

Abstract
N-Methyl-D-Aspartate receptors (NMDARs) are ion channels comprising of tetrameric assemblies of GluN1 and GluN2 receptor subunits that mediate excitatory neurotransmission in the central nervous system. Of the four different GluN2 subunits, the GluN2D subunit-containing NMDARs have been suggested as a target for antiparkinsonian therapy due to their expression pattern in some of the basal ganglia nuclei that show abnormal firing patterns in the parkinsonian state, specifically the subthalamic nucleus (STN). In this study, we describe that blockade of NMDARs alters spike firing in the STN in a male non-human primate which had been rendered parkinsonian by treatment with the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). In accompanying experiments in male rodents, we found that GluN2D-NMDAR expression in the STN is reduced in acutely or chronically dopamine-depleted animals. Taken together, our data suggests that blockade of NMDARs in the STN may be a viable antiparkinsonian strategy, but that the ultimate success of this approach may be complicated by parkinsonism-associated changes in NMDAR expression in the STN.
Keywords: NMDA receptors, Subthalamic neurons, spike firing, Parkinson’s Disease, male primate, male mice
Graphical Abstract
Introduction
N-Methyl-D-Aspartate receptors (NMDARs) are tetrameric assemblies comprising GluN1 and GluN2 subunits that mediate a slow, Ca2+-permeable component of excitatory synaptic transmission (Traynelis 2010). Of the four different GluN2 subunits (A–D), the GluN2D subunit has been suggested to be a potential target for antiparkinsonian therapies due to its expression in multiple basal ganglia nuclei (Hallett 2004, Zhang 2014), specifically the subthalamic nucleus (STN, Standaert 1994). In the STN, NMDARs play an important role in mediating synaptic transmission in the cortico-subthalamic projection, among others (Chu 2015, Swanger 2015). In individuals with Parkinson’s Disease (PD) and in animal models of parkinsonism, altered activity along this pathway may contribute to firing pattern changes in the STN, specifically the well-described changes of increased firing, increased bursting, and abnormal synchronized oscillatory activity that are typical for the parkinsonian state (Nambu 2005, Baudrexel 2011, Brunenberg 2012, Kita 2004, Rubin 2012, Wichmann 2011, Gatev 2008, Soares 2006, Bergman 1994, Miller and DeLong 1987, Galvan and Wichmann 2008).
Our understanding of the potential therapeutic use of NMDAR blocking agents in parkinsonism has slowly emerged. Decades ago, administration of the broad-spectrum NMDAR antagonists like MK801 was shown to ameliorate some of the motor abnormalities in dopamine-depleted rodents (JT Greenamyre 1993, Klockgether T and Turski L 1989, Blandini 2001, Allers 2005), but the use of broad-spectrum NMDAR blockers was considered unlikely to be clinically useful, as they would have many severe cognitive and other side effects. Studies have further shown administration of APV into the STN corrected locomotor activity, burst firing, and cortex-STN synchronization in a 6-OHDA rat model (Pan 2014). However, the use of NMDAR subunit-selective antagonists remains an attractive strategy to treat PD, based on the possibility that selective blockade of NMDA receptors in the STN, either through local administration of drugs, or through the use of compounds that act at the STN level with some specificity, such as GluN2D antagonists, may have potential as an antiparkinsonian therapy (Hallett and Standaert 2004).
To further explore the idea that local blockade of NMDAR-mediated glutamatergic transmission in the STN may have antiparkinsonian properties, we studied the neuronal effects of local injections of an NMDAR antagonist in the STN of a parkinsonian monkey (supported by in silico modeling of drug diffusion at the local injection sites), and used immuno-labeling to determine the expression of GluN2D NMDARs in the STN, the substantia nigra pars compacta (SNc), and the striatum in rodent models of acute or chronic loss of dopamine.
Methods
Non-human primate
The primate experiments were performed in accordance with the United States Public Health Service Policy on the Humane Care And Use Of Laboratory Animals, including the provisions of the “Guide for the Care and Use of Laboratory Animals” (Garber 2011), and were approved by the Biosafety Committee and the Animal Care and Use Committee of Emory University. The studies were carried out in a male Rhesus monkey (Macaca mulatta, 4.7kg, 5 years old). The animal was pair-housed with an animal used in other studies, and had ad libitum access to food and water. For the first weeks of the experiment, the monkey was slowly conditioned to being handled by the experimenter and to sit in a primate chair, using positive reinforcement methods (as detailed in McMillan 2014).
The animal was then rendered parkinsonian by weekly administration of small doses of MPTP (0.2–0.6 mg/kg i.m.). The animal was kept in quarantine (with a companion animal) for 72 hours after each injection. The animal received a total of 39.3 mg MPTP (8.2 mg/kg), in 15 individual injections, 0.4–0.6 mg/kg each. The total duration of treatment was 19 weeks. After the final MPTP treatment, we waited 8 weeks before recordings to let the parkinsonian signs stabilize. To assess the degree and stability of the MPTP-induced motor disability, we scored ten aspects of motor function (bradykinesia, freezing, extremity posture, trunk posture, action tremor, the frequency of arm and leg movements, finger dexterity, home cage activity, and balance), each on a 0-to-3 scale, as done in multiple other studies before (Galvan 2016, Devergnas 2014, Kammermeier 2016). The animal eventually reached a state of stable moderate parkinsonism, scoring 19/30 on the above scale.
The monkey then underwent an aseptic surgical procedure under isoflurane anesthesia (1–3%) for placement of two stainless steel recording chambers (Crist Instruments, Hagerstown, MD; inner chamber diameter 16mm). The chambers were stereotactically positioned over trephine holes in the skull targeting the STN (A=10, L=6, D=1.5, based on the atlas by Winters 1969) on each side with a coronal approach, 40° from the vertical. The chambers were embedded into an acrylic skull ‘cap’, along with a stainless-steel head holder and multiple bone screws. After the surgery, the monkey received analgesics and prophylactic antibiotic treatment for one week. Recordings started after the end of this post-procedural treatment.
Electrophysiological recordings
The monkey was seated in a primate chair with its head immobilized, leaving body and limbs free to move. Recordings were performed while the monkey was awake, as determined by continued visual observations. We lowered tungsten microelectrodes (FHC, Bowdoinham, ME; Z=0.5–1.0MΩ at 1kHz) into the brain with a microdrive (MO-95B; Narishige, Tokyo, Japan). The electrical signals were amplified (DAM-80 amplifier; WPI, Sarasota, FL), filtered (400−6000 Hz; Krohn-Hite, Brockton, MA), displayed on a digital oscilloscope (DL1540; Yokogawa, Tokyo, Japan), and made audible via an audio amplifier. The location and borders of the STN were defined based on the single unit extracellular recordings (Soares 2006, Devergnas 2016, Kliem 2010) and later confirmed by histology (see below).
