human hepatocytes is most notably inhibited by CMPF and p-cresol (400µM) [70]. P-cresol (300µM), indoxyl sulfate (400µM), and CMPF (400µM) significantly inhibit OATP1B1 and OATP1B3 uptake of estrone sulfate in transfected HEK293 cells [71]. The uptake of erythromycin by OATPs in rat hepatocytes is significantly inhibited by CMPF (50 µM) [56]. Inhibition of hepatic OATP transporters by microbiota-derived uremic toxins likely alter intracellular drug transport and subsequent hepatic metabolism. CMPF most consistently decreases OATP transporter function, but kynurenic acid and p-cresol also elicit inhibitory effects (Table 4). In patients with end-stage renal disease, decreased OATP transport was implicated in a 63% decrease in oral clearance of fexofenadine [15]. Increased demethylation of C14-erythromycin immediately post-hemodialysis also suggests that inhibitory uremic toxins are cleared by dialysis, thereby improving OATP uptake and intracellular exposure to drug substrates [72]. Therefore, microbiota-derived uremic toxins likely perpetrate clinically relevant interactions with OATP transporters.
7.2. Kidney Organic Anion Transporters (OATs)
Organic anion transporters (OAT) secrete solutes from the basolateral membrane (blood) into the proximal tubules to facilitate secretion into the urine. These transporters, especially OAT1 and OAT3, contribute to the tubular secretion of indoxyl sulfate, p-cresol sulfate, kynurenic acid, CMPF, hippuric acid and indole-3-acetic acid [73–75]. In fact, OAT1 and OAT3 are high-capacity transporters with high affinity for kynurenic acid and p-cresol sulfate. Microbial toxins might act as competitive inhibitors of OAT tubular secretion of both endogenous solutes and drugs, and vice versa, thereby increasing systemic exposure. For example, OAT1 and OAT3 mediated tubular secretion of indoxyl sulfate is competitively inhibited by non-steroidal anti-inflammatory drugs (NSAIDs), quinapril and probenecid [76,77], which may increase systemic exposure of drugs and/or indoxyl sulfate. Indoxyl sulfate, CMPF and hippuric acid also have inhibitory effects on OAT1 and OAT3 activity [78–80]. In rat kidney slices, hippuric acid and indoxyl sulfate in combination and independently decrease OAT1 and OAT3 uptake of morinidazole-metabolites [81]. Also, CMPF, indoxyl sulfate, hippuric acid and indole-3-acetic acid each independently decrease OAT1 and OAT3 uptake of morinidazole-metabolites in HEK293 cells [81].
7.3. Kidney Breast Cancer Resistant Protein (BCRP), Multidrug Resistance Protein 4 (MRP4) and Organic Anion Transporting Polypeptides (OATP4C1)
BCRP and MRP4 are important kidney efflux transporters located on the apical membrane of the proximal tubule epithelial cell. They are responsible for movement of solutes from within the cell into the tubular lumen. These proteins are dysregulated by microbial toxins [82]. Alterations of these transporters have significant implications for tubular secretion of several endogenous substrates and uremic toxins (e.g. urate) [82]. Hippuric acid, indoxyl sulfate and kynurenic acid inhibit these efflux transporters [82]. OATP4C1, the only OATP in human kidneys, eliminates several drugs (e.g., digoxin) and uremic toxins [83]. Indoxyl sulfate directly inhibits the function and decreases mRNA and protein expression of OATP4C1 [84].
In summary, microbiota-derived toxins interact with the function and expression of drug transporters in the kidneys and liver. Several microbial toxins inhibit the function and expression of renal OAT1, OAT3, BCRP, MRP4 and OATP4C1 at concentrations observed in kidney disease. Microbial toxins also interact with expression and function of hepatic OATP1B1, OATP1B3 and OATP2B1. These drug transporter-microbial toxin interactions likely contribute to accumulation of uremic toxins and/or drugs in patients with impaired kidney function.
8. Effects of Microbial Toxins Beyond Drug Metabolism and Transport
Effects of microbial toxins extend beyond interactions with hepatic drug metabolizing enzymes and transporters. The microbiota also impacts drug exposure and toxicity in patients with kidney disease by altering serum protein binding and enterohepatic recirculation (Figure 3). Changes in the disposition of the chemotherapy drug irinotecan illustrates each of these points. For example, irinotecan undergoes biotransformation by carboxylesterase to the active metabolite SN-38, which is a substrate of hepatic OATP1B1 and is detoxified by glucuronidation. CMPF, indoxyl sulfate, hippuric acid and indole-3-acetic acid competitively inhibit OATP-mediated transport of SN-38 in human hepatocytes and HEK 293 cells [69,85]. Decreased hepatic uptake leads to increased systemic exposure to SN-38 and toxicity. In addition, CMPF is capable of competitively binding to plasma proteins (e.g., albumin) and displacing SN-38. This increases the unbound fraction and leads to systemic toxicity. In fact, exposure to the unbound fraction of SN-38 is increased by 2.6-fold in patients with advanced kidney disease [86,87]. Impaired kidney function appears to be a risk factor for irinotecan-induced neutropenia, indicating increased toxicity [88], even though SN-38 is primarily eliminated by the liver.
Figure 3.
