

Abstract
Inflammation in asthma, sepsis, transplant rejection, and many neurodegenerative diseases associates an up-regulation of NO synthesis with increased protein nitration at tyrosine. Nitration can cause protein dysfunction and is implicated in pathogenesis, but few proteins that appear nitrated in vivo have been identified. To understand how this modification impacts physiology and disease, we used a proteomic approach toward targets of protein nitration in both in vivo and cell culture inflammatory disease models. This approach identified more than 40 nitrotyrosine-immunopositive proteins, including 30 not previously identified, that became modified as a consequence of the inflammatory response. These targets include proteins involved in oxidative stress, apoptosis, ATP production, and other metabolic functions. Our approach provides a means toward obtaining a comprehensive view of the nitroproteome and promises to broaden understanding of how NO regulates cellular processes.
Biologic nitration of protein tyrosine (to form 3-nitrotyrosine) is a phenomenon that is associated with over 50 diseases including transplant rejection, lung infection, central nervous system and ocular inflammation, shock, cancer, and neurological disorders (e.g., amylotrophic lateral sclerosis, Alzheimer's disease, Parkinson's disease, and stroke) (1–3). Protein nitration may be unique among posttranslational modifications in its dependency on reactivity of tyrosine residues in the protein target instead of specific sequence motifs or protein–protein interactions. Tyrosine nitration can cause both gain or loss of protein function (4). Nitric oxide (NO) synthases provide the biological precursor for nitrating agents that perform this modification in vivo. NO can form nitrating agents in a number of ways including reacting with superoxide to make peroxynitrite (HOONO) and through enzymatic oxidation of nitrite (the oxidation product of NO) to generate NO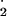 (1).
(1).
To date all protein nitration targets have been identified exclusively by antibodies directed against nitrotyrosine. Although these antibodies have linked this modification to many diseases, very few proteins that appear nitrated in vivo have been identified (1, 3). This has restricted our understanding of what roles tyrosine nitration might play in normal physiology and disease and the selectivity, kinetics, and potential repair of the modification in vivo (5, 6). We are pursuing a proteomic approach to identify nitrated proteins in biological samples that utilizes a commercially available well characterized monoclonal antibody directed against nitrotyrosine (7). This antibody has indicated biological tyrosine nitration in numerous studies and was shown to recognize a variety of proteins subjected to chemical nitration, the nitrated tyrosine residues of which were confirmed later by mass spectrometric sequence analysis (8, 9). Here we identify over 40 different proteins that appear to undergo nitration during inflammatory challenge in vivo. Our results provide a platform to examine mechanistic and biologic aspects of protein nitration.
Materials and Methods
Sample Preparation.
Rat tissue extract.
Sprague–Dawley rats (350 g) were injected with either saline or lipopolysaccharide (LPS, E. coli B120:001) at 10 mg/kg. After 18 h the rats were killed, and various tissues were extracted and frozen rapidly in liquid nitrogen. These were then stored at −80°C until needed. Frozen tissues were ground in a mortar and pestle, and weighed aliquots were added to lysis buffer {7 M urea, 2 M thiourea, 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 0.5% Triton X-100, 2% immobilized pH gradient (IPG)-ampholytes, pH 3–10, and 1% DTT; Amersham Pharmacia}. Samples were sonicated, and then debris was removed by centrifugation.
A549 cell extract.
A549 cells were grown to 70% confluency in F12R medium, and fresh medium was added either alone (unstimulated) or with medium in the presence of cytokines [IFN-γ, 1,000 units/ml; tumor necrosis factor-α, 10 units/ml; IL1-β, 10 units/ml (BioSource International, Camarillo, CA)]. After 72 h, the medium was collected, and the cells were washed in PBS three times, scraped into Eppendorf tubes, and spun-down. For samples run on two-dimensional gels, a lysis buffer (8 M Urea, 4% CHAPS, 1% DTT, and 2% IPG-ampholytes, pH 3–10) then was added to the cell pellets, and the samples were isoelectric-focused on a nonlinear pH 3–10 immobilized pH gradient strip. For samples run on SDS/PAGE, cells were lysed directly in SDS/PAGE sample loading buffer.
Nitrite/Nitrate Determination.
Nitrite/nitrate was determined by using a modification of the procedure of Schmidt (10). Cell tissue culture medium (150 μl) was used for all assays. Nitrate was reduced first to nitrite by using 0.1 units/ml nitrate reductase (from Aspergillus species, Roche Molecular Biochemicals), 50 μM NADPH, and 5 μM FAD to a final volume of 160 μl at 37°C for 2 h. Excess NADPH then was removed by the addition of 10 units/ml lactate dehydrogenase and 10 mM pyruvate and incubation at 37°C for 30 min. Total nitrite then was determined by using the Greiss reagent. A standard curve using nitrate was used to determine the concentration of nitrate/nitrite produced. Nitrate/nitrite levels were determined by a minimum of three independent determinations.
