

Significance
Microbiologists typically use laboratory systems to study the bacteria that infect humans. Over time, this has created a gap between what researchers understand about bacteria growing in the laboratory and those growing in humans. It is well-known that the behavior of bacteria is shaped by their environment, but how this behavior differs in laboratory models compared with human infections is poorly understood. We compared transcription data from a variety of human infections with data from a range of in vitro samples. We found important differences in expression of genes involved in antibiotic resistance, cell–cell communication, and metabolism. Understanding the bacterial expression patterns in human patients is a necessary step toward improved therapy and the development of more accurate laboratory models.
Keywords: Pseudomonas aeruginosa, cystic fibrosis, chronic wounds, human transcriptome, machine learning
Abstract
Laboratory experiments have uncovered many basic aspects of bacterial physiology and behavior. After the past century of mostly in vitro experiments, we now have detailed knowledge of bacterial behavior in standard laboratory conditions, but only a superficial understanding of bacterial functions and behaviors during human infection. It is well-known that the growth and behavior of bacteria are largely dictated by their environment, but how bacterial physiology differs in laboratory models compared with human infections is not known. To address this question, we compared the transcriptome of Pseudomonas aeruginosa during human infection to that of P. aeruginosa in a variety of laboratory conditions. Several pathways, including the bacterium’s primary quorum sensing system, had significantly lower expression in human infections than in many laboratory conditions. On the other hand, multiple genes known to confer antibiotic resistance had substantially higher expression in human infection than in laboratory conditions, potentially explaining why antibiotic resistance assays in the clinical laboratory frequently underestimate resistance in patients. Using a standard machine learning technique known as support vector machines, we identified a set of genes whose expression reliably distinguished in vitro conditions from human infections. Finally, we used these support vector machines with binary classification to force P. aeruginosa mouse infection transcriptomes to be classified as human or in vitro. Determining what differentiates our current models from clinical infections is important to better understand bacterial infections and will be necessary to create model systems that more accurately capture the biology of infection.
Since the earliest days of microbiology, researchers have relied on in vitro culture methods to grow pathogenic bacteria in the laboratory. Blood and beef extracts were initially used to grow bacteria to high densities, and soon researchers were using in vitro systems to understand basic biological principles, such as DNA replication, as well as clinically important questions, such as antibiotic tolerance (1). Blood and beef extracts were partially replaced by more consistent and reproducible media, and in vitro systems have since become a cornerstone of modern microbiology.
The use of in vitro models has clear benefits. In vitro experiments are typically inexpensive and allow for a relatively high degree of control and reproducibility. However, it is unclear how well these models mimic bacterial growth conditions during human infection. We know that there are fundamental differences between how a bacterium behaves in a test tube and in a human infection, but we often do not understand what causes these differences. What are the defining features of bacterial growth in a human infection that distinguish it from growth in common laboratory models?
This is a difficult question, as there are obvious limitations to human experimentation. We and others have previously used RNA-sequencing approaches to assess gene expression of human-associated bacterial communities, with a focus on predicting functional traits from gene-expression data (2–7). However, these approaches have been limited to microbial communities that contain large numbers of microbes or samples from which microbes can be easily harvested, such as the human oral cavity and urine collected from urinary tract infections. Here, we developed methodology to perform RNA-seq of the opportunistic bacterial pathogen Pseudomonas aeruginosa during human infection of soft-tissue wounds and cystic fibrosis (CF) lungs. Using machine learning approaches, we systematically identified a transcriptomic signature of P. aeruginosa during human infection that distinguishes it from in vitro laboratory transcriptomes. Specifically, we defined a limited number of genes that were sufficient to differentiate a human infection transcriptome from an in vitro culture transcriptome. Functionally, many of these genes are involved in iron acquisition and central metabolism. Genes important for antibiotic tolerance were also highly induced during human infection, potentially explaining the common occurrence of bacteria with low in vitro antibiotic susceptibility displaying high tolerance in humans. We propose that the framework provided here is an important step toward not only identifying important features of bacterial infection, but also in developing more biologically relevant in vitro models.
Results
In this work, we utilized P. aeruginosa RNA-seq results from three types of samples: human infections, mouse infections, and in vitro cultures. The human infections, which are the focus of this paper, included CF expectorated sputum samples from Denmark and soft-tissue infections from Denmark and the United States. Soft-tissue infections included chronic wound infection samples from Denmark and the United States and burn wound infection samples from the United States. Each sample was from a different patient, with the exception of the burn samples, which were acquired on two separate days from different areas on the same patient (Supporting Information and Dataset S1). These human infection samples were chosen not only because they came from distinct types of infections from different clinics, but also because the patients were treated with a variety of therapeutic regimens and had a wide-array of coinfecting microbes. This diversity among samples provided the opportunity to define the most basic ways P. aeruginosa human infection transcriptomes differ from in vitro transcriptomes independent of the infection location, therapeutic regimen, or coinfecting microbes. We also examined five P. aeruginosa transcriptomes from mouse models of infection: two from surgical wound models and two from burn wound infections from previous publications in our laboratory, and a pneumonia infection from another laboratory (8–12). Our in vitro data were composed of 87 RNA-seq datasets from our and other laboratories using laboratory, clinical, and environmental P. aeruginosa strains, 27 of which have not been published previously (Dataset S1) (8–11). The main technical obstacle of this project was obtaining sufficient P. aeruginosa reads from the human samples to analyze them. Of 28 human samples sequenced, we chose 15 for further analysis based on genome coverage (Table 1).
Table 1.
The human samples used in this study
| Sample | Location of collection | Total reads | Reads mapped to PAO1 orthologous genes |
| Human_Sputum_A | Denmark | 71,926,942 | 492,733 |
| Human_Sputum_B | Denmark | 105,647,965 | 2,047,185 |
| Human_Sputum_C | Denmark | 61,212,590 | 1,958,877 |
| Human_Sputum_D | Denmark | 124,266,260 | 414,990 |
| Human_Sputum_E | Denmark | 62,985,339 | 453,389 |
| Human_Sputum_F | Denmark | 68,022,834 | 184,178 |
| Human_Sputum_G | Denmark | 78,869,961 | 10,213,756 |
| Lubbock_CW_15 | Texas, USA | 109,403,011 | 2,251,929 |
| Lubbock_CW_16 | Texas, USA | 94,788,090 | 191,274 |
| Lubbock_CW_18 | Texas, USA | 91,993,624 | 713,108 |
| Lubbock_CW_21 | Texas, USA | 109,252,642 | 30,836 |
| Lubbock _Burn_2_3 | Texas, USA | 46,780,195 | 682,982 |
| Lubbock_ Burn_2_4 | Texas, USA | 34,878,564 | 105,278 |
| Danish_Chronic_Wound_1* | Denmark | 106,188,256 | 5,112 |
| Danish_Chronic_Wound_2* | Denmark | 99,773,828 | 10,177 |
These samples were analyzed separately from the other samples in our study due to their low P. aeruginosa read counts and were only used in Fig. 1B.