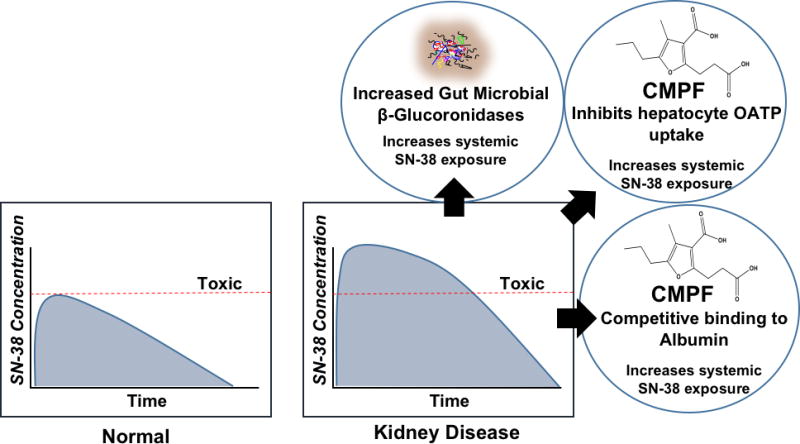
Increased presence of microbial β-glucuronidases leads to de-conjugation and subsequent enterohepatic recirculation of SN-38. In addition, CMPF may perpetrate interactions with hepatic OATP uptake of SN-38 leading to decreased clearance, and decreases SN-38 plasma protein binding leading to increased exposure to the active moiety.
The increased unbound fraction and decreased OATP-mediated uptake of SN-38 into hepatocytes are further complicated by potential increases in the abundance of intestinal β-glucuronidase producing bacteria Enterobacteriaceae [12,89]. SN-38 undergoes hepatic glucuronidation and biliary excretion into the intestines [90], but increased β-glucuronidase activity by the microbiota can lead to increased intestinal mucosal toxicity from deconjugated SN-38. Moreover, the deconjugated SN-38 may undergo enterohepatic recirculation, thereby increasing SN-38 systemic exposure and toxicity [87]. Collectively, these microbial toxin interactions in kidney disease lead to increased systemic exposure of a drug that undergoes predominantly nonrenal clearance. Microbial toxin interactions with drug metabolizing enzymes, transporters, plasma proteins or microbial de-conjugating activities can significantly influence drug exposure and response. Such interactions can occur independently or in concert to alter normal pharmacokinetics and pharmacodynamics of drugs in patients with kidney disease.
9. Therapeutic Strategies to Target Microbial Toxins
Several therapeutic strategies specifically targeting microbial toxins are possible and deserve exploration. These include antibiotics, pre- and probiotics, adsorbents, drug metabolism or transporter modulators, and enhanced dialytic elimination.
9.1. Targeted Antibiotics
Selective use of targeted antibiotics might beneficially alter intestinal microbiota communities [91–93]. Lethal or non-lethal antibiotic approaches can decrease microbial production of specific metabolites. For example, a non-lethal approach is targeting the TMA-lyase enzymes with a small molecule inhibitor that results in decreased microbial production of trimethylamine [94]. Trimethylamine is a gut-derived uremic toxin that is oxygenated into trimethylamine-N-oxide (TMAO). TMAO is associated with cardiovascular disease by potentially promoting atherosclerosis formation [6]. Therefore, this non-lethal approach may potentially improve cardiovascular outcomes in kidney disease patients by decreasing systemic TMAO concentrations. Genetic alteration of bacterial tryptophanase, which represents another non-lethal approach, may result in lower overall formation of indoxyl sulfate [95]. On the other hand, rifaximin is a lethal antibiotic that provides at least an acute decrease of TMAO concentrations in patients [96]. Both lethal and non-lethal antibiotics are appealing options that might reduce microbiota-derived uremic toxins.
9.2. Pre- & Probiotics
Prebiotics (i.e., non-digestible fiber) and probiotics (i.e., beneficial live bacteria) used alone or in combination (i.e., synbiotics) may decrease microbial toxin concentrations. A combination of pre- and probiotics has been evaluated for use in decreasing systemic exposure to p-cresol sulfate and indoxyl sulfate in kidney disease patients [97]. After controlling for oral antibiotics, significant decreases in p-cresol sulfate and indoxyl sulfate were observed. However in a separate study, prebiotic indigestible starches did not generate sustained decreases of microbiota-derived uremic toxins [98]. Lastly, fecal transplants from healthy individuals may be performed to re-populate the diseased microbiome (i.e., dysbiosis) with healthy microorganisms [99].
9.3. Adsorbents
AST-120 (spherical carbon adsorbent), sevelamer (phosphate binder) and other adsorbents might be therapeutic options to bind microbial toxins in the intestinal lumen and limit absorption [100]. Mixed results have been reported from prospective clinical studies evaluating the effects of AST-120 and sevelamer on systemic microbial toxin concentrations and clinical outcomes [101,102]. Other binding resins are used in kidney disease but further research is required to determine their efficacy in binding microbial toxins. The potential for drug interactions with adsorbents may also be an important consideration.
9.4. Drug Metabolism and Transporter Modulators
Drug metabolism and transport modulators might directly mitigate microbiota-derived uremic toxin effects. For example, hepatic SULT inhibitors (e.g., resveratrol, quercetin and meclofenamate) may protect the kidneys from indoxyl sulfate induced oxidative damage stemming from downregulation OAT1 and OAT3 renal transporter expression [79,80]. In fact, SULT inhibitors decreased hepatic formation of indoxyl sulfate in an animal model of acute kidney injury, thereby lowering oxidative stress and restoring OAT1 and OAT3 expression [79]. Also, statins may counteract microbial toxin induced downregulation of OATP4C1 in the kidney and thereby enhance subsequent secretion of uremic toxins into the urine [83]. Increasing the tubular secretion of microbiota-derived toxins might be an effective strategy to decrease systemic concentrations in kidney disease patients.