Two-Dimensional Gel Electrophoresis.
Two-dimensional gel electrophoresis was performed with the Amersham Pharmacia IPGphor/IsoDalt systems as described by West et al. (11). The first dimension used lysis buffer (above) and 18-cm nonlinear pH 3–10 immobilized pH gradient strips. IPG strips were rehydrated with sample at 30 V/6 h, and then isoelectric focusing was performed at 300 V/1 min, linear increase to 3,500 V/8 h, held at 3,500 V/1 h, and then at 8,000 V to reach a total of 90 kVh. For the second dimension, the IPG strips were equilibrated for 12 min in 50 mM Tris-Cl, pH 8.8, 6 M urea, 30% glycerol, 2% SDS, 1% DTT, and bromophenol blue, and then 12 min in 50 mM Tris-Cl, pH 8.8, 6 M urea, 30% glycerol, 2% SDS, 2% iodoacetamide, and bromophenol blue. The strips then were embedded in 0.7% (wt/vol) agarose on the top of 12% acrylamide slab gels (23.5 × 18 × 0.1 cm) containing a 4% stacking gel. The second dimension SDS/PAGE (overnight at 30–60 mA/gel) was performed essentially according to Laemmli (12). After completion of the run, the acrylamide gels were soaked in transfer buffer (Tris-HCl/glycine/20% methanol) and then partially transferred onto a PVDF membrane (Millipore) by using a semidry transfer apparatus as suggested by the manufacturer. The gels then were stained with colloidal Coomassie blue (gel code blue, Pierce), and Western analysis was performed on the PVDF membrane.
Western Analysis.
PVDF membranes were blocked for at least 4 h by using blocking buffer (20 mM Tris, 150 mM NaCl, pH 7.5, 0.2% Tween 20, and 1% BSA). Membranes then were probed overnight at 4°C with a monoclonal antibody against nitrotyrosine (1:4,000; clone 1A6, Upstate Biotechnology, Lake Placid, NY) in blocking buffer. The membranes then were washed three times in washing buffer (20 mM Tris, 150 mM NaCl, pH 7.5, and 0.2% Tween 20). The membrane then was probed with a goat anti-mouse horseradish peroxidase conjugate (1:10,000, Sigma). After washing the membrane three times in wash buffer, the immunopositive spots were visualized by using ECL-Plus (Amersham Pharmacia) as directed by the manufacturer.
Protein Identification.
Proteins were identified by matrix-assisted laser desorption ionization/time-of-flight mass spectrometric analysis of in-gel tryptic digest of immunopositive spots (11). The two-dimensional gel spots were excised and cut into ≈1-mm3 cubes, and Coomassie blue was washed away with 250-μl aliquots of 60% acetonitrile (1 × 30 min), 50% acetonitrile in 50 mM ammonium bicarbonate (1 × 30 min), and 50% acetonitrile in 15 mM N-ethylmorpholine (3 × 15 min). After destaining, the gel pieces were dried in a Speed Vac and then rehydrated in 15 μl of 15 mM N-ethylmorpholine containing 0.1 μg of modified trypsin (Promega) and incubated overnight at 37°C. The in-gel tryptic digest was extracted with 60% acetonitrile containing 0.1% trifluoroacetic acid. The extract was either used directly or extracted by using a C18 Zip-Tip (Millipore), and the extract was subjected to matrix-assisted laser desorption ionization/time-of-flight mass spectrometric analysis by using a Perkin–Elmer Voyager DE Pro instrument equipped with a nitrogen laser (337 nm) and operated in the delayed extraction and reflector mode with a matrix of a-cyano-4-hydroxy-cinnamic acid [5 mg/ml in acetonitrile/water/3% trifluoroacetic acid, 5:4:1 (vol/vol/vol)]. Internal standards were used for calibration and included two of the following three synthetic peptides: G9I (MH+ 1015.579), K13L (MH+ 1543.859), and L20R (MH+ 2474.630). One microliter of sample was mixed with 1 μl of matrix and 0.5 μl of internal standard mix; then 1.5 μl was applied to the sample plate and allowed to dry. Each spectrum was accumulated for ≈250 laser shots. Measured peptide masses were used to search the Swiss-Prot, TrEMBL, and NCBI sequence databases for protein identifications. Each peptide map data set was searched by using either MS-Fit (http://prospector.ucsf.edu/ucsfhtml3.4/msfit.htm) or Mascot (http://www.matrixscience.com). All searches were performed with a mass tolerance of 0.005% error (50 ppm).
Results
Protein nitration typically is indicated in diseases that have an inflammatory component, possibly caused by up-regulation of the inducible NO synthase (iNOS) (13). We therefore investigated tyrosine nitration in the human lung adenocarcinoma cell line A549, which expresses iNOS and undergoes protein nitration in response to cytokines (14, 15), and in an LPS-treated rat inflammatory disease model (13, 16).
