

Abstract
The ATP-dependent allosteric regulation of the ring-shaped Group II chaperonins remains ill-defined. Their complex oligomeric topology limited the success of structural techniques in suggesting allosteric determinants. Further, their high sequence conservation has hindered prediction of allosteric networks using mathematical covariation approaches, as they cannot be applied to conserved proteins. Here, we develop an information theoretic strategy robust to residue conservation and apply it to group II chaperonins. We identify a contiguous network of covarying residues that connects all nucleotide binding pockets within each chaperonin ring. An interfacial residue between the networks of neighboring subunits controls positive cooperativity by communicating nucleotide occupancy within each ring. Strikingly, chaperonin allostery is tunable through single mutations at this position. Naturally occurring variants that double the extent of positive cooperativity are less prevalent in nature. We propose that being less cooperative that attainable allows the chaperonins to support robust folding over a wider range of metabolic conditions.
Introduction
Allostery, critical for regulating protein activity in nearly all cellular processes, allows for coordinated conformational transitions in oligomeric complexes and modulates protein function in response to changing environmental or metabolic conditions [1–4]. Unlike other easily identifiable regulation strategies, such as post-translational modifications, allostery is defined by specific protein conformational changes in response to ligand concentration [5]. Defining the mechanism of an allosteric system therefore requires identifying the residues responsible for communicating conformational changes, often between distant regions in a protein or complex. Although described over 50 years ago, there remains no systematic strategy to determine allosteric mechanisms.
Detailed structures of relevant ligand states, if available, may suggest allosteric residues within a protein. Structural perturbation approaches [6,7] and network analyses [8] when applied to these structures may suggest allosteric mechanisms. However, uncovering key allosteric positions is often limited by the structural intuition of the investigator. In an effort to augment available structural data, computational frameworks have sought to predict allosteric networks from multiple sequence alignments [9] (Figure 1A). Such techniques are based on the notion that positions engaged in functionally or structurally important contacts will coevolve (Fig. 1A), producing patterns of covariation not seen between unlinked positions.
Figure 1. Predicting coevolved residues in the group II chaperonins.
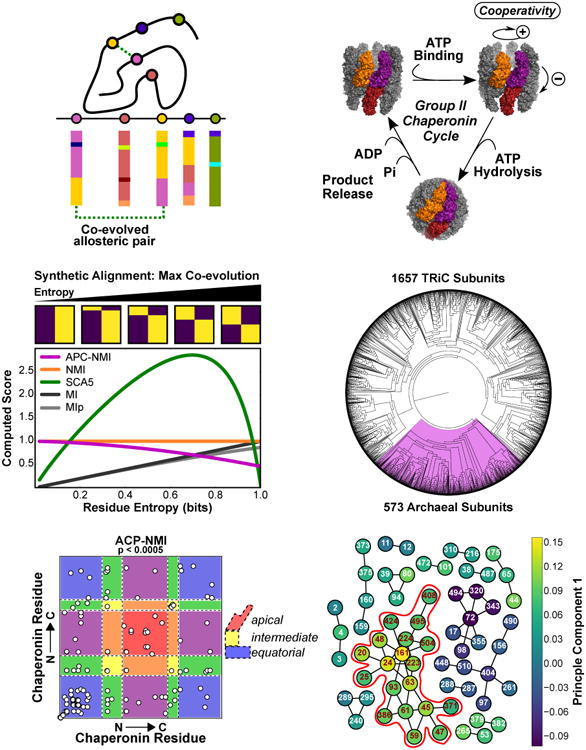
(A) Coevolution prediction theory. An important allosteric interaction exists between two residues. A sequence alignment for this protein family reveals a pattern of coevolution between these positions.
(B) Group II chaperonins undergo a nucleotide dependent conformational cycle. Three subunits are highlighted to show intra- and inter-ring pairs. For simplicity, proposed cooperative transitions are schematically indicated in the symmetric complex, however asymmetric intermediates have been proposed [47,62].
(C) The entropy dependence of various measures for two residue alignments with two residue types in the case of maximal covariation. Sample alignments are shown above the plot and move from nearly perfect conservation (left) to 1.0 bit of entropy (right). The pairwise score is computed for the indicated measure as a function of shannon entropy. We do not evaluate APC-NMI at zero entropy as it is undefined there.
(D) A phylogenetic tree of the group II chaperonin sequences found in the CpnDB. Residues with very low entropies are pruned from our alignments before analysis.
(E) Predicted covarying positions identified via empirical thresholding of the APC-NMI scores, p values were generated by bootstrapping the distribution of pairwise APC-NMI scores. Background colored according to the domain architecture of the chaperonin subunit. Blue: equatorial; Yellow, intermediate; Red apical domains. Colors blended accordingly for couples between domains.
(F) Interconnected networks formed from coupled residue pairs in F; colored according to the first component of the APC-NMI matrix decomposition. (Red outline) The largest interconnected network can also be seen to strongly correlate with the first component.
The most frequently used measures for contact prediction and covariation, mutual information (MI) and Statistical Coupling Analysis (SCA), have been wielded to great effect in biochemical studies of protein allostery [10]. As these predictions are based on detecting correlated mutations on a background of random sampling in extant sequences, they are also constrained by a number of biases. For instance, the underlying phylogeny of sequences can influence observed residue covariance [11]. Uneven sampling from the sequence space of a given protein family also introduces bias into the estimation of covariation. Finally, most existing algorithms also are strongly biased by residue conservation and are thus not applicable to highly conserved protein families. The existence of these biases has not precluded the successful application of covariation-type methods as tools for hypothesis generation in protein structure and function studies [10,12–15] [16,17][17,18]. Many essential macromolecular complexes are both difficult structural targets and well conserved across evolution, leaving few viable strategies for uncovering their mechanisms of allosteric control. Among such conserved cellular machines, group II chaperonins stand out for their structural complexity and intriguing allosteric regulation.
Group II chaperonins consist of two eight membered rings stacked back-to-back and are obligately required to folding many essential archaeal and eukaryotic proteins [19–21]. Upon ATP hydrolysis, extended helices from each subunit coalesce to form an iris-like lid, thus creating a central chamber that encapsulates bound polypeptides and provides an isolated folding environment. The ATP-driven conformational cycle of chaperonins is controlled by a system of nested cooperativity conserved from archaea to eukaryotes [22],[23,24] (Fig. 1B). Subunits in one ring exhibit positive cooperativity increasing nucleotide turnover while negative cooperativity between the two rings slows cycling at high [ATP] (Figure 1B) [23,24]. The allosteric determinants underlying both positive and negative cooperativity have yet to be identified.
