

Abstract
Voltage‐gated sodium channels (NaV channels) are essential for the initiation and propagation of action potentials that critically influence our ability to respond to a diverse range of stimuli. Physiological and pharmacological studies have linked abnormal function of NaV channels to many human disorders, including chronic neuropathic pain. These findings, along with the description of the functional properties and expression pattern of NaV channel subtypes, are helping to uncover subtype specific roles in acute and chronic pain and revealing potential opportunities to target these with selective inhibitors. High‐throughput screens and automated electrophysiology platforms have identified natural toxins as a promising group of molecules for the development of target‐specific analgesics. In this review, the role of toxins in defining the contribution of NaV channels in acute and chronic pain states and their potential to be used as analgesic therapies are discussed.
Linked Articles
This article is part of a themed section on Recent Advances in Targeting Ion Channels to Treat Chronic Pain. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.12/issuetoc
Abbreviations
- CIP
congenital insensitivity to pain
- DRG
dorsal root ganglion
- NaV
voltage‐gated sodium channel
- NGF
nerve grow factor
- PNS
peripheral nervous system
- TTX
tetrodotoxin
Introduction
http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=82 (NaV channels) are key signalling molecules that underlie the action potential upswing in electrically excitable cells as well as being involved in biological processes in non‐excitable cells. They are membrane‐spanning glycosylated proteins that rapidly activate in response to cell membrane depolarization to generate an influx of Na+ ions before rapidly inactivating. NaV channels contribute to the transmission of a broad range of somatosensory signals, including temperature, touch, smell, proprioception and pain, from the periphery to the brain (Vranken, 2012). They also underlie the release of catecholamines by neuroendocrine cells (Yamamoto et al., 1997), angiogenesis (Andrikopoulos et al., 2011), contraction of smooth, skeletal and cardiac muscle (Seda et al., 2007), melanogenesis (Ekmehag et al., 1994; Huh et al., 2010) and the invasion (Carrithers et al., 2009), phagocytosis (Carrithers et al., 2007; Black et al., 2013) and maturation (Zsiros et al., 2009) of immune cells. Thus, NaV channels are a family of molecules in which physiological roles are broad and vital for normal physiology.
The NaV family comprises nine homologous α‐subunits (NaV1.1–NaV1.9) that form the ion conducting pore plus the auxiliary β‐subunits (β1 to β4) that modulate channel gating and trafficking. The overall molecular architecture of the α‐subunit is highly conserved amongst NaV channels (Figure 1). It comprises four domains I–IV containing the transmembrane segments S1 to S6, in which S4 contributes to the voltage sensor and S5 and S6 form the sodium selective pore (Stuhmer et al., 1989; Catterall et al., 2005; Alabi et al., 2007; Zhang et al., 2012; Shen et al., 2017) (Figure 1A–C). Cell depolarization alters the electrical field across the membrane resulting in the rapid movement of the S4 segments and conformational changes that opens the ion channel pore, followed by fast inactivation that occurs as channel block by the DIII–DIV intracellular linker. Single and multiple variations in the amino acids sequence of the α‐subunit as well as alternative splicing and post‐translational modifications such as glycosylation are associated with channel modulation and their in vivo physiological signatures (Rizzo et al., 1994; Laedermann et al., 2015). This structural variability and subtype‐selective distribution help define channel function in neurons and non‐neuronal cells. Table 1 summarizes the distribution of NaV channels in the central and peripheral nervous system (PNS). http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=581 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=582 channels are mostly expressed in skeletal muscle and the heart, respectively, and will not be discussed in this review.
Figure 1.
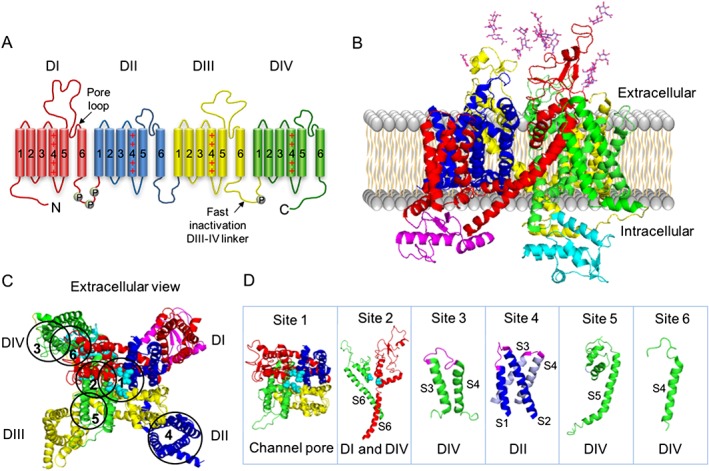
NaV channel structure. (A) Topology of the eukaryotic NaV channel α‐subunit. Each domain of the tetramer, DI (red), DII (blue), DIII (yellow) and DIV (green), contains six transmembrane helixes, a pore loop and a voltage sensor located in the S4 region, which is positively charged. The pore loops connect S5 and S6 extracellularly, while loops in the intracellular region participate in the regulation of fast inactivation (linker DIII–DIV) and regulation by endogenous molecules, with phosphorylation sites labelled P. (B) Cryo‐EM (cryogenic‐electron microscopy) structure of the eukaryotic NaVPaS channel α‐subunit (Shen et al., 2017). The ΝaV channel domains DI (red), DII (blue), DIII (yellow) and DIV (green) are shown with N‐ and C‐terminal domains coloured in magenta and cyan respectively. Glycosylations located in the extracellular loops S5S6 of DI and DIII are represented by sticks. (C) The extracellular view of the NaV channel showing the selective Na+ pore surrounded by the P‐loops, with key residues involved in the ion selectivity (asparagine in DI, glutamate in DII, lysine in DIII and alanine in DIV) represented by cyan spheres. Toxins binding sites 1–6 are indicated by circles. (D) Structural features of binding sites 1–6. Site 1 is located within the pore of the channel, site 2 within S6 of DI and DIV, site 3 within the extracellular loop connecting S3–S4 of DIV (magenta), site 4 within the extracellular loops connecting S1–S2 and S3–S4 of DII (magenta), site 5 at S5 of DIV and site 6 at S4 of DIV. Three‐dimensional structure was obtained from the RCSB Protein Data Bank and prepared in PyMol (DeLano, 2002).
Table 1.
Distribution of voltage‐gated sodium channels in the nervous system
| Sodium channel | Current type | Tissue expression | Subcellular localization | |
|---|---|---|---|---|
| NaV1.1 | TTX‐S, fast | CNS | Cerebellum, striatum, hippocampus and thalamus (Brysch et al., 1991; Black et al., 1994) | Neuronal soma and proximal dendrites (Whitaker et al., 2001) |
| PNS | DRG large cells, Aδ‐fibres and Aαβ‐fibres and outer plexiform layer (Black and Waxman, 1996; Mojumder et al., 2007; Fukuoka et al., 2008) | |||
| NaV1.2 | TTX‐S, fast | CNS | Cerebellum and hippocampus (Gong et al., 1999; Jarnot and Corbett, 2006) | Unmyelinated axons, presynaptic terminals and immature nodes of Ranvier (Westenbroek et al., 1989; Boiko et al., 2001) |
| PNS | Cochlear ganglion and nerves, myenteric neurons and outer plexiform layer (Hossain et al., 2005; Bartoo et al., 2006; Mojumder et al., 2007) | |||
| NaV1.3 | TTX‐S, fast | CNS | Neocortex, hippocampus and dentate gyrus (Westenbroek et al., 1992; Whitaker et al., 2000) | Neuronal soma and proximal dendrites |
| PNS | Absent | |||
| NaV1.6 | TTX‐S, fast | CNS | Cerebellum and hippocampus (Schaller and Caldwell, 2000) | Node of Ranvier and axon initial segment (Caldwell et al., 2000; Boiko et al., 2001) |
| PNS | DRG large and medium cells, Aδ‐fibres, Aαβ‐fibres, myenteric neurons, cochlear ganglion and nerves and outer plexiform layer (Black and Waxman, 1996; Hossain et al., 2005; Bartoo et al., 2006; Mojumder et al., 2007; Fukuoka et al., 2008) | |||
| NaV1.7 | TTX‐S, fast | CNS | Absent | Axons |
| PNS | All DRG cell sizes, C‐fibres, Aδ‐fibres and Aαβ‐fibres (Black and Waxman, 1996; Djouhri et al., 2003b; Fukuoka et al., 2008; Rupasinghe et al., 2012) | |||
| NaV1.8 | TTX‐R, slow | CNS | Absent | Axons |
| PNS | All DRG cells sizes, C‐fibres, Aδ‐fibres, Aαβ‐fibres and trigeminal and nodosal ganglia (Black and Waxman, 1996; Bongenhielm et al., 2000; Coward et al., 2000; Djouhri et al., 2003a; Stirling et al., 2005; Fukuoka et al., 2008) | |||
| NaV1.9 | TTX‐R, very slow | CNS | Absent | Soma and axons |
| PNS | DRG small and medium cells and fibres, enteric neurons and trigeminal ganglia (Dib‐Hajj et al., 1998b; Coward et al., 2000; Padilla et al., 2007; Fukuoka et al., 2008) | |||
The importance of NaV channels in therapeutics is highlighted by the clinical use of anticonvulsants, antiarrhythmic, local anaesthetics and analgesics that block NaV channels, usually non‐selectively. In addition, human molecular genetics studies have proven that inherited disorders such as cardiac arrhythmias (Wang et al., 1995; Kotta et al., 2010), epilepsy (Wallace et al., 2001; Escayg and Goldin, 2010), paralysis and myotonias (Jurkat‐Rott and Lehmann‐Horn, 2007; Zhao et al., 2012) and loss and gain of pain sensation, including congenital insensitivity to pain (CIP) (Cox et al., 2006; Danziger and Willer, 2009; Staud et al., 2011; Phatarakijnirund et al., 2016), erythromelalgia (Sheets et al., 2007; Skeik et al., 2012), idiopathic small nerve fibre neuropathy (Faber et al., 2012a) and paroxysmal extreme pain disorder (Fertleman et al., 2007; Choi et al., 2011) can be directly linked to mutations of genes encoding specific sodium channel subtypes. These insights generate singular opportunities for using selective NaV channel modulators to treat such complex disorders and have attracted considerable attention from both the academic and industry research sectors. Amongst these NaV channel modulators, natural toxins have shown remarkable modulatory properties by binding at specific sites of these channels (Figure 1C, D) and inducing various pharmacological effects.
In the light of the critical role of NaV channels in pain processes and the therapeutic potential of NaV channel modulators, it is essential to understand the distribution and function of these channels to guide the development of novel therapies targeting specific channel family members. This review describes the role of toxins in defining the distribution of these channel subtypes and how their role changes in pathological pain conditions. In addition, the therapeutic potential of natural NaV‐targeting toxins will be discussed.
Alterations in NaV channels during chronic neuropathic pain
Pain or noxious sensation is a natural defence mechanism associated with a distressing sensory experience due to actual or potential tissue damage. This mechanism comprises primary sensory neurons and axons in the PNS that detect noxious stimuli through ion channels and receptors, with NaV channels playing a key role in this nociceptive signalling. More recently, research has demonstrated in more detail the participation of some NaV channel subtypes in pain pathways. The http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=578 was revealed to be involved in mechanical but not thermal pain (Osteen et al., 2016), while http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=583 was found to be involved in multiple peripheral pain pathways including thermal pain (Deuis et al., 2013), and individuals lacking http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=584 or http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=586 function exhibit a congenital insensitivity to pain (CIP), with no other sensory abnormalities apart from anosmia (Cox et al., 2006; Phatarakijnirund et al., 2016). Besides their involvement in normally functioning acute pain pathways, NaV channels are also implicated in chronic pain disorders.
Pain disorders
NaV channels are involved in several painful chronic neuropathies as shown in Table 2. These conditions are characterized by sensory neurons membrane remodelling, comprising alterations in expression, trafficking and kinetics of NaV channel subtypes. Consequently, neurons generate action potentials spontaneously or under subthreshold stimulus, inducing ectopic pain characterized by hyperalgesia, allodynia and spontaneous pain. Chronic pain can be classified in two major types, neuropathic pain (nerve injury pain) and nociceptive pain (tissue injury and inflammatory pain). Although classified into two distinct conditions, there is clearly an overlap in these processes, with nerve injury inducing local inflammation, and inflammation leading to nerve damage.
Table 2.