A549 cells normally produced little NO and do not express iNOS, but after stimulation with tumor necrosis factor-α, IL1-β, and IFN-γ, increased NO breakdown products are detected in the medium as well as iNOS mRNA and protein within the cell. Protein nitration also increases with NO synthesis (Fig. 1). Unstimulated A549 cells contain some nitrated proteins, possibly because of the constitutive production of NO from endothelial or neuronal NOS. After stimulation, a general increase in protein nitration is observed as well as a change in the spectrum of proteins that are nitrated (Fig. 1, lanes 2 and 3). An example of this change in profile is illustrated by the appearance of an intense immunopositive band at 22 kDa that is only present in the cytokine-stimulated A549 cells. By using aminoguanidine, a partially selective inhibitor of iNOS, reduced protein nitration is observed (Fig. 1, lanes 2–7), supporting the association of NOS activity with the nitration process and the specificity of the monoclonal antibody used. Reduction in nitration of specific proteins occurs to different degrees at various aminoguanidine concentrations consistent with differential sensitivities and susceptibilities toward modification (17).
Figure 1.

Western blot analysis of the effects of aminoguanidine on anti-nitrotyrosine immunopositive proteins in A549 cells. A549 cells were uninduced (lane 1) or stimulated with cytokines (lanes 2–7: IFN-γ 1,000 units/ml; tumor necrosis factor-α 10 units/ml; IL1-β 10 units/ml). Cytokine-stimulated cells were incubated with aminoguanidine at the time of induction (0, .5, 1, 1.5, 2, and 3 mM in lanes 2–7, respectively). After 3 days, the cells were lysed immediately by using SDS/PAGE loading buffer and 20 μg of protein extracts loaded onto a 12% polyacrylamide gel. Nitrated proteins then were identified by Western blotting using an anti-nitrotyrosine monoclonal antibody.
To understand the physiological consequences of nitration, it is important to identify the proteins that are modified. To determine them we have used a proteomic approach. As outlined in Fig. 2, proteins from cell lysates or tissue homogenates were separated by two-dimensional gel electrophoresis (isoelectric focusing plus SDS/PAGE). This process generated ≈1,000 protein spots per gel. Proteins underwent partial transfer onto PVDF membranes followed by immunodetection of nitrotyrosine by using the well characterized monoclonal antibody developed by Beckman and co-workers (7). Corresponding immunopositive proteins were excised from the parent acrylamide gel and digested in-gel with trypsin, and tryptic peptides were analyzed by matrix-assisted laser desorption ionization/time-of-flight mass spectroscopy. Database searching with the peptide masses then identified the immunopositive proteins.
Figure 2.

Overview of the proteomic approach to analyze and identify proteins by using a high throughput system (see Materials and Methods for details).
Unstimulated A549 cells produced little NO as assessed by measurement of nitrate/nitrite, its stable breakdown byproducts (0.27 μmol of nitrate/nitrite per mg of protein), and exhibited only faint anti-nitrotyrosine immunoreactivity by Western analysis (Fig. 3B). Cytokine treatment induced iNOS expression in the A549 cells as judged by their NO synthesis (23.6 μmol of nitrate/nitrite per mg of protein) and caused increased, selective anti-nitrotyrosine immunoreactivity (Fig. 3D). This treatment also altered the protein expression pattern as observed by the Coomassie-stained gel (Fig. 3 A and C). MnSOD was identified to be the predominant immunoreactive protein. This essential mitochondrial protein, the expression of which was increased in the cytokine-treated cells, has been shown previously to be nitrated in renal transplants undergoing rejection (8). Adding ascorbate plus tetrahydrobiopterin to the cells during cytokine induction caused a reduction in anti-nitrotyrosine immunoreactivity (Fig. 3F) without affecting NO production (28.3 μmol of nitrate/nitrite per mg of protein). A loss of tyrosine nitration was observed also in the presence of ascorbate only (data not shown), suggesting that an ascorbate-sensitive component is involved in the nitration process and demonstrating epitope specificity by the monoclonal antibody. The persistent immunoreactivity of MnSOD in the ascorbate-treated sample may reflect a high sensitivity toward nitration and/or a close proximity to the oxidant source.
Figure 3.

