

Abstract
Objectives
Genome-wide association studies have reported that DNA polymorphisms at the CDKN2A locus modulate fasting glucose in human and contribute to type 2 diabetes (T2D) risk. Yet the causal relationship between this gene and defective energy homeostasis remains elusive. Here we sought to understand the contribution of Cdkn2a to metabolic homeostasis.
Methods
We first analyzed glucose and energy homeostasis from Cdkn2a-deficient mice subjected to normal or high fat diets. Subsequently Cdkn2a-deficient primary adipose cells and human-induced pluripotent stem differentiated into adipocytes were further characterized for their capacity to promote browning of adipose tissue. Finally CDKN2A levels were studied in adipocytes from lean and obese patients.
Results
We report that Cdkn2a deficiency protects mice against high fat diet-induced obesity, increases energy expenditure and modulates adaptive thermogenesis, in addition to improving insulin sensitivity. Disruption of Cdkn2a associates with increased expression of brown-like/beige fat markers in inguinal adipose tissue and enhances respiration in primary adipose cells. Kinase activity profiling and RNA-sequencing analysis of primary adipose cells further demonstrate that Cdkn2a modulates gene networks involved in energy production and lipid metabolism, through the activation of the Protein Kinase A (PKA), PKG, PPARGC1A and PRDM16 signaling pathways, key regulators of adipocyte beiging. Importantly, CDKN2A expression is increased in adipocytes from obese compared to lean subjects. Moreover silencing CDKN2A expression during human-induced pluripotent stem cells adipogenic differentiation promoted UCP1 expression.
Conclusion
Our results offer novel insight into brown/beige adipocyte functions, which has recently emerged as an attractive therapeutic strategy for obesity and T2D. Modulating Cdkn2a-regulated signaling cascades may be of interest for the treatment of metabolic disorders.
Keywords: Obesity, Type 2 diabetes, Insulin sensitivity, Energy expenditure, cdkn2a, Genome-wide association study, Adipose tissue browning
Highlights
-
•
Cdkn2a deficiency protects mice against high fat diet-induced obesity.
-
•
Cdkn2a modulates brown-like/beige fat gene networks involved in energy production and lipid metabolism.
-
•
Increased CDKN2A expression in human obese adipocytes.
-
•
Increased UCP1 levels in adipocytes differentiated from CDKN2A-silenced hiPS cells.
1. Introduction
Obesity is the main risk factor for type 2 diabetes (T2D) and is due to an imbalance between energy intake and energy expenditure. Current anti-obesity drugs affecting energy intake or intestinal lipid absorption cause important and troublesome side effects for the patient, which limits their use. On the other hand, bariatric surgery has proven efficient for obesity and for diabetes remission with a dramatic effect on insulin resistance, although long-term complications and obesity relapse may arise. Therefore, understanding the signaling pathways that control fat storage and energy expenditure may open alternative avenues against obesity and linked T2D.
In animal models, genes such as E2f1 and Sertad2 (TRIP-Br2) have been shown to prevent fat accumulation and protect against diet-induced obesity (DIO), but they also improve insulin action in metabolic organs including adipose tissue [1], [2], [3]. Two major types of adipose tissues exist that are anatomically and functionally distinct: white (WAT) and brown (BAT) adipose tissues. Recent studies have revealed a new distinct type of thermogenic adipocyte intermingled within WAT, named beige cells (also known as brite cells). White adipocytes store excess energy as triacylglycerol and release free fatty acids (FFAs) as energy substrate when required. On the other hand, the catabolic capability of BAT and beige adipocytes to burn fat contributes to reduce circulating FFAs. Indeed, BAT expresses high levels of uncoupling protein 1 (UCP1), leading to substrate oxidation and subsequent heat production and energy expenditure (for review, see Ref. [4]). Beige adipocytes also express UCP1 and develop under cold exposure. This adaptive process is called WAT beiging/browning [5]. In pathological conditions such as obesity, elevated circulating FFAs are associated with insulin resistance and T2D. Since energy balance is modulated by food intake, physical activity, as well as non-shivering thermogenesis in BAT, increasing energy expenditure by classical BAT activation or by promoting browning of WAT have recently emerged as new putative therapy to alleviate the effects of obesity and prevent insulin resistance and T2D [4], [6]. Recent reports in humans further confirmed the important function of brown/beige adipocytes in enhancing glucose tolerance and insulin sensitivity [7], [8]. Therefore, the identification of selective molecular pathways for beige adipocyte biogenesis would represent a first step towards innovative therapeutic options.
Genome-wide association studies (GWASs) have established the CDKN2A/B locus as a hotspot influencing genetic risk for different cardio-metabolic diseases including T2D [9], [10]. CDKN2A/B single nucleotide polymorphisms associated with T2D also modulate fasting insulin and insulin sensitivity in non-diabetic subjects [11]. In particular, rs10757278-G and rs10811661-T SNPs are situated near the CDKN2A/B locus on chromosome 9p21.3 and are respectively associated with the risk for coronary artery disease (CAD) and T2D [12]. The genomic regions containing those SNPs are also in close vicinity of the long non-coding RNA ANRIL (Antisense Non-coding RNA in the Inhibitor of CDK4 (INK4) Locus). The function of ANRIL is still unknown but it seems to be involved in the epigenetic repression of the CDKN2A/B locus [13]. Although rs10757278 and rs10811661 are less than 10 kb away from CDKN2A/B genes, those SNPs may affect dynamic chromosome structures and genome activity. Indeed, rs10811661 is negatively associated with a trans-eQTL, presenilin 1 (PSEN1), a gene associated in mice with glucose intolerance and insulin resistance [14]. In addition, Lillycrop et al. recently demonstrated that decreased DNA methylation at the CDKN2A gene is associated with a higher risk for a child to develop obesity during adulthood [15]. Furthermore, rare heterozygous loss-of-function mutations in CDKN2A affect glucose homeostasis through its metabolic role in pancreatic beta cells and liver [16]. Altogether, those results pinpoint the importance of studying the impact of loss-of-function of CDKN2A in metabolic tissues.
The human CDKN2A locus encodes for p16INK4a a CDK inhibitory (CDKI) protein and the p53 regulatory protein p14ARF (p19ARF in mice), whereas the CDKN2B gene encodes another CDKI, p15INK4b. Products of the CDKN2A/B locus are involved in apoptosis, senescence, and aging [9]. Whilst p16 and p15 are potent CDKI preventing binding of CDK4/6 to Cyclin D, p14ARF mainly exerts it anti-proliferative activity via the inhibition of MDM2, an ubiquitin-ligase that modulates the activity of the tumor suppressor p53. The CDKN2A/B pathway is involved in metabolic control through several ways including adipocyte differentiation [17], [18], [19], macrophage polarization [20], fasted–fed transition [21], [22], muscle metabolism [1], and pancreatic beta cell proliferation, regeneration, and function [23], [24], [25].
While rodent and human studies have highlighted the potential role of CDKN2A on insulin secretion [16], [26], the contribution of CDKN2A to the control of insulin sensitivity is still elusive.
To decipher the link between Cdkn2a, obesity and insulin resistance, we analyzed the effect of Cdkn2a deficiency in mice on metabolic homeostasis [27]. Here, we demonstrate that the genetic deletion of Cdkn2a promotes energy expenditure and improves insulin sensitivity during DIO. We found that the Cdkn2a locus modulates signaling pathways in inguinal WAT (ingWAT), where it controls beige adipocyte development. Importantly, we observed that CDKN2A expression is increased in adipocytes from obese patients. CDKN2A silencing during human-induced pluripotent stem cells adipogenic differentiation promoted UCP1 expression. These data suggest that Cdkn2a plays a key role in energy metabolism by regulating a white-to-brown fat transition.
2. Material and methods
2.1. Materials and oligonucleotides
Chemicals, unless stated otherwise, were purchased from Sigma–Aldrich. Anti-UCP1 antibody was purchased from Abcam, and anti-PKA substrate antibody was from Cell Signaling Technology. The oligonucleotides sequences used for various experiments are listed in Table S5.
