

Abstract
Despite the high efficiency of tyrosine kinase inhibitors (TKI), some patients with chronic myeloid leukemia (CML) will display residual disease that can become resistant to treatment, indicating intraclonal heterogeneity in chronic‐phase CML (CP‐CML). To determine the basis of this heterogeneity, we conducted the first exhaustive characterization of the DNA methylation pattern of sorted CP‐CML CD34+CD15− (immature) and CD34−CD15+ (mature) cells at diagnosis (prior to any treatment) and compared it to that of CD34+CD15− and CD34−CD15+ cells isolated from healthy donors (HD). In both cell types, we identified several hundreds of differentially methylated regions (DMRs) showing DNA methylation changes between CP‐CML and HD samples, with only a subset of them in common between CD34+CD15− and CD34−CD15+ cells. This suggested DNA methylation variability within the same CML clone. We also identified 70 genes that could be aberrantly repressed upon hypermethylation and 171 genes that could be aberrantly expressed upon hypomethylation of some of these DMRs in CP‐CML cells, among which 18 and 81, respectively, were in CP‐CML CD34+CD15− cells only. We then validated the DNA methylation and expression defects of selected candidate genes. Specifically, we identified GAS2, a candidate oncogene, as a new example of gene the hypomethylation of which is associated with robust overexpression in CP‐CML cells. Altogether, we demonstrated that DNA methylation abnormalities exist at early stages of CML and can affect the transcriptional landscape of malignant cells. These observations could lead to the development of combination treatments with epigenetic drugs and TKI for CP‐CML.
Keywords: CML, DNA methylation, epigenetics, leukemic stem cells, transcriptional defects
Abbreviation
- CGI
CpG island
- CIMP
CpG‐island methylator phenotype
- DMR
differentially methylated region
- Ph
Philadelphia chromosome
- TKI
tyrosine kinase inhibitor
1. Introduction
Chronic myeloid leukemia (CML) is one of the few examples of malignant transformation attributed to a single event, the translocation t(9; 22)(q34;q11) leading to the fusion of the ABL and BCR genes and known as Philadelphia chromosome (Ph). The resulting hybrid gene produces BCR‐ABL1, a chimeric oncoprotein with constitutive tyrosine kinase activity that promotes CML by aberrantly phosphorylating target proteins. Targeted treatments based on tyrosine kinase inhibitors (TKI) have shown considerable therapeutic effect (Gambacorti‐Passerini et al., 2011; Hochhaus et al., 2017).
However, despite the high efficiency of TKI‐based approaches, different observations highlight the intraclonal heterogeneity of chronic‐phase CML (CP‐CML) and the post‐treatment survival of a CML cell subpopulation. Specifically, residual disease can be detected even after treatment for several years (Hochhaus et al., 2017), and BCR‐ABL‐expressing leukemic stem cells (LSCs) can be observed in the bone marrow (BM) of patients with undetectable molecular residual disease. Moreover, CML relapses, due to the survival of a small number of LSCs in the BM (Chomel et al., 2016), are observed in about half of patients after treatment discontinuation (Etienne et al., 2017). In the case of secondary resistance to TKI, partial efficacy was reported before CML relapse due to a selected subclone (Druker, 2006).
The mechanisms of in vivo persistence of CML subclone(s) remain poorly understood. In the clinic, investigations have focused mainly on the occurrence of a BCR‐ABL1 mutation and insufficient plasma level of TKI. However, most cases of CP‐CML resistance are not explained by these two situations (Cortes et al., 2009; Soverini et al., 2006). Besides genetic alterations, epigenetic deregulations and more specifically abnormal DNA methylation patterns also are attractive mechanisms that could contribute to explain LSC survival. Indeed, aberrant DNA methylation pattern is a hallmark of cancer (Kim and Costello, 2017). Specifically, cancer cells are characterized by genomewide DNA hypomethylation and hypermethylation of CpG islands (CGIs). CGIs are key regulatory regions that are often localized in promoter areas and that are constitutively unmethylated in normal cells.
DNA methylation also plays an important role in normal hematopoiesis and controls the fate of hematopoietic stem cells (HSC). For instance, deletion or functional loss of DNA (cytosine‐5)‐methyltransferase 1 (DNMT1) in murine models alters HSC self‐renewal and leads to their progressive loss (Trowbridge et al., 2009). The loss of the de novo methyltransferases DNMT3a and 3b favors HSC self‐renewal and blocks their differentiation (Challen et al., 2014). Furthermore, the TET enzymes, which are involved in the active DNA demethylation process, also influence cellular functions essential for HSC homeostasis (Guillamot et al., 2016).
However, DNA methylation alterations in CML and specifically in LSCs are poorly characterized. There are only few studies on DNA methylation alterations in CML and most of them focused on a small number of genes (Dunwell et al., 2010; Janssen et al., 2010; Jelinek et al., 2011; Qian et al., 2009; Strathdee et al., 2007; Sun et al., 2001; Uehara et al., 2012). They show that DNA methylation abnormalities increase with CML progression toward the accelerated phase and blast crisis (Dunwell et al., 2010; Janssen et al., 2010; Jelinek et al., 2011; Uehara et al., 2012). DNA methylation changes during CML progression have been confirmed by the only genomewide DNA methylation analysis (to our knowledge) of CML samples (Heller et al., 2016). However, this study is only partially informative, because it was carried out using mononuclear cells isolated from PB (PB) and BM. On the other hand, to understand the molecular basis of intraclonal heterogeneity, large‐scale DNA methylation analyses should be performed using defined CML clone subsets, including some enriched for LSCs. Currently, the existence and extent of DNA methylation alterations in these key cell populations and the DNA methylation profile differences in these various cell populations from the same CP‐CML clone remain to be determined.
To address this key issue, we carried out a large methylome analysis using sorted CD34+CD15− (immature) and CD34−CD15+ (mature) cells from patients with CP‐CML at diagnosis, before any treatment and from healthy donors (HD). This allowed identifying specific disease‐related DNA methylation abnormalities and taking into account the methylation profile changes linked to cell differentiation (Bocker et al., 2011).
Our results provide the first exhaustive characterization of the DNA methylation pattern in CP‐CML, confirm the intraclonal heterogeneity of DNA methylation profiles, and suggest a functional impact of these methylation alterations on gene expression.
2. Methods
2.1. Patients, healthy donors, and human primary cells
A total of 13 patients with chronic‐phase CML (9M/4F; median age: 48 years (range: 24–90); Sokal score: 5 low, 5 intermediate, 3 high) (Table S1) were enrolled in the study after obtaining their written informed consent. This study was approved by the local ethics committee on human experimentation. Blood samples were collected in EDTA tubes at diagnosis, before any exposure to TKIs or any other treatment, and sent to the Clermont‐Ferrand center. All the sorting experiments were carried out with fresh cells, within 24 h of sampling.
To obtain the normal equivalent of circulating CML cells, we chose to use the leftovers of the samples used to evaluate the quality of G‐CSF‐mobilized peripheral blood progenitor cells (PBPCs) collected by apheresis from healthy donors (HD) [n = 10; 6 M/4 F; median age: 34 years (range: 13–67)]. These samples could be used for research because donors were informed and did not verbally express any disagreement, as stipulated by French law.
2.2. Flow cytometry
All flow cytometry experiments were carried out using a BD FACS ARIA‐SORP flow cytometer equipped with five lasers (BD Biosciences, Le Pont de Claix, France). BD FACSDivaTM CS&T Research calibrated beads were used to track the cytometer performance each day and to generate reproducible data. Compensation settings were performed using BD CompBeads and the automatic compensation setup tool in the bd facs diva 7.0 software (BD Biosciences, Le Pont de Claix, France).
To sort target cell subsets, nucleated cells from patients with CP‐CML were isolated by collecting the buffy coat, followed by erythrocyte lysis using ammonium chloride (Stemcell Technologies Inc., Vancouver, Canada). PBPCs were directly used.