Anti-nitrotyrosine immunopositive proteins in A549 cells. Cell lysates (300 μg) were run onto two-dimensional gels, and immunoreactive proteins were identified. A, B, and C are the two-dimensional acrylamide gels stained with gel code blue, and C, D, and E are the corresponding Western blots. Stimulation was carried out by using cytokines (C and D: IFN-γ, 1,000 units/ml; tumor necrosis factor-α 10 units/ml; IL1-β 10 units/ml) or with cytokines plus 100 μM H4B in 300 μM ascorbate (E and F). Western blots are exposed for 3 (B and D) and 15 (F) sec. On the 3-sec exposure of F, the intensity of manganese superoxide dismutase (MnSOD) and actin was much lower compared with D (data not shown). Identified proteins are indicated on the Western blots. Gels D and E show similar expression of all of the proteins that were detected by immunoreactivity; therefore the change in signal intensity is caused by decreased protein nitration.
To identify proteins nitrated in vivo we analyzed lung (Fig. 4A) and liver (Fig. 4C) proteins from rats killed 18 h after they received an i.p. injection of LPS. This stimulation generates widespread induction of iNOS within 2–4 h and a sustained increase in whole-animal NO synthesis (13). Corresponding Western blots are shown in Fig. 4 B and D, respectively. In both lung and liver samples there was an increase in nitrotyrosine immununoreactivity compared with PBS-injected control rats (data not shown). Comparing the distribution of immunoreactive proteins in Fig. 4B or 4D with their corresponding protein expression patterns in Fig. 4A or 4C shows that immunoreactivity was highly selective in both lung and liver. Some differences in immunoreactivity between organs correspond with their different protein expression profiles but could also reflect differences in inflammatory response or exposure to nitrating agent(s) or differential clearance. The immunoreactive proteins identified are listed in Table 1.
Figure 4.

Anti-nitrotyrosine immunopositive proteins in lung and liver of rats induced with LPS. Rats were induced with 10 mg/kg LPS and killed after 18 h. A (lung lysate, 400 μg) and C (liver lysate, 500 μg) are two-dimensional acrylamide gels stained with gel code blue, and B and D are the corresponding Western blots probed with an anti-nitrotyrosine monoclonal antibody. Identified proteins are listed on lung Western blot (B) but numbered on the liver Western blot (D) with the key in Table 1.
Table 1.
Identified anti-nitrotyrosine immunopositive proteins in livers of LPS-treated rats from Fig. 4
| Protein | Coverage | Peptide match | Function | Location | |
|---|---|---|---|---|---|
| 1 | Voltage-dependent anion channel-1 | 49% | 9 | Mitochondrial anion channel. Part of PTP. Involved in apoptosis | Mit |
| 2 | Mitochondrial aconitase | 24% | 13 | Metabolic enzyme—Citrate to isocitrate (citric acid cycle) (iron) | Mit |
| 3 | ATP synthase α-chain F1 ATPase | 14% | 5 | ATP production | Mit |
| 4 | Glutathione S-transferase (μ type) | 43% | 9 | Detoxification, steroid-selective, and oxidative stress | Cyto |
| 5 | Catalase | 43% | 18 | Breakdown of hydrogen peroxide and oxidative stress (iron) | Cyto |
| 6 | MnSOD | 19% | 4 | Breakdown of superoxide and oxidative stress (manganese) | Mit |
| 7 | Carbonic anhydrase III | 53% | 11 | Hydratation of CO2. Protection to oxidative damage (zinc) | Cyto |
| 8 | hnRNP A2/B1 | 34% | 7 | Vitamin D-binding protein? An early indicator of some cancers | Cyto |
| 9 | RACK-1 guanine nucleotide-binding protein | 28% | 7 | Activated protein kinase C receptor | Cyto |
| 10 | Annexin IV | 20% | 6 | Involved in exocytosis-binds Ca2+/phospholipid (calcium) | Cyto |
| 11 | Purine nucleoside phosphorylase | 22% | 5 | Nucleoside metabolism-implicated in apoptosis | Cyto |
| 12 | β-Tubulin chain 15 | 21% | 8 | Structural protein | Cyto |
| 13 | Sensescene marker protein 30 (Regucalcin) | 37% | 10 | Calcium-binding protein. Sensescene marker. (calcium) | Cyto |
| 14 | Aldolase B-fructose bisphosphatase | 32% | 11 | Fructose metabolism: glycolysis | Cyto |
| 15 | Mit. glutamate oxaloacetate transaminase-2 | 28% | 11 | Aspartate aminotransferase transaminase family | Mit |
| 16 | Transketolase | 15% | 5 | Involved in glycolysis | Cyto |
| 17 | Hydroxymethylglutaryl-CoA synthase | 18% | 10 | glutaryl-CoA synthesis-ketogenesis | Mit |
| 18 | 4-trimethylamino butyrataldehyde dehydrogenase | 26% | 9 | Carnitine synthesis (NAD+) | Mit |
| 19 | Glycine methyl transferase | 21% | 5 | Methyltransferase | Cyto |
| 20 | Glyceraldehyde phosphate dehydrogenase | 18% | 5 | Key enzyme in glycolysis (NAD+) | Cyto |
| 21 | 3-α-OH steroid dehydrogenase | 50% | 13 | Quinone reduction, aromatic alcohol and 15-OH prostaglandin dehydrogenase. Inhibited by NSAID (NADP+) | Cyto |
