

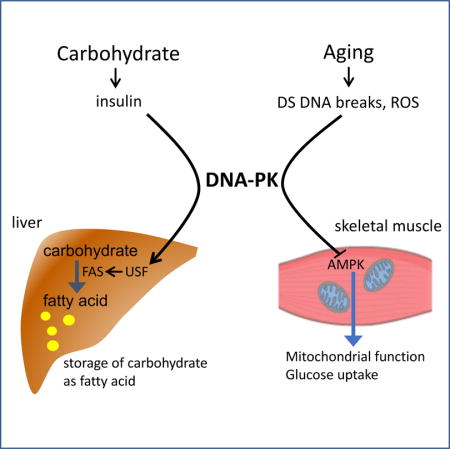
Abstract
DNA-dependent protein kinase (DNA-PK) is a very large holoenzyme comprised of the p470 kDa DNA-PK catalytic subunit (DNA-PKcs) and the Ku heterodimer consisting of the p86 (Ku 80) and p70 (Ku 70) subunits. It is best known for its non-homologous end joining (NHEJ) activity, which repairs double-strand DNA (dsDNA) breaks (DSBs). As expected, the absence of DNA-PK activity results in sensitivity to ionizing radiation, which generates DSBs and defect in lymphocyte development, which requires NHEJ of the V(D)J region in the immunoglobulin and T cell receptor loci. DNA-PK also has been reported to have functions seemingly unrelated to NHEJ. For example, DNA-PK responds to insulin signaling to facilitate the conversion of carbohydrates to fatty acids in the liver. More recent evidence indicates that DNA-PK activity increases with age in skeletal muscle, promoting mitochondrial loss and weight gain. These discoveries suggest that our understanding of DNA-PK is far from complete. As many excellent reviews have already been written about the role of DNA-PK in NHEJ, here we will review the non-NHEJ role of DNA-PK with a focus on its role in aging and energy metabolism.
Keywords: DNA-PK, DNA damage; aging; obesity; energy metabolism
Graphical abstract
DNA-dependent protein kinase (DNA-PK) is best known for its non-homologous end joining (NHEJ) activity, which repairs double-strand DNA breaks. Recent evidence indicates that DNA-PK activity increases with age in skeletal muscle promoting mitochondrial loss and weight gain. Here we will review the non-NHEJ role of DNA-PK with a focus on its role in aging and energy metabolism.
Introduction
Average life expectancy has increased dramatically, leading to a rapid graying of the population across the globe [1]. Although increased life expectancy in itself is a positive development, it comes with significant public health challenges, particularly increased frailty and risk for a diverse array of chronic diseases. Mounting evidence suggests that processes intrinsic to aging promote age-related diseases [2] but the pathway that links aging to the increase in disease risk is poorly understood.
Significant strides have been made in understanding the potential promoters of aging phenotypes and susceptibility to diseases [3, 4]. These age-related changes include mitochondrial dysfunction, reactive oxygen species (ROS), DNA damage and declines in protein quality control, cellular regenerative capacity and signaling control. These alterations disrupt metabolic and inflammatory homeostases, which create the tissue microenvironments permissive for development of diseases.
DNA damage is one well-known promoters of the aging phenotype [5]. Causes of DNA damage can range from single- and double-strand breaks produced by abortive topoisomerase actions, chemical reactions, to ionizing radiation to DNA-base mismatches introduced during DNA replication, or damage generated by chemicals or exposure to ultraviolet light [6]. DNA damage, which can cause cellular senescence or even death [7], is particularly consequential if it occurs in stem cells because it can diminish the regenerative capacity of organs and tissues [8].
To maintain genome stability, organisms have evolved mechanisms to sense the various forms of DNA damage and to initiate signaling pathways that repair them [9, 10]. As with most signaling pathways, the DNA damage signaling pathway is initiated by kinases [11]. One of the best characterized among these is DNA-PK, which is activated by dsDNA breaks [11, 12]. The existence of a kinase that is stimulated by dsDNA was first described by Ohtsuki et al. [13], although whether this activity corresponded to DNA-PK is not known. Subsequently, three groups independently detected a dsDNA binding kinase in a variety of mammalian cellular extracts that required dsDNA for its activity and called it DNA-PK [14–17]. Both Carter et al. [15] and Jackson et al. [16] observed that the enzyme functions efficiently in the presence of linear but not supercoiled DNA, indicating that DNA-PK is activated by dsDNA ends, which are absent in supercoiled DNA. Work by Lees-Miller et al. [18] indicated that a high resolution chromatographic step intended to purify the kinase dramatically reduced its catalytic activity. This curious observation was subsequently explained by the discovery that the catalytic subunit of DNA-PK (DNA-PKcs) required interaction with the DNA end-binding Ku heterodimer consisting of p86 (Ku 80) and p70 (Ku 70) for full catalytic activity [19, 20]. In 1995, cloning of the DNA-PKcs cDNA revealed that it is an approximately 470 kD polypeptide, with the kinase domain in the carboxy-terminal approximately 500 residues [21]. Interestingly, the kinase domain [21, 22] has homology to the phosphatidylinositol 3 (PI 3)-kinase family which phosphorylates inositol phospholipids [23], but DNA-PK is a pure serine/threonine protein kinase and has no lipid kinase activity [21, 24]. Ataxia-telangiectasia-mutated (ATM) [25, 26] and ATM- and Rad3-Related (ATR), two kinases that, together with DNA-PK, make up the triumvirate of DNA-damage sensing kinases also have homology to PI 3-kinase. Interestingly, while both ATM [25–28] and ATR [29, 30] are present in all eukaryotes, DNA-PKcs is only present in higher eukaryotes; vertebrates have clear homologs [31–34], but invertebrates do not.
