

Abstract
Inhibitors targeting BCL-2 apoptotic proteins have significant potential for the treatment of acute myeloid leukemia (AML); however, complete responses are observed in only 20% of patients suggesting targeting BCL-2 alone is insufficient to yield durable responses. Here we assessed the efficacy of co-administration of the PI3K/mTOR inhibitor GDC-0980 or the p110β-sparing PI3K inhibitor taselisib with the selective BCL-2 antagonist venetoclax in AML cells. Tetracycline-inducible downregulation of BCL-2 significantly sensitized MV4-11 and MOLM-13 AML cells to PI3K inhibition. Venetoclax/GDC-0980 co-administration induced rapid and pronounced BAX mitochondrial translocation, cytochrome c release, and apoptosis in various AML cell lines in association with AKT/mTOR inactivation and MCL-1 downregulation; ectopic expression of MCL-1 significantly protected cells from this regimen. Combined treatment was also effective against primary AML blasts from 17 patients, including those bearing various genetic abnormalities. Venetoclax/GDC-0980 markedly induced apoptosis in primitive CD34+/38−/123+ AML cell populations but not in normal hematopoietic progenitor CD34+ cells. The regimen was also active against AML cells displaying intrinsic or acquired venetoclax resistance or tumor microenvironment-associated resistance. Either combinatorial treatment markedly reduced AML growth and prolonged survival in a systemic AML xenograft mouse model and diminished AML growth in two patient-derived xenograft models. Venetoclax/GDC-0980 activity was partially diminished in BAK−/− cells and failed to induce apoptosis in BAX−/− and BAX−/−BAK−/− cells, whereas BIM−/− cells were fully sensitive. Similar results were observed with venetoclax alone in in vitro and in vivo systemic xenograft models. Collectively, these studies demonstrate that venetoclax/GDC-0980 exhibits potent anti-AML activity primarily through BAX and, to a lesser extent, BAK. These findings argue that dual BCL-2 and PI3K inhibition warrants further evaluation in AML.
Keywords: Venetoclax, GDC-0980, BAX/BAK, BIM, AML
Introduction
Acute myeloid leukemia (AML) cells, like other tumor cells, frequently exhibit dysregulation of BCL-2 family members, leading to defective apoptosis, enhanced survival, and therapeutic resistance (1). This prompted development of inhibitors targeting BCL-2 survival proteins. ABT-737 and its clinical analogue ABT-263, are selective BH3 mimetics that specifically target BCL-2 and BCL-XL but spare MCL-1 (2,3). While ABT-263 was well tolerated and showed activity in patients with various hematological malignancies, a limitation has been thrombocytopenia (4–6), reflecting effects of BCL-XL inhibition on platelet survival (4,7). To circumvent this problem, venetoclax (ABT-199) was designed to target selectively BCL-2, which is dispensable for platelet survival (8), but not BCL-XL and MCL-1 (9). Indeed, venetoclax spares platelets (9) and does not cause significant thrombocytopenia (10,11).
The observations that a subset of AML cells are highly dependent on BCL-2 for survival (12,13) raises the possibility of venetoclax efficacy and selectivity in this disease. Notably, venetoclax has shown significant pre-clinical activity in various tumor cell types including AML models, both in vitro and in vivo (9,10,14). Moreover, clinical trials demonstrated significant venetoclax activity in CLL and AML (11,15). Notably, venetoclax was recently approved by the FDA for relapsed/refractory CLL with chromosome 17p deletion (16) and received breakthrough status (with low-dose ara-C) in elderly AML patients (17).
Despite initial evidence of activity in AML (15), complete responses occur in only approximately 20% of patients. This and the potential emergence of drug resistance suggest that single-agent administration is unlikely to yield durable responses in most cases. However, venetoclax represents a highly attractive platform for rational combination strategies. Findings from our group and others implicating MCL-1 in AML cell resistance to other BH3 mimetics (e.g., ABT737/ABT263) (18,19) argue that agents that downregulate MCL-1 are logical candidates for combinations with venetoclax. In this context, we and others have shown that PI3K/mTOR pathway inhibition significantly diminishes MCL-1 protein levels through dephosphorylation/activation of GSK3α/β, triggering MCL-1 degradation (19,20) as well as through translation inhibition (21). Furthermore, we have found that dual PI3K/mTOR and BCL-2/BCL-XL (e.g., by ABT-737) inhibition exerts potent anti-AML activity both in vitro and in vivo (19,22). However, as BCL-XL and MCL-1 cooperate to inactivate BAK (23), it is uncertain whether similar interactions would occur with BCL-XL-sparing venetoclax. The purpose of the present studies was to determine whether a selective BCL-2 inhibitor would cooperate with PI3K inhibition (e.g., by the PI3K/mTOR inhibitor GDC-0980 or the beta-sparing PI3Kα/δ inhibitor taselisib (GDC-0032) (24) to kill AML cells, and to elucidate the molecular mechanism(s) underlying this phenomenon.
Methods
Cells
Human AML cell lines U937, MV4-11, EOL-1, and THP-1, RS4,11, were purchased from American Type Culture Collection (ATCC). MOLM-13 and OCI-AML3 cells were purchased from DSMZ (Braunschweig, Germany). MLL-ENL cells were as previously reported (25). All cell lines with the exception of MLL/ENL were authenticated and tested for mycoplasma by their suppliers. U937, MV4-11, MOLM-13, EOL-1, OCI-AML3 cells were also authenticated by ATCC (basic short tandem repeat profiling) during this study. All cell lines were tested for mycoplasma contamination one or multiple times during this study using the MycoAlert™ mycoplasma detection kit (Lonza).
MV4-11 and MOLM-13 cells exhibiting inducible knock-down of BCL-2 were generated by lentiviral infection as previously described for other AML cells (19). U937 cells ectopically expressing MCL-1 were as described (19). AML cells lacking expression of BAX, BAK, BAX/BAK, or BIM were generated by transducing cells with lentiviral particles carrying both Cas9 and specific guide RNA (gRNA) constructs for these genes or non-targeting control gRNA constructs. BAK and BAX CRISPR constructs were purchased from transOMIC Technologies Inc. (Huntsville, AL). A BIM CRISPR Construct was purchased from GeneCopoeia (Rockville, MD). Stables clones displaying no detectable protein of interest were isolated and pooled.
Venetoclax-resistant MV4-11 and MOLM-13 cells were obtained by culturing in the presence of increasing venetoclax concentrations over a period of 3 months.
Patient-derived leukemic blasts and normal CD34+ cells
Bone marrow or peripheral blood from patients with acute myeloblastic leukemia (AML) were obtained with written informed consent from the patients. These studies were conducted in accordance with the Helsinki declaration. Mononuclear cells were isolated as previously described (26). Normal hematopoietic CD34+ cells were isolated from human umbilical cord blood obtained from patients undergoing normal deliveries. All studies were sanctioned by the Virginia Commonwealth University Investigational Review Board.
Stromal studies
Human bone marrow stromal HS-5 cells were as previously described (27). MV4-11 cells were co-cultured with human bone marrow derived HS-5 cells expressing a GFP marker for 48 hours, treated for 5 hours, after which apoptosis was assessed in MV4-11 cells (GFP-negative population) and in HS-5 cells (GFP-positive population) using an Annexin-APC staining assay. Alternatively, cells were stained with 7-AAD for 30 minutes and images obtained using an IX71 Olympus microscope.
Mutation analysis
Mutation analysis was performed on genomic DNA extracted from primary blasts as previously described (22).
Reagents
Venetoclax and A-1210477 were provided by AbbVie (North Chicago, IL). GDC-0980, taselisib, and GDC-0491 were obtained from Genentech (South San Francisco CA).
Assessment of apoptosis
Apoptosis was assessed by Annexin V analysis as previously described (19).
Cell growth and viability
Cell growth and viability were assessed by the CellTiter-Glo Luminescent Assay (Promega) (19).