Voltage‐gated sodium channels involved in chronic neuropathic pain and their distribution during neuropathy
| Sodium channel | Neuropathy type and references | Altered expression, distribution and function |
|---|---|---|
| NaV1.3 |
DRG axotomy (Waxman et al., 1994; Xiao et al., 2002) CCI (Dib‐Hajj et al., 1999; Chen et al., 2014) SNL (Boucher et al., 2000; Kim et al., 2001; Yin et al., 2016) Sciatic nerve CCI (Hains et al., 2004) Diabetic neuropathy (Hong et al., 2004) Inflammatory pain (Black et al., 2004) Spared nerve injury (Lindia et al., 2005) Post‐herpetic neuralgia (Garry et al., 2005) Trigeminal neuralgia (Xu et al., 2016) |
Up‐regulation in C‐fibres, Aδ‐fibres and Aαβ‐fibres Up‐regulation in DRG and trigeminal ganglion |
| NaV1.6 |
Infraorbital nerve injury (Henry et al., 2007) Diabetic neuropathy (Ren et al., 2012) CCI (Tseng et al., 2014) SNL (Xie et al., 2015) Trigeminal neuralgia (Grasso et al., 2016; Tanaka et al., 2016) |
Accumulation in nerve injury sites Up‐regulation in DRG neurons in diabetic neuropathy Gain of function mutation in trigeminal neuralgia |
| NaV1.7 |
Diabetic neuropathy (Hong et al., 2004) Human neuroma (Kretschmer et al., 2002) Inflammatory pain (Black et al., 2004; Nassar et al., 2004; Yeomans et al., 2005) IEM and PEPD (Fertleman et al., 2006; Drenth and Waxman, 2007) CIP (Cox et al., 2006) Trigeminal neuralgia (Xu et al., 2016) |
Accumulation in nerve injury sites Up‐regulation in DRG neurons in diabetic neuropathy and inflammatory pain Down‐regulation in trigeminal ganglia during neuralgia Gain of function mutations in IEM and PEPD, and loss of function in CIP |
| NaV1.8 |
Axotomy (Dib‐Hajj et al., 1996; Okuse et al., 1997) SNL (Okuse et al., 1997; Boucher et al., 2000) Human neuroma (Kretschmer et al., 2002) Diabetic neuropathy (Hong et al., 2004) Post‐herpetic neuralgia (Garry et al., 2005) Mechanical allodynia (Dong et al., 2007) Visceral pain (King et al., 2009) Chronic nerve compression (Frieboes et al., 2010) Inflammatory pain (Zhang et al., 2002; Bielefeldt et al., 2003; Lin et al., 2017) Painful neuropathy (Faber et al., 2012b) Trigeminal neuralgia (Xu et al., 2016) |
Accumulation in nerve injury sites Up‐regulation in DRG neurons in post‐herpetic neuralgia Down‐regulation in diabetic neuropathy, axotomy, SNL and in trigeminal ganglia during neuralgia Gain of function mutation in painful neuropathy |
| NaV1.9 |
Axotomy (Dib‐Hajj et al., 1998a) SNL (Boucher et al., 2000) Inflammatory pain (Priest et al., 2005) CCI (Tseng et al., 2014) Painful neuropathy (Huang et al., 2014) Trigeminal neuralgia (Xu et al., 2016) CIP (Phatarakijnirund et al., 2016) |
Accumulation in nerve injury sites Down‐regulation in axotomy, SNL and in trigeminal ganglia during neuralgia Gain of function mutation in painful neuropathy and loss of function in CIP |
CCI, chronic constriction injury; IEM, erythromelalgia; PEPD, paroxysmal extreme pain disorder; SNL, spinal nerve ligation.
Neuropathic pain
Neuropathic pain caused by nerve injury has diverse origins. Once the synthesis and transport of NaV channels is altered, an accumulation and/or displacement of NaV channels across the axon occurs, leading to membrane remodelling and changes in intrinsic electrical excitability. For example, NaV1.7, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=585 and NaV1.9 were shown to accumulate in the neuroma endings and in patches of demyelination in both animals and humans with neuropathic pain (Coward et al., 2000; Kretschmer et al., 2002). Interestingly, the remodelling of these channels is often not accompanied by gene expression alterations at the mRNA and protein levels, as is the case for NaV1.8. The expression of this channel is altered in a few conditions such as diabetic neuropathy and post‐herpetic neuralgia (Novakovic et al., 1998; Sleeper et al., 2000; Hong et al., 2004; Garry et al., 2005). Nerve injury and demyelination can also markedly alter the gene expression of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=580, which is observed in various neuropathy models (Waxman et al., 1994; Dib‐Hajj et al., 1999; Kim et al., 2001; Hong et al., 2004; Garry et al., 2005). The fast activation and inactivation kinetics of NaV1.3 associated with its rapid re‐priming kinetics and persistent current component is thought to contribute to the ectopic discharges and sustained firing rates in injured sensory neurons (Cummins et al., 2001), although knockout of NaV1.3 produces a mouse with normal pain (Nassar et al., 2006). Table 2 summarizes the alterations in NaV subtypes in chronic neuropathic pain conditions.
Nociceptive pain
In contrast to nerve injury, nociceptive pain can emerge from the modulation of NaV channels by inflammatory mediators. Tissue injury results in local inflammation, which induces pain by increasing the excitability of afferent neurons innervating the injured area. Multiple inflammatory mediators can give rise to pain by promoting the release of secondary mediators that exert a direct action on nociceptors.
The inflammatory mediator http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1883 sensitizes neurons and decreases pain thresholds in mechanical, thermal and chemical stimuli (Taiwo and Levine, 1991; Aley and Levine, 1999; Fitzgerald et al., 1999), probably through a direct effect on neurons as observed in vitro (Khasar et al., 1998). http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=286&familyType=ENZYME, a kinase induced by PGE2, sensitizes nociceptors by increasing tetrodotoxin‐resistant (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2616‐R) currents (Gold et al., 1998). Such kinases can also be activated by pronociceptive cytokines such as TNF‐α and nerve grow factor (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5026) that are also associated with hyperalgesia (Lewin et al., 1993; Shu and Mendell, 1999; Jin and Gereau, 2006; Pezet and McMahon, 2006; Fischer et al., 2017). NGF enhances TTX‐R currents by up‐regulating NaV1.8 expression and increasing excitability (Zhang et al., 2002; Bielefeldt et al., 2003; Lin et al., 2017). NaV1.9 currents are also increased by inflammatory agents, and knockout mice display attenuated inflammatory pain, including diabetic neuropathic pain (Craner et al., 2002; Rush and Waxman, 2004; Baker, 2005; Amaya et al., 2006; Binshtok et al., 2008). The pivotal role of NaV1.9 in chronic visceral pain has been shown in gut‐projecting dorsal root ganglion (DRG) neurons, with excitatory abnormal responses mediated by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1713 and PGE2 (Hockley et al., 2014; Hockley et al., 2016). Furthermore, both NaV1.8 and NaV1.9 were shown to be up‐regulated by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4940 (GDNF) in axotomized DRG neurons (Cummins et al., 2000).
Inflammatory mediators also affect TTX‐sensitive (TTX‐S) currents as an up‐regulation of NaV1.3 and NaV1.7 has been demonstrated in a carrageenan‐induced inflammatory pain model (Black et al., 2004). Knockdown of NaV1.7 also attenuates inflammatory pain (Nassar et al., 2004; Yeomans et al., 2005). Other mediators such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2844, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=649, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=989, plasma http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=479 and the pro‐inflammatory cytokine http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974 have also been demonstrated to directly modulate the excitability of sensory neurons through an action on NaV channels (Cui and Nicol, 1995; Gold et al., 1996; Griswold et al., 1999; Cardenas et al., 2001; Zhou et al., 2002; Binshtok et al., 2008). These inflammatory mediators along with the NaV subtypes altered in ectopic pain define a group of potential targets for the development of therapeutics.
Natural toxins targeting NaV channels and applications in pain therapies
Venoms are a rich and complex mixture of biologically active molecules, also called toxins, that have a variety of targets and functions. These have evolved into potent neurotoxins able to induce excitatory and inhibitory effects in the nervous system to disable preys and also to defend against predators (Klint et al., 2012; King and Hardy, 2013). The molecular targets of these neurotoxins include voltage‐gated ion channels, with NaV channels being remarkably modulated (Figures 2 and 3).
Figure 2.
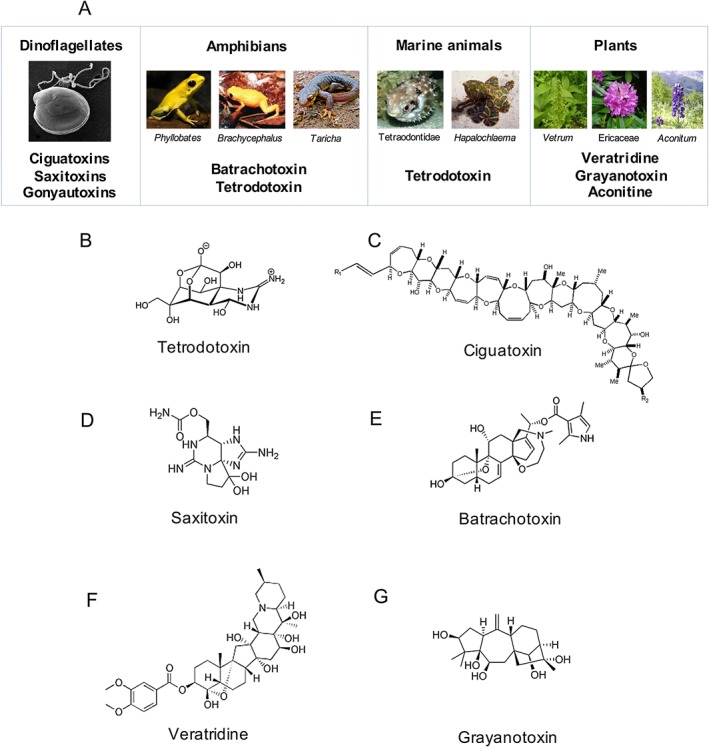
Natural small molecules toxins targeting NaV channels. (A) Ciguatoxins are guanidine‐based polyether ladder toxins, and saxitoxins and goyautoxins are also guanidine‐based toxins produced by dinoflagellates (Frace et al., 1986; Strachan et al., 1999; Thottumkara et al., 2014). Bratrachotoxins are isolated from the skin secretions of frog (Daly et al., 1965), TTX is isolated from salamanders such as of the genus Taricha, frogs such as of the genus Brachycephalus, puffer fish belonging to order Tetraodontidae and octopus of the species Hapalochlaema lunata (Hanifin, 2010; Williams et al., 2011; Lago et al., 2015) and veratridine, grayanotoxin and aconitine are isolated from plants of the genus Veratrum (Krayer and Acheson, 1946), family Ericaceae (Maejima et al., 2002) and genus Aconitum (Borcsa et al., 2014) respectively. (B–G) Structure of TTX, ciguatoxins, saxitoxins, batrachotoxin , veratridine and grayanotoxin is shown.
Figure 3.
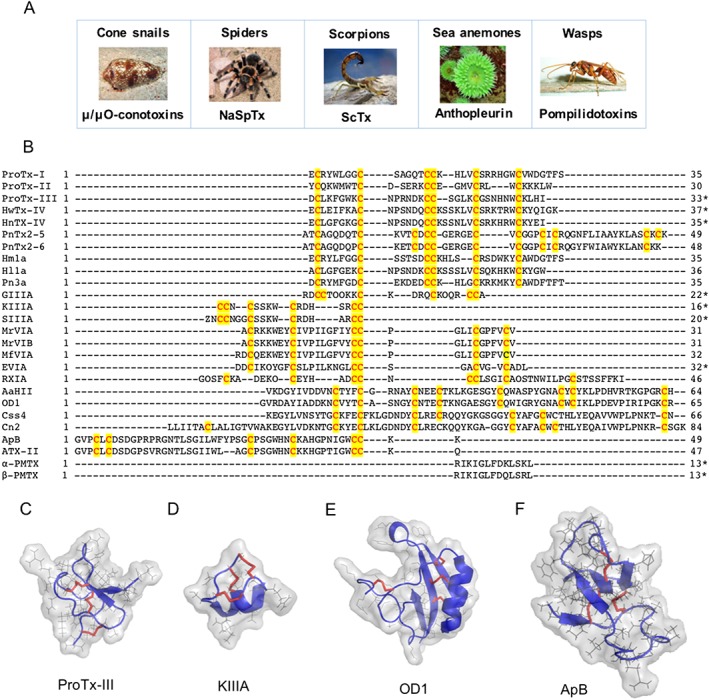
Natural peptide toxins targeting NaV channels. (A) μ/μO‐Conotoxins are a large group of peptidic toxins isolated from cone snails venoms (Lewis et al., 2012), NaSpTx is a large group of peptidic toxins isolated from spider venoms (Klint et al., 2012), ScTx is a group of toxins isolated from the venom of scorpions, anthopleurins are peptidic toxins isolated from sea anemone (Schweitz et al., 1981) and pompilidotoxins are peptides isolated from wasps (Konno et al., 1997; Konno et al., 1998). (B) Amino acid sequences alignment of the peptidic toxins discussed in this review, with highly conserved cysteines highlighted in red and yellow, and C‐terminal amidation denoted by an asterisk (*). Sequences were obtained from Arachnoserver, Conoserver and UniProt databases. (C–F) Structure of major classes of natural peptide toxins targeting NaV channels, including NaSpTx ProTx‐III isolated from the spider T. pruriens (Cardoso et al., 2015), μ‐conotoxin KIIIA isolated from Conus kinoshitai (Khoo et al., 2012), ScTx OD1 isolated from the scorpion Odonthobuthus doriae (Durek et al., 2013) and anthopleurin B (ApB) isolated from the sea anemone A. xanthogrammica (Monks et al., 1995). Disulfide bonds are represented as red sticks, amino acids side chains as grey lines and amino acids core chains as blue cartoon. Three‐dimensional structures were obtained from the RCSB Protein Data Bank and prepared in PyMol (DeLano, 2002).
To date, NaV modulators have been characterized from spiders, cone snails, scorpions, wasps, sea anemones, dinoflagellates, frogs, salamanders, puffer fish, octopus and plants such as from genera Vetrum and Aconitum (Figures 2 and 3; Table 3) (Herzig et al., 2011; Kaas et al., 2012; Pedraza Escalona and Possani, 2013; de O Beleboni et al., 2004; Wanke et al., 2009; Lehane and Lewis, 2000; Moczydlowski, 2013; Daly et al., 1965; Hanifin, 2010; Williams et al., 2011). These molecules display diverse chemical structures and modulatory properties and are capable of distinguishing amongst NaV subtypes through specific interactions with binding sites in these channels (Figure 1C, D). Their modulatory mechanisms have divided them into two major classes, pore blockers that bind to the ion conduction pore to inhibit Na+ influx and gating modifiers that interact with the segments S3, S4, S5 and S6 to modulate opening and inactivation.
Table 3.
