

Abstract
Over the past four decades, the patch clamp technique and nicotinic ACh (nACh) receptors have established an enduring partnership. Like all good partnerships, each partner has proven significant in its own right, while their union has spurred innumerable advances in life science research. A member and prototype of the superfamily of pentameric ligand‐gated ion channels, the nACh receptor is a chemo‐electric transducer, binding ACh released from nerves and rapidly opening its channel to cation flow to elicit cellular excitation. A subject of a Nobel Prize in Physiology or Medicine, the patch clamp technique provides unprecedented resolution of currents through single ion channels in their native cellular environments. Here, focusing on muscle and α7 nACh receptors, we describe the extraordinary contribution of the patch clamp technique towards understanding how they activate in response to neurotransmitter, how subtle structural and mechanistic differences among nACh receptor subtypes translate into significant physiological differences, and how nACh receptors are being exploited as therapeutic drug targets.
Linked Articles
This article is part of a themed section on Nicotinic Acetylcholine Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.11/issuetoc/
Abbreviations
- 4bp‐TQS
4‐(4‐bromophenyl)‐3a,4,5,9b‐tetrahydro‐3H‐cyclopenta[c]quinoline‐8‐sulfonamide
- 5‐HI
5‐hydroxyindole
- NAM
negative allosteric modulator
- NS‐1738
N‐(5‐chloro‐2‐hydroxyphenyl)‐N′‐[2‐chloro‐5‐(trifluoromethyl)phenyl]urea
- PAM
positive allosteric modulator
- PNU‐120596
N‐(5‐chloro‐2,4‐dimethoxyphenyl)‐N′‐(5‐methyl‐3‐isoxazolyl)‐urea
- TMD
transmembrane domain
Tables of Links
| TARGETS |
|---|
| Ligand‐gated ion channels |
| http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=76 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Several years after the patch clamp technique was introduced, it became clear that it was more useful than anyone had expected (Sigworth, 1983, 1986). Not only could currents through single ion channels be registered but also currents could be registered in a wide variety of configurations, including unitary currents from isolated membrane patches and macroscopic currents from the whole cell. The nicotinic acetylcholine (nACh) receptor from skeletal muscle holds a special place in the history of application of the patch clamp, as it was the first biological ion channel from which unitary currents were registered with sub‐millisecond resolution (Colquhoun and Sakmann, 1981). Now, in the fourth decade of patch clamp application, it is equally clear that the technique has become even more useful than was expected in the early years following its introduction. Major advances that complemented the patch clamp include molecular cloning technology, which allows structural modifications anywhere in an ion channel, and heterologous expression systems, which allow study of currents through virtually any ion channel. Here, we survey advances from application of the patch clamp technique to investigate the function and drug modulation of the nACh receptor, a prototypical neurotransmitter‐gated ion channel (Alexander et al., 2015).
Patch clamp technique
The patch clamp technique essentially solved two problems simultaneously: a current capturing problem and a signal to noise problem. A fire‐polished glass pipette filled with electrolyte solution is pressed against a cell, and following gentle suction, a giga‐ohm seal is formed between the glass and the cell membrane (Hamill et al., 1981). The high‐resistance seal directs virtually all current across the membrane into the patch clamp electronics. Moreover, because background noise decreases as seal resistance increases, single channel currents stand out as rectangular pulses superimposed upon relatively small background fluctuations. Although background noise inherent to the patch clamp increases with increasing frequency, with present day technology, it is possible to detect sub‐pico‐ampere single channel currents at bandwidths of several hundred Hz, or currents of tens of pico‐amperes at several thousand kHz. In the field of electrophysiology, such signal to noise capability was unheard of prior to 1980.
The patch clamp technique also spurred advances in mathematical descriptions of single channel dwell times based on discrete state kinetic schemes (Colquhoun and Hawkes, 1981), development of software to digitize and filter the long temporal sequences of current pulses, detection of transitions between closing and opening events (Colquhoun and Sigworth, 1983; Sigworth, 1983), and analysis of dwell time sequences according to kinetic schemes (Horn and Lange, 1983). In order to fit a kinetic scheme to the data, the dwell time omission problem had to be solved to account for the system dead time resulting from a finite recording bandwidth. This was done with both exact and approximate mathematical solutions (Roux and Sauve, 1985; Crouzy and Sigworth, 1990; Hawkes et al., 1990; Qin et al., 1996), or with Markov simulation followed by direct imposition of the dead time (Blatz and Magleby, 1986). Given present day technology, it is practical to fit a kinetic scheme with multiple closed and open states to single channel dwell time data, and estimate transition rate constants as rapid as 100 000 s−1. In the case of the muscle nACh receptor, rate constants have been estimated for agonist association and dissociation, transition between successive closed states, closed and open states, and block and unblock of the channel (Mukhtasimova et al., 2016). The ability to estimate rate constants, combined with changes in nACh receptor structure, will continue to offer a means to understand relationships between structure and function.
nACh receptor structure
Current understanding of nACh receptor structure was achieved through application of a range of technologies. The first was in biochemistry where tissues naturally rich in nACh receptors from the motor endplate were subjected to affinity purification, and the constituent subunits resolved on polyacrylamide gels (Meunier et al., 1974). The four subunits, α, β, γ and δ, were shown to form a pentameric assembly, with the α‐subunit present in two copies (Reynolds and Karlin, 1978; Lindstrom et al., 1979). The second was in amino acid sequencing technology, which yielded the amino terminal partial sequences of isolated subunits (Raftery et al., 1980). The third was molecular cloning where, given the amino terminal sequences, polynucleotide probes were generated to screen a cDNA library, and each subunit was ultimately cloned and sequenced (Noda et al., 1983). Analyses of the sequences established a receptor gene family and yielded models of how the subunit chains thread through the cell membrane. Genetic reconstitution of nACh receptor subunits in heterologous expression systems, combined with assessment of receptor function, gave ultimate proof of the composition of the nACh receptor (Methfessel et al., 1986; Claudio et al., 1987).
Near atomic resolution structural insight was achieved by electron microscopy applied to the Torpedo nACh receptor, initially yielding structures from 20 to 9 Å resolution (Toyoshima and Unwin, 1988; Mitra et al., 1989; Unwin, 1993), and culminating in a 4 Å structure (Unwin, 2005). The first atomic resolution insight was achieved by the crystal structure of the ACh binding protein (AChBP), a water‐soluble homo‐pentamer that mirrored the nACh receptor extracellular region with some 24% sequence identity to the extracellular region of the homomeric α7 nACh receptor (Brejc et al., 2001). The structure confirmed previous mutagenesis and site‐directed labelling studies, showing that the binding site is composed of multiple loops, each from separate sections of the primary sequence, which converge at the subunit interface. Over the subsequent decade, x‐ray crystallography yielded structures of bacterial homologues of the nACh receptor (Hilf and Dutzler, 2008, 2009; Bocquet et al., 2009), then structures of homomeric eukaryotic relatives of the nACh receptor (Hibbs and Gouaux, 2011; Hassaine et al., 2014; Miller and Aricescu, 2014; Du et al., 2015; Haung et al., 2015), and most recently, a hetero‐pentameric neuronal nACh receptor (Morales‐Perez et al., 2016). The collective crystal structures depict, in vivid atomic detail, the extracellular and transmembrane domains (TMD), spatial relationships between the domains and between subunits, and residue‐residue contacts within and between subunits (Figure 1).
Figure 1.
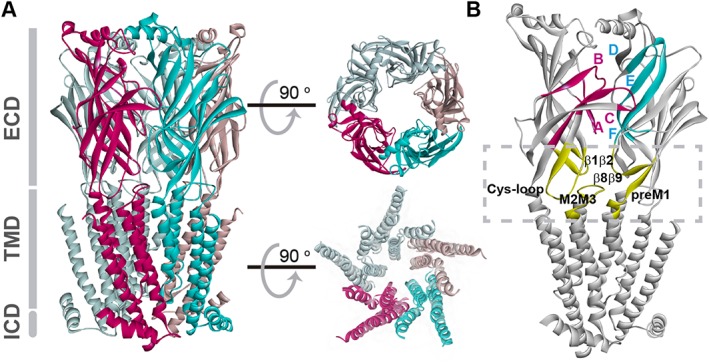
Structural model of nACh receptor. The structure corresponds to the α4β2 plus nicotine (PDB 5KXI) (Morales‐Perez et al., 2016). (A) View parallel to the plasma membrane with colouring to highlight α (purple) and β subunits (cyan). Functional domains include the extracellular domain (ECD), which carries the binding sites at subunit interfaces; the TMD, which contains the ion pore and the gate and is formed by four α‐helices from each monomer (M1–M4); and the intracellular domain (ICD) between M3 and M4 domains, that contains determinants of channel conductance and sites for regulation. Most of the ICD is not shown since it was removed to obtain well‐diffracting crystals (Morales‐Perez et al., 2016). Views of the ECD and the TMD perpendicular to the membrane are shown on the right, upper and lower parts respectively. The TMD contains intra‐subunit and inter‐subunit transmembrane cavities involved in allosteric modulation. (B) Adjacent subunits showing the loops contributing to the binding site and the coupling region (squared). Each nACh receptor ECD monomer consists of an N‐terminal α‐helix and a core of ten β‐strands that form a β‐sandwich structure. Each agonist‐binding site is found at an interface between two adjacent subunits. The principal face, provided by the α subunit, contributes three loops that span β strands and harbour predominantly key aromatic residues, named as Loop A (β4β5 loop), Loop B (β7β8 loop) and Loop C (β9β10 loop). The adjacent subunit, which forms the complementary face, contributes three β strands with residues clustered in segments called Loops D–F. The interface between the ECD and TMD, named as the coupling region, is important for coupling agonist binding to channel opening as well as for determining open channel lifetime and rate of desensitization (Bouzat et al., 2004; Lee and Sine, 2005; Bouzat et al., 2008; Bartos et al., 2009; Yan et al., 2015). The main loops are shown in yellow.
Activation of muscle nACh receptor
Development of kinetic schemes describing nACh receptor activation has been closely linked to advances in pharmacology. More than 50 years ago, del Castillo and Katz (1957) applied intracellular microelectrode recording to the frog motor endplate and found that full and partial agonists produced different sized depolarizing responses when applied at saturating concentrations. They reasoned that binding of the agonist (A) to the receptor (R) initially produced an inactive complex (AR), which then isomerized to an active complex (AO):
Full and partial agonists thus differed in the efficiency with which the inactive agonist‐receptor complex isomerized to the active complex.
