

Abstract
In this contribution, we report studies of the primary dynamics of the drug–protein complexes of daunomycin with apo riboflavin-binding protein. With femtosecond resolution, we observed the ultrafast charge separation between daunomycin and aromatic amino acid residues of the protein, tryptophan(s). Electron transfer occurs from tryptophan(s) to daunomycin with two reaction times, 1 ps and 6 ps, depending on the local complex structure. The formation of anionic daunomycin radical is crucial for triggering a series of chemical reactions in redox cycling. One of the subsequent reactions is the reduction of dioxygen to form active superoxide by the reduced daunomycin. This catalytic process was found to occur within 10 ps. In the absence of dioxygen, charge recombination takes a much longer time, more than 100 ps. These results, along with similar findings in DNA and nucleotides, elucidate that the ultrafast generation of reduced daunomycin radicals by photoactivation is a primary step for the observed photoenhancement of drug cytotoxicity by several orders of magnitude. We also studied the dependence of the dynamics on protein conformations at different ionic strengths and denaturant concentrations. We observe a sharp transition from the tertiary structure to the unfolding state at 2 M of denaturant concentration.
Daunomycin (DM, Fig. 1), an anthracycline antibiotic, is one of the effective drugs used for cancer chemotherapy. It has been shown that the cytotoxicity of DM is enhanced by several orders of magnitude with irradiation of light, but the mechanism by photoactivation is still unknown (1–3). In recent studies from this group (X. Qu, C.W., H.-C. Becker, D.Z., and A.H.Z., unpublished work), we reported the striking observation of the primary dynamics of DM when it intercalates into GC base pairs of DNA or binds to nucleotides. Ultrafast electron-transfer (ET) reactions between DM and the base G were found to occur on the femtosecond to picosecond time scales (4). The reduced DM radicals can catalyze superoxide formation from dioxygen (O2), which then triggers redox cycling. Thus, molecular oxygen becomes an integral part of the drug function.
Figure 1.

Ribbon diagram of the x-ray structure of riboflavin-binding protein (6) and a view of the hydrophobic cleft. Also shown are the molecular structure of DM and the stick presentation of Trp-156 amino acid residue (yellow) of the protein.
Here, we report our studies of the dynamics of DM with a protein. Many proteins have hydrophobic domains and can interact with DM molecules. Aromatic or sulfur-containing amino acid residues in proteins, such as Trp, Tyr, or cysteine, can often function as reductants and donate electrons. One of the flavoproteins, apo riboflavin-binding protein (apoRfBP) and a riboflavin carrier in plasma, has several Trp residues in its hydrophobic cleft (20 Å wide and 15 Å deep). This protein, a single polypeptide chain of 219 amino acids, was chosen as a prototype (Fig. 1) and the complex of DM–apoRfBP as a model system.
As mentioned in the preceding paper (5), the x-ray crystal structure reveals a compact π stack of riboflavin (vitamin B2) sandwiched between Trp-156 and Tyr-75 at an equal interplanar distance of 3.7 Å in the hydrophobic cleft of apoRfBP (6). Ultrafast ET between riboflavin and Trp-156 is dominant and occurs in ≈100 fs, a very favorable process (5, 7). DM and riboflavin have similar hydrophobic chromophores with a planar three-ring structure (anthraquinone-type), and both have a catalytic redox property. Thus, the drug DM inserts into the hydrophobic cleft of the binding site, and the DM-apoRfBP complex exhibits a similar ET reaction on photoinitiation.
Methodology.
Femtosecond-resolved fluorescence up-conversion and transient absorption techniques were used for all experimental measurements. The experimental setup is described in detail elsewhere (8). The pump pulse from an optical parametric amplifier (OPA) was attenuated to 0.1–0.4 μJ and fixed at 480 nm, whereas for some experiments, the pump pulse was tuned to 510 nm. The probe wavelength was set at 795 nm as the gating pulse for up-conversion measurements. For transient absorption experiments, another OPA was used, and the probe pulse was tuned from 315 to 700 nm. Most transients were taken at the magic angle (54.7°) of the pump polarization, relative to that of the probe pulse (transient absorption) or the fluorescence (determined by the nonlinear crystal). The pump polarization was also adjusted to be parallel or perpendicular, and the resulting anisotropy was measured carefully: r(t) = (I∥ − I⊥)/(I∥ + 2I⊥).