Because our analysis contained samples with different strains of P. aeruginosa, we did not have a reference genome in which to map the sequencing reads. Additionally, many samples contained bacterial species other than P. aeruginosa that could impact our mapping due to orthologous genes with high sequence similarity. To deal with these issues, we first mapped all RNA-seq reads to 54 bacterial species that had been identified as present in our samples by CLARK (Dataset S1) (13). We took one or more genomes among these “decoy” species (101 in total) to initially map against. We focused on those reads that did not map to these other microbes to avoid falsely ascribing differences in transcript frequencies to P. aeruginosa when another bacterium was potentially responsible. Our approach was conservative because P. aeruginosa reads that closely resemble other species were discarded. However, it had the advantage that we could be confident that the differences we observed were due to differences in P. aeruginosa RNA levels rather than RNA levels in other bacteria (see flowchart in Fig. S1). We first mapped all remaining reads to the well-studied strain P. aeruginosa PAO1, then to a pangenome containing 28 additional P. aeruginosa strains. At this point, the reads were collapsed onto P. aeruginosa PAO1 orthologs (Fig. S1) (the list of such orthologs of the strains is curated on Pseudomonas.com). This approach was reasonable because the genome of P. aeruginosa is highly conserved between strains (14). Throughout this paper, we focus on this collapsed genome to make comparisons between samples.
P. aeruginosa in Human Samples and in Vitro Samples Have Distinct Transcriptomes.
After mapping all reads, most human infection transcriptomes had several genes with zero reads that mapped (Fig. S2). This was due to a combination of sequencing depth and strain differences. To make preliminary comparisons between samples without biasing results due to the presence/absence of particular genes across strains, we restricted our analysis to samples with a common set of 1,707 genes that had at least one read in a large number of samples (Fig. S3A). A principle component analysis (PCA) of these data (Fig. 1A) shows that human transcriptomes tend to cluster near one another, with CF sputum transcriptomes most distinct from the other samples. The number of genes used in Fig. 1A was too high to include two chronic wound transcriptomes from Denmark, and we wanted to determine if these samples would cluster with chronic wound samples obtained from the United States. To determine this, we used a smaller set of 761 genes in order to include these samples (Fig. S3B). Our results revealed that the Danish chronic wound transcriptomes clustered along with the chronic wounds from patients in the United States (Fig. 1B). These data indicate that the P. aeruginosa transcriptome during human infection clusters distinctly from in vitro transcriptomes, despite differences in site of infection, coinfecting microbes, therapeutic regimen, and geographical locale. Additional PCs are shown in Fig. S4, and we included a PCA with subsampled reads in Fig. S5 to show that clustering is not a consequence of having fewer mapped reads in our human samples.
Fig. 1.

PCA of P. aeruginosa RNA-seq results. This includes human samples listed in Table 1, as well as mouse and in vitro experiments from our laboratory and others (Dataset S1). (A) Analysis was performed with 1,707 genes that were expressed (i.e., contained at least 1 RNA-seq read) in all samples. (B) To include two chronic wound samples from Denmark with low-read coverage (labeled in figure), analysis was performed with 761 genes that were expressed in all samples.
Several classes of genes differed substantially in RNA transcript levels between in vitro and human samples, and some were verified with quantitative PCR (Supporting Information). Fig. 2 shows an overview of the categories enriched for differentially expressed genes. In each of these categories, which include the TCA cycle as well as the biosynthesis of amino acids, carbohydrates, and nucleosides and nucleotides, most genes were expressed at a higher level in vitro than in human infections. The majority of genes involved in the TCA cycle—including sdhC, sdhD, sdhA, sucC, sucD, and acnB—were expressed at lower levels in the human transcriptomes compared with in vitro conditions. Additionally, several important regulons showed significant differences in transcript levels between in vitro and human samples, including the SOS stress response (under control of lexA), which was more highly expressed in humans. Furthermore, the σ-factor algU, which controls genes involved in alginate synthesis, was expressed at substantially higher levels in human samples. A previous paper from our laboratory identified long-chain fatty acid catabolism, and in particular faoA and faoB, as being required during both the P. aeruginosa murine surgical wound and burn wound infection, and induced in the burn infection (12). These genes were induced in our human samples as well; faoA was expressed higher in human samples than the in vitro ones (1.6-fold higher, P = 0.06), and faoB was also more highly expressed in our human samples (2.1-fold higher, P = 0.002).
Fig. 2.

Gene categories that are significantly different in in vitro transcriptomes compared with human transcriptomes. Analysis was performed with 1,707 genes that were expressed (i.e., contained at least 1 RNA-seq read) in all samples. Categories and enrichment calculation were obtained from the BioCyc database using Grossmann’s parent–child-union variation of the Fisher’s exact test with a P value cut-off of 0.05 (48). Plotted are the genes with a P-adjusted value of <0.05. “Considered” genes indicates the number of genes within that category that were analyzed (i.e., included in the 1,707 genes).
Quorum-Sensing Genes Are Expressed at Lower Levels in Human Infection.
Another basic question is the degree to which social behaviors are active in infecting bacteria. The last three decades have seen an explosion of work demonstrating the importance of microbial social behaviors during infection including cooperation and competition (15). Among these behaviors, cell-to-cell signaling, and in particular quorum sensing (QS), has received special attention (16). Although considerable work has gone into elucidating the properties of QS in vitro, little is known about its activity in human infection. One of the main QS systems in P. aeruginosa (referred to as the las system) regulates transcription of over 5% of the organism’s genome (17). The las system is composed of LasI, which synthesizes the AHL signal molecule 3-oxo-dodecanoyl homoserine lactone (3OC12-HSL), which then binds to the transcriptional regulator LasR, enabling transcriptional regulation of a variety of genes.
A previous paper identified a core set of 42 genes that were regulated by the P. aeruginosa las system across seven P. aeruginosa isolates (18). We performed a differential expression analysis of all genes to determine whether this “core las QS regulon” was differentially induced in human infections and in vitro conditions. In doing this comparison, for each of the 42 genes, we ignored transcriptomes that did not have at least three reads for that gene to ensure the presence of the gene in the human samples. Our rationale was to avoid falsely classifying clinical isolates that may lack a particular QS-regulated gene, instead having very low expression of it. We chose the three read cut-off as a conservative compromise between ensuring that a given gene was not incorrectly mapped by a single read and not having to discard samples. The median number of excluded samples was 3 for human samples and 1 for in vitro samples. We found that mRNA levels for genes controlled by the las QS system were considerably lower in the human transcriptomes than in vitro transcriptomes (Fig. 3A). The QS regulon is enriched for decreased gene expression in humans (Fisher’s exact test, P = 3.1e-6, odds ratio = 5.1). We looked further to see if there were in vitro conditions that most contributed to this difference in gene expression and discovered that on average, in vitro transcriptomes in which P. aeruginosa was grown as biofilms or aggregates expressed the core QS regulon the highest. When the in vitro data are split into biofilm/aggregate samples versus planktonic (Fig. 3B), the genes in general have higher expression among biofilm/aggregates compared with human samples (Fisher’s exact test, P = 1.8e-9, odds ratio = 18.1), but with this comparison approach there is no such difference with the planktonic samples (Fisher’s exact test, P > 0.05).
Fig. 3.

The expression of genes in the las core QS regulon in human samples compared with in vitro samples (18). (A) Average relative expression of 42 core QS genes in vitro and in humans. Fold-change in expression was calculated as a ratio of the geometric mean of relative expressions for each gene among the in vitro transcriptomes to the geometric mean of relative expressions of each gene among the human transcriptomes. Values above 0 indicate higher gene expression in vitro. Samples with fewer than three reads for a gene were removed from the analysis. (B) Average relative expression of 42 core QS genes within in vitro biofilm/aggregate transcriptomes or in vitro planktonic transcriptomes, compared with human transcriptomes. Fold-change in expression of each gene was calculated as above. Bars represent SEM.
