

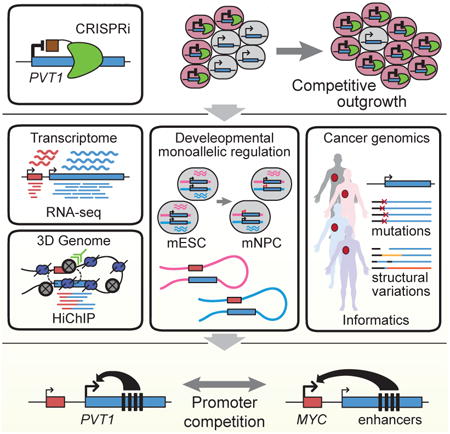
Summary
Noncoding mutations in cancer genomes are frequent but challenging to interpret. PVT1 encodes an oncogenic lncRNA, but recurrent translocations and deletions in human cancers suggest alternative mechanisms. Here we show that PVT1 promoter has tumor suppressor function independent of PVT1 lncRNA. CRISPR interference of PVT1 promoter enhances breast cancer cell competition and growth in vivo. The promoters of PVT1 and MYC oncogene, located 55 kilobases apart on chromosome 8q24, compete for engagement with four intragenic enhancers in the PVT1 locus, thereby allowing PVT1 promoter to regulate pause release of MYC transcription. PVT1 undergoes developmentally regulated monoallelic expression, and PVT1 promoter inhibits MYC expression only from the same chromosome via promoter competition. Cancer genome sequencing identifies recurrent mutations encompassing human PVT1 promoter, and genome editing verified that PVT1 promoter mutation promotes cancer cell growth. These results highlight regulatory sequences of lncRNA genes as potential disease-associated DNA elements.
Keywords: PVT1, MYC, lncRNA, CRISPRi, topological domains, promoter, enhancer, transcriptional regulation, tumor suppressor
Graphical abstract
Recurrent mutations in human cancer are found in the promotor for the lncRNA gene PVT1 regulates, which regulate MYC transcription via promoter competition for a shared set of enhancers.
Introduction
The human genome encodes tens of thousands of long noncoding RNA (lncRNA) genes (Derrien et al., 2012). While some lncRNAs have emerged as central regulators of transcriptional networks in development or disease (Fatica and Bozzoni, 2014; Schmitt and Chang, 2016), the functions and regulation of most lncRNA genes are not known. Plasmacytoma variant translocation 1 (PVT1) was the first lncRNA gene identified in human cancer translocations as a recurrent breakpoint in Burkitt's lymphoma (Graham and Adams, 1986; Shtivelman et al., 1989). Prior studies suggest PVT1-encoded lncRNA or microRNAs have oncogenic functions. The genomic region of 8q24 harboring PVT1 is one of the most frequently amplified regions in breast cancers (Curtis et al., 2012). Indeed, PVT1 lncRNA has an oncogenic function itself by stabilizing MYC protein (Tseng et al., 2014). On the other hand, cancer genomes show recurrent structural rearrangements of PVT1 locus that disrupt PVT1 transcription, suggesting unknown regulatory functions of the PVT1 locus.
In mammalian cells, each chromosome is hierarchically organized into hundreds of megabase-sized topologically associated domains (TADs) (Dixon et al., 2012). Cell type-specific enhancer-promoter contacts take place within the TAD scaffold, leading to regulated gene expression (Pombo and Dillon, 2015). Disruption of DNA boundary elements that separate neighborhoods of gene regulation has recently been recognized as an important cause of human diseases, including developmental malformations and cancer (Flavahan et al., 2016; Lupianez et al., 2015). Deletions, duplications, insertions, inversions, and translocations of genomic DNA, collectively termed structural variations, can alter the 3D chromatin conformation of the genome and cause pathogenic rewiring of enhancer-promoter contacts. PVT1 locus harbors several intragenic enhancers that regulate transcription at target promoters by chromosome looping (Fulco et al., 2016; Zhang et al., 2016). The role of PVT1 DNA regulatory elements in cancer is poorly understood at present.
Engineered CRISPR with transcription factor domains, called CRISPR-interference (CRISPRi) or CRISPR-activation (CRISPRa), allows control of gene expression at targeted promoters (Gilbert et al., 2013). Functional screens employing engineered transcription factors targeting random sequences in the genome were pioneered 15 years ago (Blancafort et al., 2003; Park et al., 2003), but expanding the technology to genome-wide screens for defined promoters was not feasible until recently. Recent development of CRISPRi screening technologies has enabled systematic functional studies of regulatory DNA elements and lncRNA genes in the human genome (Gilbert et al., 2014; Liu et al., 2017). We recently found that CRISPRi silencing of PVT1 unexpectedly enhanced cell proliferation in several cell types, including two breast cancer cell lines, glioblastoma cells, and induced pluripotent stem cells (Liu et al., 2017).
Here we show that a single DNA element in the PVT1 gene--the PVT1 promoter--is a tumor suppressor DNA element that limits MYC oncogene expression. We further show that promoter competition in cis endows PVT1 promoter with the function of a DNA boundary element, blocking MYC oncogene from accessing cell-type specific enhancers.
Results
CRISPR interference of PVT1 enhances proliferation and cell competition
We recently established genome-scale CRISPRi to interrogate the function of lncRNA loci (Liu et al., 2017). We used single guide CRISPR RNAs (sgRNA) to direct catalytically dead Cas9 (dCas9) protein fused to KRAB repressor domain to the transcriptional start sites of lncRNA loci; each sgRNA can silence lncRNA expression in a targeted manner. The PVT1 locus encodes tens of annotated transcripts with at least six distinct transcriptional start sites (TSS1-6 from 5′ to 3′) (Figures 1A and S1A). The most 5′ TSS (TSS1) is located 53 kilobases (kb) downstream of the MYC gene, followed closely by TSS2 (+1.4 kb from TSS1). The other four TSS are scattered at ∼100 kb intervals throughout the >300 kb locus, with TSS3 (+ 96.3 kb), TSS4 (+194.2 kb), TSS5 (+194.7kb), and TSS6 (+215.2 kb). Although PVT1 has been reported as an oncogenic lncRNA, we found that CRISPRi of the PVT1 locus surprisingly enhanced cell proliferation in multiple human cancer cell types (Liu et al., 2017) (Figure S1B). We note that it is highly unusual to observe pro-growth effect in cancer cell lines, and the pro-growth effect of PVT1 sgRNAs was comparable to the largest effect on growth enhancement observed for silencing any protein coding gene (Gilbert et al., 2014). Among the 60 sgRNAs targeting all 6 TSS, only CRISPRi of the 5′ TSS1 or TSS2 of PVT1 increased cancer cell proliferation (Figure S1C). CRISPRi of TSS1 was more potent than TSS2, similar results observed in both MDA-MB-231 and MCF-7 breast cancer cell lines (Figure 1B). Eight of ten different sgRNAs targeting TSS1 enhanced cell proliferation, indicating a robust and reproducible effect (Figures S1C and S1D). Therefore, we focus on TSS1 and hereafter refer to its flanking sequence as “PVT1 promoter”.
Figure 1. CRISPRi-PVT1 enhanced cancer cell proliferation.
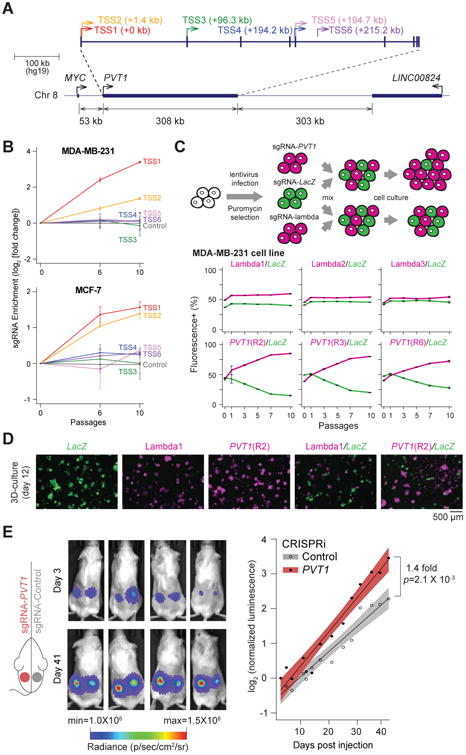
A, Schematic representing the PVT1 locus and six TSS of PVT1. B, Enrichment of sgRNAs targeting PVT1 in two breast cancer cell lines at indicated passages. Error bars indicate mean ± SD for top 3 sgRNAs targeting each TSS. C, Cell growth competition assay for CRISPRi-PVT1 in MDA-MB-231 cell line. Schematic representation of the assay (upper). Plots showing percentage of mCherry+ or GFP+ cells at indicated passages (bottom). Error bars indicate mean ± SD (n=3, technical replicates). D, Fluorescence microscopy image of cell growth competition assay by 3D cell culture of MDA-MB-231 cell line. For C and D, target genes of sgRNA are shown in the same color as of the fluorescent marker above each panel. E, Tumor growth of subcutaneous xenografted MDA-MB-231 with CRISPRi-Control or CRISPRi-PVT1 (R2). Luminescence images at day 3 or day 41 post injection(left). Fitted growth curve of injected tumor cells (right). p-value was calculated by paired Mann-Whitney U-test (n=12 for each). Shaded region indicates 95% confidence intervals.
A critical element of tumor evolution is selection, wherein selectively advantageous mutations confers a fitness benefit to cells, resulting in their competitive outgrowth. In a heterogeneous population of cancer cells, individual clones grow at the expense of each other until one clone reaches dominance (Miller et al., 1988). We designed a cell competition assay to investigate the effect of noncoding genes in a heterogeneous population (Figure 1C). Notably, cells with CRISPRi-PVT1 (mCherry+) outcompeted CRISPRi-LacZ control (GFP+), drastically expanded up to 75% of the total cell population within 3 passages and reached up to 85% within 10 passages. Control CRISPRi did not show any difference in growth rate with or without CRISPRi-PVT1 in the two breast cancer cell lines (Figures 1C and S2). In 3D cell culture, CRISPRi-PVT1 cells generated both larger colonies and many more satellite colonies with stellate morphology than control cells in the same well (Figure 1D), suggestive of an invasive phenotype (Kenny et al., 2007). These results suggest cells with CRISPRi-PVT1 have the potential to become dominant in heterogeneous tumor population. Moreover, CRISPRi-mediated silencing of PVT1 promoter enhanced breast tumor growth in subcutaneous xenografts in vivo, conferring a ∼40% increase in tumor growth rate compared to control xenografts harboring a non-targeting sgRNA (p=2.1 × 10-3, paired two-tailed Mann-Whitney U-test; Figures 1E and S1E). Because silencing PVT1 promoter enhances cancer cell proliferation, these results demonstrate that PVT1 promoter has an unexpected tumor suppressor function.
CRISPRi-PVT1 increases MYC expression
To investigate how silencing of PVT1 promoter enhances cell proliferation, we conducted gene expression analysis via RNA-seq. CRISPRi-PVT1 by two independent sgRNAs each effectively reduced PVT1 lncRNA level as expected. Interestingly, both sgRNAs yielded MYC mRNA as the most prominently increased RNA in the entire transcriptome (Figure 2A) without induction of differential MYC promoter usage or isoform switch (Figures S3A and S3B). MYC is a well-known oncogene requiring precise control; small quantitative gains in MYC expression can promote dramatic cell competition and proliferation (Kress et al., 2015). For example, cells with 3 copies of MYC gene grew at the expense of cells with 2 copies of MYC in the developing embryo (Claveria et al., 2013). MYC overexpression is implicated in over 50% of human cancers, and the MYC gene is the adjacent chromosomal neighbor of PVT1 promoter, making MYC a plausible effector gene. We observed that CRISPRi-PVT1 also increased MYC protein level by two to three-fold, which suggests MYC protein could contribute to the pro-growth phenotype (Figure 2B).
Figure 2. Pro-growth phenotype was induced by increased MYC expression in MDA-MB-231 cell line.

A, RNA-seq results comparing CRISPRi-Control and CRISPRi-PVT1 cell line (n=2, biological replicates). B, Western blot analysis for MYC protein changed by CRISPRi-PVT1 (top) and relative quantification of MYC protein (bottom). Error bars indicate mean ± SD. (n=3, biological replicates). C, Comparison of MYC and PVT1 RNA level between CRISPRi targeting upstream and downstream of PVT1-TSS1. p-values were calculated by unpaired Mann-Whitney U-test. D and E, qRT-PCR for relative RNA levels of MYC and PVT1 (D) or relative cell counts at day 4 post transfection (E) with siRNA targeting MYC with CRISPRi-Control or CRISPRi-PVT1 (R2). Error bars indicate mean ± SEM (n=3, biological replicates). F, Correlation between relative cell counts and MYC (left) or PVT1 (right) RNA level described in Figure 2D and 2E. Spearman's coefficient (R) and p-value are shown. *p<0.05, using unpaired t-test compared with control (dark grey).
Next, we systematically compared CRISPRi of 19 sgRNAs tiling across the PVT1 promoter (Figures S3C and S3D, and Table S1), and investigated each sgRNA's impact on steady state PVT1 lncRNA and MYC mRNA levels. CRISPRi with sgRNAs targeting downstream of PVT1 TSS1 showed coordinately reduced PVT1 and increased MYC transcript levels while sgRNAs targeting upstream of TSS1 did not alter expression of either PVT1 or MYC (Figure 2C). This result suggests that silencing of PVT1 transcription is linked with increased MYC expression; the lack of effect when targeting CRISPRi upstream of TSS1 also argues against potential divergent antisense transcripts from PVT1 promoter affecting MYC. The ability of CRISPRi-PVT1 to confer a competitive growth advantage was highly correlated with each sgRNA's potency to increased MYC mRNA level (R=0.752, Spearman's coefficient, p=1.3 × 10-4), more so than the level of PVT1 lncRNA silencing (R=0.658 with p=1.5 × 10-3) (Figure S3E). A few CRISPRi results deviated from the overall inverse trend between PVT1 and MYC expression; thus elements beyond the core promoters may also be involved in this phenomenon. Furthermore, siRNA-mediated knockdown of MYC mRNA reverted the proliferative advantage conferred by CRISPRi-PVT1 (Figures 2D and 2E), indicating that cell growth in PVT1-silenced cells requires increased MYC expression, similar to control cells (Figure 2F). Altogether, these results indicate MYC is a prominent effector of the pro-growth phenotype induced by CRISPRi-PVT1.