Intracerebral injections
The intracerebral microinjections in primates utilized a custom-built device (“injectrode”), consisting of a tungsten microelectrode that was glued to a thin silica tube (Kliem and Wichmann 2004). The tip of the electrode protruded 175–200 μm beyond the tip of the silica tubing. The tubing was connected to a 1 ml gas-tight syringe (CMA Microdialysis, Solna, Sweden) driven by a computer-controlled injection pump (model 102, CMA). The injectrode was lowered into the STN with a microdrive, and positioned to record single neuron activity. Recordings of individual neurons started with a baseline recording period, lasting at least 60 s, followed by recordings during the drug injections (0.5 μl at 0.1–0.2 μl/min) and post-injection (up to 10 min). Only one injection was made in each experimental session.
We used injections of D-APV or saline. The drug solution was prepared by dissolving 5 mM APV into sterile saline. The drug injections were done by an investigator blinded to the injected compound, using randomly coded matched tubes of vehicle or APV.
Termination of primate experiments
To terminate the series of experiments, the monkey was injected with an overdose of sodium pentobarbital (100 mg/kg, i.v.) and then transcardially perfused with cold oxygenated Ringer’s solution, followed by a fixative containing 4% paraformaldehyde and 0.1% glutaraldehyde in a phosphate buffer (PB) solution. After perfusion, the brains were removed from the skull, cut coronally into 10 mm thick blocks, and post-fixed overnight in 4% paraformaldehyde. The blocks were then cut into 60 μm-thick coronal sections using a vibrating microtome and stored at −20 °C in an anti-freeze solution, containing 30% ethylene glycol and 30% glycerol in PB, until ready for immunohistochemistry. Sections containing the STN were stained for Nissl substance to visualize electrode penetrations, while alternate sections were immuno-labeled for the neuronal marker microtubule associated protein 2 (mouse anti-MAP2, 1:1000, Millipore) to assess the extent of neuronal damage induced by the electrode tracks and to localize the borders of the thalamus. Injectrode tracks were visualized using images of the MAP2-stained sections. Using depth readings obtained during the injectrode recordings sessions, individual recording sites were verified to be in the STN.
Analysis of electrophysiological data
We used off line waveform-matching spike sorting (Spike2; CED, Cambridge, UK) to detect spikes in records of neuronal activity. The spike sorting was performed by an investigator unaware of the injected compound. Principal component analysis and an analysis of the distribution of inter-spike intervals (ISIs) were used to verify the quality of the spike sorting. All subsequent data analysis steps were carried out in MATLAB (MATLAB version 9.1, The Mathworks, Natick, MA, USA), using the ISI information. To analyze the effects of microinjections of saline or APV on the firing rates and pattern of neurons, we compared in each cell a “control”, and an “effect” period. The control epoch consisted of the entire (usable) baseline segment of data. The effect period was a segment of post-injection data, starting at least 30 s after the start of the injection. To determine the onset and offset of the drug effect period, we generated second-by-second readouts of firing rates (based on a sliding 30 s window), which were subsequently smoothed with a 20-point moving averaging technique. The onset and offset of “effects” were defined as the time during which the post-injection firing rate first moved beyond or below the mean ± 2SD of the baseline period. In case no significant firing rate effect was detected, we used the entire post-injection data window for the analysis.
Several parameters of neuronal firing were calculated in the analysis of the extracted baseline and effect ISI sequences, including the average firing rate (number of spikes/s within the analyzed segment), the coefficient of variation of the ISIs (ISI CV), the power spectra of neuronal firing, as well as parameters describing bursts in discharge. The power spectral analysis utilized the Neurospec 2.0 Matlab functions for frequency domain analyses of neuronal spiking data (Nielsen 2005, Halliday 2008). For each neuron, raw spectra were integrated in the 1–3 Hz, 3–8 Hz, 8–13 Hz, 13–30 Hz, and 30–100 Hz ranges, and the resultant values expressed as a fraction of the power in the entire 1–100 Hz band. Similar methods have been used in our previous publications (Legendy 1985, Wichmann 2006, Galvan 2010). To detect bursts in firing, we used the method described by Legendy and Salcman (Legendy and Salcman 1985, see also Soares 2006, Sanders 2013, Devergnas 2014), with a ‘surprise’ value of 3. We calculated the frequency of occurrence, the mean intra-burst firing rate, the proportion of spikes in bursts (compared to the total number of spikes), and the average number of spikes per burst. Comparisons between control and effect periods (for saline and APV) were carried out using paired t-tests.
Simulation of D-APV diffusion in the STN
The space-time concentration profile of D-APV (5 mM), pressure applied in the STN (500 nl), was modelled according to a modified version of the classic solution of the diffusion equation in an isotropic medium:
| (Eq. 1) |
Here, Q represents the total amount of diffusing D-APV molecules (in moles), D* is the apparent diffusion coefficient of D-APV, r and t represent the distance from and time after release, respectively. The equation below represents the modified version of the classic solution of the diffusion equation used in our simulations:
| (Eq. 2) |
Here, C0 represents the initial concentration of diffusing D-APV injected in the neuropil (5 mM). The apparent diffusion coefficient for was calculated using the free diffusion coefficient of Alexa Fluor 350 (AF350) in aqueous solution at room temperature (Dfree=0.51 μm2/ms, Sweeney 2017) adjusted to physiological temperature using the temperature dependence of diffusion (Q10=1.3), and the tortuosity of the rat striatum (λ=1.54, Rice and Nicholson 1991). The diffusion coefficient of AF350 was used because its molecular weight (350 Da) is similar to that of D-APV (197 Da). In separate models, we simulated prolonged injections of D-APV at a rate of 0.1–0.2 μl/min (i.e. 300 s or 150 s, respectively), to mimic more closely the injection rates of D-APV used in the experiments. The term C0 was normalized by the number of time steps in the simulation. At each time step, a new release event was simulated and added to the to the previously evoked concentration profile of D-APV. All calculations were performed using custom-made codes written in IgorPro 6.37 (Wavemetrics, Lake Oswego, OR).