Sample Preparation.
apoRfBP from chicken egg white (lyophilized powder), l-tryptophan, antitumor drug of DM, and other solvents (spectroscopy-grade) were purchased from Sigma. Ultrapure guanidine hydrochloride (Gdn⋅HCl) was obtained from Baker, and sodium chloride (NaCl) from Mallinckrodt. All chemicals were used as supplied.
For most experiments, the lyophilized apoRfBP powder was dissolved in 30 mM phosphate buffer at pH=7 with a concentration of 400 μM. DM was added into the solution with a concentration of 50 μM, reaching a ratio of 8:1 for apoRfBP to DM. We found that more than 98% of DM molecules bind to the protein. The association constant was previously reported to be 2 × 106 M−1 (9). The binding constant of DM to tryptophan in buffer solution is much smaller, and we estimated it to be ≈25 M−1. For experiments involving the addition of the salt NaCl to increase the ionic strength or the denaturing agent Gdn⋅HCl to unfold the protein, the mixed samples were allowed to equilibrate for more than 3 h before use. For experiments of free drug DM in buffer alone, a concentration of 10 μM DM was used to minimize dimer formation (10). All samples were measured at room temperature in a fused silicon cell with 1- to 3-mm path length, and no photobleaching was observed.
The steady-state absorption and fluorescence emission spectra of DM were studied in detail recently in this laboratory. After the complexation of DM with apoRfBP, no apparent changes were found in the absorption spectrum and fluorescence emission profile, except that the total fluorescence intensity of drug–protein complexes (at 480 nm excitation) decreases by several orders of magnitude.
Results and Discussion
Charge Separation and DM Reduction.
The femtosecond-resolved fluorescence transients of DM in the hydrophobic cleft of apoRfBP are shown in Fig. 2A for a systematic series of fluorescence wavelength detection. It is clear that all transients show ultrafast dynamics on the femtosecond to picosecond time scale with only a minor component (2–3%) in the nanosecond range. The free DM molecule in water has been well characterized (work in this group; refs. 11 and 12). Basically, the first excited state has ≈1 ns lifetime. At 480 nm excitation, vibrational relaxation was found to occur in 200 fs and up to 1 ps: ultrafast decay components at shorter wavelength (530 nm) and the corresponding rising components at longer wavelength (670 nm) were observed, in addition to the nanosecond component. At 610 nm, we only observed the relaxed-state emission with the nanosecond component, as also shown in Fig. 2B. Little solvation dynamics was observed, because no apparent changes of the dynamical transients as well as the static emission peaks were found when DM dissolves in solvents with different polarity. Thus, the observed minor nanosecond component in the drug–protein complexes results from the free, or nearly free, DM molecules in solution, indicating that almost all DM molecules insert into the protein.
Figure 2.

(A) Femtosecond, normalized fluorescence up-conversion decays of the drug DM (50 μM) with apoRfBP (400 μM) for different-wavelength detection at pH = 7. (B) Comparison of the normalized fluorescence decays, detected at 610-nm emission, for DM in buffer water solution; with tryptophan (40 mM); and with apoRfBP.
As shown in Fig. 2A, above 600 nm, all transients have the same temporal behavior and decay in 1 ps (73%) and 6 ps (24%); at 560 nm, the decay time constants are 0.8 ps (75%) and 5.8 ps (22%); at 530 nm, they become 0.5 ps (83%) and 3.7 ps (15%). The result at 610 nm gives accurate decay times for DM* in the protein, because the contribution of vibrational relaxation is negligible (Fig. 2B). At the blue side (530 nm and 560 nm), the transients result from a superposition of decay dynamics and vibrational relaxation. Thus, the decay of DM* in the protein has two components (1 ps and 6 ps) with a ratio of 3:1.