Over the succeeding years, to reconcile positive cooperativity in the dose–response relationship (Dionne et al., 1978), a second agonist binding step was added (A2R), and to account for brief channel openings at low but not high agonist concentrations (Jackson, 1988), opening by singly occupied receptors was added. The observation that channels could open even in the absence of agonist (Jackson, 1986) completed a framework that mirrored the MWC model for an allosteric protein with two ligand binding sites (Monod et al., 1965):
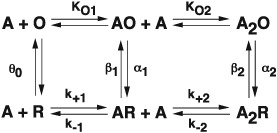
The subset of the MWC mechanism, shown with rate constants rather than equilibrium constants, has been fitted to sequences of single channel dwell times recorded over a range of agonist concentrations, yielding estimates for all the microscopic rate constants (Sine et al., 1990; Ohno et al., 1996). The agonist binding steps depended on the number of bound agonists, differing mainly in the rate constants for dissociation, which could be rationalized because the complementary subunits at the two binding sites differed (γ vs. δ). Moreover, measurements of opening and closing rate constants showed that a full agonist gated the channel efficiently, whereas a partial agonist gated inefficiently (Colquhoun and Sakmann, 1985; Marshall et al., 1991; Grosman et al., 2000). The origin of efficacy thus appeared to arise from a variable rate of channel opening with a nearly invariant rate of channel closing.
Once again, however, advances in pharmacology led to deeper insight into the activation mechanism of nACh receptors. Focusing on very brief single channel dwell times, it was realized that for low concentrations of several agonists, openings from a single nACh receptor channel were interrupted by closings belonging to two rather than one kinetic class, at variance with the MWC mechanism (Lape et al., 2008; Mukhtasimova et al., 2009; reviewed in Sine, 2012). The class with briefest mean duration was common to openings regardless of agonist efficacy, whereas the succeeding class was variable both in its mean duration and relative weight. Thus, schemes with a closed state intermediate between the resting (R) and open channel (O) states emerged, with the transition between the resting and intermediate states, called flipped or primed (P), depending on the efficacy of the agonist:
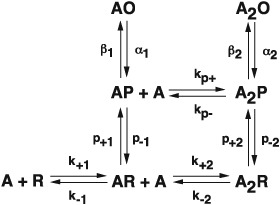
By implementing procedures that improved temporal resolution of dwell times, a recent study obtained estimates of all the rate constants in this scheme (Mukhtasimova et al., 2016; see commentary by Sivilotti and Colquhoun, 2016). The estimates were determined with sufficient accuracy to assess relationships between rate and equilibrium constants, and how they depended on the number of bound agonists. The rate and equilibrium constants for the priming steps increased with successive agonist occupancy, and priming was more efficient for a full than a partial agonist. The priming rate relative to the priming equilibrium constant increased with successive agonist occupancy, and for a full agonist, the change in log rate constant relative to the change in log equilibrium constant was greater than one, suggesting agonist occupancy affected not only the stability of the ground states but also those of the transition states. Relationships between rate and equilibrium constants and how they are expected to depend on the extent of agonist occupancy are described by Fersht et al. (1986) and Edelstein et al. (1996) respectively. By contrast, for a partial agonist, the change in log rate constant relative to the change in log equilibrium constant approached unity, suggesting agonist occupancy solely affected the ground states. Thus, priming depends on both the degree of agonist occupancy and efficacy of the agonist. The rate and equilibrium constants for the gating step also increased with successive agonist occupancy, but unlike priming, the gating step was similar for a full and a partial agonist. The gating rate constant relative to the gating equilibrium constant increased with successive occupancy, but the change in log rate constant relative to the change in log equilibrium constant was less than one, suggesting agonist occupancy solely affected the stability of the ground states. Thus, whereas channel gating depends on the extent of agonist occupancy, it does not depend on agonist efficacy. Overall, single‐channel kinetic analysis has added in‐depth insight into the concept of activation and partial agonism in the Cys‐loop receptor family (Burzomato et al., 2004; Lape et al., 2008; Mukthasimova et al., 2009; Corradi and Bouzat, 2014; Indurthi et al., 2016).
Although stable intermediates between closed and open states are not present in the MWC mechanism, the flip and primed mechanisms retains a key tenet of the MWC mechanism. In particular, the affinity of agonist for intermediate and open states is greater than affinity for the resting closed state, so that tighter binding of agonist drives entry to those states and thus promotes activation (Sine, 2012).
Before multiple closed states in advance of the open state were described, a comprehensive body of work showed that mutations of residues in the ligand binding domain affected a step that appeared to be channel opening, whereas mutations in the pore affected a step that appeared to be channel gating (Grosman et al., 2000). In light of pre‐open closed states, the correlation between structure and kinetics could be reconciled by a change in a residue's contribution to the priming step versus the gating step. Thus, residues in the binding domain may contribute mainly to priming, whereas those in the pore domain may contribute mainly to gating. In recent work by Auerbach and colleagues, a reaction mechanism with closed state intermediates was explicitly considered (Gupta et al., 2017).
Activation of homomeric α7 nACh receptors
In deciphering mechanisms of nACh receptor activation, an essential requirement is to identify sequences of channel openings and closings from the same receptor channel. In this way, desensitization has been pivotal in defining the activation mechanism of the muscle nACh receptor. When a patch of membrane containing muscle nACh receptors is exposed to high agonist concentrations, most of the time all receptors are desensitized and the current remains at baseline. On occasion, however, one receptor recovers from desensitization and then opens and closes repeatedly until it returns to the desensitized state (Sakmann et al., 1980) (Figure 2). For the muscle nACh receptor, the time for desensitization onset is long compared to the time it takes to close and reopen, and trains of many openings and closings from the same receptor channel can be unambiguously identified; data from such trains enable global fitting just described for the muscle nACh receptor. By contrast, for the neuronal α7 receptor, the time for desensitization onset approaches the open channel lifetime, so that desensitization determines the rate of channel closing as opposed to reversal of the channel opening step (Bouzat et al., 2008) (Figure 2). As a result, trains of events from the same receptor channel are not observed, sequences of openings and closings show little dependence on the agonist concentration, and rate constants for kinetic steps that precede channel opening, such as agonist association and dissociation, cannot be estimated. Thus, whereas desensitization is a blessing for studying the kinetics of muscle nACh receptor activation, it is an obstacle to studying the kinetics of α7 nACh receptor activation.
Figure 2.
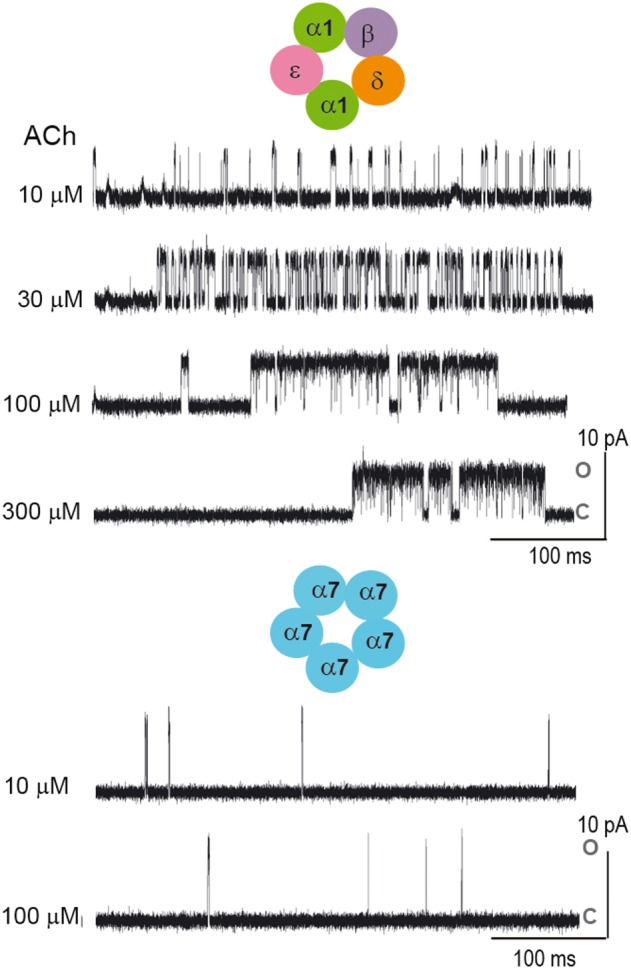
Single‐channel recordings as a function of ACh concentration for muscle and α7 nACh receptors. For the muscle nACh receptor, single‐channel activity appears as bursts separated from each other by relatively long silent periods in which the receptor is desensitized. Bursts are composed of successive opening events corresponding to the same individual channel. The main closed time within a burst reflects the transitions between unliganded closed and diliganded open states and becomes progressively briefer with increasing ACh concentration. In contrast, the temporal pattern of α7 channel currents does not show any concentration dependence due to the fact that desensitization determines the rate of channel closing. Channel activity appears as brief (~0.1 ms) isolated events. Openings are shown as upward deflections. C and O correspond to closed and open states respectively. Membrane potential: −70 mV. Filter: 9 kHz.
Control of open channel lifetime by desensitization may be important for inter‐response latency at a synapse. Desensitized α7 receptors expressed in HEK cells recover with a time constant of ~1 s (Bouzat et al., 2008). Thus, for α7 receptors, after a brief response, a latency of several seconds would be required to generate another response of full amplitude. Because the α7 receptor is highly permeable to calcium, a brief open duration may avoid cell toxicity during overstimulation.
Subunit type and stoichiometry
The many types of nACh receptor subunits assemble in various combinations, enabling a wide diversity of functional and pharmacological profiles (Alexander et al., 2015). In a classical confluence of molecular biology and patch clamp, the γ subunit of the muscle nACh receptor was shown to confer low conductance and long open time, mimicking nACh receptor from embryonic and denervated muscle, whereas the ε subunit increased conductance and shortened the open time, mimicking nACh receptors from adult innervated muscle (Mishina et al., 1986). Combining the patch clamp with site‐directed mutant or chimeric subunits revealed that the conductance difference arose from differences in charged residues at the extracellular end of the M2 TMD (Imoto et al., 1988; Herlitze et al., 1996), whereas the mean open time difference arose from differences in residues in the cytoplasmic (M3‐M4) and M4 domains (Bouzat et al., 1994). Thus, differences in functional signatures between the two subunits could be traced to amino acid differences in distinct structural domains.