Briefly, 100–150 × 106 cells were incubated in the dark with antibodies against 7‐AAD, CD45‐V500, CD34‐PC7, and CD15‐PerCpCy5.5 (BD Biosciences, Le Pont de Claix, France) for 20 min. Samples were then washed twice with PBS‐1% human AB serum and resuspended in PBS. Cells were sorted in two fractions, based on CD15 and CD34 markers to distinguish differentiated from undifferentiated cell subset: 7‐AAD−/CD45+/CD34−/CD15+ (CD34+CD15− subset) and 7‐AAD−/CD45+/CD34+/CD15− (CD34−CD15+ subset). Note that because of an overlap of expression between the CD34+ and the CD15+ populations (Fig. S1B), we defined the CD34+CD15− and CD34−CD15+ subsets as cell subpopulations of interest and we excluded the ‘gray area’ of cells coexpressing both antigens.
The gating strategy is shown in Fig. S1. Side scatter (SSC) and forward scatter (FSC) parameters were used to eliminate cell doublets. Apoptotic (7‐AAD+) cells were excluded.
2.3. DNA extraction
DNA was extracted from CD34+CD15− and CD34−CD15+ cells using the Nucleospin Tissue XS Kit (Macherey Nagel) following the manufacturer's protocol.
2.4. HM450K DNA methylation analyses
The HM450K array allows interrogating individually more than 485 000 CpG sites per sample, distributed in 99% of the annotated genes of the human genome. DNA bisulfite conversion and array hybridization were performed by Integragen, SA (Evry, France), using the Illumina Infinium HD methylation protocol. β‐values were normalized using the GenomeStudio control interplate normalization and background subtraction (version 2011.1). For each sample, β‐values with a detection P‐value >0.01 were excluded. All probes with a detection P‐value >0.01 or lacking signal in more than 5% of our cohort were rejected for the analysis. Finally, 26 957 probes containing common SNPs (dbSNP 138) in their last 5 bp or in the CpG sites were discarded. As our patient cohort included both men and women, probes on the X and Y chromosomes were also removed for the analysis.
Following above‐described quality filters, a total of 443 857 CpG methylation values were considered suitable for the downstream analysis. Differential methylation analyses were performed using the limma R package (Court et al., 2014). HM450K probes were considered differentially methylated when FDR was <0.05 and when the β‐value difference between groups was >0.15.
The β‐values for human embryonic stem cells (hESC), hematopoietic cells, acute myeloid leukemia (AML) CD34+CD38− cells, and a panel of solid cancer samples were extracted from publicly available methylation data, in details:
-
(a)
hESC: GSM1589944, GSM1589945, GSM1589946 and GSM999379.
-
(b)
Hematopoietic cells: GSE35069.
-
(c)
Acute myeloid leukemia CD34+CD38− cells and normal hematopoietic stem cells: GSE63409.
Methylation data for solid cancers and their matched normal controls were extracted from the TCGA database as described in Court and Arnaud (2017).
2.5. Gene‐specific bisulfite sequencing and combined bisulfite restriction analysis
Bisulfite conversion, PCR amplification, digestion for combined bisulfite restriction analysis (COBRA), cloning, and sequencing were performed as previously described (Arnaud et al., 2006). Details on the primers and restriction enzymes are in Table S2.
2.6. Annotation and data mining‐associated approaches
2.6.1. Comparison of HM450K β‐values and whole‐genome bisulfite‐sequencing data for CD34+ CD15− cells
Whole‐genome bisulfite‐sequencing (WGBS) coverage and methylation values for PB‐CD34+ cells were obtained from the GEO database (accession number GSM791828). Only the methylation values of CpG sites that were covered by at least 5 reads in the WGBS experiment were analyzed (25 027 707 CpGs retained). The mean β‐values of the five HD‐CD34+CD15− controls (HM450K data) were used for comparison. A total of 409 155 methylation values were compared between WGBS and HM450K.
2.6.2. CD34+ cell gene expression and chromatin state
RNA‐seq data of PB‐CD34+ cells were downloaded from the GEO database (GSM1256827). Fastq files were mapped to the hg19 genome using tophat2 (version 2.1.0). Reads were aligned with a gene annotation reference (Gencode Release 19). Gene expression level was estimated with the Cufflinks tools using Gencode gene annotations (release 19). The chromatin state of PB‐CD34+ cells was evaluated using Input (GSM537662, GSM621414, GSM621456, GSM621689), H3K4me3 (GSM537652, GSM621437, GSM621439, GSM621665), and H3K27me3 data (GSM537649, GSM621431, GSM621664, GSM669945), as described in Court and Arnaud (2017).
2.6.3. Genomic features and annotations
Gene and transcript positions in Gencode release 19 in GTF format for RNA‐seq data analysis were downloaded from the Gencode Web site. The positions of genes, SNPs, and CpG islands were downloaded from UCSC: Gencode basic V19, Common SNPs (138), and CpG‐island tracks, respectively. For each gene, promoter regions were defined as TSS ± 1 kbp. Their classification into high‐CpG‐density promoters (HCP), intermediate‐CpG‐density promoters (ICP), and low‐CpG‐density promoters (LCP) was performed using criteria defined by (Weber et al., 2007). Enhancer positions were determined with human_permissive_enhancers_phase_1_and_2.bed from the Fantom5 project. Chromatin state in hESC was from Court and Arnaud (2017).
2.6.4. Functional analyses and gene annotations
Gene enrichment analyses were conducted with the functional annotation tools in the DAVID 6.8 Web site. The tumor‐suppressor gene or oncogene lists were obtained from http://www.ongene.bioinfo-minzhao.org, http://www.cta.lncc.br, and http://www.uniprot.org with the keywords tumor suppressor (KW‐0043) or proto‐oncogene (KW‐0656).
2.7. RNA isolation and RT/PCR expression analysis
RNA was extracted using TRIzol (Fisher Scientific, Illkirch, France) according to the manufacturer's recommendations. After treatment with RQ1 RNase‐free DNase I (Promega, Charbonnières‐les‐Bains, France), RNA (100–250 ng) was reverse transcribed (RT) using the Superscript IV kit (Fisher Scientific) and random primers. Three independent experiments were conducted for each sample (except for the WT1 and PRAME transcripts, where two independent experiments were conducted). For each RNA sample, one RT was without reverse transcriptase to detect undesired amplification from DNA contamination. Real‐time PCR analyses were performed using the SYBR Green mixture (Roche, Meylan, France) and a LightCycler® 480II (Roche) apparatus. Primers and amplification conditions are summarized in Table S2.
The relative expression level was quantified as follows: E−Ct(Transcript)/geometrical mean(E−Ct(HK genes)), based on the −2ddCt methods (E: efficiency of amplification, Ct: cycle threshold, HK: housekeeping). The housekeeping genes GUSB, SDHA, and HMBS were used to normalize transcript expression. The presented data are the mean ± standard deviation of two or three independent experiments, each in duplicate.
2.8. Data accessibility
The HM450K DNA methylation data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE106600.
3. Results
3.1. Progressive hypomethylation of healthy donor CD34+CD15− and CD34−CD15+ cells
We first characterized the DNA methylation pattern of CD34+CD15− and CD34−CD15+ cells from five HDs using the HM450K array. After quality filtration, we could assign a β‐value comprised between 0 (i.e., unmethylated position) and 1 (i.e., fully methylated position) to 443 857 CpG sites for each sample. We then compared the DNA methylation data of HD CD34+CD15− cells with those obtained by whole‐genome bisulfite sequencing of PB CD34+ (PB‐CD34+) cells (GSM791828) (Hodges et al., 2011). The good correlation (R 2 = 0.89) validated the quality of the data obtained with the HM450K array (Fig. S2A). The specificity of our sorting approach was further supported using publicly available HM450K profiles of eight hematological cell types, PB mononuclear cells, and whole blood. Based on their methylation profile, HD CD34−CD15+ cells clustered in the myeloid subgroup, while HD CD34+CD15− cells formed an independent group from both myeloid and lymphoid cells (Fig. S2B).