9.5. Dialytic Clearance
Increased elimination by various dialytic modalities is an important therapeutic option. Although some microbial toxins are highly protein bound and are poorly cleared with hemodialysis, many other small, water soluble metabolites such as TMAO are highly cleared by dialysis. The dialysis modality, filter type, and treatment time should be evaluated to optimize elimination of these microbial toxin solutes [103,104].
10. Summary
The gut microbiota is of increased importance in kidney disease and dysbiosis is linked to altered nonrenal clearance. Microbiota-derived uremic toxins interact with and lead to changes in the function of important phase I and II drug metabolizing enzymes and transporters, including inhibition of CYP3A4, SULT, OATP, OAT, MRP4, and BCRP activities. Alterations to these pathways can effect the pharmacokinetics and pharmacodynamics of commonly prescribed drug substrates. Dysbiosis potentially leads to an increase in the abundance of organisms expressing β-glucuronidase, which may facilitate deconjugation and enterohepatic recirculation of drugs. Limitations of uremic toxin studies, such as use of p-cresol and not the final p-cresol sulfate metabolite and inconsistent use of clinically relevant toxin concentrations, should be considered in the design and interpretation of related work. The gut microbiota or its metabolites may prove to be therapeutic targets for future research.
11. Expert Commentary
Clinical pharmacologists are uniquely positioned to investigate the functional effects of microbial toxins on pharmacokinetics and pharmacodynamics. Indoxyl sulfate, p-cresol sulfate, hippuric acid, indole-3-acetic acid and CMPF are important microbial toxins that warrant increased attention in clinical and translational research [105]. In addition, future systematic studies of the impact of individual and combined microbial toxins on drug metabolism and transport are needed because large interindividual variability in clinically observed microbial toxin concentrations may explain some differences in drug-related adverse events and efficacy. Important considerations include use of clinically relevant microbial toxin concentrations (Table 3), as well as use of microbial toxin precursors (e.g., p-cresol) and the most accumulated metabolites (e.g., p-cresol sulfate). In our opinion, hepatic-gut-kidney crosstalk will be an area of significant scientific research and discovery. Crosstalk individually impacts both hepatic diseases (e.g.. steatohepatitis [106]) and potentially the progression of kidney disease [107]. Moreover, the implications extend to metabolic diseases, inflammatory diseases, and cardiovascular disease.
Microbial toxin interactions with metabolizing enzymes and transporters may lead to altered drug exposure, response, and drug-related adverse events. High interindividual variability of microbial toxin concentrations may explain some variability in responses to treatment. At this point, clinical monitoring of microbiota-derived uremic toxin concentrations is not available to guide drug dosing. It is possible that current drug dosing recommendations have inherently accounted for interindividual differences in toxin concentrations. Future clinical research is needed to determine effects of microbiota-derived toxins on drug disposition and response to facilitate personalized drug dosing.
The human microbiome presents unique challenges due to complex diversity, constant evolution and metabolically active microorganisms. It currently lacks informed clinical interventions, which could have several important therapeutic benefits. For example, microbiota-targeted interventions may decrease rates of cardiovascular morbidity and mortality observed in kidney disease patients. In addition, lowering microbial toxin concentrations is important because alterations to hepatic drug metabolism and transport pathways may impact therapeutic effectiveness and increase the risk for drug-related adverse events. Although several potential therapeutic targets are under investigation, to date no individual intervention has proved to be a viable therapeutic option for sustained decreases in microbial toxin production and/or systemic exposure and it remains an important research opportunity. In summary, microbiota-derived uremic toxins influence drug metabolism and transport and may be therapeutic targets in patients with kidney disease.
12. Five Year View
Extensive opportunities exist in research surrounding the effects of the microbiota on drug exposure and response. The importance of the intestinal microbiota extends from intestinal microbial metabolism to hepatic metabolism and transport, corresponding systemic drug exposure, and finally overall influence on pharmacodynamics. Human microbiome research is quickly progressing through discovery phases and investigators are poised to test clinical interventions. For instance, there are numerous clinical studies with prebiotics, probiotics, oral adsorbents, and antibiotics. One example of notable scientific interest is a novel non-lethal inhibitor of specific microbial enzymes [94]. In the next five years, there will be an exponential growth of human microbiome research in clinical pharmacology.
The intestinal microbiome itself is not only a therapeutic target, but also a potential source of novel therapeutic compounds to improve human health. Intestinal microorganisms generate unique compounds to communicate amongst community members and with human systems, these unique compounds might be a source for novel drug discoveries. Future research will also focus on positive intestinal microbiome-host interactions. For example, there will be increases in discussion of the benefits of microbiota-derived short chain fatty acids [108]. The long-term therapeutic objective will be to harness the beneficial attributes of our microbiome to improve human health. This represents a potential shift for clinical pharmacology from strictly bactericidal antibiotic therapies to innovative and novel microbiota-centered therapeutic strategies.
13. Key Issues
There are known alterations to hepatic drug metabolism and transport pathways in patients with kidney disease that place this population at increased risk of drug-related adverse events.
Microbial imbalances, known as dysbiosis, in kidney disease potentially increases the production and the absorption of microbiota-derived uremic toxins.
Microbiota-derived uremic toxins accumulate in advancing stages of kidney disease and directly affect Phase I and II drug metabolizing enzymes and drug transport pathways.
Microbiota-derived uremic toxin research in kidney disease patients presents unique opportunities for clinical pharmacologists. Identifying druggable microbiota-centered targets is a challenge for future research.
Acknowledgments
Funding
AJ Prokopienko is supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number TL1TR001858. TD Nolin is supported in part by the National Institutes of Health, National Institute of General Medical Sciences (R01-GM107122). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We gratefully acknowledge Fanuel Hagos, M.S. and Stefan Lukianov, M.S. for their critical review of the manuscript.