Immunoprecipitation and immunoblotting
Immunoprecipitation and immunoblotting were performed as described (26). Primary antibodies employed were: Polyclonal BAX, BCL-2, and MCL-1 (PharMingen; San Diego, CA). Cleaved Poly(ADP-ribose) Polymerase PARP, cleaved caspase-3, ERK1/2, phospho-AKT (Ser473), phospho-p70S6K, and BIM (Cell Signaling Technology; Beverly, MA). AIF, cytochrome c, polyclonal BAK, BCL-XL, AKT, and p70S6K (Santa Cruz Biotechnology, Santa Cruz, CA). α-tubulin (Calbiochem).
BAX and BAK conformational change
BAX and BAK conformational change was assessed as previously described (28).
Subcellular fractionation and BAX translocation
Cytosolic and membrane fractions were separated as previously described (26). BAX mitochondrial translocation was assessed by immunofluorescence staining. Briefly, 30 min prior to BAX translocation assay, cells were incubated with Mitotracker (Life technologies) for 30 min, cytospun, labeled with anti-BAX poyclonal antibodies (BD Pharmingen) followed by Alexa Fluor 488-conjugated secondary antibodies (Molecular Probes). Fluorescence was visualized by Zeiss LSM 700 confocal or Olympus IX71 microscopes.
In vivo studies
Animal studies were conducted under an approved protocol by the Virginia Commonwealth University Institutional Animal Care and Use Committee (IACUC). NOD/SCID-gamma mice obtained from Jackson laboratories were injected intravenously via tail vein with 3 × 106 luciferase-expressing MV4-11cells. One week after cell injection, mice were exposed to either venetoclax, GDC-0980, or taselisib as single agents or to combined treatment with venetoclax/GDC-0980 or venetoclax/taselisib, or to vehicle alone. Agents were administered orally once a day for 6 days per week. Mice were monitored for AML growth using the IVIS 200 imaging system (Xenogen Corporation, Alameda, CA) as before (22). For patient-derived xenografts, NOD/SCID-γ mice were inoculated with 5 × 106 primary AML blasts. After 2 weeks, mice were treated for 3 weeks and then untreated for an additional 3 months, after which they were sacrificed and the percentage of HCD45+ bone marrow cells assessed.
Statistical analysis
Statistical significance for in vitro studies was determined using the Student’s t test. Drug synergism was determined by Median Dose-Effect analysis using Calcusyn software (Biosoft, Ferguson, MO) (29). Survival rates were determined by Kaplan-Meyer analysis and comparisons of survival curves and median survival were analyzed by log-rank test.
Results
Dual BCL-2 and PI3K pathway inhibition induces apoptosis in both venetoclax-sensitive or -insensitive AML cells in association with AKT/mTOR inactivation, and MCL-1 down-regulation
To determine whether selective BCL-2 inhibition sensitizes AML cells to PI3K inhibition-mediated cell death, two AML cell lines (MV4-11 and MOLM-13) exhibiting inducible knock-down of BCL-2 after doxycycline administration were generated using a tet-inducible shRNA lentiviral system. Exposure to 1 μg/ml doxycycline induced time-dependent BCL-2 down-regulation (Fig. 1A). Notably, BCL-2 knock-down rendered these cells significantly more sensitive to two PI3K inhibitors, GDC-0980 or GDC-0941 (Fig. 1B). Furthermore, combined treatment with very low venetoclax concentrations (10 nM) and GDC-0980 (500 nM) led to rapid and pronounced induction of apoptosis in MV4-11 and MOLM-13, reflected by Annexin V positivity (Fig. 1C). Within 4 hr of treatment, more than 80% of MV4-11 and 48% of MOLM-13 cells were apoptotic (Fig. 1C). These effects were associated with profound mitochondrial injury e.g., marked release of cytochrome c and AIF into the cytosol, and increased cleavage of caspase-3 and PARP (Supplementary Fig. S1A).
Figure 1. Dual inhibition of BCL-2 and PI3K pathway potently induced apoptosis in AML cells in association with a pronounced change in BAX conformation.
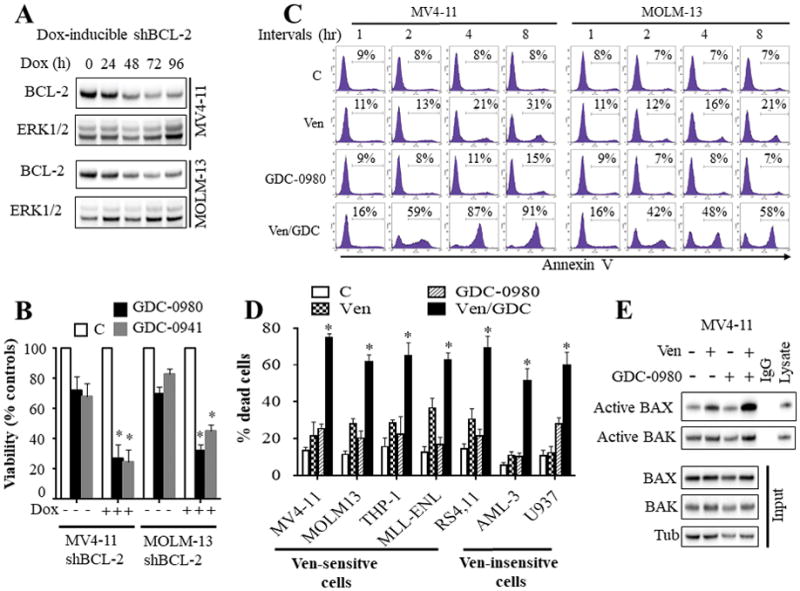
A) western blot analysis in MV4-11 and MOLM-13 cells displaying inducible knock-down of BCL-2 following exposure to 1 μg/ml doxycycline (Dox). B) These cells were left untreated or treated with doxycycline for 48 hr, and then exposed to the PI3K inhibitors GDC-0980 (500 nM) or GDC-0941 (1 μM) for 24 hr after which cell growth and viability was assessed by a CellTiter-Glo luminescent assay. Error Bars: S.D of 3 independent experiments; *, P < 0.05 in each case for values obtained in the presence vs absence of doxycycline. C) MV4-11 and MOLM-13 cells were exposed to venetoclax (10 nM) and GDC-0980 (500 nM) alone or together for the designated intervals after which the extent of apoptosis was determined by Annexin V staining. D) Various AML cell lines were exposed to ABT-199 and/or GDC-0980 for 24 hr after which the extent of cell death was determined employing Annexin V/PI staining. The agent concentrations used were: ABT-199: 2.5 nM for RS4,11; 10 nM for MV4-11 and MOLM-13; 50 nM for MLL-ENL; 100 nM for THP-1; and 500 nM for U937 and OCI-AML3 cells. GDC-0980: 1000 nM for U937 and RS4,11 cells; 500 nM for all other cell lines. Error Bars, S.D for at least 3 independent experiments; P < 0.003 for combined treatment compared to either agent alone for each cells. E) MV4-11 cells were exposed to venetoclax and/or GDC-0980 for 2 hr after which BAX and BAK conformational changes were assessed (top panel). Western blot analysis was also performed on input lysates (bottom panel).
Additional studies in a panel of acute myeloid leukemia cell lines exhibiting disparate sensitivities to venetoclax alone (Supplementary Fig. S1B) revealed that co-administration of very low concentrations of venetoclax (5 – 100 nM) with GDC-0980 (500 nM) effectively induced cell death in venetoclax-sensitive cell lines, including MV4-11, MOLM-13, THP-1, MLL-ENL, and RS4,11 (Fig. 1D). Interestingly, combined treatment was also effective in AML cells intrinsically resistant to up to 5 μM venetoclax concentrations alone e.g, U937 and OCI-AML3 cells, reflected by enhanced Annexin V staining (Fig. 1D) and caspase-3 and PARP cleavage (Supplementary Fig. S1C). Notably, these cells required higher but clinically achievable (30) venetoclax concentrations (500 nM). Similar results were obtained when cell growth and viability were monitored by a CellTiter-Glo luminescent assay (Supplementary Fig. S1D). Significantly, median dose effect analysis demonstrated that venetoclax/GDC-0980 interactions were highly synergistic, reflected by combination index values < 1.0 in both venetoclax-sensitive and venetoclax-resistant AML cells (Supplementary Fig. S2 A–D). Furthermore, similar results were obtained with venetoclax/taselisib in MV4-11 and MOLM-13 cells (Supplementary Fig. S2 E–F). Combined treatment triggered a rapid and pronounced change in BAX conformation but only modest changes in BAK (Fig. 1E) in association with pronounced BAX mitochondrial translocation (Fig. 2A). In contrast, single agents exhibited minimal or no effects, suggesting that BAX may play a key functional role in venetoclax/GDC-0980 lethality.