P. aeruginosa Antibiotic Tolerance Determinants Are Induced During Human Infection.
Our initial analysis indicated that two antibiotic efflux genes (mexX and mexY) were expressed highly in our human transcriptomes, so we decided to perform a more rigorous test to identify other antibiotic resistance determinants that were induced in these samples. To answer this question, we first identified genes that were both transcribed in humans and had previously been shown to be important for P. aeruginosa antibiotic tolerance when mutated (19). To determine which genes increased in expression among the human samples compared with in vitro samples, in contrast to our first differential expression analysis (Fig. 2), we examined all genes, even those that had no mapped reads in some transcriptomes. Our rationale was that this approach may give false positives for genes that have reduced expression in the human samples, but it should still be conservative in identifying genes that had increased expression. Table 2 shows, for each infection type, the genes that were both induced in the human samples compared with in vitro conditions and also important for growth in the presence of antimicrobials (19). We also verified that these differences were significant even if we eliminated in vitro samples with zero counts for any of these genes. Our results reveal that efflux pumps associated with tolerance to aminoglycosides and other antimicrobial classes, in addition to SOS response genes, were induced in multiple human infections. mexX, mexR, and mexY were induced in the sputum samples; mexX and mexY were induced in the chronic wounds; and armZ (a mexZ antirepressor) was induced in the burn wound (Table 2). The sputum and burn wound transcriptomes included induction of recN and lexA, indicating activation of the SOS DNA-damage response. Other antibiotic tolerance-associated genes, from a range of functions, were induced as well. Among the CF sputum samples, ftsH, which encodes a membrane protease known to be important in aminoglycoside resistance, was induced (20). Also in the CF sputum, znuA, znuB, and znuC were induced. This is consistent with a previous microarray study on human CF samples, and further, mutating these genes reduces bacterial growth in animal models (21, 22). The clinically important β-lactamase ampC, was also induced in the sputum samples, as well as various other resistance genes (Table 2). In addition to the efflux pumps, in the chronic wound samples, hudR, a virulence factor that also confers tolerance to gentamicin was induced (23). The human burn wound showed induction of mvaT, which is a resistance determinant for gentamicin (19). Several of the resistance determinants induced in human infections were hypothetical proteins (Table 2). For example, hypothetical protein PA1414 was highly induced in all three infection types (between 5- and 34-fold higher in human samples) and was shown to be important for fitness in the presence of gentamicin (19). PA5469 was highly induced in two infection types as well and was also a resistance determinant for gentamicin (19). Both of these genes were also induced in at least one mouse model (24). It is also worth noting that the genes oprD and oprF, which encode for porins known to facilitate antibiotic uptake, had significantly decreased expression across our human samples.
Table 2.
P. aeruginosa genes induced in human samples that are also important for antimicrobial tolerance (19)
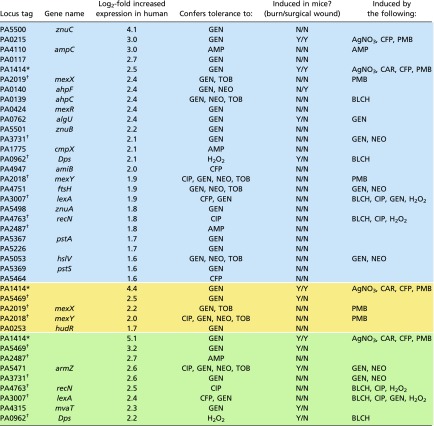 |
The above genes were both expressed higher in our human infections than in vitro samples (log2 fold-change > 1.5, FDR < 0.05) and were fitness determinants when grown in the presence of antimicrobials [log2 fold-change < −1.5, FDR < 0.05 (19)]. Blue boxes shade genes up-regulated in human CF sputum, yellow shade genes up-regulated in human chronic wounds, and green shade genes up-regulated in human burn wounds. Also included is whether these genes were induced in mouse burn or surgical wounds [fold-change > 2, P value < 0.05 (12)] and induced by any of 14 antimicrobials in vitro [log2 fold-change > 0, FDR < 0.05 (19)]. AMP, ampicillin; BLCH, bleach; CAR, carbenicillin; CFP, cefoperazone; CIP, ciprofloxacin; GEN, gentamicin; NEO, neomycin; PMB, polymyxin B; TOB, tobramycin.
Gene appears in all three infection types.
Gene appears in two of the three human infection types in the table.
We also asked the question of whether any of these genes were induced in the reference P. aeruginosa strains PA14 or PAO1 in mouse infection models in which no antibiotics had been administered (12), or if they were induced in vitro by the addition of any of 14 antimicrobials (19). This information is revealing as it provides clues as to whether increased expression of these genes in humans was a result of the growth environment or due to therapeutics administered to the patient. For example, mexX and mexY are critical determinants of aminoglycoside tolerance that were highly induced in human samples but not mouse infection models; they are, however, induced by the antibiotic polymixin B (Table 2). Because most of the patients included in this study were administered polymixin E (colistin) (Dataset S1), it is likely that induction of these genes was due to antibiotic treatment. Four genes, including PA5469 and mvaT, were highly induced in human and mouse infection but not by antimicrobial addition (Table 2), suggesting that induction of these genes may be a response to the in vivo infection environment. Finally, 12 genes including znuABC are induced in human infection but not in mouse infection models or by antimicrobial addition (Table 2), thus these genes may be responding to a human infection-specific cue.
A P. aeruginosa Transcriptional Signature of Human Infection.
Examining differences in average gene expression between human and in vitro samples provides an initial glimpse into how these environments impact P. aeruginosa physiology and behavior; however, machine learning approaches are better suited to determining what signatures most reliably differentiate these groups. To initiate these studies, we reduced the dimensions of our dataset by removing redundant information and noise. We used the DaMirSeq R package to determine a set of genes whose PCs best correlated with in vitro or human growth, by performing backward variable elimination with partial least-squares regression, and removing redundant features by eliminating those that were very highly correlated (25). Repeating this process 20 times, we chose the top 30 genes identified here as input into our support vector machine (SVM) models. We then used an SVM with a linear kernel in the Caret R package to determine how accurately simple rules could distinguish human from in vitro transcriptomes (26). We trained our model with human and in vitro transcriptome data, excluding the mouse samples. We conducted a leave-one-out cross-validation of both the feature selection and SVM training processes, repeating 20 times to account for stochasticity in the feature selection step, which yielded a mean 98.9% accuracy and a Cohen’s κ of 95.4%, indicating that this approach is highly accurate for differentiating human from in vitro transcriptomes. When we trained the model with in vitro data excluding all samples from any one of the four other laboratories that generated the data, the SVM correctly identified all samples from the omitted laboratory as being in vitro, except in the case of one laboratory (8), all of whose experiments were conducted in media specifically designed to mimic CF sputum in differing oxygen levels (27); four of these samples were designated as “human.” When we trained the SVM with all of the United States chronic wounds omitted, or all of the Denmark transcriptomes omitted, the SVM correctly identified the left-out transcriptomes as being human.