The ability to vary subunit stoichiometry within the receptor pentamer further increases the diversity in functional and pharmacological profiles. In neuronal heteromeric α4β2 nACh receptors, changes in the stoichiometry of α to β subunits not only alter the ACh sensitivity, but also the selectivity for different agonists and antagonists (Nelson et al., 2003; Moroni et al., 2006). The stoichiometry‐dependent differences are likely to arise from changes in the number of agonist binding sites, as well as changes in the interactions between subunits. Two general approaches have been developed to study how changes in binding site number or inter‐subunit interactions affect receptor function. In the first, receptors are generated by covalently linking the subunits so that the stoichiometry and positioning of the subunits are predetermined (Zhou et al., 2003; Carbone et al., 2009). These receptor concatemers have been largely studied through measurements of macroscopic currents, though recently, single channel currents from pentameric concatemers composed of α4 and β2 subunits showed that subunit stoichiometry determined conductance, kinetics and susceptibility to drug potentiation (Mazzaferro et al., 2017). In the second, called electrical fingerprinting, receptors are generated using combinations of unlinked subunits where one subunit of the combination alters the receptor's electrical signature (Rayes et al., 2009; Andersen et al., 2011, 2013).
In electrical fingerprinting, a mutant and a wild type subunit are co‐expressed to yield a population of receptors with a range of subunit stoichiometry. To determine the stoichiometry of an individual receptor, one of the subunits is tagged with a mutation that alters the single‐channel current amplitude (Figure 3). A good site for a conductance mutation is the intracellular domain between M3 and M4, which forms portals through which permeating cations pass; if the portals are lined with arginine residues, the contribution of that subunit to the single channel current amplitude approaches zero (Kelley et al., 2003). In an experiment in which high and low conductance subunits are co‐expressed, single channel current recordings reveal five distinct current amplitudes spaced at a constant increment of the unitary current. Inspection of individual channel openings reveals the stoichiometry of the receptor that elicited each opening. Segregation of the channel openings according to current amplitude then allows determination of the mean open or burst duration for each subunit stoichiometry. A crucial control is to show that the different current amplitudes are kinetically indistinguishable, and thus the conductance mutations are kinetically silent.
Figure 3.
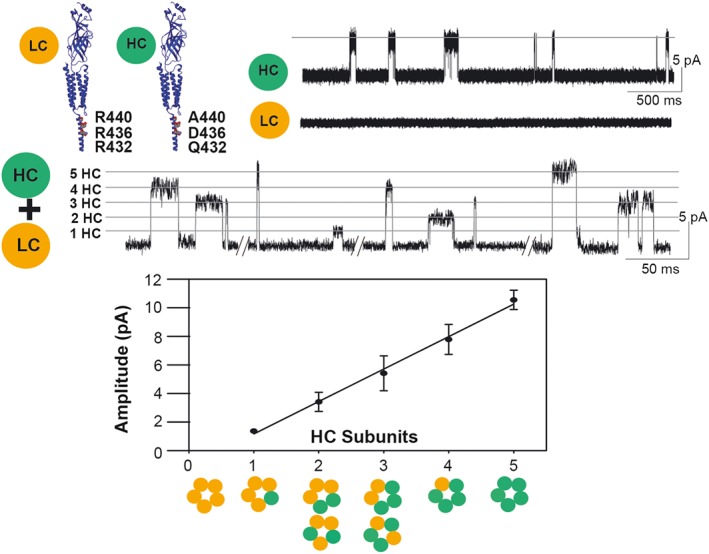
Electrical fingerprinting strategy to determine receptor stoichiometry. To determine receptor stoichiometry, a subunit with a reporter mutation that alters unitary conductance is generated, and mutant and non‐mutant subunits are co‐expressed. Although receptors with a range of different subunit compositions are produced, patch clamp recordings reveal that the amplitude of each single channel opening event reports the number of mutant subunits in the receptor that originated that event. Due to its low conductance, single‐channel currents of the α7‐5HT3A chimeric receptor (LC) are not resolved from cell‐attached patches. The triple mutation increases the conductance (HC) and single‐channel openings appear as a uniform population of 10 pA at −120 mV. Recordings from cells expressing HC and LC subunits show discrete classes of channel amplitudes, each one corresponding to a receptor containing a given number of HC subunits. Thus, the amplitude of the opening event is the signature of the stoichiometry of the receptor that originated the event.
Electrical fingerprinting was applied to the homomeric α7‐5HT3A chimeric receptor in which the extracellular domain contained the α7 sequence and the pore and cytoplasmic domains contained the 5‐HT3A sequence (Rayes et al., 2009). Given that homomeric receptors contain five identical binding sites, the study aimed to determine the number of agonist binding sites required to maximize the open duration. To vary the number of binding sites, one of the subunits contained a mutation that prevented agonist binding, and either that subunit or the non‐mutant subunit included arginine substitutions within the intracellular portal. The results revealed that maximal open duration was achieved with three functional binding sites, two of which were consecutive within the pentamer. In receptors with two non‐consecutive binding sites, the open duration was reduced to half of the maximum, and in receptors with a single site to about a tenth. Thus, binding of agonist to each of the first three sites on the α7‐5HT3A receptor contributes an equal increment of free energy to stabilize the open state, but binding of agonist to a fourth or fifth site provides no further stabilization.
Subsequently, electrical fingerprinting was applied to study the agonist stoichiometry question for native α7 receptors (Andersen et al., 2013), but the conclusion diverged from that for the α7‐5HT3A receptor. Because only a small fraction of channel openings from native α7 receptors last long enough to reach full amplitude, the experiments were done in the presence of the potentiator 5‐hydroxyindole (5‐HI) to prolong the openings, or with an α7 mutant with prolonged open time. Both methods showed that agonist occupancy of a single site produced maximal open duration. To corroborate this conclusion, α7 subunits were co‐expressed with a threefold excess of a subunit with an inactivated agonist binding‐site. Channel open‐time was the same as that obtained for α7 alone, even though the vast majority of receptors contained one or two functional binding sites. Thus in native α7 receptors, occupancy of a single site is transduced into the maximally stable open state structure, whereas in the chimeric receptor, structural mismatches impair transduction and reduce the stabilization provided by the occupancy of fewer than three binding sites.
In an alternative approach to the agonist stoichiometry question, the stoichiometry of α‐bungarotoxin binding required to prevent channel opening was determined using electrical fingerprinting (daCosta et al., 2015). The first step was to generate an α‐bungarotoxin‐resistant α7 subunit by replacing two residues at the binding site with equivalent residues from the toxin‐insensitive α4 subunit (Sine et al., 2013). The toxin‐resistant subunit was then co‐expressed with the native α7 subunit, and one of the subunits was tagged with intracellular arginine substitutions. Before α‐bungarotoxin application, recordings revealed multiple amplitude classes of channel openings reflecting receptors with multiple subunit compositions. However, after α‐bungarotoxin application, only one class of openings was observed corresponding to receptors with five high conductance, α‐bungarotoxin insensitive subunits. Thus, occupancy of a single site by α‐bungarotoxin was enough to prevent channel opening. Given that occupancy of one site by agonist is enough to open the channel in the absence of the toxin, the findings suggested binding of one α‐bungarotoxin molecule immobilizes the four remaining sites so they either cannot bind agonist or cannot transduce agonist binding into channel opening. These findings show that whereas α‐bungarotoxin competes with ACh for the binding sites, functional antagonism is achieved by a conformational arrest mechanism.
Allosteric modulation of nACh receptor
The ACh‐elicited response can be modulated by a broad spectrum of structurally diverse molecules that bind to sites distinct from the orthosteric site. These allosteric ligands are likely to alter energy barriers for transitions between conformational states, and therefore potentiate or inhibit receptor function (Figure 4). nACh receptors are emerging as therapeutic targets for pain, epilepsy, Alzheimer's disease, Parkinson's disease, Tourette's syndrome, schizophrenia, anxiety, depression and inflammatory processes (Changeux and Taly, 2008; Egea et al., 2015; Zanetti et al., 2016). Therefore, understanding the basis of allosteric modulation is essential for rational design of novel therapeutic agents.
Figure 4.
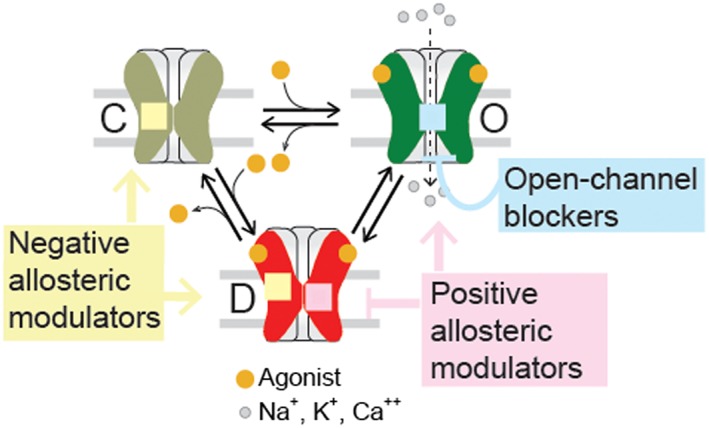
Schematic allosteric scheme for activation and drug modulation. The resting (C), open (O) and desensitized (D) states can be positively or negatively modulated by allosteric compounds. Open‐channel blockers bind to the ion channel and inhibit the flux of ions. NAMs stabilize resting or desensitized states. PAMs stabilize open states or decrease desensitization.
From a functional perspective, allosteric ligands are classified into four groups: (i) negative allosteric modulators (NAMs); (ii) positive allosteric modulators (PAMs), which potentiate agonist‐elicited responses; (iii) allosteric agonists, which activate receptors from non‐orthosteric sites; and (iv) silent allosteric modulators (SAMs), which have no effect on orthosteric agonist responses but block allosteric modulation (Figure 4). Single‐channel recordings have been invaluable in probing the mechanistic bases of these actions.
Mechanistic actions and sites of NAMs
NAMs comprise a wide range of structurally different compounds that inhibit receptor function. The negative modulation probably occurs via several different molecular mechanisms and sites.
