

Abstract
Aiming at the design of an allosteric modulator of the neonatal Fc receptor (FcRn)–Immunoglobulin G (IgG) interaction, we developed a new methodology including NMR fragment screening, X-ray crystallography, and magic-angle-spinning (MAS) NMR at 100 kHz after sedimentation, exploiting very fast spinning of the nondeuterated soluble 42 kDa receptor construct to obtain resolved proton-detected 2D and 3D NMR spectra. FcRn plays a crucial role in regulation of IgG and serum albumin catabolism. It is a clinically validated drug target for the treatment of autoimmune diseases caused by pathogenic antibodies via the inhibition of its interaction with IgG. We herein present the discovery of a small molecule that binds into a conserved cavity of the heterodimeric, extracellular domain composed of an α-chain and β2-microglobulin (β2m) (FcRnECD, 373 residues). X-ray crystallography was used alongside NMR at 100 kHz MAS with sedimented soluble protein to explore possibilities for refining the compound as an allosteric modulator. Proton-detected MAS NMR experiments on fully protonated [13C,15N]-labeled FcRnECD yielded ligand-induced chemical-shift perturbations (CSPs) for residues in the binding pocket and allosteric changes close to the interface of the two receptor heterodimers present in the asymmetric unit as well as potentially in the albumin interaction site. X-ray structures with and without ligand suggest the need for an optimized ligand to displace the α-chain with respect to β2m, both of which participate in the FcRnECD–IgG interaction site. Our investigation establishes a method to characterize structurally small molecule binding to nondeuterated large proteins by NMR, even in their glycosylated form, which may prove highly valuable for structure-based drug discovery campaigns.
Author summary
In drug design, a detailed characterization of structural changes induced by drug binding is useful for further optimizing lead compounds. In many cases, structural alterations are distant from the compound binding site, potentially acting through allosteric effects. These allosteric effects are often difficult to observe by static methods, i.e., X-ray crystallography, but can be monitored by NMR spectroscopy. The latter method, however, has size-limitations when investigating the protein backbone structure in solution-state. To overcome this, we present an innovative approach employing ultrafast magic-angle-spinning (MAS) NMR on the extracellular domain of the neonatal Fc receptor (FcRnECD). This is a validated drug target in autoimmune diseases, and we aim to identify and characterize novel compounds to serve as starting points to develop allosteric inhibitors of this receptor. After sedimentation, we could record well-resolved proton-detected MAS NMR spectra of the fully protonated [13C,15N]-labeled protein, enabling the observation of structural changes. In combination with computational methods, X-ray crystallography, and other biophysical tools, we present new compounds that may be used as allosteric modulators of FcRn after further optimization. The introduced MAS NMR approach can be applied to a large variety of proteins to support structure-based drug design, facilitating the detection of allosteric effects.
Introduction
In order to discover new chemical drugs, fragment screening followed by structure-based design is an efficient way to sample chemical space and find hits for challenging target classes such as protein-protein interactions [1–3]. In addition to discovering orthosteric ligands, fragment screening has the potential to locate secondary binding sites on a protein that may be exploited for allosteric regulation [4]. In the development process, a methodology that includes detection of allosteric effects is highly welcome. Magic-angle-spinning (MAS) NMR has the potential to contribute via the detection of long-range chemical-shift changes when the investigated protein is too large for solution-state NMR and can even not be deuterated. It is applied here to a soluble 42 kDa construct of the neonatal Fc receptor (FcRn) within a search for allosteric regulators, employing very fast MAS (100 kHz).
FcRn facilitates new-born humoral immunity by regulating Immunoglobulin (IgG) transport across the epithelium [5]. In addition, it has been shown to bind to IgG and Human Serum Albumin (HSA) at nonoverlapping sites in a pH-dependent manner (Fig 1) [6,7]. This allows maintenance of IgG and HSA homeostasis, accounting for the long serum half-life of both proteins [8–11]. At low pH, the interaction of FcRn with IgG occurs through protonation of ionizable residues, located at the CH2–CH3 hinge of the IgG Fc, which produces transient, intermolecular salt bridges with negatively charged residues on FcRn [12]. The interaction of FcRn with IgG and HSA occurs in acidified early endosomes, diverting the proteins from catabolism and carrying them back to the neutral pH environment of the extracellular compartment. At near-neutral pH, the affinity of the interaction decreases, and the complex dissociates [10,13].
Fig 1. FcRn allows maintenance of protein homeostasis.

The soluble extracellular domain of neonatal Fc receptor (FcRnECD, PDB code 1EXU) is a heterodimer composed of β2m (green) and α-chain (blue) with a cavity at the interface between the two proteins. FcRn is involved in the regulation of HSA (orange) and IgG (red) levels. The binding of both HSA and IgG to FcRn is pH dependent, which provides a mechanism for protein homeostasis through endosomal trafficking. β2m, β2-microglobulin; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor; HSA, Human Serum Albumin; IgG, Immunoglobulin G; PDB, Protein Data Bank.
Existing as a heterodimer composed of β2-microglobulin (β2m) and a membrane-anchored α-chain (Fig 1), FcRn is homologous to the class I major histocompatibility complex (MHC1) [14]. MHC1 presents antigenic peptide fragments to the T cell receptor (TCR) in complex with cluster of differentiation 8 (CD8) [14]. In contrast, FcRn is not involved in endogenous peptide presentation to the TCR; its peptide-binding groove is closed and nonfunctional [14].
The effect of FcRn loss has been studied using β2m-deficient mice, which develop normally but are defective in T cell–mediated cytotoxicity [15]. Deletion of β2m precluded FcRn expression, resulting in a reduction in IgG half-life [16–18]. Additionally, random mutation of residues proximal to the IgG–FcRn binding site allowed selection of Fc variants with increased affinity for FcRn at pH 6. These Fc constructs maintained pH-dependent binding and showed an extended half-life relative to wild-type IgG [19].
FcRn has been proposed as a drug target in the treatment of autoimmune diseases, in which pathogenic autoantibodies are detrimental to health [20]. Examples of such diseases include, but are not restricted to, myasthenia gravis, Guillain–Barré syndrome, and dermatomyositis [21–24]. Antibodies produced against the FcRn heavy chain ameliorated myasthenia gravis symptoms in rats, and it has been shown that mice without FcRn are resistant to autoimmune disease [25–27]. The current treatment for these conditions is intravenous application of immunoglobulin, which increases the turnover of pathogenic IgG by saturating FcRn [26]. Recently, rozanolixizumab, an antihuman FcRn monoclonal antibody, reduced the serum IgG concentration in a randomized phase 1 study, providing clinical evidence for the potential of an anti-FcRn therapeutic [28].
A chemical inhibitor to control FcRn trafficking is therapeutically desirable for potential treatment of autoimmune disorders. Orthosteric peptide inhibitors with in vivo efficacy have previously been reported [29–33]. Additionally, structure–activity relationships of small molecule antagonists using ELISA assays have also been described [34]. However, to date, no allosteric modulators of FcRn–IgG or FcRn–HSA interactions have been reported. Should such molecules be found, they could be used therapeutically or as tools in biomedical investigations.
In this study, we present the discovery of a compound that binds to the soluble extracellular domain of FcRn, abbreviated here as FcRnECD. In the initial screening process, complementary in silico methods were used to predict binding sites on the basis of charge and topography or sequence conservation, alone and in combination [35–37]. The interaction of the ligand with FcRnECD is investigated by a combination of X-ray crystallography and proton-detected, ultrafast MAS NMR, revealing its localization in an evolutionarily conserved binding pocket. At high MAS frequencies of 100 kHz, we could acquire well-resolved proton-detected NMR spectra on sedimented, fully protonated FcRnECD, allowing de-novo chemical-shift assignments of residues in the binding pocket and in β2m through establishing sequential connections. MAS NMR pinpoints chemical-shift changes upon ligand binding close to the binding site and in regions distant to it. In this context, MAS NMR helps to exclude the influence of crystal packing effects by making use of protein solutions. Both crystal structures with and without ligand were available, but structural changes were not obvious when using global fitting procedures, with the molecules possibly “locked” into a conformation by crystal contacts. To identify sections of the structure not affected by ligand binding for a fit that is also sensitive to small allosteric effects, residues that did not show chemical-shift changes were used to produce “chemical-shift–informed” overlays of FcRnECD X-ray structures with and without ligand. Our findings suggest that therapeutic intervention in autoimmune diseases may be achieved through allosteric small molecules that bind to FcRnECD. In addition, such compounds could potentially be used as chemical probes to study FcRn trafficking. The presented approach highlights further the use of MAS NMR for detecting structural changes in nondeuterated proteins expressed in mammalian cells upon ligand binding.
Results and discussion
X-ray crystallography of FcRnECD and ligandability assessments
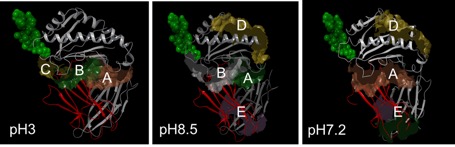
Small molecule binding sites on proteins can be identified based on surface shape, charge, and functionality. We used SiteMap software to identify binding sites on the FcRnECD [35]. To obtain structural data for our analysis, diffraction data for FcRnECD crystals, at pH 3 and pH 8.5, were collected at cryogenic temperatures and structures solved to 2.0 Å and 2.45 Å, respectively, by molecular replacement. Two copies of FcRnECD were found in the asymmetric unit. In addition to the structures at acidic and basic pH, a pH 7.2 structure was generated using molecular dynamics simulation (S1 Fig and S1 Text).
The SiteMap software detected a number of regions with a druggability score >1.0, a nominal quantifier indicating that nM binding might be achieved with a conventional small molecule of <500 Da molecular weight. In particular, SiteMap identified a ligandable site at the interface between the α-chain and β2m (S1 Fig and S1 Text). The respective boundaries were predicted to vary with pH, with the area described as either one large or three distinct cavities.
No regions near the IgG binding site were found by SiteMap. Based on our analysis, should an orthosteric pocket for an IgG blocking small molecule exist, it is likely to be transient in nature and not stabilized in the crystal lattice.
Evolutionary conservation of FcRnECD
On the premise that evolutionarily conserved cavities may have an associated function, we evaluated such conservation of the cavities found by SiteMap (S2 Fig). We identified ortholog sequences using OrthoDB and performed sequence alignments using Clustal Omega [38,39]. A homology model was then created for each sequence using MEDELLER that enabled us to visualize mutations according to the evolutionary conservation in PyMOL [40,41].
Our sequence analysis broadly supports the notion that regions of the protein important for structural integrity or function are conserved, in particular the interface between the α-chain and β2m. It contains key contacts, such as those between D53β2m, Q34α-chain, and S37α-chain, and conservation of these contacts preserves the structural integrity of the noncovalently linked heterodimer.
Additionally, motifs within the HSA binding site were also conserved. One of them has previously been identified as being critical for albumin binding and was the site of a protein contact in our pH 3 and pH 8.5 crystal structures, between the two copies of FcRnECD in the asymmetric unit [8]. We were initially surprised to see the IgG binding site is poorly conserved with the exception of key residues, such as D130α-chain. This may be attributed to the heterogeneity in Fc moieties between mammalian orthologues, which presumably reduces species’ cross-reactivity. Of the regions identified by SiteMap, the central cavity was highly conserved, potentially due to the proximity to the dimer interface.
Fragment screening to identify small molecules
Our in silico analysis suggests the presence of an evolutionarily conserved binding site at the interface between the α-chain and β2m. To find ligands for this cavity and, potentially, other sites, a solution-state NMR fragment screen was performed. For this purpose, we used an in-house library of approximately 1,100 molecules containing fluorine atoms to enable ligand interrogation by 19F Carr-Purcell-Meiboom-Gill NMR [42,43]. This screen detected 143 potential binders. In order to select compounds for crystallographic studies, active molecules were further tested in Saturated Transfer Difference NMR experiments and by Surface Plasmon Resonance (SPR), yielding an estimated KD of 80 μM for the highest affinity fragment, the racemate UCB-FcRn-84 (S3 Fig and S1 Data) [44,45]. Chiral separation showed preferential binding of the R enantiomer. Competition of UCB-FcRn-84 with IgG was tested in a FcRnECD–IgG FRET assay; however, no measurable inhibition was observed.
Crystallographic studies of ligand-bound FcRnECD
Confirmed hits from the fragment-screen were soaked into FcRnECD crystals that had been grown under acidic conditions. Based on the derived crystal structures, we aimed to further improve both affinity and solubility of the ligand.