Figure 2. Venetoclax/GDC-0980 inactivates AKT/mTOR/p70S6K, down-regulates MCL-1, triggers BAX mitochondrial translocation, and effectively kills primary AML blasts including CD34+/38−/123+ AML progenitor but not normal CD34+ cells.
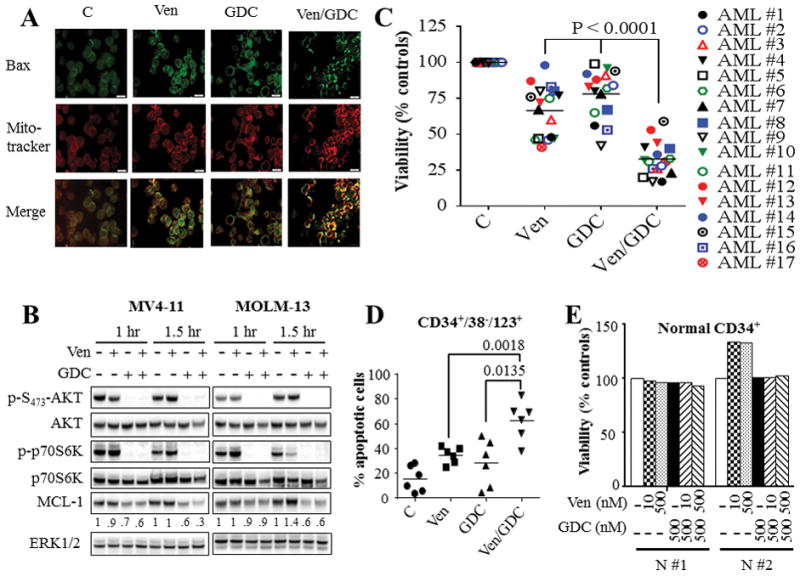
(A) MV4-11 cells were exposed to 10 nM venetoclax (Ven) and/or 500 nM GDC-0980 (GDC) for 2 hr after which BAX translocation to the mitochondria was assessed using IX71 Olympus microscope. (B) MV4-11 and MOLM-13 cells were treated with 10 nM venetoclax (Ven) and/or 500 nM GDC-0980 (GDC) for the indicated intervals, after which cells were lysed and the lysates were subjected to western blot analysis. Densitometry analysis was performed on MCL-1 blots using Image Studio lite Software (Li-Cor Biosciences), and values were normalized for ERK1/2 loading controls. (C) Assessment of cell viability using Annexin V/7-AAD or CellTiter-Glo luminescent assays for 17 primary AML specimens with a preponderance of blasts (≥ 80%) following 16 hr treatment with venetoclax and GDC-0980 alone or together. As in the case of cell lines, primary AML samples exhibited heterogeneous responses to venetoclax. Concentrations of venetoclax were selected based upon marginal toxicity when administered alone and clinical relevance. Venetoclax concentrations varied between 10 – 100 nM for patients #1–10, and between 200 – 2000 nM for patients #11 – 17. GDC-0980 was administered at concentrations varying between 200 nM and 1000 nM. The median values for combined treatment were significantly lower than values for either agent alone (P < 0.0001 in each case). (D) Primary AML blasts from 6 patients were treated as in (C) for 16 hr after which the extent of the cell death was assessed selectively in AML progenitor CD34+/CD38−/CD123+ cells by Annexin V/7-AAD staining. The median values were significantly lower for combined treatment compared to either agent alone (P < 0.0018; P < 0.0135 for venetoclax and GDC-0980 respectively). (E) Normal hematopoietic mononuclear cells were isolated from umbilical cord blood of 2 subjects (N #1 and N #2) and were exposed to the designated concentrations of venetoclax and GDC-0980 alone or in combination for 16 hr after which viability was assessed selectively in the CD34+ cell population by Annexin V/7-AAD staining.
Finally, studies in venetoclax-sensitive MV4-11 and MOLM-13 cell lines revealed that GDC-0980 or venetoclax/GDC-0980 exposure induced rapid (within 1 hr) and marked decreases in phosphorylation of AKT and its downstream signaling target p70S6K, and MCL-1 down-regulation (Fig. 2B). No major changes were observed in BCL-XL, BCL-2, BAX, BAK, or BIM protein levels (Supplementary Fig. S3A). Similar observations were made in venetoclax-resistant U937 and OCI-AML3 cells (Supplementary Fig. S3B).
Venetoclax/GDC-0980 effectively kills primary AML blasts but not normal CD34+ cells
Studies performed in primary blasts isolated from AML patients (N = 17) revealed that combined treatment led to greater diminution of cell viability compared to either agent alone in both venetoclax-sensitive (e.g., requiring 10–100 nM venetoclax) and venetoclax-resistant cells (e.g., requiring 200–2000 nM venetoclax) (Fig. 2C). Genetic analysis using Next Generation sequencing (NGS) revealed that several of these specimens carried diverse genetic aberrations including mutations in FLT3, NPM1, IDH1, IDH2, Nras, Kras, and c-Kit, among others (Supplementary Fig. S4), indicating that combined treatment is active across a spectrum of AML sub-types. Furthermore, combined treatment significantly increased apoptosis in CD34+/CD38−/CD123+ AML progenitor cells (14,31) (Fig. 2D), but not in normal hematopoietic CD34+ cells (Fig. 2E), consistent with selective AML cell killing.
Venetoclax/GDC-0980 inhibits AML growth and enhances survival in in vivo AML xenograft and PDX models
To test the in vivo activity of combined venetoclax/GDC-0980 treatment, mice bearing systemic MV4-11 cell-derived xenografts were employed. Co-administration of venetoclax (80 mg/kg) and GDC-0980 (5 – 10 mg/kg) significantly reduced in vivo tumor growth (Figure 3A). Agents alone also reduced tumor growth but to a considerably lesser extent than combined treatment. Kaplan-Meier analysis (Fig. 3B) revealed that co-treatment significantly prolonged mouse survival (P values 0.0057 and 0.0248 for combined treatment vs GDC-0980 or venetoclax alone respectively; log-rank test). Median survival was extended from 36 days (control) to 70 days for combined treatment, versus 55 or 43 days for GDC-0980 or venetoclax respectively. Treatment was discontinued at day 55 due to weight loss in 2 mice. No significant changes in mouse weight were observed before day 51 (Supplementary Fig. S5A). Additional studies using lower doses of GDC-0980 (5 mg/kg) with venetoclax (80 mg/kg) over 24 days yielded similar results (Supplementary Fig. S5B) with no significant changes in mouse body weights (Supplementary Fig. S5C). Notably, co-administration of venetoclax and low doses (1.5 – 2.5 mg/kg) of the clinically relevant p110β-sparing PI3K inhibitor taselisib was also significantly more effective than either single agent alone in suppressing tumor growth (Fig. 3C and Supplementary Fig. S5D). In addition, combined treatment with venetoclax and 1.5 or 2.5 mg/kg taselisib significantly extended mouse survival from 33 days (control) to 60 and 76 days respectively (Figure 3D and Supplementary Fig. S5E). Of note, 2.5 mg/kg taselisib alone extended mouse survival to 40.5 days (Figure 3D) whereas lower doses (1.5 mg/kg) did not (Supplementary Fig. S5E). Importantly, this regimen was well tolerated as indicated by the absence of changes in animal weight (Supplemental Fig. S5F), or other signs of toxicity (e.g, hair loss, decreased mobility).