Electrophysiology, mutagenesis and photoaffinity labelling have shown that the ion channel is one of the sites for receptor inhibition. Several compounds, called open‐channel blockers, bind within the channel when the receptor is in the open state, thereby physically blocking ion permeation and inhibiting the receptor non‐competitively. They can bind between the −2′ and 20′ positions of the M2 domains (Leonard et al., 1991). By studying the action of local anaesthetics on nACh receptors, Neher and Steinbach (1978) were the first to show the remarkable power of single‐channel recording in elucidating kinetic mechanisms for drug action (Jackson, 2010). They analysed the data using a sequential model for a pure open channel blocker (B), from which they calculated rate constants for blocking and unblocking (k+b and k‐b):
Open‐channel block of single nACh receptor channels has been observed for a wide spectrum of compounds, including, among others, local anaesthetics (Neher and Steinbach (1978), antiepileptic drugs (Vallés et al., 2007), and pyrantel (Rayes et al., 2001). In general, there is a relationship between rate constants in the sequential mechanism and the appearance of channel opening events: openings can appear as sequences of incompletely resolved spikes or as resolved but brief pulses, depending on the concentration of the drug and the blocking and unblocking rate constants (Dilger et al., 1991; Figure 5). At high concentrations, ACh and other agonists also act as open‐channel blockers (Sine and Steinbach, 1984; Zhang et al., 1995; Rayes et al., 2007). Most agonists act as low‐affinity blockers, binding and unbinding very quickly and leading to unresolved openings and closings that reduce the apparent current amplitude.
Figure 5.
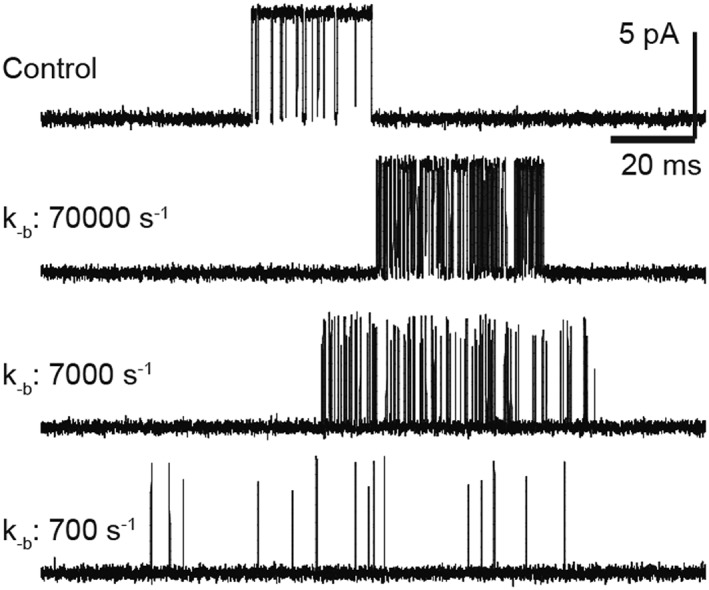
Effects of open‐channel blockers on nACh receptor activity as a function of the unblocking rate. Single‐channel currents for muscle nACh receptor activated by 100 μM ACh were simulated on the basis of the linear open‐channel blocking scheme, including a closed (C), an open (O) and a blocked state (OB). A desensitized state was also included to allow the termination of the bursts in the simulations. Rate constants are β = 50 000 s−1 (opening rate), d + = 100 s−1 (desensitization rate), d‐ = 1 s−1 (recovery from desensitization), k+b = 1 × 107 M−1 s−1, [B] = 5 mM, k‐b = 700, 7000 or 70 000 s−1.
Some compounds are not pure open‐channel blockers, but instead allow the channel to close while still bound, or block the receptor channel in the resting state. Their actions can be described by an expanded linear or cyclic scheme:
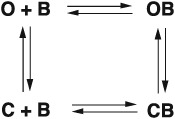
These schemes describe the actions of many channel blockers, including isoflurane and general anaesthetics (Dilger et al., 1991), chlorpromazine and phencyclidine (Changeux et al., 1986), ephedrine (Milone and Engel, 1996; Bouzat, 1996), MK‐801 (Amador and Dani, 1991), and amphetamine (Spitzmaul et al., 1999). The ability of blockers of nACh receptors to become trapped by closure of the channel has been described for the first time for methonium compounds (Gurney and Rang, 1984) and chlorisondamine (Neely and Lingle, 1986).
For some cases, NAMs can be therapeutically useful. For example, long‐lived open‐channel blockers, such as quinidine and fluoxetine, which do not allow rapid unblocking, are therapeutically effective in shortening abnormally prolonged channel openings of the muscle nACh receptor in slow‐channel congenital myasthenic syndromes (Fukudome et al., 1998; Harper et al., 2003; Engel, 2007).
In addition to the channel block mechanism, NAMs may inhibit receptor function by preferentially stabilizing the nACh receptor in a non‐conducting state (resting or desensitized state), or by increasing the rate and extent of desensitization (Figure 4). For example, tricyclic antidepressants and adiphenine produce a concentration‐dependent decrease in the number of openings per activation episode without changing open and closed durations, indicating acceleration of desensitization onset (Gumilar et al., 2003; Spitzmaul et al., 2009).
Given its transmembrane nature, the nACh receptor establishes close physical contact with lipids, which modulate function (Bouzat et al., 1998, 2000; Antollini and Barrantes, 1998; Tamamizu et al., 2000; Baenziger et al., 2015; Barrantes, 2015). The annular (surrounding the perimeter of the receptor) and non‐annular lipid domains (between transmembrane helices) are sites for a great variety of hydrophobic NAMs that by different mechanisms inhibit function. These compounds include, among others, quinacrine, which decreases the frequency and duration of nACh receptor activation episodes due to increased desensitization (Spitzmaul et al., 2001), fatty acids, which reduce open duration and open probability (Bouzat and Barrantes, 1993; Antollini and Barrantes, 2016), and steroids, which produce slow channel blockade (Bouzat and Barrantes, 1996).
Positive allosteric modulators of α7 nACh receptors
PAMs neither activate nACh receptors nor compete with ACh binding, but instead potentiate agonist‐evoked responses. PAMs of α7 receptors are emerging as novel therapeutic agents for neurological and inflammatory disorders. When compared with exogenous agonists, PAMs are promising therapeutics because they maintain the temporal and spatial characteristics of endogenous activation processes, are more target selective, and reduce tolerance due to desensitization (Uteshev, 2014; Corradi and Bouzat, 2016).
α7 PAMs have been classified as type I or II on the basis of their effects on macroscopic currents. Type I PAMs, which include ivermectin (Krause et al., 1998), genistein (Grønlien et al., 2007), N‐(5‐chloro‐2‐hydroxyphenyl)‐N′‐[2‐chloro‐5‐(trifluoromethyl)phenyl]urea (NS‐1738) (Bertrand et al., 2008) and 5‐HI (Thinschmidt et al., 2008), enhance agonist‐induced peak currents without significantly affecting current decay, whereas type II PAMs slow the onset of desensitization and reactivate desensitized receptors (Bertrand and Gopalakrishnan, 2007; Williams et al., 2011; Corradi and Bouzat, 2016).
The most efficacious α7 PAM to date is N‐(5‐chloro‐2,4‐dimethoxyphenyl)‐N′‐(5‐methyl‐3‐isoxazolyl)‐urea (PNU‐120596), a type II PAM (Hurst et al., 2005). When analysed at the single‐channel level, PNU‐120596 prolongs channel openings approximately a thousand‐fold, and the openings coalesce into long clusters lasting several seconds (daCosta et al., 2011; Pałczyńska et al., 2012; Andersen et al., 2016) (Figure 6). Type II PAMs may increase the energetic barrier for desensitization onset, accounting for the long clusters (daCosta et al., 2011), and reduce the barrier for desensitization recovery, which allows reversal of agonist‐induced desensitization (Williams et al., 2011). Alternatively, they may induce conformations for which channel opening is energetically favoured (Szabo et al., 2014). Based on the macroscopic observations that type I PAMs only increase the peak of ACh‐elicited currents, it has been postulated that these PAMs act by decreasing the energetic barrier for opening (Williams et al., 2011; Hurst et al., 2013). However, single‐channel recordings show that type I PAMs prolong α7 receptor openings and activation episodes, which reveals that they do affect the closing and opening rates of α7 receptors (Andersen et al., 2016) (Figure 6). Alternatively, as seen with type II PAMs, the increase in the open duration could be due to changes in desensitization rate, which in turn, could be too slight to be detected from whole‐cell macroscopic currents. However, this may not be the case because for some type II PAMs, the increase in open and burst durations is similar to that mediated by type I PAMs, but the effects on the decay of macroscopic currents are different between both types (Andersen et al., 2016). Thus, PAMs potentiate α7 receptors through multiple mechanisms.
Figure 6.
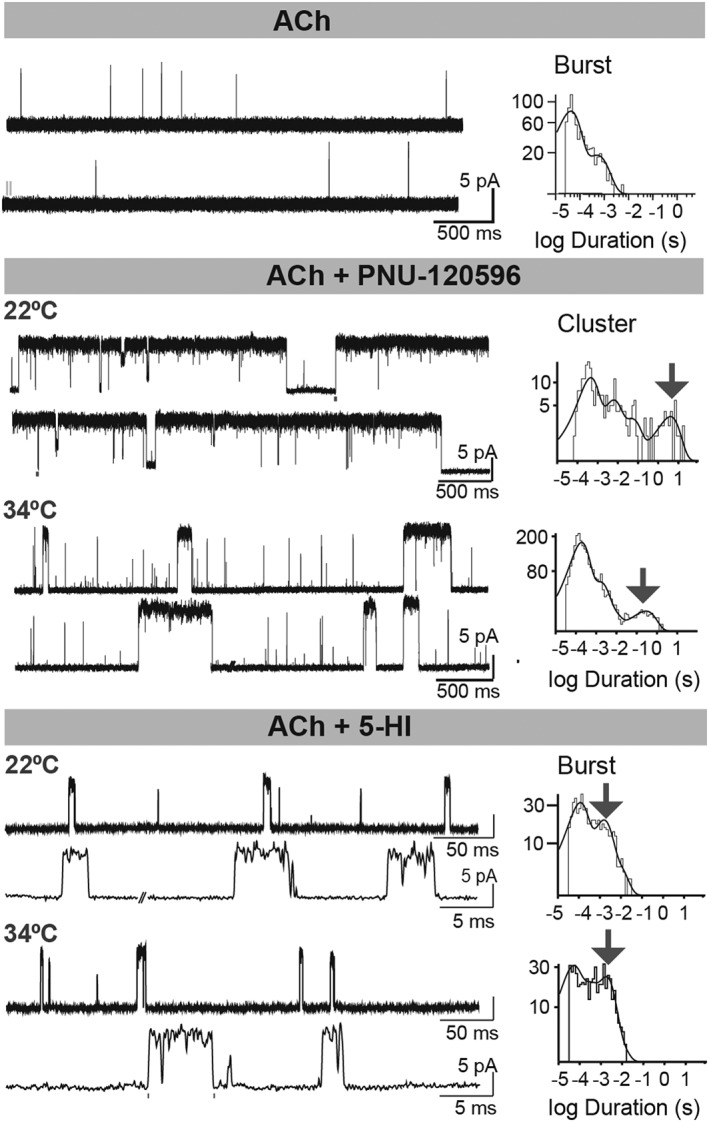
Potentiation of human α7 receptors by type I and type II PAMs as a function of temperature. Single‐channel currents of human α7 receptors activated by 100 μM ACh in the absence and presence of potentiators. The increase of temperature reduces the long clusters induced by PNU‐120596 but does not affect significantly the duration of bursts induced by 5‐HI. Channel openings are shown as upward deflections. Membrane potential: −70 mV. ACh: 100 μM, PNU‐120596: 1 μM, 5‐HI: 2 mM.