Uncoupling PVT1 promoter vs PVT1 lncRNA in MYC expression and proliferation
To understand how CRISPRi-PVT1 induces MYC transcription, we investigated the role of PVT1 lncRNA in MYC expression and the pro-growth phenotype. Three independent strategies showed that PVT1 lncRNA is dispensable for the tumor suppressor function of PVT1 promoter. First, we positioned dCas9 to create a transcription block and truncated the PVT1 transcript without heterochromatin formation (Gilbert et al., 2013). dCas9-mediated interference reduced full-length PVT1 lncRNA to the same level as dCas9-KRAB, but did not increase MYC expression or cell proliferation (Figure 3A and S3D). Second, we transfected antisense oligonucleotide (ASO), which induce RNase H-mediated degradation of nuclear lncRNAs. ASO targeting PVT1 reduced PVT1 lncRNA levels, but failed to increase MYC expression or cell growth (Figure 3B). In fact, cell viability was reduced by ASOs targeting PVT1 lncRNA, consistent with a previous study (Tseng et al., 2014). Third, siRNA-mediated knockdown through the RNA interference pathway again reduced PVT1 lncRNA levels without affecting MYC expression (Figure 3C). Altogether, these results suggest an lncRNA-independent mechanism by which PVT1 promoter impacts MYC expression and cell growth.
Figure 3. PVT1 lncRNA independent mechanism for MYC induction in MDA-MB-231 cell line.

A, qRT-PCR for relative RNA level of MYC or PVT1 (left) or relative cell growth at day 4 post plating (right) for sgRNA-Control or sgRNA-PVT1 with dCas9-KRAB or dCas9. Relative cell growth was measured by MTT assay. B, qRT-PCR for relative RNA levels of MYC and PVT1 (left) or relative cell count at day 4 post transfection (right) with ASO targeting PVT1. C, qRT-PCR for relative RNA levels of MYC and PVT1 with siRNA targeting PVT1. Schematic representation for each experiment is shown above each plot. Error bars indicate mean ± SEM (n=3, biological replicates). *p<0.05, using unpaired t-test compared with control (dark grey).
Promoter competition underlies antagonism between PVT1 and MYC
We identified a chromatin-based mechanism for PVT1 regulation of MYC transcription. The MYC gene is controlled by a multitude of long-range cis-regulatory elements (Sotelo et al., 2010). Chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) confirmed that dCas9-KRAB protein bound specifically to PVT1 promoter, and induced histone H3 lysine 9 trimethylation (H3K9me3), a signature of KRAB-mediated gene repression (Liu et al., 2017), at PVT1 promoter but not at MYC (Figure S4). We used HiChIP, a method for protein-directed chromosome conformation, targeting the enhancer-associated mark histone H3 lysine 27 acetylation (H3K27ac) to map the enhancer connectome in control and CRISPRi-PVT1 cells (Mumbach et al., 2016; Mumbach et al., 2017). H3K27ac HiChIP revealed that the PVT1 promoter and MYC promoter, located 58 kb apart on the linear genome, exhibit high contact frequency in 3D within a small but dense topologically associated domain (Figures 4A and S5A). Notably, four intragenic enhancers within the PVT1 locus, herein named 822E, 866E, 912E, and 919E based on their genomic coordinates, normally contacted with PVT1 promoter preferentially over the MYC locus in control cells. CRISPRi-PVT1 decreased contacts of these four enhancers with the PVT1 promoter but increased contacts with the MYC promoter and a MYC 3′-enhancer (Fulco et al., 2016; Figures 4A and S5B). Similar results demonstrating competitive interaction of PVT1 intragenic enhancer between PVT1 promoter and MYC 3′-enahancers were obtained in MCF-7 cells as measured by UMI-4C, an orthogonal method of targeted chromosome conformation capture with unique molecular identifiers (Schwartzman et al., 2016) (Figures 4B and S5C). These results suggest that PVT1 promoter acts as a boundary element in breast cancer cells: PVT1 promoter interacts with PVT1 intragenic enhancers and blocks the latter from inducing MYC expression.
Figure 4. PVT1 promoter suppresses MYC transcription by competing for PVT1 intragenic enhancers.

A, PVT1 promoter functions as a boundary element in MDA-MB-231 cell line. Top: Heat-map representing chromatin conformation around PVT1 locus measured by H3K27ac HiChIP (n=2, biological replicates). ATAC-seq peaks, H3K27ac ChIP-seq peaks and ChromHMM diagram are shown to indicate enhancer elements. Middle: Virtual 4C plots showing H3K27ac HiChIP contact frequency at 1 kb resolution anchored at each indicated genomic position (solid black lines) with CRISPRi-Control or CRISPRi-PVT1. Bottom: Schematic diagram showing changes in chromosome interaction induced by CRISRPi-PVT1. Shaded region indicates mean ± SEM (n=2, biological replicates). p-value of each interaction between MYC 3′-enhancer and PVT1 intragenic enhancer was calculated by Fisher's exact test. B, Boxplots showing chromosome contact frequency at 5 kb resolution measured by UMI-4C in MCF-7 cell line with CRISPRi-Control or CRISPRi-PVT1 (R2). p-value was calculated by un-paired Mann-Whitney U-test (n=8). C, qRT-PCR for relative RNA level of MYC and PVT1 with dual CRISPRi targeting PVT1 promoter or intragenic enhancers in MDA-MB-231 cell line. The difference between single CRISPRi-PVT1 and dual CRISPRi-PVT1 with CRISPRi-enhancer (822E, 866E or 919E) is not significant (p>0.05). D to F, ChIP-qPCR representing relative PVT1 or MYC promoter DNA bound to BRD4 (D), Pol II-S5P (E), Pol II-S2P (F) in MDA-MB-231 cell line with CRISPRi-control or CRISPRi-PVT1. G, qRT-PCR for the relative level of 4sU-labeled nascent transcripts of PVT1 or MYC in MDA-MB-231 cell line with CRISPRi-control or CRISPRi-PVT1. Error bars indicate mean ± SEM (n=3, biological replicates). *p<0.05, using unpaired t-test compared with control (dark grey). n.s., not significant (p>0.05); n.d., not detected. See also STAR METHOD for details.
The PVT1 intragenic enhancers proved to be important for PVT1 promoter function. First, the four intragenic PVT1 DNA elements each demonstrate bona fide enhancer activity when cloned upstream of a minimal promoter in a luciferase reporter assay (Figure S5D). Second, CRISPRi of these enhancers in MDA-MB-231 cells lowered MYC and PVT1 mRNA levels, indicating that the endogenous PVT1 enhancers regulate both genes (Figure 4C). Third and importantly, we developed a dual CRISPRi strategy to assess the function of the PVT1 enhancers concomitant with PVT1 promoter silencing. Dual CRISPRi of PVT1 enhancers and PVT1 promoter showed that silencing of 3 of 4 PVT1 enhancers abrogated the increase in MYC mRNA level after PVT1 promoter silencing (Figure 4C). Enhancer 822E is particularly intriguing because it is required for MYC activation only upon PVT1 promoter silencing, but not in control cells. Together, this functional epistasis supports the idea that increased interaction between MYC and PVT1 enhancers is the basis of the induction of MYC transcription upon silencing of PVT1 promoter.
The model of promoter competition raises the hypothesis that PVT1 promoter specifically inhibits MYC promoter firing. In metazoan genomes, RNA polymerase II (Pol II) initiates transcription for 25-60 nucleotides at most genes, and then pauses before release for productive elongation (Scruggs and Adelman, 2015). The bromodomain protein BRD4 is a key regulator of pause release, both by controlling the phosphorylation state of Pol II C-terminal domain and interaction with enhancers (Devaiah et al., 2012). MYC transcription is known to be highly sensitive to the bromodomain inhibitor JQ1 (Filippakopoulos et al., 2010), and we found that JQ1 treatment similarly reduced PVT1 transcript level, suggesting that both MYC and PVT1 are regulated by bromodomain proteins (Figure S5E). Moreover, we found that CRISPRi-PVT1 decreased BRD4 occupancy at PVT1 promoter, but significantly increased BRD4 occupancy at MYC promoter as shown by ChIP-qPCR (Figure 4D). CRISPRi-PVT1 decreased Pol II initiation at the PVT1 locus but did not alter pol II initiation at MYC, as shown by serine 5-phosphorylated polymerase (Pol II-S5P) occupancy (Figure 4E). In contrast, CRISPRi-PVT1 decreased elongating serine 2-phoshorylated RNA polymerase (pol II-S2P) at PVT1 but increased polymerase elongation for MYC (Figure 4F). Moreover, interrogation of nascent transcription by 4-thiouridine (4sU) labeling (Cleary et al., 2005) showed that PVT1 silencing decreased nascent PVT1 transcript levels but nearly doubled MYC mRNA synthesis (Figure 4G). These results suggest that PVT1 promoter is a brake for MYC gene transcriptional elongation.
The model of PVT1-MYC promoter competition also potentially explains the cell-type specificity of PVT1 function. CRISPRi-PVT1 did not induce MYC transcription in some cell types, such HCT116 colon cancer cells or HeLa cervical carcinoma cells. (Figure S5F). We found that in these cell types, MYC loops to the CCAT1 enhancer instead of PVT1 (Figures S5G to S5J); the colon cancer result is consistent with a prior study (Xiang et al., 2014). These observations provide additional evidence that the chromosome conformation landscape is an important determinant of the dynamic interplay between PVT1 and MYC.
PVT1 promoter reversibly regulates MYC transcription
Conversely, enforced transcription from PVT1 promoter inhibited MYC transcriptional elongation. We engineered MDA-MB-231 cells to stably express dCas9 fused to potent transcription activation domains (dCas9-VP64) (Konermann et al., 2015); expression of specific sgRNAs allowed us to activate transcription of endogenous loci (Figure 5A). CRISPR activation (CRISPRa) of PVT1 increased PVT1 lncRNA but significantly decreased MYC mRNA level (Figure 5B). Moreover, CRISPRa of PVT1 reduced BRD4 occupancy at MYC promoter, reduced Pol II elongation at MYC, and reduced MYC nascent transcript (Figures 5C to 5F). These effects were highly specific as Pol II initiation at MYC was not affected. Moreover, CRISPRi-MYC promoter increased PVT1 transcription (Figure 5G). These results further distinguish the RNA products as separable from the DNA element competition of the two promoters. Altogether, these results support a model of bi-directional regulation of PVT1 and MYC genes via promoter competition (Figure 5H).
Figure 5. PVT1 reversibly regulates MYC transcription in MDA-MB-231 cell line.

A, Schematic representing CRISPRa at PVT1 promoter. B, qRT-PCR for PVT1 or MYC with CRISPRa-control or CRISPRa-PVT1. C to E, ChIP-qPCR for PVT1 or MYC TSS for BRD4 (C), Pol II-S5P (D) or Pol II-S2P (E) with CRISPRa-Control or CRISPRa-PVT1. F, qRT-PCR for relative level of 4sU-labeled nascent transcripts with CRISPRa-Control or CRISPRa-PVT1. G, qRT-PCR for PVT1 or MYC with CRISPRi-Control or CRISPRi-MYC. H, Schematic representing the model of promoter-enhancer competition between PVT1 and MYC. Error bars indicate mean ± SEM (n=3, biological replicates). *p<0.05, using un-paired t-test compared with control (grey). n.s., not significant (p>0.05); n.d., not detected.
Monoallelic regulation of PVT1 promoter controls MYC expression in cis
A strong prediction arising from the promoter competition model is that PVT1 promoter should regulate MYC promoter on the same chromosome (i.e. the allele in cis). PVT1 lncRNA is known to diffuse throughout the nucleus and cytoplasm. The previously published models of PVT1 lncRNA exerting oncogenic function by interaction with RNA binding proteins or other regulatory RNAs throughout the cell predicts effect on both homologous chromosomes (i.e. action in trans) (Tseng et al., 2014; Wang et al., 2014a; Xu et al., 2017b). We and others recently described the recognition of wide-spread random monoallelic chromatin accessibility where one of two alleles in a diploid cell is active in a heritable fashion (Xu et al., 2017a). Using cells derived from an F1 hybrid mouse of two highly divergent parental genome, 129S1 (here referred to as 129) and Castaneous (Cast), we isolated individual cell clones and measured chromatin accessibility, histone modifications, and RNA transcripts in an allele-specific fashion (Figure 6A). In the mouse embryonic stem cells (mESC), Pvt1 showed bi-allelic transcription and no difference in chromatin status between two alleles. After differentiation into the neural precursor cells (mNPC), however, some clones showed monoallelic transcription of Pvt1 (Figure 6B). A histone mark for active promoters, histone H3 lysine 4 trimethylation (H3K4me3), was observed only on the transcribed allele whereas heterochromatic H3K9me3 was only present on the silent allele at Pvt1 promoter. Importantly, monoallelic Pvt1 promoter activation led to specific reduction of Myc chromatin accessibility and mRNA level from the same chromosome, but not of the homologous chromosome in trans in the same nucleus (Figure 6B).
Figure 6. PVT1 promoter regulates MYC expression in cis and allele-specific manner.

A, Schematic representing experiments for allele-specific regulation. B, ATAC-seq, RNA-seq, H3K9me3 or H3K4me3 signals around PVT1-MYC locus measured in mESC or mNPC derived from a hybrid mouse. Reads with SNPs corresponding to the 129 or Cast allele are shown as pink or blue, respectively. C, Box plot showing MYC mRNA level according to the number of active PVT1 alleles. p-value was calculated by unpaired Mann-Whitney U-test. D, Correlation between the d-score for accessibility of the PVT1 locus from ATAC-seq and MYC transcription level from RNA-seq. For C and D, n=15 from 11 clones and 4 additional technical replicates. E, Correlation between d-score of RNA level measured by targeted RNA-seq (n=35). For D and E, Spearman's coefficient (R) and p-value are shown.
We found mono- or bi- allelic transcription of Pvt1 is associated with reduction of Myc transcription in cis across numerous independent mNPC clones (Figure 6C). Based on SNPs from each allele, we calculated the allele bias of histone marks, chromatin accessibility or RNA transcription from 11 clones, represented by d-score. A positive or negative d-score indicates allelic bias towards 129 or Cast allele, respectively (Figure 6A). As expected, the d-score of chromatin accessibility at Pvt1 promoter is highly correlated with the d-score of Pvt1 lncRNA level (R=0.990, Spearman's coefficient, p=9.0 × 10-11) (Figure S6A). Importantly, the d-score of Pvt1 promoter chromatin accessibility is inversely correlated with the d-score of Myc mRNA level (R=-0.724 with p=2.3 × 10-3) (Figure 6D). Indeed, the d-scores of Pvt1 and Myc transcript level are also inversely correlated (R=-0.733 with p=4.4 × 10-3) (Figure S6B). We expanded this analysis with additional independent clones via targeted reverse transcription followed by amplicon sequencing (Figure S6C). From 35 mNPC clones, the d-scores of Pvt1 and Myc transcript levels are inversely correlated (R<-0.427 with p<1.0 × 10-2) whereas the control gene (Tbp) showed no correlation with Pvt1 (Figures 6E and S6D). Altogether, these results indicate that PVT1 promoter directly represses MYC transcription in cis, consistent with a DNA-element based mechanism.