The genes used by the SVM to differentiate human and in vitro P. aeruginosa transcriptomes in a single iteration are shown in Table 3. It is important to note that these are not necessarily all of the important genes that can differentiate sample type, but rather a subset of them selected using a nondeterministic algorithm. Furthermore, they are not necessarily the most differentially expressed (Dataset S2), but they are effective in discriminating human from in vitro transcriptomes when used together. These genes encode proteins that localize in a range of cell compartments, but are predominantly in the cytoplasm and cytoplasmic membrane (28). Table 3 also shows the fold-change, calculated by DESeq2 (29). Most of the features that distinguish human infections from in vitro transcriptomes, like the efflux pump mexY, pathways for Psl exopolysaccharide production, siderophores (pvdF, pchR), heme uptake (phuR, phuS), and lldA (l-lactate dehydrogenase), were transcribed more highly in human samples. However, a few of the strongest signature genes showed reduced mRNA levels in humans, including gcbA (involved in flagellar motility and biofilm formation) (30, 31). In principle, a gene may be important to distinguish human from in vitro transcriptomes even if it only can distinguish between in vitro growth and one of our two classes of infection (CF sputum or soft tissue). However, we see in Table 3 that the majority of the highest-ranking genes are indeed substantially different in the in vitro samples compared with both human infection types. Some genes though, like phuR, were clearly more distinct between in vitro conditions and CF sputum in comparison with in vitro versus soft tissue.
Table 3.
Genes used to distinguish P. aeruginosa human infection transcriptome from in vitro transcriptome
| Locus tag | Gene name | Human infection vs. in vitro (log2 fold-change) | Soft tissue infection vs. in vitro (log2 fold-change) | CF sputum vs. in vitro (log2 fold-change) |
| PA2911 | 4.1 | 3.8 | 4.2 | |
| PA2914 | 3.2 | 3.1 | 3.2 | |
| PA5535 | 4.1 | 3.7 | 4.4 | |
| PA1414 | 4.6 | 5.4 | 3.2 | |
| PA0781 | 2.9 | 2.6 | 3.2 | |
| PA2382 | lldA | 4.9 | 2.1 | 5.7 |
| PA4835 | 2.8 | 2.2 | 3.1 | |
| PA2943 | 3.2 | 1.8 | 3.7 | |
| PA3598 | 3.5 | 2.3 | 4.0 | |
| PA4063 | 2.9 | 2.0 | 3.4 | |
| PA1797 | 2.8 | 3.2 | 2.5 | |
| PA4570 | 2.8 | 2.0 | 3.2 | |
| PA3237 | 7.0 | 4.0 | 7.9 | |
| PA2018 | mexY | 2.3 | 2.0 | 2.5 |
| PA4495 | 4.0 | 1.5 | 4.8 | |
| PA4709 | phuS | 2.4 | 1.9 | 2.8 |
| PA4710 | phuR | 3.4 | 0.8 | 4.2 |
| PA2662 | 3.7 | 1.7 | 4.4 | |
| PA4843 | gcbA | −3.2 | −2.6 | −3.9 |
| PA4470 | fumC1 | 1.8 | 0.6 | 2.3 |
| PA2931 | cifR | 2.2 | 1.7 | 2.6 |
| PA2562 | 1.7 | 0.3 | 2.3 | |
| PA2396 | pvdF | 1.7 | 1.1 | 2.1 |
| PA4227 | pchR | 1.6 | 0.5 | 2.1 |
| PA2386 | pvdA | 1.0 | −0.1 | 1.5 |
| PA3418 | ldh | −0.5 | −0.1 | −0.9 |
| PA0865 | hpd | −2.6 | −3.5 | −2.1 |
| PA3691 | −0.1 | 0.8 | −2.6 | |
| PA2291 | −2.8 | −2.6 | −2.9 | |
| PA2553 | −2.3 | −2.8 | −2.0 |
Genes were ranked according to their ROC importance score in a trained SVM model (26). Also shown is the DESeq2 log2 fold-changes when in vitro samples were compared with all human infections (human infection vs. in vitro), human soft-tissue infections (soft tissue infection vs. in vitro) and CF sputum (CF sputum vs. in vitro) (29). Positive values indicate genes up-regulated in human infection.
Surgical Wound Infection Mouse Models Are Classified as “Human,” While Other Mouse Models Are Classified as “in Vitro.”
In the previous section, we trained an SVM model to distinguish between human infections and in vitro transcriptomes. In doing so, we omitted the five mouse transcriptomes. As an example of the type of question one can begin to address with our comparative methods, we next asked the simple question: Where do the mice fall in this classification—as human infections or as in vitro models? Because the feature (gene) selection process of our SVM model is stochastic, we ran it 50 times and took the average of these runs. Because we used an SVM with binary classification, each mouse sample was forced into one of the two categories in each run. Table 4 shows the average assigned “probability” score calculated by the Caret package from the distance to the SVM decision boundary (32). We found that the P. aeruginosa murine surgical wound transcriptomes tended to be classified as human rather than in vitro, while murine burn transcriptomes and the murine pneumonia transcriptome were more strongly classified as in vitro. We next asked where these mice would be categorized when classified by an SVM trained to reliably distinguish soft-tissue infection transcriptomes (chronic wounds, burn) from CF sputum transcriptomes. All of the murine transcriptomes were classified as soft-tissue infections.
Table 4.
Classification of P. aeruginosa transcriptomes from mouse models of infection as: human or in vitro, and human soft tissue or CF sputum
| Sample | Human vs. in vitro | Soft tissue vs. CF sputum | ||
| Mean probability human | SD | Mean probability soft tissue | SD | |
| Mouse burn 1 | 0.07 | 0.04 | 0.73 | 0.17 |
| Mouse burn 2 | 0.15 | 0.08 | 0.72 | 0.19 |
| Mouse surgical 1 | 0.72 | 0.15 | 0.68 | 0.19 |
| Mouse surgical 2 | 0.75 | 0.12 | 0.68 | 0.21 |
| Mouse pneumonia | 0.08 | 0.08 | 0.67 | 0.25 |
For both distinctions (human vs. in vitro and soft tissue vs. CF sputum), feature (gene) selection and model training were repeated 50 times. The mean probability according to the SVM model and its SD were calculated using the Caret package (26). Mean probability is the likelihood that a mouse transcriptome is human (for the human vs. in vitro) or soft tissue (for the soft tissue vs. CF sputum) based on the SVM model as calculated by the Caret package. For example, a mean probability of 1 would indicate that the sample is unambiguously categorized as human for the human vs. in vitro.
Discussion
Determining the behavior of bacteria during human infection is one of the most basic and important questions in infectious disease microbiology (12, 24). To address this question, we performed RNA-seq analysis on a diverse set of 15 human infection samples containing P. aeruginosa and analyzed these data in the context of 87 in vitro transcriptomes and 5 murine infection transcriptomes (Table 1 and Dataset S1). The in vitro samples differed widely from one another, while human soft-tissue wound infections (chronic wound and burn) were more similar to each other, and human CF sputum samples were varied but clearly distinct from the other samples (Fig. 1). Several metabolic gene categories were significantly down-regulated in human transcriptomes compared with in vitro conditions (Fig. 2), as well as the core QS regulon (Fig. 3). We also found that a small set of genes (Table 3) could be used to reliably determine which transcriptomes are from human infections and which are from in vitro samples. Using this signature of human growth, some mouse models were classified as human and others in vitro.