Figure 3. Co-treatment with venetoclax and GDC-0980 exhibits potent in vivo anti-AML activity.
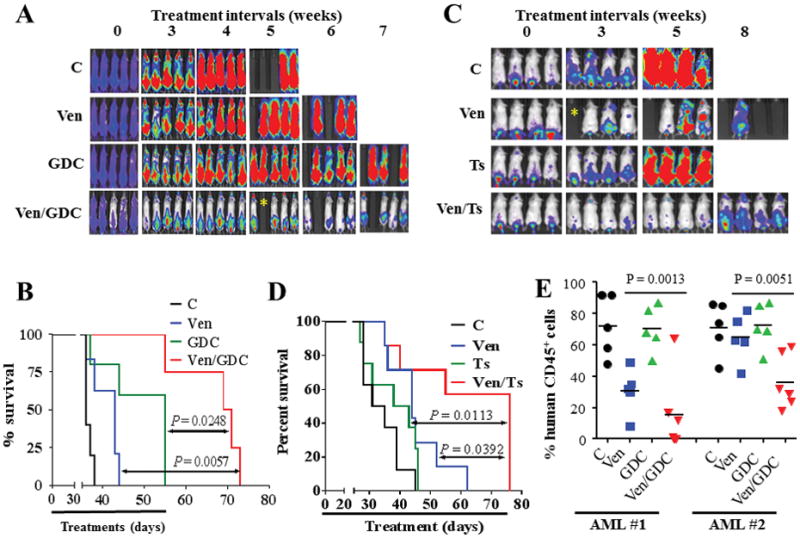
NOD/SCID-γ mice were inoculated with 3 × 106 luciferase-expressing MV4-11 cells via tail vein injection. 1 week later, mice were treated with regimens consisting of oral administration of venetoclax, GDC-0980, or taselisib once every day, 6 days a week as follow. (A) Mice were treated with venetoclax (80 mg/kg), GDC-0980 (5 mg/kg for 2 weeks, then 10 mg/kg until day 55) alone or in combination, and AML progression was monitored by the IVIS 200 imaging system. (B) Survival analysis of mice using Kaplan-Meier survival plot. These studies involved 5–6 mice/condition; combined treatment significantly prolonged mouse survival compared to either agent alone (P = 0.0248, and P = 0.0057 for combination versus GDC-0980 and venetoclax respectively, log-rank test). (C) Mice (8 mice/condition) were treated with venetoclax (80 mg/kg) and/or taselisib (2.5 mg/kg), and subjected to IVIS 200 imaging. (D) Kaplan-Meier survival analysis. Venetoclax/taselisib significantly prolonged mouse survival compared to either agent alone (P = 0.0113, and P = 0.0392 for combination versus taselisib and venetoclax respectively, log-rank test). * indicate mouse accidental death; these mice were excluded from survival analysis. (E) NOD/SCID-γ mice were inoculated with primary AML blasts (5 × 106 cells) isolated from 2 patients with AML. 2 weeks later, mice were treated with venetoclax (80 mg/kg) and/or GDC-0980 (5 mg/kg) po qd 6 days/week for 3 weeks. Mice were left untreated for additional 3 months after which they were sacrificed and the percentages of HCD45+ cells in the bone marrow were assessed. Each dot represents an individual mouse (n = 5 – 6 per group). The means of different groups are statistically different for both patients AML1; P = 0.0013; AML2; P = 0.0051; One-way ANOVA.
Finally, parallel in vivo studies using 2 separate patient-derived xenograft models revealed that combined treatment markedly attenuated AML growth, reflected by diminished percentages of human CD45+ cells in the bone marrows of recipient mice (Fig. 3E).
Anti-AML activity of venetoclax or venetoclax/GDC-0980 primarily involves BAX and to a lesser extent BAK but does not require BIM
To define the functional roles of BAX, BAK, and BIM in cell death mediated by venetoclax, MV4-11 cells were engineered using CRISPR technology to exhibit deletion of BAX or BAK individually, dual BAX/BAK, or BIM (Fig. 4A). Notably, knockout of BAX and particularly BAX/BAK completely abrogated venetoclax activity, manifested by cytochrome c and AIF release into the cytosol, caspase activation (Fig. 4B), and apoptosis (Fig. 4C). However, BAK deletion alone only modestly attenuated these actions (Fig. 4B–C). In contrast, none of these events was affected by BIM knockout (Fig 4B–C). Moreover, 100 nM venetoclax induced extensive BAX mitochondrial translocation in both BIM−/− and control cells equivalently (Supplementary Fig. S6A).
Figure 4. Functional roles of BAX, BAK, and BIM in venetoclax activity in vitro and in vivo.
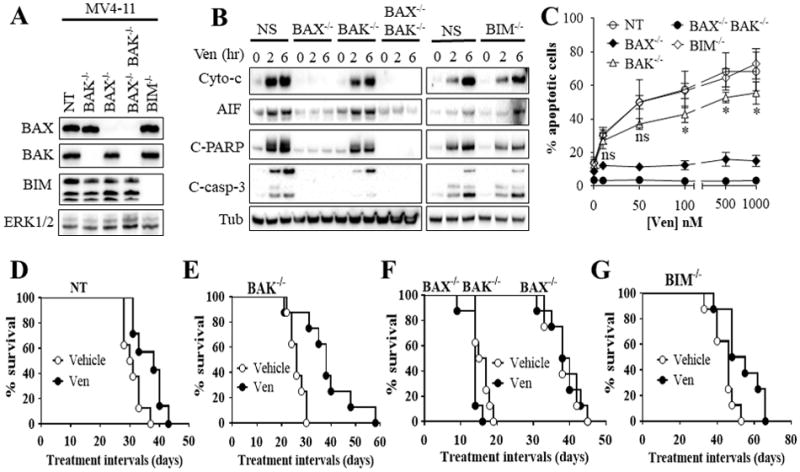
(A) western blot analysis in untreated MV4-11 cells in which BAX (BAX−/−), BAK (BAK−/−), double BAX/BAK (BAX−/−BAK−/−), or BIM (BIM−/−) were knocked out using CRISPR technology as described in Methods or non-targeting (NT) control cells. (B) These cell were exposed to 100 nM venetoclax (Ven) for 2 or 6 hr after which cytosolic fractions were isolated and subjected to western blot analysis. (C) Cells were treated with the designated concentrations of venetoclax for 24 hr after which the extent of apoptosis was determined using Annexin V staining. Error Bars, S.D for at least 3 independent experiments.*, P < 0.05; ns = non-significant for BAK−/− versus NT cells. Values obtained for BAX−/− or BAX−/−BAK−/− cells before versus after venetoclax treatment were not significantly different, P > 0.05 in each case. (D–G) NOD/SCID-γ mice were inoculated with 3 × 106 BAX−/−, BAK−/−, BAX−/−BAK−/−, BIM−/−, or NT control MV4-11 cells via tail vein. One week later, mice were treated orally with 75 mg/kg venetoclax (Ven) once every day, 6 days a week. Mouse survival was analyzed by Kaplan-Meier survival plot. Survival of mice with Bax−/− or Bax−/−Bak−/−-derived xenografts are both included in Figure 4F. These studies involved 8 mice/condition. Venetoclax significantly prolonged mouse survival in mice with NT, BAK−/−, or BIM−/− cells, P = 0.0138, P = 0.0015, and P = 0.0117 respectively. In contrast, no survival benefit was observed in mice with BAX−/− or BAX−/−BAK−/− cells.