Recurrent mutations encompassing PVT1 promoter in cancer patients
Finally, somatic mutations in human cancers support a tumor suppressive role of the PVT1 promoter. The DNA sequence of PVT1 promoter, especially between TSS1 and TSS2, exhibits a higher degree of evolutionary conservation than other portions of the PVT1 lncRNA, and noncoding variations in this region are predicted to be especially damaging (Figures 7A and S7A). We analyzed published cancer genomes from a variety of human tumors, and annotated the frequency of somatic mutations in the PVT1 promoter versus mutations in other annotated lncRNA promoters on human chromosome 8. PVT1 promoter exhibited recurrent point mutations and indels in human breast cancer and malignant lymphoma, but not other cancer types (Figure S7B). The 8q24 region spanning MYC and PVT1 was recently reported to be a hotspot for somatic structural variants (SV) in breast cancer (Nik-Zainal et al., 2016). Further analysis of these data indicates that the PVT1 promoter region is significantly enriched for SVs as compared to other lncRNA promoters (Figure 7B, p-value=1 × 10-5, FDR<0.03). Human breast tumors exhibited diverse SVs, including deletions, inversions, and duplications that overlapped the PVT1 promoter (Figure 7C), which are expected to inactivate the tumor suppressive function of PVT1 promoter. We also identified rare but illustrative intra-chromosomal inversion in an ER- HER2+ human breast tumor that breaks precisely in intron 1 of PVT1, separating PVT1 promoter from intragenic enhancers and creating a PVT1 fusion transcript with genes located more than 40 megabases away (Figures 7D and S7C). The chromosomal inversion and PVT1 fusion transcripts are supported by both DNA and RNA sequencing, and is present in the majority of tumor cells, suggesting that this event occurred early or was strongly selected. PVT1 translocations with at least 15 different fusion partners have been reported in diverse types of human cancers; these translocations invariably occur in intron 1 of PVT1 gene, separating PVT1 promoter from its genomic context (Chinen et al., 2014; Kim et al., 2014a; Nagoshi et al., 2012; Northcott et al., 2012; Pleasance et al., 2010; Shtivelman et al., 1989) (Table S2). These diverse translocations share the property of altering the chromatin and TAD environment of PVT1 promoter, suggesting inactivation of repressive function of PVT1 promoter as a unifying mechanism.
Figure 7. PVT1 promoter functions as a tumor suppressor.

A, Scatter plot representing C-scores for variants at PVT1-TSS ± 5 kb region from CADD analysis (upper) and conservation plot (bottom). Grey line indicates C-score=10, top 10%. TSS1 or TSS2 of PVT1 is shown as a red or yellow line, respectively. B, Number of SVs including duplications, deletions and inversions overlapping with lncRNA promoter in the BRCA-EU cohort (y-axis). Basal SV frequency was measured in adjacent windows within 5 megabase (x-axis). See also STAR METHOD. C, SVs encompassing the PVT1 promoter in breast tumor patients. The patient ID is shown on the left. D, Genomic structure showing a recurrent intra-chromosomal inversion within PVT1 intron 1 in breast tumor patients. Ideogram of Chr. 8, heat-map of chromatin conformation within indicated region (top), inversion in genomic context (middle) and fusion transcription found in this tumor (bottom) were shown. E, Footprinting for regulatory element of PVT1 promoter. Fold changes of each base or alleles after 10 passages (for p<0.01) were listed. F, Relative MYC mRNA level compared between MDA-MB-231 cells harboring unmodified or mutant allele of PVT1 promoter. p-value was calculated by using unpaired Mann-Whitney U-test (n=6 for each).
To demonstrate that mutations in PVT1 promoter suffice to promote cell proliferation, we used CRISPR-mediated genome editing to mutate the PVT1 promoter. After genome editing, we passaged the edited and unedited cells as a pool in a competitive growth assay. Indeed, alleles with small deletions at PVT1 promoter were significantly enriched up to 3-fold after 10 passages, indicating that PVT1 mutations confer a distinct growth advantage (p<0.01 for each allele, Figure 7E). Mutant alleles of PVT1 promoter were further enriched longitudinally over 30 passages, suggesting their effects persist and dominate over time (Figure S7D). Further, we generated clonal MDA-MD-231 cells harboring small deletions at PVT1-TSS. 5 out of 6 clones showed increased MYC mRNA level up to 1.5-fold over cells harboring wild type PVT1 promoter (Figures 7F and S7E). These experimental results corroborate the findings from human cancer genomes, and suggest that the PVT1 promoter is a candidate tumor suppressor DNA element.
Discussion
Our studies may shed light on a long-standing puzzle in cancer genetics. PVT1 was the first lncRNA gene associated with cancer, and likely also the first lncRNA associated with human disease. Frequent translocations, retroviral insertions, and interstitial deletions that disrupt the PVT1 transcription unit have been known for more than 25 years (Shtivelman et al., 1989). In 1990, J. Michael Bishop and colleagues hypothesized that PVT1 translocations activate MYC transcription (Shtivelman and Bishop, 1990), but the potential mechanism has remained obscure. Our results suggest that the PVT1 locus harbors a series of DNA regulatory elements for MYC; activity of the PVT1 promoter gates the ability of MYC to access these PVT1 intragenic enhancers. Hence, the PVT1 promoter is a tumor suppressor DNA element. Silencing PVT1 promoter increases MYC transcription and cell proliferation in a manner independent of PVT1 lncRNA. Developmental monoallelic regulation of PVT1 may also regulate somatic stem cell competition, a possibility that will be addressed elsewhere. The topography of the MYC-PVT1 interval may underlie the tissue specificity of PVT1 promoter action. PVT1 promoter is located 3′ of MYC and regulates downstream enhancers of MYC, such as those active in breast cancer cells. In contrast in HeLa and HCT116 cells where PVT1 promoter does not regulate MYC, upstream MYC enhancers are dominant, such as the multicopy insertion of human papillomavirus sequence 500 kb upstream of MYC in HeLa (Adey et al., 2013) or colon cancer –specific enhancer 300 kb upstream of MYC in HCT116 cells (Sur et al., 2012).
A prior study using chromosome engineering in mice suggested that PVT1 lncRNA acts as an oncogene through post-translational regulation of MYC protein (Tseng et al., 2014). In fact, our model is fully compatible with and may better explain the prior results. First, Tseng et al. engineered different tandem duplications of the MYC-PVT1 interval, and found that the MYC-PVT1 genomic DNA need to be co-amplified in cis to promote breast tumor growth in vivo. Amplification of either MYC or PVT1 alone had no detectable tumorigenic activity. The cis requirement is readily explained by regulated enhancer-promoter contact in the same TAD, but difficult to explain for trans-acting lncRNA and protein products. Second, to evaluate the requirement of PVT1 lncRNA, Tseng et al. generated a ∼300 kb deletion of PVT1 locus that also removed all four PVT1 intragenic enhancers. This PVT1 deletion lowered MYC level and abrogated tumorigenic activity in vivo, exactly as our model would predict. The recognition of PVT1 promoter as a MYC transcriptional repressor suggests that PVT1 maintains rheostatic control of MYC expression. The PVT1 promoter, as a DNA element, limits MYC transcription while PVT1 RNA product sustains MYC protein level. These two strategies of control are expected to constrain MYC protein level within a narrow range. The function of PVT1 promoter is likely dominant over that of PVT1 lncRNA as evidenced by our experimental data and the pattern of human cancer mutations. PVT1 joins other lncRNA loci as dual DNA and RNA regulatory elements with opposing function, such as Haunt and linc-p21 (Groff et al., 2016; Yin et al., 2015). Although we have shown that PVT1 promoter inactivation enhances cell proliferation in established cancer cells, it remains to be seen whether PVT1 promoter mutation alone can cause cancer in vivo.
Our study suggests lncRNA gene promoter as a new class of DNA boundary elements that ensure proper allocation of enhancer-promoter interactions. We posit that lncRNA promoters (and indeed any gene promoter) may serve this function due to (i) their participation in long-range 3D chromatin interactions; (ii) promoter-promoter competition to engage the same set of enhancers. MYC and PVT1 transcription units do not overlap and are separated by 55 kb; hence 3D chromosome conformation is essential for promoter competition to take place. Promoter competition differs from currently known mechanisms of lncRNA-based regulation in cis, where transcription through the target DNA element mediates the effect (Anderson et al., 2016; Csorba et al., 2014). We show that when promoter competition is abrogated by silencing or mutation of the PVT1 promoter, pathogenic rewiring of enhancer-promoter contact occurs between intragenic PVT1 enhancers and the MYC promoter, resulting increased MYC expression. LncRNA promoters are more evolutionarily conserved than their gene bodies (Derrien et al., 2012), and hence this mechanism could be general. Transcriptome analyses of targeted mutations (Engreitz et al., 2016) or CRISPRi (Liu et al., 2017) of lncRNA promoters suggest approximately 10% of lncRNA promoters may regulate nearby genes by competition. 4 out of 36 lncRNA promoters silenced by CRISPRi (Liu et al., 2017) consequently activated genes within a 1 megabase window, which possibly under promoter competition or other 3D conformation-mediated regulation (Figure S7F). The advent of powerful technologies to edit the genome, epigenome, and map enhancer connectomes now permits systematic testing of these concepts. We note that even from a DNA-centric viewpoint, the lncRNA transcript is hardly an irrelevant molecule, but is the evidence of successful competition of the lncRNA promoter at the expense of target gene promoters. With the precise mapping of tens of thousands of precise lncRNA promoters in the human genome (Hon et al., 2017), future investigations using strategies reported herein will likely illuminate novel function of lncRNA loci in cancer and other disease states.
Star Method
KEY RESOURCES TABLE is provided as a separated file.
Key Resources Table.
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-H3 acetyl-K27 | Abcam | Cat#ab4729; RRID: AB_2118291 |
| Rabbit polyclonal anti-H3 trimethyl-K9 | Abcam | Cat#ab8898; RRID: AB_306848 |
| Rabbit polyclonal anti-H3 trimethyl-K4 | Active Motif | Cat#39159; RRID: AB_2615077 |
| Rabbit polyclonal anti-BRD4 | Bethyl Laboratories | Cat#A301-985A; RRID: AB_1576498 |
| Mouse monoclonal anti-HA | Cell Signaling | Cat#2367; RRID: AB_10691311 |
| Rabbit polyclonal anti-Pol II phospho-Ser2 | Abcam | Cat#ab5095; RRID: AB_304749 |
| Rabbit polyclonal anti-Pol II phospho-Ser5 | Abcam | Cat#ab5131; RRID: AB_449369 |
| Rabbit polyclonal IgG | Abcam | Cat#ab171870; RRID: AB_2687657 |
| Mouse polyclonal IgG | EMD Millipore | Cat#12-371; RRID: AB_145840 |
| Mouse monoclonal anti-GAPDH (7B) | Santa Cruz | Cat#sc69778; RRID: AB_1124759 |
| Rabbit monoclonal anti-MYC (D84C12) | Cell Signaling | Cat#5605S; RRID: AB_1903938 |
| IRDye 800CW Goat anti-mouse IgG | LI-CORBioScience | Cat#926-32211; RRID: AB_621843 |
| IRdye 680RD Goat anti-rabbitIgG | LI-CORBioScience | Cat#926-68070; RRID: AB_10956588 |
| Bacterial and Virus Strains | ||
| One Shot Stbl3 Chemically Competent E.coli | Thermo Fisher | Cat#C737303 |
| NEB Turbo Competent E.coli | NEB | Cat#C2984H |
| One Shot BL21(DE3) pLysS Chemically Competent E. coli | Thermo Fisher | Cat#C606003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Proteinase K, Molecular Biology Grade | NEB | Cat#P8107S |
| Quick Ligation Kit | NEB | Cat#M2200L |
| Klenow Fragment (3′-5′exo-) | NEB | Cat#M0212L |
| NEBNext End Repair Module | NEB | Cat#E6050L |
| Adenosine 5′-Triphosphate | NEB | Cat#P0756S |
| T4 DNA Ligase | NEB | Cat#M0202L |
| MboI restriction enzyme | NEB | Cat#R0147M |
| Alkaline Phosphatase, Calf Intestinal | NEB | Cat#M0290L |
| Hot Start Taq DNA Polymerase | NEB | Cat#M0495L |
| NEBNext® High-Fidelity 2X PCR Master Mix | NEB | Cat#M0541L |
| Deoxynucleotide (dNTP) Solution Set | NEB | Cat#N0446S |
| NEBuilder® HiFi DNA Assembly Master Mix | NEB | Cat#E2621L |
| Recombinant Human FGF-basic | PeproTech | Cat#100-18B |
| Epidermal Growth Factor | PeproTech | Cat#315-09 |
| MEGAshortscript T7 Transcription kit | Thermo Fisher | Cat#AM1354 |
| MEGAclearTransription Clean-Up kit | Thermo Fisher | Cat#AM1908 |
| Lipofectamine 3000 transfection Reagent | Thermo Fisher | Cat#L3000-015 |
| Lipofectamine 2000 transfection Reagent | Thermo Fisher | Cat#11668-019 |
| Hygromycin B | Thermo Fisher | Cat#10687010 |