Natural toxins targeting NaV channels
| Organism | Species | Toxin and references | NaV channel activity (order of potency) | Mode of action/potential binding sites |
|---|---|---|---|---|
| Spider | T. pruriens | ProTx‐I (Middleton et al., 2002) | NaV1.8 > NaV1.7 inhibitor | Gating modifier/site 4 or multiple sites |
| ProTx‐II (Middleton et al., 2002) | NaV1.7 > NaV1.6 > NaV1.2 > NaV1.5 > NaV1.3 > NaV1.8 inhibitor | |||
| ProTx‐III (Cardoso et al., 2015) | NaV1.7 > NaV1.6 > NaV1.2 > NaV1.1 > NaV1.3 inhibitor | |||
| H. huwenum | HwTx‐IV (Xiao et al., 2008a) | NaV1.7 > NaV1.2 > NaV1.3 > NaV1.4 inhibitor (Minassian et al., 2013) | ||
| H. haianum | HnTX‐IV (Liu et al., 2003) | NaV1.7 > NaV1.2 > NaV1.3 inhibitor | ||
| P. nigriventer | PnTx2–5 and PnTx2–6 (Matavel et al., 2002; Matavel et al., 2009) | NaVs activator | Gating modifier/site 3 | |
| Cone snail | C. geographus | GIIIA (Cruz et al., 1985) | NaV1.4 > NaV1.6 inhibitor | Pore blocker/site 1 |
| C. kinoshitai | KIIIA (Zhang et al., 2007) | NaV1.2 > NaV1.4 > NaV1.6 > NaV1.1 = NaV1.7 inhibitor | ||
| C. striatus | SIIIA (Wang et al., 2006) | NaV1.4 > NaV1.2 = NaV1.6 inhibitor | ||
| C. marmoreus | MrVIA/B (McIntosh et al., 1995) | NaV1.7 > NaV1.4 > NaV1.2 inhibitor | Gating modifier/sites 1 and 4 | |
| MrVIB (Ekberg et al., 2006) | NaV1.8 selective inhibitor | |||
| C. magnificus | MfIVA (Vetter et al., 2012a) | NaV1.4 and NaV1.8 inhibitor | ||
| C. ermineus | EVIA (Barbier et al., 2004) | NaV1.2 = NaV1.3 = NaV1.6 activator | Gating modifier/site 6 | |
| C. radiatus | RXIA (Buczek et al., 2007) | NaV1.2 > NaV1.6 > NaV1.7 activator | Not described | |
| Scorpion | A. australis | AaHII (Martin and Rochat, 1986) | NaV1.7 activator (Abbas et al., 2013) | Gating modifier/site 3 |
| O. doriae | OD1 (Jalali et al., 2005) | NaV1.7 > NaV1.4 > NaV1.6 activator (Durek et al., 2013) | ||
| C. suffusus | Css4 (Martin et al., 1987) | NaV activator (Cestele et al., 1998) | Gating modifier/site 4 | |
| C. noxius | Cn2 (Pintar et al., 1999) | NaV1.6 activator (Schiavon et al., 2006) | ||
| Sea anemone | A. xanthogramica | ApB (Schweitz et al., 1981) | NaV activator | Gating modifier/site 3 |
| A. sulcata | ATX‐II (Romey et al., 1976) | NaV1.1 = NaV1.2 > NaV1.5 > NaV1.4 > NaV1.6 activator (Oliveira et al., 2004) | ||
| Dinoflagellate | G. toxicus | CTX‐1 (Strachan et al., 1999) | TTX‐S, TTX‐R, NaV1.2, and NaV1.3 and NaV1.8 activator (Yamaoka et al., 2009; Zimmermann et al., 2013) | Gating modifier/site 5 |
| Alexandrium sp. | Saxitoxin (Thottumkara et al., 2014); Neosaxitoxin (Penzotti et al., 2001) | NaV inhibitors | Pore blocker/site 1 | |
| Gonyaulax sp. | Gonyautoxin (Frace et al., 1986) | |||
| Cyanobacteria | L. majuscula | Antillatoxin (Cao et al., 2010) | NaV activator | Not described |
| Puffer fish | Family Tetraodontidae | NaV1.1–1.4, NaV1.6–1.7 inhibitor | Pore blocker/site 1 | |
| Salamander | Genus Taricha | TTX (Hanifin, 2010; Williams et al., 2011; Lago et al., 2015) | ||
| Frog | Genus Brachycephalus | |||
| Octopus | H. lunata | |||
| Wasp | A. samariensis | α‐PMTX (Konno et al., 1997) | NaV1.1‐NaV1.3, NaV1.6 and NaV1.7 activators (Schiavon et al., 2010) | Not described |
| B. maculifrons | β‐PMTX (Konno et al., 1998) | |||
| Frog | Genus Phyllobates | BTX (Daly et al., 1965) | NaV activator | Gating modifier/site 2 |
| Plant | Genus Vetrum | Veratridine (Krayer and Acheson, 1946) | NaV activators | Gating modifier/site 2 |
| Family Ericaceae | Grayanotoxin (Maejima et al., 2002) | |||
| Genus Aconitum | Aconitine (Borcsa et al., 2014) |
The table shows representatives of major venomous species and respective toxins with highest potencies and diversity in modulatory mechanisms. Relative potencies are from references listed in the table, or from the database http://www.conoserver.org/ for cone snail toxins (Kaas et al., 2012) and from the database http://www.arachnoserver.org/ for spider toxins (Herzig et al., 2011)
Natural small molecules toxins
Small molecules were the first group of NaV modulators described (Figure 2; Table 3). The discovery of TTX, which is produced by microorganisms and bioaccumulated in the food chain by salamanders, frogs, puffer fish and octopus, further classified NaV channels in TTX‐R and TTX‐S channels (Hanifin, 2010; Williams et al., 2011; Lago et al., 2015). TTX incorporates a guanidinium group that blocks NaV1.1–NaV1.4 and NaV1.6–NaV1.7 with IC50 values in the single nanomolar range (Figure 2B). Similarly, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2625 (Thottumkara et al., 2014) (Figure 2D), neosaxitoxin (Penzotti et al., 2001) and gonyautoxin (Frace et al., 1986) are potent sodium channel inhibitors produced by aquatic microorganisms and bioaccumulated in the food chain. Guanidinium‐based toxins bind to the site 1 of NaV channels to plug the ion conducting pore (Chen and Chung, 2014). Extensive studies with TTX analogue have revealed a new metabolite 4,9 anhydro‐TTX with great selectivity for NaV1.6 (Rosker et al., 2007). Furthermore, TTX has shown in vivo efficacy in preclinical pain models of inflammatory and neuropathic pain (Beloeil et al., 2006; Marcil et al., 2006; Alvarez and Levine, 2015; Salas et al., 2015) (Table 4). These results suggest TTX is a potential lead for further development of NaV‐specific blockers.
Table 4.
Current toxins in in vivo preclinical and human clinical trials showing promising therapeutic efficacy for treating chronic pain
| Toxin |
Organism Species |
NaV subtypes targeted | Preclinical/clinical studies showing therapeutic efficacy | Reference |
|---|---|---|---|---|
| TTX |
Puffer fish Family Tetraodontidae |
NaV1.1–NaV1.4, NaV1.6 and NaV1.7 | Inflammatory and neuropathic muscle mechanical hyperalgesia | Alvarez and Levine (2015) |
| Inflammatory thermal and mechanical pain | Beloeil et al. (2006) | |||
| Inflammatory, visceral and neuropathic pain | Marcil et al. (2006) | |||
| Burn‐associated neuropathic pain | Salas et al. (2015) | |||
| Chemotherapy‐induced neuropathic pain (clinical trials) | Wex Pharmaceutical Inc. | |||
| Neosaxitoxin | Dinoflagellates | NaVs | Bladder pain syndrome (clinical trials) | Manriquez et al. (2015) |
| Gonyautoxin | Dinoflagellates | NaVs | Chronic tension‐type headache (clinical trials) | Lattes et al. (2009) |
| ProTx‐II |
Spider T. pruriens |
NaV1.7 | Painful diabetic neuropathy | Tanaka et al. (2015) |
| Inflammatory pain | Flinspach et al. (2017), Patent US20150099705 A1 | |||
| HnTX‐IV |
Spider H. haianum |
NaV1.2, NaV1.3 and NaV1.7 | SNI‐induced neuropathic pain and formalin‐induced inflammatory pain | Liu et al. (2014a) |
| μ‐TRTX‐Hl1a |
Spider H. lividium |
NaV1.8 | Inflammatory and neuropathic pain | Meng et al. (2016) |
| HwTx‐IV |
Spider O. huwena |
NaV1.7 | Inflammatory pain and SNI‐induced neuropathic pain | Liu et al. (2014b) |
| μ‐TRTX‐Pn3a |
Spider P. nigricolor |
NaV1.7 | Inflammatory pain, co‐administrated with opioid | Deuis et al. (2017) |
| μO‐MrVIB |
Cone snail C. marmoreus |
NaV1.8 | Allodynia and hyperalgesia associate to neuropathic and chronic inflammatory pain | Ekberg et al. (2006) |
| Post‐incision allodynia | Bulaj et al. (2006) | |||
| μO‐MfVIA |
Cone snail C. magnificus |
NaV1.4 and NaV1.8 | Inflammatory pain (analogue E5K and E8K MfVIA) | Deuis et al. (2016a) |
| μ‐KIIIA |
Cone snail C. kinoshitai |
NaV1.2, NaV1.4, NaV1.6 and NaV1.7 | Inflammatory pain | Zhang et al. (2007) and Han et al. (2009) |
| μ‐SIIIA |
Cone snail C. striatus |
NaV1.2, NaV1.4 and NaV1.6 | Inflammatory pain | Green et al. (2007) |
Another remarkable group of toxins that modulate NaV channels comprise the ciguatoxins and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2619 (Daly et al., 1965; Strachan et al., 1999) (Figure 2). These molecules are capable of activating the influx of Na+ through interactions with distinct domains of the NaV channels. Ciguatoxins are a ladder‐frame polyether toxin produced by marine dinoflagellates circum‐tropically (Figure 2A, C). They are bioaccumulated through the food chain and responsible for the ciguatera caused by the consumption of ciguateric reef fish. Ciguatoxins bind to site 5 of the NaV channel located on DIV to induce channel opening and increase Na+ permeability (Lombet et al., 1987). These molecules have played a key role in elucidating the mechanism associated with cold pain, revealing NaV1.8 and TTX‐S NaV channels in specific subsets of nerves containing http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=485 as main effectors in ciguatoxin‐induced cold allodynia (Vetter et al., 2012c).
Batrachotoxin is a steroidal alkaloid isolated from skin secretions of the frog genus Phyllobates present in South and Central America (Figure 2A, E). It is also believed batrachotoxin is bioaccumulated in the food chain, similar to TTX and ciguatoxins. It induces alterations in NaV channels through its interactions with site 2 located at DI and DIV, leading to channel depolarization and persistent activation at hyperpolarized potentials (Trainer et al., 1994; Trainer et al., 1996). Interestingly, batrachotoxins are also found in the integument of birds from the genus Pitohui in New Guinea (Weldon, 2000). Finally, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2626 is another NaV activator alkaloid that is produced by plants of the genus Veratrum (Krayer and Acheson, 1946) (Figure 2A, F). It binds to site 2 at DI and DIV and to site 6 at DIV S4 and leads to partial channel activation and stabilization of the open conformation for persistent opening (Wang and Wang, 2003; Yoshinaka‐Niitsu et al., 2012). Similarly, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2628 produced by plants from the family Ericaceae (Maejima et al., 2002) (Figure 2G), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2617 from the plant Aconitum (Borcsa et al., 2014) and antillatoxin produced by marine cyanobacterium (Cao et al., 2010) are potent NaV channel activators. Veratridine and antillatoxin in particular have been used in programmes for the discovery of NaV channel modulators due to its ability to specifically activate these channels in neuroblastomas and recombinant cells lines and facilitate assay development (Vetter et al., 2012b; Cardoso et al., 2015; Zhao et al., 2016). In addition, veratridine and grayanotoxin have been used in in vivo studies of pain mediated by NaV channels (Gingras et al., 2014; Cardoso et al., 2015).
Natural peptide toxins
Peptide toxins often target NaV channel subtypes selectively, making these attractive starting points to find better research probes with some showing potential as leads to improved pain treatments (Figure 3; Table 3). While finding the most promising amongst the millions of unique disulfide‐rich venom peptides, advances in high‐throughput screening, transcriptomic and proteomic studies have accelerated the rapid identification and isolation of new activators and inhibitors of NaV channels (Pineda et al., 2014; Prashanth et al., 2014; Cardoso et al., 2015; Klint et al., 2015; Deuis et al., 2017). Perhaps the most promising are the highly stable disulfide‐rich inhibitory cysteine knot (ICK) scaffold peptides common in spider and cone snail venoms, although the more complex disulfide‐rich structures are found in scorpions and sea anemones toxins (Figure 3C–F). Those venom peptides showing promising efficacy in reversing chronic pain in preclinical in vivo models of pain are shown in Table 4 and discussed in more detail in this review.
Pore‐blocker peptides
Sodium channel toxins present in cone snail venoms comprise a diverse array of small ICK peptides for prey capture and defence. While there are five families (μ‐conotoxin, μΟ‐conotoxin, δ‐conotoxin and ι‐conotoxin), only the μ‐conotoxins are pore blockers. μ‐Conotoxins bind to site 1 of the NaV channel and display potent subtype‐selective inhibition of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=579 and/or NaV1.4 as reported for http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2630 (Sato et al., 2015), KIIIA (Zhang et al., 2007) and SIIIA (Schroeder et al., 2008). These conotoxins have been used as part of a constellation pharmacology approach to determine the functional sodium channels found in native neuronal tissue such as the DRG (Teichert et al., 2012), as well as in the research for the discovery of new analgesics targeting NaV channels (Munasinghe and Christie, 2015).