In vivo studies using a systemic xenograft mouse model with MV4-11 cells bearing BAX−/−, BAK−/−, BAX−/−BAK−/−, BIM−/− or non-targeting control (NT) cells largely recapitulated the previous in vitro data. Specifically, while the engraftment capacity differed between the various cell types, treatment of mice with 75 mg/kg/d of venetoclax orally extended the median survival by 7.5 days for control cells (Fig. 4D) and 12 days for BAK−/− (Fig. 4E; P < 0.05 versus control). However only a 1-day survival advantage was observed for mice inoculated with BAX−/− cells, and no survival advantage was observed for BAX−/−BAK−/− mice (Fig. 4F). Finally, as in the case of control and BAK−/− cells, venetoclax treatment increased survival by 5.5 days for BIM−/− cells (Fig. 4G, P < 0.05). Collectively, these findings demonstrate that direct BCL-2 targeting by venetoclax kills human AML cells primarily through BAX, but does not require BIM for activity both in vitro and in vivo. In addition, BAK plays a modest role in venetoclax anti-AML activity in vitro, but not in vivo.
Consistently, BAX−/− or BAX−/−BAK−/− cells were completely resistant to venetoclax/GDC-0980-mediated cytochrome c and AIF release, caspase activation (Fig. 5A), and apoptosis (Fig. 5B), whereas BAK−/− knockout only slightly reduced these effects (Fig 5A–B). In contrast, venetoclax/GDC-0980-mediated cytochrome c and AIF release into the cytosol, caspase activation (Fig. 5C), and apoptosis (Fig. 5D) were identical in BIM−/− and NT cells.
Figure 5. Role of BAX, BAK, and BIM in venetoclax/GDC-0980 lethality in venetoclax-sensitive MV4-11 cells.
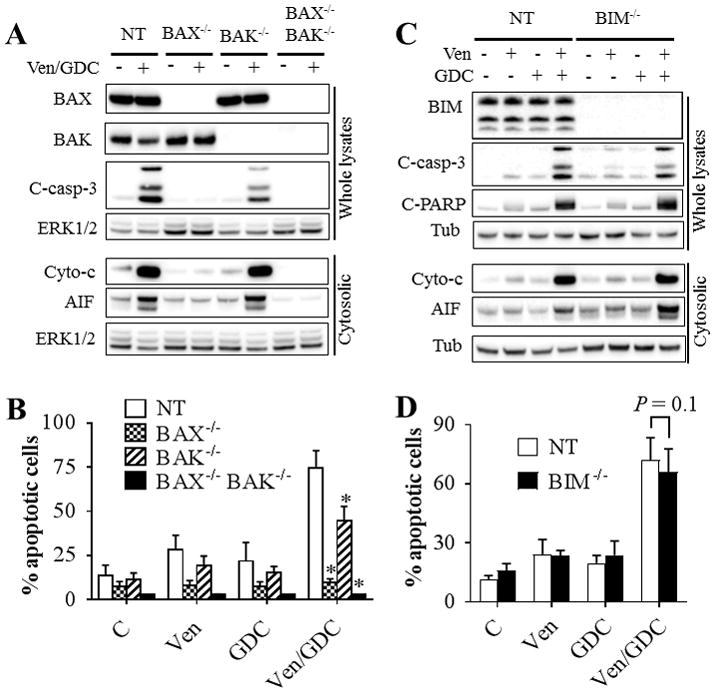
Cells in which BAX, BAK, double BAX/BAK (A–B), or BIM (C–D) were knocked out using CRISPR or non-targeting (NT) control cells were exposed to 10 nM venetoclax and/or 500 nM GDC-0980. Cells were either lysed after 2 hr and subjected to western blot analysis (A–C), or analyzed for the extent of apoptosis at 5 hr using Annexin V staining (B–D). Error Bars, S.D for at least 4 independent experiments. For B: P = 0.0004; P = 0.0027; P = 0.0002 for BAX−/−, BAK−/−, or BAX−/−BAK−/− cells respectively compared to NT cells; for D: P = 0.1307 for BIM−/− cells vs NT cells.
To determine whether similar mechanisms occurred in cells intrinsically resistant to venetoclax but sensitive to the GDC-0980 combination, knockout of BAX, BAK, or BAX/BAK, was carried out in U937 cells (Fig. 6A). Notably, as in MV4-11 cells, BAX−/−BAK−/− U937 cells were insensitive to venetoclax/GDC-0980-mediated apoptosis and BAX−/− cells exhibited only minimal lethality (Fig. 6B). BAK knockout modestly but significantly diminished venetoclax/GDC-0980-induced apoptosis (Fig. 6B). In contrast, BIM knockout failed to confer protection (Fig. 6C–D) or prevent BAX conformational change (Supplementary Fig. S6B). Finally, western blot analysis revealed that BIM knockout did not induce major changes in protein levels of other BCL-2 members (Supplementary Fig. S6C). Collectively, these findings demonstrate that venetoclax and venetoclax/GDC-0980 anti-AML activity primarily involves BAX and to a lesser extent BAK but not BIM.
Figure 6. Role of BAX, BAK, and BIM in venetoclax/GDC-0980 lethality in venetoclax-insensitive U937 cells.
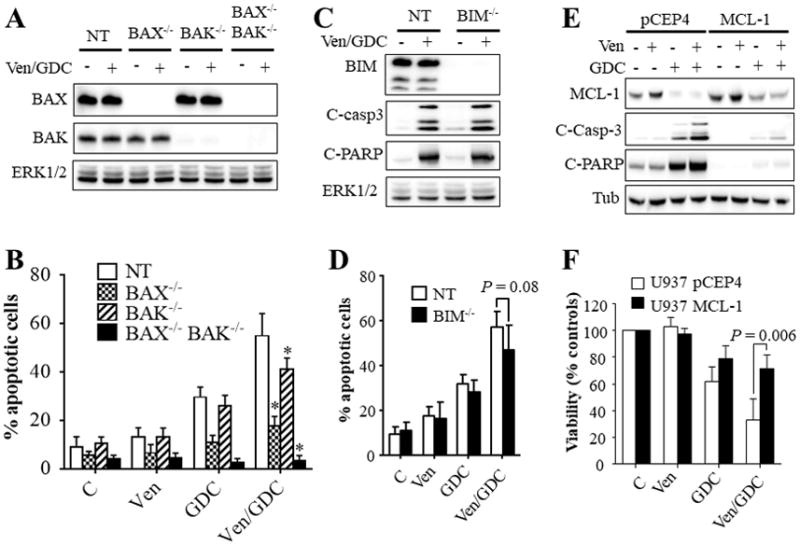
Cells exhibiting knockout of BAX, BAK, double BAX/BAK (A–B), or BIM (C–D), or non-targeting (NT) control cells were exposed to venetoclax (500 nM) and/or GDC-0980 (1.5 μM) for 16 hr. The cells were then lysed, and the lysates subjected to western blot analysis (A, C). Alternatively, the extent of apoptosis was determined using the Annexin V staining (B, D). Error Bars, S.D of at least 3 independent experiments. For B: P = 0.0005; P = 0.0223; P < 0.0001 for BAX−/−, BAK−/−, or BAX−/−BAK−/− cells respectively compared to NT cells; for D: P = 0.08. (E–F) western blot analysis (E) and CellTiter-Glo luminescent viability assay (F) on U937 cells ectopically expressing MCL-1 or the empty vector pCEP4 following exposure to venetoclax (500 nM) and/or GDC-0980 (1.5 μM). For (F), Error Bars, S.D for 4 independent experiments; P = 0.006.
MCL-1 down-regulation plays a functional role in venetoclax/GDC-0980 lethality
The finding that venetoclax/GDC-0980 downregulates MCL-1 protein levels in AML cells is consistent with previous studies by our group and others involving other PI3K/mTOR inhibitors (19,21,22). Interestingly, this also occurred in BAX−/− or BAX−/−BAK−/− cells in the absence of apoptosis (Supplementary Fig. S6D), demonstrating that this phenomenon is not a consequence of caspase activation or other apoptotic processes.