Potentiation of α7 receptors by PAMs depends strongly on temperature and type of PAM. In the presence of PNU‐120596, the enhancement of macroscopic peak currents (Williams et al., 2012), and of cluster duration (Andersen et al., 2016) is markedly reduced as the temperature increases from 22 to 34°C. In contrast, the increase in burst duration elicited by 5‐HI does not change significantly over the same temperature range, although the proportion of potentiated bursts at 34°C decreases with respect to that at room temperature (Andersen et al., 2016) (Figure 6). Previous studies have demonstrated that the desensitization rate increases at higher temperatures in the absence or presence of PNU‐120596 (Gupta and Auerbach, 2011; Sitzia et al., 2011; Jindrichova et al., 2012). Thus, if the effect of temperature were mainly on desensitization, increasing temperature would affect mainly type II PAM modulation. This could explain the reduction in the duration of the longest bursts or clusters in the presence of type II PAMs, while the maximal duration of bursts elicited by 5‐HI remains more constant when the temperature is increased. These differences suggest that bursts/clusters have a different mechanistic origin in type I (5‐HI) or type II PAMs. Nonetheless, more PAMs should be tested to confirm this hypothesis. Alternatively, it could be possible that dissociation of PAMs from their binding sites responds differently to temperature depending on both the PAM and the binding‐site structures.
Thus, to better mimic the in vivo situation, in vitro studies should also be assessed at physiological temperatures. That potentiation is reduced at physiological temperatures may be beneficial in attenuating potential toxicity due to calcium influx in the case of very efficacious PAMs (Hu et al., 2009; Guerra‐Álvarez et al., 2015; Uteshev, 2016).
Modulatory sites for nACh receptor PAMs
Electrophysiological studies on α7 receptors have identified amino acids within the transmembrane α‐helices of a single subunit that, when mutated individually, significantly reduced potentiation of macroscopic α7 receptor responses by PNU‐120596, LY2087101 and ivermectin (Young et al., 2008; Collins and Millar, 2010). Particularly, two individual point mutations (A226D in M1 and M254L in M2, numbering of human α7) reduced PNU‐120596 potentiation of macroscopic currents by 90%. Given the sensitivity of single‐channel recordings, potentiated clusters of the single mutants were still evident. However, the simultaneous mutation of these two residues with three other ones, one of which was also previously identified by Young et al. (2008) (S223 at α7M1), abolished the long ACh‐elicited clusters typical of PNU‐120596 potentiation, thus defining structural determinants of potentiation (daCosta et al., 2011).
Reference to homology models of α7 receptors based on the 4 Å structure of the Torpedo nACh receptor suggested that the amino acids involved in PNU‐120596 potentiation may be part of an intra‐subunit transmembrane cavity (Young et al., 2008). A cavity in the upper part of the TMD between the four transmembrane helices of each subunit was also identified from structural studies on the prokaryotic homologue GLIC as the binding site of general anaesthetics (Nury et al., 2011; Sauguet et al., 2013, 2014) and from photolabeling studies on Torpedo nACh receptor as the site of propofol (Jayakar et al., 2013). Although it is tempting to speculate that this cavity is also the site for PNU‐120596 in α7 receptors, it should be taken into account that mutagenesis experiments cannot rule out indirect allosteric effects, and that docking studies were carried out using a homology model. Thus, the unequivocal localization of the PNU‐120596 binding site to an intra‐subunit cavity requires structural studies of the complex with α7 subunits.
The quintuple mutant subunit that prevents PNU‐120596 potentiation was used to determine the stoichiometry required for potentiation. To this end, the electrical fingerprinting strategy was applied by generating α7 receptors composed of normal and PNU‐resistant subunits, one of which contained arginine substitutions of the intracellular portal, and patch clamp recording monitored PNU‐120596 potentiation of α7 receptors with defined stoichiometry (daCosta and Sine, 2013). Potentiation was found to depend steeply on the number of PNU‐120596‐resistant subunits in the pentamer, and at least four and probably five subunits must be sensitive to PNU‐120596 for α7 potentiation to occur.
The quintuple mutant receptor insensitive to PNU‐120596 is also insensitive to another type II PAM, PAM‐2, and to the type I PAM, NS‐1738, indicating common structural determinants for their actions (Andersen et al., 2016). In contrast, 5‐HI potentiates the PNU‐insensitive mutant α7 receptors when evaluated at the single‐channel level, thus indicating that not all PAMs bind to the same site, interact with the same residues, or require the same determinants for transduction.
In α7 receptors, the intrasubunit transmembrane cavity has also been proposed as the binding site for the allosteric agonist, 4‐(4‐bromophenyl)‐3a,4,5,9b‐tetrahydro‐3H‐cyclopenta[c]quinoline‐8‐sulfonamide (4bp‐TQS), which elicits long clusters of channel openings in the absence of ACh (Gill et al., 2011, 2012; Pałczyńska et al., 2012). It was proposed that, in addition to occupancy of this transmembrane site, allosteric activation by the active isomer of 4bp‐TQS requires simultaneous occupancy of an extracellular binding site (Horenstein et al., 2016), which is analogous to a vestibular binding pocket previously reported from structural studies in an α7‐AChBP chimeric receptor (Spurny et al., 2015).
In addition to being the site of PAMs and allosteric agonists, the intrasubunit transmembrane site has been proposed to be the site for some NAMs and silent allosteric modulators of α7 receptors (Gill et al., 2013; Gill‐Thind et al., 2015).
An inter‐subunit transmembrane site between transmembrane helices of adjacent subunits has been identified as a modulatory site for ethanol, general anaesthetics and ivermectin in pentameric ligand‐gated ion channels (Hibbs and Gouaux, 2011; Lynagh and Lynch, 2012; Sauguet et al., 2013, 2015). Photoreactive anaesthetics label homologous residues of the α/γ interface in Torpedo nACh receptors (Husain et al., 2006; Nirthanan et al., 2008; Forman et al., 2015). This site has been proposed for binding of short chain alcohols that positively modulate Torpedo and neuronal nACh receptors by stabilizing the open state (Nagata et al., 1996; Forman and Zhou, 1999; Zuo et al., 2001).
Other PAMs act through different sites and by different mechanisms. Galantamine, an inhibitor of AChE, acts as a low‐efficacy agonist of the muscle nACh receptor and a potentiator of α7 receptors by binding to a site at subunit interfaces close to the ACh‐binding site (Akk and Steinbach, 2005; Hansen and Taylor, 2007; Ludwig et al., 2010). 17‐β‐estradiol increases open probability of neuronal α4β2 receptors; a site at the C‐terminal tail of the α4 subunit is required for the potentiation (Paradiso et al., 2001; Curtis et al., 2002; Jin and Steinbach, 2011). Three different allosteric sites in the extracellular domain of an α7‐AChBP chimera were also identified by an innovative fragment‐library screening in combination with X‐ray (Spurny et al., 2015). Although all the allosteric binders tested behaved on human α7 receptors as NAMs, it was proposed that their chemical modification could lead to a change in functional activity.
In conclusion, multiple binding sites and domains may be involved in the conformational changes associated with potentiation of α7 receptors. This is in line with structural studies in prokaryotic pLGICs proposing that the different allosteric binding sites present in Cys‐loop receptors form an almost continuous path stretching from top to bottom of the receptor (Nys et al., 2016).
Concluding remarks
Investigators remain as stunned by the extraordinary power of patch clamp as when Neher and Sakmann first observed currents through single ion channels. Single‐channel recording has provided molecular insights that are unattainable from macroscopic measurements, and has made invaluable contributions to understanding nACh receptor function, the molecular bases of human diseases and the mechanisms of drug modulation. Kinetic analysis has provided deep insight into activation of ligand‐gated channels by describing how the receptor moves through different conformations in response to the neurotransmitter. Still, there is a long way to go. New challenges include improvement of patch clamp temporal resolution, although we are approaching the limit of present technology. The combination of high‐resolution crystal structures at defined conformational states with single‐channel electrophysiological characterization of drug action on mutant nACh receptors emerges as a means towards defining sites and mechanisms of drug action essential to rational drug design for disorders involving nACh receptors.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by grants from Agencia Nacional de Promoción Científica y Tecnológica de Argentina, Consejo Nacional de Investigaciones Científicas y Técnicas Argentina (CONICET), Universidad Nacional del Sur, and Bill and Melinda Gates Foundation to CB. Research in the Sine laboratory is supported in part by NIH grants NS31744 and NS94124. We are grateful to Dr. Corradi and Mr. Lasala for the help with the figures.
Bouzat, C. , and Sine, S. M. (2018) Nicotinic acetylcholine receptors at the single‐channel level. British Journal of Pharmacology, 175: 1789–1804. doi: 10.1111/bph.13770.