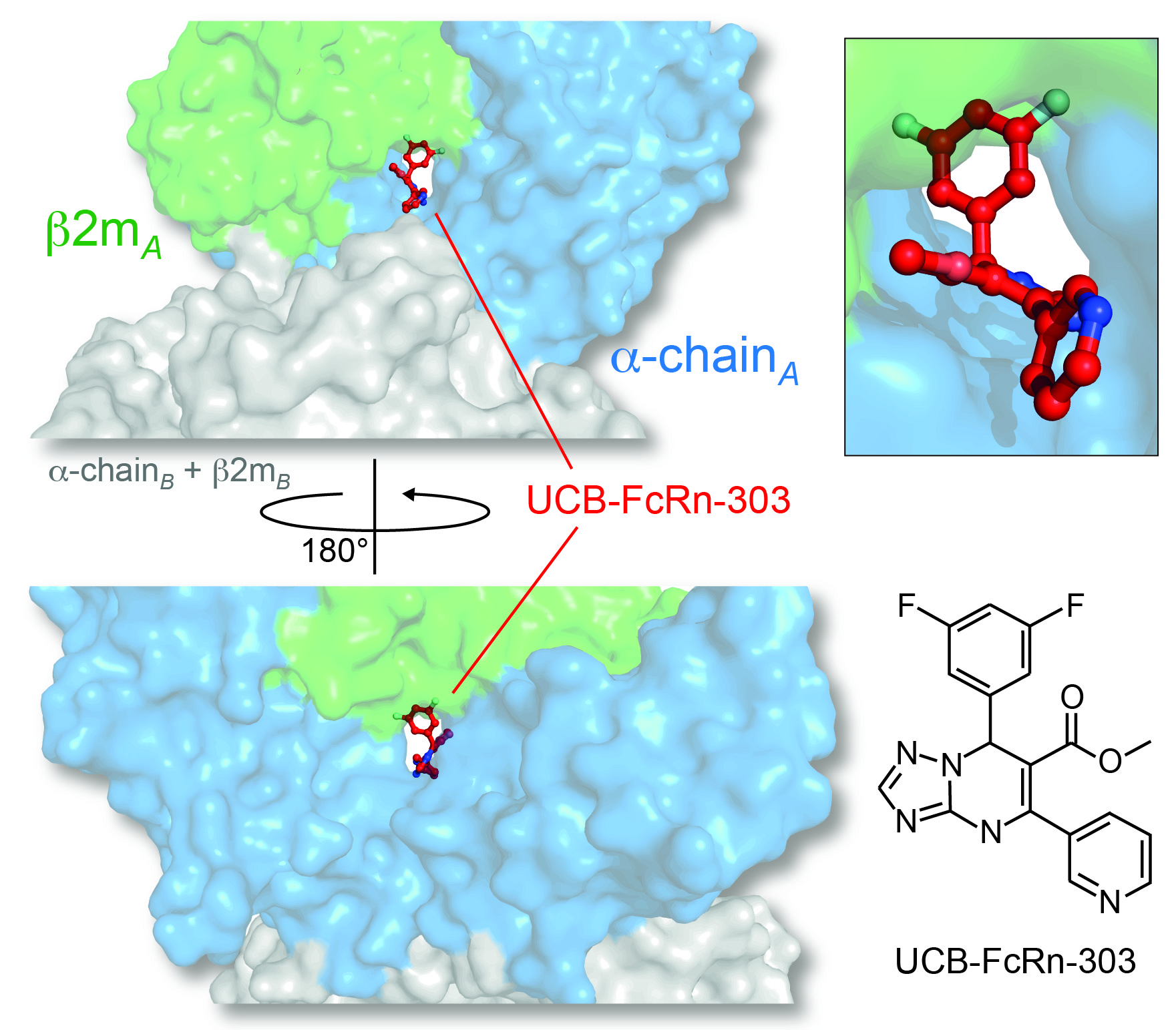
UCB-FcRn-84 was found in the conserved cavity at the interface of β2m and the α-chain (S4 Fig and S2 Text), as predicted by SiteMap. Key properties of binding interactions include burial of the 3-fluorophenyl group in a hydrophobic cavity composed of Y26β2m, S52β2m, Y63β2m, L65β2m, W29α-chain and P228α-chain. The most visually striking feature of the binding pocket is a tunnel-like cavity that extends through the middle of the protein (S5 Fig and S2 Text). The bicyclic ring of UCB-FcRn-84 occupies this cavity, being involved in one direct and one water-mediated hydrogen bond to the main-chain of Q34α-chain.
As the fragment was centrally located in this region, it afforded us a tractable chemical platform to explore the binding site further (S4 Fig and S2 Text). We attempted to improve the affinity of UCB-FcRn-84 by better occupying space around the 3-fluorophenyl group, which was nested in a well-defined hydrophobic pocket. Notably, we identified a small area available for growth adjacent to the unsubstituted meta position of the 3-fluorophenyl group. After scanning a range of disubstituted phenyl derivatives, we identified 3,5 difluorophenyl as the best option, yielding a ligand with 2.4 μM affinity for the racemate. Moreover, adding a 3-pyridile group in position 5 led to an equipotent compound (UCB-FcRn-303) but with improved solubility (S5 and S6 Figs, S1 Data and S2 Text).
The crystal structure of UCB-FcRn-303 bound to FcRnECD displays the R enantiomer (Fig 2 and S5 Fig). The binding mode is consistent with the position observed for UCB-FcRn-84 (Fig 2A and 2B, S4 and S5 Figs). The di-substituted phenyl ring better fills the hydrophobic pocket in the α-chain/β2m interface, while hydrogen bond interactions are maintained with the α-chain and the local water network.
Fig 2. Crystal structure of the compound UCB-FcRn-303 (R enantiomer) bound to FcRnECD.

(A) The protein crystallized as a dimer composed of two β2m (dark grey and green) and two α-chain (light grey and blue) molecules. (B) At the interface of β2m and the α-chain, UCB-FcRn-303 (grey) occupies a binding pocket with Glycine, Cysteine, hydrophobic (Leucine), charged (Histidine, Aspartate), and polar uncharged (Serine, Glutamine) residues. β2m, β2-microglobulin; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor.
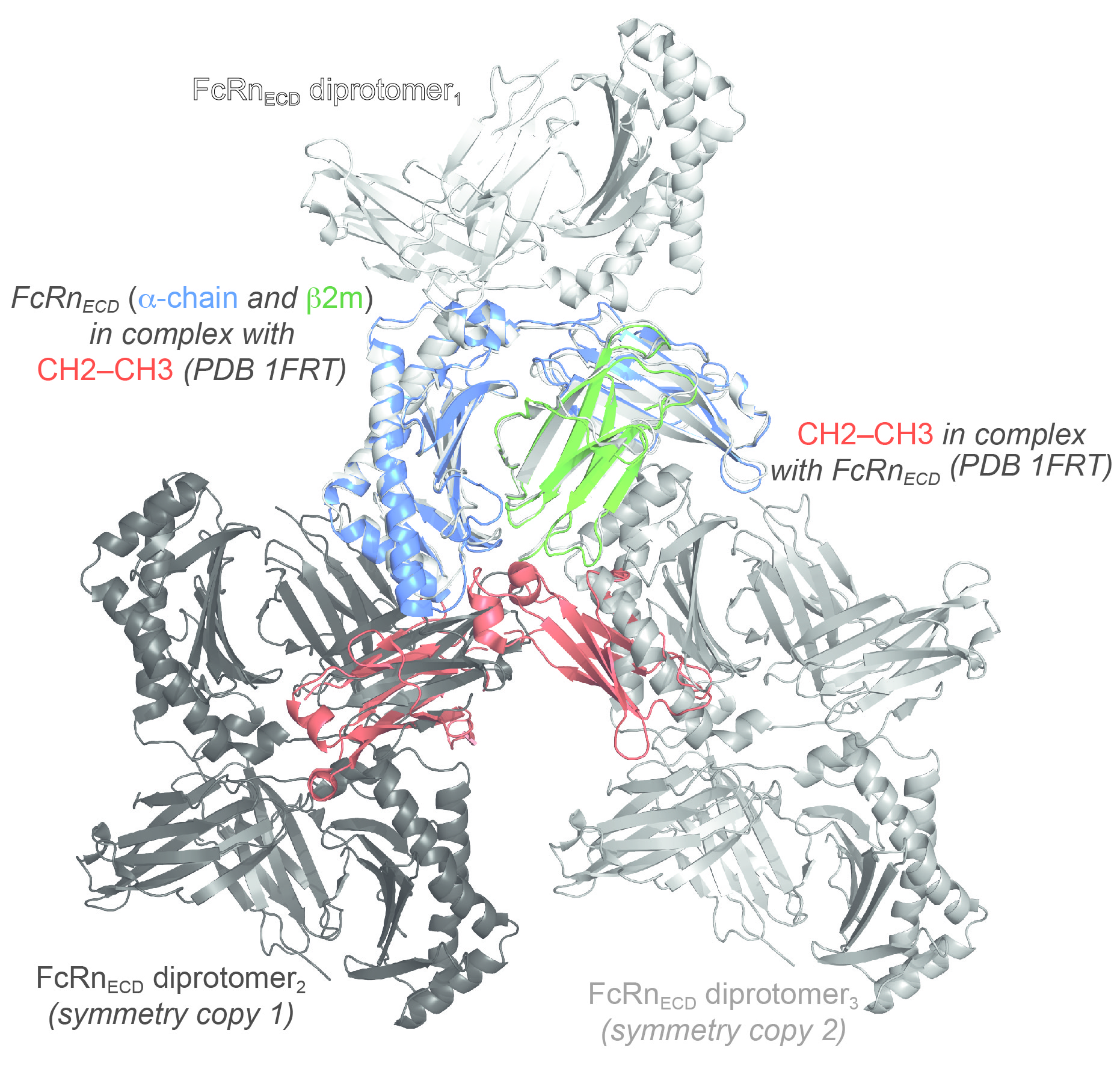
In biochemical experiments molecules from our series did not inhibit IgG binding when tested in a FcRnECD–IgG FRET competition assay. This was consistent with our crystallographic studies which showed no discernible change in the IgG binding site, although accurate interpretation of small changes may be hampered by crystal contacts. The FcRnECD crystallized with two copies of the heterodimer present in the asymmetric unit, packing in an anti-parallel fashion (Fig 2A). Overlay of the IgG heavy chain–FcRnECD complex (Protein Data Bank [PDB] code 1FRT) with the ligand-free crystal structure shows significant overlap between CH2 and CH3 domains of IgG and symmetry related copies of FcRnECD (S7 Fig) [46].
In order to determine whether the ligand might induce small conformational and/or dynamic changes in regions beyond the direct binding site, we have used orthogonal solution-based techniques, which will avoid conformational restrictions imposed by crystal packing.