Here, we develop a novel framework for predicting allosteric networks in conserved proteins and apply it to archaeal group II chaperonins. To correct the bias introduced by residue conservation we combined the average product correction (APC) with normalized mutual information (NMI). We show that our approach outperforms SCA5 and MI with respect to residue conservation and sensitivity to noise. Using this approach, we identify a spatially contiguous network of residues that controls chaperonin allostery. This network comprises the ATP binding pockets of all subunits of a given ring, linked through a single interfacial residue. We show that this residue, Met47, is involved in the assembly of the chaperonin complex and regulates the allostery of nucleotide cycling.
Our analyses suggests that evolution has limited the extent of cooperativity of nucleotide cycling to be lower than attainable by the complex, likely to broaden the range of conditions over which the chaperonin can function. Thus, complexes maintaining cellular proteostasis have tuned allostery to ensure the robustness of the folding network. The successful identification of allosteric determinants in the group II chaperonins suggests this framework may also be useful for understanding allosteric networks in other conserved protein machines.
Results
An entropy-independent coevolution analysis framework
Statistical coupling analysis (SCA) and mutual information (MI) are the most commonly used covariation measures for inferring allosteric residues from protein multiple sequence alignments. These measures are known to depend strongly upon the conservation of residues in an alignment [16,17] (Figure 1C, S1; Supplemental Methods). As such, SCA and MI perform poorly with well conserved protein families such as the group II chaperonin (not shown)[16,17]. The conservation dependence of mutual information is ameliorated by normalizing the mutual information between each residue pair by dividing by the joint entropy of the pair yielding a measure known as normalized mutual information (NMI; see Supplemental Material for detailed description) [25]. We combined NMI with the average product correction (APC) which corrects to some extent for the effects of phylogeny and sampling bias in the extant sequence pool [26]. Average product corrected mutual information (MIp) is regarded as the best performing information theoretic measure of amino acid covariation which does not rely on an optimization routine [27]. Nevertheless, MIp has similar entropy dependence to MI and is thus not applicable to conserved protein complexes (Figure 1C, S1). We constructed a measure that combines the favorable properties of MIp with the reduced entropy sensitivity of NMI. We call this new measure average product corrected normalized mutual information (APC-NMI; see Supplemental Methods). For a multiple sequence alignment of a protein with n residues and m sequences, M ∈ Rn×m, the average product corrected normalized mutual information (APC-NMI) between any pair of residue positions, i and j, is given as:
wherein I(Mi, Mj) denotes the mutual information between positions i and j and H(Mi, Mj is the Shannon entropy, i.e. the sequence diversity, of the joint probability distribution for residue i and j. The average product correction (APC) is given as:
which is the product of the means of the mutual information vectors for residues i and j (see Supplementary Material for detailed description).
To test the performance of our approach, we first compared it to published methods, SCA5, MI and NMI, using synthetic alignments of a model protein with only two residues and two residue types (Fig. 1C, Fig S1). This test scenario demonstrated that our new framework has reduced dependence on conservation as compared to MIp [26] or to the previously described coevolution metric SCA5 [9] (Figure 1C). Our APC-NMI analysis also has the desirable feature of assigning higher raw scores to residue pairs with similar Shannon entropies (Figure S1A). This makes intuitive sense inasmuch as covarying residues would be expected to have similar degrees of conservation. However, unlike uncorrected NMI, the analysis using APC-NMI is not biased towards highly conserved positions (Figure S1B). As in previous work, predicted coupled or covarying residues may be subjected to an empirical significance threshold and inspected directly [25,26] or may be decomposed into components as in statistical coupling analysis [9].
We utilized our new approach to define an allosteric network within the archaeal group II chaperonins [19,20] (Figure 1B, see detailed description in Supplemental Material). We began our inquiry by generating a high quality alignment and phylogenetic tree of the curated chaperonin peptide sequences found in the cpnDB [28,29]. Of the 2225 annotated group II sequences, 573 resided in the archaeal clade (Fig. 1D). We aligned the archaeal clade with Clustal Omega [29] and computed the APC-NMI matrix (Figure S2C,D). The APC-NMI scores were converted into p-values by bootstrapping the distribution of pairwise scores. We retained 63 residues participating in 64 couples below a threshold of p=0.005. The residues communicate across all three domains of the chaperonin subunit (Figure 1E) creating interconnected networks, the largest consisting of 19 residues (red highlight, Fig. 1F). Most importantly, this network strongly associated with the first component of the APC-NMI matrix (Figure 1F, S3).
A spatially contiguous allosteric network for intra-ring communication
Mapping the larger network of covarying residues from Fig 1F onto the structure of an archaeal chaperonin [30] revealed contiguous contacts throughout all subunits of each chaperonin ring (Figure 2A, left). Centered around the nucleotide binding pocket of each subunit, this network suggests a pathway through which allosteric information is communicated around the ring to regulate ATP cycling (Figure 2A, right). The other, smaller networks uncovered in our approach also mapped to spatially contiguous sets of residues (e.g. Figure S4) suggesting that for these covariation likely arises from structural, or perhaps also functional, constraints. Given our interest in allosteric regulation of ATP hydrolysis, we focused on the large network communicating the ATP binding sites and the smaller networks were not investigated further.
Figure 2. Contiguous network connecting all nucleotide binding sites of the group II chaperonin.
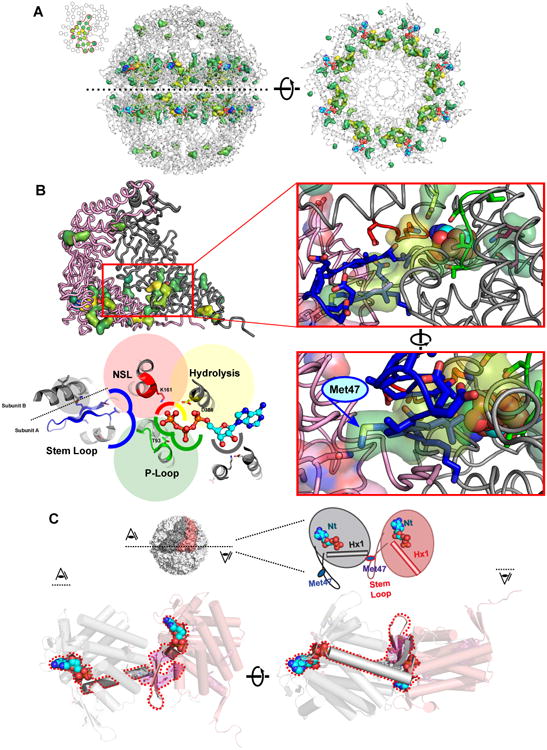
(A) (Left) Surface rendering of the residues of the largest network identified from empirical thresholding in Fig 1F are shown on the structure of the closed Cpn (PDBID: 3RUW) and colored by the first component of the APC-NMI matrix. (Right) Same network viewed on a single ring looking at the base of the equatorial domain towards the chamber lid.