We then evaluated changes in DNA methylation patterns during blood cell differentiation, from hESC to CD34+CD15− hematopoietic stem/progenitor cells and finally to differentiated CD34−CD15+ cells. Most changes were between hESC and HD CD34+CD15− cells (68 164 differentially methylated probes). Conversely, differentially methylated probes were five times less numerous between CD34+CD15− and CD34−CD15+ cells. These changes were mainly associated with loss of DNA methylation. Specifically, 68% (46 379/68 164) of differentially methylated probes were hypomethylated in CD34+CD15− cells compared with hESC. This trend was further accentuated upon hematopoietic differentiation, because 98% of DNA methylation changes between CD34+CD15− and CD34−CD15+ cells corresponded to hypomethylation events (Fig. 1A,B). Similarly, myeloid cell types were globally unmethylated compared with HD CD34+CD15− cells (Fig. S2C). These changes were underrepresented in CGIs (Fig. S2D), whereas they were widespread throughout the genome, including promoter and enhancer regions (Fig. 1A,B). Ontology analyses revealed that inflammatory and immune response genes were over‐represented among the genes with a hypomethylated promoter (p value < 10−4) (Figs 1C and S2E).
Figure 1.

DNA methylation changes between hESCs, CD34+ CD15−, and CD34− CD15+ cells from healthy donors. Heatmaps of differentially methylated probes between (A) hESC and HD CD34+ CD15− cells and (B) HD CD34+ CD15− and HD CD34− CD15+ cells, respectively. The methylation level varies from low (β‐value close to 0; in green) to high (β‐value close to 1; in red). Hyper‐ and hypomethylated probes are shown on the upper and lower panels, respectively, and their number is indicated. The CpG‐island status and genomic features of the probes are indicated by color codes. (C) Gene ontology terms (GO database) in promoters with hyper‐ (red bars) and hypomethylated (blue bars) probes (n ≥ 2) between hESC and HD CD34 + CD15‐ and between HD CD34+ CD15− and HD CD34− CD15+ cells.
3.2. DNA methylation alterations in CP‐CML CD34+CD15− and CD34−CD15+ cells
We next investigated the DNA methylation profiles of CD34+CD15− and CD34−CD15+ cells from six patients with CP‐CML. Compared with HD cells, we observed DNA methylation alterations in both cell types. The extent of such defects was similar to what observed in AML cells, but more limited (up to 8 times) than in the representative panel of solid cancers, regardless of their CpG‐island methylator phenotype (CIMP) status (Fig. 2A,B).
Figure 2.

DNA methylation alterations in CP‐CML cells. (A) Heatmaps of differentially methylated probes in CP‐CML and HD CD34+ CD15− (left panel) and CD34− CD15+ (right panel) cells. Hypo‐ and hypermethylated probes are shown on the upper and lower panels, respectively, and their number is indicated. The CpG‐island status and genomic features of the probes are indicated by color codes. (B) Distribution of hyper‐ and hypomethylated probes in CP‐CML cells, AML CD34+ cells, and eight types of solid tumors. For each solid tumor type, CpG‐island methylator phenotype (CIMP)‐positive (+) and CIMP‐negative (−) samples were analyzed separately. BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; HNSC, head–neck squamous cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LIHC, liver hepatocellular carcinoma; LUSC, lung squamous cell carcinoma; LUAD, lung adenocarcinoma. (C) Circular karyotype showing the position of hyper‐ (in red) and hypomethylated (in blue) DMRs in CP‐CML CD34+ CD15− (inner circle) and CD34− CD15+ (outer circle) cells. Some hotspot regions are named. (D) Detail of the WT1 locus showing a cluster of hypermethylated probes in the two promoter regions in CP‐CML CD34+ CD15− and CD34− CD15+ cells compared with HD cells.
We identified both hyper‐ and hypomethylated probes, with a majority of hypomethylation events (74%) in CP‐CML CD34+CD15− cells. Overall, these changes were widespread throughout the genome and affected enhancer, promoter, gene body, and intergenic areas. In agreement with the hypomethylated status of CGIs in normal cells, most hypomethylation events observed in CP‐CML samples were in non‐CGI regions (Fig. 2A).
Specifically, 11 251 probes were differentially methylated in CP‐CML CD34+CD15− cells and 6345 in CP‐CML CD34−CD15+ cells compared with the relevant HD samples. To further delineate the affected genomic loci, we mapped the regions that contained at least two consecutive hyper‐ or hypomethylated probes (absolute methylation difference >15%) in a maximal window of 1Kb between CP‐CML and the relevant HD cell samples. In CP‐CML CD34+CD15− cells, we identified 1857 windows that could be merged into 400 hyper‐ and 742 hypomethylated differentially methylated regions (DMRs) of up to 2.3Kbp in size, respectively (Figs 2C, S3A and Table S3). In CP‐CML CD34−CD15+ cells, we identified 396 hyper‐ and 285 hypomethylated DMRs (Table S3). Only a subset of them (225 and 218, respectively) was in common with those identified in CD34+CD15− cells, suggesting DNA methylation variability within the same CML clone (Fig. S3B). In both CP‐CML cell types, hypermethylated DMRs tended to be enriched at CGIs, specifically at CGIs located in the gene body, enhancer and intergenic areas, but not in promoter regions. Conversely, hypomethylated DMRs were enriched in enhancer and non‐CGI promoter regions, and more specifically in those with a low and, to a lesser extent, an intermediate‐CpG density (i.e., LCP and ICP promoters (Weber et al., 2007), respectively) (Fig. S3C,D).
By considering regions with at least five consecutive affected probes, we defined 33 highly affected genomic areas in CP‐CML CD34+CD15− cells (Table S4). Such dense clusters of hypermethylation included the translocated ABL1 promoter, the promoter regions of Wilms tumor 1 (WT1), a transcription factor overexpressed in myeloid malignancies (Rosenfeld et al., 2003), and the promoter of the ZNF577 gene that encodes a zinc finger protein (Figs 2C,D, S4A). On chromosome 6, hypomethylated clusters overlapped with the G6B (C6orf25) gene, a member of the immunoglobulin superfamily, and the promoter of TRIM15 that encodes a focal adhesion protein (Figs 2C, S4A). Most of but not all these ‘hotspot’ areas were also present in CD34−CD15+ cells (Table S4). For example, some regions, including the VARS, EGFL8, and EIF4E promoters, were specifically hypomethylated only in CP‐CML CD34+CD15− cells (Fig. S4B and Table S4).
Moreover, the 33 highly affected genomic areas found in CP‐CML CD34+CD15− cells displayed methylation patterns different not only from those of HD CD34+CD15− and CD34−CD15+ cells, but also from those of various hematological cell types and blood cells. This suggests that these methylation defects constitute a pathological signature (Fig. S4C).
3.3. The promoters of genes repressed in PB CD34+ cells are more prone to aberrant hypermethylation in CP‐CML CD34+CD15− cells
Aberrant DNA hypermethylation of CGIs is a well‐defined feature of cancer cells. In agreement, about 58% of hypermethylated DMRs in CP‐CML CD34+CD15− cells were localized in CGI regions.
Studies on CGI hypermethylation in solid cancer and AML revealed that it is observed mainly at CGIs targeted by polycomb group proteins in embryonic or adult stem cells and particularly at CGIs that are concomitantly marked by the ‘active’ H3K4me3 and the ‘repressive’ polycomb‐derived H3K27me3 marks, constituting a bivalent chromatin signature (Court and Arnaud, 2017; Deneberg et al., 2011; Ohm et al., 2007). We did not observe this feature in CP‐CML CD34+CD15− cells (Fig. 3A). CGIs with bivalent chromatin in hESC and in PB‐CD34+ cells were not preferentially hypermethylated in CP‐CML CD34+CD15− cells. Rather, the 231 CGI‐associated hypermethylated DMRs were strongly enriched in CGIs depleted of both H3K4me3 and H3K27me3 in hESC or PB‐CD34+ cells (referred to as ‘non‐CGI’) and, to a lesser extent, in CGIs marked only by H3K27me3 in PB‐CD34+ cells. For instance, 65% and 55% of the probes localized at hypermethylated CGI‐associated DMRs were in non‐CGIs in hESC and PB‐CD34+ cells, respectively (compared with 34% and 30% of probes at non‐CGIs in the HM450K array).