| Puromycin dihydrochloride | Thermo Fisher | Cat#A11138-03 |
| Blasticidin S HCl | Thermo Fisher | Cat#A1113903 |
| Neurobasal Medium | Thermo Fisher | Cat#21103-049 |
| DMEM/f12 | Thermo Fisher | Cat#11320-082 |
| B-27 Supplemnet (50X), serum free | Thermo Fisher | Cat#17504044 |
| EGF Recombinant Human Protein | Thermo Fisher | Cat#PHG0311 |
| DynabeadsMyOne Streptavidin C1 | Thermo Fisher | Cat#65002 |
| DynabeadsProtein A for Immunoprecipitation | Thermo Fisher | Cat#10002D |
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher | Cat#10004D |
| 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher | Cat#28908 |
| TrypLE Express Enzyme (1X) | Thermo Fisher | Cat#12604039 |
| Pierce Detergent Compatible Bradford Assay Kit | Thermo Fisher | Cat#23246 |
| SuperScript III Reverse Transcriptase | Thermo Fisher | Cat#18080-044 |
| DMEM | Thermo Fisher | Cat#11995 |
| Penicillin-Streptomycin | Thermo Fisher | Cat#15140163 |
| McCoy's 5A | Thermo Fisher | Cat#16600 |
| Horse serum | Thermo Fisher | Cat#16050122 |
| Click-IT Biotin DIBO Alkyne | Thermo Fisher | Cat#C10412 |
| NorthernMax™ Formaldehyde Load Dye | Thermo Fisher | Cat#AM8550 |
| BrightStar™-Plus Positively Charged Nylon Membrane | Thermo Fisher | Cat#AM10104 |
| NorthernMax™ One-Hour Transfer Buffer | Thermo Fisher | Cat#AM8672 |
| ULTRAhyb™-Oligo Buffer | Thermo Fisher | Cat#AM8663 |
| NorthernMax™ Low Stringency Wash Buffer | Thermo Fisher | Cat#AM8673 |
| NorthernMax™ High Stringency Wash Buffer | Thermo Fisher | Cat#AM8674 |
| Insulin from bovine pancreas | Sigma Aldrich | Cat#I1882 |
| Cholera Toxin from Vibrio cholerae | Sigma Aldrich | Cat#C8052 |
| 4-Thiouridine | Sigma Aldrich | Cat#T4509 |
| (+)-JQ1 | Sigma Aldrich | Cat#SML1524 |
| Hydrocortisone | Sigma Aldrich | Cat#H0888 |
| Hexadimethrine bromide | Sigma Aldrich | Cat#107689 |
| Odyssey Blocking Buffer (PBS) | LI-COR BioScience | Cat#927-40003 |
| Streptavidin-IRDye 800CW | LI-COR BioScience | Cat#926-32230 |
| Corning Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix, Phenol Red-Free | Corning | Cat#356231 |
| Stratagene Brilliant II SYBR Green QRT-PCR Master Mix | Agilent | Cat#600825 |
| MTSEA biotin-XX | Biotium | Cat#90066 |
| SPRI select reagent kit | Beckman Coulter | Cat#B23318 |
| Agencourt AMPure XP | Beckman Coulter | Cat#A63881 |
| NDiff Neuro-2 Medium Supplement (200X) | EMD Millipore | Cat#SCM012 |
| Accutase | EMD Millipore | Cat#SCR005 |
| LightCycler 480 SYBR Green I Master mix | Roche | Cat#0407516001 |
| cOplete proteinase inhibitor cocktail | Roche | Cat#11697498001 |
| Biotin-16-dUTP | Roche | Cat#11093070910 |
| Lenti-X concentrator | Clontech | Cat#631232 |
| Expand Long Template PCR kit | Roche | Cat#11681834001 |
| DharmaFECT 4 Transfection Reagent | Dharmacon | Cat#T-2004 |
| Cell Line Nucleofector Kit V | Lonza | Cat#VVCA-1003 |
| TransIT LT1 Transfection Reagent | Mirus | Cat#MIR2300 |
| HyClone fetal bovine serum | GE Healthcare | Cat#11995 |
| Proteinase K | MacheryNagel | Cat#740506 |
| Critical Commercial Assays | ||
| TruSeq Stranded Total RNA LT - (with Ribo-Zero Human/Mouse/Rat) | Illumina | Cat#RS-122-2201 |
| Nextera DNA Sample Preparation Kit | Illumina | Cat#Fc-121-1030 |
| Cell proliferation kit (MTT) | Roche | Cat#11465007001 |
| Luciferase reporter assay system | Promega | Cat#E1500 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | NEB | Cat#E7645S |
| MycoAlert PLUS mycoplasma detection kit | Lonza | Cat#LT07-705 |
| Deposited Data | ||
| Raw and analyzed data of human cell line | this study | GEO: GSE97669 |
| Raw and analyzed data of mouse cell line | this study, Xu et al., 2017 | GEO: GSE84646, GSE97669 |
| ICGC (release 23) | N/A | https://dcc.icgc.org/releases |
| METABRIC | Curtis et al., 2012 | EGA:EGAD00001003342 |
| Experimental Models: Cell Lines | ||
| Human: MDA-MB-231 cell line | ATCC | HTB-26 |
| Human: MDA-MB-231 CRISPRi clones | Liu et al., 2017 | N/A |
| Human: MCF-7 cell line | ATCC | HTB-22 |
| Human: MCF-7 CRISPRi clones | Liu et al., 2017 | N/A |
| Human: HEK-293 cell line | ATCC | CRL-1573 |
| Human: 293T cell line | ATCC | CRL-3216 |
| Human: MCF10A cell line | ATCC | CRL-10317 |
| Human: SKBR3 cell line | ATCC | HTB-30 |
| Human: HCT116 cell line | ATCC | CCL-247 |
| Human: HeLa cell line | ATCC | CCL-2 |
| Mouse: neuronal progenitor cell line | Xu et al., 2017 | N/A |
| Mouse: neuronal progenitor cell clones | Xu et al., 2017 | N/A |
| Mouse: embryonic stem cell line | Xu et al., 2017 | N/A |
| Mouse: embryonic stem cell line clones | Xu et al., 2017 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ | Jackson laboratories | Cat#005557 |
| Oligonucleotides | ||
| sgRNA sequences, see Table S1 | this paper; Liu et al., 2017 | N/A |
| ASO sequences, see Table S1 | this paper | see Table S1 |
| siRNA sequences, see Table S1 | this paper; Tseng et al., 2014 | see Table S1 |
| qRT-PCR primers, see Table S1 | this paper; Tseng et al., 2014 | N/A |
| qPCR primers, see Table S1 | this paper | N/A |
| primers used for northern blotting | this paper | N/A |
| primers used for plasmid cloning | this paper | N/A |
| primers used in UMI-4C library preparation, see Table S1 | this paper | N/A |
| primers used in genotyping, see Table S1 | this paper | N/A |
| primers used in targeted RNA-seq, see Table S1 | this paper | N/A |
| Recombinant DNA | ||
| pHR-SFFV-dCas9-BFP | Gilbert et al., 2014 | Addgene#46910 |
| pHR-SFFV-dCas9-BFP-KRAB | Gilbert et al., 2014 | Addgene#46911 |
| pHR-SFFV-dCas9-BFP-KRAB-2A-Blast | this paper | N/A |
| lenti-mU6-sgRNA-Puro-mCherry | this paper | N/A |
| lenti-mU6-sgRNA-Puro-EGFP | this paper | N/A |
| lenti-dCas9-VP64-2A-Blast | Konermann et al., 2015 | Addgene#61425 |
| lenti-MS2-P65-HSF1-Hygro | Konermann et al., 2015 | Addgene#61426 |
| lenti-sgRNA2.0-Puro-mCherry | this paper | N/A |
| lenti-EF1a-luciferase-2A-Hygro | this paper | N/A |
| pET-Cas9 | this paper | N/A |
| pGL4.23[luc2/minP] | Promega | Cat#E8411 |
| lenti-bovine U6 promoter-sgRNA, pMJ114 | Adamson et al., 2016 | Addgene#85995 |
| lenti-human U6 promoter-sgRNA, pMJ117 | Adamson et al., 2016 | Addgene#85997 |
| lenti-mouse U6 promoter-sgRNA, pMJ179 | Adamson et al., 2016 | Addgene#85996 |
| Software and Algorithms | ||
| LivingImagesoftware (v4.4) | Perkin Elmer | N/A |
| Image Studio (v1.0.11) | LI-COA BioScience | N/A |
| Tophat2 (v2.1.1) | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat/index.shtml |
| DEseq2 (v1.16.1) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie2 (v2.2.8) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Picard (v.1.79) | N/A | http://broadinstitute.github.io/picard/ |
| MACS2 (v2.1.0) | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| Samtools (v1.3.1) | Li et al., 2009 | http://samtools.sourceforge.net |
| Bedtools (v2.17.0) | Quinlan and Hall, 2010 | http://bedtools.readthedocs.io/en/latest/ |
| HiC-pro (v.2.7.8) | Servant et al., 2015 | https://github.com/nservant/HiC-Pro |
| Fastax-collapser (Fastx Toolkit v0.0.14) | N/A | http://hannonlab.cshl.edu/fastx_toolkit/ |
| CADD | Kircher et al., 2014 | http://cadd.gs.washington.edu/score |
| CRISPResso (v1.0.0) | Pinello et al., 2016 | http://crispresso.rocks |
| HISAT2 (v2.0.1) | Kim et al., 2015 | https://ccb.jhu.edu/software/hisat2/index.shtml |
| R Statistical Programming Language and Bioconductor | Gentleman et al., 2004 | https://www.r-project.org |
| Other | ||
Contact for Reagent and Resource Sharing
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Howard Y. Chang (howchang@stanford.edu)
Experimental Model and Subject Details
Cell lines
MDA-MB-231, MCF-7, HEK293, HEK293T and HeLa cells were maintained with DMEM (Thermo Fisher, Cat#11995) supplemented with 10% fetal bovine serum (GE Healthcare, Cat#SH30396.03) and 1% pen-strep (Thermo Fisher, Cat#15140). SKBR3 and HCT116 cells were maintained with modified McCoy's 5A (Thermo Fisher, Cat#16600) supplemented with 10% fetal bovine serum and 1% pen-strep. MCF-10A cells were maintained with DMEM:F12(1:1) (Thermo Fisher, Cat#11320) supplemented with 5% horse serum (Thermo Fisher, Cat#16050122), 10 mg/ml insulin (Sigma Aldrich, Cat#I0516), 20 ng/ml epidermal growth factor (Thermo Fisher, Cat#PHG0311), 100 ng/ml cholera toxin (Sigma Aldrich, Cat#C8052), 0.5 μg/ml hydrocortisone (Sigma Aldrich, Cat#H0888) and 1% pen-strep. mESCs were cultured in serum (Fisher Scientific, SH30071.03) and LIF-containing (EMD Millipore, Cat#ESG1107) media on 0.2% gelatin-coated plates. mNPC cells were maintained on plates which were coated with 0.2% gelatin with N2B27 media which is composed of 1:1 mixed media of DMEM:F12(1:1) and Neurobasal media (Thermo Fisher, Cat#21103049) supplemented with 1× NDiff Neuro2 Medium Supplement (EMD Millipore, Cat#SCM012), 0.5× B-27 Supplement (Thermo Fisher, Cat#17504044), 1× Glutamax Supplement (Thermo Fisher, Cat#35050061), 0.1 mM of β-mercaptoethanol (Thermo Fisher, Cat#31350010), 10 ng/μl of epidermal growth factor (EGF, Peprotech, Cat#31509) and 10 ng/μl of fibroblast growth factor-basic (FGF, Peprotech, Cat#100-18B). In order to differentiate into mNPCs from mESC, mESCs were plated on gelatin-coated plates in N2B27 media for 7 days. On day 7, cells were dissociated with Accutase and cultured in suspension in N2B27 media supplemented with 10 ng/ml of FGF and 10 ng/ml of EGF. On day 10 cells embryoid bodies were plated onto 0.2% gelatin-coated plates and allowed to grow for 3 passages before sub-cloning single cells. For routine sub-culture, 0.25% Trypsin-EDTA (Thermo Fisher, Cat25200-114) were used for detaching of most cell lines; TrypLE Express Enzyme (Thermo Fisher, Cat#12604039) for MCF-10A or Accutase (EMD Millipore, Cat#SCR005) for mNPC. Puromycin (Thermo Fisher, Cat#A11138) was used at final concentration 1 μg/ml for all cell lines except SKBR3 (final 0.5 μg/ml) for 2 to 4 days. Blasticidin (Thermo Fisher, Cat#A1113903) was used at 4 μg/ml for MDA-MB-231, MCF-7, MCF-10A or HeLa, 2 μg/ml for HEK293 or SKBR3, or 16 μg/ml for HCT116 for 8 days. Hygromycin (Thermo Fisher, Cat#10687010) was used at 200 μg/ml for MDA-MB-231 for 8 days. All human cell lines were obtained from ATCC (American Type Culture Collection) and were tested for mycoplasma contamination.
Mouse subcutaneous xenograft
All animal experimentation was conducted according to protocols approved by the Stanford University Institutional Animal Care and Use Committee (IACUC). Six-week old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (Jackson laboratories) were housed at the animal care facility at Stanford University School of Medicine, kept under standard temperature, humidity, and timed lighting conditions and were provided with mouse chow and water ad libitum. For imaging, MDA-MB-231 cells expressing dCas9-KRAB were infected with lentivirus harboring EF1a-luciferase-2A-Hygro cassette. After hygromycin selection, luciferase activity was validated using Luciferase Reporter Assay System (Promega, Cat#E1500). Non-targeting sgRNA or sgRNA targeting PVT1 (R2) was transduced by lentivirus infection followed by puromycin selection for 4 days. Luciferase-labeled MDA-MB-231 cell line expressing dCas9-KRAB with non-targeting sgRNA or sgRNA targeting PVT1 was injected subcutaneously and bilaterally on the flank of the mice in 0.1 ml of sterile PBS (3 × 106 cells/animal) mixed with 50% Matrigel (BD Biosciences, Cat#356231). For bioluminescence imaging, mice received luciferin (150 mg/kg, 10 minutes prior to imaging) and were anesthetized and imaged in an IVIS 100 imaging system (Perkin Elmer). Images were analyzed with Living Image software (Perkin Elmer). Bioluminescent flux (photons/sec/cm2/sr) was measured for the primary tumors for up to 6 weeks. J.K. performed cell injection, animal maintenance, imaging, and quantification and was blinded for the animal experiments.
Method Details
Lentivirus production and infection
For virus production, 5 × 106 or 1 × 107 of HEK293T cells were plated per 10 cm or 15 cm plate, respectively. The following day, plasmid encoding lentivirus was co-transfected with pMD2.G and psPAX2 into the cells using Lipofectamine 3000 (Thermo Fisher. Cat#L3000) or TransIT LT1 (Mirus, Cat#MIR2300) according to the manufacturer's instructions. Supernatant containing viral particles was collected 48 hours after transfection and filtered. For lentivirus encoding individual sgRNAs, virus was concentrated using Lenti-X concentrator (Clontech, Cat#631232) and stored at -80°C. Viral titer was verified by adding 10 μl to 640 μl of supernatant containing virus (1/10 for 10-fold concentrated virus) to the cells. For virus infection, virus and polybrene (final 4 mg/ml, Sigma Aldrich, Cat#107689) was added to 25% confluent cells. Fresh media was added 24 hours after infection. Media was changed with media containing appropriate antibiotics 48 hours after infection. After drug selection, cells were maintained for at least one day without drug for further experiments.