The μ‐conotoxins KIIIA and SIIIA have shown promising in vivo therapeutic efficacy in preclinical inflammatory pain models (Green et al., 2007; Zhang et al., 2007; Han et al., 2009) (Table 4). KIIIA also potently inhibits NaV1.6 and NaV1.7, which are implicated in pain pathways and ectopic pain (Cox et al., 2006; Deuis et al., 2013) (Table 2), and potentially contribute to the analgesia observed. Alanine scan and related analogue studies revealed that K7 in KIIIA was an essential residue for the inhibition of NaV1.4 and NaV1.7, but not the inhibition of NaV1.2 and NaV1.6, with structurally minimized disulfide‐deficient KIIIA analogues providing an alternative scaffold for NaV inhibitors (Han et al., 2009). SIIIA shares 74% identity with KIIIA (Figure 3B), and besides inhibiting NaV1.2 and NaV1.4, it also inhibits NaV1.6 (Wang et al., 2006). Polyethylene glycol (PEG)‐SIIIA had improved efficacy compared to SIIIA in inflammatory pain (Green et al., 2007), showing the potential of this peptide class to be modified to improve pharmacodynamic properties, presumably by reducing renal clearance.
Gating modifier peptides
Gating modifiers are the most diverse group of NaV modulators venom peptides. They are present in venoms of cone snails, spiders, scorpions and sea anemones (Figure 3; Table 3) and have evolved to modify the gating properties of opening and inactivation of ion channels, causing alterations that either excite or inhibit channel function.
Conotoxins
The family of cone snail toxins that inhibit NaV channels as gating modifiers are the μO‐conotoxins. The toxins MrVIA and MrVIB were the first peptides belonging to this family to be characterized (McIntosh et al., 1995). These μO‐conotoxins bind to the NaV channel pore loop, but only residues present in the DIII seem to the involved in these interactions (Zorn et al., 2006). MrVIA also interacts with site 4 in the DII, sharing similar mechanisms with β‐scorpions toxins (Leipold et al., 2007). Curiously, the μO‐conotoxins ability to alter NaV gating properties is strongly regulated by the presence of β auxiliary subunits (Wilson et al., 2011). MrVIB was tested in models of neuropathic and inflammatory pain in rats (Table 4), where i.t. infusions of this peptide reversed neuropathic pain caused by partial ligation of the sciatic nerve and inflammatory pain induced by intraplantar injection of CFA (Ekberg et al., 2006). In a different study, local infusion of MrVIB into the area prior to a surgical incision produced long‐lasting reduction of post‐incision allodynia in rats (Bulaj et al., 2006), but effects on Nav1.4 may have interfered with the assessment of analgesia. More recently, a μO‐MfVIA (E5K and E8K‐MfVIA) analogue was developed with improved NaV1.8 activity that reduced inflammatory pain induced by intraplantar injection of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4196 (Deuis et al., 2016a) (Table 4).
Conotoxins belonging to the pharmacological families δ and ι can induce channel activation through interactions with site 6 in the DIV (Figure 3B; Table 3). EVIA was the first δ‐conotoxin described and shows preferential activation of NaV1.2, NaV1.3 and NaV1.6 (Barbier et al., 2004). The structure of EVIA revealed a typical ICK motif from the O superfamily of conotoxins (Volpon et al., 2004). Finally, RXIA is an ι‐conotoxin showing preference for activation of NaV1.2, NaV1.6 and NaV1.7 (Buczek et al., 2007). Although still poorly studied, this family of conotoxins revealed the presence of a d‐phenylalanine amino acid at position 44 of RXIA, which is essential for its excitatory function (Buczek et al., 2005). While not useful as analgesics, subtype‐specific activator toxins can be used to elucidate the role of different NaV subtypes in driving different pain pathways, as well as for the establishment of NaV‐specific pain models.
Spider toxins
To date, more than 40 000 species of spiders have been described, making this the largest group of venomous animals (Klint et al., 2012). Impressively, these venoms are also the richest source of NaV modulators, which are classified into 12 distinct families (NaSpTx1–12). NaSpTx produces a diverse array of modulatory effects on NaV channels. The venom of the spider Thrixopelma pruriens contains peptides amongst the most potent NaV inhibitors described to date, with ProTx‐I, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7571 and ProTx‐III showing remarkable potency to inhibit NaV1.7 (Middleton et al., 2002; Priest et al., 2007; Cardoso et al., 2015) (Table 3; Figure 3B, C). Although belonging in the same spider venom, these peptides contain distinct primary structures and are classified into three distinct NaSpTx families. ProTx‐I is a NaSpTx family 2 toxin firstly isolated in a NaV1.8 screen (Middleton et al., 2002). ProTx‐I also has http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=80 and http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=81 inhibitory activity, highlighting the challenge to find gating modifier toxins that are truly selective for the target of interest. In the same study, ProTx‐II, a NaSpTx family 3 toxin, showed similar activity at NaV1.8 and CaV3 channels but no activity against KV channels. The remarkable potency of ProTx‐II at NaV1.7 (IC50 0.3 nM) makes it the most potent NaSpTx peptide reported to date (Priest et al., 2007). Mechanism of action studies on ProTx‐II revealed interactions with DI, DII and DIV of NaV1.2 and DII and DIV of NaV1.7 that cause a shift in the voltage dependence of activation to more positive potentials and a slowing of fast inactivation to produce a persistent Na+ current (Bosmans et al., 2008; Xiao et al., 2010). ProTx‐III was identified in a NaV1.7‐targeting screen and belongs to the NaSpTx family 1. It has shown improved selectivity for NaV subtypes and a C‐terminal amidation that enhances NaV inhibitory activity by up to ninefold (Cardoso et al., 2015).
The unique potency of ProTx‐II for NaV1.7 makes it an attractive lead for the development of new pain therapies. Evaluation in a preclinical model of neuropathic pain showed ProTx‐II was able to reverse painful diabetic neuropathy through a reduction of thermal hyperalgesia (Tanaka et al., 2015) (Table 4). ProTx‐II analogues studies developed a highly specific and potent NaV1.7 inhibitor with very low affinity for NaV1.6 and no detectable activity for other NaV subtypes at relevant therapeutic concentrations (Flinspach et al., 2017). This peptide (JNJ63955918) differed from the wild‐type ProTx‐II at three positions, W7Q, W30L and N‐terminal addition of G(−1) and P(0). ProTx‐II and JNJ63955918 were evaluated in a formalin model of inflammatory pain, and both produced a significant reduction in phases I and II thermal pain, with no significant effect on motor function. JNJ63955918 and ProTxII bind preferentially to the closed state to inhibit NaV1.7 gating.
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7570 is another NaSpTx that belongs to the family 1. This toxin was isolated from the spider Haplopelma huwenum and has preferential inhibition for NaV1.7 and NaV1.2, followed by NaV1.3 and NaV1.4 (Xiao et al., 2008b; Minassian et al., 2013). HwTx‐IV binds to DII and traps the S4 voltage sensor in the closed configuration. Consistent with its inhibitory effect on NaV1.7, HwTx‐IV reversed hyperalgesia in in vivo models of inflammatory and neuropathic pain (Liu et al., 2014b) (Table 4). Mutants of HwTx‐IV were developed to enhance its inhibitory activity over NaV1.7, with (E1G,E4G,Y33W)HwTx‐IV showing a remarkable improvement in IC50 from 27 to 0.4 nM in electrophysiology assays (Revell et al., 2013). More recently, (E1G,E4G,F6W,Y33W)HwTx‐IV, a derivative of (E1G,E4G,Y33W)HwTx‐IV, was developed to enhance its binding to the lipidic membrane and this increased its potency at NaV1.7 compared with native HwTx‐IV from IC50 values of 32 to 7.5 nM in fluorescence imaging assays (Agwa et al., 2017).
HnTX‐IV isolated from the spider Haplopelma hainanum also belongs to the NaSpTx family 1. It has preferential inhibition for NaV1.7, followed by NaV1.2 and NaV1.3 (Liu et al., 2003), binding to DII to trap the S4 voltage sensor in the closed configuration (Cai et al., 2015). Structure–activity relationship studies revealed a mixed positively charged and hydrophobic surface essential for its inhibitory activity (Li et al., 2004; Liu et al., 2012). Consistent with its mode of action, HnTX‐IV is efficacious in various preclinical models of pain, alleviating inflammatory pain induced by acetic acid or formalin and allodynia induced by spared nerve injury in a neuropathic pain model (Liu et al., 2014a). In contrast, another likely NaSpTx family 10 toxin, μ‐TRTX‐Hl1a recently identified in the venom of Haplopelma lividium, preferentially inhibited NaV1.8 (Meng et al., 2016). This toxin significantly reversed inflammatory pain induced by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1058 and formalin, with an exceptional reversal of the paw‐licking response observed during phase II. Finally, the most recently identified NaSpTx family 2 toxin Pn3a was isolated from in the venom of the spider Pamphobeteus nigricolor. Pn3a was found to be a remarkably selective inhibitor of NaV1.7 and reversed pain behaviours in preclinical models of inflammatory pain induced by formalin, carrageenan and Freund's Complete Adjuvant, but only in the presence of subtherapeutic doses of opioids (Deuis et al., 2017).
In addition to the inhibitory NaSpTx described above, a large group of spider toxins preferentially activate NaV channels through interactions with site 3 in DIV. Classical examples are NaSpTx family 6 toxins PnTx2‐5 and PnTx2‐6 isolated from the spider Phoneutria nigriventer (Matavel et al., 2002), and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9553, NaSpTx family 2, isolated from the spider Heteroscodra maculata (Osteen et al., 2016). These toxins alter the voltage‐dependence of activation and inactivation of NaV channels and probably account for most of the symptoms of envenomation caused by P. nigriventer (Matavel et al., 2009). Interestingly, Hm1a displays specific activation of NaV1.1 and has been a unique pharmacological tool to characterize the involvement of NaV1.1 in pain, as previously discussed.
Scorpion toxins
Scorpions peptide toxins (ScTxs) acting on NaV channels are mostly activators that elicit strong pain. These long‐chain peptides are classified into two major groups, α‐ScTxs and β‐ScTxs. α‐ScTxs interact with site 3 at the NaV α‐subunit to slow fast inactivation and prolong channel opening, while β‐ScTxs interact with site 4 to alter the voltage‐dependence of activation and induce repetitive firing (Bosmans and Tytgat, 2007; Zhang et al., 2011). Within the α‐ScTxs, AaHII and OD1 are amongst the best characterized toxins (Figure 3; Table 3). AaHII was isolated from the scorpion Androctonus australis and has remarkable preference for NaV1.7 (Martin and Rochat, 1986; Abbas et al., 2013). AaHII alters the voltage‐dependence of activation with little effect on the voltage‐dependence of inactivation. OD1 is another potent α‐ScTx with preference for NaV1.4, NaV1.6 and NaV1.7 (Jalali et al., 2005; Durek et al., 2013). This particular toxin has been used to develop NaV1.7 in vitro and in vivo assays to rapidly identify and characterize peptides reversing pain behaviours induced by its injection (Cardoso et al., 2015; Deuis et al., 2016b). β‐ScTxs elicit similar changes in the NaV channels as those observed for the α‐ScTxs, although with preferential binding to DII loops S1S2 and S3S4. Amongst these toxins, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2620, isolated from the scorpion Centruroides suffusus, alters Na+ currents by altering the voltage‐dependence of activation and trapping the DII S4 voltage sensor in its outward position (Cestele et al., 1998). Finally, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2632, purified from the venom of Centruroides noxius, preferentially activates NaV1.6 (Schiavon et al., 2006) and has helped unravel the role of NaV1.6 channel in different pain pathways (Deuis et al., 2013).
Other peptide NaV toxins
Sea anemone venoms are another source of NaV toxins that enhance Na+ currents by inhibiting channel inactivation and prolonging action potential duration. These toxins share an overlapping binding site with α‐ScTxs in the DIV S3S4 loop associated with site 3 (Catterall and Beress, 1978). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2614 from Anemonia sulcata (Romey et al., 1976) preferentially activates NaV1.1 and NaV1.2 (Oliveira et al., 2004) and produces persistent and resurgent sodium currents in DRG neurons, but only in the presence of the β4 auxiliary subunit (Klinger et al., 2012). In contrast, anthopleurin B from Anthopleura xanthogrammica preferentially activates NaV1.5 (Khera et al., 1995) through an Arg12, Leu18 and Lys49 pharmacophore essential for activity (Gallagher and Blumenthal, 1994; Dias‐Kadambi et al., 1996). Finally, solitary wasps belonging to the family Pompilidae produce neurotoxic peptides in their venoms (Konno et al., 1997; Konno et al., 1998). Amongst these peptides, α‐PMTX and β‐PMTX isolated from Anopolis samariensis and Batozonellus maculifrons, respectively, are small linear peptides that are structurally distinct from most natural peptidic NaV toxins (Figure 3B). Interestingly, these peptides activate Na+ currents by increasing the steady‐state currents at NaV1.6 or by slowing NaV1.1, NaV1.2, NaV1.3 and NaV1.7 inactivation currents (Schiavon et al., 2010).
Conclusions and future directions
Ranging from inhibitors to activators of NaV channels, natural toxins have proven to be a powerful group of molecules for unravelling the role of NaV channels in pain pathways and as leads towards the development of novel therapies for treating chronic pain. The discovery that NaV1.1, NaV1.3, NaV1.6, NaV1.7, NaV1.8 and NaV1.9 are involved in pain pathways commenced a new era of research for therapeutics targeting these channels. The revelation that humans with mutations causing loss of NaV1.7 function were pain free but otherwise normal (except for loss of olfaction) has led to an expanded effort to find specific inhibitors of this channel with therapeutic potential. TTX has now advanced into human clinical trials for treating chemotherapy‐induced neuropathic pain (Wex Pharmaceuticals Inc.), while neosaxitoxin, tested as a blocker of bladder pain syndrome, and gonyautoxin, as blocking chronic tension‐type headache, have been shown to be safe and effective in humans (Lattes et al., 2009; Manriquez et al., 2015) (Table 4). In addition, a considerable number of active programmes in pharmaceutical companies have used natural NaV toxins such as marine toxins (e.g. SiteOne Therapeutics) and spider toxins (e.g. Amgen Inc.) in the development of novel analgesics to treat, previously untreatable, chronic pain conditions. Gating modifiers are arguably the most promising source of NaV inhibitors for pain because the binding sites are on the voltage sensors of these channels, which vary amongst NaV subtypes. In addition to their value in dissecting the role of sodium channels in different types of pain, NaV modulators have the potential to be a new class of drugs targeting NaV channels for the treatment of chronic pain.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the Australian National Health & Medical Research Council (Programme Grant APP1072113 to R.J.L. and Principal Research Fellowship to R.J.L.).