MPTP treatment and dopamine transporter (DAT) immunoreactivity in mice
All procedures using mice were approved by the Institutional Animal Care and Use Committee of Emory University. Male C57Bl/6J mice (6 months old) received five subcutaneous injections of 20 mg/kg MPTP (Sigma, cat. M0896) with an inter-injection interval of 24 hr (Lohr 2014). While it is important to incorporate gender differences into studies, this model is only suitable for male mice because of the increased mortality of female animals (Jackson-Lewis and Przedborski, 2007). The animals were housed according to standard guidelines, with ad libitum access to food and water, and under standard light and dark cycle. The animals were monitored by trained staff at the animal housing facility at Emory University. In two separate groups of MPTP-treated animals (n=4 per condition), the lesion was allowed to stabilize for either 21 or 42 days following the final injection. No animal was excluded from the analyses. The assignment of the animals to their experimental group was done at random. The experiments were performed between two research groups, one performing the DAT immunoreactivity experiments, and the other group performing the GluN subunit detection experiments. At the termination of the in vivo portion of the experiment, the animals were sacrificed by rapid decapitation. Their brains were immediately removed, and the striatum dissected and frozen on dry ice. The frozen striatal samples were then homogenized in buffer (640mM sucrose + 10mM HEPES) using a TissueTearor and differentially centrifuged to generate a crude synaptosomal fraction. The striatal DAT immunoreactivity was assessed by immunoblot and normalized to the β-actin immunoreactivity to determine the extent of the dopaminergic lesion. The following antibodies and dilutions were used for immunoblotting: rat anti-DAT (Millipore Cat# MAB369, RRID:AB_2190413, 1:1,000), mouse anti-β-actin (Sigma-Aldrich Cat# A5441, RRID:AB_476744, 1:10,000), HRP-conjugated goat anti-rat (Jackson ImmunoResearch Labs Cat# 112-035-003, RRID:AB_2338128, 1:5,000), and HRP-conjugated goat anti-mouse (Jackson ImmunoResearch Labs Cat# 115-035-003, RRID:AB_10015289 1:10,000). Immunoreactivity was detected using a chemiluminescent substrate (SuperSignal, Thermo Fisher) and visualized using a BioRad UV imager.
Micro-punching and NMDAR immunoblotting from mouse tissue
Brains obtained from rapidly decapitated animals (separate from those used for the aforementioned DAT immunoreactivity studies) were quickly removed and 250–300 μm sagittal slices containing the STN, SNc and striatum were prepared in ice-cold PBS using a vibrating microtome (VT1200S, Leica). Tissue discs from the above-mentioned three nuclei were removed using a 0.5 mm tissue punch under a dissecting light microscope (Stoelting, Wood Dale, IL) and frozen immediately on dry ice (Swanger 2015). The tissue was homogenized in lysis buffer (pH 7.4) containing (in mM) 150 NaCl, 50 Tris, 50 NaF, 5 EDTA, 5 EGTA, 1% Triton, 1% SDS, and protease inhibitor cocktail obtained from Sigma Aldrich (Cat# P8340) using a Model CL-18 tissue homogenizer with a setting below 10 for the amplitude to minimize protein degradation (Fisher Scientific). We used 4 mice per group (one group per age i.e. 21 and 42 days, one group per treatment). Animals were randomly selected for assignment to different groups. The investigators were not blinded to these group assignments.
Equal amounts of protein were loaded for each sample quantified by Bradford assay and diluted with lysis buffer (1μg/μl or as possible), and normalized to α-tubulin as loading control. Samples were then either stored at −80 or −20°C for later use or heated at 95°C for 5 min, and loaded on a 12% SDS-PAGE gel (Bio-Rad). Samples were run at 60 mV for 15 minutes and then at a constant voltage of 80 V after crossing the stacking gel. Proteins were then transferred to PVDF membranes (Immun-Blot, Bio-Rad, 0.2 μM pore size) for Western blotting for 3 h at 90 V. The antibodies used for immunoblotting were mouse anti-GluN2D (Millipore Cat# MAB5578, RRID:AB_838227, 1:5000, incubated for 4 hr at room temperature, Swanger 2015), rabbit anti- GluN2B (Millipore Cat# AB1557, RRID:AB_2112907, 1:1000, incubated for 1 hr at room temperature, Swanger 2016), mouse anti-α-tubulin (Sigma-Aldrich, RRID: AB_477593, 1: 50,000, incubated for 1 hr at room temperature), and HRP-conjugated goat anti-mouse (Thermo Fisher Scientific Cat# 31430, RRID:AB_228307, 1:10,000, incubated for 1 hr at room temperature) and anti-rabbit secondary (Jackson ImmunoResearch Labs Cat# 111-035-144, RRID:AB_2307391, 1: 10,000, incubated for 1 hr at room temperature). Details of antibodies are listed in Table 1. Blocking of non-specific binding at membranes was performed with 5% milk in TTBS for all antibodies except for GluN2B (2% non-fat milk used for blocking and 1% for antibody incubation). To re-evaluate the same membrane, they were exposed to Restore Stripping Buffer (Pierce, Cat# 21059) for 15 min, washed with PBS and blocked with 4% milk again before being re-blotted with the next primary antibodies. Signals from antibodies were imaged with raw films, using a Bio-Rad Gel Doc Imager. The blots were analyzed using Alpha View immunoblotting analysis software (Bhattacharya 2017).
Table 1.
Detailed information for each antibody used (human: h, mouse: m, rat: r, bovine: b, and monkey: mk, RRID: Research Resource Identification)
| Antibodyf | RRID | Full name | Specificity | Citation | Type | Host |
|---|---|---|---|---|---|---|
| GluN2D | AB_838227 | Mouse Anti-NMDAR2D, Unconjugated antibody | NMDAR2D h, m, r | Millipore Cat# MAB5578, RRID:AB_838227 | monoclonal antibody | mouse |
| GluN2B | AB_2112907 | Anti-NMDAR2B, pain antibody | NMDAR2B h, m, r | Millipore Cat# AB1557, RRID:AB_2112907 | polyclonal antibody | rabbit |
| Dopamine Transporter | AB_2190413 | Dopamine Transporter N-terminus clone DAT-Nt | h, m, mk r, non-human primate | Millipore Cat# MAB369, RRID:AB_2190413 | monoclonal antibody | rat |
| α-tubulin | AB_477593 | Mouse Anti-alpha-Tubulin Monoclonal Antibody, Unconjugated, Clone DM1A | Tubulin, alpha, b, h, m, r | Sigma-Aldrich Cat# T9026, RRID:AB_477593 | monoclonal antibody | mouse |
| β-actin | AB_476744 | Mouse Anti-beta-Actin Monoclonal Antibody, Unconjugated, Clone AC-15 | Actin, beta b, h, m, r | Sigma-Aldrich Cat# A5441, RRID:AB_476744 | monoclonal antibody | mouse |
| Secondary AB anti-mouse | AB_228307 | Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Mouse IgG (H+L) mouse | Thermo Fisher Scientific Cat# 31430, RRID:AB_228307 | polyclonal antibody | goat |
| Secondary AB anti-rabbit | AB_2307391 | Peroxidase-AffiniPure Goat Anti-Rabbit IgG (H+L) (min X Hu, Ms, Rat Sr Prot) antibody | Rabbit IgG (H+L) rabbit | Jackson ImmunoResearch Labs Cat# 111-035-144, RRID:AB_2307391 | polyclonal antibody | goat |
| Secondary AB antimouse | AB_10015289 | Peroxidase-AffiniPure Goat Anti-Mouse | IgG (H + L) antibody | Jackson ImmunoResearch Labs Cat# 115-035-003, RRID:AB_10015289 | polyclonal antibody | goat |
| Secondary AB anti-rat | AB_2338128 | Peroxidase-AffiniPure Goat Anti-Rat | IgG (H+L) antibody | Jackson ImmunoResearch Labs Cat# 112-035-003, RRID:AB_2338128 | polyclonal antibody | goat |
Statistics
For each cell in primate recording, the values for the different descriptor of firing patterns (such as global firing rates, intra-burst firing rates, etc.) obtained from the effect periods were divided by the values obtained during the control period from the same cells. We then used independent-sample t-tests, comparing the ratios obtained in the D-APV injection experiments to those obtained with saline injections. A p-value of less than 0.05 was accepted as indicating a significant difference. For all other experiments statistical tests are mentioned in the individual results section or figure legends.