| 22 | Glutamate dehydrogenase | 21% | 6 | Citric acid cycle—malate to oxaloacetate (NAD(P)+) | Mit |
| 23 | 3-oxo-5 β steroid dehydrogenase | 16% | 5 | Bile acid and steroid hormone metabolism (NADP+) | Cyto |
| 24 | Short chain 3-OH acyl CoA dehydrogenase | 28% | 5 | Mitochondrial β-oxidation of short chain fatty acids (NAD+) | Mit |
| 25 | D-β-OH butyrate dehydrogenase | 15% | 4 | β-oxidation of fatty acids (NAD+) | Mit |
| 26 | 3-ketoacyl CoA thiolase | 36% | 15 | Involved in β oxidation of fatty acids | Mit |
| 27 | Lamin A | 15% | 5 | Structural protein | Nuc |
| 28 | Homogenisate 1,2 dioxygenase | 18% | 7 | Key enzyme in alkaptonuria in mice | Cyto |
| 29 | Betaine homocysteine S-transferase | 32% | 9 | Regulation of homocysteine metabolism (zinc) | Cyto |
| 30 | 3 Mercaptopyruvate sulfur transferase | 29% | 7 | Cyanide degradation and thiosulfate biosynthesis | Cyto |
| 31 | Rhodanese (thiosulfate sulfurtransferase) | 25% | 10 | Mitochondrial thiosulfate sulfurtransferase | Mit |
Identified nitrated protein in the liver of LPS-treated rats are shown with the number of peptide that matched the protein and percentage coverage of the protein. Cofactors required for activity are indicated in bold. Location of the protein is also indicated: mit, mitochondrial; Cyto, cytoplasmic; Nuc, nuclear.
Discussion
Our proteomic approach for identifying nitrated proteins offers advantages for the analysis of cells and tissues. The precision of the method stems from using two-dimensional gels to separate cellular macromolecules and the ability to obtain highly accurate peptide molecular mass values to identify immunoreactive proteins. Importantly, the method is sensitive enough to detect proteins that apparently undergo endogenous nitration in cells and tissues. This helps restrict identification to proteins that become modified because of natural processes and eliminates identification artifacts that may arise when exposing cells or tissues to exogenous NO or nitrating agents. Thus, the method promises to be a sensitive means to initiate studies on mechanisms, selectivity, and consequences of biological tyrosine nitration.
This investigation may be the first comprehensive study to identify proteins nitrated in vivo. Fig. 5 illustrates the functional classifications of the target proteins identified here. Many diseases in which nitration has been implicated, especially neurodegenerative diseases, involve defects in mitochondria (18, 19). These diseases include Alzheimer's disease (20, 21), Parkinson's disease (22, 23), and amyotrophic lateral sclerosis (24, 25). Over a third of the proteins we identified are located in the mitochondria, and its altered function may lead to cell death. Mitochondria are essential for energy production and apoptosis, and both of these functions are affected by NO and related oxides (26–28). The electron transport chain is a major cellular source of superoxide (29, 30) and is expected to be a major site of peroxynitrite formation when NO is present. NO can be generated in the same or adjacent cells by cytosolic or mitochondrial NOS (31). In our case a cytosolic iNOS was induced by cytokines and likely served as the predominant NO source. The local generation of a nitrating species via combination of superoxide and NO may preferentially sensitize mitochondrial proteins to nitration. Some of the target mitochondrial proteins are key enzymes in energy production; these include enzymes that are involved directly or indirectly in the citric acid cycle (e.g., glutamate dehydrogenase, aconitase, and glyceraldehyde phosphate dehydrogenase) and are involved in the electron transport chain (e.g., cytochrome oxidase V and F1 ATPase) and energy distribution (e.g., creatine kinase). Aconitase catalyses a crucial step in the citric acid cycle, and its inhibition affects energy production. Previously data have suggested that peroxynitrite inhibits aconitase activity in isolated mitochondria (32). Although the mechanism is thought to involve disruption of the enzyme's 4Fe-4S cluster, our finding suggests that tyrosine nitration may also play a role. Radi et al. (33) have demonstrated that exposing rat heart mitochondria to peroxynitrite resulted in significant inactivation of mitochondrial ATPase. Our observation that the α-chain of F1 ATPase was nitrated links this modification to the effect observed and demonstrates that it can occur during inflammation. Because this protein helps couple the proton gradient to ATP generation, its nitration could alter energy balance in the cell.
Figure 5.

Functional classification of anti-nitrotyrosine immunoreactive proteins.