Sedimented, fully protonated FcRnECD yielded well resolved proton-detected MAS NMR spectra
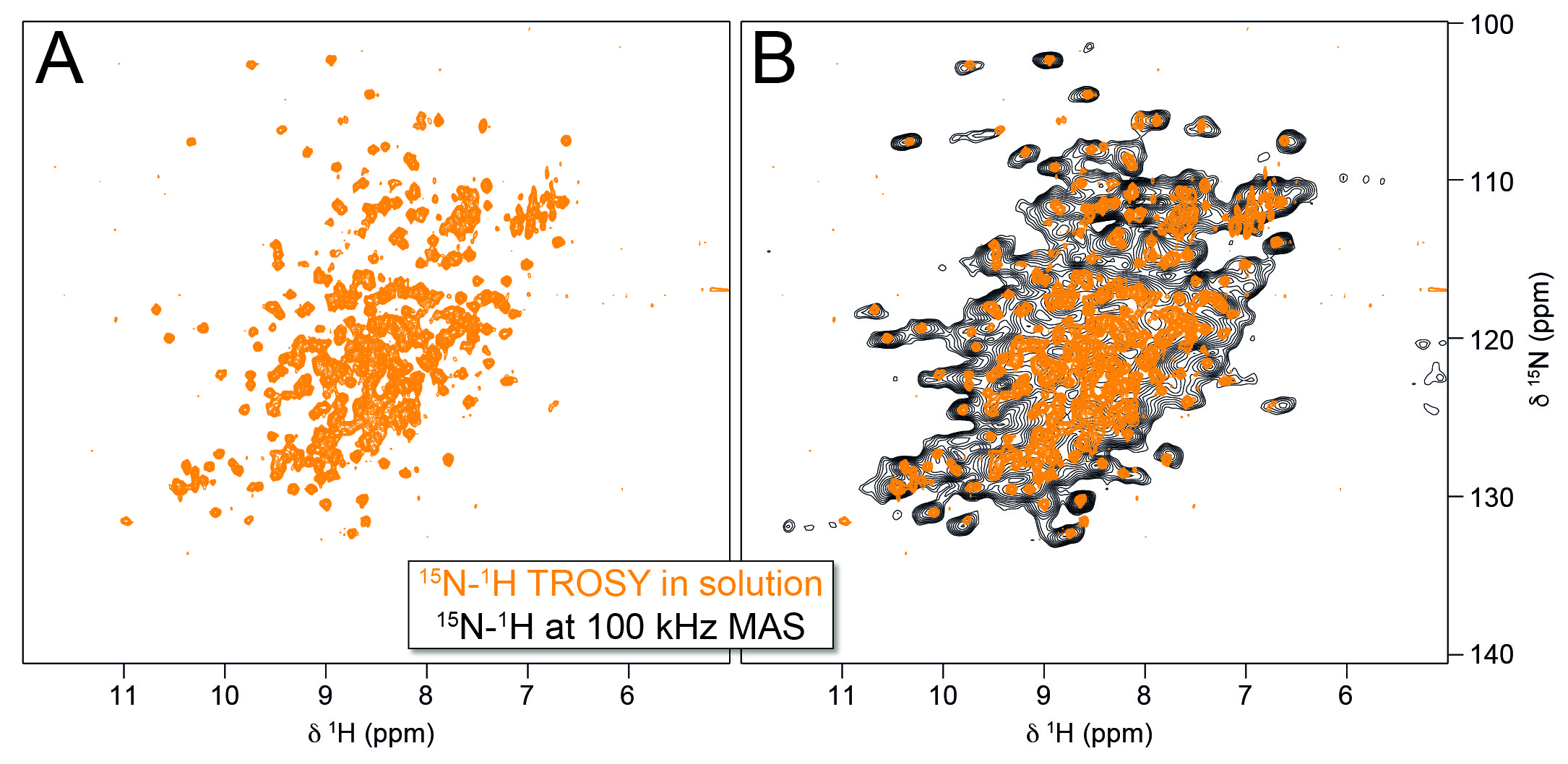
Until recently, solution-state and proton-detected MAS NMR typically required deuteration of proteins in the size-range of the 42 kDa FcRnECD, as the signal-to-noise ratios obtained in triple-resonance experiments involving 13Cβ and 13Cα chemical-shifts critically depend on the T2 of 13C coherences [47,48]. Since we were unable to produce deuterated FcRnECD in sufficient amounts it was not possible to acquire 3D solution-state NMR spectra that would allow sequential assignments. The spectral quality of a 2D 15N-1H correlation spectrum recorded in solution, however, is remarkably high (S8 Fig) [49]. It indicates that FcRnECD most likely does not form a stable dimer of heterodimers in solution at the applied concentrations, which agrees with biochemical data and earlier studies [50]. At least 227 of the expected 349 backbone amide signals could be observed, with the remainder of the signals most probably obscured by overlap.
To circumvent these experimental limitations, we applied MAS NMR. Progress in recent years, in particular ever faster sample spinning, enables the acquisition of proton-detected MAS NMR spectra on fully protonated samples which increases sensitivity and facilitates resonance assignments through triple-resonance experiments [51–54]. At the highest MAS frequency routinely available (110 kHz), well resolved proton spectra have been reported for fully protonated microcrystalline and membrane proteins, sedimented assemblies and fibrillar proteins [55–57].
In the present case, we have investigated soluble FcRnECD by MAS NMR experiments via sedimentation. For this purpose, the availability of dedicated filling tools is a prerequisite [58,59]. With the help of such appliances, we sedimented the 42 kDa soluble, fully protonated [13C,15N]-labeled FcRnECD by ultracentrifugation directly into a 0.7 mm MAS rotor (Fig 3A).
Fig 3. Proton-detected MAS NMR on fully protonated FcRnECD.

(A) The soluble FcRnECD (42 kDa) was sedimented by ultracentrifugation at 100,000 x g directly into a 0.7 mm MAS NMR rotor using a home-made filling tool. (B) 2D 15N-1H correlation spectrum recorded at 100 kHz MAS of fully protonated [13C,15N]-labeled FcRnECD. (C) Typical linewidths of 1H (1) and 15N (2) at full-width-half-maximum (FWHM) of a selected cross peak from the 15N-1H spectrum. β2m, β2-microglobulin; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor.
The 2D 15N-1H correlation spectrum measured at 100 kHz MAS is shown in Fig 3B. The observed linewidths demonstrate a remarkable spectral quality by MAS NMR standards (Fig 3C). Recorded at a magnetic field of about 20 T, the 1H and 15N linewidths of a selected typical cross peak are 184 Hz and 41 Hz, respectively. The amide 1H linewidth of FcRnECD is still higher than is observed in the model protein GB1, by a factor of approximately 1.8, but similar to the precipitated viral capsid coat protein AP205 [60]. The spectrum displays a large number of unresolved signals and a considerable fraction of signals that are well dispersed, both of which may be assigned on the basis of suitable triple-resonance spectra.
In order to monitor the folding state of FcRnECD in the sedimented sample, we compared the MAS 15N-1H spectrum to the 15N-1H correlation recorded in solution (S8 Fig and S3 Text). The agreement of the spectra indicates that the structure of FcRnECD in solution is very similar to the one in the sedimented sample. FcRnECD did not form the stable dimers of heterodimers in solution as had been observed in the crystal structure. Analytical ultracentrifugation, however, reveals a concentration-dependent protomer–diprotomer equilibrium of the FcRnECD heterodimer in solution, with a very low populated diprotomer fraction (S9 Fig and S4 Text). It is possible that this equilibrium is changed in the sedimentation process which may explain small chemical-shift differences. However, due to the signal overlap in the 15N-1H MAS NMR spectrum, a more detailed chemical-shift comparison is not possible.
Resonance assignments in proton-detected MAS NMR spectra of FcRnECD
To allow interpretation of chemical-shift perturbations (CSPs) upon binding of UCB-FcRn-303 to FcRnECD, resonance assignments are critical. The high MAS frequencies now available facilitate an assignment procedure based on triple-resonance MAS NMR experiments as in solution-state NMR. These include (H)CANH, (H)CBCANH, (H)CA(CO)NH, and (H)CBCA(CO)NH spectra, yielding assignments of 15N, 1HN, 13Cα0, and 13Cβ chemical-shifts [61,62]. If it is not possible to obtain a (H)CBCA(CO)NH spectrum with sufficient signal-to-noise due to too short 13Cα and 13CO relaxation times, the first two experiments allow for identification of amino acids that, in a following step, can be sequentially connected according to the protein sequence by using a (H)CA(CO)NH spectrum. In this study, such spectra were acquired on the fully protonated [13C,15N]-labeled FcRnECD enabling assignments of 25 α-chain residues close to the binding pocket and of 73% of all β2m residues.
To ease the assignment of β2m resonances, we made use of data in Beerbaum and colleagues, in which [2H,13C,15N]-labeled β2m was investigated in complex with unlabeled MHC1 [63]. However, the final assignments were achieved by establishing sequential connections along the protein backbone, as shown for the sequence from K41β2m to R45β2m (Fig 4). All chemical-shift assignments are listed in S1 Table and S2 Data and are deposited in the Biological Magnetic Resonance Data Bank (BMRB) (accession number 27437). In total, signals of 98 residues were assigned this way (S1 Table, S2 Data, and S5 Text). The more sensitive (H)CANH spectrum contained 84 additional resolved cross peaks that could not be assigned further due to overlap or lacking correlations in the less sensitive (H)CA(CO)NH and (H)CBCANH spectra.
Fig 4. Triple-resonance MAS NMR spectra enable assignments of chemical-shifts.

Sequential resonance assignments using the experiments (H)CANH (blue), (H)CA(CO)NH (red), and (H)CBCANH (green) recorded on fully protonated [13C,15N]-labeled FcRnECD at 100 kHz MAS. As an example, the sequential connections from K41β2m to R45β2m in β2m are indicated by dashed lines. All assigned chemical-shifts can be found in S1 Table, S2 Data, and in the BMRB (accession number 27437). β2m, β2-microglobulin; BMRB, Biological Magnetic Resonance Data Bank; FcRnECD, extracellular domain of the neonatal Fc receptor; MAS, magic-angle-spinning.
Interestingly, a large number of the measured 13Cα, 13Cβ, 15N, and 1H chemical-shifts of β2m in the FcRnECD heterodimer match the observed solution-state NMR resonances for deuterated β2m in MHC1 complexes of the previous study (S1 Table and S5 Text) [63]. Differences in chemical-shifts can be explained by 1H/2H isotope effects, the limited number of amino acid substitutions in MHC1, potential dimers of FcRnECD heterodimers in the sedimented sample, and differences in buffer and temperature [64]. The similarity of the chemical-shifts supports the notion that β2m adopts its globular fold in the sedimented sample of FcRnECD. Based on these findings and on the similarity between the solution-state 15N-1H and MAS 15N-1H spectra, we assume the overall FcRnECD structure to be highly similar in solution and in sedimented samples (S8 Fig, S3 Text, S1 Table and S5 Text).
In summary, (H)CANH, (H)CA(CO)NH, and (H)CBCANH spectra represent an acceptable basis for obtaining backbone resonance assignments and allowed us to exploit CSPs as monitors for structural alterations in FcRnECD upon UCB-FcRn-303 binding.
Structural changes in FcRnECD upon binding of UCB-FcRn-303
CSPs (Δδ) are probes for both direct effects of ligand binding and concomitant long-range structural changes in receptor proteins that may hint at allosteric effects. Since differences between X-ray structures with and without ligand may be masked by crystal contacts, we analyzed this possibility by comparing 3D (H)CANH spectra recorded on samples of FcRnECD with and without UCB-FcRn-303 (Fig 5A and 5B). The introduction of a third dimension compared to a 2D 15N-1H correlation leads to better spectral resolution. The observed minimal chemical-shift differences are displayed in Fig 5A. Overall, many of the assigned signals show very similar chemical-shifts in both spectra (Δδ < 0.02 ppm, white in Fig 5C and 5D), whereas signals that shift significantly (0.02 ppm < Δδ < 0.03 ppm, cyan; 0.03 ppm < Δδ < 0.04 ppm, marine; 0.04 ppm < Δδ, dark blue in Fig 5C and 5D) appear well clustered, suggesting a conformational/dynamic change extending from the ligand binding site. In the following discussions, we consider all residues with Δδ < 0.02 ppm as not perturbed. Selected cross peaks of strongly affected residues (0.04 ppm < Δδ) are shown in 2D planes of the (H)CANH spectra with and without ligand (Fig 5B). The calculated CSPs shown in Fig 5A are based on the chemical-shift changes in all three spectral dimensions. Large CSPs displayed in Fig 5A may therefore not be obvious from the 2D planes shown in Fig 5B, see D96β2m.
Fig 5. CSPs (Δδ) indicate structural changes in FcRnECD upon ligand binding.

(A) CSPs of all assigned amino acids in FcRnECD upon binding to UCB-FcRn-303, calculated with Δδ = MIN{SQRT[(Δδ(1H))2 + (Δδ(13C)/10)2 + (Δδ(15N)/5)2]} (standard deviation = 0.015 ppm). The changes were measured in 3D (H)CANH MAS NMR spectra. (B) CSPs upon UCB-FcRn-303 binding to FcRnECD in 2D planes of 3D (H)CANH spectra with (black) and without (blue) the ligand, both recorded in the presence of 3% DMSO. (C) Structural view of UCB-FcRn-303 (red) bound to FcRnECD with assigned residues in stick representation color-coded according to their CSP (Δδ < 0.02, white; 0.02 < Δδ < 0.03, cyan; 0.03 < Δδ < 0.04, marine; 0.04 < Δδ, dark blue). (D) Structural view of FcRnECD in complex with UCB-FcRn-303 (red) with the same color-coding of changes in chemical-shifts as in (C). All chemical-shifts can be found in S1 Table, S2 Data, and in the BMRB (accession number 27437). BMRB, Biological Magnetic Resonance Data Bank; CSP, chemical-shift perturbation; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor.
As expected, strong effects are seen in the vicinity of the binding pocket of UCB-FcRn-303 observed by X-ray crystallography (G232α-chain, C48α-chain, G49α-chain and S52β2m, Fig 5A and 5C) since the chemical environment of these residues is altered upon binding.