CRISPR construct
pHR-SFFV-dCas9-BFP (Addgene, Cat#46910) and pHR-SFFV-dCas9-BFP-KRAB (Addgene, Cat#46911) were used for making clonal MDA-MB-231 or MCF-7 cell lines expressing dCas9 or dCas9-KRAB. For multiple cell line experiments, dCas9-BFP-KRAB-2A-Blast construct was used which was generated by inserting 2A-Blast cassette into dCas9-BFP-KRAB vector. sgRNAs were cloned into mU6(modified)-sgRNA-Puromycin-mCherry vector, which was modified from Addgene-Cat#46914. For competitive cell growth assay, mCherry ORF was replaced with EGFP. For CRISPR-mediated gene activation experiments, dCas9-VP64-2A-Blast (Addgene, Cat#61425) and MS2-P65-HSF1-Hygro (Addgene, Cat#61426) vectors were used and sgRNA was inserted into sgRNA2.0-puro-mCherry vector which was modified from Addgene-Cat#61427. For purifying recombinant Cas9 protein, pET28-Cas9 expression vector was generated from p3s-Cas9 (Addgene, Cat#43935). sgRNA sequences not included in CRiNCL library (Liu et al., 2017) were chosen considering number of their potential off-target sites (Cho et al., 2014). All sgRNAs used in this study are listed in Table S1.
Cell growth assay
3 × 103 cells or 1 × 105 cells were plated per 96-well or 12-well plates. For MTT assay, MTT labeling reagent (Roche, Cat#11465007001) was added and incubated at 37°C for 4 hours. Then solubilization solution was added and incubated at 37°C for overnight. Cell proliferation was measured by using spectrometer (Molecular Devices) as A550nm-A690nm. The absorbance at day 4 post plating was to the signal at day 1 post plating. For MTT assay, mean was averaged from two technical replicates. For cell counting, cells were detached 4 days post plating and counted by using Countess II FL (Thermo Fisher) with Trypan blue staining. Mean counts from triplicate measurements were used to calculate cell growth rate.
siRNA or ASO
One day before transfection, 2 × 105 cells were plated on a 12-well plate. siRNA (Qiagen) or ASO (Exiqon) was transfected into the cells using DharmaFECT4 (Dharmacon, Cat#T-2004) or Lipofectamin 2000 (Thermo Fisher, Cat#11668), respectively, with final 50 nM in 1 mL media. Media was refreshed 24 hours post transfection and cells were harvested or sub-cultured 48 hours post transfection. siRNA or ASO sequences are listed in Table S1.
qRT-PCR
RNA was prepared using RNeasy Plus mini Kit (Qiagen, Cat#74136). Purified RNA was quantified by Nanodrop (Thermo Fisher). For qRT-PCR, 60 ng of RNA and Brilliant qRT-PCR mastermix (Agilent, Cat#600825) were used. Each Ct value was measured using Lightcycler 480 (Roche) and each mean dCt was averaged from triplicate qRT-PCR reaction. Relative RNA level was calculated by ddCt method compared to GAPDH control. The mean dCt value of each replicate was used to calculate p-value. Primer sequences are listed in Table S1.
RNA-seq
50 ng of total RNA was used to RNA-seq. RNA-seq libraries were prepared using Truseq Stranded total RNA LT kit (Illumina, Cat#RS-122-2201) as according to manufacturer's instructions. Paired-end reads were obtained on HiSeq 4000. Reads were mapped by using Tophat2 (Kim et al., 2013) and analyzed by using DESeq2 (Love et al., 2014).
Western blotting
After puromycin selection, cells were harvested and lysed using RIPA buffer (50 mM Tris pH 8.0, 1mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1 % SDS, 0.1% sodium doxycholate) supplemented with cOmplete proteinase inhibitor cocktail (Roche, Cat# 11697498001). Lysate was homogenized using sonication and quantified using BCA reaction (Thermo Fisher, Cat#23227). 20 μg of lysate was resolved on NuPAGE 4-12% Bis-Tris gel (Thermo Fisher, Cat# NP0322) and transferred to nitrocellulose membrane (GE Healthcare, Cat#10600002). After blocking using Odyssey blocking buffer-PBS (LI-COR BioSciences, Cat#927-40003), membrane was incubated for overnight at 4°C with antibody against to MYC (Cell Signaling, Cat#5605S, 1/1,000) or GAPDH (Santa Cruz, Cat#sc69778, 1/5,000). Afterward, the membrane was incubated for 1 hour at room temperature with IRDye 800CW goat anti-rabbit secondary antibody (LI-COR BioSciences, Cat#926-32211, 1/10,000) and IRDye 680RD goat anti-mouse secondary antiboby (LI-COR BioSciences, Cat#926-68070, 1/10,000). Protein bands were visualized and quantified using Odyssey CLx and Image Studio 1.0.11 (LI-COA BioSciences).
Cell growth competition assay
Cells with CRISPRi-Control or CRISPRi-PVT1 were marked with different fluorescent proteins encoded in lentiviral vector and then co-cultured. Following puromycin selection for lentivirus infection with sgRNA, cells were counted and mixed GFP+ cells (for control sgRNA) and mCherry+ cells (for control sgRNA or sgRNA-PVT1). For 2D-culture, 1 × 105 cells were plated on 12-well plate and maintained by routine sub-culture every other day at 1:4 ratio and analyzed by FACSAria II (BD Biosciences). For 3D-culture, 2.5 × 105 cells in 2.5 ml of media containing 2% Matrigel were plated on 6-well plate which was coated with 450 μl of Matrigel. Media containing 2% Matrigel was refreshed at every 4 days. Fluorescence microscope images were taken by 5× magnification of Axiovert 200M (Carl Zeiss).
Northern blotting
10 mg of RNA was dissolved on a denaturing 1.2% MOPS agarose gel using formaldehyde load dye (Thermo Fisher, Cat#AM8550) followed by transferring to BrigtStar-Plus nylon membrane (Thermo Fisher, Cat#AM10104) using NorthernMax Transfer Buffer (Themo Fisher, Cat#AM8672) for overnight. RNA transferred to membrane was crosslinked by UV Stratalinker 2400 (Stratagene) with auto-crosslink protocol and baked at 80°C for 30 minutes. Membrane was incubated in ULTRAhyb-Oligo buffer (Thermo Fisher, Cat#AM8663) for 1 hour at 42°C. Then, membrane and 500 ng of denatured probe was incubated with ULTRAhyb-Oligo buffer for overnight at 55°C. After hybridization, membrane was washed twice with NorthernMax Low-Stringency Wash Solution (Thermo Fisher, Cat#AM8673) for 5 min followed by washed twice NorthernMax High-Stringency Wash Solution for 15 min at 42°C (Thermo Fisher, Cat#AM8674). Membrane was blocked using Odyssey Blocking Buffer-PBS containing 1% SDS for 1 hour at room temperature. Then, membrane was incubated with Streptavidin-IRDye 800CW (LI-COR BioSciences, Cat#926-32230, 1/10,000) in Odyssey Blocking Buffer-PBS containing 1% SDS for 30 min at room temperature followed by wash three times using PBS containing 0.1% Tween-20 for 5 min. RNA bands were visualized and quantified using Odyssey CLx and Image Studio 1.0.11. Templates of probes were amplified using reverse-transcriptase and purified on agarose gel. Then, biotin incorporated probe was generated by PCR using Hot Start Taq DNA polymerase (NEB, Cat#M0495L) and standard Taq reaction buffer (NEB, Cat#B9014S) supplemented with 125 μM of dTTP, 125 μM of biotin-16-dUTP (Roche, Cat#11093070910) and 250 μM of each dATP, dCTP and dGTP (NEB, Cat#N0446S). Probe DNA was dissolved on an 1.2% agarose gel to confirm of appropriate incorporation of biotin and purified by gel extraction. Probes were denatured prior to hybridize by incubation for 10 min at 95°C. Primer sequences used to make probes are listed in Table S1.
ATAC-seq
For ATAC-seq, 5 × 104 cells were harvested and washed by PBS. Cells were resuspended in lysis buffer (10 mM Tris-HCl pH 7.5, 10 mM NaCl2, 3 mM MgCl2, 0.05% NP40) and pelleted. Cell pellet was resuspended in TD buffer (10 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10% dimethlyformamide). 2.5 μl of Tn5 (Illumina, Cat#Fc-121-1030) was added to permeabilized cells and incubated at 37°C for 30 minutes. Fragmented DNA was purified using MinElute kit (Qiagen, Cat#28206) and library was generated by PCR using NEBnext PCR mastermix (New England Biolabs (NEB), Cat#M0541). Appropriate number of PCR cycles was determined by qPCR as described (Buenrostro et al., 2015). Libraries were sequenced on HiSeq 4000 with paired-end reads. Reads were mapped using Bowtie2 (Langmead and Salzberg, 2012) followed by removing PCR duplicates using Picard. Peaks were called using MACS2 (Zhang et al., 2008). Peaks from two biological replicates were merged using Samtools (Li et al., 2009) and Bedtools (Quinlan and Hall, 2010) and subjected to DESeq2 or calculating differences between non-targeting sgRNA or sgRNA targeting PVT1.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed as described (Ram et al., 2011) with minor modification. Briefly, an appropriate number of MDA-MB-231 cells [2 × 106 cells for H3K27Ac (Abcam, Cat#ab4729) or H3K9me3 (Abcam, Cat#ab8898), 5 × 106 cells for Pol II-S5P (Abcam, Cat#ab5131), 1 × 107 cells for BRD4 (Bethyl Laboratories, Cat#A301-985A100), 2 × 107 cells for HA [(Cell Signaling, Cat#2367S) or Pol II-S2P (Abcam, Cat#ab5095)] or mNPC [5 × 106 cells for H3K9me3 or H3K4me3 (Active Motif, Cat#39159)] was harvested and crosslinked with 1% formaldehyde (Thermo Fisher, Cat#28908) for 10 min and subsequently quenched with final 0.125 M glycine. Chromatin was sheared using Covaris ultra sonicator to around 200 bp followed by centrifugation for clearing. Soluble fraction of sheared chromatin was diluted 5-fold with ChIP-dilution buffer (16.7 mM Tris pH 7.5, 127 mM NaCl, 1.2 mM EDTA, 1.1% Triton X-100 and 0.01% SDS) incubated with pre-mixed Dynabead of A-protein and G-protein (Thermo Fisher, #10002D or #10004D) without antibody at 4°C for 1 hour. After removing the bead, an appropriate antibody or negative control IgG was added to chromatin and incubated for overnight at 4°C. Antibody-bound chromatins were captured by incubating 2 hours with pre-mixed Dynabead of A-protein and G-protein. Chromatin- captured bead was washed twice with low-salt RIPA (50 mM Tris pH 8.0, 1 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1% SDS and 0.1% DOC), twice with high-salt RIPA (50 mM Tris pH 8.0, 1 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.1% SDS and 0.1% DOC), twice with LiCl wash buffer (50 mM Tris pH 8.0 250 mM LiCl, 0.5% NP-40 and 0.5% DOC) and twice with TE (10 mM Tris pH 8.0 and 1 mM EDTA). Bead was resuspended in elution buffer (10 mM Tris pH 8.0, 5 mM EDTA, 300 mM NaCl and 0.5% SDS) and bead was removed by magnet. Proteinase K (NEB, Cat#P8107) was added to eluted DNA and incubated at 55°C for 2 hours followed by incubating at 65°C overnight and at 37°C for 30 minutes with RNaseA (Thermo Fisher, Cat#12091). Reverse-crosslinked DNA was purified by using MinElute column. Eluted DNA was used to qPCR or sequencing library preparation with NEBnext ultra-II kit (for human cell line, NEB, Cat#E7645) or tagmentation using Nextera enzyme (for mouse cell line). qPCR was performed using qPCR master mix (Roche, #04707516001). Each Ct value was measured using Lightcycler 480 (Roche) and each mean dCt was averaged from triplicate qPCR reaction. Ct values were normalized to relative amount of 2 to 5% of input DNA and the mean dCt value from each replicate was used for calculating p-value. qPCR primers are listed in Table S1. Sequencing libraries were sequenced on HiSeq 4000 with paired-end reads. Reads were mapped by using Bowtie2 followed by removing PCR duplicates using Picard. Peaks were called using MACS2 with subtraction of input signal. Peaks from two biological replicates were merged using Samtools and Bedtools. Differences between non-targeting sgRNA or sgRNA targeting PVT1 were analyzed by DESeq2. Enrichment of any IgG control between CRISPRi-Control and CRISPRi-PVT1 is not significantly different (p>0.05).
JQ1 treatment
4 × 106 cells were plated on 6-well plate. JQ1 (Sigma Aldrich, Cat#SML1524) in DMSO was added final 1 μM or 5 μM 1 day post plating. After 6 hours, cells were harvested and RNA was extracted as decribed in qRT-PCR method. Equal volume of DMSO without JQ1 was added as a negative control.
Hi-ChIP
Hi-ChIP was performed as described (Mumbach et al., 2016). Briefly, 1 × 107 cells for each biological replicate were collected and crosslinked by using 1% formaldehyde for 10 minutes. Chromatin was digested using MboI restriction enzyme (NEB, Cat#R0147) followed by end-repair, ligation and sonication. Sheared chromatin was cleared and 3-fold diluted as described in ChIP method and then incubated with anti-H3K27ac antibody at 4°C for overnight. Chromatin-antibody complex was captured by Dynabead Protein-A bead. Biotin was incorporated by adding DIBO-biotin (Thermo Fisher, Cat#C-10412) followed by capture with Streptavidin C-1 bead (Thermo Fisher, Cat#65002). Captured DNA was quantified using Qubit (Thermo Fisher) and an appropriate amount of Tn5 enzyme was added to captured DNA to generate sequencing library. Sequencing was performed on HiSeq 4000 with paired-end read and analyzed by using HiC-pro (Servant et al., 2015).
UMI-4C
UMI-4C was performed as described (Schwartzman et al., 2016) with minor modification. 5 × 106 cells were harvested and crosslinked with 1% formaldehyde for 10 minutes. Pellet was resuspended in ice-cold lysis buffer (10 mM Tris pH 8.0, 10 mM NaCl, 0.2% NP-40) and incubated at 4°C for 30 min. Nuclei was pelleted and resuspended in 0.5% SDS followed by incubation at 62°C for 10 min. Chromatin was digested by MboI restriction enzyme and ligated by T4 DNA ligase (NEB, Cat#M0202) at room temperature for 4 hrs. Reaction was cleared by centrifugation. Pellet was resuspended in Proteinase K buffer (10 mM Tris pH. 7.5, 100 mM NaCl, 1 mM EDTA, 0.5% SDS) and incubated 55°C for 2 hours with Proteinase K and then incubated 68°C for 2 hours. After incubating with RNaseA, DNA was purified by ethanol precipitation. DNA was resolved in 10 mM Tris pH 7.5 and sonicated by Bioruptor followed by sequential incubation with end repair mix (NEB, Cat#E6050), Klenow exo- (NEB, Cat#M0212) and CIP (NEB, Cat#M0290). Reaction was cleared by SPRI selection (2×, Beckman Coulter, Cat#B23318). Illumina forked adaptors was added to DNA and incubated with Quick Ligase (NEB, Cat#M2200) at room temperature for 15 minutes. To release the non-ligated strand of the adaptor, DNA was denatured at 95°C for 2 minutes and then purified by SPRI (1×). Denatured DNA was quantified by Qubit. 200 ng of DNA was used for library preparation by nested PCR. Sequencing was performed on MiSeq or HiSeq 4000 with paired-end reads and analyzed by using HiC-pro. The interaction frequency was measured from two biological replicates each of which comprises two technical replicates with two different primers. Mean was normalized to CRISPRi-Control for each interaction. PCR primers used in UMI-4C are listed in Table S1.