Cardoso, F. C. , and Lewis, R. J. (2018) Sodium channels and pain: from toxins to therapies. British Journal of Pharmacology, 175: 2138–2157. doi: 10.1111/bph.13962.
References
- Abbas N, Gaudioso‐Tyzra C, Bonnet C, Gabriac M, Amsalem M, Lonigro A et al (2013). The scorpion toxin Amm VIII induces pain hypersensitivity through gain‐of‐function of TTX‐sensitive Na(+) channels. Pain 154: 1204–1215. [DOI] [PubMed] [Google Scholar]
- Agwa AJ, Lawrence N, Deplazes E, Cheneval O, Chen R, Craik DJ et al (2017). Spider peptide toxin HwTx‐IV engineered to bind to lipid membranes has an increased inhibitory potency at human voltage‐gated sodium channel hNaV1.7. Biochim Biophys Acta . [DOI] [PubMed] [Google Scholar]
- Alabi AA, Bahamonde MI, Jung HJ, Kim JI, Swartz KJ (2007). Portability of paddle motif function and pharmacology in voltage sensors. Nature 450: 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Levine JD (1999). Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci 19: 2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez P, Levine JD (2015). Antihyperalgesic effect of tetrodotoxin in rat models of persistent muscle pain. Neuroscience 311: 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya F, Wang H, Costigan M, Allchorne AJ, Hatcher JP, Egerton J et al (2006). The voltage‐gated sodium channel NaV1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci 26: 12852–12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrikopoulos P, Fraser SP, Patterson L, Ahmad Z, Burcu H, Ottaviani D et al (2011). Angiogenic functions of voltage‐gated Na+ Channels in human endothelial cells: modulation of vascular endothelial growth factor (VEGF) signaling. J Biol Chem 286: 16846–16860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MD (2005). Protein kinase C mediates up‐regulation of tetrodotoxin‐resistant, persistent Na+ current in rat and mouse sensory neurones. J Physiol 567: 851–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier J, Lamthanh H, Le Gall F, Favreau P, Benoit E, Chen H et al (2004). A δ‐conotoxin from Conus ermineus venom inhibits inactivation in vertebrate neuronal Na+ channels but not in skeletal and cardiac muscles. J Biol Chem 279: 4680–4685. [DOI] [PubMed] [Google Scholar]
- Bartoo AC, Sprunger LK, Schneider DA (2006). Expression of sodium channel NaV1.6 in cholinergic myenteric neurons of guinea pig proximal colon. Cell Tissue Res 325: 203–209. [DOI] [PubMed] [Google Scholar]
- Beloeil H, Ababneh Z, Chung R, Zurakowski D, Mulkern RV, Berde CB (2006). Effects of bupivacaine and tetrodotoxin on carrageenan‐induced hind paw inflammation in rats (part 1): hyperalgesia, edema, and systemic cytokines. Anesthesiology 105: 128–138. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Ozaki N, Gebhart GF (2003). Role of nerve growth factor in modulation of gastric afferent neurons in the rat. Am J Physiol Gastrointest Liver Physiol 284: G499–G507. [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L et al (2008). Nociceptors are interleukin‐1β sensors. J Neurosci 28: 14062–14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JA, Yokoyama S, Higashida H, Ransom BR, Waxman SG (1994). Sodium channel mRNAs I, II and III in the CNS: cell‐specific expression. Brain Res Mol Brain Res 22: 275–289. [DOI] [PubMed] [Google Scholar]
- Black JA, Waxman SG (1996). Sodium channel expression: a dynamic process in neurons and non‐neuronal cells. Dev Neurosci 18: 139–152. [DOI] [PubMed] [Google Scholar]
- Black JA, Liu S, Tanaka M, Cummins TR, Waxman SG (2004). Changes in the expression of tetrodotoxin‐sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108: 237–247. [DOI] [PubMed] [Google Scholar]
- Black JA, Newcombe J, Waxman SG (2013). NaV1.5 sodium channels in macrophages in multiple sclerosis lesions. Mult Scler 19: 532–542. [DOI] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS et al (2001). Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30: 91–104. [DOI] [PubMed] [Google Scholar]
- Bongenhielm U, Nosrat CA, Nosrat I, Eriksson J, Fjell J, Fried K (2000). Expression of sodium channel SNS/PN3 and ankyrin(G) mRNAs in the trigeminal ganglion after inferior alveolar nerve injury in the rat. Exp Neurol 164: 384–395. [DOI] [PubMed] [Google Scholar]
- Borcsa B, Fodor L, Csupor D, Forgo P, Molnar A, Hohmann J (2014). Diterpene alkaloids from the roots of Aconitum moldavicum and assessment of NaV1.2 sodium channel activity of aconitum alkaloids. Planta Med 80: 231–236. [DOI] [PubMed] [Google Scholar]
- Bosmans F, Tytgat J (2007). Voltage‐gated sodium channel modulation by scorpion α‐toxins. Toxicon 49: 142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosmans F, Martin‐Eauclaire MF, Swartz KJ (2008). Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456: 202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB (2000). Potent analgesic effects of GDNF in neuropathic pain states. Science 290: 124–127. [DOI] [PubMed] [Google Scholar]
- Brysch W, Creutzfeldt OD, Luno K, Schlingensiepen R, Schlingensiepen KH (1991). Regional and temporal expression of sodium channel messenger RNAs in the rat brain during development. Exp Brain Res 86: 562–567. [DOI] [PubMed] [Google Scholar]
- Buczek O, Yoshikami D, Bulaj G, Jimenez EC, Olivera BM (2005). Post‐translational amino acid isomerization: a functionally important D‐amino acid in an excitatory peptide. J Biol Chem 280: 4247–4253. [DOI] [PubMed] [Google Scholar]
- Buczek O, Wei D, Babon JJ, Yang X, Fiedler B, Chen P et al (2007). Structure and sodium channel activity of an excitatory I1‐superfamily conotoxin. Biochemistry 46: 9929–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulaj G, Zhang MM, Green BR, Fiedler B, Layer RT, Wei S et al (2006). Synthetic μO‐conotoxin MrVIB blocks TTX‐resistant sodium channel NaV1.8 and has a long‐lasting analgesic activity. Biochemistry 45: 7404–7414. [DOI] [PubMed] [Google Scholar]
- Cai T, Luo J, Meng E, Ding J, Liang S, Wang S et al (2015). Mapping the interaction site for the tarantula toxin hainantoxin‐IV (β‐TRTX‐Hn2a) in the voltage sensor module of domain II of voltage‐gated sodium channels. Peptides 68: 148–156. [DOI] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR (2000). Sodium channel NaV1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A 97: 5616–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Gerwick WH, Murray TF (2010). Antillatoxin is a sodium channel activator that displays unique efficacy in heterologously expressed rNaV1.2, rNaV1.4 and rNaV1.5 α subunits. BMC Neurosci 11: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas LM, Cardenas CG, Scroggs RS (2001). 5HT increases excitability of nociceptor‐like rat dorsal root ganglion neurons via cAMP‐coupled TTX‐resistant Na(+) channels. J Neurophysiol 86: 241–248. [DOI] [PubMed] [Google Scholar]
- Cardoso FC, Dekan Z, Rosengren KJ, Erickson A, Vetter I, Deuis J et al (2015). Identification and characterization of ProTx‐III [μ‐TRTX‐Tp1a], a new voltage‐gated sodium channel inhibitor from venom of the tarantula Thrixopelma pruriens . Mol Pharmacol 88: 291–303. [DOI] [PubMed] [Google Scholar]
- Carrithers MD, Dib‐Hajj S, Carrithers LM, Tokmoulina G, Pypaert M, Jonas EA et al (2007). Expression of the voltage‐gated sodium channel NaV1.5 in the macrophage late endosome regulates endosomal acidification. J Immunol 178: 7822–7832. [DOI] [PubMed] [Google Scholar]
- Carrithers MD, Chatterjee G, Carrithers LM, Offoha R, Iheagwara U, Rahner C et al (2009). Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A. J Biol Chem 284: 8114–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Beress L (1978). Sea anemone toxin and scorpion toxin share a common receptor site associated with the action potential sodium ionophore. J Biol Chem 253: 7393–7396. [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG (2005). International Union of Pharmacology. XLVII. Nomenclature and structure‐function relationships of voltage‐gated sodium channels. Pharmacol Rev 57: 397–409. [DOI] [PubMed] [Google Scholar]
- Cestele S, Qu Y, Rogers JC, Rochat H, Scheuer T, Catterall WA (1998). Voltage sensor‐trapping: enhanced activation of sodium channels by β‐scorpion toxin bound to the S3‐S4 loop in domain II. Neuron 21: 919–931. [DOI] [PubMed] [Google Scholar]
- Chen HP, Zhou W, Kang LM, Yan H, Zhang L, Xu BH et al (2014). Intrathecal miR‐96 inhibits NaV1.3 expression and alleviates neuropathic pain in rat following chronic construction injury. Neurochem Res 39: 76–83. [DOI] [PubMed] [Google Scholar]
- Chen R, Chung SH (2014). Mechanism of tetrodotoxin block and resistance in sodium channels. Biochem Biophys Res Commun 446: 370–374. [DOI] [PubMed] [Google Scholar]
- Choi JS, Boralevi F, Brissaud O, Sanchez‐Martin J, Te Morsche RH, Dib‐Hajj SD et al (2011). Paroxysmal extreme pain disorder: a molecular lesion of peripheral neurons. Nat Rev Neurol 7: 51–55. [DOI] [PubMed] [Google Scholar]
- Coward K, Plumpton C, Facer P, Birch R, Carlstedt T, Tate S et al (2000). Immunolocalization of SNS/PN3 and NaN/SNS2 sodium channels in human pain states. Pain 85: 41–50. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K et al (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444: 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craner MJ, Klein JP, Renganathan M, Black JA, Waxman SG (2002). Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann Neurol 52: 786–792. [DOI] [PubMed] [Google Scholar]
- Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D et al (1985). Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J Biol Chem 260: 9280–9288. [PubMed] [Google Scholar]
- Cui M, Nicol GD (1995). Cyclic AMP mediates the prostaglandin E2‐induced potentiation of bradykinin excitation in rat sensory neurons. Neuroscience 66: 459–466. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Black JA, Dib‐Hajj SD, Waxman SG (2000). Glial‐derived neurotrophic factor upregulates expression of functional SNS and NaN sodium channels and their currents in axotomized dorsal root ganglion neurons. J Neurosci 20: 8754–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Aglieco F, Renganathan M, Herzog RI, Dib‐Hajj SD, Waxman SG (2001). NaV1.3 sodium channels: rapid repriming and slow closed‐state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci 21: 5952–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly JW, Witkop B, Bommer P, Biemann K (1965). Batrachotoxin. The active principle of the Colombian arrow poison frog, Phyllobates bicolor . J Am Chem Soc 87: 124–126. [DOI] [PubMed] [Google Scholar]
- Danziger N, Willer JC (2009). Congenital insensitivity to pain. Rev Neurol (Paris) 165: 129–136. [DOI] [PubMed] [Google Scholar]
- de O Beleboni R, Pizzo AB, Fontana AC, de O G Carolino R, Coutinho‐Netto J, Dos Santos WF (2004). Spider and wasp neurotoxins: pharmacological and biochemical aspects. Eur J Pharmacol 493: 1–17. [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002). The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.