Footnotes
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Contributor Information
Alexander J. Prokopienko, Clinical and Translational Postdoctoral Fellow, Clinical Pharmaceutical Sciences PhD Candidate, University of Pittsburgh School of Pharmacy, Center for Clinical Pharmaceutical Sciences, 335 Sutherland Drive, 222 Salk Hall Pavilion
Thomas D. Nolin, Associate Professor of Pharmacy and Therapeutics, University of Pittsburgh School of Pharmacy, Center for Clinical Pharmaceutical Sciences, 335 Sutherland Drive, 208 Salk Hall Pavilion, Phone: 412-624-4683, nolin@pitt.edu
References
- 1.Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292(5519):1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 2.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 3.Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016;14(8):e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu IW, Hsu KH, Lee CC, et al. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol. Dial. Transplant. 2011;26(3):938–947. doi: 10.1093/ndt/gfq580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stubbs JR, House JA, Ocque AJ, et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016;27(1):305–313. doi: 10.1681/ASN.2014111063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moradi H, Sica DA, Kalantar-Zadeh K. Cardiovascular burden associated with uremic toxins in patients with chronic kidney disease. Am. J. Nephrol. 2013;38(2):136–148. doi: 10.1159/000351758. [DOI] [PubMed] [Google Scholar]
- 8.Johnson CH, Spilker ME, Goetz L, Peterson SN, Siuzdak G. Metabolite and Microbiome Interplay in Cancer Immunotherapy. Cancer Res. 2016;76(21):6146–6152. doi: 10.1158/0008-5472.CAN-16-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012;13(10):701–712. doi: 10.1038/nrn3346. [DOI] [PubMed] [Google Scholar]
- 10.Marchesi JR, Adams DH, Fava F, et al. The gut microbiota and host health: a new clinical frontier. Gut. 2016;65(2):330–339. doi: 10.1136/gutjnl-2015-309990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaziri ND. CKD impairs barrier function and alters microbial flora of the intestine: a major link to inflammation and uremic toxicity. Curr. Opin. Nephrol. Hypertens. 2012;21(6):587–592. doi: 10.1097/MNH.0b013e328358c8d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaziri ND, Wong J, Pahl M, et al. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013;83(2):308–315. doi: 10.1038/ki.2012.345. [DOI] [PubMed] [Google Scholar]
- 13.Ramezani A, Massy ZA, Meijers B, Evenepoel P, Vanholder R, Raj DS. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am. J. Kidney Dis. 2016;67(3):483–498. doi: 10.1053/j.ajkd.2015.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nolin TD. Altered nonrenal drug clearance in ESRD. Curr. Opin. Nephrol. Hypertens. 2008;17(6):555–559. doi: 10.1097/MNH.0b013e3283136732. [DOI] [PubMed] [Google Scholar]
- 15.Nolin TD, Frye RF, Le P, et al. ESRD impairs nonrenal clearance of fexofenadine but not midazolam. J. Am. Soc. Nephrol. 2009;20(10):2269–2276. doi: 10.1681/ASN.2009010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomson BK, Nolin TD, Velenosi TJ, et al. Effect of CKD and dialysis modality on exposure to drugs cleared by nonrenal mechanisms. Am. J. Kidney Dis. 2015;65(4):574–582. doi: 10.1053/j.ajkd.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Joy MS, Frye RF, Nolin TD, et al. In vivo alterations in drug metabolism and transport pathways in patients with chronic kidney diseases. Pharmacotherapy. 2014;34(2):114–122. doi: 10.1002/phar.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nolin TD, Naud J, Leblond FA, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin. Pharmacol. Ther. 2008;83(6):898–903. doi: 10.1038/clpt.2008.59. [DOI] [PubMed] [Google Scholar]
- 19.Nolin TD, Frye RF, Matzke GR. Hepatic drug metabolism and transport in patients with kidney disease. Am. J. Kidney Dis. 2003;42(5):906–925. doi: 10.1016/j.ajkd.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 20.Yeung CK, Shen DD, Thummel KE, Himmelfarb J. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int. 2014;85(3):522–528. doi: 10.1038/ki.2013.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun H, Frassetto L, Benet LZ. Effects of renal failure on drug transport and metabolism. Pharmacol. Ther. 2006;109(1–2):1–11. doi: 10.1016/j.pharmthera.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 22.Leblond F, Guevin C, Demers C, Pellerin I, Gascon-Barre M, Pichette V. Downregulation of hepatic cytochrome P450 in chronic renal failure. J. Am. Soc. Nephrol. 2001;12(2):326–332. doi: 10.1681/ASN.V122326. [DOI] [PubMed] [Google Scholar]
- 23.Dani M, Boisvert C, Michaud J, et al. Down-regulation of liver drug-metabolizing enzymes in a murine model of chronic renal failure. Drug Metab. Dispos. 2010;38(3):357–360. doi: 10.1124/dmd.109.029991. [DOI] [PubMed] [Google Scholar]
- 24.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plichta DR, Juncker AS, Bertalan M, et al. Transcriptional interactions suggest niche segregation among microorganisms in the human gut. Nat Microbiol. 2016;1(11):16152. doi: 10.1038/nmicrobiol.2016.152. [DOI] [PubMed] [Google Scholar]