4C-Seq
Chromosome conformation capture combined with high-throughput sequencing (4C-Seq) was performed according to described (Stadhouders et al., 2013) with modifications. 5 × 106 cells were fixed by adding 37% formaldehyde to a final concentration of 1% and incubating for 10 minutes at room temperature while tumbling, followed by addition of 1M glycine to a final concentration of 0.125M to quench. Samples were washed with cold PBS and resuspended in 5 mL ice-cold lysis buffer (10 mM Tris-HCl (pH 8.0), 10 mM NaCl, 0.2% NP-40 supplied with proteinase inhibitor), followed by 10 min incubation on ice. Samples were re-resuspended in 500 μL 1.2× CutSmart restriction digest buffer (NEB). 20% SDS was added to a final concentration of 0.3%, and samples were incubated for 1 hr at 37°C while shaking at 900 rpm on a thermomixer. 20% Triton X-100 was added to a final concentration of 2%, followed by incubation for 1 hr at 37°C while shaking at 900 rpm on a thermomixer. NlaIII (NEB, #R0125) was added to each sample and incubated 20 hr at 37°C while shaking at 900 rpm on a thermomixer. To inactivate enzyme, 20% SDS was added to a final concentration of 1.6% SDS, followed by incubation for 25 min at 65°C while shaking at 900 rpm on a thermomixer. T4 DNA Ligase buffer (NEB) and T4 DNA Ligase (NEB) were added, and incubated for 18 hr at 16°C. Proteinase K (Machery Nagel, #740506) and incubating for 16 hr at 65°C. RNase was added and incubated for 30 min at 37°C. DNA was purified by phenol:chloroform extraction followed by ethanol precipitation. The second 4C-Seq digest was then performed by digestion using DpnII (NEB, #R0543) for 16 hr at 37°C. Digests were then purified using phenol:chloroform and resuspended in distillated water. Second ligation was performed by adding 10× T4 DNA Ligase buffer (NEB) and T4 DNA Ligase (NEB), followed by incubation for 18 hr at 16°C. Circularized fragments were purified by phenol:chloroform and resuspended in 10 mM Tris-HCl (pH 7.5), and then purified again using NucleoSpin PCR Clean-up (Machery Nagel, #740609). 1.2 μg of each template was amplified using the Expand Long Template PCR kit (Roche, #11681834001) for 30 cycles. PCR primers used for 4C-seq were described in Table S1. PCR amplicons were purified using Agencourt Ampure XP beads. Sequencing libraries were generated using the KAPA HyperPlus kit according to manufacturer's recommendations with modifications. 4C-Seq libraries were validated using the Agilent 2200 High Sensitivity DNA ScreenTape, Qubit, and ddPCR (Bio-Rad). Cluster generation and sequencing was performed on a HiSeq 4000 with single end read. Reads were aligned to the human genome using the spliced read aligner HISAT2 (Kim et al., 2015).
Luciferase reporter assay
Genomic region harboring PVT1 intragenic enhancers was amplified by nested-PCR and cloned into pGL4.23[luc2/minP] vector (Promega, Cat#E8411) digeted by XhoI and HindIII using NEBuilder Hifi DNA Assembly Master Mix (NEB, Cat#E2621L). Luciferase activity was measured using Luciferase Assay System (Promega, Cat#E1500). One day prior to transfection, 1 × 105 of MDA-MB-231 cells plated in 24-well plate. 500 ng of plasmid was transfected into the cells using Lipofectamine 3000. Fresh media was replaced at post 24 hours transfection. At post 48 hours transfection, media was removed and the plate washed by PBS briefly. 200 μl of 1× Cell Culture Lysis Reagent was added to plates directly and transferred to new tubes. After brief centrifugation, 20 μl of cell lysate was mixed with 100 μl of Luciferase Assay Reagent. Luminescence was measured using pre-set protocol of SpectraMax M5 and SoftMax Pro 6.3 software (Molecular Devices).
CRISPRi targeting enhancers
In order to target enhancer region efficiently which long around 1 kb, we used three sgRNA in one vector system (Adamson et al., 2016). In order to select cells infected both sgRNA targeting PVT1 and enhancer, lentiviral vector backbone was modified by replacing Puromycin-mCherry cassette to Hygromycin resistance gene. Three sgRNAs were designed for each enhancer and cloned into pMJ114 (bovine U6, Addgene, Cat#85995), pMJ117 (human U6, Addgene, Cat#85997) or pMJ179 (mouse U6, Addgene, Cat#85996) individually. Then, U6 promoter and sgRNA sequences were amplified by PCR and combined into lentiviral vector using NEBuilder Hifi DNA Assembly Master Mix, which digested with XbaI and XhoI. Cells infected lentivirus containing sgRNA targeting PVT1 or LacZ (control) first followed by Puromycin selection for 4 days as described above. Then, cells were re-plated by 1:8 ratio to 6 well plate and infected with lentivirus harboring three sgRNAs targeting each enhancer and selected with Hygromycin for 8 days. After one day recovery without antibiotics, RNAs were extracted and used for qRT-PCR. Sequences of sgRNAs and primers are listed in Table S1.
4sU-labeling
Nascent RNA labelling with 4sU was performed as described with minor modification (Duffy et al., 2015). 1 × 106 cells were plated on 15 cm plate 1 day prior to labelling. 4-thiouridine (Sigma Aldrich, Cat#T4509) dissolved in DMSO or equal volume of DMSO was added to media at final 200 μM and incubated at 37°C for 20 minutes. After incubation, cells were harvested and stored at -80°C for overnight. Total RNA was purified by adding TRIzol (Thermo Fisher, Cat#15596018) and 0.2 volume of chloroform. 1 volume of ethanol was added to aqueous phase and then transferred to RNeasy midi column (Qiagen, Cat#75144). RNA was eluted in RNase free water and quantified by Nanodrop. 5 μg of MTSEA biotin-XX (Biotium, Cat#90066) was added to 20 μg of RNA and then incubated for 2 hours in final 400 μl of binding buffer (10 mM HEPES pH 7.5, 1 mM EDTA and 20% dimethylformamide). After biotinylation, free biotin was washed out by purifying with RNeasy MinElute column (Qiagen, Cat#74204). 100 μl of Streptavidin C-1 bead was added to RNA and then incubated with rotating for 1 hour in binding buffer (10 mM Tris pH 7.5, 1 mM EDTA, 100 mM NaCl and 0.1% tween-20). Bead was washed 3 times with binding buffer. RNA was eluted sequentially by 5% of β-mercaptoethanol at room temperature and 100% of β-mercaptoethanol at 50°C followed by purification using RNeasy MinElute column. Purified RNA was used to qRT-PCR for quantification and normalized to relative abundance of ACTB RNA in 5% of input RNA which was measured by qRT-PCR. Relative abundance of nascent transcripts was calculated by ddCt method compared to non-targeting sgRNA control. Mean dCt value of each replicate was used to calculate p-value. Enrichment of any biotinylated-control GAPDH mRNA is not significantly different (p>0.05). Primer sequences are listed in Table S1.
PVT1 promoter mutagenesis
For footprinting of regulatory element affecting cell growth (Vierstra et al., 2015), Cas9 endonuclease and sgRNA were delivered in ribonucleotide to avoid accumulation of indels by residual nuclease activity. pET28-Cas9 plasmid was transduced into BL21-DE3 (pLysS) strain (Thermo Fisher, Cat#C6060) and Cas9 protein and sgRNA were prepared and transduced as described (Cho et al., 2014). Pre-assembled CRISPR-ribonucleoprotein complex consisting of 100 μg of Cas9 and 100 μg of sgRNA was transfected into 1 × 106 of MDA-MB-231 cells by using Amaxa Nucleofetor kit V (Lonza, Cat#VVCA-1003) with program X-013. Cells were plated on 6-well plate 3 days post-transfection (P0). Cells were passaged by 1:4 ratio every 2 days. At P10, genomic DNA was purified and 5 μg of DNA was used for PCR amplification of PVT1 promoter region. Parallel PCR reactions were mixed and purified using AMPure beads (Beckman Coulter, #A63881) at concentrations ranging from 0.6× to 1×. Libraries were generated by nested PCR and sequenced on HiSeq 4000 with paired-end reads. Sequencing read was analyzed using CRISPresso (Pinello et al., 2016). Any variation without overlap with expected cleavage site ± 3 bp was ignored. Difference between P0 and P10 was calculated by DESeq2. To generate mutant clones, cells were diluted and plated as 0.2 cells in 96-well plate 2 days post ribonucleoprotein transfection. Genomic DNA was extracted using DNeasy 96 Blood & Tissue Kit (Qiagen, Cat#69581) and 5 to 20 μl of eluted DNA was used to PCR. Target regions were amplified from each clone and sequenced with MiSeq for genotyping. Reads from each clones were counted using Fastax-collapser. For qRT-PCR, RNA was extracted from clones at multiple passages. Primer sequences are listed in Table S1.
Targeted RNA-seq
50 ng of RNA extracted from each mNPC clone was converted to cDNA by 30 cycles of qRT-PCR reaction. Then, the region with defined SNP was amplified using 1 μl of diluted PCR product (1/20) and 15 cycles of PCR. Sequencing library was generated by 15 cycles of nested PCR using 1 μl of diluted PCR product (1/20). DNA was extracted from 1% agarose gel and sequenced on MiSeq. Reads were counted using Fastax-collapser. Reads with exactly expected sequences were considered for calculating d-score. The transcription level of each allele was averaged from 3 technical replicates. Primer sequences are listed in Table S1.
Allelic specific data analysis
All the sequencing data from mouse cell line were processed as described (Xu et al., 2017). The d-score for PVT1 expression was calculated using the first exon instead of the whole transcripts to better present the allelic different isoform of PVT1. The relative fold change of MYC was normalized to the average expression from clones with two active alleles of PVT1.
Deleterious score for PVT1 promoter regions
To evaluate whether variants in the PVT1 promoter region were potentially deleterious in the human genome, we employed the UCSC Table Browser and specified “Variation” group, “1000G ph3 Vars” track and a 5 kb window around PVT1 transcription start sites (chr8: 128,801,802-128,811,802) to obtain VCF files for all variants. VCF files were analyzed using CADD (Kircher et al., 2014) (http://cadd.gs.washington.edu/score) to generate a C-score corresponding to the predicted deleteriousness of genome-wide variants. The CADD C-score was used to illustrate the predicted deleterious effect for each variant, which is PHRED-like (-10 × log10(rank/total)) scaled, ranking a variant relative to all possible substitutions of the human genome (8.6 × 109). On this scale, variants with a C-score of greater than or equal to 10 are predicted to be amongst the top 10% most deleterious substitutions in the human genome.
Somatic mutations around PVT1 promoter regions
Somatic mutations (SNVs) for different cancer types were obtained from the International Cancer Genome Consortium (ICGC) (Release 23). Mutations within a 5 kb window of the TSS from a set of annotated lncRNAs (ENSEMBL, release 70) were evaluated where lncRNA promoter regions from Chr 1 or Chr 8 were employed as controls. The number of somatic mutations within each lncRNA promoter region for each cancer type was used to generate a background (control) distribution against which the frequency of PVT1 promoter mutations was evaluated.
Significance of SV overlaps in PVT1 promoter region
Somatic structural variant (SV) calls for the ICGC BRCA-EU cohort were obtained from the original publication (Glodzik et al., 2017). The number of SVs (less than 1 million base pair in length, for duplications, deletions or inversions) overlapping with 3,826 lncRNA promoters (ENSEMBL, release 70, with gene length > 5000 bp) was calculated. Promoters were defined as [-1000, 2414] bp of the corresponding transcription start site (TSS), keeping with the definition used in this study for PVT1. Local background SV overlap rates were computed based on the mean overlap for windows in adjacent regions (5 million bp). Negative binomial regression was performed using the local background rate as the offset and SV overlap in each promoter as the observed variable (Nik-Zainal et al., 2016). The estimated over dispersion parameter was then used as input to a negative binomial test to evaluate the significance of SV overlap in each promoter. FDR adjusted p-values were employed to correct for multiple testing.
Rearrangement analysis
Fusion transcripts and their exact breakpoints were identified in RNA-seq data (Illumina, 30×) of an ER-, HER2+ breast cancer using breakpoint assembly for candidate sites with an enrichment of chimeric reads or clusters of abnormal read pairs that map to unexpectedly distant sites (Fernandez-Cuesta et al., 2015). DNA-level breakpoints associated with the fusion events were determined based on abnormal read pairs in the vicinity of the detected fusion points by analyzing whole genome sequencing (WGS) data (30×) from the same donor. The RNA-seq and WGS data for this case were generated on the Illumina platform using standard protocols. The patient (MB-0152) derives from the METABRIC cohort (Curtis et al., 2012) for which array based copy number and gene expression data were previously described.
Quantification and Statistical Analysis
In relevant figures, Figure Legends denote the statistical details of experiments including statistical tests used, kind of replicates and the value of n. Asterisks define degree of significance as described in the Figure Legends. All Student's t-test and Mann-Whitney U-test were analyzed in two-sided. All the sequencing data were aligned to human genome (GRCh37/hg19) or mouse genome (NCBI37/mm9) unless indicated specifically. All statistical analyses and graphics were performed using the R Statistical Programming Language and Bioconductor (Gentleman et al., 2004).
Data and Software Availability
All software used in this study is listed in KEY RESOURSES TABLE. ChromHMM was imported from Breast vHMEC from Roadmap Epigenomics Consortium. Conservation plot is imported from100 vertebrates Basewise Conservation by PhyloP at UCSC genome browser. All raw sequencing reads and raw count matrices generated in this study are available through Gene Expression Omnibus (GEO) with accession number GSE97669. Data for human cancer patient is available through European Genome-Phenome Archive (EGA) with accession number EGAD00001003342.