The observed decay of DM* results from ET from Trp residues in the protein to DM* for several reasons. Resonance energy transfer is ruled out because of no spectral overlap between DM* emission (>500 nm) and apoRfBP absorption (<300 nm). Collision quenching by diffusion control occurs on much longer time scale (>ns). Finally, a proton-transfer quenching mechanism is also excluded because no intermolecular proton transfer was observed in D2O, H2O, and methanol solutions and at different pH.
The redox properties of DM and Trp give a favorable driving force for the ET reaction: the potential for DM/DM⋅− is ≈−0.4 V vs. NHE (ref. 13 and refs. therein) and for Trp/Trp⋅+, it is ≈+1.15 V vs. NHE (14). Knowing the 0–0 transition (S1←S0) energy of 2.3 eV, we obtained a net ΔG° of −0.75 eV. Ultrafast ET between Trp-156 and riboflavin has been observed to occur in ≈100 fs (85%) and 700 fs (14%) at 480 nm excitation (5). Here, we observed the same ET reaction with longer times (1 ps and 6 ps). The big difference in ET rates is due to their binding structures in the hydrophobic cleft, although the net ΔG° is only −0.35 eV for riboflavin in the protein. The chromophore part of DM was shown to be mobile in the binding site by circular dichroism (15). The perfect intercalation of DM in DNA (16) also accelerates the ET reaction (290 fs and 1.7 ps) although the net ΔG° (−0.4 eV) is smaller than that of DM*-Trp complexes in the protein.
We studied the decay dynamics of DM* in a tryptophan solution (at the maximum Trp solubility of ≈50 mM). To minimize the formation of DM dimer, we used as low as 10 μM concentration of DM. The observed transient is shown in Fig. 2B. Similar ET reaction time was observed as in the protein for the small fraction that is not free in solution. The observed two ET rates of DM in apoRfBP, as in DNA and nucleotides, are attributed to two types of binding structures. Two ground-state isomers of DM have been observed by NMR measurements (17) and also proposed by theoretical calculations (18). Both isomers are present in solution in a thermal equilibrium, and fluorescence studies also confirm these two conformational states.
To fully elucidate the ET mechanism, we performed transient absorption measurements to study the dynamics of the charge-separated state and the ground-state recovery of DM. Fig. 3 shows five transients probed in the range of 315–630 nm. All transients have a positive absorption, except that at 480 nm we observed a negative absorption. Specifically, at 630 and 510 nm, the two transients are nearly identical and have a rise component of ≈900 fs and a decay component of 4.8 ps. At 545 nm, the transient decays in 1 ps and 4.8 ps. But at 315 nm, the transient is dominated by a 6-ps decay component. These observations are very similar to those obtained for DM-nucleotides and DM in DNA, and all transients reflect the excited-state DM*-Trp dynamics at these wavelengths: the observed 1 ps and 4.8–6 ps decay components are consistent with the results obtained by up-conversion fluorescence decay measurements. Note that the probe wavelengths here are more sensitive to the longer-time component (≈6 ps). The initial rise component (<1 ps) is attributed to vibrational relaxation, consistent with the result observed for the free DM molecules in bulk solvents.
Figure 3.

Femtosecond transient absorption profiles of DM (50 μM) with apoRfBP (400 μM) probed at different wavelengths, from 315 to 630 nm.
To probe the recovery of DM or the ET product, we tuned the pump wavelength to 510 nm and set the probe wavelength at 480 nm for the maximum ground-state absorption, detecting only the 480 nm transmission. At t = 0, we observed a net negative signal, which is the superposition of the ground-state depletion (negative) and the excited-state absorption (positive). After t = 0, we observed a dominant rise signal, reflecting either the formation dynamics of the ET state or the ground-state DM recovery. To distinguish these two processes, we have to understand the decay dynamics of the ET state and the DM recovering process, which are given below.
Role of Molecular Oxygen.