To determine whether MCL-1 down-regulation contributes functionally to venetoclax/GDC-0980 lethality, U937 cells ectopically expressing MCL-1 were employed. MCL-1-overexpressing cells were significantly more resistant to combined treatment than control cells reflected by reduced caspase-3/PARP cleavage (Fig. 6E) and attenuation of diminished cell growth and viability (Fig. 6F). Additionally, co-administration of the MCL-1 inhibitor A-1210477 (32) (e.g., 2 μM) and venetoclax (10 nM) led to a profound induction of cell death in MV4-11 and MOLM-13 cells (Supplementary Figure S6E). Collectively, these findings support a significant functional role for MCL-1 down-regulation in venetoclax/GDC-0980 anti-AML activity.
Venetoclax/GDC-0980 kills AML cells exhibiting various forms of venetoclax resistance
To test whether venetoclax/GDC-0980 circumvents acquired resistance to venetoclax, MV4-11 and MOLM-13 cells made resistant to venetoclax were employed. MV4-11R or MOLM-13R cells were fully resistant to 500 nM venetoclax (Fig. 7A–B). However, addition of GDC-0980 to venetoclax markedly diminished cell viability, although not to the extent observed in sensitive cells (MV4-11S or MOLM-13S; Fig 7A–B). Notably, GDC-0980 alone or in combination equally downregulated MCL-1 protein levels in ABT-resistant or parental cells (Supplementary Fig. S6F). In contrast to parental cells, venetoclax alone failed to trigger BAX mitochondrial translocation in MV4-11R, whereas combined treatment was very effective in this regard (Supplementary Fig. S7).
Figure 7. Venetoclax/GDC-0980 is effective in AML cells exhibiting various forms of venetoclax resistance.
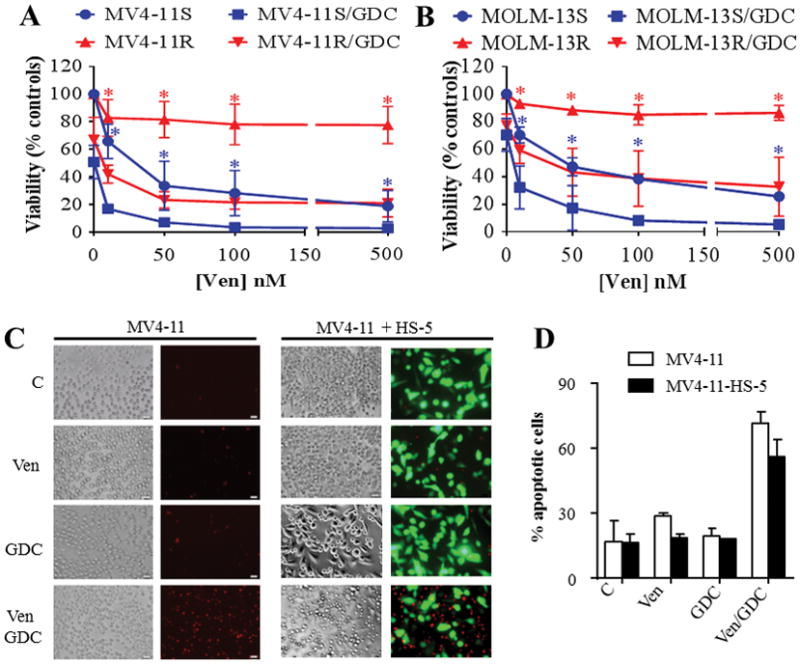
(A–B) venetoclax-sensitive (MV4-11S, MOLM-13S) or venetoclax-resistant (MV4-11R, MOLM-13R) cells were exposed to the designated concentrations of venetoclax alone or in combination with 500 nM GDC-0980 for 24 hr after which cell growth and viability was assessed using the CellTiter-Glo luminescent assay. Error Bars, S.D for at least 3 independent experiments. P < 0.01 for combined treatment compared to venetoclax alone for each concentration in each cell line. (C) MV4-11 cells were cultured in the presence or absence of GFP-labeled HS-5 stromal cells for 48 hr then exposed to venetoclax ± GDC-0980 (10 and 500 nM respectively) for 5 hr after which, cells were stained with 7-AAD for 30 min and photographed as described in Methods. Alternatively, the extent of apoptosis was determined in MV4-11 cells (GFP-negative population) using the Annexin V-APC staining (Fig. 7D). Error Bars, S.D for 4 independent experiments.
Parallel studies assessed whether stromal cells, a postulated mechanism of AML resistance to chemotherapy (33,34), confer resistance to venetoclax/GDC-0980. To this end, MV4-11 cells were co-cultured with bone marrow stroma-derived HS-5 cells 48 hr prior to treatment. Venetoclax/GDC-0980 effectively induced apoptosis in MV4-11 cells in the presence of HS-5 cells (Fig. 7C–D). In contrast, this regimen was non-toxic to HS-5 cells as reflected by the absence of 7-AAD staining (Figure 7C). Together, these findings indicate that combined treatment with venetoclax and GDC-0980 is effective against AML cells exhibiting acquired resistance to venetoclax as well as in cells cultured in the presence of a protective stromal microenvironment.
Discussion
We reported that dual inhibition of BCL-2 and BCL-XL (e.g., by the BH3 mimetic ABT-737) and PI3K (e.g., by BEZ235) robustly induced cell death in AML cells (19). Here we sought to determine whether selective inhibition of BCL-2 by venetoclax would interact similarly and if so, to elucidate the molecular mechanisms underlying this phenomenon. Unlike ABT-737, venetoclax does not neutralize BCL-XL,(9) which cooperates with MCL-1 to tether BAK (23). Nevertheless, both genetic and pharmacologic inactivation of BCL-2 sharply increased the anti-leukemic activity of the PI3K inhibitor GDC-0980, demonstrating that BCL-XL disruption is not required for synergistic anti-leukemic interactions in AML cells.
Although venetoclax has shown early evidence of activity in AML (15,17), it is unlikely that single-agent therapy will be durably effective in the broad spectrum of this disease. Previous studies revealed that agents that down-regulate MCL-1 e.g., CDK9 or deubiquitinase inhibitors potentiate the activity of BH3-mimetics such as ABT-737 and venetoclax (10,35). The ability of PI3K inhibitors to downregulate MCL-1 (19,21,22), and target primitive AML progenitors (36), makes such agents attractive for venetoclax combination therapy in AML. These findings, along with evidence that venetoclax also targets AML stem cell-like cells (14), may explain the effectiveness of the regimen against AML cell populations enriched for leukemia progenitor cells (CD34+/CD38−/CD123+). Notably, co-treatment of primary AML blasts or AML cell lines with venetoclax and GDC-0980 or induced rapid (2 hr) and pronounced apoptosis in venetoclax-sensitive cells. However, the regimen was also effective against relatively venetoclax-resistant cells, although higher concentrations and longer treatment intervals were required. Importantly, intrinsically venetoclax resistant AML cells were sensitized to pharmacologically achievable venetoclax concentrations (≤ 1 μM) (30) by GDC-0980. Finally, in vivo studies using AML cell line-derived or AML patient-derived xenograft models demonstrated that combined treatment with venetoclax and the clinically relevant p110β-sparing taselisib or GDC-0980 effectively inhibited AML growth and prolonged survival with minimal toxicity. These findings, along with the lack of toxicity of the venetoclax/GDC-0980 regimen in normal hematopoietic cells, raise the possibility of therapeutic selectivity for this strategy. The ability of taselisib, which does not target mTOR (24) to recapitulate the activity of GDC-0980 also argues, albeit indirectly, that mTOR direct inhibition is dispensable for venetoclax interactions.
Although the number of primary samples assayed for genetic abnormalities was limited, the regimen was also active against primary AML cells carrying diverse genetic aberrations, including MLL translocations, mutations in FLT3, Kras, Nras, NPM1, and IDH1/IDH2, among others. The molecular mechanism(s) determining AML sensitivity to venetoclax or to the venetoclax/GDC-0980 regimen remains to be determined, and will require analysis of a considerably larger number of samples. However, the present data raise the possibility that cells displaying MLL translocations or IDH1/2 mutations may be particularly sensitive to such regimens. In this context, previous studies revealed that AML cells bearing IDH1/2 mutations or MLL translocations are highly sensitive to venetoclax alone (12,13). Moreover, the latter cells are also highly sensitive to PI3K inhibitors (37). Efforts are currently underway to assess more definitively the susceptibility of these genetic sub-types to PI3K inhibitor/venetoclax regimens.