(B) Close inspection of network reveals it surrounds the nucleotide binding sites. (Upper left) Network displayed on pair of intra-ring neighbors, nucleotide binding site highlighted with red outline. (Upper right) Enhanced view of boxed area. Members of key functional motifs involved in nucleotide cycling shown in sticks; the identified network residues shown in transparent surface: yellow - catalytic residue Asp386; red - Nucleotide Sensing Loop; green - P-Loop; blue - Stem Loop; magenta - Ribose binding Glu408 and Lys495. (Lower right) Rotated view of same area reveals how a residue, Met47 (yellow highlight) in the stem loop (blue) spans the intersubunit interface to interact with network residue Asn24 of neighboring subunit. (Lower left) Cartoon representation highlighting identified functional motifs (in the same colors).
(C) Structural role of Met47 in the Stem Loop and Helix 1 in communicating nucleotide binding sites in neighboring subunits. Schematic and cut-aways of the equatorial domain viewed from above (left) and below (right) highlight how adjacent nucleotide binding sites are allosterically linked via the stem-loop region and helix 1.
Almost all residues in the largest network we identified surround the bound nucleotide, supporting the notion that our approach uncovered a bona fide allosteric network in group II chaperonins. Only two residues in this network are located outside the equatorial domain, apical residues Met223 and Pro224. Notably, the mutation M223I allows the archeal group II chaperonin to replace the essential group I chaperonin, GroE, to support bacterial growth, suggesting this position may also influence the chaperonin folding cycle [31]. In the equatorial domain, this network encompasses key elements known to participate in the chaperonin nucleotide cycle (Fig. 2B). Such motifs include the P-loop, key for ATP binding (Thr93, green, Fig. 2B); the Nucleotide Sensing Loop (NSL), which senses whether the γ-phosphate in ATP has been hydrolyzed to ADP (Lys161, red, Figure 2B) and the catalytic residue, Asp386 (yellow, Figure 2B) [30,32]. Further examination of the network revealed two as yet unstudied residues, Glu408 and Lys495, contacting the ribose of bound nucleotide (magenta, Fig. 2B). Multiple residues of the Stem Loop, a β-hairpin at the interface of adjacent equatorial domains, are also identified (blue, Figure 2B). One of these, Met47, spans the subunit interface and interacts with the first helix of its neighbor (Figure 2B, C).
Met47 is well positioned to allosterically link the nucleotide binding sites of neighboring subunits (Figure 2C). Starting from the ATP binding site, the base of the Stem Loop interacts with the nucleotide and extends towards the central axis of the complex. Met47 then extends from the stem loop across the intra-ring subunit interface to residues Asn24 and Ala20 at the N-terminal end of helix H1 in the neighboring subunit (Hx1 in Figure 2C) [30,32]. The C-terminal end of helix H1 contacts the nucleotide in this neighboring subunit and then forms the base of its Stem Loop. We hypothesized that this repeating pattern of communication could connect all intra-ring nucleotide binding sites and regulate allostery.
Steric compatibility at stem-loop interface is required for chaperonin assembly
The single interfacial residue of the network, Met47, was a clear candidate to mediate communication between adjacent subunits. We tested the relevance of this residue using the homo-oligomeric archaeal group II from Methanococcus maripaludis, herein Cpn. Cpn has served as a versatile model system for investigating the group II chaperonins [33,34] and has proven particularly amenable to structural [30,35–37] and biochemical studies [23,38–40].
Over 95% of archaeal sequences, including Cpn, contain methionine at position 47, with leucine and isoleucine being the only alternative residues (Figure 3A, left). Half of the subunits in eukaryotic chaperonin TRiC also favor methionine at this position, with the other four favoring leucine or isoleucine (Figure 3A, right). To test the functional significance of these variants, we introduced these substitutions in Cpn (herein Met47). While M47L-Cpn (herein Leu47) assembles into the full 1 MDa complex, M47I-Cpn (herein Ile47) is monomeric, as determined by SEC-MALS (Figure 3B). This assembly phenotype can be rationalized by the architecture of the inter-subunit interface. Met47 interacts with a narrow pocket on the adjacent subunit, which appears incapable of accommodating β-branched amino acids (Figure S5A). Indeed, additional mutants predicted to disrupt this tight interface, also yield stable monomers (Figure S5C, S6A-B).
Figure 3. Allosteric signals controlling nucleotide cycling are communicated through Met47.

(A) Sequence logos of Cpn-47 equivalent positions in archaeal and eukaryotic chaperonins, arrangement of the TRiC subunits indicated by inner numbers.
(B) Size exclusion chromatography of TRiC-like Cpn mutants. Purified proteins were separated using a 500Å gel filtration column upstream of MALLS/QELS detection. Calculated molecular masses were obtained by SEC-MALLS separations shown.
(C) Nucleotide hydrolysis rates of WT and M47L Cpn over a range of [ATP]. Rates shown were measured utilizing an enzyme coupled assay for ADP generated calculated by monitoring NADH oxidation at 340 nm (see Supplemental Methods). Data shown as the mean ± 95% confidence intervals of three independent replicates.
(D) Enhanced view of first allosteric transition highlighted in (C).
The tunable allosteric regulation of chaperonin cooperativity
We next examined the effect of these variants on the allostery of ATP hydrolysis. As all chaperonins, Cpn has nested cooperativity for ATP hydrolysis, whereby subunits in one ring exhibit positive cooperativity at low and intermediate ATP concentrations, while negative cooperativity between the rings slows turnover at high ATP concentrations [23]. Of note, the monomeric Ile47 chaperonin hydrolyzes ATP at roughly twice the rate of WT (Figure S7A). This likely arises due to the lack of negative cooperativity, which limits cycling in the assembled complex at high [ATP] (Figure S7A) [23]. In contrast, Leu47 displays nested cooperativity as observed for Met47; nucleotide hydrolysis increases cooperatively over low [ATP] and decreases at physiological levels of ATP (Figure 3C). Strikingly, Leu47 alters the allosteric regulation of ATP cycling. The initial transition at low [ATP] became more cooperative, with a Hill coefficient for nearly double that of Met47 (Figure 3D). The decrease in turnover at high [ATP] is largely unchanged in Leu47 with respect to the Met47, suggesting this variant affects specifically intra-ring positive cooperativity (Figure S7B). We conclude that the residue at position 47 relays allosteric information between neighboring intra-ring subunits.
Mechanistic basis of positive cooperativity in group II chaperonins
The fact that the single Leu47 mutation can double the Hill coefficient in Cpn affords an opportunity to understand allosteric regulation in chaperonins. To examine the allosteric changes introduced by Leu47, we compared discrete steps of the nucleotide cycle. Total ATP binding was monitored through α-32P-ATP recovery, whereas ATP hydrolysis was monitored by comparing the levels of bound α- and γ-32P radiolabeled ATP, which define the amount of ATP hydrolyzed by the complex (Figure 4A). The hydrolysis-deficient mutant D386A serves as a control, as it should bind equal levels of both α- and γ-32P ATP. Despite cycling ATP faster than Met47, Leu47 binds only modestly more nucleotide than the WT (Figure 4Bi-ii). Further, the difference between total and non-hydrolyzed ATP is unchanged (Figure S8). This indicates that the increase in nucleotide turnover at high ATP concentrations is not due to Leu47 complexes hydrolyzing more nucleotides per cycle.