Several pathways, including the pyoverdine synthesis pathway, several efflux systems, and the SOS response regulon were induced more in human infections compared with in vitro conditions. Consistent with previous work, mRNA for the important P. aeruginosa fatty-acid catabolism genes faoA and faoB were increased in human samples (12). Several other metabolic pathways, including the TCA cycle, had lower expression in human samples than in the in vitro samples. The fact that efflux systems and DNA damage stress-response regulons were expressed significantly higher in the human infections motivated us to look for other genes with high expression in human infections that may also be antibiotic-resistance determinants. In addition to several efflux-related and DNA damage-response genes, many other P. aeruginosa resistance genes were induced as well, including those involved in zinc transport, the heat-shock response, and other virulence factors (Table 2). Furthermore, two porins (oprF and oprD) implicated in antibiotic import into the cell were down-regulated. Some of these genes, including mexXY, appear to be induced by the therapeutics being administered to the patient, although many have not been shown to be induced by antimicrobials (Table 2). Three genes (PA0140, PA5469, PA4315) were induced in human and mouse infections but not by antimicrobial addition, and 13 other genes were induced in human infection but not in mouse infections or by antimicrobial addition. The prevalence of genes that were induced in human infections but not inducible by antibiotics suggests a possible explanation for why clinical antimicrobial susceptibilities often overestimate the drug efficacy seen in patients. Standard media may not induce several important resistance factors that are induced in humans, and therefore may overestimate the bacterium’s sensitivity to an antibiotic. It is worth noting that the above tally of genes is in some ways conservative because it includes a larger set of antimicrobials than any particular patient was exposed to, and also two genes were induced only by bleach, which is not administered to patients.
We were also able to address the important question of QS gene expression during human infection. Although considerable work has gone into characterizing P. aeruginosa QS, almost nothing is known about its activity during human infection. The P. aeruginosa las QS system is known to regulate a suite of virulence factors and over 5% of the bacterium’s genome (20). To explore this issue, we focused on a set of 42 genes that were previously shown to be conserved across a wide variety of isolates (18). Our data indicate that overall, genes in the core “QS regulon” of P. aeruginosa had a considerably lower expression in human transcriptomes compared with in vitro transcriptomes (Fig. 3A). However, when we dug deeper, we discovered that the in vitro samples that were most highly induced were those growing in colony biofilms and media that promotes aggregation (Fig. 3B). These results suggest that some laboratory growth environments may inflate the expression of QS and other social traits compared with human infection. It is important to remember that our data represent a single snapshot of gene expression; so even though these genes are relatively low in expression during human infection, they may still be important for bacterial fitness and could have been expressed at higher levels in earlier stages of infection. In addition, bacteria in chronic infections may acquire mutations that lower expression of genes encoding social behaviors.
In an effort to determine the rules for distinguishing human infection transcriptomes from in vitro transcriptomes, we trained an SVM to classify the samples. Our SVM performed well in cross-validation, which is somewhat surprising given that the human samples and in vitro samples contained different P. aeruginosa strains and the human infections contain a wide array of coinfecting microbes. When we extracted the genes most responsible for the differentiation, many were either involved in nutrient acquisition and metabolism or were hypothetical proteins (Table 3). This suggests that human infections are a distinct growth environment that likely require a unique suite of metabolic pathways. Although the mexY gene was also shown to be important for discriminating in vitro from human (Table 3), it is likely that the transcriptional response to antibiotic administration in human infections has a minor impact on the discriminating genes as only four other genes of the 30 are inducible by antimicrobials (PA4063, PA1797, PA1414, PA2883) (19).
For over 80 y, laboratory mice have been used as a model to understand human infection (33). Recently, researchers have begun asking to what degree laboratory mice recapitulate the immune system of wild mice (33). With our human and mouse transcriptomes, we have begun to ask the next question: to what extent do mouse infection models recapitulate the bacterial physiology of that in human infections? Not only are mice fundamentally different from humans biologically, but the P. aeruginosa infection models themselves, particularly inoculation procedures, have a significant in vitro component. A first hint about the relation between P. aeruginosa mouse infections and our other categories is given in the PCA in Fig. 1, wherein the mice cluster between the human and in vitro samples. To address this question more directly, after training our machine learning model to distinguish human from in vitro transcriptomes, we asked whether our mouse transcriptomes would be classified as human or in vitro transcriptomes. We found that P. aeruginosa murine surgical wound transcriptomes were classified as human infections rather than as in vitro samples, while the murine pneumonia and burn wound transcriptomes were classified as in vitro rather than human (Table 4). It is worth cautioning that the SVM classification we used inherently forces the mouse transcriptomes into one of two categories (e.g., in vitro or human) based on the genes it used to differentiate between those categories. As a result, it is difficult to know whether mice were in some sense intermediate between the two categories, or if they are very distinct and were being forced into a dissimilar class due to the nature of binary classification. This said, the mice, on average, have transcription levels that are intermediate between in vitro and human samples among the SVM genes in Table 3 (Fig. S6), with burn mice consistently closer to in vitro samples among our SVM gene set, but there is more ambiguity for the pneumonia and surgical wound samples. With the above caveat in mind, it seems reasonable that the P. aeruginosa surgical wound transcriptome, which is from a self-limiting infection model intended to mimic chronic infection, would be more similar to our human transcriptomes since all but the burn samples were from chronic infections. In contrast, the murine burn and pneumonia transcriptomes are more acute mouse infections and likely involve a rapid proliferation of P. aeruginosa, which may in some ways resemble typical in vitro growth conditions. It is also interesting that the murine pneumonia transcriptome tended to be classified as a human chronic wound transcriptome more than as a transcriptome from human lungs (CF sputum) (Table 4).
Performing RNA-seq on clinical samples is a powerful approach to study natural infections because it does not require special preparation before sampling that would disturb the bacterial community. This allows researchers to examine bacterial behaviors and functions in a relatively undisturbed environment. However, in contrast to laboratory models, neither the genetics of the infecting bacterium nor the genetics of the host can be manipulated. For example, although we expect most of the shifts in expression to be caused by environmental differences, some expression differences may be caused by bacterial mutations. The alternative σ factor encoding gene algU, for example, was induced in many CF sputum transcriptomes, and this could be because mutations that induce its expression are common in long-term infections (34). This said, many of the effects are clearly not entirely genetically determined as chronic wound transcriptomes from Denmark samples clustered closely with chronic wound samples from the United States (Fig. 1B). Additionally, the same strain often clustered differently on the PCA (Fig. S7A), and media seemed to have a substantial impact on clustering (Fig. S7B). Future work should begin to uncover the interplay between bacterial genetics and environmental effects. Although the lack of manipulative control makes teasing out particular features of gene expression difficult, it also demonstrates the robustness of the “human biomarkers” that we discovered. Even with large differences between strains, differing coinfecting bacteria, and variations in patient genetics, comorbidities, drugs, and diet, there were clear patterns across samples. Similarly, our in vitro data came from different laboratories using different protocols for sample preparation before sequencing, which might impact results (Fig. S7C); however, this is difficult to know with our data because many of the experimental conditions differed by laboratory as well. However, even though the samples we used were from different experiments conducted in different laboratories, there were still underlying similarities across the in vitro samples that made it possible to distinguish sample types from one another. So the approach we used here is a double-edged sword: while providing less control over particular genetic or environmental factors, it provides a more robust assessment of the most salient features in human infections.