2.2. Animals experiments
Cdkn2a+/+ and Cdkn2a−/− mice were previously described [27] and were maintained on a mixed background according to European Union guidelines for use of laboratory animals. In vivo experiments were performed in compliance with the French ethical guidelines for studies on experimental animal (Animal house agreement no. B 59-35015, Authorization for Animal Experimentation no. 59-350294, project approval by our local ethical committee no. CEEA 482012). All experiments were performed with healthy male mice. Animals with detectable tumors were excluded from the study. Mice were housed under a 12 h light–dark cycle and given a regular chow (A04; Safe). For HFD studies, 8-week old mice were placed on D12492 diet (60% cal/fat; Research Diet Inc.) for 13 weeks. Metabolic phenotyping experiments were performed according to the EMPRESS protocols (http://empress.har.mrc.ac.uk). Briefly, intraperitoneal glucose (2 g of glucose per kg of body weight) and insulin (0.75 U of insulin per kg of body weight) tolerance tests were performed as previously described [28], [29] on 16-h-fasted animals for IPGTT and 5-h-fasted animals for ITT. Glycemia was measured before and at different time after glucose and insulin injections using the Accu-Check Performa (Roche Diagnostics). Circulating insulin levels were measured using the Ultrasensitive Insulin Elisa kit (Mercodia). For cold experiments, mice were maintained at +23 °C or placed at +4 °C for 5h and fasted during the time of the experiment. Body temperature was measured using a rectal probe as described previously [1]. Metabolic rate was measured by indirect calorimetry using the Phenomaster metabolic cage system (TSE Systems). Mice were housed individually and maintained at 23 °C under a 12 h light/12 h dark cycle. Food and water were available ad libitum. Mice were sacrificed by cervical dislocation and tissues were harvested and snap-frozen for further processing.
2.3. RNA extraction, measurements, and profiling
Total RNA was extracted from rodent cells and tissues using trizol reagent (Life Technologies) as described previously [1], [28]. mRNA expression was measured after reverse transcription by quantitative real-time PCR (qRT-PCR) with FastStart SYBR Green master mix (Roche) according to the manufacturer's recommendations and gene-specific oligonucleotides. Mouse qRT-PCR results were normalized to endogenous cyclophilin reference mRNA levels. The results are expressed as the relative mRNA level of a specific gene expression using the formula 2−ΔCt. For human cells, RNAs were purified on RNeasy Plus Universal columns (Qiagen, France) according to the manufacturer's instructions. Total RNA sample concentrations were determined using a Nanodrop spectrophotometer (Thermo Scientific, Waltham, MA, USA). 1 μg RNA was used for reverse transcription. Real-time PCR assays were run on a OneStep real-time PCR machine (Applied Life Sciences) and SYBR green was from Applied Biosystems. Normalization was performed using the geometric averages of the housekeeping gene TBP. Delta Ct values were used to achieve relative quantification of gene expression when “expression relative to TBP” is indicated.
2.4. Histology, immunofluorescence (IF), and immunohistochemistry (IHC)
IF and IHC were performed exactly as described previously [1], [28]. Briefly, deparaffinized adipose tissues sections (5 μm) were subjected to heat-induced antigen retrieval. The slides were incubated with 0.3% H2O2 for 10 min to quench the endogenous peroxidase. The sections were then incubated in 1% BSA/PBS for 10 min, followed by overnight incubation with primary (Rabbit-UCP1) antibodies at 4 °C. Afterward, the slides were incubated at room temperature for 2 h with horseradish peroxidase (HRP)-conjugated (for IHC) or Alexa-conjugated anti-rabbit secondary antibodies (for IF, Fischer Scientific). The IHC staining was developed using fast 3,3′-diaminobenzidine (DAB) tablet sets (D4293; Sigma, St. Louis, MO, USA). The sections were counterstained with hematoxylin and examined by light microscopy. H&E staining was performed using classical protocols. For IF, nuclei were stained with Hoechst. Images were processed using ImageJ software by two observers blinded to experimental groups.
2.5. Protein extracts, PKA activity measurement and immunoblot analysis
Immunoblot was performed as described previously [1], [29]. Tissues and cells were washed using cold PBS, and lysis was performed by using 50 mM Tris–HCl pH 8, 137 mM NaCl, 10% glycerol, 1% NP-40 and phosphatase, protease, deacetylase inhibitors (Sigma–Aldrich) on ice. Western blotting was performed using 30 μg of proteins loaded on SDS-PAGE precast gel (Biorad). After electrotransfer, the membrane was blocked for 1 h at room temperature with 5% nonfat milk in 0.1% Tween–Tris-buffered saline (TTBS) buffer. Membranes were then incubated overnight at 4 °C with primary antibodies as indicated in blocking buffer containing 5% nonfat milk or 0.5% bovine serum albumin at the dilution specified by the manufacturers. Subsequently, membranes were incubated with the secondary antibody conjugated with the horseradish peroxidase enzyme. The visualization of immunoreactive bands was performed using the enhanced chemiluminescence plus western blotting detection system (GE Healthcare). hiPSCs-BAP cells were rinsed with PBS and solubilized in stop buffer containing 50 mM Hepes, pH 7.2, 150 mM NaCl, 10 mM EDTA, 10 mM Na4P2O7, 2 mM Na3VO4, and 1% Triton X-100 supplemented with Protease Inhibitor Cocktail (Roche). The primary antibodies and dilutions used were: UCP1 (1/500, Abcam), Tubulin (1/10,000, Sigma); secondary horseradish peroxidase-conjugated antibodies were purchased from Sigma. Quantitation of protein signal intensity was performed by volume densitometry using the ImageJ 1.47t software (National Institutes of Health). PKA activity was measured on tissue homogenates using the non-radioactive PKA Kinase Activity assay kit following manufacturers' instructions (Abcam).
2.6. Primary adipocyte cell culture
The stromal vascular fraction (SVF) of inguinal adipose or BAT tissues of 8-week old mice was isolated by collagenase (ROCHE) digestion followed by two alternative filtration steps (using 100 and 40 μM strainers) and centrifugations for 5 min at 700 × rpm. Cells were then plated and differentiated upon confluence in DMEM/F-12 GlutaMAX with adipogenic cocktail (0.5 mM 3-isobutyl-1-methylxanthine [IBMX], 2.5 μM dexamethasone, 1 μM rosiglitazone, 0.02 μM insulin for inguinal cells and 0.02 μM insulin, 0.125 μM indomethacin, 0.5 mM 3-isobutyl-1-methylxanthine [IBMX], 2.5 μM dexamethasone and 1 nM T3, for BAT cells) for 48 h. Inguinal cells were maintained in 0.02 μM insulin and BAT cells in 1 nM T3 and 0.02 μM insulin. Cells were harvested at days 8 post-differentiation. To label lipid droplets, fully differentiated primary adipocytes cells were fixed in 4% PFA then labeled with BODIPY 493/503 lipid probe solution (Molecular Probes/Life Technologies) for 30 min. After PBS wash the staining was examined by fluorescence microscopy.
2.7. Cell culture and siRNA experiments
Brown-like adipose progenitors (BAPs) were derived from human-induced pluripotent stem cells (hiPSCs) and induced to undergo adipocyte differentiation as described in Mohsen-Kanson et al. [65] and Hafner et al. [66]. For more details see http://www.nature.com/protocolexchange/protocols/5175. hiPSC-BAPs were transfected with small interfering RNA duplexes (Human CDKN2A siRNA-SMART pool, GEHealth Bio-Sciences, Rosersberg, Sweden) using HIPerfect reagent (Qiagen, France) during the exponential growth phase as described by the supplier.
2.8. Pamgene experiments
For kinome analysis, STK microarrays were purchased from PamGene International BV (STK pamchips, Ref. [32]). Each array contained 140 phosphorylatable peptides as well as 4 control peptides. Sample incubation, detection, and analysis were performed in a PamStation 12 according to the manufacturer's instructions. Briefly, extracts from fully differentiated day 8 (D8) primary adipocytes from Cdkn2a+/+ and Cdkn2a−/− mice were freshly prepared using M-PER mammalian extraction buffer (Thermo Scientific) containing 1:50 Halt phosphatase inhibitor cocktail (Thermo Scientific) and 1:50 Halt protease inhibitor cocktail, EDTA-free (Thermo Scientific) for 20 min on ice. The lysates were then centrifuged at 15,871 g for 20 min to remove all debris. The supernatant was aliquoted, snap-frozen in liquid nitrogen, and stored at −80 °C until further processing. Prior to incubation with the kinase reaction mix, the arrays were blocked with 2% BSA in water for 30 cycles and washed 3 times with the PK assay buffer. Kinase reactions were performed for 1 h with 2.5 μg of total extract of the mature adipocyte and 400 μM ATP at 30 °C. Phosphorylated peptides were detected with an anti-rabbit-FITC antibody that recognizes a pool of anti-phospho serine/threonine antibodies. The instrument contains a 12-bit CCD camera suitable for imaging of FITC-labeled arrays. The images obtained from the phosphorylated arrays were quantified using the BioNavigator software (PamGene International BV), and the list of peptides whose phosphorylation was significantly different between Cdkn2a+/+ and Cdkn2a−/− was uploaded to IPA for pathway analysis. The BioNavigator software was used to perform the HPRD based upstream kinase determination that is shown in Figure 4C.
Figure 4.