Figure 4. Chaperonin cycling is defined by nucleotide occupancy.

(A) A nucleotide recovery assay utilizing both α- and γ- labeled 32P-ATP allows for differentiating between the phosphorylation state of bound nucleotide and calculating the amount of hydrolyzed nucleotide.
(B) Nucleotide bound to the chaperonin at the indicated concentrations was separated utilizing HA-nitrocellulose filter membranes prior to liquid scintillation. The hydrolysis deficient mutant, D386A, is shown as a control. Data shown as the mean ± s.e.m. of three independent samples. A t-test was performed
(C) State of the chaperonin monitored by protease sensitivity. Complexes were incubated with 40 ng/uL Proteinase K at indicated nucleotide conditions. Fraction uncut determined by SYPRO Ruby fluorescence after separation by SDS-PAGE. Data is the mean ± s.e.m of three independent replicates.
(D) Allosteric differences between WT and M47L reveal how the folding cycle of the group II chaperonins is regulated under different nucleotide concentration regimes.
At low ATP concentrations, Met47 and Leu47 also bound equivalent amounts of nucleotide (Figure 4Biii). However, unlike Met47, Leu47 did not hydrolyse detectable amounts of bound nucleotide (Figure S8). Thus, the slower cycling of M47L at low [ATP] is linked to a lack of nucleotide hydrolysis rather than lower nucleotide binding. Thus, the network centered around Met47 senses nucleotide occupancy and establishes a minimal threshold required to trigger the conformational change that accompanies ATP hydrolysis.
Our data indicate that Leu47 raises the nucleotide occupancy requirement for ATP hydrolysis, which manifests as no hydrolysis at low [ATP]. Since Leu47 has higher ATPase rates at high [ATP], we next tested whether M47L changes the dynamic equilibrium between the open and closed populations. To this end, we exploited the observation that open and closed chaperonins have distinct sensitivities to Proteinase K digestion [23]. Upon closure, the unstructured, protease sensitive apical domain protrusions coalesce to form a protease-resistant lid over the folding chamber. Accordingly, protease sensitivity can assess the kinetics of lid closure of the complex (Figure 4C). Leu47 shows slower protease digestion (Figure 4C) indicating that, at steady state, a greater proportion of complexes exist in the closed conformation. Given equivalent nucleotide occupancy and faster overall turnover, we can conclude that the transition from the open to the closed conformations, driven by ATP hydrolysis, is occurring faster in Leu47 (Figure 4D).
Taken together, the above experiments establish how the network centered on residue 47 establishes allosteric communication between subunits. First, it allows subunits to sense nucleotide occupancy in their intra-ring neighbors, thereby setting the threshold of occupancy required to trigger ATP hydrolysis. In turn, this modulates the rate of lid closure (Figure 4D). Allostery is tunable by a single point mutation, which can enhance cooperativity by raising the nucleotide occupancy threshold, thereby resulting in faster cycling.
Linking chaperonin cooperativity to substrate folding
We next examined the role of chaperonin oligomerization state and cooperativity on misfolded protein binding and folding (Figure 5). Whether the oligomeric structure of chaperonins is required to suppress protein aggregation and promote folding has been a matter of debate [41–44]. We first compared the ability of monomeric and oligomeric forms of the chaperonin to suppress aggregation (Fig. 5A and S9). We used the aggregation-prone RadB, an endogenous Cpn substrate in M. maripaludis (Fig 5B) (Lopez et al. unpublished). RadB readily aggregates when diluted from denaturant, but Met47 Cpn efficiently suppresses aggregate formation. Leu47 Cpn similarly suppressed RadB aggregation, indicating both oligomeric chaperonins have similar affinity for the unfolded polypeptide (Fig. 5B, 5C). Surprisingly, the monomeric chaperonin Ile47 also suppressed RadB aggregation at similar substrate-Cpn monomer molar ratios as in the fully assembled complexes (Figure 5B, C, S9). Thus, the oligomeric architecture is not required for efficient suppression of aggregation. While previous findings showed isolated substrate binding apical domains of GroEL and CCT subunits can prevent aggregation in vitro and in vivo, the apical domains always exhibited lower efficiency than the assembled complex [41–44]. This finding, that cpn monomers are as efficient as the full oligomer, suggests that avidity derived from polyvalent interaction does not play a substantial role in the binding of at least some proteins.
Figure 5. Influence of the extent of cooperativity on folding activity of a group II chaperonin.

(A) Comparing oligomeric and monomeric ability to suppress substrate aggregation.
(B) Time course of aggregation suppression by Cpn. Archaeal substrate RadB was diluted from 6M guanidinium into Cpn (shown for Met47 Cpn) at indicated molar ratios and aggregation kinetics measured by absorbance at 320 nm. Trace shown is the average of independent duplicates.
(C) Comparing suppression of substrate aggregation by monomeric (Ile47) and oligomeric (Met47; Leu47) Cpn variants. RadB aggregation was assessed as in (B) at the indicated molar ratios and A320 values normalized to a no Cpn control (-Cpn). Independent duplicates are shown.
(D) Ability of residue 47 variants of Cpn to promote rhodanese folding at 1 mM ATP in the presence or absence of AlFx. Data is the mean ± s.e.m of three independent replicates.
(E) Hypothetical curves highlight how the indicated Hill coefficient tunes activity without altering affinity. The group II chaperonin transitions from the open to closed conformation only in the presence of sufficient ATP.
(F) (G) Rhodanese folding by Met47 and Leu47 variants at (F) intermediate [ATP] and (G) low [ATP]. Nucleotide levels were maintained using a pyruvate kinase ATP regenerating system. Data is the mean ± s.e.m of three independent replicates.
Efficient substrate binding does not necessarily predict efficient folding. We compared the ability of Met47, Leu47 and Ile47 to promote nucleotide-dependent folding of the model substrate, rhodanese. Folding was examined under two regimes. With ATP, the chaperonin cycles between the open, substrate binding, state and the closed, substrate folding, state allowing for iterative cycles of substrate binding and release [20,45,46]. ATP-AlFx creates a trapped, closed, state following one round of hydrolysis, as the AlFx mimics the trigonal-bipyramidal state of ATP-hydrolysis [47]. Under these conditions the substrate is encapsulated within the chamber and folds without re-opening (Fig. 5C) [38]. Under both nucleotide regimes, Met47 and Leu47 efficiently and comparably folded rhodanese in an ATP-dependent manner (Fig. 5D). In contrast, despite rapid nucleotide hydrolysis, Ile47 monomers failed to promote ATP-dependent substrate folding (Figure 5D). Thus, productive folding of proteins such as rhodanese requires encapsulation and release within the oligomeric chaperonin chamber [38].. However, monomers retain basic chaperone functionality, suppression of aggregation and ATP hydrolysis, that may serve a role in the proteostasis network. This could be particularly useful in archaea, which lack the diverse chaperone machinery common to higher organisms [48].