Figure 3.
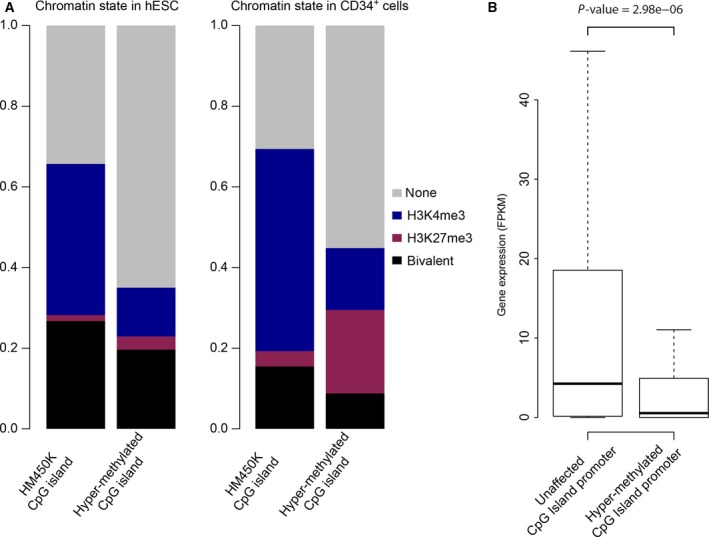
CpG island/promoters of genes repressed in PB CD34+ cells tend to be hypermethylated in CP‐CML CD34+ CD15− cells. (A) Distribution of probes in hypermethylated CGI‐associated DMRs according to their chromatin signatures in hESCs (left panel) and PB (PB) CD34+ cells (right panel), respectively. The distribution of all HM450K probes at CGIs according to their chromatin signature in hESC and PB‐CD34+ cells, respectively, is shown as reference. (B) Genes with aberrantly hypermethylated CGI/promoter in CP‐CML CD34+ CD15− cells tend to be repressed in PB‐CD34+ cells. Boxplot representation of the expression levels in PB‐CD34+ cells for genes with unaffected and hypermethylated CGI/promoter‐associated DMRs, respectively, in CML CD34+ CD15− cells (Mann–Whitney test).
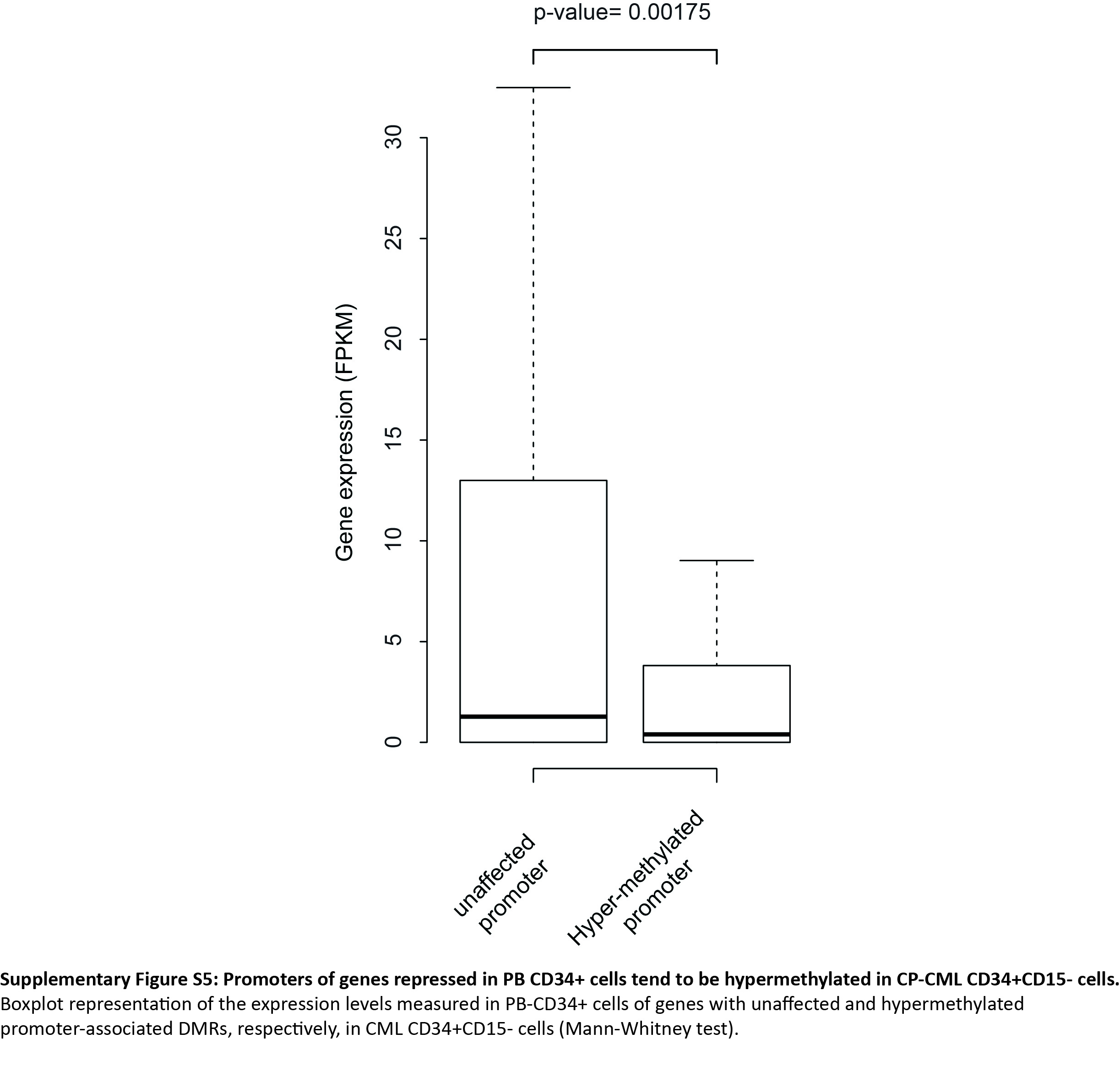
Besides the chromatin signature in stem cells, the propensity of CGI/promoters to be aberrantly methylated in cancer cells has also been linked to the transcriptional activity in the matched normal tissues (Court and Arnaud, 2017; Sproul et al., 2011, 2012). We thus collected publicly available RNA sequencing data for PB‐CD34+ cells to determine whether the gene transcriptional status influenced the CGI/promoter methylation status in CP‐CML CD34+CD15− cells. Overall, we observed a significantly lower expression in normal tissues of genes with promoter‐associated hypermethylated DMRs in tumor cells, independently of the presence/absence of CGIs in the promoter, compared with gene with unaltered DNA methylation pattern (Figs 3B, S5). This suggests that promoters of poorly expressed or repressed genes in healthy CD34+ cells are more likely to be aberrantly methylated in CML.
3.4. Lists of genes candidate to be affected by aberrant DNA methylation in CP‐CML CD34+CD15− cells
The observation that hypermethylation mainly affects the promoter of genes that are already repressed in HD cells questions the impact of DNA methylation alterations on the transcriptional landscape of CP‐CML cells. To address this key point, and in the absence of publicly available RNA‐seq data for CML cells, we used RNA‐seq data from PB‐CD34+ cells to determine a list of candidate genes the expression of which could be repressed by aberrant DNA methylation in CP‐CML CD34+CD15− cells (i.e., genes that are expressed in PB‐CD34+ cells (rpkm > 1) and the promoter of which is a hypermethylated DMR in CML). We found that 280 promoters of 177 genes (some genes have several promoters) were hypermethylated in CP‐CML CD34+CD15− cells. Among them, 70 (39.5%) were expressed in PB‐CD34+ and consequently could be aberrantly repressed in CP‐CML CD34+CD15− cells (red dots in Fig. 4; Table S5). They included the tumor‐suppressor TP53INP1 (tumor protein P53‐inducible nuclear protein), ZAP70 (zeta‐chain‐associated protein kinase 70) and HOXB3.