Results
Effect of D-APV on spike firing in an MPTP-treated primate
We performed diffusion simulations to estimate the time course of the drug concentration in the brain parenchyma, following microinjections of D-APV into the STN (Figure 1A). In these simulations, we estimated the apparent diffusion coefficient (D* = 0.28 μm2/ms) using a value for tortuosity (λ=1.54) that matched the one measured experimentally in the rat striatum (Rice and Nicholson 1991). We first modeled the release of 0.5 μl D-APV (5 mM) at a rate of 0.1–0.2 μl/min using a modified version of the classic solution of the diffusion equation (see Methods) to mimic closely the injection rate of D-APV used in the experiments (Figure 1B–C). The diffusion analysis showed that in the first 5 min after the pressure-application, there is a 10,000-fold dilution of D-APV at 175–200 μm from the injection point (the distance at which the recording electrode was located). Therefore, the concentration of D-APV is 0.6–0.7 μM at 175–200 μm away from the injection site, 5 min after an intra-parenchymal application of 500 nl of 5 mM D-APV. Similar estimates were also obtained when we modelled the release of D-APV from the injectrode as an instantaneous release event from a point source (Eq. 1; Figure 1D). These simulations show that slight variations in the location of the recording site with respect to the position of the injectrode lead to modest changes in the effective concentration of D-APV in the neuropil (Figure 1E). In contrast, slight variations in the time at which the recordings are made lead to profound changes in the concentration of D-APV in the neuropil. Accordingly, the concentration of D-APV at the recording site varies over 4 orders of magnitude within the first 5 min after the injection. Based on these findings, in our recordings, we made sure that our electrophysiology measures were performed consistently 5 min after the end of the D-APV injection (see Figure 2). We then performed a series of simulations of NMDAR-EPSCs using a kinetic scheme described by Clements and Westbrook (1994) following pre-equilibration with different D-APV concentrations and instantaneous release of 1 mM glutamate (Figure 1F–G). The glutamate transient decayed exponentially with a time constant of 1 ms (Clements 1992, Figure 1D). The EC50 for block by pre-equilibrated D-APV was ~0.5 μM, similar to the concentration predicted to be reached by the diffusion analysis. The analysis thus indicates that APV blocked >50% NMDARs near the recording site within the time period analyzed in this report.
Figure 1.

Diffusion of D-APV from the injection site. A. Analytical simulations were used to evaluate the D-APV concentration at different distances from the injection site. B, C. Diffusion analysis demonstrates that, after a single D-APV injection at 0.1 μl/min (B) or 0.2 μl/min (C), there is a 10-fold dilution of D-APV at 175–200 μm from the point of injection. D. Time course of the D-APV concentration obtained when approximating the release of D-APV from the injectrode as an instantaneous event from a point source. E. Distribution of D-APV concentration within a 400 μm radius from the injection site, at different times after the injection. Slight inaccuracies in the distance between the location of the injectrode and the recording site do not confound our estimates of the effective D-APV concentration in the neuropil, measured 10 min after the injection. F. Simulations of NMDAR-EPSCs using a kinetic scheme after pre657 equilibration with different D-APV concentrations (0 nM to 10 μM) with instantaneous release of 1 mM glutamate. Glutamate transient decays exponentially with a time constant of 1 ms; G. The EC50 for blockade of NMDARs by D-APV was approximately 0.5 mM as shown in the relative response vs. concentration of D-APV curve. Because the EC50 was lower than the concentration predicted to be reached by the diffusion analysis within the stipulated time window, the chosen dose of D-APV can be assumed to effectively block >50% of the NMDARs during this time.
Figure 2.

Effects of D-APV on spike firing in the primate subthalamic nucleus (STN). The top row shows representative extracellular in vivo recordings of the spiking activity of STN neurons before, during and after the microinjection of 0.5 μl of saline/0.1% DMSO (left) or D-APV 5 mM/0.1% DMSO (right). The bar graph on the left shows the average responses of firing rates to injection of saline or 5 mM D-APV, expressed as a ratio of the pre-injection firing rate in individual cells. The bar graph on the right shows the average intra-burst frequencies following the injections. Data are from 11 experiments in which the vehicle was injected, and 4 cells recorded after exposure to D-APV, respectively. *, p < 0.05, t-tests, examining differences between saline vehicle and D-APV experiments. Lower row shows Nissl stained coronal section of the brain at approximately A10. The outlines of relevant surrounding brain structures are marked with dashed lines. An injection system tract is visible (arrows). Abbreviations: STN, subthalamic nucleus; SN, substantia nigra; ZI, zona incerta; GPe, external pallidal segment; GPi, internal pallidal segment; RN, reticular nucleus of the thalamus.
In the in vivo portion of these experiments, we injected vehicle or D-APV into the STN of a parkinsonian monkey while simultaneously recording neuronal spike firing in this nucleus. As shown in Figure 2, we identified neurons with a stable baseline rate of spontaneous spike firing. We then injected 500 nl of either vehicle or 5 mM D-APV directly into the STN. The results are shown in Table 2. We found that both the spike firing rate and the intra-burst firing rate were significantly reduced. The proportion of spikes in bursts (out of all spikes recorded) and the number of spikes per burst remained the same for both vehicle and D-APV microinjection, suggesting that the cells continued to fire bursts, but that the bursting was less intense after the D-APV injections. D-APV did not alter power spectral measures of oscillatory spiking activity (Table 2).
Table 2.
Effects of D-APV or saline on firing properties of STN neurons recorded during the control and effect periods. Data are presented as mean ± SEM of post-injection values, normalized to the pre-injection baseline of the respective cells. The significance was assessed using independent-sample t-tests, comparing the ratios obtained with D-APV to those obtained with saline injections.
| D-APV (4) | Saline (11) | |||
|---|---|---|---|---|
| Normalized values | Normalized values | P | ||
| Firing rate | 0.30 ± 0.06 | 1.14 ± 0.14 | <0.001 | |
| CV of inter-spike intervals | 1.55 ± 0.48 | 1.33 ± 0.28 | 0.714 | |
| Integrated spectral power | 1–3 Hz | 0.95 ± 0.36 | 1.12 ± 0.15 | 0.681 |
| 3–8 Hz | 0.84 ± 0.13 | 1.06 ± 0.03 | 0.182 | |
| 8–13 Hz | 1.01 ± 0.09 | 1.02 ± 0.04 | 0.882 | |
| 13–30 Hz | 1.39 ± 0.20 | 1.08 ± 0.04 | 0.224 | |
| 30–100 Hz | 1.15 ± 0.15 | 0.98 ± 0.01 | 0.332 | |
| Proportion spikes in bursts | 1.15 ± 0.06 | 1.03 ± 0.06 | 0.198 | |
| Number of spikes per burst | 1.12 ± 0.21 | 1.00 ± 0.04 | 0.623 | |
| Intra-burst firing rate | 0.66 ± 0.05 | 1.10 ± 0.07 | <0.001 | |
Subsequent post mortem analysis of the STN revealed that the location of all recording electrodes and microinjections were in the STN (Figure 2B). These data provide the first evidence of a contribution of NMDAR activity to STN spike firing in a non-human primate model of parkinsonism.