That single substitutions are capable of eliciting such dramatic changes in allostery, raises the questions of why Met47 is the dominant variant in archaea and why this specific cooperative setpoint has become prevalent across group II chaperonin evolution. The chaperonins use ATP hydrolysis to cycle between two conformational states. In the open state they bind non-native proteins and suppress aggregation, independent of nucleotide concentration. Folding occurs only when the bound polypeptide is encapsulated and released into the closed chamber [38]. The hydrolysis cycle drives the transit between these states and any allosteric change will impact the folding activities of the chaperonin (Figure 5E).
In principle, it may appear beneficial to increase the extent of cooperativity and maximize folding activity with minimal ATP. To test this, we compared substrate folding rates for the Met47 and Leu47 Cpn variants at intermediate and low [ATP]. Despite faster cycling at intermediate [ATP], substrate folding by Leu47 shows no rate or yield enhancement compared to the less-cooperative Met47 (Figure 5F). Of note, at very low [ATP] the more cooperative Leu47 shows a defect in substrate folding (Figure 5G). These results suggest that group II chaperonins have tuned this allosteric network to maximize the range of [ATP] at which folding can occur over a mechanism which allows faster cycling. Increasing cooperativity appears to undermine chaperonin folding activity when ATP is limiting, without providing any benefit when it is abundant.
Discussion
The complex allosteric regulation of group II chaperonins, key to their activity in vivo, has remained elusive. Since chaperonin conservation precluded application of current mathematical approaches to predict networks of covarying residues, we developed a method for predicting covarying residues in highly conserved protein families based on a corrected form of mutual information. This approach revealed a contiguous allosteric network that comprises residues involved in nucleotide cycling and connects all nucleotide binding pockets across the chaperonin ring. Focusing on the interfacial residue in this network defined how intra-ring positive cooperativity is regulated in group II chaperonins. In particular, this residue, located in the stem loop connecting adjacent subunits determines the setpoint for allosteric regulation.
We find that the extent of chaperonin cooperativity is not defined by the overall architecture of the complex or the large scale domain movements of the individual subunits. Instead, it is guided by a network of residues communicating the earliest steps of the hydrolysis cycle throughout the ring. Nucleotide occupancy within a ring is sensed and communicated through this network by a residue in the β-sheet formed by the stem loop of one subunit and the N- and C-terminal tails of its neighbor..
Our analysis highlights the way in which cycling is controlled under different nucleotide concentration regimes (Figure 4D). At low ATP concentrations, only those complexes that have bound a sufficient number of ATP are able to close their lid. That a single mutation of the network increases this occupancy threshold indicates that the chaperonin does not require full nucleotide occupancy for closure. In the case of Cpn, all the subunits are chemically identical and thus sub-stoichiometric occupancy cannot be attributed to distinct intrinsic affinities. This is not the case for the hetero-oligomeric eukaryotic chaperonin TRiC/CCT. TRiC subunits have markedly distinct affinities for ATP and nucleotide binding to half of its eight distinct subunits is dispensable for viability [49]. The evolution of TRiC subunits with distinct nucleotide affinity likely arose from the fact that archaeal chaperonins, even those that are homo-oligomeric, cycle with fewer than eight bound nucleotides.
The interface between the stem loop and neighboring subunit is also important for complex assembly. The analysis of the geometry at this interface (Figure S5) provides an intriguing possibility for the unique architecture of TRiC, where the eight paralogous subunits assemble into a sole functional arrangement [50,51]. Perhaps eukaryotes exploit this interface to limit the number of possible subunit arrangements within TRiC. Incompatibilities between subunits, such as those that exist between narrow binding sites and β-branched side chains at this interface, would lead to a dramatic decrease in the number of allowable arrangements. Given the eight CCT subunits, there exist 7! (5040) possible ring arrangements. Ile47 is highly conserved in subunits CCT5 and CCT8. Requiring that these subunits abut one of the two compatible binding partners results in the number of possible arrangements being 2*5! (240); a greater than 95% reduction. It is tempting to suggest that subunit-specific modulation of this interface architecture helped give rise to the precise and evolutionarily conserved arrangement of the hetero-oligomeric TRiC/CCT complex [50,51].
Our findings underscore how complexes evolve to tune cooperativity to resolve the trade-offs required to maintain protein homeostasis under all growth conditions. The ATP-dependent enzymes responsible must be tuned to balance folding speed during rapid growth with functionality under energy-limited conditions. While single point mutations can increase chaperonin cooperativity to produce maximal substrate turnover at lower ligand concentrations, this creates a less versatile complex that has little to no activity below a given ligand threshold (Figure 6). Group II chaperonins tune their allosteric networks to be less cooperative than attainable likely to support folding over the widest range of cellular nucleotide concentrations. Interestingly, this may be a general feature of ATPases involved in proteostasis. Other ATP-dependent oligomeric complexes involved in proteostasis, including the eukaryotic chaperonin TRiC, also exhibit Hill coefficients in the 1.2-1.8 range, similar to that of the chaperonin [52–54]. Complexes do not evolve to maximize cooperativity, but rather finely tune allosteric networks to achieve optimal activity over the range of necessary ligand concentrations. Where any given complex falls on this continuum will rely on factors such as substrate availability and how crucial function is for survival.
Figure 6. Optimizing multisubunit enzyme cooperativity for robust proteostasis activity.

Activity of a complex is a product of both substrate availability and extent of cooperativity. The Hill coefficient is an evolvable characteristic that can be tuned to achieve function over a range of ligand concentrations. Since proteostasis is essential and must be maintained under all growth conditions, ATP-dependent machines tune their cooperativity to optimize robustness over speed.
Our analysis of chaperonin allostery, historically refractory to many methods, highlights the predictive power of our information theoretic approach. Our approach may be useful for the analysis of other conserved protein complexes, although it does not explicitly account for other biases which plague co-evolution studies, such as the phylogenetic relationship between sequences in the MSA which is known to introduce artifactual covariation between taxa [11],[55]. Indeed, covariation techniques are without a doubt useful aids to hypothesis generation [10,12–15,56,57]. Contemporary algorithms relying on pseudo-likelihoods and/or sparse priors [58–60] are already performing well enough to inform ab initio fold prediction [61]. Despite their favorable performance, these unsupervised algorithms will likely give way in coming years to supervised deep learning-based methods with the potential to account for the biases discussed here and those of which we remain unaware. With the rapid expansion of available sequence data, these approaches could help define coevolving functional residue networks that regulate activity in other highly conserved protein families.