Activation of the Protein Kinase A pathway in Cdkn2a−/− adipocytes induces white adipose tissue browning. (A) mRNA levels for adipogenic and thermogenic genes in ingWAT primary differentiated adipocytes isolated from Cdkn2a+/+ and Cdkn2a−/− mice (n = 3). (B) Heatmap showing differences in peptide phosphorylation between primary differentiated adipocyte from Cdkn2a+/+ and Cdkn2a−/− inguinal fat depots (n = 3). Upstream kinases were identified using the Human Protein Reference Database. (C) Pamgene results were analyzed using Ingenuity Pathway Analysis (IPA) to identify the top activated canonical pathways in Cdkn2a−/− primary differentiated adipocyte compared to Cdkn2a+/+. (D) PKA activity measurement in tissue homogenates from BAT and ingWAT isolated from Cdkn2a+/+ and Cdkn2a−/− mice (n = 3) treated with DMSO or the PKA inhibitor H89 at 10 μM. (E, F) Oxygen consumption rate (OCR) was measured using the Seahorse XFe24 platform. After 8 days of differentiation (D8), OCR was measured in Cdkn2a+/+ and Cdkn2a−/− differentiated adipocytes (E) and quantification of basal respiration, basal proton leak (by blocking ATP synthase activity with oligomycin), maximal respiration by stimulating uncoupling process with FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) and non-mitochondrial respiration (with rotenone) is represented (F, n = 5). Cdkn2a−/− cells were treated 1 h with 10 μM of PKA inhibitor H89 before the experiment (n = 5). All values are expressed as means ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001.
2.9. RNA sequencing
Total RNA was extracted from primary adipocytes 8 days after differentiation using the RNeasy Microkit (Qiagen) following manufacturer's instructions. RNA quality was verified using RNA 6000 nanochips on the Agilent 2100 bioanalyzer. Purified RNA (200 ng) with RNA integrity number ≥8 was subsequently used for library preparation (TruSeq Stranded mRNA Library Preparation Kit, Illumina) and sequenced on a HiSeq2500 (Illumina). Three biological replicates per condition were sequenced in paired-end. A mean of 54 million paired-end reads of 75 bp were generated for each sample. After initial checks and validation of sequence quality, RNA-seq reads were aligned to the mouse reference genomes (mm10) using TopHat2 [33]. Subsequently, both quantification and annotation of the reads were performed using Bioconductor package Rsubread [34]. Finally, the differential gene expression analyses were performed using Bioconductor package DESeq2 [35]. Using a p < 0.05 adjusted for multiple comparisons as threshold, we then performed pathway analysis using Ingenuity Pathway Analysis (Ingenuity Sytems).
2.10. Seahorse experiments
Oxygen consumption rate (OCR) was performed using the XF24e platform from Seahorse Biosciences. Primary adipocytes from Cdkn2a+/+ and Cdkn2a−/− mice were grown and differentiated in a Seahorse plate. Growth medium was replaced with 37 °C seahorse DMEM 1 h before the measurement to allow equilibrium. Measurement was performed at 37 °C, using 3–3–3-min intervals. Basal respiration was measured then uncoupled, and maximal OCR were determined using oligomycin and carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP, 1 μM each), respectively. Complex I-dependent respiration was inhibited with rotenone (3 μM). OCR data were normalized using total protein or DNA content as indicated.
2.11. Human adipocyte expression data
Gene Expression Omnibus (GEO) analysis from the NCBI was performed using “adipocytes obese human” as keywords, filtered with “published in the last year” and sorted by the number of samples “high to low”. The dataset GSE94753 [36] was selected and analyzed using GEO2R. The platform GPL11532 was selected and groups were defined using the disease status (lean, WAT, control versus obese resistant, WAT OIR). Expression level data were extracted from “profile graph” using CDKN2A ID 8160441.
2.12. Statistical analysis
Data are presented as mean ± SEM. Statistical analyses were performed using unpaired two-tailed Student's t-test, one-way ANOVA with least significant difference Bonferroni post hoc test or two-way ANOVA with Bonferroni post hoc tests as appropriate using GraphPad Prism software. Differences were considered statistically significant at p < 0.05 (*p < 0.05; **p < 0.01 and ***p < 0.001).
3. Results
3.1. Germline Cdkn2a deficiency improves insulin sensitivity in mice
We first analyzed the metabolic profile of Cdkn2a-deficient mice [27]. 9- to 12-week old Cdkn2a+/+ and Cdkn2a−/− mice fed a chow diet (CD) displayed similar body weight (Figure 1A), glucose tolerance (Figure 1B, C) and fasting glycemia (Figure S1A). In vivo glucose stimulated insulin secretion (GSIS) was similar in both genotypes under CD (Figure 1D). After 13 weeks of high fat diet (HFD) feeding, body weight of Cdkn2a−/− mice was lower than that of age-matched control mice (Figure 1E), with comparable fasting glucose levels (Figure S1B) and similar glucose clearance during ipGTT tests (Figure 1F, G). As expected, upon HFD feeding Cdkn2a+/+ had increased basal insulin secretion to maintain normal glucose levels, indicating an insulin-resistant state (Figure 1H). By contrast, HFD Cdkn2a-deficient mice displayed lower basal insulin levels and were still responsive to a glucose load (Figure 1H), suggesting that Cdkn2a deficiency may affect insulin sensitivity rather than insulin production by the pancreatic beta cells. Consistently, insulin sensitivity was significantly improved in Cdkn2a−/− mice under CD (Figure 1I, J) and even more under HFD (Figure 1K, L). These results indicate that Cdkn2a deficiency prevents weight gain and improves insulin sensitivity during metabolic stress.
Figure 1.

Cdkn2a-deficient mice are insulin sensitive during DIO. (A) Body weight of 9- to 12-week old control (Cdkn2a+/+) and mutant (Cdkn2a−/−) mice fed with chow diet (CD, n = 30–32). (B, C) Intraperitoneal glucose tolerance test (ipGTT, B) in Cdkn2a+/+ and Cdkn2a−/− mice fed with CD (n = 11–12) and the corresponding area under the curve (AUC, C) of ipGTT. (D) Blood insulin levels as measured during ipGTT in CD-fed mice. (E) Body weight of high fat diet (HFD, n = 7–16) fed mice. (F, G) ipGTT in HFD mice (F, n = 7) and its AUC (G). (H) Blood insulin levels during ipGTT in HFD-fed mice. (I) Intraperitoneal insulin tolerance test (ipITT) in 8 weeks mice fed with CD (n = 7). (J) AUC of ipITT. (K, L) ipITT in HFD-fed mice (K, n = 7) and the corresponding AUC of ipITT (L). All values are expressed as means ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001.
3.2. Increased energy expenditure in Cdkn2a−/− mice
To characterize the effect of Cdkn2a deficiency on insulin sensitivity and body weight, we monitored energy expenditure using indirect calorimetry measurements. Under CD, Cdkn2a−/− mice showed increased food intake only during the second (1AM to 7AM) part of the dark phase compared to controls (Figure 2A and Figure S2A), but this was doubled under HFD during the entire dark phase (Figure 2B and Figure S2B). The reduced body weight gain under HFD (Figure 1) despite an increase in food intake suggests enhanced energy expenditure. The ambulatory activity was similar among all genotypes either under CD (Figure S2C, D) or HFD (Figure S2E, F). Interestingly, Cdkn2a−/− mice showed increased energy expenditure measured by heat production, under both CD (Figure 2C and Figure S2G) and HFD (Figure 2D and Figure S2H). This increased metabolic rate was not due to substrate utilization preference (glucose or free fatty acids), since no significant difference in respiratory exchange ratio (RER) was observed between Cdkn2a+/+ and Cdkn2a−/− mice (Figure 2E, F and Figure S2I, J). We then investigated adaptive thermogenesis by either performing a cold test or subjecting mice to 6 h of fasting. During an acute cold stress or under fasting conditions Cdkn2a−/− mice maintained a higher body temperature as compared to control animals (Figure 2G, H). Altogether, these findings suggest that the global genetic deletion of Cdkn2a locus in mice enhances energy expenditure and adaptive thermogenesis.
Figure 2.

Increased energy expenditure in Cdkn2a−/− mice. (A, B) Cumulative food intake in (A) CD and (B) HFD-fed control (Cdkn2a+/+) and mutant (Cdkn2a−/−) mice (n = 4) measured during the dark (7PM to 1AM and 1AM to 7AM) and the light (7AM to 1PM and 1PM to 7PM) periods. (C, D) Heat production in (C) chow diet and (D) HFD-fed mice. (E, F) Respiratory exchange ratio (RER) in (E) CD and (F) HFD-fed mice. (G, H) Body temperature in CD-fed Cdkn2a+/+ and Cdkn2a−/− mice after 5 h fasting (G, n = 4) and 5 h of cold exposure (H, n = 4). All values are expressed as means ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001.