A second important function of mitochondria is their involvement in apoptosis. Exposing mitochondria to NO or oxidative stress opens the permeability transition pore (PTP) through a process thought to involve peroxynitrite (34, 35). This then causes collapse of the proton gradient, mitochondrial swelling, calcium uptake, and release of death-promoting factors such as cytochrome c. The latter then initiates cascades that lead to apoptosis or necrosis depending on the energy status of the cell. We identified the protein voltage-dependent anion channel (VDAC) as being nitrated. This channel protein is a component of the PTP. It is conceivable that protein nitration is the signal that causes PTP to open. Many proteins are associated with the PTP (34). One such protein, mitochondrial creatine kinase, was identified also as being nitrated during inflammation. This enzyme acts to maintain the intracellular ATP pool and thus allows an energy distribution in the cell.
In addition to these mitochondrial proteins, four enzymes that protect cells against oxidative damage appear to be nitrated: MnSOD, catalase, glutathione S-transferase, and carbonic anhydrase III (36). These proteins exist in different cell compartments, but because they protect against oxidative damage one would expect that they are located near sites at which oxidants become present. It is established that in vitro chemical nitration of MnSOD causes selective modification at Tyr-34 and inactivation (9, 37), but how the other enzymes are affected is not known. It is interesting to speculate that in catalase two important tyrosines may get nitrated. Tyr-358 is the proximal heme ligand and is believed to be part of a conserved charge relay system that spans Tyr-358, Arg-354, His-218, and Asp-348 in human catalase (38). Prostacyclin synthase (PGI2) is another heme protein that is nitrated and inactivated by very low levels of peroxynitrite (39). Zou et al. (40) have proposed a mechanism in which peroxynitrite directly interacts with the heme in prostacyclin synthase. Its binding causes the production of oxyferryl complex and an NO2 radical, which then can react with a tyrosine radical leading to nitration. This mechanism may also occur with catalase. Our finding that key mitochondrial and antioxidant proteins both are targets for nitration in the inflammatory models gives new insight into how NO or other nitrating intermediates impact energy production and apoptotic events in vivo.
On average, proteins are composed of 4% tyrosine residues, but only a subset of total proteins was nitrated in our biological samples. This suggests that an innate property of the target protein or its location may predispose it toward nitration (17). Chemical nitration of isolated proteins also modifies only a subset of tyrosine residues, and the basis for this selectivity is not fully understood, although solvent accessibility certainly plays a role. Such modifications may alter protein function in ways that may be physiologically meaningful. For example, consider glutamate dehydrogenase, which is one of several dehydrogenases that appeared to be nitrated in this study. Mitochondrial glutamate dehydrogenase plays a central role in mammalian nitrogen metabolism by interconverting α-amino groups and ammonia. In vitro chemical nitration of the bovine enzyme occurred at only two tyrosine residues with more rapid nitration at Tyr-412 (41). This nitration resulted in the loss of its allosteric inhibition by GTP and consequently increased activity. Because Tyr-412 is conserved between bovine and rat glutamate dehydrogenases, the apparent in vivo nitration of the rat enzyme in the present study suggests that biological nitration could also result in a gain in enzyme activity. We are currently pursuing this possibility. Other work using chemical agents to nitrate tyrosines in other dehydrogenases have also demonstrated that nitration occurs at a restricted number of residues and leads to enzyme inhibition, loss of allosteric inhibition, disrupted cofactor binding, or subunit dissociation (42–47).
In this study we have identified over 40 proteins that are potentially nitrated based on antibody recognition. To date, all protein nitration targets have been identified exclusively by antibodies directed against nitrotyrosine. However, the possibilities that antibodies may be biased toward certain epitopes, or may generate some false positives, cannot be ruled out. We therefore are working to develop independent methods to confirm and identify the protein residues that undergo nitration in vivo by using specific chemical modifying reagents and enrichment procedures combined with MS/MS sequence analysis. Our current proteomic approach provides a means toward obtaining a comprehensive view of the nitroproteome and thus complements recent studies that have begun to create a phosphoproteome (48, 49) and identify proteins undergoing S-nitrosylation in vivo (50). In addition to its potential for identifying novel targets of tyrosine nitration in cells and tissues, the method opens doors for future studies that elucidate common features of protein targets, specificity and consequences of nitration, and the complex effect of NO on biological systems.
Acknowledgments
We thank Drs. Serpil Erzurum and Stanley L. Hazen and members of the Stuehr laboratory for their comments and support. This work was supported by National Institutes of Health Grants CA53914, GM51491, NS41644, and EY06603, a research center grant from the foundation to fight blindness, American Heart Association Grant AHA 01601598, and the funds from the Cleveland Clinic Foundation.