Materials and Methods
Mutant purification
All Cpn mutants were generated using site-directed mutagenesis. Assembled complexes were purified as described previously [23]. Monomeric Cpn mutants were expressed off pET21a containing E. coli Rosetta DE3 pLys (EMD Millipore). After harvesting, cells were lysed by pulsed sonication on ice in MQ-A with fresh protease inhibitors. Lysate was clarified by 20,000g centrifugation for 20. Lysates were precipitated using 35% ammonium sulfate followed by pelleting at 20,000g for 20 minutes. The resulting supernatant was then brought to 55% ammonium sulfate and pelleted again. The pellet was dissolved in 50 mL of MQ-A and dialyzed against 2 L of MQ-A overnight prior to loading onto Q Sepharose FF. Proteins were eluted using a linear gradient from 0 to 1.0 M NaCl. Fractions containing Cpn were diluted 1:1 with MQ-A before loading onto Hi-Trap Heparin equilibrated in MQ-A with 0.2 M NaCl and eluted with a linear gradient to 1.0 M NaCl. Chaperonin containing fractions were concentrated using an Amicon 10K concentrator (Millipore) before sizing by Superdex200. Fractions analyzed by SDS-PAGE and cpn containing fractions concentrated using Amicon 10K concentrators.
HPLC SEC-MALS/QELS
A 500 Å analytical sizing column (Agilent) was placed upstream of a Wyatt DAWN HELIOS, Optilab T-rEx, and QELS detectors. Column and detectors were allowed to equilibrate in 30 mM TRIS (pH 7.4), 100 mM KCl, 5 mM MgCl2, 5% (v/v) glycerol, and 0.05% sodium azide. Purified protein was diluted to 1.0 mg/mL in the mobile phase and 125 μL analyzed.
RadB aggregation suppression
RadB purified from inclusion bodies was solubilized in 6 M guanidinium with 1 mM DTT and diluted to 100 μM. RadB was diluted 100 fold with Cpn at indicated molar ratios in ATPase buffer (30 mM TRIS (pH 7.4), 100 mM KCl, 5 mM MgCl2, 10% (v/v) glycerol). Reactions were monitored at 320 nm using an 8453 spectrophotometer (Agilent) while being held at 37 ° C.
Rhodanese folding
Rhodanese folding was assayed as described [63]. In brief, 0.25 μM Cpn protein was incubated in ATPase buffer supplemented with 5 mM sodium thiosulfate. Purified rhodanese was denatured in 6 M guanidinium/HCl containing 5 mM DTT. Binding to the chaperonin was achieved by 100-fold dilution into Cpn in ATPase buffer to a final concentration of 1 μM. Endpoint measurements were taken after 60 minute incubation at 37 ° C. Rhodanese either bound to Cpn or aggregated. The yield of folded rhodanese was calculated following an enzymatic assay by comparing with the activity of an equivalent amount of native rhodanese.
Enzyme coupled ATPase
Pyruvate kinase (PK) and lactate dehydrogenase (LDH) were used to couple ADP production to NADH oxidation which can be monitored spectroscopically. Chaperonin was incubated in ATPase buffer supplemented with 1.0 mM PEP, 0.15 mM NADH, and 5 units each of PK and LDH (Sigma-Aldrich). Reactions were added onto ATP at indicated concentrations and monitored at 340 nm using an 8453 spectrophotometer (Agilent) while being held at 37 ◦.
Radiolabeled ATP recovery
Purified chaperonin was diluted to 1 μM in ATPase buffer and warmed to 37 ◦. Stocks of nucleotide were made at 10× the indicated concentrations for both radio-labeled variant (Perkin-Elmer). Chaperonin was added to ATP and incubated for 90″ before vacuum filtration through HA-nitrocellulose (EMD Millipore). Filter membranes were immediately washed with 1 mL of cold ATPase buffer before drying and counting by liquid scintillation (Beckman-Coulter).
Protease sensitivity time course
Modified from [23]. Concentration of proteinase K was increased to 40 ng/μL for one set of experiments. Large digest reactions were made such that time points could be removed as indicated. Reactions were stopped with 10 mM PMSF and placed on ice to halt protease activity. Samples were separated by SDS-PAGE and gels were stained with SYPRO Ruby (Invitrogen). Quantitation was performed using ImageQuant TL (GE Lifesciences).
Low [ATP] rhodanese folding time course
As described from [63]. The indicated concentrations of ATP were maintained throughout the course of the reaction using 1 mM PEP and 0.025 units of PK. A -ATP condition for each mutant was utilized to subtract spontaneous background refolding.
Data Availability
Python libraries and example code are available through the Stanford Digital Repository at: https://purl.stanford.edu/bq811xh5964 Data for biochemistry panels available in CSV format and upon request.
Supplementary Material
Acknowledgments
We thank Dr. Nick Douglas for advice and manuscript comments, Richard Pfuetzner and Axel Brunger for help with the SEC-MALS experiments, Alex Colavin and KC Huang for discussions of the coupling algorithms and members of the Frydman lab for helpful discussions. Research was supported by the NIH awards GM074074 (JF), GM062868 (VP) and by award DE-SC0008504 from the Department of Energy. KD was a recipient of a Stanford Graduate Fellowship and TL was supported by T32GM007276.
Footnotes
Authors Contributions: TL did all experiments, KD developed algorithm and did analyses, AT assisted with light scattering measurements, VP directed algorithm development and JF directed all aspects of work. All authors contributed to final MS.