More interestingly, several residues that experienced changes in their chemical-shifts are distant from the binding pocket. These include, for example, E54α-chain at the C-terminal end of the short α-helix close to the binding region (Fig 5C and 5D). Such an effect could potentially be explained by a small movement of the α-helix. Furthermore, strong CSPs can be observed in a rather remote region composed of loops in β2m, involving D96β2m, G18β2m, K19β2m, S20β2m, and H13β2m (Fig 5C and 5D). These residues cluster at the surface close to or at the interface of β2m and the α-chain of two different heterodimers seen in the asymmetric unit (Fig 6A and 6B). A second ligand binding site in this region of FcRnECD can be excluded since stoichiometric ratios >1 were not observed in SPR experiments or by X-ray crystallography (Fig 2A, S3, S4, S5, S6 Figs, and S1 Data). Interestingly, strong shift changes were observed at comparably long distances from the small molecule binding site. Those strong CSPs may be direct effects of ligand binding, such as introducing a slight displacement of the β2m subunit with respect to the α-chain. Alternatively, it is possible that changes in the protomer–diprotomer equilibrium occur, and thus the chemical-shifts of residues at a potential diprotomer interface are affected. Although we do not observe a stable dimer of heterodimers in solution, a small fraction is present as seen in analytical ultracentrifugation (AUC) experiments (S9 Fig and S4 Text). A diprotomer may also be more highly populated at very high protein concentrations after the applied sedimentation through ultracentrifugation in preparation of 100 kHz MAS experiments. However, the samples with and without ligand have been prepared under identical conditions, making structural effects unrelated to ligand binding unlikely. For a ligand-induced alteration of the equilibrium, long-range structural changes towards the potential diprotomer interface are required, highlighting again the possibility for the occurrence of allosteric effects upon ligand binding.
Fig 6. CSPs cluster at the potential FcRnECD diprotomer interface.

(A) CSPs in surface representation of the FcRnECD diprotomer crystal structure in complex with UCB-FcRn-303 (red), with the same color-coding as in Fig 5. (B) For orientation, the FcRnECD crystal structure is shown in cartoon representation with β2m in green and dark grey and the α-chain molecules in blue and light grey. (C) The IgG and HSA interaction sites are depicted in purple and orange, respectively. The highlighted residues are discussed in the text. CSP, chemical-shift perturbation; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor; HSA, Human Serum Albumin; IgG, Immunoglobulin G.
In the HSA interaction site, a number of strongly affected residues could be found. These included R12β2m, H13β2m, C48α-chain, G49α-chain, A50α-chain, and E54α-chain with CSPs around 0.04 ppm or larger (Figs 5A, 6C and S2 Data). Some of these amino acids are close to the binding site of UCB-FcRn-303, but especially R12β2m, H13β2m, and E54α-chain are distant to it and may be therefore structurally altered through allosteric effects (Figs 5C and 6C). It is possible that the FcRn–HSA interaction can be modulated with UCB-FcRn-303. The binding site to HSA, however, is located at the possible diprotomer interface and CSPs in this region may be caused by potential ligand-induced changes in the protomer–diprotomer equilibrium of FcRnECD as discussed above.
Of the NMR assigned residues, only a few from β2m are found in the IgG binding area, such as R3β2m and T86β2m (Fig 6C), for which negligible chemical-shift changes occur (Fig 5A) [46]. In general, residues in the β-sheet region of β2m distant from the binding site or the interface between heterodimers do not exhibit notable CSPs (Fig 5D).
In order to further investigate the potential for allosteric interference with IgG binding, we generated chemical-shift–informed overlays of the two FcRnECD crystal structures, with and without UCB-FcRn-303. All residues with Δδ < 0.02 ppm (Fig 5A) were used for fitting, including the area around R3β2m, T86β2m, and L87β2m in β2m that is involved in IgG interaction (Fig 6C), yielding a root mean square deviation (r.m.s.d.) of 0.095 Å. If a movement of the α-helical region of the α-chain could be induced upon ligand binding, it may disseminate towards the loop from D130α-chain to E133α-chain, which is part of the binding site to IgG (Fig 6C) [46]. Unfortunately, the overlay does not reveal any significant structural changes of α-chain residues close to this loop, except for subtle differences, which we are unable to isolate as a compound-induced effect due to its proximity to crystal packing interactions. Still, the CSPs observed distant to the ligand cavity reveal the potential for allosteric effects in FcRnECD induced by UCB-FcRn-303 by affecting the HSA interaction site. This provides evidence that binding of an optimized ligand could be aimed at displacing the β2m and α-chain subunits and distally disrupting the IgG binding interface of FcRnECD.
Conclusions
In the context of a druggability study, we identified a compound of <500 Da molecular weight, UCB-FcRn-303, that binds to the extracellular domain of FcRn with low μM affinity. The conserved binding site is located at the interface of β2m and the α-chain, featuring a tunnel-like cavity with solvent access from two different sides. The pocket was identified by computational chemistry methods in a theoretical druggability examination and was corroborated by fragment screening, X-ray crystallography, and NMR spectroscopy. The respective crystal structures show a dimer of heterodimers, with a 1:1 stoichiometry for the ligand/protein complex.
Due to the distance of about 35 Å between the small molecule binding pocket and the site of IgG interaction (Fig 6C), substantial allosteric effects would be required for pharmacologically relevant interference with IgG binding [46]. Such allosteric effects can be monitored by comparing X-ray structures with and without ligand and by NMR spectroscopy via the analysis of ligand-induced CSPs. We therefore established a new approach for testing ligand binding effects on soluble proteins, employing proton-detected 100 kHz MAS NMR spectroscopy on fully protonated FcRnECD. After successful sedimentation of the protein by ultracentrifugation using dedicated filling tools, remarkably well-resolved proton-detected MAS NMR spectra could be obtained and partially assigned. CSPs upon UCB-FcRn-303 binding are observed around the small molecule binding pocket but also distant residues are affected, however, overlapping with the HSA interaction site.
Since a large fraction of residues in β2m towards the IgG binding site and some in the α-chain do not show substantial CSPs (Figs 5A, 6A and 6C), we generated alignments of the two X-ray structures with and without ligand, superposing the nonshifting residues. This approach revealed no substantial changes of residues close to the IgG interaction site upon FcRn-UCB-303 binding, which could modulate the FcRnECD–IgG interaction. It may be envisaged that an optimized ligand could produce a shift of the α-helical region in the α-chain, with potential effects on IgG interaction. Such an optimization could include derivatives with additional functional groups that enter the α-chain/β2m interface and thus produce a slight displacement of the two with respect to each other. Strong CSPs could be observed in the HSA interaction site of FcRnECD, highlighting the potential to achieve a functional modulation of FcRn with UCB-FcRn-303. However, it cannot be fully excluded that changes in oligomeric state of FcRnECD caused by the ligand under MAS conditions lead to the observed CSPs in this region.
The presented MAS NMR methodology provides an appealing approach for structural investigations of large, soluble proteins expressed in mammalian or other types of eukaryotic cells in which deuteration is challenging, especially in cases when glycosylation is crucial. Moreover, it extends the range of NMR applications to pharmacologically relevant targets, which are often inaccessible by solution-state NMR methods due to size. The MAS NMR approach works with high molecular weight targets and is independent of the physical state of the sample, providing spectral complexity can be handled, e.g., by applying appropriate labelling concepts. This facilitates protein-ligand interaction studies by NMR for any type of protein or biomolecular complex, making previously intractable pharmacological targets accessible, such as the ribosome, G protein-coupled receptors, and the like. Our investigation shows that MAS NMR complements X-ray crystallography in structure-based drug discovery campaigns. It is particularly useful to explore allosteric changes beyond small molecule binding sites, which may be difficult to observe by X-ray crystallography.
Materials and methods
FcRnECD vector generation
The coding sequences of extracellular domain (ECD) (amino acids 1–297) of human FcRn α-chain and human β2m were synthesized by Entelechon (Entelechon, Regensburg, Germany). The α-chain fragment was cloned into the expression vector pMH (UCB) and the β2m sequence was cloned into pMK (UCB) as HindIII and EcoRI fragments. Both vectors were digested with SalI and NotI and the relevant fragments excised and ligated to generate a vector containing both the α-chain and β2m genes (pM-ECDFcRn-B2M).
The vector was further digested with SalI and a neomycin cassette was ligated in to generate a double gene vector with antibiotic selection (pMFcRnECD-B2M-Neo).
Generation of a FcRnECD stable mammalian cell line
HEK293 cells were transfected with pMFcRnECD-B2M-Neo using 293fectin (Thermofisher) according to the manufacturer’s instructions. Transfected cells were incubated in a static incubator at 37 °C and 8% CO2 for 24 hours. The cells were then diluted to the required concentration with medium supplemented with 0.5 mg/L G418 (Invitrogen) and subsequently divided into 1-mL pools in 24-well plates and incubated in a static incubator at 37 °C with 8% CO2. Every 7 days, the medium was removed from each well and replaced with fresh medium. After a further 14 days, the pools that exhibited cell growth were transferred to 6-well plates. These were expanded up to 50-mL cultures in E250 flasks in shaking incubators. To determine total expression of the FcRnECD heterodimer composed of β2m and α-chain, batch overgrows were set up and incubated for 10 days.
Samples of the 50-mL batch overgrow supernatants were analyzed for FcRnECD expression using western blotting. 15 μL of each supernatant was run on a (denatured Tris/Gly gel western blot) alongside known concentrations of purified human FcRn as a control.
The highest expressing pool was expanded (without Neomycin selection) up to 10-L scale in a Wave Bioreactor 20/50 EHT at 37 °C, 8% CO2 for 4 days, at which point the temperature was reduced to 32 °C and incubated for a further 6 days. The cell culture was harvested by centrifugation (1,000x g for 1 hour) and supernatant put through a 0.22-μm filter.
Purification of FcRnECD expressed in HEK293 cells
Mammalian cell culture supernatant containing the heterodimer FcRnECD was concentrated with a 10,000 MWCO membrane using tangential flow filtration (Centramate, Pall) or Amicon stirred cell (Millipore), depending on scale. The sample buffer was exchanged into 50 mM sodium phosphate, pH 5.8, 30 mM NaCl by diluting and concentrating. FcRnECD was loaded onto a KappaSelect (GE Healthcare) column, which had been preloaded with an IgG4 monoclonal antibody to bind FcRnECD, before washing with 50 mM sodium phosphate, pH 5.8, 30 mM NaCl, and eluting with 50 mM sodium phosphate, pH 8.0, 30 mM NaCl. Elution fractions were analyzed by SDS-PAGE and relevant fractions pooled.
The sample was concentrated using an Amicon spin concentrator (Millipore), 10,000 MWCO membrane. The protein was purified by Gel Filtration with a Superdex 200 (GE Healthcare) column using 25 mM sodium phosphate, pH 7.4, 100 mM NaCl. Peak fractions were analyzed by SDS-PAGE before pooling and concentrating if required.
SPR analysis
SPR was carried out using BIAcore 4000 instruments (GE Healthcare). Reagents including CM5 sensor chips, N-hydroxysuccinimide (NHS), N-ethyl-N-(3-dimethylaminopropyl) carbodiimide (EDC), and ethanolamine HCl, 10 mM sodium acetate buffers (pH 5.0, pH 4.5) and HBS-P (10x buffer) were obtained from GE Healthcare.
FcRnECD was diluted into 10 mM sodium acetate buffer, pH 5.0 and immobilized on a CM5 Sensor Chip via amine coupling chemistry to a capture level of approxmimately 4,000 response units. Compounds were screened in a 10-point titration from 200 μM, at 2% DMSO, pH 6.0, and the surface was re-immobilized after each 384-well plate. Injections were performed at a flow rate of 10 μL/min.
All data were double-referenced for blank injections and reference surface, following standard procedures. Both data processing and fitting were performed using Activity Base template protocols developed in house.
Fragment library screening using 19F NMR
A library of approximately 1,100 fluorine-containing fragments were cocktailed into groups of 12 ensuring no overlap of 19F signals. Cocktails were initially prepared at a concentration of 4.2 mM in d6-DMSO and diluted to 800 μM in PBS pH 7.4 before a final dilution to 40 μM ligand concentration (1% d6-DMSO) in either PBS containing 10% D2O (for control samples) or 20 μM FcRnECD containing 10% D2O (for protein samples). NMR spectra were acquired at 25 °C on a Bruker 600 MHz AVIII-HD spectrometer equipped with a QCI-F cryoprobe and a SampleJet autosampler. Data were collected using a CPMG pulse sequence with a total echo time of 160 ms across a sweep width of 126 ppm with an acquisition time of 1 s. All spectra were processed using TopSpin 3.2. Fragments were considered binders to FcRnECD when the 19F signal intensity was significantly reduced in the spectra with FcRnECD present compared to the spectra recorded in the absence of protein.