Figure 4.

Candidate genes to aberrant expression following DNA methylation alterations in CP‐CML CD34+ CD15− cells. Genes with DMRs in their promoter are classified according to the methylation variation between HD CD34+ CD15− and CP‐CML CD34+ CD15− cells (y‐axis) and level of expression in PB‐CD34+ cells (x‐axis). Blue and red dots represent genes that could be aberrantly expressed and repressed, respectively. Genes with four or more consecutive differentially methylated probes in their promoter are indicated.
Besides focal CGI/promoter hypermethylation, cancer cells are also characterized by concomitant widespread DNA hypomethylation that contributes to genome instability and could lead to ectopic expression by affecting promoter activity. However, this last event is rare and documented for a limited number of genes, such as cancer‐germline genes (Van Tongelen et al., 2017) and inserted viral oncogenes. To determine whether some genes are reactivated following hypomethylation, we investigated genes that are repressed in PB‐CD34+ cells (rpkm < 1) and display hypomethylated DMRs in the promoter in CP‐CML CD34+CD15− cells. Unexpectedly, this list included much more candidate genes than the one for genes with hypermethylated promoters. Among the 403 genes with hypomethylated promoters (n = 802) in CP‐CML cells, 171 (42.3%) could be aberrantly expressed (blue dots in Fig. 4; Table S5). This list included several oncogenes, namely MIR544A, MIR146B, FAM83A (also known as tumor antigen BJ‐TSA‐9), the cancer‐germline genes PBK (PDZ binding kinase) and PRAME (preferentially expressed antigen in melanoma), and GCKR that, with PRAME, is a downstream target of the BCR‐ABL protein (Shi et al., 1999; Watari et al., 2000).
Except for the over‐representation for genes subject to alternatives splicing among hypermethylated genes (P value <10−2), no specific ontology term was associated with these two candidate gene lists (Fig. S6A).
To restrict the list to genes that could explain the specific features of CP‐CML CD34+CD15− cells, we considered only promoters that were aberrantly methylated specifically in this cell subset and identified 81 and 18 genes that could be abnormally expressed and repressed, respectively, only in CP‐CML CD34+CD15− cells (Fig. S6B and Table S6).
3.5. Methylation alterations can correlate with modified expression of candidate genes
To determine whether the expression of the genes in these two lists was affected in CP‐CML CD34+CD15− cells and to validate the HM450K data in an independent cohort, we performed DNA methylation and expression analyses of selected genes using independent CP‐CML and HD CD34+CD15− samples (n = 7 and n = 5, respectively).
Expression data analysis identified ZAP70 and INPP5F‐V2 as genes that could be repressed by aberrant DNA methylation. The COBRA approach indicated that the CGI/promoter governing the main ZAP70 isoform gained DNA methylation in CP‐CML samples, validating the HM450K data. This hypermethylation correlated with a decrease in gene expression in the same samples (Fig. 5A). The same analyses confirmed a gain of methylation on the CGI/promoter of the isoform‐V2 of the imprinted gene INPP5F, which encodes a phosphatidylinositide phosphatase, in CP‐CML samples. This gain correlated with aberrant INPP5F silencing (Fig. S7A).
Figure 5.
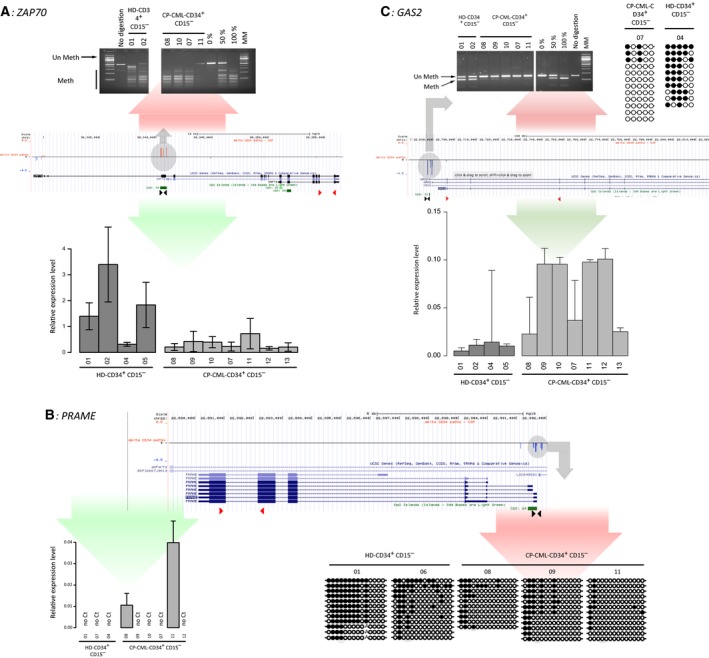
Gene expression and DNA methylation analyses at selected candidate genes. Results of the gene expression and DNA methylation analyses (COBRA) for the ZAP70 (A), PRAME (B), and GAS2 (C) genes in independent CP‐CML and HD cell samples. For each gene, the central panel indicates areas with differences in DNA methylation levels between CP‐CML and HD CD34+ CD15− cells (HM450K array data). Red, methylation gain; blue, methylation decrease. The upper panel shows bisulfite‐based DNA methylation analyses by COBRA and/or sequencing in HD and CP‐CML samples. For each sequenced region, the methylation patterns are symbolized by lollipops (black: methylated CpG; white: unmethylated CpG). Expression data (quantitative PCR) are in the lower panel.
We also observed a hotspot of aberrant hypermethylation at the promoter regions of WT1 (Fig. 2C), a gene that is overexpressed in CML cells (Gerber et al., 2011; Rosenfeld et al., 2003). A more detailed analysis showed that this methylation gain affected the whole downstream alternative CpG island/promoter, but only the 3′ edge of the upstream CpG‐island promoter, while the TSS area remained free of methylation (Figs 2C, S7B). Accordingly, promoter‐specific RT/PCR analysis showed that WT1 overexpression in CP‐CML CD34+CD15− cells initiated mainly from the upstream hypomethylated CpG‐island promoter, because the transcript that initiates from the alternative hypermethylated promoter was not detected (Fig. S7B).
It is well documented that in cancer, promoter hypomethylation is associated with reactivation of cancer‐germline genes. In agreement, we observed that PRAME overexpression was correlated with its promoter hypomethylation, but only in a subset of CP‐CML samples (e.g., CP‐CML samples 8 and 11 but not CP‐CML sample 9) (Fig. 5B). Outside this class of genes, this phenomenon is a rare event. We then assessed the expression of GAS2 (growth arrest‐specific 2) because it contributes to CML cell growth (Zhou et al., 2014) and thus could be an oncogene. COBRA and bisulfite‐sequencing approaches confirmed the results of the HM450K array data analysis: The GAS2 promoter region was hypomethylated in all CP‐CML samples compared with HD cells. In all cases, this correlated with a robust, though variable, gain of GAS2 expression in CP‐CML CD34+CD15− cells (Fig. 5C).
4. Discussion
We report here the first exhaustive DNA methylation analysis of cell subsets from patients with CP‐CML at diagnosis before any treatment. To ensure the identification of DNA methylation differences only related to the pathology, we compared the DNA methylation profiles of immature CD34+CD15− and mature CD34−CD15+ cells from patients with CP‐CML and HDs. Moreover, as the hematopoietic tissue origin could influence the cell methylation profile, we compared circulating CP‐CML cells and HD PBPCs that are believed to be equivalent to circulating CML cells (Farlik et al., 2016).