References
- 1.Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature. 2014;508:331–9. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hilser VJ, Wrabl JO, Motlagh HN. Structural and energetic basis of allostery. Annu Rev Biophys. 2012;41:585–609. doi: 10.1146/annurev-biophys-050511-102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai CJ, Nussinov R. A unified view of “how allostery works. PLoS Comput Biol. 2014;10:e1003394. doi: 10.1371/journal.pcbi.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16:102–8. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Monod J, Wyman J, Changeux JP. On The Nature of Allosteric Transitions: A Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 6.Zheng W, Brooks BR, Thirumalai D. Low-frequency normal modes that describe allosteric transitions in biological nanomachines are robust to sequence variations. Proc Natl Acad Sci U S A. 2006;103:7664–9. doi: 10.1073/pnas.0510426103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng W, Brooks BR, Doniach S, Thirumalai D. Network of dynamically important residues in the open/closed transition in polymerases is strongly conserved. Structure. 2005;13:565–77. doi: 10.1016/j.str.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 8.Lee Y, Choi S, Hyeon C. Mapping the intramolecular signal transduction of G-protein coupled receptors. Proteins. 2014;82:727–43. doi: 10.1002/prot.24451. [DOI] [PubMed] [Google Scholar]
- 9.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–9. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 10.Smock RG, Rivoire O, Russ WP, Swain JF, Leibler S, Ranganathan R, et al. An interdomain sector mediating allostery in Hsp70 molecular chaperones. Mol Syst Biol. 2010;6:414. doi: 10.1038/msb.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Felsenstein J. Phylogenies and the Comparative Method. Am Nat. 1985;125:1–15. [Google Scholar]
- 12.Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, et al. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci U S A. 2011;108:E1293–301. doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malinverni D, Marsili S, Barducci A, De Los Rios P. Large-Scale Conformational Transitions and Dimerization Are Encoded in the Amino-Acid Sequences of Hsp70 Chaperones. PLoS Comput Biol. 2015;11:e1004262. doi: 10.1371/journal.pcbi.1004262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M, et al. Rewiring the specificity of two-component signal transduction systems. Cell. 2008;133:1043–54. doi: 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aakre CD, Herrou J, Phung TN, Perchuk BS, Crosson S, Laub MT. Evolving new protein-protein interaction specificity through promiscuous intermediates. Cell. 2015;163:594–606. doi: 10.1016/j.cell.2015.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fodor AA, Aldrich RW. Influence of conservation on calculations of amino acid covariance in multiple sequence alignments. Proteins. 2004;56:211–21. doi: 10.1002/prot.20098. [DOI] [PubMed] [Google Scholar]
- 17.Teşileanu T, Colwell LJ, Leibler S. Protein sectors: statistical coupling analysis versus conservation. PLoS Comput Biol. 2015;11:e1004091. doi: 10.1371/journal.pcbi.1004091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fodor AA, Aldrich RW. Influence of conservation on calculations of amino acid covariance in multiple sequence alignments. Proteins. 2004;56:211–21. doi: 10.1002/prot.20098. [DOI] [PubMed] [Google Scholar]
- 19.Lopez T, Dalton K, Frydman J. The Mechanism and Function of Group II Chaperonins. J Mol Biol. 2015;427:2919–30. doi: 10.1016/j.jmb.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayer-Hartl M, Bracher A, Hartl FU. The GroEL-GroES Chaperonin Machine: A Nano-Cage for Protein Folding. Trends Biochem Sci. 2016;41:62–76. doi: 10.1016/j.tibs.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Yam AY, Xia Y, Lin HTJ, Burlingame A, Gerstein M, Frydman J. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol. 2008;15:1255–62. doi: 10.1038/nsmb.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bigotti MG, Clarke AR. Chaperonins: The hunt for the Group II mechanism. Arch Biochem Biophys. 2008;474:331–9. doi: 10.1016/j.abb.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 23.Reissmann S, Parnot C, Booth CR, Chiu W, Frydman J. Essential function of the built-in lid in the allosteric regulation of eukaryotic and archaeal chaperonins. Nat Struct Mol Biol. 2007;14:432–40. doi: 10.1038/nsmb1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yifrach O, Horovitz A. Two lines of allosteric communication in the oligomeric chaperonin GroEL are revealed by the single mutation Arg196–> Ala. J Mol Biol. 1994;243:397–401. doi: 10.1006/jmbi.1994.1667. [DOI] [PubMed] [Google Scholar]
- 25.Martin LC, Gloor GB, Dunn SD, Wahl LM. Using information theory to search for co-evolving residues in proteins. Bioinformatics. 2005;21:4116–24. doi: 10.1093/bioinformatics/bti671. [DOI] [PubMed] [Google Scholar]
- 26.Dunn SD, Wahl LM, Gloor GB. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics. 2008;24:333–40. doi: 10.1093/bioinformatics/btm604. [DOI] [PubMed] [Google Scholar]
- 27.Mao W, Kaya C, Dutta A, Horovitz A, Bahar I. Comparative study of the effectiveness and limitations of current methods for detecting sequence coevolution. Bioinformatics. 2015;31:1929–37. doi: 10.1093/bioinformatics/btv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hill JE, Penny SL, Crowell KG, Goh SH, Hemmingsen SM. cpnDB: a chaperonin sequence database. Genome Res. 2004;14:1669–75. doi: 10.1101/gr.2649204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pereira JH, Ralston CY, Douglas NR, Kumar R, Lopez T, McAndrew RP, et al. Mechanism of nucleotide sensing in group II chaperonins. EMBO J. 2012;31:731–40. doi: 10.1038/emboj.2011.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah R, Large AT, Ursinus A, Lin B, Gowrinathan P, Martin J, et al. Replacement of GroEL in Escherichia coli by the Group II Chaperonin from the Archaeon Methanococcus maripaludis. J Bacteriol. 2016;198:2692–700. doi: 10.1128/JB.00317-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ditzel L, Löwe J, Stock D, Stetter KO, Huber H, Huber R, et al. Crystal structure of the thermosome, the archaeal chaperonin and homolog of CCT. Cell. 1998;93:125–38. doi: 10.1016/s0092-8674(00)81152-6. [DOI] [PubMed] [Google Scholar]
- 33.Bigotti MG, Bellamy SRW, Clarke AR. The asymmetric ATPase cycle of the thermosome: elucidation of the binding, hydrolysis and product-release steps. J Mol Biol. 2006;362:835–43. doi: 10.1016/j.jmb.2006.07.064. [DOI] [PubMed] [Google Scholar]