References
- Akk G, Steinbach JH (2005). Galantamine activates muscle‐type nicotinic acetylcholine receptors without binding to the acetylcholine‐binding site. J Neurosci 25: 1992–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amador M, Dani JA (1991). MK‐801 inhibition of nicotinic acetylcholine receptor channels. Synapse 7: 207–215. [DOI] [PubMed] [Google Scholar]
- Andersen N, Corradi J, Bartos M, Sine SM, Bouzat C (2011). Functional relationships between agonist binding sites and coupling regions of homomeric Cys‐loop receptors. J Neurosci 31: 3662–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen N, Corradi J, Sine SM, Bouzat C (2013). Stoichiometry for activation of neuronal α7 nicotinic receptors. Proc Natl Acad Sci U S A 110: 20819–20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen ND, Nielsen BE, Corradi J, Tolosa MF, Feuerbach D, Arias HR et al. (2016). Exploring the positive allosteric modulation of human α7 nicotinic receptors from a single‐channel perspective. Neuropharmacology 107: 189–200. [DOI] [PubMed] [Google Scholar]
- Antollini SS, Barrantes FJ (1998). Disclosure of discrete sites for phospholipid and sterols at the protein–lipid interface in native acetylcholine receptor‐rich membrane. Biochemistry 37: 16653–16662. [DOI] [PubMed] [Google Scholar]
- Antollini SS, Barrantes FJ (2016). Fatty acid regulation of voltage‐ and ligand‐gated ion channel function. Front Physiol 7: 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baenziger JE, Hénault CM, Therien JP, Sun J (2015). Nicotinic acetylcholine receptor‐lipid interactions: mechanistic insight and biological function. Biochim Biophys Acta 1848: 1806–1817. [DOI] [PubMed] [Google Scholar]
- Barrantes FJ (2015). Phylogenetic conservation of protein‐lipid motifs in pentameric ligand‐gated ion channels. Biochim Biophys Acta 1848: 1796–1805. [DOI] [PubMed] [Google Scholar]
- Bartos M, Corradi J, Bouzat C (2009). Structural basis of activation of cys‐loop receptors: the extracellular‐transmembrane interface as a coupling region. Mol Neurobiol 40: 236–252. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M (2007). Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol 74: 1155–1163. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Bertrand S, Cassar S, Gubbins E, Li J, Gopalakrishnan M (2008). Positive allosteric modulation of the α7 nicotinic acetylcholine receptor: ligand interactions with distinct binding sites and evidence for a prominent role of the M2‐M3 segment. Mol Pharmacol 74: 1407–1416. [DOI] [PubMed] [Google Scholar]
- Blatz AL, Magleby KL (1986). Correcting single channel data for missed events. Biophys J 49: 967–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M et al. (2009). X‐ray structure of a pentameric ligand gated ion channel in an apparently open conformation. Nature 457: 111–114. [DOI] [PubMed] [Google Scholar]
- Bouzat C (1996). Ephedrine blocks wild‐type and long‐lived mutant acetylcholine receptor channels. Neuroreport 8: 317–321. [DOI] [PubMed] [Google Scholar]
- Bouzat CB, Barrantes FJ (1993). Effects of long‐chain fatty acids on the channel activity of the nicotinic acetylcholine receptor. Receptors Channels 1: 251–258. [PubMed] [Google Scholar]
- Bouzat CB, Barrantes FJ (1996). Modulation of muscle nicotinic acetylcholine receptors by the glucocorticoid hydrocortisone. Possible allosteric mechanism of channel blockade. J Biol Chem 271: 25835–25841. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Bren N, Sine SM (1994). Structural basis of the different gating kinetics of fetal and adult acetylcholine receptors. Neuron 13: 1395–1402. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Roccamo AM, Garbus I, Barrantes FJ (1998). Mutations at lipid‐exposed residues of the acetylcholine receptor affect its gating kinetics. Mol Pharmacol 54: 146–153. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Barrantes FJ, Sine S (2000). Nicotinic receptor fourth transmembrane domain: hydrogen bonding by conserved threonine contributes to channel gating kinetics. J Gen Physiol 115: 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB et al. (2004). Coupling of agonist binding to channel gating in an ACh‐binding protein linked to an ion channel. Nature 430: 896–900. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Bartos M, Corradi J, Sine SM (2008). The interface between extracellular and transmembrane domains of homomeric Cys‐loop receptors governs open‐channel lifetime and rate of desensitization. J Neurosci 28: 7808–7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB et al. (2001). Crystal structure of an ACh‐binding protein reveals the ligand binding domain of nicotinic receptors. Nature 411: 269–276. [DOI] [PubMed] [Google Scholar]
- Burzomato V, Beato M, Groot‐Kormelink PJ, Colquhoun D, Sivilotti LG (2004). Single‐channel behavior of heteromeric alpha1beta glycine receptors: an attempt to detect a conformational change before the channel opens. J Neurosci 24: 10924–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone L, Moroni M, Groot‐Kormelink PJ, Bermudez I (2009). Pentameric concatenated (alpha4)(2)(beta2)(3) and (alpha4)(3)(beta2)(2) nicotinic acetylcholine receptors: subunit arrangement determines functional expression. Br J Pharmacol 156: 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux JP, Taly A (2008). Nicotinic receptors, allosteric proteins and medicine. Trends Mol Med 14: 93–102. [DOI] [PubMed] [Google Scholar]
- Changeux JP, Pinset C, Ribera AB (1986). Effects of chlorpromazine and phencyclidine on mouse C2 acetylcholine receptor kinetics. J Physiol 378: 497–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio T, Green W, Hartman D, Hayden D, Paulson HL, Sigworth FJ et al. (1987). Genetic reconstitution of functional acetylcholine receptor channels in mouse fibroblasts. Science 238: 1688–1694. [DOI] [PubMed] [Google Scholar]
- Collins T, Millar NS (2010). Nicotinic acetylcholine receptor transmembrane mutations convert ivermectin from a positive to a negative allosteric modulator. Mol Pharmacol 78: 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes A (1981). On the stochastic properties of single ion channels. Proc R Soc Lond B Biol Sci 211: 205–235. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B (1981). Fluctuations in the microsecond time range of the current through single acetylcholine receptor ion channels. Nature 294: 464–466. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B (1985). Fast events in single channel currents activated by acetylcholine and its analogs at the frog muscle endplate. J Physiol 369: 501–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth FJ (1983). Fitting and statistical analysis of single‐channel records In: Sakmann B, Neher E. (eds). Single‐Channel Recording, 1st edn. Plenum Press: NY. [Google Scholar]
- Corradi J, Bouzat C (2014). Unraveling mechanisms underlying partial agonism in 5‐HT3A receptors. J Neurosci 34: 16865–16876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi J, Bouzat C (2016). Understanding the bases of function and modulation of α7 nicotinic receptors: implications for drug discovery. Mol Pharmacol 90: 288–299. [DOI] [PubMed] [Google Scholar]
- Crouzy SC, Sigworth FJ (1990). Yet another approach to the dwell‐time omission problem of single channel analysis. Biophys J 58: 731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis L, Buisson B, Bertrand S, Bertrand D (2002). Potentiation of human alpha4beta2 neuronal nicotinic acetylcholine receptor by estradiol. Mol Pharmacol 61: 127–135. [DOI] [PubMed] [Google Scholar]
- daCosta CJB, Sine SM (2013). Stoichiometry for drug potentiation of a pentameric ion channel. Proc Natl Acad Sci U S A 110: 6595–6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- daCosta CJB, Free CR, Corradi J, Bouzat C, Sine SM (2011). Single‐channel and structural foundations of neuronal α7 acetylcholine receptor potentiation. J Neurosci 31: 13870–13879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- daCosta CJB, Free CR, Sine SM (2015). Stoichiometry for α‐bungarotoxin block of α7 acetylcholine receptors. Nat Commun 6: 8057 https://doi.org/10.1038/ncomms9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Castillo J, Katz B (1957). Interaction at end‐plate receptors of different choline derivatives. Proc R Soc Lond B Biol Sci 146: 369–381. [DOI] [PubMed] [Google Scholar]
- Dilger JP, Brett RS, Lesko LA (1991). Effects of isoflurane on acetylcholine receptor channels. 1. Single‐channel currents. Mol Pharmacol 41: 127–133. [PubMed] [Google Scholar]
- Dionne VE, Steinbach JH, Stevens CF (1978). An analysis of the dose‐response relationship at voltage‐clamped frog neuromuscular junctions. J Physiol 281: 421–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Lu W, Wu S, Cheng Y, Gouaux E (2015). Glycine receptor mechanism elucidated by electron cryo‐microscopy. Nature 526: 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein SJ, Schaad O, Henry H, Bertrand D, Changeux JP (1996). A kinetic mechanism for nicotinic acetylcholine receptors based on multiple allosteric transitions. Biol Cybern 75: 361–379. [DOI] [PubMed] [Google Scholar]
- Egea J, Buendia I, Parada E, Navarro E, León R, Lopez MG (2015). Anti‐inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol 97: 463–472. [DOI] [PubMed] [Google Scholar]
- Engel AG (2007). The therapy of congenital myasthenic syndromes. Neurotherapeutics 4: 252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht AR, Leatherbarrow RJ, Wells TNC (1986). Quantitative analysis of structure‐activity relationships in engineered proteins by linear free‐energy relationships. Nature 322: 284–286. [DOI] [PubMed] [Google Scholar]
- Forman SA, Zhou Q (1999). Novel modulation of a nicotinic receptor channel mutant reveals that the open state is stabilized by ethanol. Mol Pharmacol 55: 102–108. [DOI] [PubMed] [Google Scholar]
- Forman SA, Chiara DC, Miller KW (2015). Anesthetics target interfacial transmembrane sites in nicotinic acetylcholine receptors. Neuropharmacology 96: 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukudome T, Ohno K, Brengman JM, Engel AG (1998). AChR channel blockade by quinidine sulfate reduces channel open duration in the slow‐channel congenital myasthenic syndrome. Ann N Y Acad Sci 841: 199–202. [DOI] [PubMed] [Google Scholar]