Our study confirmed and extended the results of previous works (Dunwell et al., 2010; Heller et al., 2016; Janssen et al., 2010; Jelinek et al., 2011; Qian et al., 2009; Strathdee et al., 2007; Sun et al., 2001; Uehara et al., 2012) showing that the DNA methylation landscape is altered at CP‐CML diagnosis. To better characterize these alterations, we identified the affected loci and DMRs. Only a subset of these regions were affected in both CD34+CD15− and CD34−CD15+ cells, indicating that DNA methylation could contribute to the clonal heterogeneity of the early CML phase.
The DNA methylation defects at the 33 ‘strongest’ DMRs constitute a pathological signature and could be considered as candidate biomarkers of Ph clones. These DMRs included the ABL1 and WT1 genes the aberrant methylation of which has already been reported in chronic phase (Sun et al., 2001) and blast crisis (Janssen et al., 2010), respectively.
In immature CP‐CML CD34+CD15− cells, we identified about two times more hypomethylated than hypermethylated DMRs (742 vs 400), suggesting that the trend toward hypomethylation observed during normal differentiation into the myeloid lineage (Farlik et al., 2016) is accentuated during malignant transformation. As observed in other cancers, hypermethylated DMRs are mainly localized in CGI regions. However, and unlike other cancers (Court and Arnaud, 2017; Deneberg et al., 2011; Ohm et al., 2007), the hypermethylation gain appears to be only partially predefined by a polycomb‐derived chromatin signature in healthy embryonic or adult stem cells. Although our study supports that CGIs marked by H3K27me3 alone in PB‐CD34+ are more prone to hypermethylation, it also highlights that CGIs with bivalent chromatin are not preferentially methylated in CP‐CML CD34+CD15− cells. The reason for this discrepancy is unclear and could result from the limited number of hypermethylated DMRs we identified. It could also result from a protection brought by the TET‐derived 5 hydroxymethylcytosine (5hmC) modification. TET proteins, particularly TET2, are key regulators of human HSC biology (Langlois et al., 2014; Pronier et al., 2011). Specifically, by catalyzing the conversion of 5 methylcytosine (5mc) to 5hmC throughout the genome, TET2 might contribute to the marked trend toward hypomethylation during normal hematopoiesis (Tekpli et al., 2016). A recent study conducted in colorectal cancer revealed that promoters marked by 5hmC in normal tissue are resistant to DNA methylation gain in tumor cells. This study also highlighted that a relevant subset of these promoters overlaps with those marked by a bivalent signature in stem cells (Uribe‐Lewis et al., 2015). Therefore, high level of TET2‐mediated 5hmC in a subset of CGI/promoters with bivalent chromatin in healthy CD34+ cells could protect them against aberrant methylation in CP‐CML cells. In agreement, we observed that hypermethylation mainly affects nonpromoter CGIs in CP‐CML CD34+CD15− cells, and CGI promoters in solid cancers (Court and Arnaud, 2017).
To which extent alterations in the DNA methylation landscape affect the transcriptional profile in CP‐CML CD34+CD15− cells remains a pending question. To address this key question, and in the absence of publicly available RNA‐seq data on HD and CP‐CML CD34+CD15− cells, we evaluated the transcriptional status of PB CD34+ cells to identify candidate genes the expression of which could be affected by aberrant DNA methylation of their promoter in CP‐CML CD34+CD15− cells. In agreement with what observed in other cancers (Court and Arnaud, 2017; Sproul et al., 2011, 2012), DNA hypermethylation mainly affected the promoter of genes already repressed in healthy cells. This phenomenon probably restricts the overall functional impact of this defect. Not exclusively, this stable repression could also affect the tumor biology by limiting its epigenetic plasticity and, for instance, its ability to adapt following environmental changes, such as metastasis formation or treatment (Sproul and Meehan, 2013).
Nonetheless, we identified 70 genes that could be aberrantly repressed by DNA methylation in CP‐CML CD34−CD15+ cells and confirmed the repression of two of them in an independent cohort. Among these genes, ZAP70 encodes a kinase essential for thymopoiesis, T‐cell receptor‐dependent survival, and proliferation (Schim van der Loeff et al., 2014) and is aberrantly expressed in B‐cell malignancies, such as acute lymphoblastic leukemia and chronic lymphocytic leukemia (Rassenti et al., 2004). This repression could be related to the preferential commitment to the myeloid lineage in CML.
DNA hypermethylation could also be associated with differential promoter usage or gene overexpression, as documented in prostate cancer (Bert et al., 2013). Although our strategy was not designed to identify such candidate genes, we observed that DNA hypermethylation of the inner WT1 CGI/promoter is associated with overexpression from the upstream promoter. A causal effect between these events remains to be evidenced.
Promoter hypomethylation leading to ectopic expression in cancer cells is a rare event. Therefore, the identification of 170 genes that could be aberrantly expressed in CP‐CML CD34+CD15− cells was unexpected. In breast cancer, DNA methylation loss at promoters can be compensated by a gain of the repressive H3K27me3 mark (Hon et al., 2012); therefore, many of these candidate genes might remain repressed in cancer cells. For instance, we observed the maintenance of a DNA methylation‐independent repressed state at the GP6 and TRPV4 genes (data not shown). Nonetheless, our validation experiments and literature data indicate that several genes of this list are aberrantly expressed, for example IL‐3 that is produced by CML cells and a therapeutic target (Frolova et al., 2014; Jiang et al., 1999). Similarly, the cancer‐germline gene PRAME is overexpressed in various cancer and leukemia types as well as in CML CD34+ cells (Gerber et al., 2011). Although it has already been shown that DNA hypomethylation at PRAME can lead to its overexpression (Schenk et al., 2007), the variable effect we observed in CP‐CML samples suggests that the decrease in DNA methylation at the PRAME promoter is not sufficient to induce ectopic expression. Among other candidates, GAS2 deserves a specific attention. This gene is an indirect target of the BCR‐ABL chimeric protein (Hjort et al., 2016), and a candidate oncogene because of its involvement in CML cell growth and survival, particularly via the Wnt/β‐catenin pathway (Huang et al., 2017; Zhou et al., 2014). We confirmed in an independent cohort that GAS2 is robustly hypomethylated and overexpressed in all the studied cell samples, thus emerging as one of the rare genes in which promoter hypomethylation is associated with expression reactivation in cancer cells.
Interestingly, it has been reported that WT1, PRAME, and GAS2 are overexpressed during CML progression toward the blast crisis phase (Radich et al., 2006). This observation suggests that the subpopulation of cells in which these genes are aberrantly methylated is selected during CML progression.
5. Conclusions
In conclusion, we showed that DNA methylation abnormalities exist at early stages of CP‐CML and can affect the transcriptional profile of malignant cells. Although their role in TKI resistance and CML progression remains to be determined, these alterations emerge as relevant candidates for disease progression or resistance of subclones.
The characterization of DNA methylation alterations specifically in immature CD34+CD15− cells and the identification of several candidate genes implicated in CML progenitor survival or proliferation open new avenues for developing combination therapies with epigenetic drugs and TKIs for the treatment of CP‐CML.
Authors contributions
SMM, FC, CB, PA, and MGB designed the study and analyzed the data. PCM, HJ, GB, PR, DG, EH, AJ provided patient samples. CB performed the immunophenotyping, cell sorting, and DNA extraction. JB prepared primary samples and stored the sorted subpopulations in the CRB‐Auvergne, an NF96‐900‐labeled structure. SS collected the associated patients’ data and verified the presence of informed consents. SMM, FC, and CB performed the experiments and realized the figures. FC did the data mining and bioinformatics analyses. PA and MGB coordinated the study and wrote the manuscript.
Supporting information
Fig. S1. CD34+CD15− and CD34−CD15+ cells sorting.
{kind=link}
Fig. S2. Characterization of the methylation profile of HD CD34+CD15− and HD CD34−CD15+ cells.
{kind=link}
Fig. S3. Characterization of DNA methylation alterations in CP‐CML cells.