Abbreviations
- LPS
lipopolysaccharide
- IPG
immobilized pH gradient
- iNOS
inducible NO synthase
- MnSOD
manganese superoxide dismutase
- PTP
permeability transition pore
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Beckman J S, Koppenol W H. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 2.Halliwell B, Zhao K, Whiteman M. Free Radical Res. 1999;31:651–669. doi: 10.1080/10715769900301221. [DOI] [PubMed] [Google Scholar]
- 3.Ischiropoulos H. Arch Biochem Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- 4.Gole M D, Souza J M, Choi I, Hertkorn C, Malcolm S, Foust R F, III, Finkel B, Lanken P N, Ischiropoulos H. Am J Physiol. 2000;278:L961–L967. doi: 10.1152/ajplung.2000.278.5.L961. [DOI] [PubMed] [Google Scholar]
- 5.Kamisaki Y, Wada K, Bian K, Balabanli B, Davis K, Martin E, Behbod F, Lee Y C, Murad F. Proc Natl Acad Sci USA. 1998;95:11584–11589. doi: 10.1073/pnas.95.20.11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Souza J M, Choi I, Chen Q, Weisse M, Daikhin E, Yudkoff M, Obin M, Ara J, Horwitz J, Ischiropoulos H. Arch Biochem Biophys. 2000;380:360–366. doi: 10.1006/abbi.2000.1940. [DOI] [PubMed] [Google Scholar]
- 7.Ye Y Z, Strong M, Huang Z Q, Beckman J S. Methods Enzymol. 1996;269:201–209. doi: 10.1016/s0076-6879(96)69022-3. [DOI] [PubMed] [Google Scholar]
- 8.MacMillan-Crow L A, Crow J P, Kerby J D, Beckman J S, Thompson J A. Proc Natl Acad Sci USA. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamakura F, Taka H, Fujimura T, Murayama K. J Biol Chem. 1998;273:14085–14089. doi: 10.1074/jbc.273.23.14085. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt H H W. Biochemica. 1995;2:22. [Google Scholar]
- 11.West, K. A., Yan, L., Miyagi, M., Crabb, J. S., Murmorstein, A. D. Murmorstein, L. & Crabb, J. W. (2001) Exp. Eye Res., in press. [DOI] [PubMed]
- 12.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 13.Liu S, Adcock I M, Old R W, Barnes P J, Evans T W. Biochem Biophys Res Commun. 1993;196:1208–1213. doi: 10.1006/bbrc.1993.2380. [DOI] [PubMed] [Google Scholar]
- 14.Guo F H, Uetani K, Haque S J, Williams B R, Dweik R A, Thunnissen F B, Calhoun W, Erzurum S C. J Clin Invest. 1997;100:829–838. doi: 10.1172/JCI119598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dweik R A, Comhair S A, Gaston B, Thunnissen F B, Farver C, Thomassen M J, Kavuru M, Hammel J, Abu-Soud H M, Erzurum S C. Proc Natl Acad Sci USA. 2001;98:2622–2627. doi: 10.1073/pnas.051629498. . (First Published February 20, 2001; 10.1073/pnas.051629498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adcock I M, Brown C R, Kwon O, Barnes P J. Biochem Biophys Res Commun. 1994;199:1518–1524. doi: 10.1006/bbrc.1994.1403. [DOI] [PubMed] [Google Scholar]
- 17.Souza J M, Daikhin E, Yudkoff M, Raman C S, Ischiropoulos H. Arch Biochem Biophys. 1999;371:169–178. doi: 10.1006/abbi.1999.1480. [DOI] [PubMed] [Google Scholar]
- 18.Schapira A H, Cooper J M. Mutat Res. 1992;275:133–143. doi: 10.1016/0921-8734(92)90018-k. [DOI] [PubMed] [Google Scholar]
- 19.Beal M F. Biochem Soc Symp. 1999;66:43–54. doi: 10.1042/bss0660043. [DOI] [PubMed] [Google Scholar]
- 20.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell R L, Atwood C S, Johnson A B, Kress Y, Vinters H V, Tabaton M, et al. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ojaimi J, Byrne E. Biol Signals Recept. 2001;10:254–262. doi: 10.1159/000046890. [DOI] [PubMed] [Google Scholar]
- 22.Greenamyre J T, MacKenzie G, Peng T I, Stephans S E. Biochem Soc Symp. 1999;66:85–97. doi: 10.1042/bss0660085. [DOI] [PubMed] [Google Scholar]
- 23.Kosel S, Hofhaus G, Maassen A, Vieregge P, Graeber M B. Biol Chem. 1999;380:865–870. doi: 10.1515/BC.1999.106. [DOI] [PubMed] [Google Scholar]