Oxygen is present in solution (≈1 mM in air-exposed water), and its reduced form (O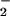 ) or singlet-excited state (1O*2) plays an important role in biological functions (19). All above transients were taken in aqueous solution in presence of O2 (without degassing of O2). Fig. 4 shows the transient absorption signals taken without O2 and under exposure to the air at 480-nm probe wavelength (λpu = 510 nm). A clear difference was observed for the positive absorption (ΔA>0). However, the excited-state DM*-Trp dynamics measured by the transient absorption at 630 nm (Fig. 4B) does not show any noticeable change under these two conditions. Thus, the presence of O2 in close proximity does not influence the forward charge-separation rate but does affect the ET-state decay dynamics, as observed in nucleotides and in DNA (X. Qu, C.W., H.-C. Becker, D.Z., and A.H.Z., unpublished work).
) or singlet-excited state (1O*2) plays an important role in biological functions (19). All above transients were taken in aqueous solution in presence of O2 (without degassing of O2). Fig. 4 shows the transient absorption signals taken without O2 and under exposure to the air at 480-nm probe wavelength (λpu = 510 nm). A clear difference was observed for the positive absorption (ΔA>0). However, the excited-state DM*-Trp dynamics measured by the transient absorption at 630 nm (Fig. 4B) does not show any noticeable change under these two conditions. Thus, the presence of O2 in close proximity does not influence the forward charge-separation rate but does affect the ET-state decay dynamics, as observed in nucleotides and in DNA (X. Qu, C.W., H.-C. Becker, D.Z., and A.H.Z., unpublished work).
Figure 4.

(A) Femtosecond transient absorption profiles of DM-apoRfBP complexes in the absence of dioxygen and under exposure to the air. The transient under the aerated condition is best represented by two components with (53%) and without dioxygen (47%); see Inset. (B) Comparison of transient absorption profiles without dioxygen and under the aerated condition at different probe wavelengths of 480 and 630 nm (see Inset). Note that the transients under two conditions at 630-nm probe are identical, showing that excited-state dynamics of DM*-apoRfBP are the same; see text for details.
Upon purging with argon to remove O2, the transient initially rises from a negative absorption to a positive one in 3.6 ps (66%) and 7 ps (34%), and then decays in 135 ps (74%) and ≈950 ps (26%). Note that the small net positive signal is sensitive to the absorption difference of the ET state and the ground state, and the charge-recombination rate. The observed rise times are consistent with the excited-state DM*-Trp decay dynamics (≈1–6 ps), reflecting the formation of the DM⋅−-Trp⋅+ state. The long decay component (>100 ps) represents the charge recombination without involvement of O2, that is, the electron recombination to from the final ground state of DM.
When the sample was exposed to air, the amplitude of the negative absorption shows nearly no change but the positive signal decreases by a factor of ≈2. The rise components apparently become longer but the decay components keep the same. In buffer solution, there exists equilibrium between O2 and the DM–Trp complexes in the hydrophobic cleft. Thus, the observed positive absorption in air is still from the DM–Trp complexes without the influence of O2. The total signal was best fitted by two components under the same forward ET rates and the same absorptions for the ET state and the ground-state DM, as shown in the Fig. 4A Inset. One component (47%) is from the DM⋅−-Trp⋅+ complexes without O2, and the other one (53%) is from the DM⋅−-Trp⋅+ (O2) complexes with ≈10 ps recovering time for the ground-state DM.
We note that if the reaction of DM⋅−-Trp⋅+ with O2 takes longer than 10 ps and less than 100 ps, the positive signal, compared with that in the absence of O2, does become smaller, but the decay times also become shorter, inconsistent with our observation. Therefore, our observed decrease of the positive absorption and invariance of the decay times support the presence of two components under aerated conditions and the reduction of O2 by DM⋅−occurs in less than 10 ps to form the active superoxide O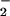 and ground-state DM. The overall mechanism is illustrated in Fig. 5, and the scheme resembles that for the drug/DNA, nucleotide systems.
and ground-state DM. The overall mechanism is illustrated in Fig. 5, and the scheme resembles that for the drug/DNA, nucleotide systems.