{kind=link}
Fig. S4. Details of the DMRs with affected methylation in CP‐CML cells.
{kind=link}
Fig. S5. Promoters of genes repressed in PB CD34+ cells tend to be hypermethylated in CP‐CML CD34+CD15− cells.
{kind=link}
Fig. S6. Candidate genes to aberrant expression following DNA methylation alterations in CP‐CML CD34+CD15− cells.
{kind=link}
Fig. S7. INPP5F‐V2 repression and WT1 overexpression in CP‐CML cells.
{kind=link}
Table S1. Details of the patients with CP‐CML.
Table S2. (XLSX): Details of the primers used in this study.
Table S3. (XLSX): List of DMRs identified in CP‐CML CD34+CD15− and CP‐CML CD34−CD15+ cells, according their methylation status.
Table S4. Hotspots of DNA methylation alteration in CP‐CML cells. Genomic positions are according to hg19.
Table S5. (XLSX): List of candidate genes to aberrant repression upon DNA hypermethylation, or to aberrant expression following DNA hypomethylation in CP‐CML CD34+CD15− cells.
Table S6. List of candidate genes to aberrant repression following DNA hypermethylation, or aberrant expression following DNA hypomethylation specifically in CP‐CML CD34+CD15− cells.
Acknowledgements
This study was funded by the Plan Cancer‐INSERM (CS14085) (PA), the Fonds de dotation Patrick Brou de Lauriére (PA), the Regional Clinical Research Hospital Projects (PHRC, CHU Clermont‐Ferrand) (MGB & PA), the Ligue Contre le Cancer (Comité de l'Allier) (MGB), and EA CHELTER (Université Clermont Auvergne) (MGB). This research is supported by the French Government IDEX‐ISITE Initiative 16‐IDEX‐0001 (CAP 20‐25).
SMM, FC and CB contributed equally to this study.
PA and MGB are co‐senior authors.
Contributor Information
Philippe Arnaud, Email: philippe.arnaud@uca.fr.
Marc G. Berger, Email: mberger@chu-clermontferrand.fr
References
- Arnaud P, Hata K, Kaneda M, Li E, Sasaki H, Feil R and Kelsey G (2006) Stochastic imprinting in the progeny of Dnmt3L‐/‐ females. Hum Mol Genet 15, 589–598. [DOI] [PubMed] [Google Scholar]
- Bert SA, Robinson MD, Strbenac D, Statham AL, Song JZ, Hulf T, Sutherland RL, Coolen MW, Stirzaker C and Clark SJ (2013) Regional activation of the cancer genome by longrange epigenetic remodeling. Cancer Cell 23, 9–22. [DOI] [PubMed] [Google Scholar]
- Bocker MT, Hellwig I, Breiling A, Eckstein V, Ho AD and Lyko F (2011) Genome‐wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood 117, e182–e189. [DOI] [PubMed] [Google Scholar]
- Challen GA, Sun D, Mayle A, Jeong M, Luo M, Rodriguez B, Mallaney C, Celik H, Yang L, Xia Z et al (2014) Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 15, 350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomel JC, Bonnet M‐L, Sorel N, Sloma I, Bennaceur‐Griscelli A, Rea D, Legros L, Marfaing‐Koka A, Bourhis JH, Ame S et al (2016) Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy discontinuation. Oncotarget 7, 35293–35301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes JE, Egorin MJ, Guilhot F, Molimard M and Mahon F‐X (2009) Pharmacokinetic/pharmacodynamic correlation and blood‐level testing in imatinib therapy for chronic myeloid leukemia. Leukemia 23, 1537–1544. [DOI] [PubMed] [Google Scholar]
- Court F and Arnaud P (2017) An annotated list of bivalent chromatin regions in human ES cells: a new tool for cancer epigenetic research. Oncotarget 8, 4110–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court F, Tayama C, Romanelli V, Martin‐Trujillo A, Iglesias‐Platas I, Okamura K, Sugahara N, Simón C, Moore H, Harness JV et al (2014) Genome‐wide parent‐of‐origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation‐independent mechanism of establishment. Genome Res 24, 554–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneberg S, Guardiola P, Lennartsson A, Qu Y, Gaidzik V, Blanchet O, Karimi M, Bengtzén S, Nahi H, Uggla B et al (2011) Prognostic DNA methylation patterns in cytogenetically normal acute myeloid leukemia are predefined by stem cell chromatin marks. Blood 118, 5573–5582. [DOI] [PubMed] [Google Scholar]
- Druker BJ (2006) Circumventing resistance to kinase‐inhibitor therapy. N Engl J Med 354, 2594–2596. [DOI] [PubMed] [Google Scholar]
- Dunwell T, Hesson L, Rauch TA, Wang L, Clark RE, Dallol A, Gentle D, Catchpoole D, Maher ER, Pfeifer GP et al (2010) A genome‐wide screen identifies frequently methylated genes in haematological and epithelial cancers. Mol Cancer 9, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne G, Guilhot J, Rea D, Rigal‐Huguet F, Nicolini F, Charbonnier A, Guerci‐Bresler A, Legros L, Varet B, Gardembas M et al (2017) Long‐term follow‐up of the French Stop Imatinib (STIM1) study in patients with chronic myeloid leukemia. J Clin Oncol 35, 298–305. [DOI] [PubMed] [Google Scholar]
- Farlik M, Halbritter F, Müller F, Choudry FA, Ebert P, Klughammer J, Farrow S, Santoro A, Ciaurro V, Mathur A et al (2016) DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell 19, 808–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova O, Benito J, Brooks C, Wang RY, Korchin B, Rowinsky EK, Cortes J, Kantarjian H, Andreeff M, Frankel AE et al (2014) SL‐401 and SL‐501, targeted therapeutics directed at the interleukin‐3 receptor, inhibit the growth of leukaemic cells and stem cells in advanced phase chronic myeloid leukaemia. Br J Haematol 166, 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambacorti‐Passerini C, Antolini L, Mahon F‐X, Guilhot F, Deininger M, Fava C, Nagler A, Della Casa CM, Morra E, Abruzzese E et al (2011) Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib. J Natl Cancer Inst 103, 553–561. [DOI] [PubMed] [Google Scholar]
- Gerber JM, Qin L, Kowalski J, Smith BD, Griffin CA, Vala MS, Collector MI, Perkins B, Zahurak M, Matsui W et al (2011) Characterization of chronic myeloid leukemia stem cells. Am J Hematol 86, 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillamot M, Cimmino L and Aifantis I (2016) The impact of DNA methylation in hematopoietic malignancies. Trends Cancer 2, 70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller G, Topakian T, Altenberger C, Cerny‐Reiterer S, Herndlhofer S, Ziegler B, Datlinger P, Byrgazov K, Bock C, Mannhalter C et al (2016) Next‐generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia 30, 1861–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjort EE, Huang W, Hu L and Eklund EA (2016) Bcr‐abl regulates Stat5 through Shp2, the interferon consensus sequence binding protein (Icsbp/Irf8), growth arrest specific 2 (Gas2) and calpain. Oncotarget 7, 77635–77650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, Baccarani M, Deininger MW, Cervantes F, Fujihara S et al (2017) Long‐term outcomes of Imatinib treatment for chronic myeloid leukemia. N Engl J Med 376, 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, Park J, Butler J, Rafii S, McCombie WR et al (2011) Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell 44, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE et al (2012) Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res 22, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Bei L, Hjort EE and Eklund EA (2017) Decreased calpain activity in chronic myeloid leukemia impairs apoptosis by increasing survivin in myeloid progenitors and xiap1 in differentiating granulocytes. Oncotarget 8, 50629–50641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen JJWM, Denkers F, Valk P, Cornelissen JJ, Schuurhuis G‐J and Ossenkoppele GJ (2010) Methylation patterns in CD34 positive chronic myeloid leukemia blast crisis cells. Haematologica 95, 1036–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinek J, Gharibyan V, Estecio MRH, Kondo K, He R, Chung W, Lu Y, Zhang N, Liang S, Kantarjian HM et al (2011) Aberrant DNA