Besides the issues common to any nonmanipulative experiment, there are a couple caveats specific to interpreting RNA-seq data. Recent work has demonstrated that within the lungs of CF patients, clonally related strains of P. aeruginosa differ functionally and genetically, depending on location in the lung (35). RNA-seq only provides an average expression of the bacteria in our sample, and so differences between subpopulations are ignored. Although we cannot know whether specific subpopulations are responsible for the differential expression we identified, we can say that at the population level, P. aeruginosa had high expression of particular genes. Also, as mentioned above, RNA-seq experiments represent only a snapshot of the population at one point in time. It may well miss important dynamics that occurred before sampling.
Laboratory models have been essential for the progress of microbiology over the last century, but there is still a large gap between our knowledge of bacterial growth in laboratory conditions and in humans. It is clear that the environment in a human infection differs fundamentally from in the laboratory. These differences include the presence of immune cells, different nutritional sources, and the presence of coinfecting bacteria (36). However, we know relatively little about how these differences impact bacterial physiology in infection. As more laboratories begin to conduct RNA-seq of human infections, we will get a better understanding of bacterial physiology and behaviors during infection. The machine learning framework we use here will become increasingly accurate as more data are published, providing an increasingly clear description of the transcriptional signature of P. aeruginosa and other species during infection. As acute infection data become available, it will be interesting to compare the characteristically induced genes of these infections to chronic infections. Future transcriptomic data will bridge the current chasm between laboratory experiments and human infection, and will help inform the development of more accurate in vitro models.
Methods
Bacterial Strains and Growth Media.
The following strains were used in this study: P. aeruginosa strains PAO1 (37), UCBPP-PA14 (PA14) (38), LESB58-SE021 (39), Acinetobacter baumanii strain 5075 (AB5075) (40), Staphylococcus aureus strains TCH70 (TCH70; GenBank accession ACHH00000000.2) and LAC* (41), Staphylococcus epidermidis (SK135; GenBank accession ADEY00000000.1), and Micrococcus luteus SK58 (SK58; GenBank accession ADCD00000000.1). S. aureus TCH70 and LAC* are methicillin resistant strains. Isolates were routinely grown on tryptic soy agar incubated at 37 °C in ambient air. SCFM2 was prepared as previously described (24). Chemically defined media (CDM) was prepared as previously described (19, 42). Planktonic cultures were grown at 37 °C with shaking at 225 rpm. For CDM agar plates, 2× CDM was combined with equal volumes of 3% Noble Agar. Mops-succinate was prepared as previously described (27).
Colony Biofilms.
Bacteria were grown on CDM supplemented with 20 mM glucose (19, 42). Isolates were grown overnight in CDM glucose at 37 °C with shaking at 250 rpm. The OD600 of the overnight culture was measured. Each isolate was adjusted to OD600 = 1 in 200 μL of overnight culture supernatant, which was prepared by centrifuging a 1-mL aliquot of overnight culture at maximum speed on a tabletop centrifuge for 2 min. Isolates were combined 1:1 in the following coculture combinations: PA14 and AB5075, PA14 and TCH70, PA14 and SK135, and PA14 and SK58. PA14 in monoculture was used as a control. Colony biofilms were formed by spreading 40 μL of coculture or monoculture onto a 0.2-μm nuclepore track-etch membrane (Whatman) placed on a CDM glucose agar plate. The combined final cell concentration was ∼2 × 107 colony forming units per strain. Cocultures were incubated at 37 °C for 5 h. Five biofilm filters were combined for each RNA-seq experiment and placed in 5 mL RNAlater (Thermo Fisher Scientific).
Growth in SCFM2.
P. aeruginosa was grown in SCFM2 as previously described (24). Briefly, 500 µL of SCFM2 in four-well microchamber slides from Nunc (900 µL per chamber) was inoculated at an OD of 0.05 of P. aeruginosa PAO1 or LESB58-SED21, grown for 7 h, and then an equal volume of RNA later was immediately added. Four technical replicates were combined for each biological replicate. The PA14 samples in SCFM2 were grown the same way, except with two technical replicates combined for each biological replicate.
Datasets from Previous Studies.
Datasets from several previously published studies from our laboratory were used, including murine surgical wounds (12), murine burn wounds (12), PAO1 planktonically grown in Mops-succinate (12), and PA14 planktonically grown in CDM (19). Additionally, we used data from other laboratories (8–11).
RNA Extraction and Preparation of Sequencing Libraries for RNA-Seq.
In vitro and human samples were prepared as previously described (19) with a few modifications for the human samples. The burn sample was removed from RNAlater and immediately placed into 1 mL RNA-Bee in bead-beating tubes containing 0.1-mm beads (MP Biomedical). For the human chronic wounds, large pieces of tissue were removed and placed in 2 mL RNA-bee in bead-beating tubes and the remaining debrided tissue was spun at 14,000 × g. The pellet of debrided tissue was resuspended in 1 mL RNA-bee and combined with the large pieces of tissue, resulting in a total of 3 mL RNA-bee. In vitro cultures stored in RNAlater were pelleted, resuspended in 1 mL RNA Bee, and transferred to bead-beating tubes. Cells were lysed by bead beating three times for 60 s, and the tubes placed on ice for 1 min between each homogenization. Amounts of 200 µL of chloroform per 1 mL of RNA-bee were added, and the tubes were shaken vigorously for 30 s and incubated on ice for 5 min. Samples were centrifuged at 12,000 × g for 15 min at 4 °C to separate the aqueous and organic phases. The top aqueous phase from each tube was transferred to a new microcentrifuge tube to which 0.5 mL isopropanol per 1 mL of RNA-bee was added, and the tubes were incubated at room temperature for 10 min. Amounts of 20 µg of linear acrylamide were added to the tubes, and the samples were centrifuged at 12,000 × g for 5 min at 4 °C. The pellets were washed with 1 mL 75% ethanol, air dried for 10 min, and resuspended in 25 µL of RNase-free water. The RNA concentration for each sample was determined with a NanoDrop spectrophotometer (Thermo Fisher Scientific). Ribosomal RNA was depleted using the RiboZero bacteria kit (Epicentre) for the in vitro samples and the RiboZero epidemiology kit (Epicentre) for the human samples and then purified by ethanol precipitation using 12.5 μg linear acrylamide to precipitate the RNA. The depleted RNA was fragmented for 4 min for in vitro samples and 2 min for human samples and cDNA libraries were prepared as described previously (39). Libraries were sequenced at the Genome Sequencing and Analysis Facility at the University of Texas at Austin on an Illumina HiSeq. 4000 50 bp, an Illumina HiSeq. 2500 100 bp, or NextSeq. 500 75 bp single-end run.
Bioinformatic Analyses.