Figure 5.

A reaction scheme for the ET from tryptophan(s) in the hydrophobic cleft of the protein to the drug DM. The solid arrows represent the observed ET processes by photoactivation. The lighter dashed line represents the thermal reaction pathway involving a barrier crossing, a much slower process. The dashed arrow indicates the final charge recombination. (Inset) The elementary steps involved in the photoinitiated ET processes.
The observed fast ET reaction of DM⋅− with O2 is energetically favorable based on the net ΔG° [−0.24 eV; DM/DM⋅−≈−0.4 V vs. NHE and O2/O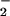 ≈−0.16 V vs. NHE (20)]. Compared with the reaction time of DM⋅−-O2 (<3 ps) in aqueous solution of nucleotides, the observed longer reaction time with O2 in the protein is probably due to the change of redox potentials in hydrophobic environments. It should be pointed out that O2 binding to the DM–Trp complex takes place already in the ground state in the protein cleft, because the diffusion-controlled reaction will occur on a much longer time scale (≈microseconds) at our concentrations.
≈−0.16 V vs. NHE (20)]. Compared with the reaction time of DM⋅−-O2 (<3 ps) in aqueous solution of nucleotides, the observed longer reaction time with O2 in the protein is probably due to the change of redox potentials in hydrophobic environments. It should be pointed out that O2 binding to the DM–Trp complex takes place already in the ground state in the protein cleft, because the diffusion-controlled reaction will occur on a much longer time scale (≈microseconds) at our concentrations.
The identification of O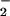 formation is important because O
formation is important because O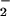 is a very active species in the biological reactions. It has been known that redox cycling, an important role for drug cytotoxicity, begins with the formation of the DM⋅− anionic radical and then the radical reacts with O2 to form superoxide O
is a very active species in the biological reactions. It has been known that redox cycling, an important role for drug cytotoxicity, begins with the formation of the DM⋅− anionic radical and then the radical reacts with O2 to form superoxide O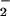 (ref. 13 and refs. therein, refs. 21 and 22). Thus, the photoinitiated ET-state formation by femtosecond pulses leads to the formation of drug radicals (anionic) on an ultrafast time scale, and these radicals could trigger a series of chemical reactions in redox cycling with much higher efficiency and enhance cell killing by the anticancer drug. With DNA and nucleotides, this concept of short-range oxygen involvement is clear, and its presence in proteins elucidates the role of hydrophobicity in both systems.
(ref. 13 and refs. therein, refs. 21 and 22). Thus, the photoinitiated ET-state formation by femtosecond pulses leads to the formation of drug radicals (anionic) on an ultrafast time scale, and these radicals could trigger a series of chemical reactions in redox cycling with much higher efficiency and enhance cell killing by the anticancer drug. With DNA and nucleotides, this concept of short-range oxygen involvement is clear, and its presence in proteins elucidates the role of hydrophobicity in both systems.
Conformation vs. ET Rate.
Because the ET rate between DM* and Trp in the protein is sensitive to their relative positions, we studied the ET reaction by changing protein conformations by adding two different salts into the protein solution. Fig. 6 shows our results. By increasing the ionic strength to 4 M NaCl, the transient (Fig. 6A) changes and gives four decay times: 1.5 ps (56%), 9.2 ps (28%), 89 ps (7%), and 1.1 ns (9%). These observations are interesting in that they contrast those obtained in RfBP, where the transient showed no significant change under the same condition (5). The long components (89 ps and 1.1 ns) account for up to 16% of the total signal, and it is only 2–3% without NaCl. These results reveal that the protein at 4 M NaCl pushes DM molecules out of the tight hydrophobic pocket.
Figure 6.

(A) Femtosecond fluorescence up-conversion decays of DM (50 μM) with apoRfBP (400 μM), detected at 610 nm, without and with the addition of the ionic salt NaCl. The Inset gives the time-resolved anisotropy [r(t)] of DM in the protein with a concentration of 4 M NaCl. Note the difference in the time scale between the fluorescence and anisotropy decay. (B) Fluorescence decays (610 nm) of DM (50 μm) bound to the apoRfBP (400 μM) with a series of concentrations of Gdn⋅HCl, from 0 M to 2 M.