Impaired GluN2D expression in STN of MPTP-treated mice
We assessed the loss of striatal dopaminergic terminals in mice treated with MPTP by measuring the DAT immunoreactivity. DAT immunoreactivity has been further shown to strongly correlate with other markers of dopamine terminal integrity, such as dopamine content and tyrosine hydroxylase expression (Caudle 2007). Mice injected with MPTP (5 daily injections of 20 mg/kg) showed an 80% loss of striatal DAT immunoreactivity 21 days after the final injection (Figure 3, left panel, n=5, ****p < 0.0001). Forty-two days after completing the MPTP injection protocol, the animals showed a 55% loss of striatal DAT (Figure 3, right panel, n=5, **p = 0.005). A 2-tailed t-test was performed to establish statistical significance in these experiments.
Figure 3.

Analysis of dopamine transporter (DAT) expression in the mouse striatum following 5 daily injections of MPTP (20 mg/kg), 21 (left) and 42 days post injection (right). The insets are representative western blots demonstrating the reduction of DAT immunoreactivity (S= Saline, M= MPTP treated sample, the numbers correspond to different samples, n=5 independent experiments, ****p < 0.0001 for 21 days post-lesion and n=5, **p = 0.005 for 42 days post-lesion, 2-tailed t-test).
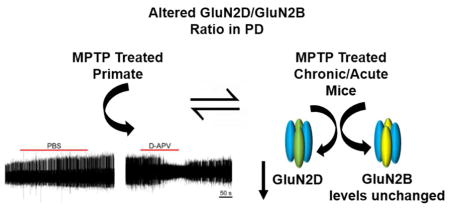
Murine STN neurons express the GluN1, GluN2A, GluN2B, and GluN2D subunits of NMDARs, which likely combine to form triheteromeric NMDARs and their altered structure is involved in disease states (Swanger 2015, Standaert 1994). GluN2D-containing NMDARs mediate synaptic transmission and control spike firing in the STN. Hence, we wanted to examine GluN2D protein expression in the parkinsonian state (Swanger 2015, Swanger 2017). To investigate if MPTP-treated mice showed altered GluN2D subtype expression, we quantified GluN2D protein expression in tissue micropunches, using western blots. We found that GluN2D immunoreactivity was significantly reduced in the STN both 21 days and 42 days after the MPTP injections as compared to their age matched controls (Figure 4A, B, and C; n=4 animals per group; * p < 0.05, ** p < 0.005; 2-tailed t-test, each sample was replicated two times). GluN2D expression was also reduced 42 days post MPTP injections in the SNc and the striatum, but not in 21 days age group, suggesting a gradual progression of the effect of MPTP on GluN2D expression in adjoining areas of the brain in our MPTP model.
Figure 4.

(A) Representative western blots showing GluN2D and GluN2B immunoreactivity and tubulin (Tub) loading controls from tissue obtained from micropunches from STN, SNc, and striatum (striatum- Str, C= Control and M= MPTP treated sample). Negative controls shown for anti-GluN2D antibody using GluN2D knock out (2D−/−690) mouse brain tissue sample compared with wild type (Wt). (B and C). Quantification of data from n= 4 animals evaluated in independent experiments, samples repeated one to two times per animal. Shown are the GluN2D expression for each animal as means of replicates, GluN2B expression for each sample tested once, with the intensity normalized to tubulin as loading control. * p < 0.05, ** p < 0.005 as compared to age-matched controls; two-tailed t-test.
We also quantified the expression of GluN2B subunits in the same mice (n = 4 animals per group, each sample measured once). There were no significant differences in GluN2B subunit expression in the three regions under investigation STN, SNc and striatum (Figure 4A, B, and C; p>0.05, 2-tailed t-test). This finding shows that not all receptor subunits are affected in MPTP-treated rodents leading to an altered ratio of different subunits. These findings might have far-reaching effects on the need of subtype-selective compounds for the treatment of PD.
Discussion
NMDARs are potentially important CNS targets for the treatment of neurological and psychiatric disorders (Zhou and Sheng 2013). In the last decade, the development of drugs targeting NMDAR subunits has gained momentum due to the fact that these receptors are expressed in a subtype-specific manner in different areas and nuclei of the CNS (Hallett and Standaert 2004, Paoletti 2007, Paoletti 2013, Zhang 2014b). While GluN2D receptors are expressed in parts of the brain outside of the basal ganglia and related structures, high levels of these receptors exist in several basal ganglia structures in the adult brain, including the globus pallidus, substantia nigra, and prominently, the subthalamic nucleus (Standaert 1994, Monyer 1994, Wenzel 1996, Clarke and Bolam 1998, Hamani 2004, Wilson and Bevan 2011, Swanger 2015). GluN2D subunit expression in these nuclei suggests that these receptors could be a useful pharmacologic target for the treatment of parkinsonism.
While parkinsonism results from the loss of the dopaminergic nigrostriatal tract, the pathophysiology of PD includes secondary changes in GABAergic and glutamatergic transmission throughout the basal ganglia (Kish 1988, DeLong 1990, Carlsson and Carlsson 1990, Fearnley 1991, Obeso 2000). The STN is a site of prominent alterations in glutamatergic transmission (Chu 2015 and Chu 2017), which may contribute to increased firing, increased burst firing, and the development of alpha- and beta-band oscillations in firing in the STN which are typical for the parkinsonian state (Galvan 2008, Pan 2014 and Pan 2016).
Our study of the effects of NMDA receptor blockade was limited because of the fact that only one animal was studied, and because only a single dose of the NMDA receptor blocker D-APV could be used. However, even given these limitations, we were able to show that NMDAR blockade with D-APV significantly reduced spike firing rate and the intensity of burst firing (as judged by the maximal number of spikes in bursts), although not affecting the overall prevalence of bursts. Importantly, the D-APV dose, and the timing of recording relative to the drug injections was clearly adequate to assess the effects of blocking NMDARs in the STN, as demonstrated through our drug diffusion simulations. Both the effects on firing rates and on bursting would counteract important components of the pathophysiology of parkinsonism, i.e., increased spiking activity, and increased bursting in the STN, but it is worth pointing out that we studied this animal only in the parkinsonian state. It is therefore not possible to say with certainty that the drug infusions ‘normalized’ STN firing in this particular animal. Studies in rodent models have previously shown the positive effects of APV in ameliorating Parkinsonian symptoms (Pan 2014). Taken together, this data is the first evidence of NMDAR control of spike firing in a non-human primate model of PD.