- 27.Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 2016;14(5):273–287. doi: 10.1038/nrmicro.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin CJ, Liou TC, Pan CF, et al. The Role of Liver in Determining Serum Colon-Derived Uremic Solutes. PLoS One. 2015;10(8):e0134590. doi: 10.1371/journal.pone.0134590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muting D, Perisoara A, Baum G, Flasshoff HJ, Bucsis L. The role of protein metabolism in 204 liver cirrhotics with, without hepatic encephalopathy II. Amino acids, free phenols and indoles. Hepatogastroenterology. 1986;33(2):66–70. [PubMed] [Google Scholar]
- 30.Muting D, Kalk JF, Fischer R, Wuzel H, Reikowski J. Hepatic detoxification and hepatic function in chronic active hepatitis with and without cirrhosis. Dig. Dis. Sci. 1988;33(1):41–46. doi: 10.1007/BF01536629. [DOI] [PubMed] [Google Scholar]
- 31.Claus SP, Ellero SL, Berger B, et al. Colonization-induced host-gut microbial metabolic interaction. MBio. 2011;2(2):e00271–00210. doi: 10.1128/mBio.00271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meinl W, Sczesny S, Brigelius-Flohe R, Blaut M, Glatt H. Impact of gut microbiota on intestinal, hepatic levels of phase 2 xenobiotic-metabolizing enzymes in the rat. Drug Metab. Dispos. 2009;37(6):1179–1186. doi: 10.1124/dmd.108.025916. [DOI] [PubMed] [Google Scholar]
- 33.Bjorkholm B, Bok CM, Lundin A, Rafter J, Hibberd ML, Pettersson S. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS One. 2009;4(9):e6958. doi: 10.1371/journal.pone.0006958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toda T, Saito N, Ikarashi N, et al. Intestinal flora induces the expression of Cyp3a in the mouse liver. Xenobiotica. 2009;39(4):323–334. doi: 10.1080/00498250802651984. [DOI] [PubMed] [Google Scholar]
- 35.Selwyn FP, Cui JY, Klaassen CD. RNA-Seq Quantification of Hepatic Drug Processing Genes in Germ-Free Mice. Drug Metab. Dispos. 2015;43(10):1572–1580. doi: 10.1124/dmd.115.063545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Selwyn FP, Cheng SL, Klaassen CD, Cui JY. Regulation of Hepatic Drug-Metabolizing Enzymes in Germ-Free Mice by Conventionalization and Probiotics. Drug Metab. Dispos. 2016;44(2):262–274. doi: 10.1124/dmd.115.067504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montagner A, Korecka A, Polizzi A, et al. Hepatic circadian clock oscillators and nuclear receptors integrate microbiome-derived signals. Sci. Rep. 2016;6:20127. doi: 10.1038/srep20127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch. Intern. Med. 1999;159(22):2647–2658. doi: 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- 39.Vanholder R, Glorieux G, Massy ZA. Intestinal metabolites, chronic kidney disease and renal transplantation: Enigma Variations? Nephrol. Dial. Transplant. 2016;31(10):1547–1551. doi: 10.1093/ndt/gfw040. [DOI] [PubMed] [Google Scholar]
- 40.Wong J, Piceno YM, Desantis TZ, Pahl M, Andersen GL, Vaziri ND. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014;39(3):230–237. doi: 10.1159/000360010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kikuchi M, Ueno M, Itoh Y, Suda W, Hattori M. Uremic Toxin-Producing Gut Microbiota in Rats with Chronic Kidney Disease. Nephron. 2017;135(1):51–60. doi: 10.1159/000450619. [DOI] [PubMed] [Google Scholar]
- 42.Lau WL, Kalantar-Zadeh K, Vaziri ND. The Gut as a Source of Inflammation in Chronic Kidney Disease. Nephron. 2015;130(2):92–98. doi: 10.1159/000381990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersen K, Kesper MS, Marschner JA, et al. Intestinal Dysbiosis, Barrier Dysfunction, and Bacterial Translocation Account for CKD-Related Systemic Inflammation. J. Am. Soc. Nephrol. 2017;28(1):76–83. doi: 10.1681/ASN.2015111285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duranton F, Cohen G, De Smet R, et al. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012;23(7):1258–1270. doi: 10.1681/ASN.2011121175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sumida K, Molnar MZ, Potukuchi PK, et al. Constipation and Incident CKD. J. Am. Soc. Nephrol. 2017;28(4):1248–1258. doi: 10.1681/ASN.2016060656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barnes KJ, Rowland A, Polasek TM, Miners JO. Inhibition of human drug-metabolising cytochrome P450 and UDP-glucuronosyltransferase enzyme activities in vitro by uremic toxins. Eur. J. Clin. Pharmacol. 2014;70(9):1097–1106. doi: 10.1007/s00228-014-1709-7. [DOI] [PubMed] [Google Scholar]
- 47.Volpe DA, Tobin GA, Tavakkoli F, Dowling TC, Light PD, Parker RJ. Effect of uremic serum and uremic toxins on drug metabolism in human microsomes. Regul. Toxicol. Pharmacol. 2014;68(2):297–303. doi: 10.1016/j.yrtph.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 48.Vanholder R, De Smet R, Lameire N. Protein-bound uremic solutes: the forgotten toxins. Kidney Int. Suppl. 2001;78:S266–270. doi: 10.1046/j.1523-1755.2001.59780266.x. [DOI] [PubMed] [Google Scholar]