To further characterize these “free” DM molecules, we measured the time-resolved anisotropy, as shown in Fig. 6A Inset. The anisotropy [r(t)] decays with a time constant of 310 ps and then remains constant at 0.061. The anisotropy decay time of free DM molecules in buffer water without protein is 200 ps. Therefore, the DM molecules, which are pushed out of the direct ET-reaction center by adding 4 M NaCl, are not totally free and still in proximity to the protein, leading to a longer anisotropy decay time and a hindered rotation motion.
The transients obtained after adding the ionic denaturant Gdn⋅HCl are shown in Fig. 6B with a series of concentrations. Clearly, when the concentration reaches 2 M, all of the DM molecules behave like free molecules (nanosecond decay), and no ultrafast ET reaction was observed. As mentioned in the preceding paper (5), it has been reported that the unfolding of the native RfBP protein occurs in two phases from 0 to 6 M Gdn⋅HCl, as measured by circular dichroism and fluorescence spectra (23). Between 0–2 M Gdn⋅HCl, the loss of its tertiary structure and binding ability to riboflavin occurs, but the protein still remains in its secondary structure. At the high concentration of 4–6 M Gdn⋅HCl, the loss of the secondary structure takes place and between 2–4 M Gdn⋅HCl, and there probably exists an intermediate conformation, a molten-globule state. The absence of the ET reaction in our transients at 2 M Gdn⋅HCl undoubtedly indicates the collapsing of the hydrophobic cleft, consistent with the loss of the tertiary structure. The complete collapse of the binding site occurs at 3 M Gdn⋅HCl (5).
Conclusion
The reported studies with femtosecond resolution of the drug–protein complex, DM with apoRfBP, elucidate the different time scales involved in charge separation, recombination, and the involvement of dioxygen. The process involving the formation of DM anionic radicals is critical to the drug function. Both the photoactivated and thermal reactions are likely to have similar transition states (24, 25), with the latter being much slower in the rate due to the involvement of a barrier crossing (see Fig. 5). The resulting anionic DM radical can trigger a series of chemical reactions in redox cycling under physiological conditions. One of the subsequent reactions is the reduction of molecular oxygen in close proximity, not diffusive, as shown here and elsewhere (X. Qu, C.W., H.-C. Becker, D.Z., and A.H.Z., unpublished work). As with DNA, this primary step is important for the observed photoenhancement of drug cytotoxicity by several orders of magnitude. Another major result of this study is the use of ET rates as a sensitive probe of the local binding structure of the protein, which is revealed by the unfolding processes observed in our transients.
Acknowledgments
We thank Prof. Hugo L. Monaco (University of Verona, Italy) for providing the riboflavin-binding protein coordinates, and Dr. Hans-Christian Becker for discussion and help. This work was supported by the National Science Foundation.