Our previous study emphasized the importance of glutamatergic transmission at GluN2D-containing NMDA receptors for STN function (Swanger 2015), suggesting that GluN2D-containing NMDARs would be an attractive target for the development of novel pharmacological tools for therapeutic approaches in PD (Hallett and Standaert 2004). Our experiments profiling NMDAR expression in the basal ganglia provide further insight in this regard. We found in both short- and long-term mouse models of dopamine depletion (21 and 42 days after the end of MPTP treatment) that the expression of GluN2D subunits was reduced in the STN, and that GluN2D expression in the SNc and striatum was significantly reduced in the chronic exposure paradigm (42 days age). Other studies using 6-OHDA model showed increased GluN2D expression in the striatum (Zhang 2015). However, the differences in results might be due to usage of different model. We also investigated changes in GluN2B subunit expression in the same regions and found that there were no significant changes across different nuclei. Data from our rodent MPTP model is congruent with our electrophysiological observations from our primate experiment. In our primate MPTP model, we observed rescue of spike firing activity after NMDAR blockade with D-APV. Matching this observation, we expectedly found that expression of at least one subunit (GluN2D) was impaired while the other subunit (GluN2B) was unaltered increasing the contribution of GluN2B containing NMDARs. This finding suggests that an altered ratio of GluN2D to GluN2B might be an important change associated with loss of dopaminergic neurons. Our current data are consistent with our previous study showing that the glutamatergic corticosubthalamic innervation of the STN was substantially reduced in MPTP-treated parkinsonian animals (Pan 2014, Mathai 2015, Sanders 2016, Sanders 2017, Chu 2017, Wang 2018), perhaps as a component of a more general glutamatergic disconnection of the basal ganglia (the cortico- and thalamo-striatal innervation also fail in this disease, Villalba and Smith 2017 and Villalba, Mathai and Smith 2015). Thus, while blockade of glutamatergic transmission in the STN, e.g., through the use of GluN2D-selective antagonists, may have antiparkinsonian effects, these effects may be limited, given the apparent loss of GluN2D expression in the STN and at other basal ganglia sites. Yet, given the finding that NMDAR blockade reduced the activity of STN neurons in a parkinsonian monkey, further exploration of the therapeutic potential of NMDA receptor antagonist therapies targeting the STN remains important.
Significance statement.
Our research shows that NMDAR blockade can ameliorate abnormalities of spike firing of STN neurons in parkinsonian non-human primates. We also show that GluN2D subunit containing NMDARs are downregulated in our parkinsonian model. Taken together, our data suggests that blockade of NMDARs in the STN could be a viable antiparkinsonian strategy. However, the ultimate success of this approach may be limited by parkinsonism-associated changes in NMDAR expression in the STN.
Acknowledgments
Funding:
This work was supported by a research grant from the Michael J. Fox Foundation, as well as NIH/NINDS grants R01-NS036654 (ST), R01-NS065371 (ST), R01ES023839 and R01ES019776 (GWM), and F31NS089242 (ARD) and P50-NS071669 (TW). The primate studies were supported in part via an NIH/ORIP infrastructure grant to the Yerkes National Primate Research Center (P51-OD011132).
Footnotes
Competing Financial Interests: S.F.T. is a consultant for Janssen Pharmaceuticals, Inc., a member of the Scientific Advisory Board for Sage Therapeutics, and a co-founder of NeurOp Inc.
Contributor Information
Subhrajit Bhattacharya, Department of Pharmacology, Emory University School of Medicine, Atlanta, GA 30322, United States, performed experiments, analyzed data, wrote manuscript.
Yuxian Ma, Yerkes National Primate Research Center, Emory University, Atlanta, GA 30322, United States, performed experiments, analyzed data, wrote manuscript.
Amy Rose Dunn, Rollins School of Public Health, Emory University, Atlanta, GA 30322, United States, performed experiments, analyzed data, wrote manuscript.
Joshua McCall Bradner, Rollins School of Public Health, Emory University, Atlanta, GA 30322, United States, performed experiments, analyzed data, wrote manuscript.
Annalisa Scimemi, Department of Biology, SUNY at Albany, Albany, NY 12222, United States performed simulations, wrote manuscript, 518-442-4367, fax- 518-442-4367.
Gary W. Miller, Rollins School of Public Health, Emory University, Atlanta, GA 30322, United States, designed experiments, wrote manuscript, 404-712-8582, fax- 404-727-8744
Thomas Wichmann, Yerkes National Primate Research Center, Emory University, Atlanta, GA 30322, United States, performed experiments, analyzed data, wrote manuscript, 404-727-3511, fax- 404-712-9294.