RNA-seq reads were trimmed using Cutadapt 1.13, using a minimum read length threshold of 25 bases (43). The non-P. aeruginosa species from our samples were identified using CLARK 1.2.3 (using an abundance cut-off of 2% in at least one human sample), and we built a metagenome by downloading from the National Center for Biotechnology Information at least one genome from each of these 53 species, in addition to S. epidermidis. This list likely overestimates the bacterial species in our samples, and some species identified are likely different, but closely related species, to what actually was in the sample. This said, non-P. aeruginosa reads should map to the similar decoy species better than to P. aeruginosa. For all samples, reads were mapped to this metagenome using Bowtie 2.2.6 with the default parameters for end-to-end alignment (44). We removed the reads that mapped from our trimmed reads files using Seqtk (45), and mapped the remaining reads to a pangenome of 28 additional P. aeruginosa strains. These strains were from the P. aeruginosa PAO1 ortholog database curated by Pseudomonas.com (46). Reads were then tallied for each gene using Rsubread 1.26.1 (47). We chose a subset of 1,707 genes such that each gene had least one read mapping to it for a large set of our samples (Fig. S2). This set of genes was used in our analyses unless stated otherwise.
The PCA plots included only the 1,707 genes mentioned above that were shared by all our selected samples, while the differential expression analysis used in the antibiotics and QS comparisons used all PAO1 genes. These data were normalized with DESeq2’s rlog function. Differential expression was then determined with DESeq2 (29). The enriched pathway analysis was conducted using the BioCyc P. aeruginosa database through the BioCyc website, with P values input without an additional multiple-comparison correction (48). This provided enriched categories based on a Fisher’s exact test, using Grossmann’s parent–child-union variation, which corrects for hierarchical effects in gene sets (49). Plotted in Fig. 2 are genes with a P-adjusted value of <0.05. In determining the differential expression of the core QS regulon of P. aeruginosa between in vitro and human transcriptomes, we calculated the ratio of the geometric means in relative expression for each gene. In calculating the mean for each QS gene, we omitted samples that had fewer than three reads, and for consistency, when calculating the relative expression of each QS gene in each sample, we omitted counts of other genes with fewer than three mapped reads.
For the machine learning component of the paper, gene selection and normalization was performed using the R package DaMIRSEq. 1.2.0 (25). For distinguishing in vitro samples from mouse samples, we used a correlation cutoff of 0.5 for the partial least-squares feature selection (FSelect), and the default correlation coefficient for the redundant feature removal (FReduct). For distinguishing sputum from soft tissue, we used a correlation threshold of 0.4 for FSelect. We chose nondefault values for the FSelect threshold because this value is limited by the correlation between PC’s and class in the data; however, this parameter did not impact our results qualitatively. The gene-selection process was repeated 20 times, and the top 30 scoring genes were used to train the SVM. We then used the R package Caret 6.0-78 for SVM model training and prediction, using SVMLinear2 (from the e1071 package implementation) for our kernel (26, 50). When determining how the mice were classified, we repeated the above process, except iterating it 50 times for each of the two comparisons in Table 4; runs where too few features were identified were discarded. Using the model fit from each trained run, we determined the probability score from the 50 classifications.
Ethical Statement.
For United States chronic wound samples, patients were enrolled in this study at the Southwest Regional Wound Care Center in Lubbock, Texas, and provided consent under a protocol that was approved by the Western Institutional Review Board (WIRB PRO NUM: 20062425). For burn wound samples, this study was approved by the Texas Tech University Health Sciences Center Institutional Review Board (IRB#L13-092). Written consent was obtained upon the patient’s admission to the Timothy A. Harnar Burn Unit at University Medical Center, Lubbock, Texas, by a staff member of the Clinical Research Institute at the Texas Tech University Health Sciences Center in compliance with ethical practices. If the patient was unable to provide written consent, written consent was obtained from designated next of kin by a Clinical Research Institute staff member according to the protocol approved by the Institutional Review Board. No children were involved in this study. For Denmark CF sputum samples, the sampling was approved by the Regional Ethics Committee of Copenhagen, Denmark (H-15008060) and with permission by signed informed consent of the patients. For Denmark chronic wound samples, the sampling was approved by the Regional Ethics Committee of Copenhagen, Denmark (H-15020632) and with permission by informed consent of the patients.
Supplementary Material
Acknowledgments
We thank Sophie Darch for help with SCFM2 cultures; Rebecca Gabrilska, Camilla Stavnsbjerg, and Blaine Fritz for help acquiring clinical samples; the contribution of the Texas Tech University Health Sciences Center Clinical Research Institute and the Burn Center of Research Excellence for their assistance with this study; and the Texas Advanced Computing Center at The University of Texas at Austin for providing HPC resources that have contributed to the research results reported within this paper. This study was funded by National Institutes of Health Grant R01GM116547-01A1 (to M.W.); a Human Frontiers Science grant (to M.W.); Cystic Fibrosis Foundation Grant WHITEL16G0 (to M.W.); Lundbeck Foundation Grant R204-2015-4205 (to T.B. and M.W.); and Lundbeck Foundation Grant R105-A9791 (to T.B.). D.M.C. and C.B.I. were supported by Cystic Fibrosis postdoctoral Fellowships CORNFO15F0 and IBBERS16F0, respectively.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequence reported in this paper has been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive, https://www.ncbi.nlm.nih.gov/sra (accession no. SRP135669).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1717525115/-/DCSupplemental.
References
- 1.Lagier J-C, et al. Current and past strategies for bacterial culture in clinical microbiology. Clin Microbiol Rev. 2015;28:208–236. doi: 10.1128/CMR.00110-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bielecki P, et al. In vivo mRNA profiling of uropathogenic Escherichia coli from diverse phylogroups reveals common and group-specific gene expression profiles. MBio. 2014;5:e01075–14. doi: 10.1128/mBio.01075-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gangaiah D, et al. Haemophilus ducreyi seeks alternative carbon sources and adapts to nutrient stress and anaerobiosis during experimental infection of human volunteers. Infect Immun. 2016;84:1514–1525. doi: 10.1128/IAI.00048-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hagan EC, Lloyd AL, Rasko DA, Faerber GJ, Mobley HL. Escherichia coli global gene expression in urine from women with urinary tract infection. PLoS Pathog. 2010;6:e1001187. doi: 10.1371/journal.ppat.1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subashchandrabose S, et al. Host-specific induction of Escherichia coli fitness genes during human urinary tract infection. Proc Natl Acad Sci USA. 2014;111:18327–18332. doi: 10.1073/pnas.1415959112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu Y, et al. In vivo gene expression in a Staphylococcus aureus prosthetic joint infection characterized by RNA sequencing and metabolomics: A pilot study. BMC Microbiol. 2016;16:80. doi: 10.1186/s12866-016-0695-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jorth P, et al. Metatranscriptomics of the human oral microbiome during health and disease. MBio. 2014;5:e01012-14. doi: 10.1128/mBio.01012-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tata M, et al. RNASeq based transcriptional profiling of Pseudomonas aeruginosa PA14 after short-and long-term anoxic cultivation in synthetic cystic fibrosis sputum medium. PLoS One. 2016;11:e0147811. doi: 10.1371/journal.pone.0147811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gifford AH, et al. Use of a multiplex transcript method for analysis of Pseudomonas aeruginosa gene expression profiles in the cystic fibrosis lung. Infect Immun. 