- 49.Velenosi TJ, Hennop A, Feere DA, et al. Untargeted plasma and tissue metabolomics in rats with chronic kidney disease given AST-120. Sci. Rep. 2016;6:22526. doi: 10.1038/srep22526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vanholder R, Bammens B, de Loor H, et al. Warning: the unfortunate end of p-cresol as a uraemic toxin. Nephrol. Dial. Transplant. 2011;26(5):1464–1467. doi: 10.1093/ndt/gfr056. [DOI] [PubMed] [Google Scholar]
- 51.Wilkinson GR. Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 2005;352(21):2211–2221. doi: 10.1056/NEJMra032424. [DOI] [PubMed] [Google Scholar]
- 52.Manley HJ, Garvin CG, Drayer DK, et al. Medication prescribing patterns in ambulatory haemodialysis patients: comparisons of USRDS to a large not-for-profit dialysis provider. Nephrol. Dial. Transplant. 2004;19(7):1842–1848. doi: 10.1093/ndt/gfh280. [DOI] [PubMed] [Google Scholar]
- 53.Tsujimoto M, Nagano Y, Hosoda S, et al. Effects of decreased vitamin D and accumulated uremic toxin on human CYP3A4 activity in patients with end-stage renal disease. Toxins (Basel) 2013;5(8):1475–1485. doi: 10.3390/toxins5081475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshitani T, Yagi H, Inotsume N, Yasuhara M. Effect of experimental renal failure on the pharmacokinetics of losartan in rats. Biol. Pharm. Bull. 2002;25(8):1077–1083. doi: 10.1248/bpb.25.1077. [DOI] [PubMed] [Google Scholar]
- 55.Hanada K, Ogawa R, Son K, et al. Effects of indoxylsulfate on the in vitro hepatic metabolism of various compounds using human liver microsomes and hepatocytes. Nephron Physiol. 2006;103(4):p179–186. doi: 10.1159/000092919. [DOI] [PubMed] [Google Scholar]
- 56.Sun H, Huang Y, Frassetto L, Benet LZ. Effects of uremic toxins on hepatic uptake and metabolism of erythromycin. Drug Metab. Dispos. 2004;32(11):1239–1246. doi: 10.1124/dmd.104.000521. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki Y, Itoh H, Fujioka T, et al. Association of plasma concentration of 4beta-hydroxycholesterol with CYP3A5 polymorphism and plasma concentration of indoxyl sulfate in stable kidney transplant recipients. Drug Metab. Dispos. 2014;42(1):105–110. doi: 10.1124/dmd.113.054171. [DOI] [PubMed] [Google Scholar]
- 58.Liu H, Narayanan R, Hoffmann M, Surapaneni S. The Uremic Toxin Indoxyl-3-Sulfate Induces CYP1A2 In Primary Human Hepatocytes. Drug Metab Lett. 2016;10(3):195–199. doi: 10.2174/1872312810666160719143703. [DOI] [PubMed] [Google Scholar]
- 59.Schroeder JC, Dinatale BC, Murray IA, et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry. 2010;49(2):393–400. doi: 10.1021/bi901786x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsujimoto M, Sugimoto S, Nagatomo M, et al. Possibility of decrease in CYP1A2 function in patients with end-stage renal disease. Ther. Apher. Dial. 2014;18(2):174–180. doi: 10.1111/1744-9987.12100. [DOI] [PubMed] [Google Scholar]
- 61.Tsujimoto M, Higuchi K, Shima D, et al. Inhibitory effects of uraemic toxins 3-indoxyl sulfate and p-cresol on losartan metabolism in vitro. J. Pharm. Pharmacol. 2010;62(1):133–138. doi: 10.1211/jpp.62.01.0015. [DOI] [PubMed] [Google Scholar]
- 62.Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc. Natl. Acad. Sci. U. S. A. 2009;106(34):14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Banoglu E, King RS. Sulfation of indoxyl by human and rat aryl (phenol) sulfotransferases to form indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 2002;27(2):135–140. doi: 10.1007/BF03190428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poesen R, Evenepoel P, de Loor H, Kuypers D, Augustijns P, Meijers B. Metabolism, Protein Binding, and Renal Clearance of Microbiota-Derived p-Cresol in Patients with CKD. Clin. J. Am. Soc. Nephrol. 2016;11(7):1136–1144. doi: 10.2215/CJN.00160116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mabuchi H, Nakahashi H. Inhibition of hepatic glutathione S-transferases by a major endogenous ligand substance present in uremic serum. Nephron. 1988;49(4):281–283. doi: 10.1159/000185076. [DOI] [PubMed] [Google Scholar]
- 66.Mutsaers HA, Wilmer MJ, Reijnders D, et al. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim. Biophys. Acta. 2013;1832(1):142–150. doi: 10.1016/j.bbadis.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 67.Tsujimoto M, Hatozaki D, Shima D, et al. Influence of serum in hemodialysis patients on the expression of intestinal and hepatic transporters for the excretion of pravastatin. Ther. Apher. Dial. 2012;16(6):580–587. doi: 10.1111/j.1744-9987.2012.01100.x. [DOI] [PubMed] [Google Scholar]
- 68.Sato T, Yamaguchi H, Kogawa T, Abe T, Mano N. Organic anion transporting polypeptides 1B1 and 1B3 play an important role in uremic toxin handling and drug-uremic toxin interactions in the liver. J. Pharm. Pharm. Sci. 2014;17(4):475–484. doi: 10.18433/j3m89q. [DOI] [PubMed] [Google Scholar]
- 69.Fujita K, Sugiura T, Okumura H, et al. Direct inhibition and down-regulation by uremic plasma components of hepatic uptake transporter for SN-38, an active metabolite of irinotecan, in humans. Pharm. Res. 2014;31(1):204–215. doi: 10.1007/s11095-013-1153-x. [DOI] [PubMed] [Google Scholar]