- Gill JK, Savolainen M, Young GT, Zwart R, Sher E, Millar NS (2011). Agonist activation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci U S A 108: 5867–5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill J, Dhankher P, Sheppard T, Sher E, Millar N (2012). A series of α7 nicotinic acetylcholine receptor allosteric modulators with close chemical similarity but diverse pharmacological properties. Mol Pharmacol 81: 710–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill JK, Chatzidaki A, Ursu D, Sher E, Millar NS (2013). Contrasting properties of α7‐selective orthosteric and allosteric agonists examined on native nicotinic acetylcholine receptors. PLoS One 8: e55047 https://doi.org/10.1371/journal.pone.0055047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill‐Thind JK, Dhankher P, D'Oyley JM, Sheppard TD, Millar NS (2015). Structurally similar allosteric modulators of α7 nicotinic acetylcholine receptors exhibit five distinct pharmacological effects. J Biol Chem 290: 3552–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grønlien JH, Håkerud M, Ween H, Thorin‐Hagene K, Briggs CA, Gopalakrishnan M et al. (2007). Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol 72: 715–724. [DOI] [PubMed] [Google Scholar]
- Grosman C, Zhou M, Auerbach A (2000). Mapping the conformational wave of acetylcholine receptor channel gating. Nature 403: 773–776. [DOI] [PubMed] [Google Scholar]
- Guerra‐Álvarez M, Moreno‐Ortega AJ, Navarro E, Fernández‐Morales JC, Egea J, López MG et al. (2015). Positive allosteric modulation of alpha‐7 nicotinic receptors promotes cell death by inducing Ca2+ release from the endoplasmic reticulum. J Neurochem 133: 309–319. [DOI] [PubMed] [Google Scholar]
- Gumilar F, Arias HR, Spitzmaul G, Bouzat C (2003). Molecular mechanisms of inhibition of nicotinic acetylcholine receptors by tricyclic antidepressants. Neuropharmacology 45: 964–976. [DOI] [PubMed] [Google Scholar]
- Gupta S, Auerbach A (2011). Temperature dependence of acetylcholine receptor channels activated by different agonists. Biophys J 100: 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Chakraborty S, Vij R, Auerbach A (2017). A mechanism for acetylcholine receptor gating based on structure, coupling, phi and flip. J Gen Physiol 149: 85–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney AM, Rang HP (1984). The channel‐blocking action of methonium compounds on rat submandibular ganglion cells. Br J Pharmacol 82: 623–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981). Improved patch‐clamp techniques for high‐resolution current recording from cells and cell‐free membrane patches. Pflugers Arch 391: 85–100. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Taylor P (2007). Galanthamine and non‐competitive inhibitor binding to ACh‐binding protein: evidence for a binding site on non‐alpha‐subunit interfaces of heteromeric neuronal nicotinic receptors. J Mol Biol 369: 895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CM, Fukodome T, Engel AG (2003). Treatment of slow‐channel congenital myasthenic syndrome with fluoxetine. Neurology 60: 1710–1713. [DOI] [PubMed] [Google Scholar]
- Hassaine G, Deluz C, Grasso L, Wyss R, Tol MB, Hovius R et al. (2014). X‐ray structure of the mouse serotonin 5‐HT3 receptor. Nature 512: 276–281. [DOI] [PubMed] [Google Scholar]
- Haung X, Chen H, Michelson K, Schneider S, Shaffer PL (2015). Crystal structure of human glycine receptor‐a3 bound to antagonist strychnine. Nature 526: 277–280. [DOI] [PubMed] [Google Scholar]
- Hawkes AG, Jalali A, Colquhoun D (1990). The distributions of the apparent open times and shut times in a single channel record when brief events cannot be detected. Philos Trans R Soc Lond A 332: 511–538. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Villarroel A, Witzemann V, Koenen M, Sakmann B (1996). Structural determinants of channel conductance in fetal and adult rat muscle acetylcholine receptors. J Physiol 492: 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E (2011). Principles of activation and permeation in an anion‐selective Cys‐loop receptor. Nature 474: 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilf RJ, Dutzler R (2008). X‐ray structure of a prokaryotic ligand‐gated ion channel. Nature 452: 375–379. [DOI] [PubMed] [Google Scholar]
- Hilf RJ, Dutzler R (2009). Structure of a potentially open state of a proton‐activated pentameric ligand gated ion channel. Nature 457: 115–118. [DOI] [PubMed] [Google Scholar]
- Horenstein NA, Papke RL, Kulkarni AR, Chaturbhuj GU, Stokes C, Manther K et al. (2016). Critical molecular determinants of α7 nicotinic acetylcholine receptor activation. J Biol Chem 291: 5049–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R, Lange K (1983). Estimating kinetic constants from single channel data. Biophys J 43: 207–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Gopalakrishnan M, Li J (2009). Positive allosteric modulation of alpha7 neuronal nicotinic acetylcholine receptors: lack of cytotoxicity in PC12 cells and rat primary cortical neurons. Br J Pharmacol 158: 1857–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst RS, Hajós M, Raggenbass M, Wall TM, Higdon NR, Lawson JA et al. (2005). A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci 25: 4396–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst R, Rollema H, Bertrand D (2013). Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther 137: 22–54. [DOI] [PubMed] [Google Scholar]
- Husain SS, Nirthanan S, Ruesch D, Solt K, Cheng Q, Li GD et al. (2006). Synthesis of trifluoromethylaryl diazirine and benzophenone derivatives of etomidate that are potent general anesthetics and effective photolabels for probing sites on ligand‐gated ion channels. J Med Chem 49: 4818–4825. [DOI] [PubMed] [Google Scholar]
- Imoto K, Busch C, Sakmann B, Mishina M, Konno T, Nakai J et al. (1988). Rings of negatively charged amino acids determine the acetylcholine receptor channel conductance. Nature 335: 645–648. [DOI] [PubMed] [Google Scholar]
- Indurthi DC, Lewis TM, Ahring PK, Balle T, Chebib M, Absalom NL (2016). Ligand binding at the α4‐α4 agonist‐binding site of the α4β2 nAChR triggers receptor activation through a pre‐activated conformational state. PLoS One 11: e0161154 https://doi.org/10.1371/journal.pone.0161154. eCollection 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB (1986). Kinetics of unliganded acetylcholine receptor gating. Biophys J 49: 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB (1988). Dependence of acetylcholine receptor channel kinetics on agonist concentration in cultured muscle fibres. J Physiol 397: 555–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB (2010). Open channel block and beyond. J Physiol 588: 553–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakar SS, Dailey WP, Eckenhoff RG, Cohen JB (2013). Identification of propofol binding sites in a nicotinic acetylcholine receptor with a photoreactive propofol analog. J Biol Chem 288: 6178–6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Steinbach JH (2011). A portable site: a binding element for 17β‐estradiol can be placed on any subunit of a nicotinic α4β2 receptor. J Neurosci 31: 5045–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindrichova M, Lansdell SJ, Millar NS (2012). Changes in temperature have opposing effects on current amplitude in alpha7 and alpha4beta2 nicotinic acetylcholine receptors. PLoS One 7: e32073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley SP, Dunlop JI, Kirkness EF, Lambert JJ, Peters JA (2003). A cytoplasmic region determines single channel conductance in 5‐HT3 receptors. Nature 424: 321–324. [DOI] [PubMed] [Google Scholar]
- Krause RM, Buisson B, Bertrand S, Corringer PJ, Galzi JL, Changeux JP et al. (1998). Ivermectin: a positive allosteric effector of the alpha7 neuronal nicotinic acetylcholine receptor. Mol Pharmacol 53: 283–294. [DOI] [PubMed] [Google Scholar]
- Lape RD, Colquhoun D, Sivilotti LG (2008). On the nature of partial agonism in the nicotinic receptor superfamily. Nature 454: 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY, Sine SM (2005). Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature 438: 243–247. [DOI] [PubMed] [Google Scholar]
- Leonard RJ, Charnet P, Labarca C, Vogelaar NJ, Czyzyk L, Gouin A et al. (1991). Reverse pharmacology of the nicotinic acetylcholine receptor. mapping the local anesthetic binding site. Ann N Y Acad Sci 625: 588–599. [DOI] [PubMed] [Google Scholar]
- Lindstrom J, Merlie J, Yogeeswaran G (1979). Biochemical properties of acetylcholine receptor subunits from Torpedo californica . Biochem 18: 4465–4470. [DOI] [PubMed] [Google Scholar]
- Ludwig J, Höffle‐Maas A, Samochocki M, Luttmann E, Albuquerque EX, Fels G et al. (2010). Localization by site‐directed mutagenesis of a galantamine binding site on α7 nicotinic acetylcholine receptor extracellular domain. J Recept Signal Transduct Res 30: 469–483. [DOI] [PubMed] [Google Scholar]
- Lynagh T, Lynch JW (2012). Ivermectin binding sites in human and invertebrate Cys‐loop receptors. Trends Pharmacol Sci 33: 432–441. [DOI] [PubMed] [Google Scholar]
- Marshall CG, Ogden D, Colquhoun D (1991). Activation of ion channels in the frog endplate by several analogs of acetylcholine. J Physiol 433: 73–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzaferro S, Bermudez I, Sine SM (2017). α4β2 Nicotinic Acetylcholine Receptors: relationships between subunit stoichiometry and function at the single channel level. J Biol Chem 292: 2729–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methfessel C, Witzemann V, Takahashi T, Mishina M, Numa S, Sakmann B (1986). Patch clamp measurements on Xenopus laevis oocytes: currents through endogenous channels and implanted acetylcholine receptor and sodium channels. Pflugers Arch 407: 577–588. [DOI] [PubMed] [Google Scholar]
- Meunier JC, Sealock R, Olsen R, Changeux JP (1974). Purification and properties of the cholinergic receptor from Electrophorus electricus electric tissue. Eur J Biochem 45: 371–394. [DOI] [PubMed] [Google Scholar]
- Miller PS, Aricescu AR (2014). Crystal structure of a human GABAA receptor. Nature 512: 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone M, Engel AG (1996). Block of the endplate acetylcholine receptor channel by the sympathomimetic agents ephedrine, pseudoephedrine, and albuterol. Brain Res : 346–352. [DOI] [PubMed] [Google Scholar]
- Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S et al. (1986). Molecular distinction between fetal and adult forms of the acetylcholine receptor. Nature 321: 406–411. [DOI] [PubMed] [Google Scholar]
- Mitra AK, McCarthy MP, Stroud RM (1989). Three‐dimensional structure of the nicotinic acetylcholine receptor and location of the major associated 43‐kD cytoskeletal protein, determined at 22 Å by low dose electron microscopy and x‐ray diffraction to 12.5 Å. J Cell Biol 109: 755–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP (1965). On the nature of allosteric transitions: a plausible model. J Mol Biol 12: 88–118. [DOI] [PubMed] [Google Scholar]
- Morales‐Perez CL, Noviello CM, Hibbs RE (2016). X‐ray structure of the human α4β2 nicotinic receptor. Nature 538: 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I (2006). alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long‐term exposure to nicotine. Mol Pharmacol 70: 755–768. [DOI] [PubMed] [Google Scholar]