As mentioned above, DNA-PK repairs DSBs, and as a result, cell lines carrying a mutation in either Ku or DNA-PKcs are sensitive to agents that induce DSBs such as ionizing radiation [35–42] (Fig. 1). DNA-PK also repairs stalled DNA replication forks [43, 44], consistent with the observation that DNA-PK deficient cells are sensitive to DNA replication fork stalling agents [45]. In addition to the DNA repair process itself, DNA-PK activates checkpoint in response to replication fork stress [43, 44, 46]. The primary mode of DSB repair by DNA-PK is NHEJ, a low-fidelity mode of repair in which the DNA ends are directly ligated without using a homologous template [47]. Programmed DSBs created during V(D)J recombination and class switching recombination in lymphocytes are joined by NHEJ [48]. Evidence that DNA-PK mediates NHEJ came from humans, animals and cell lines. Severe combined immune deficiency (SCID) mice which carry a leaky mutation in DNA-PKcs, have impaired lymphocyte development [35, 49–51]. The SCID phenotype has also been described in humans [34], dogs [52] and horses [33] carrying a mutation in DNA-PKcs.
Fig. 1.
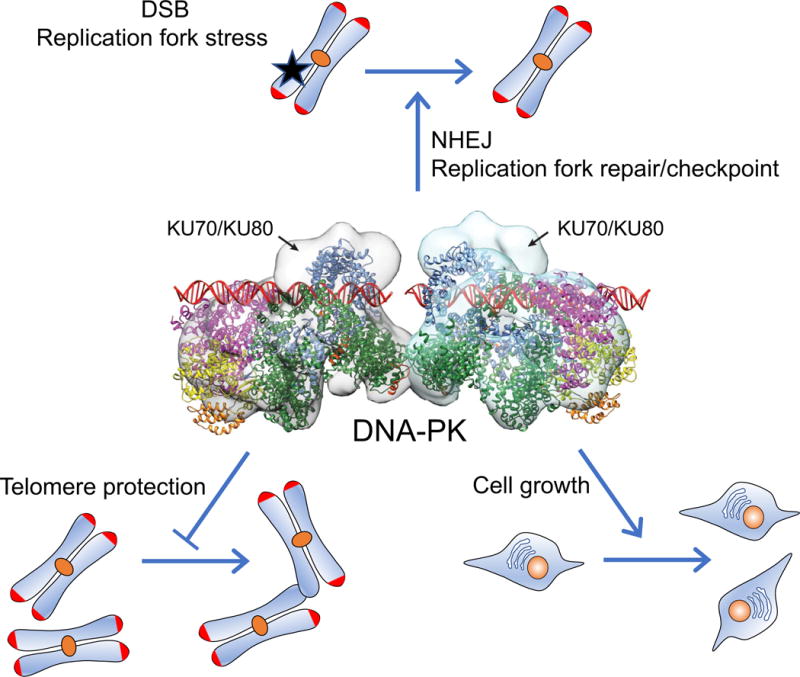
DNA-PK function in genetic stability and cell growth. Structure of DNA-PK complexed with DSBs is shown in the center [54]. Three functions of DNA-PK shown are: DSB repair, V(D)J recombination in lymphocytes via NHEJ and replication fork stress repair and checkpoint (top), binding to and protecting telomeres (lower left) and promoting cell division and growth (lower right).
As a DNA DSB sensor and mediator of NHEJ, DNA-PK has some unexpected properties. For example, DNA-PK is very abundant: it is estimated that HeLa cells contain approximately 100,000 copies of DNA-PKcs per cell [53], far in excess of what is probably needed for NHEJ as only one DNA-PKcs binds to each DSB end [54]. In addition, DNA-PKcs is present not only in the nucleus but also in the cytoplasm [55]. These features suggest that DNA-PK may have additional functions unrelated to NHEJ; indeed, there is emerging evidence that DNA-PK also regulates aging and metabolism. Here, we will focus on this new development and discuss the questions and implications that these novel functions raise about DNA-PK, aging and metabolism.