- Deuis JR, Zimmermann K, Romanovsky AA, Possani LD, Cabot PJ, Lewis RJ et al (2013). An animal model of oxaliplatin‐induced cold allodynia reveals a crucial role for NaV1.6 in peripheral pain pathways. Pain 154: 1749–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuis JR, Dekan Z, Inserra MC, Lee TH, Aguilar MI, Craik DJ et al (2016b). Development of a μO‐conotoxin analogue with improved lipid membrane interactions and potency for the analgesic sodium channel NaV1.8. J Biol Chem 291: 11829–11842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuis JR, Wingerd JS, Winter Z, Durek T, Dekan Z, Zimmermann K et al (2016a). Analgesic effects of GpTx‐1, PF‐04856264 and CNV1014802 in a mouse model of NaV1.7‐mediated pain. Toxins 8: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuis JR, Dekan Z, Wingerd JS, Smith JJ, Munasinghe NR, Bhola RF et al (2017). Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide Pn3a. Sci Rep 7: 40883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias‐Kadambi BL, Drum CL, Hanck DA, Blumenthal KM (1996). Leucine 18, a hydrophobic residue essential for high affinity binding of anthopleurin B to the voltage‐sensitive sodium channel. J Biol Chem 271: 9422–9428. [DOI] [PubMed] [Google Scholar]
- Dib‐Hajj S, Black JA, Felts P, Waxman SG (1996). Down‐regulation of transcripts for Na channel α‐SNS in spinal sensory neurons following axotomy. Proc Natl Acad Sci U S A 93: 14950–14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Black JA, Cummins TR, Kenney AM, Kocsis JD, Waxman SG (1998b). Rescue of α‐SNS sodium channel expression in small dorsal root ganglion neurons after axotomy by nerve growth factor in vivo . J Neurophysiol 79: 2668–2676. [DOI] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Tyrrell L, Black JA, Waxman SG (1998a). NaN, a novel voltage‐gated Na channel, is expressed preferentially in peripheral sensory neurons and down‐regulated after axotomy. Proc Natl Acad Sci U S A 95: 8963–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Fjell J, Cummins TR, Zheng Z, Fried K, LaMotte R et al (1999). Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 83: 591–600. [DOI] [PubMed] [Google Scholar]
- Djouhri L, Fang X, Okuse K, Wood JN, Berry CM, Lawson SN (2003b). The TTX‐resistant sodium channel NaV1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J Physiol 550: 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouhri L, Newton R, Levinson SR, Berry CM, Carruthers B, Lawson SN (2003a). Sensory and electrophysiological properties of guinea‐pig sensory neurones expressing NaV 1.7 (PN1) Na+ channel α subunit protein. J Physiol 546: 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XW, Goregoaker S, Engler H, Zhou X, Mark L, Crona J et al (2007). Small interfering RNA‐mediated selective knockdown of NaV1.8 tetrodotoxin‐resistant sodium channel reverses mechanical allodynia in neuropathic rats. Neuroscience 146: 812–821. [DOI] [PubMed] [Google Scholar]
- Drenth JP, Waxman SG (2007). Mutations in sodium‐channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest 117: 3603–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durek T, Vetter I, Wang CI, Motin L, Knapp O, Adams DJ et al (2013). Chemical engineering and structural and pharmacological characterization of the α‐scorpion toxin OD1. ACS Chem Biol 8: 1215–1222. [DOI] [PubMed] [Google Scholar]
- Ekberg J, Jayamanne A, Vaughan CW, Aslan S, Thomas L, Mould J et al (2006). μO‐conotoxin MrVIB selectively blocks NaV1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficits. Proc Natl Acad Sci U S A 103: 17030–17035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekmehag B, Persson B, Rorsman P, Rorsman H (1994). Demonstration of voltage‐dependent and TTX‐sensitive Na(+)‐channels in human melanocytes. Pigment Cell Res 7: 333–338. [DOI] [PubMed] [Google Scholar]
- Escayg A, Goldin AL (2010). Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51: 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS et al (2012a). Gain of function NaV1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 71: 26–39. [DOI] [PubMed] [Google Scholar]
- Faber CG, Lauria G, Merkies IS, Cheng X, Han C, Ahn HS et al (2012b). Gain‐of‐function NaV1.8 mutations in painful neuropathy. Proc Natl Acad Sci U S A 109: 19444–19449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B et al (2006). SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52: 767–774. [DOI] [PubMed] [Google Scholar]
- Fertleman CR, Ferrie CD, Aicardi J, Bednarek NA, Eeg‐Olofsson O, Elmslie FV et al (2007). Paroxysmal extreme pain disorder (previously familial rectal pain syndrome). Neurology 69: 586–595. [DOI] [PubMed] [Google Scholar]
- Fischer BD, Ho C, Kuzin I, Bottaro A, O'Leary ME (2017). Chronic exposure to tumor necrosis factor in vivo induces hyperalgesia, upregulates sodium channel gene expression and alters the cellular electrophysiology of dorsal root ganglion neurons. Neurosci Lett 653: 195–201. [DOI] [PubMed] [Google Scholar]
- Fitzgerald EM, Okuse K, Wood JN, Dolphin AC, Moss SJ (1999). cAMP‐dependent phosphorylation of the tetrodotoxin‐resistant voltage‐dependent sodium channel SNS. J Physiol 516 (Pt 2): 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinspach M, Xu Q, Piekarz AD, Fellows R, Hagan R, Gibbs A et al (2017). Insensitivity to pain induced by a potent selective closed‐state NaV1.7 inhibitor. Sci Rep 7: 39662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frace AM, Hall S, Brodwick MS, Eaton DC (1986). Effects of saxitoxin analogues and ligand competition on sodium currents of squid axons. Am J Physiol 251: C159–C166. [DOI] [PubMed] [Google Scholar]
- Frieboes LR, Palispis WA, Gupta R (2010). Nerve compression activates selective nociceptive pathways and upregulates peripheral sodium channel expression in Schwann cells. J Orthop Res 28: 753–761. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Kobayashi K, Yamanaka H, Obata K, Dai Y, Noguchi K (2008). Comparative study of the distribution of the α‐subunits of voltage‐gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J Comp Neurol 510: 188–206. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, Blumenthal KM (1994). Importance of the unique cationic residues arginine 12 and lysine 49 in the activity of the cardiotonic polypeptide anthopleurin B. J Biol Chem 269: 254–259. [PubMed] [Google Scholar]
- Garry EM, Delaney A, Anderson HA, Sirinathsinghji EC, Clapp RH, Martin WJ et al (2005). Varicella zoster virus induces neuropathic changes in rat dorsal root ganglia and behavioral reflex sensitisation that is attenuated by gabapentin or sodium channel blocking drugs. Pain 118: 97–111. [DOI] [PubMed] [Google Scholar]
- Gingras J, Smith S, Matson DJ, Johnson D, Nye K, Couture L et al (2014). Global NaV1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS One 9: e105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Levine JD, Correa AM (1998). Modulation of TTX‐R INa by PKC and PKA and their role in PGE2‐induced sensitization of rat sensory neurons in vitro . J Neurosci 18: 10345–10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, Levine JD (1996). Hyperalgesic agents increase a tetrodotoxin‐resistant Na+ current in nociceptors. Proc Natl Acad Sci U S A 93: 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Rhodes KJ, Bekele‐Arcuri Z, Trimmer JS (1999). Type I and type II Na(+) channel α‐subunit polypeptides exhibit distinct spatial and temporal patterning, and association with auxiliary subunits in rat brain. J Comp Neurol 412: 342–352. [PubMed] [Google Scholar]
- Grasso G, Landi A, Alafaci C (2016). A novel pathophysiological mechanism contributing to trigeminal neuralgia. Mol Med 22: 452–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green BR, Catlin P, Zhang MM, Fiedler B, Bayudan W, Morrison A et al (2007). Conotoxins containing nonnatural backbone spacers: cladistic‐based design, chemical synthesis, and improved analgesic activity. Chem Biol 14: 399–407. [DOI] [PubMed] [Google Scholar]
- Griswold DE, Douglas SA, Martin LD, Davis TG, Davis L, Ao Z et al (1999). Endothelin B receptor modulates inflammatory pain and cutaneous inflammation. Mol Pharmacol 56: 807–812. [PubMed] [Google Scholar]
- Hains BC, Saab CY, Klein JP, Craner MJ, Waxman SG (2004). Altered sodium channel expression in second‐order spinal sensory neurons contributes to pain after peripheral nerve injury. J Neurosci 24: 4832–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TS, Zhang MM, Walewska A, Gruszczynski P, Robertson CR, Cheatham TE 3rd et al (2009). Structurally minimized μ‐conotoxin analogues as sodium channel blockers: implications for designing conopeptide‐based therapeutics. Chem Med Chem 4: 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanifin CT (2010). The chemical and evolutionary ecology of tetrodotoxin (TTX) toxicity in terrestrial vertebrates. Mar Drugs 8: 577–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry MA, Freking AR, Johnson LR, Levinson SR (2007). Sodium channel NaV1.6 accumulates at the site of infraorbital nerve injury. BMC Neurosci 8: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig V, Wood DL, Newell F, Chaumeil PA, Kaas Q, Binford GJ et al (2011). Arachno Server 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res 39: D653–D657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockley JR, Boundouki G, Cibert‐Goton V, McGuire C, Yip PK, Chan C et al (2014). Multiple roles for NaV1.9 in the activation of visceral afferents by noxious inflammatory, mechanical, and human disease‐derived stimuli. Pain 155: 1962–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockley JR, Winchester WJ, Bulmer DC (2016). The voltage‐gated sodium channel NaV 1.9 in visceral pain. Neurogastroenterol Motil 28: 316–326. [DOI] [PubMed] [Google Scholar]
- Hong S, Morrow TJ, Paulson PE, Isom LL, Wiley JW (2004). Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin‐sensitive and ‐resistant sodium channels in dorsal root ganglion neurons in the rat. J Biol Chem 279: 29341–29350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain WA, Antic SD, Yang Y, Rasband MN, Morest DK (2005). Where is the spike generator of the cochlear nerve? Voltage‐gated sodium channels in the mouse cochlea. J Neurosci 25: 6857–6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Han C, Estacion M, Vasylyev D, Hoeijmakers JG, Gerrits MM et al (2014). Gain‐of‐function mutations in sodium channel NaV1.9 in painful neuropathy. Brain 137: 1627–1642. [DOI] [PubMed] [Google Scholar]
- Huh S, Jung E, Lee J, Roh K, Kim JD, Lee J et al (2010). Mechanisms of melanogenesis inhibition by propafenone. Arch Dermatol Res 302: 561–565. [DOI] [PubMed] [Google Scholar]
- Jalali A, Bosmans F, Amininasab M, Clynen E, Cuypers E, Zaremirakabadi A et al (2005). OD1, the first toxin isolated from the venom of the scorpion Odonthobuthus doriae active on voltage‐gated Na+ channels. FEBS Lett 579: 4181–4186. [DOI] [PubMed] [Google Scholar]
- Jarnot M, Corbett AM (2006). Immunolocalization of NaV1.2 channel subtypes in rat and cat brain and spinal cord with high affinity antibodies. Brain Res 1107: 1–12. [DOI] [PubMed] [Google Scholar]
- Jin X, Gereau RW (2006). Acute p38‐mediated modulation of tetrodotoxin‐resistant sodium channels in mouse sensory neurons by tumor necrosis factor‐α. J Neurosci 26: 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkat‐Rott K, Lehmann‐Horn F (2007). Genotype‐phenotype correlation and therapeutic rationale in hyperkalemic periodic paralysis. Neurotherapeutics 4: 216–224. [DOI] [PubMed] [Google Scholar]
- Kaas Q, Yu R, Jin AH, Dutertre S, Craik DJ (2012). ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res 40: D325–D330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Gold MS, Levine JD (1998). A tetrodotoxin‐resistant sodium current mediates inflammatory pain in the rat. Neurosci Lett 256: 17–20. [DOI] [PubMed] [Google Scholar]
- Khera PK, Benzinger GR, Lipkind G, Drum CL, Hanck DA, Blumenthal KM (1995). Multiple cationic residues of anthopleurin B that determine high affinity and channel isoform discrimination. Biochemistry 34: 8533–8541. [DOI] [PubMed] [Google Scholar]
- Khoo KK, Gupta K, Green BR, Zhang MM, Watkins M, Olivera BM et al (2012). Distinct disulfide isomers of μ‐conotoxins KIIIA and KIIIB block voltage‐gated sodium channels. Biochemistry 51: 9826–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Oh Y, Chung JM, Chung K (2001). The changes in expression of three subtypes of TTX sensitive sodium channels in sensory neurons after spinal nerve ligation. Brain Res Mol Brain Res 95: 153–161. [DOI] [PubMed] [Google Scholar]
- King DE, Macleod RJ, Vanner SJ (2009). Trinitrobenzenesulphonic acid colitis alters NaV 1.8 channel expression in mouse dorsal root ganglia neurons. Neurogastroenterol Motil 21: 880–e864. [DOI] [PubMed] [Google Scholar]
- King GF, Hardy MC (2013). Spider‐venom peptides: structure, pharmacology, and potential for control of insect pests. Annu Rev Entomol 58: 475–496. [DOI] [PubMed] [Google Scholar]
- Klinger AB, Eberhardt M, Link AS, Namer B, Kutsche LK, Schuy ET et al (2012). Sea‐anemone toxin ATX‐II elicits A‐fiber‐dependent pain and enhances resurgent and persistent sodium currents in large sensory neurons. Mol Pain 8: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klint JK, Senff S, Rupasinghe DB, Er SY, Herzig V, Nicholson GM et al (2012). Spider‐venom peptides that target voltage‐gated sodium channels: pharmacological tools and potential therapeutic leads. Toxicon 60: 478–491. [DOI] [PubMed] [Google Scholar]
- Klint JK, Smith JJ, Vetter I, Rupasinghe DB, Er SY, Senff S et al (2015). Seven novel modulators of the analgesic target NaV1.7 uncovered using a high‐throughput venom‐based discovery approach. Br J Pharmacol 172: 2445–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno K, Hisada M, Itagaki Y, Naoki H, Kawai N, Miwa A et al (1998). Isolation and structure of pompilidotoxins, novel peptide neurotoxins in solitary wasp venoms. Biochem Biophys Res Commun 250: 612–616. [DOI] [PubMed] [Google Scholar]