Supplementary Material
Figure S1. Pro-growth phenotype induced by CRISPRi-PVT1, related to Figure 1. A, Schematic representation of transcripts and corresponding TSS at PVT1 locus. sgRNAs used in this study are shown in the shaded box. Transcripts from each TSS are shown with the same color. B, Violin plots showing all pro-growth phenotypes across seven human cell lines except PVT1, which is represented as a dot. Significance was determined by p-value and basal transcription level in given cell line. Data was derived from (Liu et al., 2017). C, sgRNA enrichment of all individual sgRNAs targeting PVT1 TSS. Each TSS is colored as in Figure S1A. D, Comparison of sgRNA enrichment for representing reproducibility of CRISPRi screen for sgRNAs targeting PVT1 TSS. E, Images of time-course luminescence from subcutaneous xenografted MDA-MB-231 cell line with CRISPRi-Control or CRISPRi-PVT1 (R2) (upper). Luminescence from all mice with xenografts was quantified and presented as box plot (bottom). p-value was calculated by paired Mann-Whitney U-test (n=12 for each). Lines indicate mean of the luminescence at each time point.
Figure S2, CRISPRi-PVT1 cells outcompete in cell growth competition assay, related to Figure 1. A, Growth competition assay for CRISPRi-PVT1 in MCF-7 cell line, represented the same as Figure 1C. B, Fluorescence microscope images of competitive cell growth assay by 2D cell culture of MDA-MB-231 cell line. C, Density plots showing percentage of mCherry+ or EGFP+ cells measured using FACS in competitive cell growth assay of MDA-MB-231 cell line. The numbers indicate percentage of Q1 (mCherry+) or Q4 (GFP+) populations. Target genes of sgRNA are shown in the same color as of the fluorescent marker above each panel.
Figure S3. Pro-growth phenotype is induced by increased MYC expression, related to Figure 2 and Figure 3. A, Schematic representing qRT-PCR amplicon, probe, siRNA or sgRNA targeting MYC mRNA. Annotated isoforms of MYC are presented with accession numbers. Alternative promoters are shown (p0, p1 or p2). Median coverage of RNA-seq at indicated region (left) was used to calculate MYC promoter usage affected by CRISPRi-PVT1 (right). Mean percentage (n=2) of each promoter was used for statistical analysis (paired Mann-Whitney U-test). n.s., not significant (p>0.05). B, Image of northern blot for GAPDH (control) and MYC with CRISPRi-Control or CRISPRi-PVT1 in MDA-MB_231 cell line (top) and relative quantification of MYC mRNA (bottom). Error bars indicate mean ± SD (n=3, biological replicates). *p<0.05, using unpaired t-test. C, Schematic representing qRT-PCR primers, ASO or siRNA targeting PVT1 lncRNA. Arrows and bars are not scaled. PVT1-3′ primer was used for PVT1 unless indicated specifically. D, qRT-PCR for PVT1 (top) or MYC (bottom) in MDA-MB-231 cells expressing dCas9-KRAB or dCas9 with sgRNA-Control (Ctrl.), sgRNAs-PVT1 upstream (U1 to U10), TSS1(R1 to R10) or TSS2 (Y1 to Y10). Mean ± SEM (n=3, biological replicates). E, Correlation between sgRNA enrichment and relative MYC or PVT1 RNA levels, which were described in Figures S1C and S3D. Spearman's coefficient (R) and p-value are shown.
Figure S4. Epigenetic changes induced by CRISPRi targeting PVT1, related to Figure 4. A, RNA-seq, ChIP-seq or ATAC-seq signals around PVT1-MYC locus measured in MDA-MB-231 cell lines with CRISPRi-control or CRISPRi-PVT1. ChIP-seq peaks against H3K9me3 or H3K27ac at PVT1 promoter are shown enlarged in box. All sequencing was performed biological replicates. Peaks were visualized using UCSC genome browser. B to E, Fold changes comparing CRISPRi-PVT1 to CRISPRi-Control in ChIP-seq with indicated antibody (B to D) or ATAC-seq (E). Peaks within ± 1 kb of annotated transcribing region in RefSeq were compared with all other peaks by DESeq2. p-values were calculated by unpaired Mann-Whitney U-test (for n≥3). n.d., not detected.
Figure S5. PVT1 and MYC are co-regulated through chromatin contacts in a cell line specific manner, related to Figure 5. A, Enrichment of chromatin contacts comparing CRISPRi-PVT1 to CRISPRi-Control in MDA-MB-231 cell line which was measured by HiChIP-H3K27ac. left, 5 megabase; right, 500 kb region around PVT1. B, Chromosome contact frequency at 1 kb resolution anchored at MYC 3′-enhancer measured by HiChIP-H3K27ac in MDA-MB-231 cell line with CRISPRi-Control or CRISPRi-PVT1 (R3). C, Chromosome contact frequency at 5 kb resolution anchored at PVT1 promoter (top) or MYC 3′-enhancer (bottom) measured by UMI-4C in MCF-7 cell lines with CRISPRi-Control or CRISPRi-PVT1 (R2). D, Schematic representation of luciferase reporter assay (upper) and boxplots showing relative luminescence from the reporter assay. Luminescence of each biological replicate (n=3) was measured by triplicate luciferase reactions. E, qRT-PCR for PVT1 or MYC at 6 hours post JQ1 treatment in MDA-MB-231 cell line. Equal volume of DMSO was used for non-treated control. F, qRT-PCR for PVT1 or MYC transcripts levels in the indicated seven human cell lines with CRISPRi-Control or CRISPRi-PVT1. For E and F, Error bars indicate mean ± SEM (n=3, biological replicates). G, Chromatin accessibility profiles measured by ATAC-seq around PVT1-MYC locus in indicated cell lines. H, Chromosome contact frequency at 5 kb resolution anchored at PVT1 promoter (top) or MYC 3′-enhancer (bottom) measured by UMI-4C in MDA-MB-231, MCF-7 or HCT116 cell lines. I, Boxplots showing chromosome contact frequency at 5 kb resolution measured by UMI-4C in three different cell lines. Mean was normalized to MDA-MB-231 cell line for each interaction. p-value was calculated by unpaired Mann-Whitney U-test (n=8) compared to HCT116 cell line. J, Sequencing read alignment from 4C-seq anchored at MYC locus. For B, C and H, mean ± SEM from two biological replicates is represented with lines and shading. For C and H, the mean was measured from two biological replicates each of which comprises two technical replicates with two different primers. *p<0.05, using unpaired t-test compared with control (dark grey).
Figure S6, PVT1 allele-specifically regulates MYC transcription, related to Figure 6. A, Correlation between d-score of PVT1 chromatin accessibility from ATAC-seq and PVT1 transcription level from RNA-seq. B, Correlation between d-score of transcription level between PVT1 and MYC from RNA-seq. For A and B, n=15 from 11 clones and 4 additional technical replicates. C, Schematics showing location of primers used in targeted RNA-seq. Sequence of transcripts spanning defined SNPs of 129 (pink) or Cast allele (blue) are shown. D, Correlation between d-score from targeted RNA-seq with indicated primers (n=35). Spearman's coefficient (R) and p-value are shown.
Figure S7. Recurrent mutation and mutagenesis at PVT1 promoter, related to Figure 7. A, Scatter plot representing C-scores for all bases around PVT1 TSS ±5 kb region from CADD analysis. TSS1 or TSS2 of PVT1 is shown as a red or yellow line, respectively. Grey line indicates C-score=10, top 10%. Thick black line indicates mean of each base. B, Plots showing kernel density estimation of mutation frequency of 227 lncRNA on Chr 8 at TSS ±5 kb region of PVT1 TSS in all cancer types or sub-populations having high or low frequency of PVT1 mutation. Red line indicates PVT1. Grey line or dashed-grey line indicates 80th or 90th percentile, respectively. C, Circos plot illustrating genomic/transcriptomic evidence for PVT1 fusion transcripts detected in an ER- HER2+ human breast tumor. The ideogram for Chr 8 is shown in the outermost ring, followed by predicted absolute total Copy Number (Nt) and minor allele CN (Nb) from WGS data, SNP-chip data and transcriptional abundances shown on log2 scale of TPM (Transcripts Per Kilobase Million, 0-10). Links indicate breakpoints detected from RNA-seq (green) corresponding to fusion transcripts, or WGS (gray) indicating genomic translocations. The insert panel illustrates two sets of genomic breakpoints and three sets of fusion breakpoints, two of which are located in the PVT1 promoter region (chr8:128,806,984). D, Most abundant 10 alleles containing deletions at passage 30 (left) and their enrichment over passages (right). Allele sequences were ordered by their abundant at passage 30. Unmodified, unenriched or enriched sequence were shown in black, blue or red, respectively. E, qRT-PCR for MYC mRNA in clonal MDA-MB-231 cell lines having unmodified or mutant allele at PVT1 promoter. Sequences of sgRNAs used in mutant cloning and each heterozygous mutant were shown (left). Error bars indicate mean ± SEM (n=3, technical replicates). *p<0.05, using unpaired t-test comparing to non-clonal wild-type population. F, Three cases of lncRNA targeting CRISPRi upregulated genes distanced from the target locus. Genomic coordinates and their neighbor genes (left), relative RNA fold changes and adj. p-value from DESeq2 (right) are shown. Light blue, target lncRNA of CRISPRi; red, upregulated genes; triangles in green or light green, position of each sgRNA. Data was derived from (Liu et al., 2017).
Table S2. PVT1 translocation and fusion RNAs in human cancers, related to Figure 7.
Highlights.
Silencing PVT1 promoter enhances breast cancer cell competition.
PVT1 promoter inhibits MYC transcription independent of PVT1 lncRNA.
PVT1 and MYC promoters compete for enhancer contact in cis.
Mutations encompassing PVT1 promoter are recurrent in human cancers
Acknowledgments
We thank members of the Chang lab for helpful discussions and assistance. We thank Seung K. Kim for access to FACS, Hokyung K. Chung for her help in microscopy imaging, and Hye Mi Jung for advice on scientific illustration. Supported by National Institutes of Health R35-CA209919, P50-HG007735 (to H.Y.C), 1R01- NS091544 (to D.A.L.), U01-CA168370, R01-DA036858 (to J.S.W.) and S10OD018220 (to Stanford Functional Genomics Facility). J.S.W. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions: S.W.C. and H.Y.C. conceived and designed the study. S.W.C., M.R.M., A.C.C., Y.G.C., K.E.Y., S.N. and S.J.L. performed experiments. S.W.C., J.X., M.R.M., A.C.C. and S.J.L. analyzed the sequencing data. J.K. performed the animal experiment. J.X., R.S., J.H. and C.Curtis analyzed the tumor patient data. S.-F.C. and C.Caldas provided human tumor data. S.J.L, M.H., D.A.L. and J.S.W. contributed experimental methods. S.W.C. and H.Y.C. wrote the manuscript with input from all authors and H.Y.C. supervised the project.
Declaration of Interest: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamson B, Norman TM, Jost M, Cho MY, Nunez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded ProteinResponse. Cell. 2016;167:1867–1882 e1821. doi: 10.1016/j.cell.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207–211. doi: 10.1038/nature12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KM, Anderson DM, McAnally JR, Shelton JM, Bassel-Duby R, Olson EN. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature. 2016;539:433–436. doi: 10.1038/nature20128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancafort P, Magnenat L, Barbas CF., 3rd Scanning the human genome with combinatorial transcription factor libraries. Nat Biotechnol. 2003;21:269–274. doi: 10.1038/nbt794. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015;109:21 29 21–29. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinen Y, Sakamoto N, Nagoshi H, Taki T, Maegawa S, Tatekawa S, Tsukamoto T, Mizutani S, Shimura Y, Yamamoto-Sugitani M, et al. 8q24 amplified segments involve novel fusion genes between NSMCE2 and long noncoding RNAs in acute myelogenous leukemia. J Hematol Oncol. 2014;7:68. doi: 10.1186/s13045-014-0068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claveria C, Giovinazzo G, Sierra R, Torres M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature. 2013;500:39–44. doi: 10.1038/nature12389. [DOI] [PubMed] [Google Scholar]
- Cleary MD, Meiering CD, Jan E, Guymon R, Boothroyd JC. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat Biotechnol. 2005;23:232–237. doi: 10.1038/nbt1061. [DOI] [PubMed] [Google Scholar]
- Csorba T, Questa JI, Sun Q, Dean C. Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization. Proc Natl Acad Sci U S A. 2014;111:16160–16165. doi: 10.1073/pnas.1419030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, Ozato K, Sims RJ, 3rd, Singer DS. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci U S A. 2012;109:6927–6932. doi: 10.1073/pnas.1120422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, Simon MD. Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol Cell. 2015;59:858–866. doi: 10.1016/j.molcel.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539:452–455. doi: 10.1038/nature20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- Fernandez-Cuesta L, Sun R, Menon R, George J, Lorenz S, Meza-Zepeda LA, Peifer M, Plenker D, Heuckmann JM, Leenders F, et al. Identification of novel fusion genes in lung cancer using breakpoint assembly of transcriptome sequencing data. Genome Biol. 2015;16:7. doi: 10.1186/s13059-014-0558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, Engreitz JM. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354:769–773. doi: 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glodzik D, Morganella S, Davies H, Simpson PT, Li Y, Zou X, Diez-Perez J, Staaf J, Alexandrov LB, Smid M, et al. A somatic-mutational process recurrently duplicates germline susceptibility loci and tissue-specific super-enhancers in breast cancers. Nat Genet. 2017;49:341–348. doi: 10.1038/ng.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham M, Adams JM. Chromosome 8 breakpoint far 3′ of the c-myc oncogene in a Burkitt's lymphoma 2;8 variant translocation is equivalent to the murine pvt-1 locus. EMBO J. 1986;5:2845–2851. doi: 10.1002/j.1460-2075.1986.tb04578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groff AF, Sanchez-Gomez DB, Soruco MML, Gerhardinger C, Barutcu AR, Li E, Elcavage L, Plana O, Sanchez LV, Lee JC, et al. In Vivo Characterization of Linc-p21 Reveals Functional cis-Regulatory DNA Elements. Cell Rep. 2016;16:2178–2186. doi: 10.1016/j.celrep.2016.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J, et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature. 2017;543:199–204. doi: 10.1038/nature21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, Lorenz K, Lee EH, Barcellos-Hoff MH, Petersen OW, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1:84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HP, Cho GA, Han SW, Shin JY, Jeong EG, Song SH, Lee WC, Lee KH, Bang D, Seo JS, et al. Novel fusion transcripts in human gastric cancer revealed by transcriptome analysis. Oncogene. 2014a;33:5434–5441. doi: 10.1038/onc.2013.490. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress TR, Sabo A, Amati B. MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer. 2015;15:593–607. doi: 10.1038/nrc3984. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Horlbeck MA, Cho SW, Birk HS, Malatesta M, He D, Attenello FJ, Villalta JE, Cho MY, Chen Y, et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science. 2017;355 doi: 10.1126/science.aah7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BE, Miller FR, Wilburn D, Heppner GH. Dominance of a tumor subpopulation line in mixed heterogeneous mouse mammary tumors. Cancer Res. 1988;48:5747–5753. [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods. 2016;13:919–922. doi: 10.1038/nmeth.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, Nguyen ML, Rubin AJ, Granja JM, Kazane KR, et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet. 2017 doi: 10.1038/ng.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagoshi H, Taki T, Hanamura I, Nitta M, Otsuki T, Nishida K, Okuda K, Sakamoto N, Kobayashi S, Yamamoto-Sugitani M, et al. Frequent PVT1 rearrangement and novel chimeric genes PVT1-NBEA and PVT1-WWOX occur in multiple myeloma with 8q24 abnormality. Cancer Res. 2012;72:4954–4962. doi: 10.1158/0008-5472.CAN-12-0213. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Lee DK, Lee H, Lee Y, Jang YS, Kim YH, Yang HY, Lee SI, Seol W, Kim JS. Phenotypic alteration of eukaryotic cells using randomized libraries of artificial transcription factors. Nat Biotechnol. 2003;21:1208–1214. doi: 10.1038/nbt868. [DOI] [PubMed] [Google Scholar]
- Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, Yuan GC. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol. 2016;34:695–697. doi: 10.1038/nbt.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo A, Dillon N. Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol. 2015;16:245–257. doi: 10.1038/nrm3965. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, Kellis M, Gymrek M, Issner R, Coyne M, et al. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. 2011;147:1628–1639. doi: 10.1016/j.cell.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AM, Chang HY. Long Noncoding RNAs in Cancer Pathways. Cancer Cell. 2016;29:452–463. doi: 10.1016/j.ccell.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzman O, Mukamel Z, Oded-Elkayam N, Olivares-Chauvet P, Lubling Y, Landan G, Izraeli S, Tanay A. UMI-4C for quantitative and targeted chromosomal contact profiling. Nat Methods. 2016;13:685–691. doi: 10.1038/nmeth.3922. [DOI] [PubMed] [Google Scholar]
- Scruggs BS, Adelman K. The Importance of Controlling Transcription Elongation at Coding and Noncoding RNA Loci. Cold Spring Harb Symp Quant Biol. 2015;80:33–44. doi: 10.1101/sqb.2015.80.027235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtivelman E, Bishop JM. Effects of translocations on transcription from PVT. Mol Cell Biol. 1990;10:1835–1839. doi: 10.1128/mcb.10.4.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtivelman E, Henglein B, Groitl P, Lipp M, Bishop JM. Identification of a human transcription unit affected by the variant chromosomal translocations 2;8 and 8;22 of Burkitt lymphoma. Proc Natl Acad Sci U S A. 1989;86:3257–3260. doi: 10.1073/pnas.86.9.3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao H, Stephens RM, Harris TJ, Munroe DJ, Wu X. Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A. 2010;107:3001–3005. doi: 10.1073/pnas.0906067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Kolovos P, Brouwer R, Zuin J, van den Heuvel A, Kockx C, Palstra RJ, Wendt KS, Grosveld F, van Ijcken W, et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat Protoc. 2013;8:509–524. doi: 10.1038/nprot.2013.018. [DOI] [PubMed] [Google Scholar]
- Sur IK, Hallikas O, Vaharautio A, Yan J, Turunen M, Enge M, Taipale M, Karhu A, Aaltonen LA, Taipale J. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science. 2012;338:1360–1363. doi: 10.1126/science.1228606. [DOI] [PubMed] [Google Scholar]
- Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, Ronning P, Reuland B, Guenther K, Beadnell TC, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–86. doi: 10.1038/nature13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra J, Reik A, Chang KH, Stehling-Sun S, Zhou Y, Hinkley SJ, Paschon DE, Zhang L, Psatha N, Bendana YR, et al. Functional footprinting of regulatory DNA. Nat Methods. 2015;12:927–930. doi: 10.1038/nmeth.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Yuan JH, Wang SB, Yang F, Yuan SX, Ye C, Yang N, Zhou WP, Li WL, Li W, et al. Oncofetal long noncoding RNA PVT1 promotes proliferation and stem cell-like property of hepatocellular carcinoma cells by stabilizing NOP2. Hepatology. 2014a;60:1278–1290. doi: 10.1002/hep.27239. [DOI] [PubMed] [Google Scholar]
- Xiang JF, Yin QF, Chen T, Zhang Y, Zhang XO, Wu Z, Zhang S, Wang HB, Ge J, Lu X, et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014;24:513–531. doi: 10.1038/cr.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Carter AC, Gendrel AV, Attia M, Loftus J, Greenleaf WJ, Tibshirani R, Heard E, Chang HY. Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells. Nat Genet. 2017a;49:377–386. doi: 10.1038/ng.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu MD, Wang Y, Weng W, Wei P, Qi P, Zhang Q, Tan C, Ni SJ, Dong L, Yang Y, et al. A Positive Feedback Loop of lncRNA-PVT1 and FOXM1 Facilitates Gastric Cancer Growth and Invasion. Clin Cancer Res. 2017b;23:2071–2080. doi: 10.1158/1078-0432.CCR-16-0742. [DOI] [PubMed] [Google Scholar]
- Yin Y, Yan P, Lu J, Song G, Zhu Y, Li Z, Zhao Y, Shen B, Huang X, Zhu H, et al. Opposing Roles for the lncRNA Haunt and Its Genomic Locus in Regulating HOXA Gene Activation during Embryonic Stem Cell Differentiation. Cell Stem Cell. 2015;16:504–516. doi: 10.1016/j.stem.2015.03.007. [DOI] [PubMed] [Google Scholar]
- Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, Meyerson M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet. 2016;48:176–182. doi: 10.1038/ng.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Pro-growth phenotype induced by CRISPRi-PVT1, related to Figure 1. A, Schematic representation of transcripts and corresponding TSS at PVT1 locus. sgRNAs used in this study are shown in the shaded box. Transcripts from each TSS are shown with the same color. B, Violin plots showing all pro-growth phenotypes across seven human cell lines except PVT1, which is represented as a dot. Significance was determined by p-value and basal transcription level in given cell line. Data was derived from (Liu et al., 2017). C, sgRNA enrichment of all individual sgRNAs targeting PVT1 TSS. Each TSS is colored as in Figure S1A. D, Comparison of sgRNA enrichment for representing reproducibility of CRISPRi screen for sgRNAs targeting PVT1 TSS. E, Images of time-course luminescence from subcutaneous xenografted MDA-MB-231 cell line with CRISPRi-Control or CRISPRi-PVT1 (R2) (upper). Luminescence from all mice with xenografts was quantified and presented as box plot (bottom). p-value was calculated by paired Mann-Whitney U-test (n=12 for each). Lines indicate mean of the luminescence at each time point.
Figure S2, CRISPRi-PVT1 cells outcompete in cell growth competition assay, related to Figure 1. A, Growth competition assay for CRISPRi-PVT1 in MCF-7 cell line, represented the same as Figure 1C. B, Fluorescence microscope images of competitive cell growth assay by 2D cell culture of MDA-MB-231 cell line. C, Density plots showing percentage of mCherry+ or EGFP+ cells measured using FACS in competitive cell growth assay of MDA-MB-231 cell line. The numbers indicate percentage of Q1 (mCherry+) or Q4 (GFP+) populations. Target genes of sgRNA are shown in the same color as of the fluorescent marker above each panel.
Figure S3. Pro-growth phenotype is induced by increased MYC expression, related to Figure 2 and Figure 3. A, Schematic representing qRT-PCR amplicon, probe, siRNA or sgRNA targeting MYC mRNA. Annotated isoforms of MYC are presented with accession numbers. Alternative promoters are shown (p0, p1 or p2). Median coverage of RNA-seq at indicated region (left) was used to calculate MYC promoter usage affected by CRISPRi-PVT1 (right). Mean percentage (n=2) of each promoter was used for statistical analysis (paired Mann-Whitney U-test). n.s., not significant (p>0.05). B, Image of northern blot for GAPDH (control) and MYC with CRISPRi-Control or CRISPRi-PVT1 in MDA-MB_231 cell line (top) and relative quantification of MYC mRNA (bottom). Error bars indicate mean ± SD (n=3, biological replicates). *p<0.05, using unpaired t-test. C, Schematic representing qRT-PCR primers, ASO or siRNA targeting PVT1 lncRNA. Arrows and bars are not scaled. PVT1-3′ primer was used for PVT1 unless indicated specifically. D, qRT-PCR for PVT1 (top) or MYC (bottom) in MDA-MB-231 cells expressing dCas9-KRAB or dCas9 with sgRNA-Control (Ctrl.), sgRNAs-PVT1 upstream (U1 to U10), TSS1(R1 to R10) or TSS2 (Y1 to Y10). Mean ± SEM (n=3, biological replicates). E, Correlation between sgRNA enrichment and relative MYC or PVT1 RNA levels, which were described in Figures S1C and S3D. Spearman's coefficient (R) and p-value are shown.
Figure S4. Epigenetic changes induced by CRISPRi targeting PVT1, related to Figure 4. A, RNA-seq, ChIP-seq or ATAC-seq signals around PVT1-MYC locus measured in MDA-MB-231 cell lines with CRISPRi-control or CRISPRi-PVT1. ChIP-seq peaks against H3K9me3 or H3K27ac at PVT1 promoter are shown enlarged in box. All sequencing was performed biological replicates. Peaks were visualized using UCSC genome browser. B to E, Fold changes comparing CRISPRi-PVT1 to CRISPRi-Control in ChIP-seq with indicated antibody (B to D) or ATAC-seq (E). Peaks within ± 1 kb of annotated transcribing region in RefSeq were compared with all other peaks by DESeq2. p-values were calculated by unpaired Mann-Whitney U-test (for n≥3). n.d., not detected.
Figure S5. PVT1 and MYC are co-regulated through chromatin contacts in a cell line specific manner, related to Figure 5. A, Enrichment of chromatin contacts comparing CRISPRi-PVT1 to CRISPRi-Control in MDA-MB-231 cell line which was measured by HiChIP-H3K27ac. left, 5 megabase; right, 500 kb region around PVT1. B, Chromosome contact frequency at 1 kb resolution anchored at MYC 3′-enhancer measured by HiChIP-H3K27ac in MDA-MB-231 cell line with CRISPRi-Control or CRISPRi-PVT1 (R3). C, Chromosome contact frequency at 5 kb resolution anchored at PVT1 promoter (top) or MYC 3′-enhancer (bottom) measured by UMI-4C in MCF-7 cell lines with CRISPRi-Control or CRISPRi-PVT1 (R2). D, Schematic representation of luciferase reporter assay (upper) and boxplots showing relative luminescence from the reporter assay. Luminescence of each biological replicate (n=3) was measured by triplicate luciferase reactions. E, qRT-PCR for PVT1 or MYC at 6 hours post JQ1 treatment in MDA-MB-231 cell line. Equal volume of DMSO was used for non-treated control. F, qRT-PCR for PVT1 or MYC transcripts levels in the indicated seven human cell lines with CRISPRi-Control or CRISPRi-PVT1. For E and F, Error bars indicate mean ± SEM (n=3, biological replicates). G, Chromatin accessibility profiles measured by ATAC-seq around PVT1-MYC locus in indicated cell lines. H, Chromosome contact frequency at 5 kb resolution anchored at PVT1 promoter (top) or MYC 3′-enhancer (bottom) measured by UMI-4C in MDA-MB-231, MCF-7 or HCT116 cell lines. I, Boxplots showing chromosome contact frequency at 5 kb resolution measured by UMI-4C in three different cell lines. Mean was normalized to MDA-MB-231 cell line for each interaction. p-value was calculated by unpaired Mann-Whitney U-test (n=8) compared to HCT116 cell line. J, Sequencing read alignment from 4C-seq anchored at MYC locus. For B, C and H, mean ± SEM from two biological replicates is represented with lines and shading. For C and H, the mean was measured from two biological replicates each of which comprises two technical replicates with two different primers. *p<0.05, using unpaired t-test compared with control (dark grey).
Figure S6, PVT1 allele-specifically regulates MYC transcription, related to Figure 6. A, Correlation between d-score of PVT1 chromatin accessibility from ATAC-seq and PVT1 transcription level from RNA-seq. B, Correlation between d-score of transcription level between PVT1 and MYC from RNA-seq. For A and B, n=15 from 11 clones and 4 additional technical replicates. C, Schematics showing location of primers used in targeted RNA-seq. Sequence of transcripts spanning defined SNPs of 129 (pink) or Cast allele (blue) are shown. D, Correlation between d-score from targeted RNA-seq with indicated primers (n=35). Spearman's coefficient (R) and p-value are shown.
Figure S7. Recurrent mutation and mutagenesis at PVT1 promoter, related to Figure 7. A, Scatter plot representing C-scores for all bases around PVT1 TSS ±5 kb region from CADD analysis. TSS1 or TSS2 of PVT1 is shown as a red or yellow line, respectively. Grey line indicates C-score=10, top 10%. Thick black line indicates mean of each base. B, Plots showing kernel density estimation of mutation frequency of 227 lncRNA on Chr 8 at TSS ±5 kb region of PVT1 TSS in all cancer types or sub-populations having high or low frequency of PVT1 mutation. Red line indicates PVT1. Grey line or dashed-grey line indicates 80th or 90th percentile, respectively. C, Circos plot illustrating genomic/transcriptomic evidence for PVT1 fusion transcripts detected in an ER- HER2+ human breast tumor. The ideogram for Chr 8 is shown in the outermost ring, followed by predicted absolute total Copy Number (Nt) and minor allele CN (Nb) from WGS data, SNP-chip data and transcriptional abundances shown on log2 scale of TPM (Transcripts Per Kilobase Million, 0-10). Links indicate breakpoints detected from RNA-seq (green) corresponding to fusion transcripts, or WGS (gray) indicating genomic translocations. The insert panel illustrates two sets of genomic breakpoints and three sets of fusion breakpoints, two of which are located in the PVT1 promoter region (chr8:128,806,984). D, Most abundant 10 alleles containing deletions at passage 30 (left) and their enrichment over passages (right). Allele sequences were ordered by their abundant at passage 30. Unmodified, unenriched or enriched sequence were shown in black, blue or red, respectively. E, qRT-PCR for MYC mRNA in clonal MDA-MB-231 cell lines having unmodified or mutant allele at PVT1 promoter. Sequences of sgRNAs used in mutant cloning and each heterozygous mutant were shown (left). Error bars indicate mean ± SEM (n=3, technical replicates). *p<0.05, using unpaired t-test comparing to non-clonal wild-type population. F, Three cases of lncRNA targeting CRISPRi upregulated genes distanced from the target locus. Genomic coordinates and their neighbor genes (left), relative RNA fold changes and adj. p-value from DESeq2 (right) are shown. Light blue, target lncRNA of CRISPRi; red, upregulated genes; triangles in green or light green, position of each sgRNA. Data was derived from (Liu et al., 2017).
Table S2. PVT1 translocation and fusion RNAs in human cancers, related to Figure 7.