3.3. Beige fat programming is dependent on Cdkn2a
To determine if Cdkn2a regulates brown and beige adipocyte function, we first analyzed BAT thermogenic gene expression in Cdkn2a+/+ and Cdkn2a−/− mice. Ucp1 and Prdm16 expression increased in Cdkn2a-deficient BAT fed a CD or HFD (Figure 3A, B). Ppargc1a expression was similar in control and Cdkn2a-deficient BAT under CD, but raised upon HFD in Cdkn2a−/− BAT (Figure 3A, B). Mitochondrial content (Figure S3A) and Ucp1 protein levels (Figure S3B, C) were identical in Cdkn2a+/+ and Cdkn2a−/− BAT tissue homogenates. Total BAT mass was not significantly different in Cdkn2a+/+ and Cdkn2a−/− mice fed CD, but was decreased in HFD-fed Cdkn2a−/− mice (Figure S3D, E). When expressed relative to total body weight no difference between genotypes was observed (Figure S3F, G), suggesting that Cdkn2a deficiency may have a week effect on BAT functions. When measuring p16, p19, and Ucp1 mRNA levels in different mouse fat depots, we observed that p16 (Figure 3C) and p19 (Figure 3D) were mostly expressed in inguinal WAT (ingWAT) compared to epididymal WAT or BAT, whereas Ucp1 (Figure 3D) was strongly expressed in BAT depots. We then hypothesized that the Cdkn2a gene products p16 and p19 may promote the WAT beiging/browning process. To determine if Cdkn2a deficiency modifies beige adipocyte differentiation, we compared the expression levels of several adipocyte markers between control and Cdkn2a-deficient animal's ingWAT. The expression of brown fat-selective genes Ppargc1a, Ucp1, Prdm16, Tfam, Cox5a, Cox8a, and Dio2 was increased in ingWAT from CD-fed Cdkn2a−/− animals (Figure 3F). This effect was more pronounced in HFD-Cdkn2a−/− ingWAT (Figure 3G). Hematoxylin and eosin (H&E) staining showed that Cdkn2a−/− ingWAT had smaller adipocytes with multilocular lipid droplets (Figure 3H), associated with an increase in UCP1 immunostaining (Figure 3H), further confirmed through immunoblot (Figure 3I, J) and immunofluorescence (Figure S3H) experiments. Mitochondrial content was identical in Cdkn2a+/+ and Cdkn2a−/− ingWAT (Figure S3A). Altogether, these data suggest that Cdkn2a deficiency induces a white-to-brown fat conversion in ingWAT.
Figure 3.

Cdkn2a deficiency promotes the browning process in vivo. (A, B) mRNA level of thermogenic genes in BAT from Cdkn2a+/+ and Cdkn2a−/− mice fed chow (A) or HFD (B, n = 5–6). (C–E) mRNA levels of p16 (C), p19 (D) and Ucp1 (E) in epididymal WAT (epiWAT), inguinal WAT (ingWAT) and BAT. (F, G) mRNA levels for adipogenic and thermogenic genes in ingWAT from Cdkn2a+/+ and Cdkn2a−/− mice fed CD (F) or HFD (G, n = 5–6). An inset of some genes is represented. (H) Hematoxylin and eosin (H&E) staining and UCP1 immunohistochemistry of representative sections of ingWAT from Cdkn2a+/+ and Cdkn2a−/− mice fed CD. (I, J) Western blot assay (I) and quantification (J) showing UCP1 protein levels in ingWAT of Cdkn2a+/+ and Cdkn2a−/− mice. Tubulin was used as a loading control. Quantification was performed using the ImageJ software. All values are expressed as means ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001.
3.4. The effect of Cdkn2a on browning is cell-autonomous and requires the activation of the Protein Kinase A pathway
Stromal vascular cells from ingWAT and BAT can be differentiated into brown-like adipocytes. To determine the cell-autonomous requirement of Cdkn2a to the beiging process, we isolated ingWAT and BAT pre-adipocytes from control and Cdkn2a-deficient mice and differentiated them for 8 days (Figure S4A). Lipid droplet accumulation was more pronounced in Cdkn2a−/− ingWAT and BAT differentiated primary adipocytes, but not in adipocytes differentiated from epididymal pre-adipocytes (eWAT, Figure S4B). We next examined the expression of adipocytes (Fabp4) and brown/beige fat markers (Ppargc1a, Ucp1, Prdm16, Cox5a, Cidea, Elovl3, Dio2) and observed a strong induction of these genes in differentiated ingWAT primary cells upon p16 and p19 genetic deletion (Figure 4A), but not in differentiated BAT or eWAT primary cells (Figure S4C and data not shown).
The Cdkn2a products p16ink4a and p19ARF are key regulators of the activity of serine/threonine kinases (STK) involved in cell proliferation and senescence [9]. We postulated that STK may be involved in beige fat programming and performed a global kinome analysis in primary Cdkn2a+/+ and Cdkn2a−/− differentiated adipocytes. We used Pamgene arrays containing 140 serine/threonine peptides that are the substrates of one or multiple kinases. We incubated these chips with protein lysates of primary adipocytes after 8 days of differentiation (D8). We then analyzed the phosphorylation status of these peptides and identified the putative upstream kinases involved. At D8, 47 peptides with significant differences in phosphorylation levels between control and Cdkn2a-deficient adipocytes were observed (Figure 4B and Table S1). Using the Human Protein Reference Database (HPRD) [37] to identify potential upstream kinases we identified several signaling pathways that may be affected in Cdkn2a-deficient adipocytes, including Protein Kinase A (PKA) and Protein Kinase G (PKG) signaling pathways. Interestingly, PKG is an important regulator of WAT browning [38], [39]. For some phosphorylated peptides upstream kinases could not be identified using HPRD analysis (Figure 4B, NA or –). When we analyzed the diseases and/or biological functions affected by the differences in peptide phosphorylation observed upon Cdkn2a genetic deficiency through Ingenuity Pathway Analysis (IPA), we found networks related to “Cancer,” “Organismal Injury and Abnormalities,” and “Developmental Disorders,” but also to “Cardiovascular Disease” (Table S2). Then we found a significant enrichment of potential signaling pathways upstream kinases involved in “Protein Kinase A Signaling” in Cdkn2a-deficient cells (P = 1.07 × 10−15; Figure 4C), confirming HPRD analysis. In addition, “AMPK signaling” (P = 4.96 × 10−12; Figure 4C), “synaptic long-term potentiation” (P = 1.28 × 10−10; Figure 4C), “prostate cancer signaling” (P = 9.26 × 10−10; Figure 4C) or “Molecular Mechanisms of Cancer” (P = 3.92 × 10−09; Figure 4C) were canonical pathways also affected in Cdkn2a−/− primary adipocytes. These analyses prompted us to focus on dissecting the contribution of PKA to our phenotype. Increased PKA activity was confirmed in Cdkn2a−/− ingWAT homogenates, whereas no changes were found in BAT or eWAT (Figure 4D and data not shown). The increased PKA activity was further confirmed in Cdkn2a−/− ingWAT using protein homogenates and an anti-PKA phospho-substrate antibody (Figure S4D, E). The increase in PKA activity was associated with a significant raise in basal and maximal respiration, as well as basal proton leak of Cdkn2a−/− primary ingWAT cells, as measured by oxygen consumption rate (OCR, Figure 4E, F). The PKA inhibitor H89 completely abolished PKA activity in tissue homogenates (Figure 4D) as well as the increase of oxygen consumption observed in Cdkn2a−/− primary cells (Figure 4E, F). Altogether, these findings indicate that Cdkn2a modulates PKA activity in adipocytes, oxygen consumption and promotes beiging in ingWAT. Our results also suggest that Cdkn2a may regulate other pathways, such as PKG, that can further contribute to adipose beiging.
3.5. Cdkn2a modulates a regulatory gene network involved in brown adipocyte function
We next carried out RNA-sequencing (RNA-seq) analysis in Cdkn2a+/+ and Cdkn2a−/− primary adipocytes at D8 of differentiation. We found 11,855 transcripts differentially expressed (Table S3). IPA highlighted the presence of a network of 98 genes associated to Cdkn2a deletion which was involved in “energy production, lipid metabolism and small molecule biochemistry,” including Ppargc1a and Prdm16 (Figure S5A). Subsequently, we analyzed the diseases and/or functions associated to Cdkn2a deficiency. The main differences between Cdkn2a+/+ and Cdkn2a−/− primary adipocytes were included into “carbohydrate metabolism” (P = 1.67 × 10−3) or “cellular development, connective tissue development and function, tissues development” (P = 4.36 × 10−3; Table S4). Next, we used the “upstream regulator analysis” function of IPA to identify potential contributors to the phenotype of Cdkn2a+/+ and Cdkn2a−/− primary adipocytes. Among the most significant upstream regulators, tumor necrosis factor (TNF) and promyelocytic leukemia (PML) signaling pathways were activated (Table 1). Interestingly, norepinephrine, which acts on β-AR receptors in BAT [40] and WAT [41], [42], was also activated (Table 1). This analysis further revealed a potential inhibition of the NR1H2 pathway, which has been associated to severe obesity [43] (Table 1). Transcription factor network analyses revealed that PPARGC1A and PRDM16 were activated in Cdkn2a−/− cells (Table 2). The period circadian clock 2 (PER2) transcription factor was also activated (Table 2). This is consistent with the observation that PER2 is necessary for adaptation to cold temperature in mice through the regulation of Ucp1 expression [44]. Moreover, Aryl hydrocarbon receptor nuclear translocator like (ARNTL) activity is potentially inhibited in our model (Table 2), in agreement with its antagonistic role on PER2 in the control of the circadian rhythm. Finally, the marked inhibition of inhibitor of DNA (ID1) further confirmed the brown-like features of Cdkn2a−/− primary adipocytes. Indeed, germline deficient Id1 mice are protected against DIO through enhanced energy expenditure [45]. These RNA-seq data further suggest that Cdkn2a is a crucial regulator of effectors controlling the expression of brown-like markers.