- 70.Tsujimoto M, Kinoshita Y, Hirata S, Otagiri M, Ohtani H, Sawada Y. Effects of uremic serum and uremic toxins on hepatic uptake of digoxin. Ther. Drug Monit. 2008;30(5):576–582. doi: 10.1097/FTD.0b013e3181838077. [DOI] [PubMed] [Google Scholar]
- 71.Reyes M, Benet LZ. Effects of uremic toxins on transport and metabolism of different biopharmaceutics drug disposition classification system xenobiotics. J. Pharm. Sci. 2011;100(9):3831–3842. doi: 10.1002/jps.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nolin TD, Appiah K, Kendrick SA, Le P, McMonagle E, Himmelfarb J. Hemodialysis acutely improves hepatic CYP3A4 metabolic activity. J. Am. Soc. Nephrol. 2006;17(9):2363–2367. doi: 10.1681/ASN.2006060610. [DOI] [PubMed] [Google Scholar]
- 73.Deguchi T, Kusuhara H, Takadate A, Endou H, Otagiri M, Sugiyama Y. Characterization of uremic toxin transport by organic anion transporters in the kidney. Kidney Int. 2004;65(1):162–174. doi: 10.1111/j.1523-1755.2004.00354.x. [DOI] [PubMed] [Google Scholar]
- 74.Watanabe H, Sakaguchi Y, Sugimoto R, et al. Human organic anion transporters function as a high-capacity transporter for p-cresyl sulfate, a uremic toxin. Clin. Exp. Nephrol. 2014;18(5):814–820. doi: 10.1007/s10157-013-0902-9. [DOI] [PubMed] [Google Scholar]
- 75.Deguchi T, Takemoto M, Uehara N, Lindup WE, Suenaga A, Otagiri M. Renal clearance of endogenous hippurate correlates with expression levels of renal organic anion transporters in uremic rats. J. Pharmacol. Exp. Ther. 2005;314(2):932–938. doi: 10.1124/jpet.105.085613. [DOI] [PubMed] [Google Scholar]
- 76.Fujita T, Ishihara K, Yasuda S, et al. In vivo kinetics of indoxyl sulfate in humans and its renal interaction with angiotensin-converting enzyme inhibitor quinapril in rats. J. Pharmacol. Exp. Ther. 2012;341(3):626–633. doi: 10.1124/jpet.111.187732. [DOI] [PubMed] [Google Scholar]
- 77.Yu CP, Sweet DH, Peng YH, et al. Effects of nonsteroidal anti-inflammatory drugs on the renal excretion of indoxyl sulfate, a nephro-cardiovascular toxin, in rats. Eur. J. Pharm. Sci. 2017;101:66–70. doi: 10.1016/j.ejps.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 78.Deguchi T, Ohtsuki S, Otagiri M, et al. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney Int. 2002;61(5):1760–1768. doi: 10.1046/j.1523-1755.2002.00318.x. [DOI] [PubMed] [Google Scholar]
- 79.Saigo C, Nomura Y, Yamamoto Y, et al. Meclofenamate elicits a nephropreventing effect in a rat model of ischemic acute kidney injury by suppressing indoxyl sulfate production and restoring renal organic anion transporters. Drug Des. Devel. Ther. 2014;8:1073–1082. doi: 10.2147/DDDT.S67456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Saito H, Yoshimura M, Saigo C, et al. Hepatic sulfotransferase as a nephropreventing target by suppression of the uremic toxin indoxyl sulfate accumulation in ischemic acute kidney injury. Toxicol. Sci. 2014;141(1):206–217. doi: 10.1093/toxsci/kfu119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kong F, Pang X, Zhong K, et al. Increased Plasma Exposures of Conjugated Metabolites of Morinidazole in Renal Failure Patients: A Critical Role of Uremic Toxins. Drug Metab. Dispos. 2017;45(6):593–603. doi: 10.1124/dmd.116.074492. [DOI] [PubMed] [Google Scholar]
- 82.Mutsaers HA, van den Heuvel LP, Ringens LH, et al. Uremic toxins inhibit transport by breast cancer resistance protein and multidrug resistance protein 4 at clinically relevant concentrations. PLoS One. 2011;6(4):e18438. doi: 10.1371/journal.pone.0018438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Toyohara T, Suzuki T, Morimoto R, et al. SLCO4C1 transporter eliminates uremic toxins and attenuates hypertension and renal inflammation. J. Am. Soc. Nephrol. 2009;20(12):2546–2555. doi: 10.1681/ASN.2009070696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Akiyama Y, Kikuchi K, Saigusa D, et al. Indoxyl sulfate down-regulates SLCO4C1 transporter through up-regulation of GATA3. PLoS One. 2013;8(7):e66518. doi: 10.1371/journal.pone.0066518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Katsube Y, Tsujimoto M, Koide H, et al. Cooperative inhibitory effects of uremic toxins and other serum components on OATP1B1-mediated transport of SN-38. Cancer Chemother. Pharmacol. 2017;79(4):783–789. doi: 10.1007/s00280-017-3276-y. [DOI] [PubMed] [Google Scholar]
- 86.Fujita K, Sunakawa Y, Miwa K, et al. Delayed elimination of SN-38 in cancer patients with severe renal failure. Drug Metab. Dispos. 2011;39(2):161–164. doi: 10.1124/dmd.110.035451. [DOI] [PubMed] [Google Scholar]
- 87.Fujita K, Masuo Y, Okumura H, et al. Increased Plasma Concentrations of Unbound SN-38, the Active Metabolite of Irinotecan, in Cancer Patients with Severe Renal Failure. Pharm. Res. 2016;33(2):269–282. doi: 10.1007/s11095-015-1785-0. [DOI] [PubMed] [Google Scholar]
- 88.de Jong FA, van der Bol JM, Mathijssen RH, et al. Renal function as a predictor of irinotecan-induced neutropenia. Clin. Pharmacol. Ther. 2008;84(2):254–262. doi: 10.1038/sj.clpt.6100513. [DOI] [PubMed] [Google Scholar]