The observations that venetoclax/GDC-0980 rapidly down-regulated MCL-1 (within 1–1.5 hr) before apoptosis (caspase activation, or Annexin V positivity), and that MCL-1 down-regulation was pronounced in BAX−/−BAK−/− cells in the absence of apoptosis suggest that MCL-1 down-regulation represents a primary event rather than a consequence of cell death. It is also consistent with the short half-life (~30 minutes) of the MCL-1 protein (38). This notion is supported by the observation that ectopic MCL-1 expression significantly diminished venetoclax/GDC-0980-mediated lethality, consistent with our previous studies with other PI3K/mTOR inhibitors (e.g., BEZ235, or INK128) and ABT737 (19,22). Importantly, GDC-0980 alone downregulated MCL-1, presumably through mechanisms involving protein degradation as a consequence of phosphorylation by GSK3 which is activated following AKT inhibition (20). Notably, GDC-0980 minimally induced cell death, suggesting that MCL-1 down-regulation cooperates with BCL-2 inhibition by venetoclax to kill AML cells. This interpretation is consistent with studies describing venetoclax/MCL-1 inhibition interactions in non-Hodgkin’s lymphoma cells (39) as well as solid tumors (40). It is also compatible with recent evidence that in myeloma cells, sensitivity to venetoclax reflects BCL-2 and MCL-1 interplay (41,42).
The venetoclax/GDC-0980 regimen induced rapid (within 2 hr) and extensive BAX translocation to the mitochondria, an initial step in apoptosis induction (43). Significantly, individually administered agents failed to trigger this phenomenon. This may reflect release of BAX from both BCL-2 (by venetoclax) and MCL-1 (by GDC-0980). Of note, Fresquet et al., described mitochondrial BAX translocation by venetoclax in mantle cell lymphoma cells (HBL2) (44). We have observed similar findings in AML cells exposed to higher venetoclax concentrations or longer treatment intervals. The observations that venetoclax alone (at toxic concentrations) or venetoclax/GDC-0980 largely failed to induce apoptosis in BAX−/− or BAX−/−BAK−/− AML cells but triggered apoptosis in cells lacking BAK argue that BAX is a key mediator of apoptosis induction by these regimens, whereas BAK may primarily amplify this process. To the best of our knowledge, this is the first characterization of the contribution of BAX and BAK employing syngeneic AML cells lacking protein expression through genetic manipulation with CRISPR technology. Consistent with our findings, Gong et al., recently demonstrated that KMS-12-PE multiple myeloma cells lacking both BAX and BAK were insensitive to venetoclax (45). However, much of our previous understanding of the functional role of these proteins stems from knock-out mouse embryonic fibroblasts (46) or knock-down studies (e.g., siRNA, shRNA, anti-sense oligonucleotides) which diminish protein levels in only a fraction of the cell population. The significance of the BAX-dependence of venetoclax or venetoclax/GDC-0980 anti-AML activity lies in the possibility that cells displaying diminished BAX expression might be resistant to these regimens. Conversely, cells exhibiting high basal BAX protein levels may be particularly susceptible. Finally, the critical importance of BAX rather than BAK in venetoclax lethality might be explained by preferential binding of BAX to BCL-2 resulting in BAX release by pure BCL-2 antagonists (e.g., venetoclax), whereas BAK, which preferentially binds to BCL-XL, is not directly affected by this agent.
The observations that venetoclax/GDC-0980 triggered BAX conformational change in the absence of BIM, and that this regimen was equally effective in BIM−/− versus non-targeting control AML cells demonstrate that BIM is not required for venetoclax/GDC-0980 lethality in this setting. One plausible explanation for these findings is that BCL-2 neutralization (by venetoclax) combined with MCL-1 down-regulation (by GDC-0980) induces BAX and BAK release from these survival proteins, leading to BAK and particularly BAX activation, culminating in apoptosis. This is consistent with the findings that BIM loss had no impact on BAX mitochondrial translocation or apoptosis mediated by venetoclax (at toxic concentrations) as well as with recent studies in human HCT116 colon cancer cells demonstrating that the BH3 mimetic ABT-737 induces BAX/BAK activation and apoptosis in the absence of BH3-only proteins Bid, BIM, PUMA, and NOXA (47,48). In any event, these findings suggest that BIM may not be a reliable determinant of AML cell sensitivity to venetoclax or venetoclax/GDC-0980.
While the mechanism of AML cells resistance (intrinsic or acquired) to venetoclax is likely to be multifactorial, the ability of venetoclax/GDC-0980 to kill these cells is significant. Venetoclax resistance has been linked to low BCL-2/MCL-1-BCL-XL ratios presumably reflecting loss of BCL-2 dependence, and/or an increase in the protective effects of redundantly acting anti-apoptotic proteins MCL-1 or BCL-XL (42). While venetoclax alone failed to promote BAX mitochondrial translocation, GDC-0980 overcame this deficiency, although longer exposure intervals were required. As MCL-1/BAX binding is well documented (19,22,49), the possibility that MCL-1 down-regulation by GDC-0980 contributed to this phenomenon appears plausible. Fresquet et al., (44) recently described a missense mutation in the C-terminal transmembrane domain of BAX (G179E) in human lymphoma cells exhibiting in vitro venetoclax acquired resistance. This mutation triggered diminished BAX mitochondrial binding (44). However, this mutation is not universal as it was not observed in mouse-derived lymphoma cells which exhibited a BCL-2 mutation instead (44), arguing that venetoclax-resistance mechanisms may be cell type-specific. Finally, venetoclax/GDC-0980 effectively induced apoptosis in AML cells in the presence of a protective stromal microenvironment, which may contribute to leukemia progenitor cell persistence (50). Collectively, these findings suggest that the present strategy may be effective in AML cells displaying intrinsic, acquired, or micro-environmental forms of resistance.
In summary, combined treatment with venetoclax and PI3K inhibitors (e.g., GDC-0980 or taselisb) exhibited robust and broad anti-AML activity both in vitro and in vivo, as well as against multiple forms of venetoclax resistance. These findings provide a clearer understanding of the molecular mechanisms underlying venetoclax/GDC-0980 activity in AML, highlighting the important functional contributions of BAX and MCL-1, but not BIM, to this regimen’s activity. Given encouraging preliminary clinical results for venetoclax in AML (17), a combination strategy involving clinically relevant PI3K inhibitors such as taselisib warrants further consideration in this disease.
Supplementary Material
Acknowledgments
Financial support: This work was supported by UH2TR001373 (S. Grant), CA205607 (S. Grant), CA167708 (S. Grant and M. Rahmani) and an award from the Leukemia and Lymphoma Society of America #6472-15 (S. Grant).
We thank the VCU Department of Anatomy and Neurobiology Microscopy Facility for its help with confocal microscopy studies. This facility is supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Footnotes
Conflict of interest disclosures: J.L, and D.S are employees of AbbVie and Genentech respectively. All other authors have no conflict of interest to disclose.