Table 1.
Pathway analysis of RNA-sequencing data in Cdkn2a+/+ and Cdkn2a−/− primary adipocytes.
| Upstream regulator | Exp log ratio | Molecule type | Predicted activation state | Activation z-score | p-Value of overlap | Target molecules in dataset |
|---|---|---|---|---|---|---|
| CIDEC | Other | 0.786 | 6.82E−03 | ACADL,COX4I2,CYCS,DGAT2,EBF2,FOXC2,PNPLA2,PPARG,PPARGC1A,PPARGC1B | ||
| CCDC3 | Other | 9.86E−03 | ACACA,DGAT2,FASN | |||
| PPARGC1B | −1.230 | Transcription regulator | 9.86E−03 | EGLN3,ERO1A,VEGFA | ||
| Melatonin | Chemical – endogenous mammalian | 9.86E−03 | ACACA,FASN,IRS1 | |||
| ARNTL | −0.518 | Transcription regulator | 2.18E−02 | DBP,ELOVL6,PER2,PER3 | ||
| PTPN1 | Phosphatase | −1.387 | 2.62E−02 | IRS1,IRS2,PPARGC1A,PRDM16,TMEM26 | ||
| SGCB | 0.379 | Other | 3.31E−02 | DAG1,SGCD,SGCE | ||
| SGCD | 1.385 | Other | 3.31E−02 | DAG1,SGCB,SGCE | ||
| IL1B | Cytokine | 3.31E−02 | IRS1,SERPINE1,VEGFA | |||
| PNPLA2 | −0.896 | Enzyme | −1.131 | 4.23E−02 | ACADL,FASN,Gk,PPARG | |
| FFAR4 | G-protein coupled receptor | 4.60E−02 | INSR,IRS1 | |||
| TNFRSF1B | 0.504 | Transmembrane receptor | 4.60E−02 | SERPINE1,SOCS3 | ||
| STAT1 | Transcription regulator | 4.60E−02 | PPARG,SOCS3 | |||
| EGR1 | Transcription regulator | 4.60E−02 | CSF1,F3 | |||
| TGFB1 | Growth factor | 4.60E−02 | F3,SERPINE1 | |||
| TNFRSF1A | Transmembrane receptor | 4.60E−02 | SERPINE1,SOCS3 | |||
| Norepinephrine | Chemical – endogenous mammalian | 1.930 | 2.03E−01 | ELOVL6,PPARGC1A,RBP4,SLC2A1,VEGFA | ||
| TNF | Cytokine | Activated | 2.048 | 2.27E−01 | AOC3,APLN,Celf1,DBI,F3,FABP5,FASN,FOS,INSR,NRIP1,PPARG,RELA,SERPINE1,SOCS3,TALDO1 | |
| NR1H2 | 0.471 | Ligand-dependent nuclear receptor | Inhibited | −2.449 | 2.42E−01 | ELOVL6,FASN,PNPLA2,PRDM16,RB1,Saa3,SLC16A2 |
| PML | Transcription regulator | Activated | 2.000 | 3.52E−01 | ACACA,ACOX1,FASN,PPARGC1A |
Table 2.
Analysis of upstream transcription factors in RNA-sequencing data from Cdkn2a+/+ and Cdkn2a−/− primary adipocytes.
| Molecules in network | Score | Focus molecules | Top diseases and functions |
|---|---|---|---|
| CEBPA,FOXC2,ID1,KLF11,MDM2,PPARGC1A,PPARGC1B,PRDM16,RB1,SREBF1,STAT6,TP53 | 3 | 9 | Cellular Development, Connective Tissue Development and Function, Tissue Development |
| ARNTL,DBP,PER2 | 2 | 3 | Behavior, Nervous System Development and Function, Organ Morphology |
| ID3,TCF3 | 0 | 1 | Hematological System Development and Function, Lymphoid Tissue Structure and Development, Tissue Morphology |
| HDAC3,RELA | 0 | 1 | Cell Death and Survival, Neurological Disease, Organismal Injury and Abnormalities |
3.6. CDKN2A modulates adipose tissue browning in human adipocytes
We next questioned whether deregulated expression of CDKN2A could be associated to obesity in humans. Interestingly, using global transcriptome profiling of adipocytes from lean and obese subjects (GEO: GSE94752; Ref. [36]) we observed a significant increase of CDKN2A mRNA expression levels in obese insulin-resistant adipocytes compared to lean controls (Figure 5A). We next wondered whether decreasing CDKN2A levels in human adipocytes could recapitulate some of the effects we observed in mice and induce browning in human cells. Recent reports demonstrated that human-induced pluripotent stem cells (hiPSCs) could be differentiated into brown/brite adipose progenitors (hiPSCs-BAP, Refs. [30], [31]). To further analyze whether CDKN2A modulates UCP1 expression in human cells, we silenced CDKN2A expression during the exponential growth phase of hiPSCs and differentiated them into brown/brite adipose cells during 22 days as described previously [30], [31]. As shown in Figure 5B, C, p16 and p14 mRNA levels were significantly reduced after silencing of the CDKN2A gene. In addition, UCP1 mRNA levels were strongly induced in hiPSCs-BAP upon CDKN2A silencing (Figure 5D). This transcriptional effect was associated with a strong increase of UCP1 protein levels in hiPSCs-BAP CDKN2A-deficient cells (Figure 5E, F). Moreover, silencing CDKN2A increased lipid droplet accumulation and adipocyte differentiation in hiPSCs-BAP cells (Figure 5G). Altogether, our data indicate that CDKN2A is an important mediator of human adipose tissue browning.
Figure 5.

CDKN2A is increased in obese insulin-resistant adipocytes and its silencing in hiPSCs-BAP cells induces UCP1 expression. (A) Correlation between CDKN2A and human obesity. CDKN2A expression is upregulated in adipocytes from obese insulin-resistant patients. Analyses are based on the human dataset GEO: GSE94752. (B–D) Relative expression of p16 (B), p14 (C), and UCP1 (D) genes in control (siCont) or CDKN2A silenced (siCDKN2A) hiPSCs-BAPs (n = 3). (E, F) Western blot assay (E) and quantification (F) showing UCP1 protein levels in control (siCont) or CDKN2A silenced (siCDKN2A) hiPSCs-BAPs (n = 3). Tubulin was used as a loading control. Quantification was performed using the ImageJ software. (G) Representative microscopy analysis of siCont and siCDKN2A hiPSCs-BAPs after differentiation showing increased lipid droplets upon CDKN2A silencing. All values are expressed as means ± SEM; *p < 0.05 and **p < 0.01.
4. Discussion
Our results demonstrate that CDKN2A gene products p16INK4A and p19ARF are, beside their known role as cell cycle regulatory proteins, crucial regulators of adipocyte function and adaptation to metabolic stress. Previous studies have linked the CDKN2A locus to diabetes pathophysiology through its putative role on pancreatic islet biology [16], [25]. Here, we show that CDKN2A-loss may also impact energy balance and metabolic homeostasis via non-pancreatic dependent mechanisms. First, we observed that CDKN2A expression is increased in human obese adipocytes, and deletion of both p16INK4a and p19ARF protect mice from diet-induced obesity and associated insulin resistance by enhancing energy expenditure. Second, Cdkn2a invalidation enhances ingWAT plasticity, facilitating the acquisition of beige adipocytes phenotype. Increasing energy expenditure is associated with decreased blood glucose and triglycerides and improving hyperglycemia and dyslipidemia in both mice and humans [8], [46]. In humans, BAT activation by cold exposure positively affects body weight and insulin sensitivity [7], [47], [48], [49]. In addition, the implantation of human beige adipocytes in mice improves metabolic homeostasis [50]. Interestingly, besides its classical BAT feature, human adult BAT also has some beige-like characteristics [5], [51], [52]. Altogether these data suggest that beige adipocyte activation may be useful against obesity and related metabolic disorders. In this broad context, our data show that CDKN2A locus is involved in adipose tissue adaptation to metabolic challenges in mice and humans that may have potential implications for human obesity.