Stephen F. Traynelis, Department of Pharmacology, Emory University School of Medicine, Atlanta, GA 30322, United States, designed experiments, wrote manuscript
References
- Allers KA, Bergstrom DA, et al. MK801 and amantadine exert different effects on subthalamic neuronal activity in a rodent model of Parkinson’s disease. Experimental neurology. 2005;191(1):104–118. doi: 10.1016/j.expneurol.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Baudrexel S, Witte T, et al. Resting state fMRI reveals increased subthalamic nucleus–motor cortex connectivity in Parkinson’s disease. Neuroimage. 2011;55(4):1728–1738. doi: 10.1016/j.neuroimage.2011.01.017. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, et al. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72(2):507–20. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Kimble W, et al. Altered AMPA receptor expression plays an important role in inducing bidirectional synaptic plasticity during contextual fear memory reconsolidation. Neurobiology of learning and memory. 2017;139:98–108. doi: 10.1016/j.nlm.2016.12.013. [DOI] [PubMed] [Google Scholar]
- Blandini F, Nappi G, et al. Subthalamic infusion of an NMDA antagonist prevents basal ganglia metabolic changes and nigral degeneration in a rodent model of Parkinson’s disease. Annals of neurology. 2001;49(4):525–529. [PubMed] [Google Scholar]
- Brunenberg EJ, Moeskops P, et al. Structural and resting state functional connectivity of the subthalamic nucleus: identification of motor STN parts and the hyperdirect pathway. PloS one. 2012;7(6):e39061. 12. doi: 10.1371/journal.pone.0039061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia-implications for schizophrenia and Parkinson’s disease. Trends in Neurosciences. 1990;13(7):272–276. doi: 10.1016/0166-2236(90)90108-m. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. Journal of Neuroscience. 2007;27(30):8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HY, Atherton JF, et al. Heterosynaptic regulation of external globus pallidus inputs to the subthalamic nucleus by the motor cortex. Neuron. 2015;85(2):364–376. doi: 10.1016/j.neuron.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HY, McIver EL, et al. Loss of Hyperdirect Pathway Cortico-Subthalamic Inputs Following Degeneration of Midbrain Dopamine Neurons. Neuron. 2017;95(6):1306–18 e5. doi: 10.1016/j.neuron.2017.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke N, Bolam J. Distribution of glutamate receptor subunits at neurochemically characterized synapses in the entopeduncular nucleus and subthalamic nucleus of the rat. Journal of Comparative Neurology. 1998;397(3):403–420. [PubMed] [Google Scholar]
- Clements J, Westbrook G. Kinetics of AP5 dissociation from NMDA receptors: evidence for two identical cooperative binding sites. Journal of neurophysiology. 1994;71(6):2566–2569. doi: 10.1152/jn.1994.71.6.2566. [DOI] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends in Neurosciences. 1990;13(7):281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- Devergnas A, Wichmann T, et al. Electrophysiological effects of local administration of a selective T-type calcium channel blocker on thalamic neurons. Society of Neuroscience Annual Meeting Abstracts 2014 [Google Scholar]
- Devergnas A, Wichmann T, et al. Relationship between oscillatory activity in the cortico-basal ganglia network and parkinsonism in MPTP-treated monkeys. Neurobiol Dis. 2014;68:156–66. doi: 10.1016/j.nbd.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devergnas A, Chen E, et al. Anatomical localization of Cav3.1 calcium channels and electrophysiological effects of T-type calcium channel blockade in the motor thalamus of MPTP-treated monkeys. J Neurophysiol. 2016;115(1):470–85. doi: 10.1152/jn.00858.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias S, Joshua M, Goldberg JA, Heimer G, Arkadir D, Morris G, Bergman H. Statistical properties of pauses of the high-frequency discharge neurons in the external segment of the globus pallidus. J Neurosci. 2007;27(10):2525–38. doi: 10.1523/JNEUROSCI.4156-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(5):2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- Galvan A, Wichmann T. Pathophysiology of parkinsonism. Clinical Neurophysiology. 2008;119(7):1459–1474. doi: 10.1016/j.clinph.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan A, Hu X, et al. Localization and function of GABA transporters in the globus pallidus of parkinsonian monkeys. Experimental neurology. 2010;223(2):505–515. doi: 10.1016/j.expneurol.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan A, Devergnas A, et al. Lack of Antiparkinsonian Effects of Systemic Injections of the Specific T-Type Calcium Channel Blocker ML218 in MPTP-Treated Monkeys. ACS Chem Neurosci. 2016;7(11):1543–51. doi: 10.1021/acschemneuro.6b00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber J, Wurbel H, et al. Guide for the Care and Use of Laboratory Animals. 8. Washington (DC): National Academies Press (US); 2011. [PubMed] [Google Scholar]
- Gardoni F, Bellone C. Modulation of the glutamatergic transmission by Dopamine: a focus on Parkinson, Huntington and Addiction diseases. Frontiers in cellular neuroscience. 2015:9. doi: 10.3389/fncel.2015.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatev P, Wichmann T. Interactions between cortical rhythms and spiking activity of single basal ganglia neurons in the normal and parkinsonian state. Cerebral cortex. 2008;19(6):1330–1344. doi: 10.1093/cercor/bhn171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Bergman H. Computational physiology of the neural networks of the primate globus pallidus: function and dysfunction. Neuroscience. 2011;198:171–192. doi: 10.1016/j.neuroscience.2011.08.068. [DOI] [PubMed] [Google Scholar]
- Greenamyre J. Glutamate-dopamine interactions in the basal ganglia: relationship to Parkinson’s disease. Journal of neural transmission. 1993;91(2):255–269. doi: 10.1007/BF01245235. [DOI] [PubMed] [Google Scholar]
- Hallett PJ, Standaert DG. Rationale for and use of NMDA receptor antagonists in Parkinson’s disease. Pharmacology & therapeutics. 2004;102(2):155–174. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Hamani C, Saint-Cyr JA, et al. The subthalamic nucleus in the context of movement disorders. Brain. 2004;127(1):4–20. doi: 10.1093/brain/awh029. [DOI] [PubMed] [Google Scholar]
- Hammond C, Bergman H, et al. Pathological synchronization in Parkinson’s disease: networks, models and treatments. Trends in Neurosciences. 2007;30(7):357–364. doi: 10.1016/j.tins.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Kammermeier S, Pittard D, et al. Effects of high-frequency stimulation of the internal pallidal segment on neuronal activity in the thalamus in parkinsonian monkeys. J Neurophysiol. 2016;116(6):2869–81. doi: 10.1152/jn.00104.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, et al. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. New England Journal of Medicine. 1988;318(14):876–880. doi: 10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- Kita H, Nambu A, et al. Role of ionotropic glutamatergic and GABAergic inputs on the firing activity of neurons in the external pallidum in awake monkeys. Journal of neurophysiology. 2004;92(5):3069–3084. doi: 10.1152/jn.00346.2004. [DOI] [PubMed] [Google Scholar]
- Kliem MA, Wichmann T. A method to record changes in local neuronal discharge in response to infusion of small drug quantities in awake monkeys. Journal of neuroscience methods. 2004;138(1):45–49. doi: 10.1016/j.jneumeth.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Kliem MA, Pare JF, et al. Ultrastructural localization and function of dopamine D1-like receptors in the substantia nigra pars reticulata and the internal segment of the globus pallidus of parkinsonian monkeys. Eur J Neurosci. 2010;31(5):836–51. doi: 10.1111/j.1460-9568.2010.07109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether T, Turski L. Excitatory amino acids and the basal ganglia: implications for the therapy of Parkinson’s disease. Trends in Neurosciences. 1989;12(8):285–286. doi: 10.1016/0166-2236(89)90007-6. [DOI] [PubMed] [Google Scholar]
- Legendy C, Salcman M. Bursts and recurrences of bursts in the spike trains of spontaneously active striate cortex neurons. Journal of neurophysiology. 1985;53(4):926–939. doi: 10.1152/jn.1985.53.4.926. [DOI] [PubMed] [Google Scholar]