2016;84:2995–3006. doi: 10.1128/IAI.00437-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damron FH, Oglesby-Sherrouse AG, Wilks A, Barbier M. Dual-seq transcriptomics reveals the battle for iron during Pseudomonas aeruginosa acute murine pneumonia. Sci Rep. 2016;6:39172. doi: 10.1038/srep39172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dötsch A, et al. The Pseudomonas aeruginosa transcriptional landscape is shaped by environmental heterogeneity and genetic variation. MBio. 2015;6:e00749-15. doi: 10.1128/mBio.00749-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner KH, Everett J, Trivedi U, Rumbaugh KP, Whiteley M. Requirements for Pseudomonas aeruginosa acute burn and chronic surgical wound infection. PLoS Genet. 2014;10:e1004518. doi: 10.1371/journal.pgen.1004518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ounit R, Wanamaker S, Close TJ, Lonardi S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics. 2015;16:236. doi: 10.1186/s12864-015-1419-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kung VL, Ozer EA, Hauser AR. The accessory genome of Pseudomonas aeruginosa. Microbiol Mol Biol Rev. 2010;74:621–641. doi: 10.1128/MMBR.00027-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.West SA, Diggle SP, Buckling A, Gardner A, Griffin AS. The social lives of microbes. Annu Rev Ecol Evol Syst. 2007;38:53–77. [Google Scholar]
- 16.Diggle SP, Griffin AS, Campbell GS, West SA. Cooperation and conflict in quorum-sensing bacterial populations. Nature. 2007;450:411–414. doi: 10.1038/nature06279. [DOI] [PubMed] [Google Scholar]
- 17.Gilbert KB, Kim TH, Gupta R, Greenberg EP, Schuster M. Global position analysis of the Pseudomonas aeruginosa quorum-sensing transcription factor LasR. Mol Microbiol. 2009;73:1072–1085. doi: 10.1111/j.1365-2958.2009.06832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chugani S, et al. Strain-dependent diversity in the Pseudomonas aeruginosa quorum-sensing regulon. Proc Natl Acad Sci USA. 2012;109:E2823–E2831. doi: 10.1073/pnas.1214128109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray JL, Kwon T, Marcotte EM, Whiteley M. Intrinsic antimicrobial resistance determinants in the superbug Pseudomonas aeruginosa. MBio. 2015;6:e01603-15. doi: 10.1128/mBio.01603-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hinz A, Lee S, Jacoby K, Manoil C. Membrane proteases and aminoglycoside antibiotic resistance. J Bacteriol. 2011;193:4790–4797. doi: 10.1128/JB.05133-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Son MS, Matthews WJ, Jr, Kang Y, Nguyen DT, Hoang TT. In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect Immun. 2007;75:5313–5324. doi: 10.1128/IAI.01807-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cerasi M, Ammendola S, Battistoni A. Competition for zinc binding in the host-pathogen interaction. Front Cell Infect Microbiol. 2013;3:108. doi: 10.3389/fcimb.2013.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim S-H, Park S-Y, Heo Y-J, Cho Y-H. Drosophila melanogaster-based screening for multihost virulence factors of Pseudomonas aeruginosa PA14 and identification of a virulence-attenuating factor, HudA. Infect Immun. 2008;76:4152–4162. doi: 10.1128/IAI.01637-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci USA. 2015;112:4110–4115. doi: 10.1073/pnas.1419677112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiesa M, Colombo GI, Piacentini L. The DaMiRseq package-data mining for RNA-Seq data: Normalization, feature selection and classification. Bioinformatics. 2018;34:1416–1418. doi: 10.1093/bioinformatics/btx795. [DOI] [PubMed] [Google Scholar]
- 26.Kuhn M. Building predictive models in R using the caret package. J Stat Softw. 2008;28:1–26. [Google Scholar]
- 27.Palmer KL, Mashburn LM, Singh PK, Whiteley M. Cystic fibrosis sputum supports growth and cues key aspects of Pseudomonas aeruginosa physiology. J Bacteriol. 2005;187:5267–5277. doi: 10.1128/JB.187.15.5267-5277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu NY, et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26:1608–1615. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrova OE, Cherny KE, Sauer K. The diguanylate cyclase GcbA facilitates Pseudomonas aeruginosa biofilm dispersion by activating BdlA. J Bacteriol. 2015;197:174–187. doi: 10.1128/JB.02244-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrova OE, Cherny KE, Sauer K. The Pseudomonas aeruginosa diguanylate cyclase GcbA, a homolog of P. fluorescens GcbA, promotes initial attachment to surfaces, but not biofilm formation, via regulation of motility. J Bacteriol. 2014;196:2827–2841. doi: 10.1128/JB.01628-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuhn M. 2012 Variable selection using the caret package. Available at https://r-forge.r-project.org/scm/viewvc.php/*checkout*/pkg/caret/inst/doc/caretSelection.pdf?revision=77&root=caret&pathrev=90. Accessed March 2, 2018.
- 33.Yang H, et al. Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet. 2011;43:648–655. doi: 10.1038/ng.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Firoved AM, Deretic V. Microarray analysis of global gene expression in mucoid Pseudomonas aeruginosa. J Bacteriol. 2003;185:1071–1081. doi: 10.1128/JB.185.3.1071-1081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jorth P, et al. Regional isolation drives bacterial diversification within cystic fibrosis lungs. Cell Host Microbe. 2015;18:307–319. doi: 10.1016/j.chom.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ibberson CB, et al. Co-infecting microorganisms dramatically alter pathogen gene essentiality during polymicrobial infection. Nat Microbiol. 2017;2:17079. doi: 10.1038/nmicrobiol.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whiteley M, et al. Gene expression in Pseudomonas aeruginosa biofilms. Nature. 2001;413:860–864. doi: 10.1038/35101627. [DOI] [PubMed] [Google Scholar]
- 38.Rahme LG, et al. Common virulence factors for bacterial pathogenicity in plants and animals. Science. 1995;268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 39.Darch SE, et al. Recombination is a key driver of genomic and phenotypic diversity in a Pseudomonas aeruginosa population during cystic fibrosis infection. Sci Rep. 2015;5:7649. doi: 10.1038/srep07649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobs AC, et al. AB5075, a highly virulent isolate of Acinetobacter baumannii, as a model strain for the evaluation of pathogenesis and antimicrobial treatments. MBio. 2014;5:e01076-14. doi: 10.1128/mBio.01076-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boles BR, Thoendel M, Roth AJ, Horswill AR. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One. 2010;5:e10146. doi: 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Socransky SS, Dzink JL, Smith CM. Chemically defined medium for oral microorganisms. J Clin Microbiol. 1985;22:303–305. doi: 10.1128/jcm.22.2.303-305.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- 44.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li HS. 2015 A toolkit for processing sequences in FASTA/Q formats. Available at https://github.com/lh3/seqtk. Accessed March 2, 2018.
- 46.Winsor GL, et al. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016;44:D646–D653. doi: 10.1093/nar/gkv1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liao Y, Smyth GK, Shi W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caspi R, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic acids Res. 2007;36:D623–D631. doi: 10.1093/nar/gkm900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grossmann S, Bauer S, Robinson PN, Vingron M. Improved detection of overrepresentation of Gene-Ontology annotations with parent child analysis. Bioinformatics. 2007;23:3024–3031. doi: 10.1093/bioinformatics/btm440. [DOI] [PubMed] [Google Scholar]
- 50.Dimitriadou E, Hornik K, Leisch F, Meyer D, Weingessel A. 2011 e1071: Misc Functions of the Department of Statistics (e1071), TU Wien. R package Version 1.5-27. Available at CRAN.R-project.org/package=e1071. Accessed March 2, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.