- 89.McIntosh FM, Maison N, Holtrop G, et al. Phylogenetic distribution of genes encoding beta-glucuronidase activity in human colonic bacteria and the impact of diet on faecal glycosidase activities. Environ. Microbiol. 2012;14(8):1876–1887. doi: 10.1111/j.1462-2920.2012.02711.x. [DOI] [PubMed] [Google Scholar]
- 90.Wallace BD, Roberts AB, Pollet RM, et al. Structure and Inhibition of Microbiome beta-Glucuronidases Essential to the Alleviation of Cancer Drug Toxicity. Chem. Biol. 2015;22(9):1238–1249. doi: 10.1016/j.chembiol.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 92.Kang DJ, Kakiyama G, Betrapally NS, et al. Rifaximin Exerts Beneficial Effects Independent of its Ability to Alter Microbiota Composition. Clin Transl Gastroenterol. 2016;7(8):e187. doi: 10.1038/ctg.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reijnders D, Goossens GH, Hermes GD, et al. Effects of Gut Microbiota Manipulation by Antibiotics on Host Metabolism in Obese Humans: A Randomized Double-Blind Placebo-Controlled Trial. Cell Metab. 2016;24(1):63–74. doi: 10.1016/j.cmet.2016.06.016. [DOI] [PubMed] [Google Scholar]
- 94.Wang Z, Roberts AB, Buffa JA, et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell. 2015;163(7):1585–1595. doi: 10.1016/j.cell.2015.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Devlin AS, Marcobal A, Dodd D, et al. Modulation of a Circulating Uremic Solute via Rational Genetic Manipulation of the Gut Microbiota. Cell Host Microbe. 2016;20(6):709–715. doi: 10.1016/j.chom.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stremmel W, Schmidt KV, Schuhmann V, et al. Blood Trimethylamine-N-Oxide Originates from Microbiota Mediated Breakdown of Phosphatidylcholine and Absorption from Small Intestine. PLoS One. 2017;12(1):e0170742. doi: 10.1371/journal.pone.0170742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rossi M, Johnson DW, Morrison M, et al. Synbiotics Easing Renal Failure by Improving Gut Microbiology (SYNERGY): A Randomized Trial. Clin. J. Am. Soc. Nephrol. 2016;11(2):223–231. doi: 10.2215/CJN.05240515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Poesen R, Evenepoel P, de Loor H, et al. The Influence of Prebiotic Arabinoxylan Oligosaccharides on Microbiota Derived Uremic Retention Solutes in Patients with Chronic Kidney Disease: A Randomized Controlled Trial. PLoS One. 2016;11(4):e0153893. doi: 10.1371/journal.pone.0153893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Persky SE, Brandt LJ. Treatment of recurrent Clostridium difficile-associated diarrhea by administration of donated stool directly through a colonoscope. Am. J. Gastroenterol. 2000;95(11):3283–3285. doi: 10.1111/j.1572-0241.2000.03302.x. [DOI] [PubMed] [Google Scholar]
- 100.Goto S, Yoshiya K, Kita T, Fujii H, Fukagawa M. Uremic toxins and oral adsorbents. Ther. Apher. Dial. 2011;15(2):132–134. doi: 10.1111/j.1744-9987.2010.00891.x. [DOI] [PubMed] [Google Scholar]
- 101.Brandenburg VM, Schlieper G, Heussen N, et al. Serological cardiovascular and mortality risk predictors in dialysis patients receiving sevelamer: a prospective study. Nephrol. Dial. Transplant. 2010;25(8):2672–2679. doi: 10.1093/ndt/gfq053. [DOI] [PubMed] [Google Scholar]
- 102.Schulman G, Berl T, Beck GJ, et al. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015;26(7):1732–1746. doi: 10.1681/ASN.2014010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vanholder RC, Eloot S, Glorieux GL. Future Avenues to Decrease Uremic Toxin Concentration. Am. J. Kidney Dis. 2016;67(4):664–676. doi: 10.1053/j.ajkd.2015.08.029. [DOI] [PubMed] [Google Scholar]
- 104.Meijers BK, Weber V, Bammens B, et al. Removal of the uremic retention solute p-cresol using fractionated plasma separation and adsorption. Artif. Organs. 2008;32(3):214–219. doi: 10.1111/j.1525-1594.2007.00525.x. [DOI] [PubMed] [Google Scholar]
- 105.Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: a systematic review. J. Am. Soc. Nephrol. 2014;25(9):1897–1907. doi: 10.1681/ASN.2013101062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bluemel S, Williams B, Knight R, Schnabl B. Precision medicine in alcoholic and nonalcoholic fatty liver disease via modulating the gut microbiota. Am. J. Physiol. Gastrointest. Liver Physiol. 2016;311(6):G1018–G1036. doi: 10.1152/ajpgi.00245.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Targher G, Byrne CD. Non-alcoholic fatty liver disease: an emerging driving force in chronic kidney disease. Nat Rev Nephrol. 2017;13(5):297–310. doi: 10.1038/nrneph.2017.16. [DOI] [PubMed] [Google Scholar]
- 108.Goffredo M, Mass K, Parks EJ, et al. Role of Gut Microbiota and Short Chain Fatty Acids in Modulating Energy Harvest and Fat Partitioning in Youth. J. Clin. Endocrinol. Metab. 2016;101(11):4367–4376. doi: 10.1210/jc.2016-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sassa T, Matsuno H, Niwa M, et al. Measurement of furancarboxylic acid, a candidate for uremic toxin, in human serum, hair, and sweat, and analysis of pharmacological actions in vitro. Arch. Toxicol. 2000;73(12):649–654. doi: 10.1007/s002040050020. [DOI] [PubMed] [Google Scholar]