Although it is not yet clear whether short-term cold acclimation induces browning in humans [7], severe adrenergic stress does activate human WAT browning [53], [54]. In mice, beige adipocytes appear after cold exposure or other inducers [4]. In our model, we observed the presence of beige adipocytes in Cdkn2a invalidated mice in the absence of any browning inducing agent, suggesting that this locus may act as modulator of the β3-adrenergic receptor (β-AR) that controls the browning process. The link between β-AR signaling pathway, CDKN2A and the browning process remains yet to be elucidated.
Using a multiplex method for global kinase activity profiling in ingWAT, we identified the PKA pathway among the most significantly affected pathway in Cdkn2a−/− adipocytes. Furthermore we demonstrated that loss of p16INK4a and p19ARF expression is sufficient to activate the thermogenic program in ingWAT. We also confirmed that PKA activity was higher in Cdkn2a-deficient ingWAT, suggesting an in vivo activation of this pathway. Interestingly in the liver, p16INK4A controls gluconeogenesis and PKA activation in a CDK4-dependent manner [22]. In contrast in ingWAT, our kinome analysis revealed that CDK4 kinase activity, which is regulated by p16INK4A, is not modulated in the absence of Cdkn2a. Therefore, the effect on PKA signaling in ingWAT may be independent of the activation of CDK4. In agreement with this, the analysis of adipose tissues depots from mice expressing a constitutively active CDK4 form (CDK4R24C, Ref. [55]) did not show any signs of browning (data not shown). Moreover, PKA activity is increased in epididymal WAT from Cdk4-deficient mice [56]. Although we cannot rule out that other key signaling pathways, including PKG, could contribute to the phenotype observed in Cdkn2a-deficient mice, our observations suggest that the molecular mechanisms controlled by p16INK4A and p19ARF may be tissue-specific and independent of the bona fide cell cycle regulation, as previously described in the liver for other cell cycle regulators [21]. As demonstrated for p16INK4A/CDK4 and p19ARF/mdm2 in the control of cell proliferation or senescence, respectively, it is tempting to speculate that p16INK4A and p19ARF may act as direct repressors of the PKA complex to regulate its activity. The potential interaction of these proteins with different yet specific partners may subsequently dictate metabolic versus proliferative functions of p16INK4A and p19ARF.
It is noteworthy that CDKN2A loss-of-function is involved in cancer development, particularly melanoma [57]. Recent reports shed light on the contribution of adipose tissue browning process in cancer-associated cachexia in rodents and humans [58], [59], [60], [61], [62]. Here we describe a novel metabolic function for this key tumor suppressor gene in energy expenditure and adipose tissue browning. The activation of the β3-AR pathway during cachexia [63], [64], and the putative tight relationship between CDKN2A and β3-AR, further suggests that CDKN2A deficiency in cancer cells may profoundly affect energy homeostasis facilitating the development of cachexia.
In conclusion, our results demonstrate that the Cdkn2a locus is an important regulator of energy homeostasis and adipose tissue function in response to metabolic stress in mice. Mice model approaches can be useful to elucidate and further carefully dissect the molecular mechanisms underlying the pleiotropic impact of GWAS on T2D associated genes. Modulating Cdkn2a-regulated signaling cascades may constitute novel targetable pathways for treating metabolic disorders.
Authors' contribution
NR, SAH, and XG performed most of the experiments. ES, XY, CC, FO, ICL-M, and EC contributed to the in vivo and cellular experiments. ICL-M and LF performed and/or analyzed the Pamgene experiments and provided reagents and data. ED, IR, and AB performed the RNA-sequencing experiments. XY and CD performed the hiPSCs experiments and analyzed the data. PF discussed and interpreted the results from the study. J-SA designed the study, supervised the project and contributed to experiments and/or their analysis. NR, SAH, PF, and J-SA wrote the manuscript.
Acknowledgments
We thank Dr. Manuel Serrano for providing us with the Cdkn2a−/− mice. We thank Carine de Bettignies and Olivier Sand from UMR 8199 for helpful discussions and technical assistance. We thank Estelle Le Borgne for help with illustrations. We thank Dr. David Blum for sharing reagents. The authors thank the Experimental Resources platform from Lille 2 University, especially Yann Lepage, Ludovic Mercier, Kelly Timmerman, Mélanie Besegher, and Delphine Taillieu for animal care. We thank the Department of Histology from the Lille Medicine Faculty, in particular M.H. Gevaert and R.M. Siminski, for histological preparations. We are indebted to the RHEM network (Réseau d'Histologie Expérimentale de Montpellier, IFR122, France) for histology and, in particular, for tissue and slide preparations. This work was supported by grants from «European Genomic Institute for Diabetes» (EGID, ANR-10-LABX-46 to PF and J-SA), Agence Nationale pour la Recherche (BETAPLASTICITY, ANR-17-CE14-0034 to PF and J-SA; Equipex 2010 ANR-10-EQPX-07-01; ‘LIGAN-PM’ Genomics platform), European Foundation for the Study of Diabetes (EFSD, to J-SA), European Commission, European Research Council (GEPIDIAB 294785 to PF), Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Association pour la Recherche sur le Diabète (to J-SA), Lille 2 University (to NR, XG, ES, and J-SA), Conseil Régional Hauts de France and Métropole Européenne de Lille (to NR, XG, and J-SA), FEDER (Fonds Européen de Développement Régional, to NR, PF, J-SA) and Société Francophone du Diabète/Servier (to SAH and J-SA).
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.molmet.2017.11.012.
Contributor Information
Philippe Froguel, Email: p.froguel@imperial.ac.uk.
Jean-Sébastien Annicotte, Email: jean-sebastien.annicotte@inserm.fr.
Conflict of interest
The authors declare no competing financial interests.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
Figure S1.
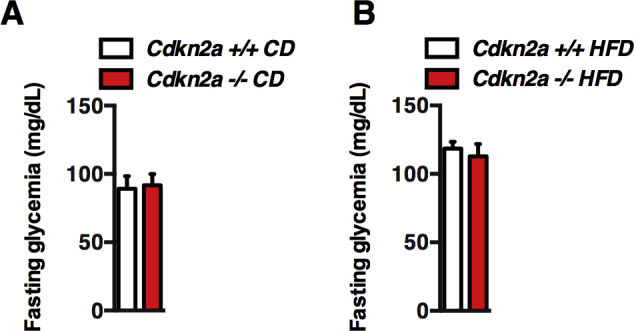
(A, B) A 16 h fasting blood glucose levels in Cdkn2a+/+ and Cdkn2a−/− chow (A) and HFD (B) fed mice (n = 4). Related to Figure 1.

Figure S2(A, B) Cumulative food intake of control (Cdkn2a+/+) and mutant (Cdkn2a−/−) mice under CD (A) and HFD (B) was measured during 3 consecutive days (n = 4). (C–F) 3 days measurement of ambulatory activity (C, E) and its quantification (D, F) of Cdkn2a+/+ and Cdkn2a−/− mice placed in metabolic cages fed a CD (C, D) or HFD (E, F) (n = 4). (G, H) 3 days measurement of heat output for Cdkn2a+/+ and Cdkn2a−/− under CD (g) and HFD (h) fed mice (n = 4). (I, J) Respiratory exchange ratio (RER) was evaluated during 3 days in Cdkn2a+/+ and Cdkn2a−/− fed a chow (I) and HFD (J) diet (n = 4). All values are expressed as means ± SEM. Related to Figure 2.

Figure S3(A) Mitochondrial DNA content (mtDNA) measured relative to nuclear DNA in BAT and in ingWAT tissues of Cdkn2a+/+ and Cdkn2a−/− mice (n = 6–10). (B, C) Western blot assays showing UCP1 levels (B) and its quantification (C) in BAT tissues of Cdkn2a+/+ and Cdkn2a−/− mice (n = 3). (D, E) Absolute BAT weight under chow (D) or HFD (E). (F, G) Relative BAT weight to total body weight under chow (D) or HFD (E). (H) Representative immunofluorescence images of inguinal WAT sections showing nuclei labeling (Hoechst, blue), IgG (negative control) and UCP1 staining (green) on Cdkn2a+/+ and Cdkn2a−/− mice. All values are expressed as means ± SEM. Related to Figure 3.

Figure S4(A) Strategy and subsequent experiments used to analyze the effect of Cdkn2a on browning. (B) Bodipy staining of differentiated ingWAT, eWAT and BAT adipocytes from Cdkn2a+/+ and Cdkn2a−/− mice. (C) mRNA levels for adipogenic and thermogenic genes in BAT primary adipocytes isolated from Cdkn2a+/+ and Cdkn2a−/− mice after 8 days induction of differentiation (n = 3). (D, E) Western blot assays (D) and their quantification (E) showing PKA substrates in Cdkn2a+/+ and Cdkn2a−/− ingWAT tissue homogenates. Tubulin was used as loading controls. All values are expressed as means ± SEM. *p < 0.05, **p < 0.01 and ***p < 0.001. Related to Figure 4.