Fragment library screening using STD NMR
STD NMR samples were prepared with a ligand to protein ratio of 50:1 (500 μM ligand, 10 μM FcRnECD) in 500 μL phosphate buffered saline, pH 7.4 (90% H2O, 10% D2O) with 5% d6-DMSO to help solubilize the ligand. STD NMR spectra were recorded using a Bruker Avance III HD 600 MHz spectrometer equipped with a 5 mm QCI-F Cryoprobe. Data were acquired and processed using the standard Bruker software and were collected at 298 K. The protein was saturated in the methyl region of the spectrum at 0 ppm, and off-resonance saturation was performed at 33 ppm. A series of 120 EBurp2 pulses (50 ms each) were applied with a 4-μs delay between each pulse, resulting in total saturation time of 6 s. Protein signals were removed by applying a spinlock of 100 ms. Interleaved on- and off-resonance data were recorded, processed separately, and then the difference spectra obtained by subtracting the on- from the off-resonance spectra. Data were zero filled once and an exponential multiplication window function applied (LB 2 Hz).
Plasmid construction of FcRnECD for expression in Sf9 insect cells
The parent construct for α-chain/β2m expresses two Open Reading Frames (ORFs) within one baculovirus multiple-target expression plasmid, pBacugs4X-1. Protein targets are based on the following amino acid sequences (both α-chain and β2m each contain their native leaders): α-chain 1–297 based on NCBI reference sequence NP_004098, β2m 1–119 based on NCBI reference sequence NP_004039. Codons for both ORFs were engineered by GeneComposer for highly expressed baculovirus genes, such that BamHI, HindIII, BglII, and EcoRI restriction sites were eliminated from the inserts to facilitate cloning [65]. Both genes, including flanking restriction sites, were synthesized at GeneArt. The ORF for α-chain was cloned behind the polyhedrin promoter via unique BamHI and HindIII sites; in this case, the BamHI site preceded the signal sequence while HindIII followed the sequence “TGAT” such that two stops are introduced after the C-terminal residue. The ORF for β2m was cloned behind the p10 promoter via unique BglII and EcoRI sites; the BglII site preceded the signal sequence, while EcoRI followed the sequence “TGATAA” such that two stops are introduced after the C-terminal residue. Using this cloning scheme, polyhedrin and p10 promoters are arranged in divergent/opposing orientations within the baculovirus transfer vector. ORFs were sequence verified prior to the commencement of expression studies.
Expression of FcRnECD in Sf9 insect cells
The α-chain/β2m construct was transfected into Sf9 insect cells (Expression Systems) using BestBac 2.0, v-cath/chiA Deleted Linearized Baculovirus DNA (Expression Systems, Cat#-91-002). Virus from each transfection was amplified through 3 rounds to produce virus stock for large-scale production. The large-scale preparations were grown in ESF921 medium (Expression Systems, Cat#96–001). Large-scale preparations were infected using the titerless infected-cells preservation and scale-up (TIPS) method [66]. Approximately 106 Tni cells (Trichopulsia ni, Expression Systems) per mL were infected using 1 mL of TIPS cells. Secreted proteins were harvested after 2–3 days by Tangential Flow Filtration (Spectrum KrosFlo, 0.2 μm filter Cat# P-NO2-E20U-05-N).
Purification of FcRnECD expressed in Sf9 insect cells
Harvested baculovirus medium (Trichoplusia ni) containing secreted FcRnECD was concentrated 10-fold and buffer exchanged (50 mM sodium phosphate pH 5.8 and 30 mM NaCl via Tangential Flow Filtration (TFF) (Spectrum Labs). The concentrated medium was centrifuged using a JA-10 rotor at 9,000 RPM for 15 minutes at 4 °C and then filtered through a 0.2 μm bottle top filter. Three complete EDTA free protease inhibitor tablets were added to the concentrated media prior to chromatography. The filtered concentrated medium was applied to 35 mL of IgG Sepharose FF resin (GE Healthcare) equilibrated with 50 mM sodium phosphate buffer, pH 5.8, and 30 mM NaCl (Sigma) and rotated end over end for one hour at 4 °C. The resin was then poured into a gravity flow column and washed with 10 column volumes of the same equilibration buffer. FcRnECD was eluted from the resin with eight column volumes of 50 mM sodium phosphate buffer pH 8.0 and 30 mM NaCl. The elution fractions containing FcRnECD were pooled and loaded onto two 5-mL HiTrap Q FF columns (GE Healthcare) and eluted over a 1 M NaCl gradient. The fractions of interest were pooled and glycerol was added to a final concentration of 10%. The pool was concentrated to 15 mg/mL via centrifugal concentration (Amicon Regenerated Cellulose, 10 kDa MWCO, Millipore) and further purified via size exclusion chromatography over a HiLoad 16/600 Superdex 200 pg (GE Healthcare) column in 50 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) pH 7.0 and 75 mM NaCl. The fractions containing FcRnECD were pooled and concentrated for crystallography via centrifugal concentration (Amicon Regenerated Cellulose, 10 kDa MWCO, Millipore) to 9.9 mg/mL prior to being aliquoted and flash frozen in liquid nitrogen for later use in crystallization experiments.
Crystallization of FcRnECD
In order to search for crystallization conditions for FcRnECD expressed in insect cells, sitting-drop vapor diffusion crystallization trials were set up at 291 K using a variety of commercial spare-matrix (Rigaku Reagents: JCSG+, Wizard 1/2, Wizard 3/4; Hampton Research: Crystal Screen HTIndex; Molecular Dimensions: PACT, Morpheus, Proplex; Microlytics: MCSG1) using 0.4 μL of protein solution at 5 mg/mL that were mixed with 0.4 μL of reservoir solution and equilibrated against 80 μL of reservoir solution. The initial crystallization trials produced small kite crystals in several conditions that contain PEG 3350, PEG 6000, or PEG 8000 at low pH. Low pH crystals of FcRnECD were produced in an optimized crystallization condition screen (Rigaku) containing 12%–16% PEG 6000, 100 mM Citric Acid/Ammonium citrate tribasic pH 3.00–3.09. Crystals of FcRnECD appeared within 24 hours and grew larger overtime, typically to 50–150 microns in size. The crystals were harvested using 20% glycerol as a cryoprotectant and flash frozen in liquid nitrogen prior to data collection.
Additionally, small rod-like crystals appeared in a single condition at a significantly higher pH (condition PACT H3, 100 mM Bis-Tris Propane/HCl pH 8.5, 200 mM NaI, and 20% PEG 3350) [67]. High/neutral pH crystals of FcRnECD were produced in optimized crystallization condition containing 100 mM Bis-Tris Propane/HCl pH 8.5, 200 mM NaI, and 20% PEG 3350. Crystals of FcRnECD appeared within 48 hours and grew larger overtime, typically greater than 150 microns in size. The crystals were harvested using 20% ethylene glycol and flash frozen in liquid nitrogen prior to data collection. To obtain compound bound crystals, apo FcRnECD crystals grown at pH 3 were soaked for three days in buffer containing 0.1 M Citric Acid/NaOH at pH 3.0, 20% w/v PEG 6000, 20% glycerol, and 12.5–20 mM compound dissolved in 100% DMSO. For crystallization of the UCB-FcRn-303 bound structure of FcRnECD, protein expressed in Sf9 insect cells was used.
Structure determination by X-ray crystallography
Datasets were collected at Canadian Light Source (CLS) on beamline 08ID-1 (CMCF) equipped with a Rayonix MX300 CCD X-ray detector and Advanced Photon Source (APS) on beamline 21-ID-F (LS-CAT) equipped with a Marmosaic 225 CCD X-ray detector. Diffraction data were reduced and scaled with XDS/XSCALE [68]. The structures of FcRnECD at low and high/neutral pH were solved by molecular replacement using a pre-existing structure of the complex. A significant portion of the structure required remodeling; therefore, Phenix.autobuild was run to generate a starting structure for further refinement. All structures were refined using iterative cycles of TLS and restrained refinement with Phenix.refine and model-building using the Crystallographic Object-Oriented Toolkit (COOT; Version 0.8.1-pre) and were validated using Molprobity prior to deposition in the PDB (IDs 6C97, 6C98, 6C99) [69–73]. Diffraction data and refinement statistics for apo FcRnECD at pH 3 and both ligand bound structures are listed in S2 Table.
Molecular dynamics simulation
The FcRnECD structures at pH 3 and pH 8.5 were used as the starting points for molecular dynamics simulation, after adding hydrogen. Protonation states and missing loops were predicted with Maestro (Schrödinger LLC). The structure was solvated in a dodecahedron such that no protein atom was within 10 Å of the edge of the solvent. Monatomic ions were added to a salt concentration of 0.15 M. All simulations were 1,000 ns in length and were carried out with GROMACS 4.6.2 [74]. Particle mesh Ewald was used for long-range electrostatics along with 10 Å cutoffs for Coulomb and Lennard–Jones potential functions.
Expression of fully protonated [13C,15N]-labeled FcRnECD for NMR experiments
FcRnECD was expressed using a stable HEK293 cell line, as described above, but the growth media was replaced with Bioexpress 6000 media (Cambridge Isotope Laboratories).
Acquisition of 2D 15N-1H TROSY solution-state NMR spectrum on fully protonated [13C,15N]-labeled FcRnECD
The solution-state 2D 15N-1H TROSY spectrum was acquired from a 0.35-mL sample of 300 μM [13C,15N]-labeled FcRnECD in a 25 mM Na2HPO4, 100 mM NaCl, 50 μM EDTA, 0.02% (w/v) NaN3 buffer at pH 7.4 containing 5% D2O/95% H2O [49]. NMR data were acquired at 35 °C on a 600 MHz Bruker AVIII HD spectrometer fitted with a cryogenically cooled probe. The spectrum was acquired for 40 minutes with acquisition times of 40 ms in F1 (15N) and 60 ms in F2 (1H). It was processed using Topspin 3.5 (Bruker Biospin Ltd) with linear prediction used to extend the effective acquisition time to 60 ms in F1.
Sedimentation of FcRnECD by ultracentrifugation for NMR experiments using fast MAS
A 0.5-mL sample of 100 μM [13C,15N]-labeled FcRnECD in a 25 mM Na2HPO4, 100 mM NaCl, 50 μM EDTA, 0.02% (w/v) NaN3 buffer at pH 7.4 containing 3% DMSO was directly ultracentrifuged into a 0.7 mm MAS NMR rotor at 100,000 x g and 4 °C for 40 hours using a swinging bucket ultracentrifuge rotor. Dedicated home-made filling tools were used for this step. Both bottom and top caps of the 0.7 mm MAS rotor were glued to avoid the loss of liquid or removal of caps during MAS. Excess protein and liquid after ultracentrifugation was removed before closing the 0.7 mm NMR MAS rotor. For experiments to detect CSPs, 3 mM UCB-FcRn-303 was added to a 0.5-mL sample of 100 μM [13C,15N]-labeled FcRnECD in a 25 mM Na2HPO4, 100 mM NaCl, 50 μM EDTA, 0.02% (w/v) NaN3 buffer at pH 7.4 containing 3% DMSO (ligand:protein ratio of 30:1). The ligand-bound sample was sedimented into a second 0.7 mm MAS rotor under the same conditions as the ligand-free sample.
Proton-detected NMR experiments using fast MAS on fully protonated [13C,15N]-labeled FcRnECD
All MAS NMR experiments were performed on a Bruker AVANCE III 850 MHz spectrometer. The spectra were recorded at 80, 90, or 100 kHz MAS with a triple-resonance 0.7 mm MAS probe (Bruker) and referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) (see S3 Table for experimental parameters). The sample temperature of 283 K was monitored by the frequency of the water resonance line. To achieve this sample temperature, the sample was cooled during MAS using nitrogen gas through a cooling unit (BCU II, Bruker) in strong mode with the gas flow set to 400 L/h. (H)NH, (H)CANH, (H)CA(CO)NH, and (H)CBCANH spectra were recorded using Cross Polarization (CP) (heteronuclear transfers) and DREAM (homonuclear transfers) magnetization transfer steps according to the procedures described in Penzel and colleagues [61,75,76]. All spectra were processed with Topspin 3.5 (Bruker) and analyzed with CCPNmr Analysis v. 2.4.2. [77]. All assigned chemical-shifts and observed CSPs are listed in S1 Table and S2 Data, and are deposited in the Biological Magnetic Resonance Data Bank (BMRB) (accession number 27437).