Abbreviations
- DM
daunomycin
- Trp
tryptophan
- apoRfBP
apo riboflavin-binding protein
- ET
electron transfer
- Gdn⋅HCl
guanidine hydrochloride
References
- 1.Andreoni A, Colasanti A, Malatesta V, Roberti G. Radiat Res. 1991;127:24–29. [PubMed] [Google Scholar]
- 2.Andreoni A, Colasanti A, Kisslinger A, Mastrocinque M, Portella G, Riccio P, Roberti G. Photochem Photobiol. 1993;57:851–855. doi: 10.1111/j.1751-1097.1993.tb09223.x. [DOI] [PubMed] [Google Scholar]
- 3.Paiva M B, Saxton R E, Graeber I, Jongewaard N, Eshraghi A A, Suh M J, Paek W H, Castro D J. Lasers Surg Med. 1998;23:33–39. doi: 10.1002/(sici)1096-9101(1998)23:1<33::aid-lsm5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 4.Wan C, Fiebig T, Schiemann O, Barton J K, Zewail A H. Proc Natl Acad Sci USA. 2000;97:14052–14055. doi: 10.1073/pnas.250483297. . (First Published December 5, 2000; 10.1073/pnas.250483297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong D, Zewail A H. Proc Natl Acad Sci USA. 2001;98:11867–11872. doi: 10.1073/pnas.211440398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monaco H L. EMBO J. 1997;16:1475–1483. doi: 10.1093/emboj/16.7.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mataga N, Chosrowjan H, Shibata Y, Tanaka F, Nishina Y, Shiga K. J Phys Chem B. 2000;104:10667–10677. [Google Scholar]
- 8.Fiebig T, Wan C, Kelley S O, Zewail A H. Proc Natl Acad Sci USA. 1999;96:1187–1192. doi: 10.1073/pnas.96.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher J, Ramakrishnan K, McLane K E. Biochemistry. 1982;21:6172–6180. doi: 10.1021/bi00267a023. [DOI] [PubMed] [Google Scholar]
- 10.Chaires J B, Dattagupta N, Crothers D M. Biochemistry. 1982;21:3927–3932. doi: 10.1021/bi00260a004. [DOI] [PubMed] [Google Scholar]
- 11.Chaires J B, Dattagupta N, Crothers D M. Biochemistry. 1982;21:3933–3940. doi: 10.1021/bi00260a005. [DOI] [PubMed] [Google Scholar]
- 12.Malatesta V, Andreoni A. Photochem Photobiol. 1988;48:409–415. doi: 10.1111/j.1751-1097.1988.tb02838.x. [DOI] [PubMed] [Google Scholar]
- 13.Priebe W, editor. Anthracycline Antibiotics New Analogues, Methods of Delivery and Mechanism of Actions. Washington, DC: Am. Chem. Soc.; 1995. [Google Scholar]
- 14.DeFelippis M R, Murthy C P, Broitman F, Weinraub D, Faraggi M, Klapper M H. J Phys Chem. 1991;95:3416–3419. [Google Scholar]
- 15.Galat A. Int J Biochem. 1988;20:1021–1029. doi: 10.1016/0020-711x(88)90191-7. [DOI] [PubMed] [Google Scholar]
- 16.Wang A H, Ughetto G, Quigley G J, Rich A. Biochemistry. 1987;26:1152–1163. doi: 10.1021/bi00378a025. [DOI] [PubMed] [Google Scholar]
- 17.Penco S, Angelucci F, Ballabio M, Barchielli G, Suarato A, Vanotti E, Vigevani A, Arcamone F. Heterocycles. 1984;21:21–28. [Google Scholar]
- 18.Tosi C, Fusco R, Ranghino G, Malatesta V. J Mol Struct (Theochem) 1986;134:341–350. [Google Scholar]
- 19.Valentine J S, Foote C, editors. Active Oxygen in Biochemistry. London: Black Academic & Professional; 1995. [Google Scholar]
- 20.Sawyer D T, editor. Oxygen Chemistry. New York: Oxford Univ. Press; 1991. [Google Scholar]
- 21.Taatjes D J, Gaudiano G, Resing K, Koch T H. J Med Chem. 1997;40:1276–1286. doi: 10.1021/jm960835d. [DOI] [PubMed] [Google Scholar]
- 22.Carmichael A J, Mossoba M M, Riesz P. FEBS Lett. 1983;164:401–405. doi: 10.1016/0014-5793(83)80325-1. [DOI] [PubMed] [Google Scholar]
- 23.Allen S, Stevens L, Duncan D, Kelly S M, Price N C. Int J Biol Macromol. 1992;14:333–337. doi: 10.1016/s0141-8130(05)80074-1. [DOI] [PubMed] [Google Scholar]
- 24.Steiger B, Baskin J S, Anson F C, Zewail A H. Angew Chem Int Ed. 2000;39:257–260. [PubMed] [Google Scholar]
- 25.Donahue N M, Clarke J S, Anderson J G. J Phys Chem A. 1998;102:3923–3933. [Google Scholar]