Figure S5(A) IPA analysis showing gene network associated to Cdkn2a deletion involved in “energy production, lipid metabolism and small molecule biochemistry”. Related to Results section.
List of peptides differentially phosphorylated in Cdkn2a+/+ and Cdkn2a−/− primary adipocytes. Related to Figure 4.
Pathway analysis of diseases and/or biological functions affected in Cdkn2a+/+ and Cdkn2a−/− primary adipocytes related to differential peptide phosphorylation. Related to Figure 4.
List of transcripts differentially regulated in Cdkn2a+/+ and Cdkn2a−/− primary adipocytes identified by RNA-sequencing (download at https://drop-good.egid.fr/index.php/s/Pu8IB6dYCFR1N6T). Related to Results section.
List of diseases and/or biological functions associated to gene expression profiling of Cdkn2a+/+ and Cdkn2a−/− primary adipocytes. Related to Results section.
Oligonucleotides sequences used in this study.
References
- 1.Blanchet E., Annicotte J.-S., Lagarrigue S., Aguilar V., Clapé C., Chavey C. E2F transcription factor-1 regulates oxidative metabolism. Nature Cell Biology. 2011;13:1146–1152. doi: 10.1038/ncb2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fajas L., Landsberg R.L., Huss-Garcia Y., Sardet C., Lees J.A., Auwerx J. E2Fs regulate adipocyte differentiation. Developmental Cell. 2002;3:39–49. doi: 10.1016/s1534-5807(02)00190-9. [DOI] [PubMed] [Google Scholar]
- 3.Liew C.W., Boucher J., Cheong J.K., Vernochet C., Koh H.J., Mallol C. Ablation of TRIP-Br2, a regulator of fat lipolysis, thermogenesis and oxidative metabolism, prevents diet-induced obesity and insulin resistance. Nature Medicine. 2013;19:217–226. doi: 10.1038/nm.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kajimura S., Spiegelman B.M., Seale P. Brown and beige fat: physiological roles beyond heat generation. Cell Metabolism. 2015;22:546–559. doi: 10.1016/j.cmet.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu J., Bostrom P., Sparks L.M., Ye L., Choi J.H., Giang A.H. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu J., Cohen P., Spiegelman B.M. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes & Development. 2013;27:234–250. doi: 10.1101/gad.211649.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanssen M.J., Hoeks J., Brans B., van der Lans A.A., Schaart G., van den Driessche J.J. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nature Medicine. 2015;21:863–865. doi: 10.1038/nm.3891. [DOI] [PubMed] [Google Scholar]
- 8.Lee P., Smith S., Linderman J., Courville A.B., Brychta R.J., Dieckmann W. Temperature-acclimated brown adipose tissue modulates insulin sensitivity in humans. Diabetes. 2014;63:3686–3698. doi: 10.2337/db14-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hannou S.A., Wouters K., Paumelle R., Staels B. Functional genomics of the CDKN2A/B locus in cardiovascular and metabolic disease: what have we learned from GWASs? Trends in Endocrinology & Metabolism. 2015;26:176–184. doi: 10.1016/j.tem.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 10.Morris A.P., Voight B.F., Teslovich T.M., Ferreira T., Segre A.V., Steinthorsdottir V. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nature Genetics. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voight B.F., Scott L.J., Steinthorsdottir V., Morris A.P., Dina C., Welch R.P. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nature Genetics. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dauriz M., Meigs J.B. Current insights into the joint genetic basis of type 2 diabetes and coronary heart disease. Current Cardiovascular Risk Reports. 2014;8:368. doi: 10.1007/s12170-013-0368-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wan G., Mathur R., Hu X., Liu Y., Zhang X., Peng G. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cellular Signalling. 2013;25:1086–1095. doi: 10.1016/j.cellsig.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mody N., Agouni A., McIlroy G.D., Platt B., Delibegovic M. Susceptibility to diet-induced obesity and glucose intolerance in the APP (SWE)/PSEN1 (A246E) mouse model of Alzheimer's disease is associated with increased brain levels of protein tyrosine phosphatase 1B (PTP1B) and retinol-binding protein 4 (RBP4), and basal phosphorylation of S6 ribosomal protein. Diabetologia. 2011;54:2143–2151. doi: 10.1007/s00125-011-2160-2. [DOI] [PubMed] [Google Scholar]
- 15.Lillycrop K., Murray R., Cheong C., Teh A.L., Clarke-Harris R., Barton S. ANRIL promoter DNA methylation: a perinatal marker for later adiposity. EBioMedicine. 2017;19:60–72. doi: 10.1016/j.ebiom.2017.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pal A., Potjer T.P., Thomsen S.K., Ng H.J., Barrett A., Scharfmann R. Loss-of-function mutations in the cell-cycle control gene CDKN2A impact on glucose homeostasis in humans. Diabetes. 2016;65:527–533. doi: 10.2337/db15-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Svensson P.A., Wahlstrand B., Olsson M., Froguel P., Falchi M., Bergman R.N. CDKN2B expression and subcutaneous adipose tissue expandability: possible influence of the 9p21 atherosclerosis locus. Biochemical and Biophysical Research Communications. 2014;446:1126–1131. doi: 10.1016/j.bbrc.2014.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horswell S.D., Fryer L.G., Hutchison C.E., Zindrou D., Speedy H.E., Town M.M. CDKN2B expression in adipose tissue of familial combined hyperlipidemia patients. Journal of Lipid Research. 2013;54:3491–3505. doi: 10.1194/jlr.M041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abella A., Dubus P., Malumbres M., Rane S.G., Kiyokawa H., Sicard A. Cdk4 promotes adipogenesis through PPARgamma activation. Cell Metabolism. 2005;2:239–249. doi: 10.1016/j.cmet.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Fuentes L., Wouters K., Hannou S.A., Cudejko C., Rigamonti E., Mayi T.H. Downregulation of the tumour suppressor p16INK4A contributes to the polarisation of human macrophages toward an adipose tissue macrophage (ATM)-like phenotype. Diabetologia. 2011;54:3150–3156. doi: 10.1007/s00125-011-2324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee Y., Dominy J.E., Choi Y.J., Jurczak M., Tolliday N., Camporez J.P. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014;510:547–551. doi: 10.1038/nature13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bantubungi K., Hannou S.A., Caron-Houde S., Vallez E., Baron M., Lucas A. Cdkn2a/p16Ink4a regulates fasting-induced hepatic gluconeogenesis through the PKA-CREB-PGC1alpha pathway. Diabetes. 2014;63:3199–3209. doi: 10.2337/db13-1921. [DOI] [PubMed] [Google Scholar]
- 23.Krishnamurthy J., Ramsey M.R., Ligon K.L., Torrice C., Koh A., Bonner-Weir S. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 24.Salas E., Rabhi N., Froguel P., Annicotte J.S. Role of Ink4a/Arf locus in beta cell mass expansion under physiological and pathological conditions. Journal of Diabetes Research. 2014;2014:873679. doi: 10.1155/2014/873679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Helman A., Klochendler A., Azazmeh N., Gabai Y., Horwitz E., Anzi S. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nature Medicine. 2016;22:412–420. doi: 10.1038/nm.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimas A.S., Lagou V., Barker A., Knowles J.W., Magi R., Hivert M.F. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes. 2014;63:2158–2171. doi: 10.2337/db13-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serrano M., Lee H., Chin L., Cordon-Cardo C., Beach D., DePinho R.A. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 28.Annicotte J.S., Blanchet E., Chavey C., Iankova I., Costes S., Assou S. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nature Cell Biology. 2009;11:1017–1023. doi: 10.1038/ncb1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabhi N., Denechaud P.D., Gromada X., Hannou S.A., Zhang H., Rashid T. KAT2B is required for pancreatic beta cell adaptation to metabolic stress by controlling the unfolded protein response. Cell Reports. 2016;15:1051–1061. doi: 10.1016/j.celrep.2016.03.079. [DOI] [PubMed] [Google Scholar]
- 30.Tyler D.S., Vappiani J., Caneque T., Lam E.Y.N., Ward A., Gilan O. Click chemistry enables preclinical evaluation of targeted epigenetic therapies. Science. 2017;356:1397–1401. doi: 10.1126/science.aal2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larrieu D., Britton S., Demir M., Rodriguez R., Jackson S.P. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science. 2014;344:527–532. doi: 10.1126/science.1252651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hilhorst R., Houkes L., Mommersteeg M., Musch J., van den Berg A., Ruijtenbeek R. Peptide microarrays for profiling of serine/threonine kinase activity of recombinant kinases and lysates of cells and tissue samples. Methods in Molecular Biology. 2013;977:259–271. doi: 10.1007/978-1-62703-284-1_21. [DOI] [PubMed] [Google Scholar]