Analytical ultracentrifugation of FcRnECD
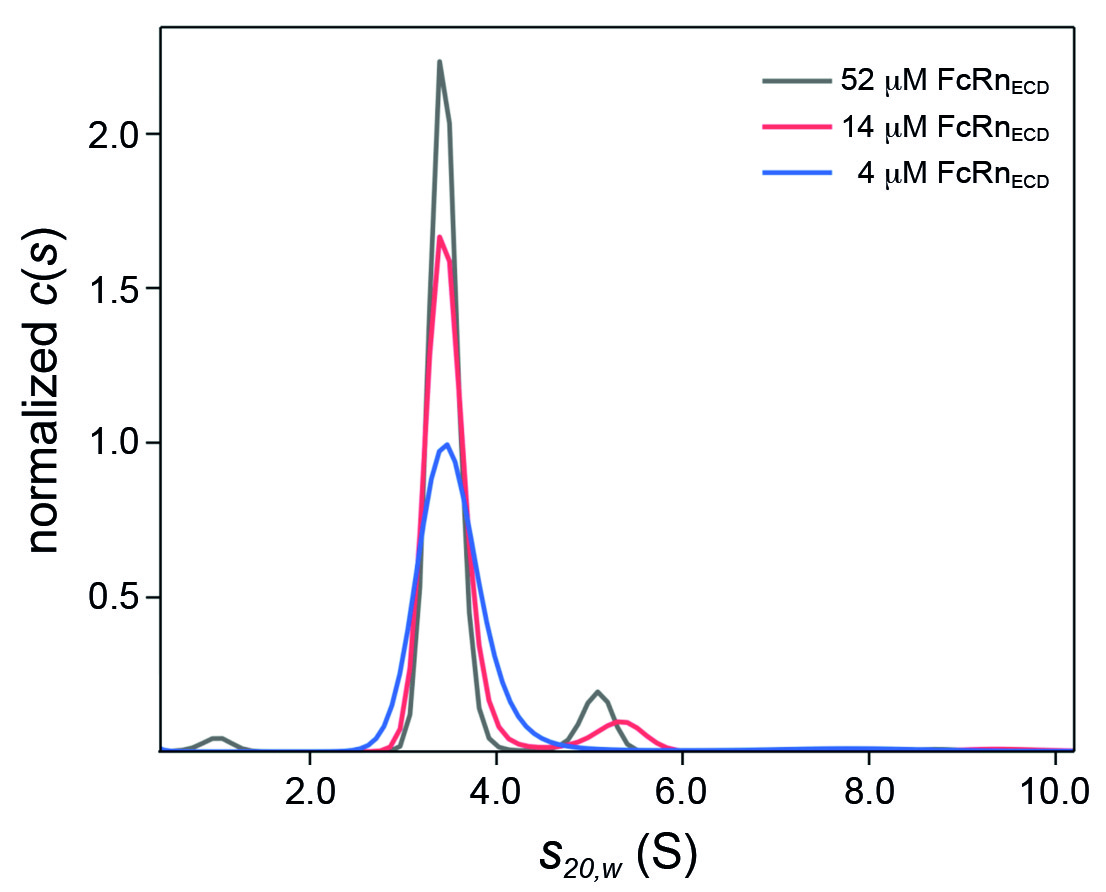
Sedimentation velocity experiments were performed at 8 °C and 35,000 rpm with an An-60Ti rotor using 12-mm Epon 2-sector centerpieces. For each of the three measurements at different FcRnECD concentrations (4 μM, 14 μM, and 52 μM), a 400-μL sample in 10 mM Na2HPO4, 137 mM NaCl, 2.7 mM KCl, 1.8 mM KH2PO4 buffer at pH 7.2 was used. The data were analyzed and plotted with the GUSSI implementation and SEDFIT [78].
Experimental and analytical details for synthetic analogues
All solvents and reagents were used as received from commercial suppliers, unless noted otherwise. The compounds were named using the Biovia Draw 2016 package (IUPAC).
NMR spectra were recorded on a Bruker Avance III HD 500 MHz or 250 MHz spectrometer.
The chemical-shifts (δ) reported are given in parts per million (ppm) and the coupling constants (J) are in Hertz (Hz). The spin multiplicities are reported as s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, ddd = doublet of doublet of doublet, dt = doublet of triplet, td = triplet of doublet, and m = multiplet.
uPLC-MS was performed on a Waters Acquity UPLC system coupled to a Waters Acquity PDA detector, an ELS detector and an MSD (Scan Positive: 150–850). Method (pH 3): Phenomenex Kinetix-XB C18 (2.1 x 100 mm, 1.7 μm) column. Elution with a linear gradient of Water + 0.1% Formic acid and Acetonitrile + 0.1% Formic acid at a flow rate of 0.6 mL/min. Chiral SFC analysis: Waters Thar 3100 SFC system connected to Waters 2998 PDA detector, Chiralcel OD-H 25 cm. Chiral SFC separation: Water Thar SFC system with a Waters Thar FDM pump, Waters Thar Alias autoinjector, Waters Thar fraction collector and a Waters 2998 PDA detector.
(R) and (S) 1-[7-(3-Fluorophenyl)-5-methyl-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl]ethanone (UCB-FcRn-84) (S10 Fig)
1-[7-(3-fluorophenyl)-5-methyl-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl]ethanone (CAS 691368-95-3) was purchased as a racemate from Life Chemicals and the mixture separated by chiral chromatography using a Chiralpak AD phase (100*500), 300 mL/min with an heptane/isopropanol (8/2) system. 1.21 G of starting material led to respectively 577 mg and 588 mg of separated isomers. Chiral analytical SFC: RT = 8.22 min, 100% ee; RT = 10.40 min, 100% ee.
1-[7-(3,5-Difluorophenyl)-5-methyl-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl]ethanone (S11 Fig)
A stirred solution of 3,5-difluorobenzaldehyde (0.1 mL, 0.946 mmol), pentane-2,4-dione (0.146 mL, 1.42 mmol, 1.5 eq.), and 4H-1,2,4-triazol-3-amine (119 mg, 1.42 mmol, 1.5 eq.) in N,N-dimethylformamide (1.5 mL) was irradiated in a microwave oven (up to 200 W) at 150 °C for 60 min. The reaction mixture was left to cool down to ambient temperature and water (6 mL) was added leading to the formation of a precipitate. The resulting solid was collected by filtration, rinsed with water (2 x 1 mL) and cyclohexane (2 x 1 mL), then triturated in hot acetonitrile (1 mL) and dried in vacuo to afford 94 mg (34% yield) of the title compound as a pale yellow solid.
The 1H NMR analysis yielded (500 MHz, DMSO-d6) δ 10.89 (s, 1H), 7.70 (s, 1H), 7.14 (t, J = 9.1 Hz, 1H), 6.98 (d, J = 6.3 Hz, 2H), 6.46 (s, 1H), 2.46 (s, 3H), 2.20 (s, 3H). uPLC-MS: [M+H]+ m/z = 291, RT = 2.38 min (99%).
(R) and (S) 1-[7-(3,5-Difluorophenyl)-5-methyl-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl]ethanone (S12 Fig)
The racemate (50 mg) was separated by chiral preparative chromatography on a Chiralpak ASV (50*490) phase, 80 mL/min with a heptane/ethanol (9/1) system to afford 24 mg and 19 mg of the pure enantiomers. Chiral analytical SFC: RT = 10.89 min, 100% ee; RT = 15.16 min, 97.8% ee.
Methyl 7-(3,5-difluorophenyl)-5-(3-pyridyl)-4,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate (UCB-FcRn-303) (S13 Fig)
To a stirred solution of 3,5-difluorobenzaldehyde (150 mg, 1.06 mmol), methyl 3-oxo-3-(pyridin-3-yl)propanoate (265 mg, 1.48 mmol, 1.4 eq.), and 4H-1,2,4-triazol-3-amine (124 mg, 1.48 mmol, 1.4 eq.) in N,N-dimethylformamide (1.5 mL) was added chloro(trimethyl)silane (0.268 mL, 2.11 mmol) dropwise. The reaction mixture was then irradiated in a microwave oven (up to 200 W) at 130 °C for 60 minutes. The reaction mixture was left to cool down to ambient temperature and water (6 mL) was added leading to the formation of a precipitate. The resulting solid was collected by filtration, rinsed with water (2 x 1 mL) and cyclohexane (2 x 1 mL) then recrystallized from acetonitrile (2 mL) and dried in vacuo to afford 238 mg (59% yield) of the title compound as a pale yellow solid. The 1H NMR analysis yielded (500 MHz, DMSO-d6) δ 11.18 (s, 1H), 8.67–8.61 (m, 2H), 7.92 (dt, J = 7.8, 1.9 Hz, 1H), 7.75 (s, 1H), 7.48 (dd, J = 7.8, 4.9 Hz, 1H), 7.20 (tt, J = 9.2, 2.2 Hz, 1H), 7.16–7.10 (m, 2H), 6.50 (s, 1H), 3.27 (s, 3H). uPLC-MS: [M+H]+ m/z = 370, RT = 2.12 min (97%).
Supporting information
At low pH binding sites with the capacity to yield high affinity binding are restricted to the dimer interface, with the region described as either one large or three distinct pockets (pockets A-C). At neutral and basic pH, transient sites arise at the albumin binding site, between the α1 and α2 helices (D), and between the β2m and the α3 domain (E). β2m, β2-microglobulin; FcRnECD, extracellular domain of the neonatal Fc receptor.
(JPG)
{kind=link}
(A) The pH 3 structure of the human FcRnECD heterodimer is colored to illustrate sequence conservation in vertebrate orthologues. Universally conserved residues are colored white; mutated residues are shown in red, with the color intensity indicating the BLOSUM62 score of the worst-matching substitution (darker red = more radical amino acid change away from the human residue). Species included in the analysis are: Pan troglodytes, Gorilla gorilla, Pongo pygmaeus, Macaca mulatta, Callithrix aurita, Microcebus murinus, Otolemur garnettii, Mus musculus, Rattus norvegicus, Cavia porcellus, Oryctolagus cuniculus, and Bos taurus. Mutations occur throughout the α-chain and β2m. Areas of clear conservation include the interface of α-chain and β2m and the central cavity that was detected in the SiteMap analysis. (B) For reference, human FcRnECD from the HSA-bound FcRn structure (PDB code 4N0F) has been colored to highlight residues that constitute the surfaces with the Fc moiety of IgG (magenta) and HSA (orange), the α-chain is shown in green and β2m in cyan. β2m, β2-microglobulin; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor; HSA, Human Serum Albumin; IgG, Immunoglobulin G; PDB, Protein Data Bank.
(TIF)
The numerical values can be found in S1 Data. FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor; SPR, Surface Plasmon Resonance.
(TIF)
The compound binds at the interface of β2m (green) and the α-chain (blue). Also in this crystal structure, a second heterodimer can be found in the asymmetric unit (grey). β2m, β2-microglobulin; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor.
(JPG)
{kind=link}
The compound occupies the same binding pocket as UCB-FcRn-84 at the interface of β2m (green) and the α-chain (blue). The binding region is a tunnel-like cavity extending through the protein. Again, a second heterodimer is found in the crystal structure (depicted in grey). β2m, β2-microglobulin; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor.
(JPG)
{kind=link}
The numerical values can be found in S1 Data. FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor; SPR, Surface Plasmon Resonance.
(TIF)
β2m, β2-microglobulin; FcRnECD, extracellular domain of the neonatal Fc receptor; IgG, Immunoglobulin G; PDB, Protein Data Bank.
(JPG)
{kind=link}
(A) 2D 15N-1H correlation using TROSY of fully protonated [13C,15N]-labeled FcRnECD measured in solution. (B) Overlay of the spectrum shown in (A) (orange) with a 2D 15N-1H spectrum of sedimented fully protonated [13C,15N]-labeled FcRnECD recorded at 100 kHz MAS (black). FcRnECD, extracellular domain of the neonatal Fc receptor; MAS, magic-angle-spinning.
(JPG)
{kind=link}
Sedimentation velocity experiments at three different concentrations (52 μM, grey; 14 μM, red; 4 μM, blue) exhibit protein concentration dependent peaks at 3.5 S, 5.1 S, and 5.3 S. FcRnECD, extracellular domain of the neonatal Fc receptor.
(JPG)
{kind=link}
FcRn, neonatal Fc receptor.
(PDF)
(PDF)
(PDF)
FcRn, neonatal Fc receptor.