methylation is associated with disease progression, resistance to imatinib and shortened survival in chronic myelogenous leukemia. PLoS One 6, e22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Lopez A, Holyoake T, Eaves A and Eaves C (1999) Autocrine production and action of IL‐3 and granulocyte colony‐stimulating factor in chronic myeloid leukemia. Proc Natl Acad Sci USA 96, 12804–12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M and Costello J (2017) DNA methylation: an epigenetic mark of cellular memory. Exp Mol Med 49, e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois T, da Costa Reis Monte‐Mor B, Lenglet G, Droin N, Marty C, Le Couédic JP, Almire C, Auger N, Mercher T, Delhommeau F et al 2014. TET2 deficiency inhibits mesoderm and hematopoietic differentiation in human embryonic stem cells. Stem Cells Dayt Ohio 32, 2084–2097. [DOI] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W et al (2007) A stem cell‐like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39, 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, Massé A, Le Couédic JP, Pendino F, Carbonne B et al 2011. Inhibition of TET2‐mediated conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood 118, 2551–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Wang Y‐L, Lin J, Yao D‐M, Xu W‐R and Wu C‐Y (2009) Aberrant methylation of the death‐associated protein kinase 1 (DAPK1) CpG island in chronic myeloid leukemia. Eur J Haematol 82, 119–123. [DOI] [PubMed] [Google Scholar]
- Radich JP, Dai H, Mao M, Oehler V, Schelter J, Druker B, Sawyers C, Shah N, Stock W, Willman CL et al (2006) Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci USA 103, 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ, Gribben JG, Neuberg DS, Flinn IW, Rai KR, Byrd JC et al (2004) ZAP‐70 compared with immunoglobulin heavy‐chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med 351, 893–901. [DOI] [PubMed] [Google Scholar]
- Rosenfeld C, Cheever MA and Gaiger A (2003) WT1 in acute leukemia, chronic myelogenous leukemia and myelodysplastic syndrome: therapeutic potential of WT1 targeted therapies. Leukemia 17, 1301–1312. [DOI] [PubMed] [Google Scholar]
- Schenk T, Stengel S, Goellner S, Steinbach D, Saluz HP. (2007). Hypomethylation of PRAME is responsible for its aberrant overexpression in human malignancies. Genes Chromosom Cancer 46, 796–804. [DOI] [PubMed] [Google Scholar]
- Schim van der Loeff I, Hsu L‐Y, Saini M, Weiss A and Seddon B (2014) Zap70 is essential for long‐term survival of naive CD8 T cells. J Immunol Baltim Md 193, 2873–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CS, Tuscano JM, Witte ON and Kehrl JH (1999) GCKR links the Bcr‐Abl oncogene and Ras to the stress‐activated protein kinase pathway. Blood 93, 1338–1345. [PubMed] [Google Scholar]
- Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, Iacobucci I, Amabile M, Abruzzese E, Orlandi E et al (2006) Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia‐positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res 12, 7374–7379. [DOI] [PubMed] [Google Scholar]
- Sproul D, Kitchen RR, Nestor CE, Dixon JM, Sims AH, Harrison DJ, Ramsahoye BH and Meehan RR (2012) Tissue of origin determines cancer‐associated CpG island promoter hypermethylation patterns. Genome Biol 13, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproul D and Meehan RR (2013) Genomic insights into cancer‐associated aberrant CpG island hypermethylation. Brief Funct Genomics 12, 174–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproul D, Nestor C, Culley J, Dickson JH, Dixon JM, Harrison DJ, Meehan RR, Sims AH and Ramsahoye BH (2011) Transcriptionally repressed genes become aberrantly methylated and distinguish tumors of different lineages in breast cancer. Proc Natl Acad Sci USA 108, 4364–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathdee G, Holyoake TL, Sim A, Parker A, Oscier DG, Melo JV, Meyer S, Eden T, Dickinson AM, Mountford JC et al (2007) Inactivation of HOXA genes by hypermethylation in myeloid and lymphoid malignancy is frequent and associated with poor prognosis. Clin Cancer Res 13, 5048–5055. [DOI] [PubMed] [Google Scholar]
- Sun B, Jiang G, Zaydan MA, La Russa VF, Safah H and Ehrlich M (2001) ABL1 promoter methylation can exist independently of BCR‐ABL transcription in chronic myeloid leukemia hematopoietic progenitors. Cancer Res 61, 6931–6937. [PubMed] [Google Scholar]
- Tekpli X, Urbanucci A, Hashim A, Vågbø CB, Lyle R, Kringen MK, Staff AC, Dybedal I, Mills IG, Klungland A et al (2016) Changes of 5‐hydroxymethylcytosine distribution during myeloid and lymphoid differentiation of CD34+ cells. Epigenetics Chromatin 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge JJ, Snow JW, Kim J and Orkin SH (2009) DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 5, 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara E, Takeuchi S, Yang Y, Fukumoto T, Matsuhashi Y, Tamura T, Matsushita M, Nagai M, Koeffler HP and Tasaka T (2012) Aberrant methylation in promoter‐associated CpG islands of multiple genes in chronic myelogenous leukemia blast crisis. Oncol Lett 3, 190–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe‐Lewis S, Stark R, Carroll T, Dunning MJ, Bachman M, Ito Y, Stojic L, Halim S, Vowler SL, Lynch AG et al (2015) 5‐hydroxymethylcytosine marks promoters in colon that resist DNA hypermethylation in cancer. Genome Biol 16, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tongelen A, Loriot A and De Smet C (2017) Oncogenic roles of DNA hypomethylation through the activation of cancer‐germline genes. Cancer Lett 396, 130–137. [DOI] [PubMed] [Google Scholar]
- Watari K, Tojo A, Nagamura‐Inoue T, Nagamura F, Takeshita A, Fukushima T, Motoji T, Tani K and Asano S (2000) Identification of a melanoma antigen, PRAME, as a BCR/ABL‐inducible gene. FEBS Lett 466, 367–371. [DOI] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M and Schübeler D (2007) Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39, 457–466. [DOI] [PubMed] [Google Scholar]
- Zhou H, Ge Y, Sun L, Ma W, Wu J, Zhang X, Hu X, Eaves CJ, Wu D and Zhao Y (2014) Growth arrest specific 2 is up‐regulated in chronic myeloid leukemia cells and required for their growth. PLoS One 9, e86195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. CD34+CD15− and CD34−CD15+ cells sorting.
Fig. S2. Characterization of the methylation profile of HD CD34+CD15− and HD CD34−CD15+ cells.
Fig. S3. Characterization of DNA methylation alterations in CP‐CML cells.
Fig. S4. Details of the DMRs with affected methylation in CP‐CML cells.
Fig. S5. Promoters of genes repressed in PB CD34+ cells tend to be hypermethylated in CP‐CML CD34+CD15− cells.
Fig. S6. Candidate genes to aberrant expression following DNA methylation alterations in CP‐CML CD34+CD15− cells.
Fig. S7. INPP5F‐V2 repression and WT1 overexpression in CP‐CML cells.
Table S1. Details of the patients with CP‐CML.
Table S2. (XLSX): Details of the primers used in this study.
Table S3. (XLSX): List of DMRs identified in CP‐CML CD34+CD15− and CP‐CML CD34−CD15+ cells, according their methylation status.
Table S4. Hotspots of DNA methylation alteration in CP‐CML cells. Genomic positions are according to hg19.
Table S5. (XLSX): List of candidate genes to aberrant repression upon DNA hypermethylation, or to aberrant expression following DNA hypomethylation in CP‐CML CD34+CD15− cells.
Table S6. List of candidate genes to aberrant repression following DNA hypermethylation, or aberrant expression following DNA hypomethylation specifically in CP‐CML CD34+CD15− cells.
Data Availability Statement
The HM450K DNA methylation data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE106600.