- 33.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao Y., Smyth G.K., Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acid Research. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kulyte A., Ehrlund A., Arner P., Dahlman I. Global transcriptome profiling identifies KLF15 and SLC25A10 as modifiers of adipocytes insulin sensitivity in obese women. PloS One. 2017;12:e0178485. doi: 10.1371/journal.pone.0178485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peri S., Navarro J.D., Kristiansen T.Z., Amanchy R., Surendranath V., Muthusamy B. Human protein reference database as a discovery resource for proteomics. Nucleic Acid Research. 2004;32:D497–D501. doi: 10.1093/nar/gkh070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jennissen K., Haas B., Mitschke M.M., Siegel F., Pfeifer A. Analysis of cGMP signaling in adipocytes. Methods in Molecular Biology. 2013;1020:175–192. doi: 10.1007/978-1-62703-459-3_11. [DOI] [PubMed] [Google Scholar]
- 39.Mitschke M.M., Hoffmann L.S., Gnad T., Scholz D., Kruithoff K., Mayer P. Increased cGMP promotes healthy expansion and browning of white adipose tissue. FASEB Journal. 2013;27:1621–1630. doi: 10.1096/fj.12-221580. [DOI] [PubMed] [Google Scholar]
- 40.Cannon B., Nedergaard J. Brown adipose tissue: function and physiological significance. Physiological Reviews. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 41.Grujic D., Susulic V.S., Harper M.E., Himms-Hagen J., Cunningham B.A., Corkey B.E. Beta3-adrenergic receptors on white and brown adipocytes mediate beta3-selective agonist-induced effects on energy expenditure, insulin secretion, and food intake. A study using transgenic and gene knockout mice. The Journal of Biological Chemistry. 1997;272:17686–17693. doi: 10.1074/jbc.272.28.17686. [DOI] [PubMed] [Google Scholar]
- 42.Jimenez M., Barbatelli G., Allevi R., Cinti S., Seydoux J., Giacobino J.P. Beta 3-adrenoceptor knockout in C57BL/6J mice depresses the occurrence of brown adipocytes in white fat. European Journal of Biochemistry. 2003;270:699–705. doi: 10.1046/j.1432-1033.2003.03422.x. [DOI] [PubMed] [Google Scholar]
- 43.Bell C.G., Benzinou M., Siddiq A., Lecoeur C., Dina C., Lemainque A. Genome-wide linkage analysis for severe obesity in French Caucasians finds significant susceptibility locus on chromosome 19q. Diabetes. 2004;53:1857–1865. doi: 10.2337/diabetes.53.7.1857. [DOI] [PubMed] [Google Scholar]
- 44.Chappuis S., Ripperger J.A., Schnell A., Rando G., Jud C., Wahli W. Role of the circadian clock gene Per2 in adaptation to cold temperature. Molecular Metabolism. 2013;2:184–193. doi: 10.1016/j.molmet.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satyanarayana A., Klarmann K.D., Gavrilova O., Keller J.R. Ablation of the transcriptional regulator Id1 enhances energy expenditure, increases insulin sensitivity, and protects against age and diet induced insulin resistance, and hepatosteatosis. FASEB Journal. 2012;26:309–323. doi: 10.1096/fj.11-190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chondronikola M., Volpi E., Borsheim E., Porter C., Annamalai P., Enerback S. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes. 2014;63:4089–4099. doi: 10.2337/db14-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blondin D.P., Labbe S.M., Tingelstad H.C., Noll C., Kunach M., Phoenix S. Increased brown adipose tissue oxidative capacity in cold-acclimated humans. The Journal of Clinical Endocrinology & Metabolism. 2014;99:E438–E446. doi: 10.1210/jc.2013-3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Lans A.A., Hoeks J., Brans B., Vijgen G.H., Visser M.G., Vosselman M.J. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. Journal of Clinical Investigation. 2013;123:3395–3403. doi: 10.1172/JCI68993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoneshiro T., Aita S., Matsushita M., Kayahara T., Kameya T., Kawai Y. Recruited brown adipose tissue as an antiobesity agent in humans. Journal of Clinical Investigation. 2013;123:3404–3408. doi: 10.1172/JCI67803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Min S.Y., Kady J., Nam M., Rojas-Rodriguez R., Berkenwald A., Kim J.H. Human ‘brite/beige’ adipocytes develop from capillary networks, and their implantation improves metabolic homeostasis in mice. Nature Medicine. 2016;22:312–318. doi: 10.1038/nm.4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jespersen N.Z., Larsen T.J., Peijs L., Daugaard S., Homoe P., Loft A. A classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metabolism. 2013;17:798–805. doi: 10.1016/j.cmet.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 52.Shinoda K., Luijten I.H., Hasegawa Y., Hong H., Sonne S.B., Kim M. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nature Medicine. 2015;21:389–394. doi: 10.1038/nm.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patsouris D., Qi P., Abdullahi A., Stanojcic M., Chen P., Parousis A. Burn induces browning of the subcutaneous white adipose tissue in mice and humans. Cell Reports. 2015;13:1538–1544. doi: 10.1016/j.celrep.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sidossis L.S., Porter C., Saraf M.K., Borsheim E., Radhakrishnan R.S., Chao T. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metabolism. 2015;22:219–227. doi: 10.1016/j.cmet.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rane S.G., Dubus P., Mettus R.V., Galbreath E.J., Boden G., Reddy E.P. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nature Genetics. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 56.Lagarrigue S., Lopez-Mejia I.C., Denechaud P.D., Escote X., Castillo-Armengol J., Jimenez V. CDK4 is an essential insulin effector in adipocytes. Journal of Clinical Investigation. 2016;126:335–348. doi: 10.1172/JCI81480. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Cannon-Albright L.A., Goldgar D.E., Meyer L.J., Lewis C.M., Anderson D.E., Fountain J.W. Assignment of a locus for familial melanoma, MLM, to chromosome 9p13-p22. Science. 1992;258:1148–1152. doi: 10.1126/science.1439824. [DOI] [PubMed] [Google Scholar]
- 58.Tsoli M., Moore M., Burg D., Painter A., Taylor R., Lockie S.H. Activation of thermogenesis in brown adipose tissue and dysregulated lipid metabolism associated with cancer cachexia in mice. Cancer Research. 2012;72:4372–4382. doi: 10.1158/0008-5472.CAN-11-3536. [DOI] [PubMed] [Google Scholar]
- 59.Shellock F.G., Riedinger M.S., Fishbein M.C. Brown adipose tissue in cancer patients: possible cause of cancer-induced cachexia. Journal of Cancer Research and Clinical Oncology. 1986;111:82–85. doi: 10.1007/BF00402783. [DOI] [PubMed] [Google Scholar]
- 60.Kir S., White J.P., Kleiner S., Kazak L., Cohen P., Baracos V.E. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature. 2014;513:100–104. doi: 10.1038/nature13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petruzzelli M., Schweiger M., Schreiber R., Campos-Olivas R., Tsoli M., Allen J. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metabolism. 2014;20:433–447. doi: 10.1016/j.cmet.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 62.Kir S., Komaba H., Garcia A.P., Economopoulos K.P., Liu W., Lanske B. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metabolism. 2016;23:315–323. doi: 10.1016/j.cmet.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Das S.K., Eder S., Schauer S., Diwoky C., Temmel H., Guertl B. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333:233–238. doi: 10.1126/science.1198973. [DOI] [PubMed] [Google Scholar]
- 64.Fearon K.C., Glass D.J., Guttridge D.C. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metabolism. 2012;16:153–166. doi: 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 65.Mohsen-Kanson T., Hafner A.L., Wdziekonski B., Takashima Y., Villageois P., Carrière A. Differentiation of human induced pluripotent stem cells into brown and white adipocytes: role of Pax3. Stem Cells. 2014 Jun;32(6):1459–1467. doi: 10.1002/stem.1607. [DOI] [PubMed] [Google Scholar]
- 66.Hafner A.L., Contet J., Ravaud C., Yao X., Villageois P., Suknuntha K. Brown-like adipose progenitors derived from human induced pluripotent stem cells: identification of critical pathways governing their adipogenic capacity. Science Reports. 2016 Aug 31;6:32490. doi: 10.1038/srep32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of peptides differentially phosphorylated in Cdkn2a+/+ and Cdkn2a−/− primary adipocytes. Related to Figure 4.
Pathway analysis of diseases and/or biological functions affected in Cdkn2a+/+ and Cdkn2a−/− primary adipocytes related to differential peptide phosphorylation. Related to Figure 4.
List of transcripts differentially regulated in Cdkn2a+/+ and Cdkn2a−/− primary adipocytes identified by RNA-sequencing (download at https://drop-good.egid.fr/index.php/s/Pu8IB6dYCFR1N6T). Related to Results section.
List of diseases and/or biological functions associated to gene expression profiling of Cdkn2a+/+ and Cdkn2a−/− primary adipocytes. Related to Results section.
Oligonucleotides sequences used in this study.