(PDF)
β2m, β2-microglobulin; CSP, chemical-shift perturbation; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor; MHC1, class I major histocompatibility complex.
(XLSX)
They are compared to the corresponding chemical-shifts (Beerbaum and colleagues) of [2H,13C,15N]-labeled β2m in MHC1 complexes measured in solution-state NMR [63]. Amino acids of the α-chain are depicted in blue, β2m residues in green. β2m, β2-microglobulin; FcRnECD, extracellular domain of the neonatal Fc receptor; MAS, magic-angle-spinning.
(PDF)
(PDF)
FcRnECD, extracellular domain of the neonatal Fc receptor; MAS, magic-angle-spinning.
(PDF)
FcRnECD, extracellular domain of the neonatal Fc receptor.
(PDF)
FcRn, neonatal Fc receptor.
(PDF)
FcRnECD, extracellular domain of the neonatal Fc receptor.
(PDF)
FcRnECD, extracellular domain of the neonatal Fc receptor.
(PDF)
FcRn, neonatal Fc receptor; MAS, magic-angle-spinning.
(PDF)
Acknowledgments
Excellent technical support by Nils Cremer is kindly acknowledged.
Abbreviations
- β2m
β2-microglobulin
- APS
Advanced Photon Source
- AUC
analytical ultracentrifugation
- BMRB
Biological Magnetic Resonance Data Bank
- CD8
cluster of differentiation 8
- CLS
Canadian Light Source
- COOT
Crystallographic Object-Oriented Toolkit
- CP
Cross Polarization
- CSP
chemical-shift perturbation
- ECD
extracellular domain
- FcRn
neonatal Fc receptor
- FcRnECD
extracellular domain of the neonatal Fc receptor
- HSA
Human Serum Albumin
- IgG
Immunoglobulin G
- MAS
magic-angle-spinning
- MHC1
class I major histocompatibility complex
- ORF
Open Reading Frame
- PDB
Protein Data Bank
- r.m.s.d.
root mean square deviation
- SPR
Surface Plasmon Resonance
- TCR
T cell receptor
- TIPS
titerless infected-cells preservation and scale-up
Data Availability
The crystallographic data are available from the Protein Data Bank (www.rcsb.org, accession numbers 6C97, 6C98, and 6C99) and the chemical-shift data from the Biological Magnetic Resonance Data Bank (www.bmrb.wisc.edu, accession number 27437). All other relevant data are within the paper and its Supporting information files.
Funding Statement
Deutsche Forschungsgemeinschaft (grant number SFB 1078 B1). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. iNEXT (grant number WP1). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Swiss National Science Foundation (grant number 200020_159707 and 200020_146757). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. European Research Council (grant number 741863, FASTER). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Verdonk ML, Hartshorn MJ. Structure-guided fragment screening for lead discovery. Curr Opin Drug Discov Devel. 2004;7: 404–410. [PubMed] [Google Scholar]
- 2.Siegal G, Ab E, Schultz J. Integration of fragment screening and library design. Drug Discov Today. 2007;12: 1032–1039. doi: 10.1016/j.drudis.2007.08.005 [DOI] [PubMed] [Google Scholar]
- 3.Magee TV. Progress in discovery of small-molecule modulators of protein-protein interactions via fragment screening. Bioorg Med Chem Lett. 2015;25: 2461–2468. doi: 10.1016/j.bmcl.2015.04.089 [DOI] [PubMed] [Google Scholar]
- 4.Ludlow RF, Verdonk ML, Saini HK, Tickle IJ, Jhoti H. Detection of secondary binding sites in proteins using fragment screening. Proc Natl Acad Sci USA. 2015;112: 15910–15915. doi: 10.1073/pnas.1518946112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brambell FW, Hemmings WA, Morris IG. A theoretical model of γ-globulin catabolism. Nature. 1964;203: 1352–1354. [DOI] [PubMed] [Google Scholar]
- 6.Raghavan M, Bonagura VR, Morrison SL, Bjorkman PJ. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry. 1995;34: 14649–14657. [DOI] [PubMed] [Google Scholar]
- 7.Raghavan M, Chen MY, Gastinel LN, Bjorkman PJ. Investigation of the interaction between the class I MHC-related Fc receptor and its immunoglobulin G ligand. Immunity. 1994;1: 303–315. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, et al. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med. 2003;197: 315–322. doi: 10.1084/jem.20021829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borvak J, Richardson J, Medesan C, Antohe F, Radu C, Simionescu M, et al. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol. 1998;10: 1289–1298. [DOI] [PubMed] [Google Scholar]
- 10.Ober RJ, Martinez C, Lai X, Zhou J, Ward ES. Exocytosis of IgG as mediated by the receptor, FcRn: an analysis at the single-molecule level. Proc Natl Acad Sci USA. 2004;101: 11076–11081. doi: 10.1073/pnas.0402970101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward ES, Martinez C, Vaccaro C, Zhou J, Tang Q, Ober RJ. From sorting endosomes to exocytosis: association of Rab4 and Rab11 GTPases with the Fc receptor, FcRn, during recycling. Mol Biol Cell. 2005;16: 2028–2038. doi: 10.1091/mbc.E04-08-0735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JK, Tsen MF, Ghetie V, Ward ES. Localization of the site of the murine IgG1 molecule that is involved in binding to the murine intestinal Fc receptor. Eur J Immunol. 1994;24: 2429–2434. doi: 10.1002/eji.1830241025 [DOI] [PubMed] [Google Scholar]
- 13.Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol. 2004;172: 2021–2029. [DOI] [PubMed] [Google Scholar]
- 14.Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ. Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature. 1994;372: 336–343. doi: 10.1038/372336a0 [DOI] [PubMed] [Google Scholar]
- 15.Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH, Jaenisch R. β2-microglobulin deficient mice lack CD4−8+ cytolytic T cells. Nature. 1990;344: 742–746. doi: 10.1038/344742a0 [DOI] [PubMed] [Google Scholar]
- 16.Israel EJ, Wilsker DF, Hayes KC, Schoenfeld D, Simister NE. Increased clearance of IgG in mice that lack β2-microglobulin: possible protective role of FcRn. Immunology. 1996;89: 573–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in β2-microglobulin-deficient mice. Eur J Immunol. 1996;26: 690–696. doi: 10.1002/eji.1830260327 [DOI] [PubMed] [Google Scholar]
- 18.Praetor A, Hunziker W. β2-microglobulin is important for cell surface expression and pH-dependent IgG binding of human FcRn. J Cell Sci. 2002;115: 2389–2397. [DOI] [PubMed] [Google Scholar]
- 19.Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, et al. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol. 1997;15: 637–640. doi: 10.1038/nbt0797-637 [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Tian Z, Thirumalai D, Zhang X. Neonatal Fc receptor (FcRn): a novel target for therapeutic antibodies and antibody engineering. J Drug Target. 2014;22: 269–278. doi: 10.3109/1061186X.2013.875030 [DOI] [PubMed] [Google Scholar]
- 21.Le Panse R, Berrih-Aknin S. Autoimmune myasthenia gravis: autoantibody mechanisms and new developments on immune regulation. Curr Opin Neurol. 2013;26: 569–576. [DOI] [PubMed] [Google Scholar]
- 22.Feltkamp TE, van den Berg-Loonen PM, Nijenhuis LE, Engelfriet CP, van Rossum AL, van Loghem JJ, et al. Myasthenia gravis, autoantibodies, and HL-A antigens. Br Med J. 1974;1: 131–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Israeli E, Agmon-Levin N, Blank M, Chapman J. Shoenfeld Y. Guillain-Barré syndrome—a classical autoimmune disease triggered by infection or vaccination. Clin Rev Allergy Immunol. 2012;42: 121–130. doi: 10.1007/s12016-010-8213-3 [DOI] [PubMed] [Google Scholar]
- 24.Curtis AC, Heckaman JH, Wheeler AH. Study of the autoimmune reaction in dermatomyositis. JAMA. 1961;178: 571–573. [DOI] [PubMed] [Google Scholar]
- 25.Liu L, Garcia AM, Santoro H, Zhang Y, McDonnell K, Dumont J, et al. Amelioration of experimental autoimmune myasthenia gravis in rats by neonatal FcR blockade. J Immunol. 2007;178: 5390–5398. [DOI] [PubMed] [Google Scholar]
- 26.Li N, Zhao M, Hilario-Vargas J, Prisayanh P, Warren S, Diaz LA, et al. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J Clin Invest. 2005;115: 3440–3450. doi: 10.1172/JCI24394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akilesh S, Petkova S, Sproule TJ, Shaffer DJ, Christianson GJ, Roopenian D. The MHC class I-like Fc receptor promotes humorally mediated autoimmune disease. J Clin Invest. 2004;113: 1328–1333. doi: 10.1172/JCI18838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiessling P, Lledo-Garcia R, Watanabe S, Langdon G, Tran D, Bari M, et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: A randomized phase 1 study. Sci Transl Med. 2017;9: eaan1208. [DOI] [PubMed] [Google Scholar]
- 29.McDonnell KA, Low SC, Hoehn T, Donnelly R, Palmieri H, Fraley C, et al. Synthesis and structure-activity relationships of dimeric peptide antagonists of the human immunoglobulin G-human neonatal Fc receptor (IgG-FcRn) interaction. J Med Chem. 2010;53: 1587–1596. doi: 10.1021/jm901128z [DOI] [PubMed] [Google Scholar]
- 30.Mezo AR, Low SC, Hoehn T, Palmieri H. PEGylation enhances the therapeutic potential of peptide antagonists of the neonatal Fc receptor, FcRn. Bioorg Med Chem Lett. 2011;21: 6332–6335. doi: 10.1016/j.bmcl.2011.08.111 [DOI] [PubMed] [Google Scholar]
- 31.Mezo AR, Sridhar V, Badger J, Sakorafas P, Nienaber V. X-ray crystal structures of monomeric and dimeric peptide inhibitors in complex with the human neonatal Fc receptor, FcRn. J Biol Chem. 2010;285: 27694–27701. doi: 10.1074/jbc.M110.120667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mezo AR, McDonnell KA, Hehir CAT, Low SC, Palombella VJ, Stattel JM, et al. Reduction of IgG in nonhuman primates by a peptide antagonist of the neonatal Fc receptor FcRn. Proc Natl Acad Sci USA. 2008;105: 2337–2342. doi: 10.1073/pnas.0708960105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mezo AR, McDonnell KA, Castro A, Fraley C. Structure-activity relationships of a peptide inhibitor of the human FcRn:human IgG interaction. Bioorg Med Chem. 2008;16: 6394–6405. doi: 10.1016/j.bmc.2008.05.004 [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, Fraley C, Mezo AR. Discovery and structure-activity relationships of small molecules that block the human immunoglobulin G-human neonatal Fc receptor (hIgG-hFcRn) protein-protein interaction. Bioorg Med Chem Lett. 2013;23: 1253–1256. doi: 10.1016/j.bmcl.2013.01.014 [DOI] [PubMed] [Google Scholar]
- 35.Halgren TA. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49: 377–389. doi: 10.1021/ci800324m [DOI] [PubMed] [Google Scholar]
- 36.Kozakov D, Grove LE, Hall DR, Bohnuud T, Mottarella SE, Luo L, et al. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc. 2015;10: 733–755. doi: 10.1038/nprot.2015.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Capra JA, Laskowski RA, Thornton JM, Singh M, Funkhouser TA. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput Biol. 2009;5: e1000585 doi: 10.1371/journal.pcbi.1000585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zdobnov EM, Tegenfeldt F, Kuznetsov D, Waterhouse RM, Simão FA, Ioannidis P, et al. OrthoDB v9.1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2017;45: 744–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Bio. 2011;7: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelm S, Shi J, Deane CM. MEDELLER: homology-based coordinate generation for membrane proteins. Bioinformatics. 2010;26: 2833–2840. doi: 10.1093/bioinformatics/btq554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delano WL. The PyMOL molecular graphics system, Version 2.0 Schrödinger, LLC. [Google Scholar]
- 42.Carr HY, Purcell EM. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys Rev. 1954;94: 630–638. [Google Scholar]
- 43.Meiboom S, Gill D. Modified spin-echo method for measuring nuclear relaxation times. Rev Sci Instrum. 1958;29: 688–691. [Google Scholar]
- 44.Forsén S, Hoffman RA. Study of moderately rapid chemical exchange reactions by means of nuclear magnetic resonance. J Chem Phys. 1963;39: 2892–2902. [Google Scholar]
- 45.Forsén S, Hoffman RA. Exchange rates by nuclear magnetic multiple resonance. III. Exchange reactions in systems with several nonequivalent sites. J Chem Phys. 1964;40: 1189–1196. [Google Scholar]