- Mukhtasimova N, daCosta CBJ, Sine SM (2016). Improved resolution of single channel dwell times reveals mechanisms of binding, priming and gating in muscle AChR. J Gen Physiol 148: 43–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N, Lee WY, Wang HL, Sine SM (2009). Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature 459: 451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Aistrup GL, Huang CS, Marszalec W, Song JH, Yeh JZ et al. (1996). Potent modulation of neuronal nicotinic acetylcholine receptor‐ channel by ethanol. Neurosci Lett 217: 189–193. [PubMed] [Google Scholar]
- Neely A, Lingle CJ (1986). Trapping of an open‐channel blocker at the frog neuromuscular acetylcholine channel. Biophys J 50: 981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Steinbach JH (1978). Local anaesthetics transiently block currents through single acetylcholine‐receptor channels. J Physiol 277: 153–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J (2003). Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol 63: 332–341. [DOI] [PubMed] [Google Scholar]
- Nirthanan S, Garcia G 3rd, Chiara DC, Husain SS, Cohen JB (2008). Identification of binding sites in the nicotinic acetylcholine receptor for TDBzl‐etomidate, a photoreactive positive allosteric effector. J Biol Chem 283: 22051–22062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Takahashi H, Tanabe T, Toyosato M, Kikyotani S, Furutani Y et al. (1983). Structural homology of Torpedo californica acetylcholine receptor subunits. Nature 302: 528–532. [DOI] [PubMed] [Google Scholar]
- Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V et al. (2011). X‐ray structures of general anaesthetics bound to a pentameric ligand‐gated ion channel. Nature 469: 428–431. [DOI] [PubMed] [Google Scholar]
- Nys M, Wijckmans E, Farinha A, Yoluk Ö, Andersson M, Brams M et al. (2016). Allosteric binding site in a Cys‐loop receptor ligand‐binding domain unveiled in the crystal structure of ELIC in complex with chlorpromazine. Proc Natl Acad Sci U S A 113: E6696–E6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Wang HL, Milone M, Bren N, Brengman JM, Nakano S et al. (1996). Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron 17: 157–170. [DOI] [PubMed] [Google Scholar]
- Pałczyńska MM, Jindrichova M, Gibb AJ, Millar NS (2012). Activation of α7 nicotinic receptors by orthosteric and allosteric agonists: influence on single‐channel kinetics and conductance. Mol Pharmacol 82: 910–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso K, Zhang J, Steinbach JH (2001). The C terminus of the human nicotinic alpha4beta2 receptor forms a binding site required for potentiation by an estrogenic steroid. J Neurosci 21: 6561–6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F (1996). Estimating single‐channel kinetic parameters from idealized patch‐clamp data containing missed events. Biophys J 70: 264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery MA, Hunkapiller MW, Strader CD, Hood LE (1980). Acetylcholine receptor: complex of homologous subunits. Science 208: 1454–1456. [DOI] [PubMed] [Google Scholar]
- Rayes D, De Rosa MJ, Spitzmaul G, Bouzat C (2001). The anthelmintic pyrantel acts as a low efficacious agonist and an open‐channel blocker of mammalian acetylcholine receptors. Neuropharmacology 41: 238–245. [DOI] [PubMed] [Google Scholar]
- Rayes D, Flamini M, Hernando G, Bouzat C (2007). Activation of single nicotinic receptor channels from Caenorhabditis elegans muscle. Mol Pharmacol 71: 1407–1415. [DOI] [PubMed] [Google Scholar]
- Rayes D, De Rosa MJ, Sine SM, Bouzat C (2009). Number and Locations of Agonist Binding Sites Required to Activate Homomeric Cys‐loop Receptors. J Neurosci 29: 6022–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JA, Karlin A (1978). Molecular weight in detergent solution of acetylcholine receptor from Torpedo californica . Biochem 17: 2035–2038. [DOI] [PubMed] [Google Scholar]
- Roux B, Sauve R (1985). A general solution to the time interval omission problem applied to single channel analysis. Biophys J 48: 149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmann B, Patlak J, Neher E (1980). Single acetylcholine‐activated channels show burst‐kinetics in presence of desensitizing concentrations of agonist. Nature 286: 71–73. [DOI] [PubMed] [Google Scholar]
- Sauguet L, Howard RJ, Malherbe L, Lee US, Corringer PJ, Harris RA et al. (2013). Structural basis for potentiation by alcohols and anaesthetics in a ligand‐gated ion channel. Nat Commun 4: 1697 https://doi.org/10.1038/ncomms2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauguet L, Shahsavar A, Poitevin F, Huon C, Menny A, Nemecz À et al. (2014). Crystal structures of a pentameric ligand‐gated ion channel provide a mechanism for activation. Proc Natl Acad Sci U S A 111: 966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauguet L, Shahsavar A, Delarue M (2015). Crystallographic studies of pharmacological sites in pentameric ligand‐gated ion channels. Biochim Biophys Acta 1850: 511–523. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ (1983). An example of analysis In: Sakmann B, Neher E. (eds). Single‐Channel Recording, 1st edn. Plenum Press: NY. [Google Scholar]
- Sigworth FJ (1986). The patch clamp is more useful than anyone had expected. Fed Proc 45: 2673–2677. [PubMed] [Google Scholar]
- Sivilotti L, Colquhoun D (2016). In praise of single channel kinetics. J Gen Physiol 148: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Steinbach JH (1984). Agonists block currents through acetylcholine receptor channels. Biophys J 46: 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Claudio T, Sigworth FJ (1990). Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts. Single channel current kinetics reveal distinct agonist binding affinities. J Gen Physiol 96: 395–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Huang S, Li SX, daCosta CJB, Chen L (2013). Inter‐residue coupling contributes to high affinity, subtype selective binding of α‐bungarotoxin to nicotinic receptors. Biochem J 454: 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM (2012). End‐plate acetylcholine receptor: structure, mechanism, pharmacology and disease. Physiol Rev 92: 1189–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitzia F, Brown JT, Randall AD, Dunlop J (2011). Voltage‐ and temperature‐dependent allosteric modulation of alpha7 nicotinic receptors by PNU120596. Front Pharmacol 2: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzmaul GF, Esandi MC, Bouzat C (1999). Amphetamine acts as a channel blocker of the acetylcholine receptor. Neuroreport 10: 2175–2181. [DOI] [PubMed] [Google Scholar]
- Spitzmaul G, Dilger JP, Bouzat C (2001). The noncompetitive inhibitor quinacrine modifies the desensitization kinetics of muscle acetylcholine receptors. Mol Pharmacol 60: 235–243. [DOI] [PubMed] [Google Scholar]
- Spitzmaul G, Gumilar F, Dilger JP, Bouzat C (2009). The local anaesthetics proadifen and adiphenine inhibit nicotinic receptors by different molecular mechanisms. Br J Pharmacol 157: 804–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurny R, Debaveye S, Farinha A, Veys K, Vos AM, Gossas T et al. (2015). Molecular blueprint of allosteric binding sites in a homologue of the agonist‐binding domain of the α7 nicotinic acetylcholine receptor. Proc Natl Acad Sci 112: E2543–E2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo AK, Pesti K, Mike A, Vizi ES (2014). Mode of action of the positive modulator PNU‐120596 on α7 nicotinic acetylcholine receptors. Neuropharmacology 81: 42–54. [DOI] [PubMed] [Google Scholar]
- Tamamizu S, Guzman GR, Santiago J, Rojas LV, McNamee MG, Lasalde Dominicci JA (2000). Functional effects of periodic tryptophan substitutions in the alpha M4 transmembrane domain of the Torpedo californica nicotinic acetylcholine receptor. Biochem 39: 4666–4673. [DOI] [PubMed] [Google Scholar]
- Thinschmidt JS, López‐Hernández GY, Ren K, King MA, Meyer EM, Papke RL (2008). Modulation of spontaneous hippocampal synaptic events with 5‐hydroxyindole, 4OH‐GTS‐21, and rAAV‐mediated alpha7 nicotinic receptor gene transfer. Brain Res 1203: 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima C, Unwin N (1988). Ion channel of acetylcholine receptor reconstructed from images of postsynaptic membranes. Nature 336: 247–250. [DOI] [PubMed] [Google Scholar]
- Uteshev VV (2014). The therapeutic promise of positive allosteric modulation of nicotinic receptors. Eur J Pharmacol 727: 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uteshev V (2016). Are positive allosteric modulators of α7 nAChRs clinically safe? J Neurochem 136: 217–219. [DOI] [PubMed] [Google Scholar]
- Unwin N (1993). Nicotinic acetylcholine receptor at 9 Å resolution. J Mol Biol 229: 1101–1124. [DOI] [PubMed] [Google Scholar]
- Unwin N (2005). Refined structure of the nicotinic receptor at 4 Å resolution. J Mol Biol 346: 967–989. [DOI] [PubMed] [Google Scholar]
- Vallés AS, Garbus I, Barrantes FJ (2007). Lamotrigine is an open‐channel blocker of the nicotinic acetylcholine receptor. Neuroreport 18: 45–50. [DOI] [PubMed] [Google Scholar]
- Williams DK, Wang J, Papke RL (2011). Positive allosteric modulators as an approach to nicotinic acetylcholine receptor‐targeted therapeutics: advantages and limitations. Biochem Pharmacol 82: 915–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DK, Peng C, Kimbrell MR, Papke RL (2012). Intrinsically low open probability of α7 nicotinic acetylcholine receptors can be overcome by positive allosteric modulation and serum factors leading to the generation of excitotoxic currents at physiological temperatures. Mol Pharmacol 82: 746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Pan N, Xue F, Zheng Y, Li C, Chang Y et al. (2015). The coupling interface and pore domain codetermine the single‐channel activity of the α7 nicotinic receptor. Neuropharmacology 95: 448–458. [DOI] [PubMed] [Google Scholar]
- Young GT, Zwart R, Walker AS, Sher E, Millar NS (2008). Potentiation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci U S A 105: 14686–14691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti SR, Ziblat A, Torres NI, Zwirner NW, Bouzat C (2016). Expression and functional role of α7 nicotinic receptor in human cytokine‐stimulated natural killer (NK) cells. J Biol Chem 291: 16541–16552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen J, Auerbach A (1995). Activation of recombinant mouse acetylcholine receptors by acetylcholine, carbamylcholine and tetramethylammonium. J Physiol 486: 189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Nelson ME, Kuryatov A, Choi C, Cooper J, Lindstrom J (2003). Human alpha4beta2 acetylcholine receptors formed from linked subunits. J Neurosci 23: 9004–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Aistrup GL, Marszalec W, Gillespie A, Chavez‐Noriega LE, Yeh JZ et al. (2001). Dual action of n‐alcohols on neuronal nicotinic acetylcholine receptors. Mol Pharmacol 60: 700–711. [PubMed] [Google Scholar]