References
- 1.Delbridge AR, Strasser A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015;22:1071–80. doi: 10.1038/cdd.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 3.Park C-M, Bruncko M, Adickes J, Bauch J, Ding H, Kunzer A, et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J Med Chem. 2008;51:6902–15. doi: 10.1021/jm800669s. [DOI] [PubMed] [Google Scholar]
- 4.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–59. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–96. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–86. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 8.Debrincat MA, Pleines I, Lebois M, Lane RM, Holmes ML, Corbin J, et al. BCL-2 is dispensable for thrombopoiesis and platelet survival. Cell Death Dis. 2015;6:e1721. doi: 10.1038/cddis.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature medicine. 2013;19:202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 10.Vandenberg CJ, Cory S. ABT-199, a new Bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood. 2013;121:2285–8. doi: 10.1182/blood-2013-01-475855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stilgenbauer S, Eichhorst B, Schetelig J, Coutre S, Seymour JF, Munir T, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17:768–78. doi: 10.1016/S1470-2045(16)30019-5. [DOI] [PubMed] [Google Scholar]
- 12.Niu X, Wang G, Wang Y, Caldwell JT, Edwards H, Xie C, et al. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia. 2014;28:1557–60. doi: 10.1038/leu.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nature medicine. 2015;21:178–84. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer discovery. 2014;4:362–75. doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer discovery. 2016;6:1106–17. doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deeks ED. Venetoclax: First Global Approval. Drugs. 2016;76:979–87. doi: 10.1007/s40265-016-0596-x. [DOI] [PubMed] [Google Scholar]
- 17.Bose P, Vachhani P, Cortes JE. Treatment of Relapsed/Refractory Acute Myeloid Leukemia. Curr Treat Options Oncol. 2017;18:17. doi: 10.1007/s11864-017-0456-2. [DOI] [PubMed] [Google Scholar]
- 18.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 19.Rahmani M, Aust MM, Attkisson E, Williams DC, Jr, Ferreira-Gonzalez A, Grant S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. 2013;73:1340–51. doi: 10.1158/0008-5472.CAN-12-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. MolCell. 2006;21:749–60. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, et al. mTORC1 promotes survival through translational control of Mcl-1. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10853–8. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rahmani M, Aust MM, Hawkins E, Parker RE, Ross M, Kmieciak M, et al. Co-administration of the mTORC1/TORC2 inhibitor INK128 and the Bcl-2/Bcl-xL antagonist ABT-737 kills human myeloid leukemia cells through Mcl-1 down-regulation and AKT inactivation. Haematologica. 2015;100:1553–63. doi: 10.3324/haematol.2015.130351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ndubaku CO, Heffron TP, Staben ST, Baumgardner M, Blaquiere N, Bradley E, et al. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2–4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a beta-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J Med Chem. 2013;56:4597–610. doi: 10.1021/jm4003632. [DOI] [PubMed] [Google Scholar]
- 25.Rahmani M, Mayo M, Dash R, Sokhi UK, Dmitriev IP, Sarkar D, et al. Melanoma differentiation associated gene-7/interleukin-24 potently induces apoptosis in human myeloid leukemia cells through a process regulated by endoplasmic reticulum stress. Mol Pharmacol. 2010;78:1096–104. doi: 10.1124/mol.110.068007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43–9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. JBiolChem. 2005;280:35217–27. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- 27.Rahmani M, Aust MM, Benson EC, Wallace L, Friedberg J, Grant S. PI3K/mTOR inhibition markedly potentiates HDAC inhibitor activity in NHL cells through BIM- and MCL-1-dependent mechanisms in vitro and in vivo. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:4849–60. doi: 10.1158/1078-0432.CCR-14-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahmani M, Anderson A, Habibi JR, Crabtree TR, Mayo M, Harada H, et al. The BH3-only protein Bim plays a critical role in leukemia cell death triggered by concomitant inhibition of the PI3K/Akt and MEK/ERK1/2 pathways. Blood. 2009;114:4507–16. doi: 10.1182/blood-2008-09-177881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. AdvEnzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 30.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374:311–22. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hauswirth AW, Florian S, Printz D, Sotlar K, Krauth MT, Fritsch G, et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur J Clin Invest. 2007;37:73–82. doi: 10.1111/j.1365-2362.2007.01746.x. [DOI] [PubMed] [Google Scholar]
- 32.Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax) Cell Death Dis. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Guo H, Duan H, Yang Y, Meng J, Liu J, et al. Improving chemotherapeutic efficiency in acute myeloid leukemia treatments by chemically synthesized peptide interfering with CXCR4/CXCL12 axis. Sci Rep. 2015;5:16228. doi: 10.1038/srep16228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen P, Huang H, Wu J, Lu R, Wu Y, Jiang X, et al. Bone marrow stromal cells protect acute myeloid leukemia cells from anti-CD44 therapy partly through regulating PI3K/Akt-p27(Kip1) axis. Mol Carcinog. 2015;54:1678–85. doi: 10.1002/mc.22239. [DOI] [PubMed] [Google Scholar]
- 35.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–7. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 36.Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, McCubrey JA. The emerging role of the phosphatidylinositol 3-kinase/akt/mammalian target of rapamycin signaling network in cancer stem cell biology. Cancers (Basel) 2010;2:1576–96. doi: 10.3390/cancers2031576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandhofer N, Metzeler KH, Rothenberg M, Herold T, Tiedt S, Groiss V, et al. Dual PI3K/mTOR inhibition shows antileukemic activity in MLL-rearranged acute myeloid leukemia. Leukemia. 2015;29:828–38. doi: 10.1038/leu.2014.305. [DOI] [PubMed] [Google Scholar]
- 38.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003 Jun 15;17(12):1475–86. doi: 10.1101/gad.1093903. Epub2003Jun3 2003;17:1475–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillips DC, Xiao Y, Lam LT, Litvinovich E, Roberts-Rapp L, Souers AJ, et al. Loss in MCL-1 function sensitizes non-Hodgkin’s lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199) Blood cancer journal. 2015;5:e368. doi: 10.1038/bcj.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bate-Eya LT, den Hartog IJ, van der Ploeg I, Schild L, Koster J, Santo EE, et al. High efficacy of the BCL-2 inhibitor ABT199 (venetoclax) in BCL-2 high-expressing neuroblastoma cell lines and xenografts and rational for combination with MCL-1 inhibition. Oncotarget. 2016;7:27946–58. doi: 10.18632/oncotarget.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morales AA, Kurtoglu M, Matulis SM, Liu J, Siefker D, Gutman DM, et al. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood. 2011;118:1329–39. doi: 10.1182/blood-2011-01-327197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Punnoose EA, Leverson JD, Peale F, Boghaert ER, Belmont LD, Tan N, et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol Cancer Ther. 2016;15:1132–44. doi: 10.1158/1535-7163.MCT-15-0730. [DOI] [PubMed] [Google Scholar]
- 43.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nature reviews Molecular cell biology. 2010;11:621–32. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 44.Fresquet V, Rieger M, Carolis C, Garcia-Barchino MJ, Martinez-Climent JA. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood. 2014;123:4111–9. doi: 10.1182/blood-2014-03-560284. [DOI] [PubMed] [Google Scholar]
- 45.Gong JN, Khong T, Segal D, Yao Y, Riffkin CD, Garnier JM, et al. Hierarchy for targeting pro-survival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood. 2016 doi: 10.1182/blood-2016-03-704908. [DOI] [PubMed] [Google Scholar]
- 46.Correia C, Lee SH, Meng XW, Vincelette ND, Knorr KL, Ding H, et al. Emerging understanding of Bcl-2 biology: Implications for neoplastic progression and treatment. Biochim Biophys Acta. 2015;1853:1658–71. doi: 10.1016/j.bbamcr.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Huang K, O’Neill KL, Pang X, Luo X. Bax/Bak activation in the absence of Bid, Bim, Puma, and p53. Cell Death Dis. 2016;7:e2266. doi: 10.1038/cddis.2016.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Neill KL, Huang K, Zhang J, Chen Y, Luo X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016;30:973–88. doi: 10.1101/gad.276725.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kazi A, Sun J, Doi K, Sung SS, Takahashi Y, Yin H, et al. The BH3 alpha-helical mimic BH3-M6 disrupts Bcl-X(L), Bcl-2, and MCL-1 protein-protein interactions with Bax, Bak, Bad, or Bim and induces apoptosis in a Bax- and Bim-dependent manner. The Journal of biological chemistry. 2011;286:9382–92. doi: 10.1074/jbc.M110.203638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riether C, Schurch CM, Ochsenbein AF. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015;22:187–98. doi: 10.1038/cdd.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.