- 34.Bigotti MG, Clarke AR. Cooperativity in the thermosome. J Mol Biol. 2005;348:13–26. doi: 10.1016/j.jmb.2005.01.066. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Ma B, DiMaio F, Douglas NR, Joachimiak LA, Baker D, et al. Cryo-EM structure of a group II chaperonin in the prehydrolysis ATP-bound state leading to lid closure. Structure. 2011;19:633–9. doi: 10.1016/j.str.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pereira JH, Ralston CY, Douglas NR, Meyer D, Knee KM, Goulet DR, et al. Crystal Structures of a Group II Chaperonin Reveal the Open and Closed States Associated with the Protein Folding Cycle. J Biol Chem. 2010;285:27958–66. doi: 10.1074/jbc.M110.125344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Baker ML, Schröder GF, Douglas NR, Reissmann S, Jakana J, et al. Mechanism of folding chamber closure in a group II chaperonin. Nature. 2010;463:379–83. doi: 10.1038/nature08701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Douglas NR, Reissmann S, Zhang J, Chen B, Jakana J, Kumar R, et al. Dual action of ATP hydrolysis couples lid closure to substrate release into the group II chaperonin chamber. Cell. 2011;144:240–52. doi: 10.1016/j.cell.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kusmierczyk AR, Martin J. Nested cooperativity and salt dependence of the ATPase activity of the archaeal chaperonin Mm-cpn. FEBS Lett. 2003;547:201–4. doi: 10.1016/s0014-5793(03)00722-1. [DOI] [PubMed] [Google Scholar]
- 40.Kusmierczyk AR, Martin J. Nucleotide-dependent protein folding in the type II chaperonin from the mesophilic archaeon Methanococcus maripaludis. Biochem J. 2003;371:669–73. doi: 10.1042/BJ20030230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hua Q, Dementieva IS, Walsh MA, Hallenga K, Weiss MA, Joachimiak A. A thermophilic mini-chaperonin contains a conserved polypeptide-binding surface: combined crystallographic and NMR studies of the GroEL apical domain with implications for substrate interactions. J Mol Biol. 2001;306:513–25. doi: 10.1006/jmbi.2000.4405. [DOI] [PubMed] [Google Scholar]
- 42.Sontag EM, Joachimiak LA, Tan Z, Tomlinson A, Housman DE, Glabe CG, et al. Exogenous delivery of chaperonin subunit fragment ApiCCT1 modulates mutant Huntingtin cellular phenotypes. Proc Natl Acad Sci U S A. 2013;110:3077–82. doi: 10.1073/pnas.1222663110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spiess C, Miller EJ, McClellan AJ, Frydman J. Identification of the TRiC/CCT substrate binding sites uncovers the function of subunit diversity in eukaryotic chaperonins. Mol Cell. 2006;24:25–37. doi: 10.1016/j.molcel.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weber F, Keppel F, Georgopoulos C, Hayer-Hartl MK, Hartl FU. The oligomeric structure of GroEL/GroES is required for biologically significant chaperonin function in protein folding. Nat Struct Biol. 1998;5:977–85. doi: 10.1038/2952. [DOI] [PubMed] [Google Scholar]
- 45.Grallert H, Rutkat K, Buchner J. Limits of protein folding inside GroE complexes. J Biol Chem. 2000;275:20424–30. doi: 10.1074/jbc.M002243200. [DOI] [PubMed] [Google Scholar]
- 46.Hyeon C, Lorimer GH, Thirumalai D. Dynamics of allosteric transitions in GroEL. Proc Natl Acad Sci U S A. 2006;103:18939–44. doi: 10.1073/pnas.0608759103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyer AS, Gillespie JR, Walther D, Millet IS, Doniach S, Frydman J. Closing the folding chamber of the eukaryotic chaperonin requires the transition state of ATP hydrolysis. Cell. 2003;113:369–81. doi: 10.1016/s0092-8674(03)00307-6. [DOI] [PubMed] [Google Scholar]
- 48.Pilak O, Harrop SJ, Siddiqui KS, Chong K, De Francisci D, Burg D, et al. Chaperonins from an Antarctic archaeon are predominantly monomeric: crystal structure of an open state monomer. Environ Microbiol. 2011;13:2232–49. doi: 10.1111/j.1462-2920.2011.02477.x. [DOI] [PubMed] [Google Scholar]
- 49.Reissmann S, Joachimiak LA, Chen B, Meyer AS, Nguyen A, Frydman J. A Gradient of ATP Affinities Generates an Asymmetric Power Stroke Driving the Chaperonin TRIC/CCT Folding Cycle. Cell Rep. 2012;2:866–77. doi: 10.1016/j.celrep.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leitner A, Joachimiak LA, Bracher A, Mönkemeyer L, Walzthoeni T, Chen B, et al. The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure. 2012;20:814–25. doi: 10.1016/j.str.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kalisman N, Adams CM, Levitt M. Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc Natl Acad Sci U S A. 2012;109:2884–9. doi: 10.1073/pnas.1119472109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishikori S, Esaki M, Yamanaka K, Sugimoto S, Ogura T. Positive cooperativity of the p97 AAA ATPase is critical for essential functions. J Biol Chem. 2011;286:15815–20. doi: 10.1074/jbc.M110.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sen M, Maillard RA, Nyquist K, Rodriguez-Aliaga P, Pressé S, Martin A, et al. The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell. 2013;155:636–46. doi: 10.1016/j.cell.2013.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song C, Wang Q, Li CCH. ATPase activity of p97-valosin-containing protein (VCP) D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity. J Biol Chem. 2003;278:3648–55. doi: 10.1074/jbc.M208422200. [DOI] [PubMed] [Google Scholar]
- 55.Talavera D, Lovell SC, Whelan S. Covariation Is a Poor Measure of Molecular Coevolution. Mol Biol Evol. 2015;32:2456–68. doi: 10.1093/molbev/msv109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lunt B, Szurmant H, Procaccini A, Hoch JA, Hwa T, Weigt M. Methods in Enzymology. Vol. 471. Academic Press; 2010. Chapter Two - Inference of Direct Residue Contacts in Two-Component Signaling; pp. 17–41. [DOI] [PubMed] [Google Scholar]
- 57.Dwyer RS, Ricci DP, Colwell LJ, Silhavy TJ, Wingreen NS. Predicting functionally informative mutations in Escherichia coli BamA using evolutionary covariance analysis. Genetics. 2013;195:443–55. doi: 10.1534/genetics.113.155861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Balakrishnan S, Kamisetty H, Carbonell JG, Lee SI, Langmead CJ. Learning generative models for protein fold families. Proteins. 2011;79:1061–78. doi: 10.1002/prot.22934. [DOI] [PubMed] [Google Scholar]
- 59.Morcos F, Hwa T, Onuchic JN, Weigt M. Direct Coupling Analysis for Protein Contact Prediction. In: Kihara D, editor. Protein Structure Prediction. Springer; New York: pp. 55–70. n.d. [DOI] [PubMed] [Google Scholar]
- 60.Jones DT, Buchan DWA, Cozzetto D, Pontil M. PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28:184–90. doi: 10.1093/bioinformatics/btr638. [DOI] [PubMed] [Google Scholar]
- 61.Ovchinnikov S, Kinch L, Park H, Liao Y, Pei J, Kim DE, et al. Large-scale determination of previously unsolved protein structures using evolutionary information. Elife. 2015;4:e09248. doi: 10.7554/eLife.09248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cong Y, Schröder GF, Meyer AS, Jakana J, Ma B, Dougherty MT, et al. Symmetry-free cryo-EM structures of the chaperonin TRiC along its ATPase-driven conformational cycle. EMBO J. 2012;31:720–30. doi: 10.1038/emboj.2011.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weber Frank, Hayer-Hartl Manajit. Refolding of bovine mitochondrial rhodanese by chaperonins GroEL and GroES. In: Schneider Christine., editor. Chaperonin Protocols, Methods in Molecular Biology TM. Springer; New York: 2000. pp. 117–126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Python libraries and example code are available through the Stanford Digital Repository at: https://purl.stanford.edu/bq811xh5964 Data for biochemistry panels available in CSV format and upon request.