- Lohr KM, Bernstein AI, et al. Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proceedings of the National Academy of Sciences. 2014;111(27):9977–9982. doi: 10.1073/pnas.1402134111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathai A, Ma Y, et al. Reduced cortical innervation of the subthalamic nucleus in MPTP-treated parkinsonian monkeys. Brain. 2015;138(4):946–962. doi: 10.1093/brain/awv018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JL, Perlman JE, et al. Refining the pole-and-collar method of restraint: emphasizing the use of positive training techniques with rhesus macaques (Macaca mulatta) Journal of the American Association for Laboratory Animal Science: JAALAS. 2014;53(1):61–8. [PMC free article] [PubMed] [Google Scholar]
- Miller WC, DeLong MR. Altered tonic activity of neurons in the globus pallidus and subthalamic nucleus in the primate MPTP model of parkinsonism. In: Carpenter MB, Jayaraman A, editors. The basal ganglia. II. Structure and function: current concepts. New York: Plenum; 1987. pp. 415–427. [Google Scholar]
- Monyer H, Burnashev N, et al. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12(3):529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Nambu A. A new approach to understand the pathophysiology of Parkinson’s disease. Journal of Neurology. 2005;252:iv1–iv4. doi: 10.1007/s00415-005-4002-y. [DOI] [PubMed] [Google Scholar]
- Nielsen JB, Conway B, et al. Organization of common synaptic drive to motor neurons during fictive locomotion in the spinal cat. The Journal of Physiology. 2005;569(1):291–304. doi: 10.1113/jphysiol.2005.091744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, et al. Pathophysiology of the basal ganglia in Parkinson’s disease. Trends in Neurosciences. 2000;23:S8–S19. doi: 10.1016/s1471-1931(00)00028-8. [DOI] [PubMed] [Google Scholar]
- Ogden KK, Chen W, et al. Molecular mechanism of disease-associated mutations in the pre-M1 helix of NMDA receptors and potential rescue pharmacology. PLoS genetics. 2017;13(1):e1006536. doi: 10.1371/journal.pgen.1006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan MK, Tai CH, et al. Deranged NMDAergic cortico-subthalamic transmission underlies parkinsonian motor deficits. J Clin Invest. 2014;124:4629–4641. doi: 10.1172/JCI75587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan MK, Kuo SH, et al. Neuronal firing patterns outweigh circuitry oscillations in parkinsonian motor control. The Journal of clinical investigation. 2016;126(12):4516. doi: 10.1172/JCI88170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Current opinion in pharmacology. 2007;7(1):39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, et al. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nature Reviews Neuroscience. 2013;14(6):383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Rice ME, Nicholson C. Diffusion characteristics and extracellular volume fraction during normoxia and hypoxia in slices of rat neostriatum. Journal of neurophysiology. 1991;65(2):264–272. doi: 10.1152/jn.1991.65.2.264. [DOI] [PubMed] [Google Scholar]
- Rubin JE, McIntyre CC, et al. Basal ganglia activity patterns in parkinsonism and computational modeling of their downstream effects. European Journal of Neuroscience. 2012;36(2):2213–2228. doi: 10.1111/j.1460-9568.2012.08108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders TH, Clements MA, et al. Parkinsonism-related features of neuronal discharge in primates. Journal of neurophysiology. 2013;110(3):720–731. doi: 10.1152/jn.00672.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders TH, Jaeger D. Optogenetic stimulation of cortico-subthalamic projections is sufficient to ameliorate bradykinesia in 6-ohda lesioned mice. Neurobiology of disease. 2016;95:225–237. doi: 10.1016/j.nbd.2016.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders TH. Stimulation of Cortico-Subthalamic Projections Amplifies Resting Motor Circuit Activity and Leads to Increased Locomotion in Dopamine-Depleted Mice. Frontiers in integrative neuroscience. 2017;11:24. doi: 10.3389/fnint.2017.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standaert DG, Testa CM, et al. Organization of N-methyl-D-aspartate glutamate receptor gene expression in the basal ganglia of the rat. Journal of Comparative Neurology. 1994;343(1):1–16. doi: 10.1002/cne.903430102. [DOI] [PubMed] [Google Scholar]
- Swanger SA, Vance KM, et al. NMDA receptors containing the GluN2D subunit control neuronal function in the subthalamic nucleus. Journal of Neuroscience. 2015;35(48):15971–15983. doi: 10.1523/JNEUROSCI.1702-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Chen W, et al. Mechanistic insight into NMDA receptor dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. The American Journal of Human Genetics. 2016;99(6):1261–1280. doi: 10.1016/j.ajhg.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Vance KM, et al. A novel negative allosteric modulator selective for GluN2C/2D-containing NMDA receptors inhibits synaptic transmission in hippocampal interneurons. ACS chemical neuroscience. 2017 doi: 10.1021/acschemneuro.7b00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney AM, Fleming KE, et al. PAR1 activation induces rapid changes in glutamate uptake and astrocyte morphology. Scientific Reports. 2017;7:43606. doi: 10.1038/srep43606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological reviews. 2010;62(3):405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Mathai A, et al. Morphological changes of glutamatergic synapses in animal models of Parkinson’s disease. Frontiers in neuroanatomy. 2015:9. doi: 10.3389/fnana.2015.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Smith Y. Loss and remodeling of striatal dendritic spines in Parkinson’s disease: from homeostasis to maladaptive plasticity? Journal of neural transmission. 2017:1–17. doi: 10.1007/s00702-017-1735-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YY, Wang Y, et al. Impaired glutamatergic projection from the motor cortex to the subthalamic nucleus in 6-hydroxydopamine-lesioned hemi-parkinsonian rats. Exp Neurol. 2018;300:135–48. doi: 10.1016/j.expneurol.2017.11.006. [DOI] [PubMed] [Google Scholar]
- Wenzel A, et al. Developmental and regional expression of NMDA receptor subtypes containing the NR2D subunit in rat brain. Journal of neurochemistry. 1996;66(3):1240–1248. doi: 10.1046/j.1471-4159.1996.66031240.x. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Soares J. Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. Journal of neurophysiology. 2006;95(4):2120–2133. doi: 10.1152/jn.01013.2005. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Dostrovsky JO. Pathological basal ganglia activity in movement disorders. Neuroscience. 2011;198:232–244. doi: 10.1016/j.neuroscience.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ, Bevan MD. Intrinsic dynamics and synaptic inputs control the activity patterns of subthalamic nucleus neurons in health and in Parkinson’s disease. Neuroscience. 2011;198:54–68. doi: 10.1016/j.neuroscience.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters WD, Kado RT, Adey WR. A stereotaxic brain atlas for Macaca Nemestrina. Berkeley California Press; 1969. [Google Scholar]
- Zhang X, Feng ZJ, et al. GluN2D-containing NMDA receptors inhibit neurotransmission in the mouse striatum through a cholinergic mechanism: implication for Parkinson’s disease. Journal of neurochemistry. 2014;129(4):581–590. doi: 10.1111/jnc.12658. [DOI] [PubMed] [Google Scholar]
- Zhang X, Feng ZJ, et al. Allosteric modulation of GluN2C/GluN2D-containing NMDA receptors bidirectionally modulates dopamine release: implication for Parkinson’s disease. British journal of pharmacology. 2014;171(16):3938–3945. doi: 10.1111/bph.12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chergui K. Dopamine depletion of the striatum causes a cell-type specific reorganization of GluN2B-and GluN2D-containing NMDA receptors. Neuropharmacology. 2015;92:108–115. doi: 10.1016/j.neuropharm.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Sheng M. NMDA receptors in nervous system diseases. Neuropharmacology. 2013;74:69–75. doi: 10.1016/j.neuropharm.2013.03.030. [DOI] [PubMed] [Google Scholar]