- 24.Swerdlow R H, Parks J K, Pattee G, Parker W D., Jr Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:185–190. doi: 10.1080/14660820050515179. [DOI] [PubMed] [Google Scholar]
- 25.Xu G P, Dave K R, Moraes C T, Busto R, Sick T J, Bradley W G, Perez-Pinzon M A. Neurosci Lett. 2001;300:141–144. doi: 10.1016/s0304-3940(01)01575-0. [DOI] [PubMed] [Google Scholar]
- 26.Brown G C, Borutaite V. Biochem Soc Symp. 1999;66:17–25. doi: 10.1042/bss0660017. [DOI] [PubMed] [Google Scholar]
- 27.Bal-Price A, Brown G C. J Neurochem. 2000;75:1455–1464. doi: 10.1046/j.1471-4159.2000.0751455.x. [DOI] [PubMed] [Google Scholar]
- 28.Chung H T, Pae H O, Choi B M, Billiar T R, Kim Y M. Biochem Biophys Res Commun. 2001;282:1075–1079. doi: 10.1006/bbrc.2001.4670. [DOI] [PubMed] [Google Scholar]
- 29.Wallace D C. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 30.Saraste M. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 31.Giulivi C. Biochem J. 1998;332:673–679. doi: 10.1042/bj3320673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castro L, Rodriguez M, Radi R. J Biol Chem. 1994;269:29409–29415. [PubMed] [Google Scholar]
- 33.Radi R, Rodriguez M, Castro L, Telleri R. Arch Biochem Biophys. 1994;308:89–95. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- 34.Crompton M, Virji S, Doyle V, Johnson N, Ward J M. Biochem Soc Symp. 1999;66:167–179. doi: 10.1042/bss0660167. [DOI] [PubMed] [Google Scholar]
- 35.Brookes P S, Salinas E P, Darley-Usmar K, Eiserich J P, Freeman B A, Darley-Usmar V M, Anderson P G. J Biol Chem. 2000;275:20474–20479. doi: 10.1074/jbc.M001077200. [DOI] [PubMed] [Google Scholar]
- 36.Raisanen S R, Lehenkari P, Tasanen M, Rahkila P, Harkonen P L, Vaananen H K. FASEB J. 1999;13:513–522. doi: 10.1096/fasebj.13.3.513. [DOI] [PubMed] [Google Scholar]
- 37.MacMillan-Crow L A, Thompson J A. Arch Biochem Biophys. 1999;366:82–88. doi: 10.1006/abbi.1999.1202. [DOI] [PubMed] [Google Scholar]
- 38.Putnam C D, Arvai A S, Bourne Y, Tainer J A. J Mol Biol. 2000;296:295–309. doi: 10.1006/jmbi.1999.3458. [DOI] [PubMed] [Google Scholar]
- 39.Zou M, Martin C, Ullrich V. Biol Chem Hoppe-Seyler. 1997;378:707–713. doi: 10.1515/bchm.1997.378.7.707. [DOI] [PubMed] [Google Scholar]
- 40.Zou M, Daiber A, Paterson J A, Shoun H, Ullrich V. Arch Biochem Biophys. 2000;376:149–155. doi: 10.1006/abbi.2000.1699. [DOI] [PubMed] [Google Scholar]
- 41.Piszkiewicz D, Landon M, Smith E L. J Biol Chem. 1971;246:1324–1329. [PubMed] [Google Scholar]
- 42.Otwell H B, Yung-Ho T A, Friedman M E. Biochim Biophys Acta. 1978;527:309–319. doi: 10.1016/0005-2744(78)90345-5. [DOI] [PubMed] [Google Scholar]
- 43.Blumenthal K M, Smith E L. J Biol Chem. 1975;250:6560–6563. [PubMed] [Google Scholar]
- 44.Jeckel D, Anders R, Pfleiderer G. Hoppe-Seyler's Z Physiol Chem. 1971;352:769–779. [PubMed] [Google Scholar]
- 45.Miyazaki K, Kadono S, Sakurai M, Moriyama H, Tanaka N, Oshima T. Protein Eng. 1994;7:99–102. doi: 10.1093/protein/7.1.99. [DOI] [PubMed] [Google Scholar]
- 46.Froschle M, Ulmer W, Jany K D. Eur J Biochem. 1984;142:533–540. doi: 10.1111/j.1432-1033.1984.tb08318.x. [DOI] [PubMed] [Google Scholar]
- 47.Buchczyk D P, Briviba K, Hartl F U, Sies H. Biol Chem Hoppe-Seyler. 2000;381:121–126. doi: 10.1515/BC.2000.017. [DOI] [PubMed] [Google Scholar]
- 48.Oda Y, Nagasu T, Chait B T. Nat Biotechnol. 2001;19:379–382. doi: 10.1038/86783. [DOI] [PubMed] [Google Scholar]
- 49.Zhou H, Watts J D, Aebersold R. Nat Biotechnol. 2001;19:375–378. doi: 10.1038/86777. [DOI] [PubMed] [Google Scholar]
- 50.Jaffrey S R, Erdjument-Bromage H, Ferris C D, Tempst P, Snyder H S. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]