- Konno K, Miwa A, Takayama H, Hisada M, Itagaki Y, Naoki H et al (1997). Alpha‐pompilidotoxin (α‐PMTX), a novel neurotoxin from the venom of a solitary wasp, facilitates transmission in the crustacean neuromuscular synapse. Neurosci Lett 238: 99–102. [DOI] [PubMed] [Google Scholar]
- Kotta CM, Anastasakis A, Gatzoulis K, Papagiannis J, Geleris P, Stefanadis C (2010). Cardiac ion channel gene mutations in Greek long QT syndrome patients. J Appl Genet 51: 515–518. [DOI] [PubMed] [Google Scholar]
- Krayer O, Acheson GH (1946). The pharmacology of the veratrum alkaloids. Physiol Rev 26: 383–446. [DOI] [PubMed] [Google Scholar]
- Kretschmer T, Happel LT, England JD, Nguyen DH, Tiel RL, Beuerman RW et al (2002). Accumulation of PN1 and PN3 sodium channels in painful human neuroma‐evidence from immunocytochemistry. Acta Neurochir 144: 803–810 discussion 810. [DOI] [PubMed] [Google Scholar]
- Laedermann CJ, Abriel H, Decosterd I (2015). Post‐translational modifications of voltage‐gated sodium channels in chronic pain syndromes. Front Pharmacol 6: 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lago J, Rodriguez LP, Blanco L, Vieites JM, Cabado AG (2015). Tetrodotoxin, an extremely potent marine neurotoxin: distribution, toxicity, origin and therapeutical uses. Mar Drugs 13: 6384–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattes K, Venegas P, Lagos N, Lagos M, Pedraza L, Rodriguez‐Navarro AJ et al (2009). Local infiltration of gonyautoxin is safe and effective in treatment of chronic tension‐type headache. Neurol Res 31: 228–233. [DOI] [PubMed] [Google Scholar]
- Lehane L, Lewis RJ (2000). Ciguatera: recent advances but the risk remains. Int J Food Microbiol 61: 91–125. [DOI] [PubMed] [Google Scholar]
- Leipold E, DeBie H, Zorn S, Borges A, Olivera BM, Terlau H et al (2007). μO‐conotoxins inhibit NaV channels by interfering with their voltage sensors in domain‐2. Channels (Austin) 1: 253–262. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Ritter AM, Mendell LM (1993). Nerve growth factor‐induced hyperalgesia in the neonatal and adult rat. J Neurosci 13: 2136–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RJ, Dutertre S, Vetter I, Christie MJ (2012). Conus venom peptide pharmacology. Pharmacol Rev 64: 259–298. [DOI] [PubMed] [Google Scholar]
- Li D, Xiao Y, Xu X, Xiong X, Lu S, Liu Z et al (2004). Structure‐activity relationships of hainantoxin‐IV and structure determination of active and inactive sodium channel blockers. J Biol Chem 279: 37734–37740. [DOI] [PubMed] [Google Scholar]
- Lin YM, Fu Y, Winston J, Radhakrishnan R, Sarna SK, Huang LM et al (2017). Pathogenesis of abdominal pain in bowel obstruction: role of mechanical stress‐induced upregulation of nerve growth factor in gut smooth muscle cells. Pain 158: 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindia JA, Kohler MG, Martin WJ, Abbadie C (2005). Relationship between sodium channel NaV1.3 expression and neuropathic pain behavior in rats. Pain 117: 145–153. [DOI] [PubMed] [Google Scholar]
- Liu Y, Li D, Wu Z, Li J, Nie D, Xiang Y et al (2012). A positively charged surface patch is important for hainantoxin‐IV binding to voltage‐gated sodium channels. J Pept Sci 18: 643–649. [DOI] [PubMed] [Google Scholar]
- Liu Y, Tang J, Zhang Y, Xun X, Tang D, Peng D et al (2014a). Synthesis and analgesic effects of μ‐TRTX‐Hhn1b on models of inflammatory and neuropathic pain. Toxins (Basel) 6: 2363–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wu Z, Tang D, Xun X, Liu L, Li X et al (2014b). Analgesic effects of Huwentoxin‐IV on animal models of inflammatory and neuropathic pain. Protein Pept Lett 21: 153–158. [DOI] [PubMed] [Google Scholar]
- Liu Z, Dai J, Chen Z, Hu W, Xiao Y, Liang S (2003). Isolation and characterization of hainantoxin‐IV, a novel antagonist of tetrodotoxin‐sensitive sodium channels from the Chinese bird spider Selenocosmia hainana . Cell Mol Life Sci 60: 972–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombet A, Bidard JN, Lazdunski M (1987). Ciguatoxin and brevetoxins share a common receptor site on the neuronal voltage‐dependent Na+ channel. FEBS Lett 219: 355–359. [DOI] [PubMed] [Google Scholar]
- Maejima H, Kinoshita E, Yuki T, Yakehiro M, Seyama I, Yamaoka K (2002). Structural determinants for the action of grayanotoxin in D1 S4‐S5 and D4 S4‐S5 intracellular linkers of sodium channel α‐subunits. Biochem Biophys Res Commun 295: 452–457. [DOI] [PubMed] [Google Scholar]
- Manriquez V, Castro Caperan D, Guzman R, Naser M, Iglesia V, Lagos N (2015). First evidence of neosaxitoxin as a long‐acting pain blocker in bladder pain syndrome. Int Urogynecol J 26: 853–858. [DOI] [PubMed] [Google Scholar]
- Marcil J, Walczak JS, Guindon J, Ngoc AH, Lu S, Beaulieu P (2006). Antinociceptive effects of tetrodotoxin (TTX) in rodents. Br J Anaesth 96: 761–768. [DOI] [PubMed] [Google Scholar]
- Martin MF, Garcia y Perez LG, el Ayeb M, Kopeyan C, Bechis G, Jover E et al (1987). Purification and chemical and biological characterizations of seven toxins from the Mexican scorpion, Centruroides suffusus suffusus . J Biol Chem 262: 4452–4459. [PubMed] [Google Scholar]
- Martin MF, Rochat H (1986). Large scale purification of toxins from the venom of the scorpion Androctonus australis Hector . Toxicon 24: 1131–1139. [DOI] [PubMed] [Google Scholar]
- Matavel A, Cruz JS, Penaforte CL, Araujo DA, Kalapothakis E, Prado VF et al (2002). Electrophysiological characterization and molecular identification of the Phoneutria nigriventer peptide toxin PnTx2‐6. FEBS Lett 523: 219–223. [DOI] [PubMed] [Google Scholar]
- Matavel A, Fleury C, Oliveira LC, Molina F, de Lima ME, Cruz JS et al (2009). Structure and activity analysis of two spider toxins that alter sodium channel inactivation kinetics. Biochemistry 48: 3078–3088. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Hasson A, Spira ME, Gray WR, Li W, Marsh M et al (1995). A new family of conotoxins that blocks voltage‐gated sodium channels. J Biol Chem 270: 16796–16802. [DOI] [PubMed] [Google Scholar]
- Meng P, Huang H, Wang G, Yang S, Lu Q, Liu J et al (2016). A novel toxin from Haplopelma lividum selectively inhibits the NaV1.8 channel and possesses potent analgesic efficacy. Toxins (Basel) 9 (1): 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton RE, Warren VA, Kraus RL, Hwang JC, Liu CJ, Dai G et al (2002). Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry 41: 14734–14747. [DOI] [PubMed] [Google Scholar]
- Minassian NA, Gibbs A, Shih AY, Liu Y, Neff RA, Sutton SW et al (2013). Analysis of the structural and molecular basis of voltage‐sensitive sodium channel inhibition by the spider toxin huwentoxin‐IV (μ‐TRTX‐Hh2a). J Biol Chem 288: 22707–22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moczydlowski EG (2013). The molecular mystique of tetrodotoxin. Toxicon 63: 165–183. [DOI] [PubMed] [Google Scholar]
- Mojumder DK, Frishman LJ, Otteson DC, Sherry DM (2007). Voltage‐gated sodium channel α‐subunits NaV1.1, NaV1.2, and NaV1.6 in the distal mammalian retina. Mol Vis 13: 2163–2182. [PubMed] [Google Scholar]
- Monks SA, Pallaghy PK, Scanlon MJ, Norton RS (1995). Solution structure of the cardiostimulant polypeptide anthopleurin‐B and comparison with anthopleurin‐A. Structure 3: 791–803. [DOI] [PubMed] [Google Scholar]
- Munasinghe NR, Christie MJ (2015). Conotoxins that could provide analgesia through voltage gated sodium channel inhibition. Toxins 7: 5386–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH et al (2004). Nociceptor‐specific gene deletion reveals a major role for NaV1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A 101: 12706–12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar MA, Baker MD, Levato A, Ingram R, Mallucci G, McMahon SB et al (2006). Nerve injury induces robust allodynia and ectopic discharges in NaV1.3 null mutant mice. Mol Pain 2: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic SD, Tzoumaka E, McGivern JG, Haraguchi M, Sangameswaran L, Gogas KR et al (1998). Distribution of the tetrodotoxin‐resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J Neurosci 18: 2174–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuse K, Chaplan SR, McMahon SB, Luo ZD, Calcutt NA, Scott BP et al (1997). Regulation of expression of the sensory neuron‐specific sodium channel SNS in inflammatory and neuropathic pain. Mol Cell Neurosci 10: 196–207. [DOI] [PubMed] [Google Scholar]
- Oliveira JS, Redaelli E, Zaharenko AJ, Cassulini RR, Konno K, Pimenta DC et al (2004). Binding specificity of sea anemone toxins to NaV1.1‐1.6 sodium channels: unexpected contributions from differences in the IV/S3‐S4 outer loop. J Biol Chem 279: 33323–33335. [DOI] [PubMed] [Google Scholar]
- Osteen JD, Herzig V, Gilchrist J, Emrick JJ, Zhang C, Wang X et al (2016). Selective spider toxins reveal a role for the NaV1.1 channel in mechanical pain. Nature 534: 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla F, Couble ML, Coste B, Maingret F, Clerc N, Crest M et al (2007). Expression and localization of the NaV1.9 sodium channel in enteric neurons and in trigeminal sensory endings: implication for intestinal reflex function and orofacial pain. Mol Cell Neurosci 35: 138–152. [DOI] [PubMed] [Google Scholar]
- Pedraza Escalona M, Possani LD (2013). Scorpion β‐toxins and voltage‐gated sodium channels: interactions and effects. Front Biosci (Landmark Ed) 18: 572–587. [DOI] [PubMed] [Google Scholar]
- Penzotti JL, Lipkind G, Fozzard HA, Dudley SC Jr (2001). Specific neosaxitoxin interactions with the Na+ channel outer vestibule determined by mutant cycle analysis. Biophys J 80: 698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezet S, McMahon SB (2006). Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci 29: 507–538. [DOI] [PubMed] [Google Scholar]
- Phatarakijnirund V, Mumm S, McAlister WH, Novack DV, Wenkert D, Clements KL et al (2016). Congenital insensitivity to pain: fracturing without apparent skeletal pathobiology caused by an autosomal dominant, second mutation in SCN11A encoding voltage‐gated sodium channel 1.9. Bone 84: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda SS, Undheim EA, Rupasinghe DB, Ikonomopoulou MP, King GF (2014). Spider venomics: implications for drug discovery. Future Med Chem 6: 1699–1714. [DOI] [PubMed] [Google Scholar]
- Pintar A, Possani LD, Delepierre M (1999). Solution structure of toxin 2 from centruroides noxius Hoffmann, a β‐scorpion neurotoxin acting on sodium channels. J Mol Biol 287: 359–367. [DOI] [PubMed] [Google Scholar]
- Prashanth JR, Brust A, Jin AH, Alewood PF, Dutertre S, Lewis RJ (2014). Cone snail venomics: from novel biology to novel therapeutics. Future Med Chem 6: 1659–1675. [DOI] [PubMed] [Google Scholar]
- Priest BT, Blumenthal KM, Smith JJ, Warren VA, Smith MM (2007). ProTx‐I and ProTx‐II: gating modifiers of voltage‐gated sodium channels. Toxicon 49: 194–201. [DOI] [PubMed] [Google Scholar]
- Priest BT, Murphy BA, Lindia JA, Diaz C, Abbadie C, Ritter AM et al (2005). Contribution of the tetrodotoxin‐resistant voltage‐gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci U S A 102: 9382–9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren YS, Qian NS, Tang Y, Liao YH, Yang YL, Dou KF et al (2012). Sodium channel NaV1.6 is up‐regulated in the dorsal root ganglia in a mouse model of type 2 diabetes. Brain Res Bull 87: 244–249. [DOI] [PubMed] [Google Scholar]
- Revell JD, Lund PE, Linley JE, Metcalfe J, Burmeister N, Sridharan S et al (2013). Potency optimization of Huwentoxin‐IV on hNaV1.7: a neurotoxin TTX‐S sodium‐channel antagonist from the venom of the Chinese bird‐eating spider Selenocosmia huwena . Peptides 44: 40–46. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Kocsis JD, Waxman SG (1994). Slow sodium conductances of dorsal root ganglion neurons: intraneuronal homogeneity and interneuronal heterogeneity. J Neurophysiol 72: 2796–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romey G, Abita JP, Schweitz H, Wunderer G, Lazdunski (1976). Sea anemone toxin: a tool to study molecular mechanisms of nerve conduction and excitation‐secretion coupling. Proc Natl Acad Sci U S A 73: 4055–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosker C, Lohberger B, Hofer D, Steinecker B, Quasthoff S, Schreibmayer W (2007). The TTX metabolite 4,9‐anhydro‐TTX is a highly specific blocker of the NaV1.6 voltage‐dependent sodium channel. Am J Physiol Cell Physiol 293: C783–C789. [DOI] [PubMed] [Google Scholar]
- Rupasinghe DB, Knapp O, Blomster LV, Schmid AB, Adams DJ, King GF et al (2012). Localization of NaV 1.7 in the normal and injured rodent olfactory system indicates a critical role in olfaction, pheromone sensing and immune function. Channels (Austin) 6: 103–110. [DOI] [PubMed] [Google Scholar]
- Rush AM, Waxman SG (2004). PGE2 increases the tetrodotoxin‐resistant NaV1.9 sodium current in mouse DRG neurons via G‐proteins. Brain Res 1023: 264–271. [DOI] [PubMed] [Google Scholar]
- Salas MM, McIntyre MK, Petz LN, Korz W, Wong D, Clifford JL (2015). Tetrodotoxin suppresses thermal hyperalgesia and mechanical allodynia in a rat full thickness thermal injury pain model. Neurosci Lett 607: 108–113. [DOI] [PubMed] [Google Scholar]
- Sato K, Yamaguchi Y, Ishida Y, Ohizumi Y (2015). Roles of basic amino acid residues in the activity of μ‐conotoxin GIIIA and GIIIB, peptide blockers of muscle sodium channels. Chem Biol Drug Des 85: 488–493. [DOI] [PubMed] [Google Scholar]
- Schaller KL, Caldwell JH (2000). Developmental and regional expression of sodium channel isoform NaCh6 in the rat central nervous system. J Comp Neurol 420: 84–97. [PubMed] [Google Scholar]