- 46.Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature. 1994;372: 379–383. doi: 10.1038/372379a0 [DOI] [PubMed] [Google Scholar]
- 47.Gardner KH, Kay LE. The use of 2H, 13C, 15N multidimensional NMR to study the structure and dynamics of proteins. Annu Rev Biophys Biomol Struct. 1998;27: 357–406. doi: 10.1146/annurev.biophys.27.1.357 [DOI] [PubMed] [Google Scholar]
- 48.Frueh DP. Practical aspects of NMR signal assignment in larger and challenging proteins. Prog Nucl Magn Reson Spectrosc. 2014;78: 47–75. doi: 10.1016/j.pnmrs.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pervushin K, Riek R, Wider G, Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94: 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gastinel LN, Simister NE, Bjorkman PJ. Expression and crystallization of a soluble and functional form of an Fc receptor related to class I histocompatibility molecules. Proc Natl Acad Sci USA. 1992;89: 638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou DH, Shah G, Cormos M, Mullen C, Sandoz D, Rienstra CM. Proton-detected solid-state NMR spectroscopy of fully protonated proteins at 40 kHz magic-angle spinning. J Am Chem Soc. 2007;129: 11791–11801. doi: 10.1021/ja073462m [DOI] [PubMed] [Google Scholar]
- 52.Linser R, Fink U, Reif B. Proton-detected scalar coupling based assignment strategies in MAS solid-state NMR spectroscopy applied to perdeuterated proteins. J Magn Reson. 2008;193: 89–93. doi: 10.1016/j.jmr.2008.04.021 [DOI] [PubMed] [Google Scholar]
- 53.Zhou DH, Nieuwkoop AJ, Berthold DA, Comellas G, Sperling LJ, Tang M, et al. Solid-state NMR analysis of membrane proteins and protein aggregates by proton detected spectroscopy. J Biomol NMR. 2012;54: 291–305. doi: 10.1007/s10858-012-9672-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbet-Massin E, Pell AJ, Retel JS, Andreas LB, Jaudzems K, Franks WT, et al. Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J Am Chem Soc. 2014;136: 12489–12497. doi: 10.1021/ja507382j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lalli D, Idso MN, Andreas LB, Hussain S, Baxter N, et al. Proton-based structural analysis of a heptahelical transmembrane protein in lipid bilayers. J Am Chem Soc. 2017;139: 13006–13012. doi: 10.1021/jacs.7b05269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Struppe J, Quinn CM, Lu M, Wang M, Hou G, Lu X, et al. Expanding the horizons for structural analysis of fully protonated protein assemblies by NMR spectroscopy at MAS frequencies above 100 kHz. Solid State Nucl Magn Reson. 2017;87: 117–125. doi: 10.1016/j.ssnmr.2017.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stanek J, Andreas LB, Jaudzems K, Cala D, Lalli D, Bertarello A, et al. NMR spectroscopic assignment of backbone and side-chain protons in fully protonated proteins: microcrystals, sedimented assemblies, and amyloid fibrils. Angew Chem Int Ed. 2016;55: 15504–15509. [DOI] [PubMed] [Google Scholar]
- 58.Böckmann A, Gardiennet C, Verel R, Hunkeler A, Loquet A, Pintacuda G, et al. Characterization of different water pools in solid-state NMR protein samples. J Biomol NMR. 2009;45: 319–327. doi: 10.1007/s10858-009-9374-3 [DOI] [PubMed] [Google Scholar]
- 59.Gardiennet C, Schütz AK, Hunkeler A, Kunert B, Terradot L, Böckmann A, et al. A sedimented sample of a 59 kDa dodecameric helicase yields high-resolution solid-state NMR spectra. Angew Chem Int Ed. 2012;51: 7855–7858. [DOI] [PubMed] [Google Scholar]
- 60.Andreas LB, Jaudzems K, Stanek J, Lalli D, Bertarello A, Le Marchand T, et al. Structure of fully protonated proteins by proton-detected magic-angle spinning NMR. Proc Natl Acad Sci USA. 2016;113: 9187–9192. doi: 10.1073/pnas.1602248113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Penzel S, Smith AA, Agarwal V, Hunkeler A, Org ML, Samoson A, et al. Protein resonance assignment at MAS frequencies approaching 100 kHz: a quantitative comparison of J-coupling and dipolar-coupling-based transfer methods. J Biomol NMR. 2015;63: 165–186. doi: 10.1007/s10858-015-9975-y [DOI] [PubMed] [Google Scholar]
- 62.Agarwal V, Penzel S, Szekely K, Cadalbert R, Testori E, Oss A, et al. De novo 3D structure determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy. Angew Chem Int Ed. 2014;53: 12253–12256. [DOI] [PubMed] [Google Scholar]
- 63.Beerbaum M, Ballaschk M, Erdmann N, Schnick C, Diehl A, Uchanska-Ziegler B, et al. NMR spectroscopy reveals unexpected structural variation at the protein–protein interface in MHC class I molecules. J Biomol NMR. 2013;57: 167–178. doi: 10.1007/s10858-013-9777-z [DOI] [PubMed] [Google Scholar]
- 64.Smith AA, Ravotti F, Testori E, Cadalbert R, Ernst M, Böckmann A, et al. Partially-deuterated samples of HET-s (218–289) fibrils: assignment and deuterium isotope effect. J Biomol NMR. 2017;67: 109–119. doi: 10.1007/s10858-016-0087-0 [DOI] [PubMed] [Google Scholar]
- 65.Lorimer D, Raymond A, Walchli J, Mixon M, Barrow A, Wallace E, et al. Gene Composer: database software for protein construct design, codon engineering, and gene synthesis. BMC Biotechnol. 2009;9: 36–58. doi: 10.1186/1472-6750-9-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wasilko DJ, Lee SE, Stutzman-Engwall KJ, Reitz BA, Emmons TL, Mathis KJ, et al. The titerless infected-cells preservation and scale-up (TIPS) method for large-scale production of NO-sensitive human soluble guanylate cyclase (sGC) from insect cells infected with recombinant baculovirus. Protein Expr Purif. 2009;65: 122–132. doi: 10.1016/j.pep.2009.01.002 [DOI] [PubMed] [Google Scholar]
- 67.Newman J, Egan D, Walter TS, Meged R, Berry I, Ben Jelloul M, et al. Towards rationalization of crystallization screening for small- to medium-sized academic laboratories: the PACT/JCSG+ strategy. Acta Cryst Section D. 2005;61: 1426–1431. [DOI] [PubMed] [Google Scholar]
- 68.Kabsch W. XDS. Acta Cryst Section D. 2010;66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst Section D. 2010;66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst Section D. 2010;66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst Section D. 2010;66: 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berman H, Henrick K, Nakamura H. Announcing the worldwide protein data bank. Nat Struct Biol. 2003;10: 980 doi: 10.1038/nsb1203-980 [DOI] [PubMed] [Google Scholar]
- 73.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26: 1701–1718. doi: 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- 75.Pines A, Gibby MG, Waugh JS. Proton-enhanced nuclear induction spectroscopy: a method for high resolution NMR of dilute spins in solids. J Chem Phys. 1972;56: 1776–1777. [Google Scholar]
- 76.Verel R, Ernst M, Meier BH. Adiabatic dipolar recoupling in solid-state NMR: the DREAM scheme. J Magn Reson. 2001;150: 81–99. doi: 10.1006/jmre.2001.2310 [DOI] [PubMed] [Google Scholar]
- 77.Stevens TJ, Fogh RH, Boucher W, Higman VA, Eisenmenger F, Bardiaux B, et al. A software framework for analysing solid-state MAS NMR data. J Biomol NMR. 2011;51: 437–447. doi: 10.1007/s10858-011-9569-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophysj. 2000;78: 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
At low pH binding sites with the capacity to yield high affinity binding are restricted to the dimer interface, with the region described as either one large or three distinct pockets (pockets A-C). At neutral and basic pH, transient sites arise at the albumin binding site, between the α1 and α2 helices (D), and between the β2m and the α3 domain (E). β2m, β2-microglobulin; FcRnECD, extracellular domain of the neonatal Fc receptor.
(JPG)
(A) The pH 3 structure of the human FcRnECD heterodimer is colored to illustrate sequence conservation in vertebrate orthologues. Universally conserved residues are colored white; mutated residues are shown in red, with the color intensity indicating the BLOSUM62 score of the worst-matching substitution (darker red = more radical amino acid change away from the human residue). Species included in the analysis are: Pan troglodytes, Gorilla gorilla, Pongo pygmaeus, Macaca mulatta, Callithrix aurita, Microcebus murinus, Otolemur garnettii, Mus musculus, Rattus norvegicus, Cavia porcellus, Oryctolagus cuniculus, and Bos taurus. Mutations occur throughout the α-chain and β2m. Areas of clear conservation include the interface of α-chain and β2m and the central cavity that was detected in the SiteMap analysis. (B) For reference, human FcRnECD from the HSA-bound FcRn structure (PDB code 4N0F) has been colored to highlight residues that constitute the surfaces with the Fc moiety of IgG (magenta) and HSA (orange), the α-chain is shown in green and β2m in cyan. β2m, β2-microglobulin; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor; HSA, Human Serum Albumin; IgG, Immunoglobulin G; PDB, Protein Data Bank.
(TIF)
The numerical values can be found in S1 Data. FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor; SPR, Surface Plasmon Resonance.
(TIF)
The compound binds at the interface of β2m (green) and the α-chain (blue). Also in this crystal structure, a second heterodimer can be found in the asymmetric unit (grey). β2m, β2-microglobulin; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor.
(JPG)
The compound occupies the same binding pocket as UCB-FcRn-84 at the interface of β2m (green) and the α-chain (blue). The binding region is a tunnel-like cavity extending through the protein. Again, a second heterodimer is found in the crystal structure (depicted in grey). β2m, β2-microglobulin; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor.
(JPG)
The numerical values can be found in S1 Data. FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor; SPR, Surface Plasmon Resonance.
(TIF)
β2m, β2-microglobulin; FcRnECD, extracellular domain of the neonatal Fc receptor; IgG, Immunoglobulin G; PDB, Protein Data Bank.
(JPG)
(A) 2D 15N-1H correlation using TROSY of fully protonated [13C,15N]-labeled FcRnECD measured in solution. (B) Overlay of the spectrum shown in (A) (orange) with a 2D 15N-1H spectrum of sedimented fully protonated [13C,15N]-labeled FcRnECD recorded at 100 kHz MAS (black). FcRnECD, extracellular domain of the neonatal Fc receptor; MAS, magic-angle-spinning.
(JPG)
Sedimentation velocity experiments at three different concentrations (52 μM, grey; 14 μM, red; 4 μM, blue) exhibit protein concentration dependent peaks at 3.5 S, 5.1 S, and 5.3 S. FcRnECD, extracellular domain of the neonatal Fc receptor.
(JPG)
FcRn, neonatal Fc receptor.
(PDF)
(PDF)
(PDF)
FcRn, neonatal Fc receptor.
(PDF)
β2m, β2-microglobulin; CSP, chemical-shift perturbation; FcRn, neonatal Fc receptor; FcRnECD, extracellular domain of the neonatal Fc receptor; MHC1, class I major histocompatibility complex.
(XLSX)
They are compared to the corresponding chemical-shifts (Beerbaum and colleagues) of [2H,13C,15N]-labeled β2m in MHC1 complexes measured in solution-state NMR [63]. Amino acids of the α-chain are depicted in blue, β2m residues in green. β2m, β2-microglobulin; FcRnECD, extracellular domain of the neonatal Fc receptor; MAS, magic-angle-spinning.
(PDF)
(PDF)
FcRnECD, extracellular domain of the neonatal Fc receptor; MAS, magic-angle-spinning.
(PDF)
FcRnECD, extracellular domain of the neonatal Fc receptor.
(PDF)
FcRn, neonatal Fc receptor.
(PDF)
FcRnECD, extracellular domain of the neonatal Fc receptor.
(PDF)
FcRnECD, extracellular domain of the neonatal Fc receptor.
(PDF)
FcRn, neonatal Fc receptor; MAS, magic-angle-spinning.
(PDF)
Data Availability Statement
The crystallographic data are available from the Protein Data Bank (www.rcsb.org, accession numbers 6C97, 6C98, and 6C99) and the chemical-shift data from the Biological Magnetic Resonance Data Bank (www.bmrb.wisc.edu, accession number 27437). All other relevant data are within the paper and its Supporting information files.