- Schiavon E, Sacco T, Cassulini RR, Gurrola G, Tempia F, Possani LD et al (2006). Resurgent current and voltage sensor trapping enhanced activation by a β‐scorpion toxin solely in NaV1.6 channel. Significance in mice Purkinje neurons. J Biol Chem 281: 20326–20337. [DOI] [PubMed] [Google Scholar]
- Schiavon E, Stevens M, Zaharenko AJ, Konno K, Tytgat J, Wanke E (2010). Voltage‐gated sodium channel isoform‐specific effects of pompilidotoxins. FEBS J 277: 918–930. [DOI] [PubMed] [Google Scholar]
- Schroeder CI, Ekberg J, Nielsen KJ, Adams D, Loughnan ML, Thomas L et al (2008). Neuronally μ‐conotoxins from Conus striatus utilize an α‐helical motif to target mammalian sodium channels. J Biol Chem 283: 21621–21628. [DOI] [PubMed] [Google Scholar]
- Schweitz H, Vincent JP, Barhanin J, Frelin C, Linden G, Hugues M et al (1981). Purification and pharmacological properties of eight sea anemone toxins from Anemonia sulcata, Anthopleura xanthogrammica, Stoichactis giganteus, and Actinodendron plumosum . Biochemistry 20: 5245–5252. [DOI] [PubMed] [Google Scholar]
- Seda M, Pinto FM, Wray S, Cintado CG, Noheda P, Buschmann H et al (2007). Functional and molecular characterization of voltage‐gated sodium channels in uteri from nonpregnant rats. Biol Reprod 77: 855–863. [DOI] [PubMed] [Google Scholar]
- Sheets PL, Jackson JO 2nd, Waxman SG, Dib‐Hajj SD, Cummins TR (2007). A NaV1.7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J Physiol 581: 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Zhou Q, Pan X, Li Z, Wu J, Yan N (2017). Structure of a eukaryotic voltage‐gated sodium channel at near‐atomic resolution. Science 355 (6328) eaal4326. [DOI] [PubMed] [Google Scholar]
- Shu X, Mendell LM (1999). Nerve growth factor acutely sensitizes the response of adult rat sensory neurons to capsaicin. Neurosci Lett 274: 159–162. [DOI] [PubMed] [Google Scholar]
- Skeik N, Rooke TW, Davis MD, Davis DM, Kalsi H, Kurth I et al (2012). Severe case and literature review of primary erythromelalgia: novel SCN9A gene mutation. Vasc Med 17: 44–49. [DOI] [PubMed] [Google Scholar]
- Sleeper AA, Cummins TR, Dib‐Hajj SD, Hormuzdiar W, Tyrrell L, Waxman SG et al (2000). Changes in expression of two tetrodotoxin‐resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J Neurosci 20: 7279–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staud R, Price DD, Janicke D, Andrade E, Hadjipanayis AG, Eaton WT et al (2011). Two novel mutations of SCN9A (NaV1.7) are associated with partial congenital insensitivity to pain. Eur J Pain 15: 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling LC, Forlani G, Baker MD, Wood JN, Matthews EA, Dickenson AH et al (2005). Nociceptor‐specific gene deletion using heterozygous NaV1.8‐Cre recombinase mice. Pain 113: 27–36. [DOI] [PubMed] [Google Scholar]
- Strachan LC, Lewis RJ, Nicholson GM (1999). Differential actions of pacific ciguatoxin‐1 on sodium channel subtypes in mammalian sensory neurons. J Pharmacol Exp Ther 288: 379–388. [PubMed] [Google Scholar]
- Stuhmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N et al (1989). Structural parts involved in activation and inactivation of the sodium channel. Nature 339: 597–603. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD (1991). Further confirmation of the role of adenyl cyclase and of cAMP‐dependent protein kinase in primary afferent hyperalgesia. Neuroscience 44: 131–135. [DOI] [PubMed] [Google Scholar]
- Tanaka BS, Zhao P, Dib‐Hajj FB, Morisset V, Tate S, Waxman SG et al (2016). A gain‐of‐function mutation in NaV1.6 in a case of trigeminal neuralgia. Mol Med 22: 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Sekino S, Ikegami M, Ikeda H, Kamei J (2015). Antihyperalgesic effects of ProTx‐II, a NaV1.7 antagonist, and A803467, a NaV1.8 antagonist, in diabetic mice. J Exp Pharmacol 7: 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichert RW, Raghuraman S, Memon T, Cox JL, Foulkes T, Rivier JE et al (2012). Characterization of two neuronal subclasses through constellation pharmacology. Proc Natl Acad Sci U S A 109: 12758–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thottumkara AP, Parsons WH, Du Bois J (2014). Saxitoxin. Angew Chem Int Ed Engl 53: 5760–5784. [DOI] [PubMed] [Google Scholar]
- Trainer VL, Baden DG, Catterall WA (1994). Identification of peptide components of the brevetoxin receptor site of rat brain sodium channels. J Biol Chem 269: 19904–19909. [PubMed] [Google Scholar]
- Trainer VL, Brown GB, Catterall WA (1996). Site of covalent labeling by a photoreactive batrachotoxin derivative near transmembrane segment IS6 of the sodium channel α subunit. J Biol Chem 271: 11261–11267. [DOI] [PubMed] [Google Scholar]
- Tseng TJ, Hsieh YL, Ko MH, Hsieh ST (2014). Redistribution of voltage‐gated sodium channels after nerve decompression contributes to relieve neuropathic pain in chronic constriction injury. Brain Res 1589: 15–25. [DOI] [PubMed] [Google Scholar]
- Vetter I, Dekan Z, Knapp O, Adams DJ, Alewood PF, Lewis RJ (2012b). Isolation, characterization and total regioselective synthesis of the novel μO‐conotoxin MfVIA from Conus magnificus that targets voltage‐gated sodium channels. Biochem Pharmacol 84: 540–548. [DOI] [PubMed] [Google Scholar]
- Vetter I, Mozar CA, Durek T, Wingerd JS, Alewood PF, Christie MJ et al (2012c). Characterisation of NaV types endogenously expressed in human SH‐SY5Y neuroblastoma cells. Biochem Pharmacol 83: 1562–1571. [DOI] [PubMed] [Google Scholar]
- Vetter I, Touska F, Hess A, Hinsbey R, Sattler S, Lampert A et al (2012a). Ciguatoxins activate specific cold pain pathways to elicit burning pain from cooling. EMBO J 31: 3795–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpon L, Lamthanh H, Barbier J, Gilles N, Molgo J, Menez A et al (2004). NMR solution structures of δ‐conotoxin EVIA from Conus ermineus that selectively acts on vertebrate neuronal Na+ channels. J Biol Chem 279: 21356–21366. [DOI] [PubMed] [Google Scholar]
- Vranken JH (2012). Elucidation of pathophysiology and treatment of neuropathic pain. Cent Nerv Syst Agents Med Chem 12: 304–314. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Scheffer IE, Barnett S, Richards M, Dibbens L, Desai RR et al (2001). Neuronal sodium‐channel α1‐subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet 68: 859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CZ, Zhang H, Jiang H, Lu W, Zhao ZQ, Chi CW (2006). A novel conotoxin from Conus striatus, μ‐SIIIA, selectively blocking rat tetrodotoxin‐resistant sodium channels. Toxicon 47: 122–132. [DOI] [PubMed] [Google Scholar]
- Wang GK, Wang SY (2003). Veratridine block of rat skeletal muscle NaV1.4 sodium channels in the inner vestibule. J Physiol 548: 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Shen J, Li Z, Timothy K, Vincent GM, Priori SG et al (1995). Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet 4: 1603–1607. [DOI] [PubMed] [Google Scholar]
- Wanke E, Zaharenko AJ, Redaelli E, Schiavon E (2009). Actions of sea anemone type 1 neurotoxins on voltage‐gated sodium channel isoforms. Toxicon 54: 1102–1111. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Kocsis JD, Black JA (1994). Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol 72: 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldon PJ (2000). Avian chemical defense: toxic birds not of a feather. Proc Natl Acad Sci U S A 97: 12948–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Merrick DK, Catterall WA (1989). Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron 3: 695–704. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Noebels JL, Catterall WA (1992). Elevated expression of type II Na+ channels in hypomyelinated axons of shiverer mouse brain. J Neurosci 12: 2259–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker WR, Clare JJ, Powell AJ, Chen YH, Faull RL, Emson PC (2000). Distribution of voltage‐gated sodium channel α‐subunit and β‐subunit mRNAs in human hippocampal formation, cortex, and cerebellum. J Comp Neurol 422: 123–139. [DOI] [PubMed] [Google Scholar]
- Whitaker WR, Faull RL, Waldvogel HJ, Plumpton CJ, Emson PC, Clare JJ (2001). Comparative distribution of voltage‐gated sodium channel proteins in human brain. Brain Res Mol Brain Res 88: 37–53. [DOI] [PubMed] [Google Scholar]
- Williams BL, Hanifin CT, Brodie ED Jr, Caldwell RL (2011). Ontogeny of tetrodotoxin levels in blue‐ringed octopuses: maternal investment and apparent independent production in offspring of Hapalochlaena lunulata . J Chem Ecol 37: 10–17. [DOI] [PubMed] [Google Scholar]
- Wilson MJ, Zhang MM, Azam L, Olivera BM, Bulaj G, Yoshikami D (2011). Navβ subunits modulate the inhibition of NaV.8 by the analgesic gating modifier μO‐conotoxin MrVIB. J Pharmacol Exp Ther 338: 687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao HS, Huang QH, Zhang FX, Bao L, Lu YJ, Guo C et al (2002). Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc Natl Acad Sci U S A 99: 8360–8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Bingham JP, Zhu W, Moczydlowski E, Liang S, Cummins TR (2008a). Tarantula huwentoxin‐IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain ii voltage sensor in the closed configuration. J Biol Chem 283: 27300–27313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Blumenthal K, Jackson JO 2nd, Liang S, Cummins TR (2010). The tarantula toxins ProTx‐II and Huwentoxin‐IV differentially interact with human NaV1.7 voltage sensors to inhibit channel activation and inactivation. Mol Pharmacol 78: 1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Luo X, Kuang F, Deng M, Wang M, Zeng X et al (2008b). Synthesis and characterization of huwentoxin‐IV, a neurotoxin inhibiting central neuronal sodium channels. Toxicon 51: 230–239. [DOI] [PubMed] [Google Scholar]
- Xie W, Strong JA, Zhang JM (2015). Local knockdown of the NaV1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience 291: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Zhang J, Wang Y, Wang L, Wang X (2016). Changes in the expression of voltage‐gated sodium channels NaV1.3, NaV1.7, NaV1.8, and NaV1.9 in rat trigeminal ganglia following chronic constriction injury. Neuroreport 27: 929–934. [DOI] [PubMed] [Google Scholar]
- Yamamoto R, Yanagita T, Kobayashi H, Yokoo H, Wada A (1997). Up‐regulation of sodium channel subunit mRNAs and their cell surface expression by antiepileptic valproic acid: activation of calcium channel and catecholamine secretion in adrenal chromaffin cells. J Neurochem 68: 1655–1662. [DOI] [PubMed] [Google Scholar]
- Yamaoka K, Inoue M, Miyazaki K, Hirama M, Kondo C, Kinoshita E et al (2009). Synthetic ciguatoxins selectively activate NaV1.8‐derived chimeric sodium channels expressed in HEK293 cells. J Biol Chem 284: 7597–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeomans DC, Levinson SR, Peters MC, Koszowski AG, Tzabazis AZ, Gilly WF et al (2005). Decrease in inflammatory hyperalgesia by herpes vector‐mediated knockdown of NaV1.7 sodium channels in primary afferents. Hum Gene Ther 16: 271–277. [DOI] [PubMed] [Google Scholar]
- Yin R, Liu D, Chhoa M, Li CM, Luo Y, Zhang M et al (2016). Voltage‐gated sodium channel function and expression in injured and uninjured rat dorsal root ganglia neurons. Int J Neurosci 126: 182–192. [DOI] [PubMed] [Google Scholar]
- Yoshinaka‐Niitsu A, Yamagaki T, Harada M, Tachibana K (2012). Solution NMR analysis of the binding mechanism of DIVS6 model peptides of voltage‐gated sodium channels and the lipid soluble alkaloid veratridine. Bioorg Med Chem 20: 2796–2802. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Yarov‐Yarovoy V, Scheuer T, Karbat I, Cohen L, Gordon D et al (2011). Structure‐function map of the receptor site for β‐scorpion toxins in domain II of voltage‐gated sodium channels. J Biol Chem 286: 33641–33651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MM, Green BR, Catlin P, Fiedler B, Azam L, Chadwick A et al (2007). Structure/function characterization of μ‐conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J Biol Chem 282: 30699–30706. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ren W, DeCaen P, Yan C, Tao X, Tang L et al (2012). Crystal structure of an orthologue of the NaChBac voltage‐gated sodium channel. Nature 486: 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD (2002). Ceramide, a putative second messenger for nerve growth factor, modulates the TTX‐resistant Na(+) current and delayed rectifier K(+) current in rat sensory neurons. J Physiol 544: 385–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Li X, Jin L, Zhang F, Inoue M, Yu B et al (2016). Development of a rapid throughput assay for identification of hNaV1.7 antagonist using unique efficacious sodium channel agonist, antillatoxin. Mar Drugs 14 (2): 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Dupre N, Puymirat J, Chahine M (2012). Biophysical characterization of M1476I, a sodium channel founder mutation associated with cold‐induced myotonia in French Canadians. J Physiol 590: 2629–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Davar G, Strichartz G (2002). Endothelin‐1 (ET‐1) selectively enhances the activation gating of slowly inactivating tetrodotoxin‐resistant sodium currents in rat sensory neurons: a mechanism for the pain‐inducing actions of ET‐1. J Neurosci 22: 6325–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann K, Deuis JR, Inserra MC, Collins LS, Namer B, Cabot PJ et al (2013). Analgesic treatment of ciguatoxin‐induced cold allodynia. Pain 154: 1999–2006. [DOI] [PubMed] [Google Scholar]
- Zorn S, Leipold E, Hansel A, Bulaj G, Olivera BM, Terlau H et al (2006). The μO‐conotoxin MrVIA inhibits voltage‐gated sodium channels by associating with domain‐3. FEBS Lett 580: 1360–1364. [DOI] [PubMed] [Google Scholar]
- Zsiros E, Kis‐Toth K, Hajdu P, Gaspar R, Bielanska J, Felipe A et al (2009). Developmental switch of the expression of ion channels in human dendritic cells. J Immunol 183: 4483–4492. [DOI] [PubMed] [Google Scholar]