DNA-damage and aging
The concept that DNA-damage is one of the drivers of the aging process is supported by numerous examples of systemic premature aging syndromes that occur due to a mutation in genes important for maintaining genomic integrity. Although there are a number of progeroid syndromes, perhaps the one that best recapitulates adult aging in an accelerated fashion is the Werner syndrome, which is caused by deficiency of WRN, a member of the RECQ family of DNA helicases [56]. Other syndromes with partial features of premature aging include Ataxia-telangiectasia, dyskeratosis congenita, Cockayne syndrome, Fanconi’s anemia and trichothiodystrophy [57, 58]. Since individuals with these syndromes have had the defects since the embryonic development, it is difficult to know what contribution, if any, that DNA damage occurring during development has on aging as an adult. However, the observation that long-term survivors of DNA-damaging chemotherapy exhibit multiple features of accelerated aging, both at the physiological and molecular levels [59], is consistent with the notion that DNA damage in adults can promote aging.
DNA-PK and aging
The first hint that DNA-PK may affect aging comes from the observation that DNA-PK plays a role in telomere maintenance. The Ku subunits can bind to telomeres [60, 61] and mouse embryonic fibroblasts (MEFs) deficient in Ku70, 80 or DNA-PKcs have a higher number of telomeric fusions, indicating that DNA-PK is important for telomere capping [62–66] (Fig. 1). Interestingly, bone marrow cells from DNA-PKcs−/− mice had no telomeric fusions. This difference between bone marrow cells and MEFs may be a reflection of different levels of extra- cellular oxygen, the source of ROS which can generate DSBs. Internal organs are likely to have lower oxygen levels than room air, the condition under which MEFs are grown, and as a result, MEFs may produce more ROS and DSBs.
DNA-PK also plays a role in the regulation of telomere length, which is controlled by the opposing effects of telomere synthesis by telomerase and telomere shortening. Inactivation of DNA-PK in the human cell line HCT116 shortened telomeres [67], indicating that DNA-PK plays a role in telomere length maintenance. Consistent with this, first generation (G1) DNA-PKcs−/− mice, telomeres were slightly shorter at older age compared with wild type mice [68, 69], but G3-G4 DNA-PKcs−/− mice had significantly shorter telomeres even at young age [70]. To determine whether the role of DNA-PKcs in telomere length control was telomerase-dependent, Espejel et al. studied telomere lengths in telomerase-deficient mice. They found that mice doubly deficient in DNA-PKcs and telomerase displayed an accelerated rate of telomere shortening when compared to mice deficient only in telomerase [70]. Whether it is due to telomere shortening or other factors, DNA-PKcs−/− mice also showed signs of accelerated aging. In keeping with the critical function of DNA-PKcs in immunity, DNA-PKcs−/− mice had higher rates of infection, including pneumonia and hepatitis and had a shorter life span [68, 69].
Like DNA-PKcs−/− mice, Ku 70−/− and Ku80−/− mice also exhibit growth retardation and accelerated aging phenotypes [71–73]. However, unlike mice doubly deficient in DNA-PKcs and telomerase, mice doubly deficient in Ku 80 and telomerase showed a similar rate of telomere shortening when compared to mice deficient in telomerase only [68]. Since the full DNA-dependent catalytic activity of DNA-PKcs requires the Ku 70/80 heterodimer, it is not clear whether the function of DNA-PKcs at telomeres is as a kinase or as a scaffold protein. These findings also suggest that the accelerated aging phenotype in DNA-PKcs−/− mice may not be directly related to telomere shortening.
Unexpectedly, Scid mice have telomeres that are 1.5-2 fold longer than those from wild-type mice [74]. Although the aging process in Scid mice has not been studied as extensively as that in DNA-PKcs−/− mice, Scid mice do not appear to exhibit significantly accelerated aging or shortened lifespan. Since the Scid mice carry a leaky mutation of DNA-PKcs, the discrepancy in telomere length and life span between Scid mice and DNA-PKcs−/− suggests that the effect of DNA-PKcs on telomere length and aging may not be simply dose-dependent and may even be indirect.
The potential role of DNA-PK in aging in humans has been reported in the context of the premature aging syndrome Hutchison-Gilford progeria syndrome (HGPS), which is caused by a single point mutation in the lamin A gene [75, 76]. The accumulation of mutated lamin A decreases expression of nuclear DNA-PKcs and Ku70/Ku80 in HGPS fibroblasts. Liu et al. found that decreasing the expression of DNA-PKcs reduced the proliferation of primary vascular smooth muscle cells and that fibroblasts isolated from normally aging individuals have reduced levels of DNA-PKcs/Ku70/Ku80 and also that decreased expression of DNA-PK is associated with reduced proliferative capacity [77] (Fig. 1).
DNA-PK promotes insulin-stimulated fatty acid synthesis
Storage of nutrients in the form of adipose tissues provides a steady energy supply despite fluctuating food intake (feast-famine cycle). After food intake, excess carbohydrates are converted to fatty acids for storage as triacylglycerol. Lipogenesis is tightly regulated by enzymes involved in fatty acid and triglyceride synthesis, such as fatty acid synthase (FAS) [78, 79] and mitochondrial glycerol-3-phosphate acyltransferase [80]. Their expressions are low during fasting and high during feeding [81]. Regulation of these enzymes occurs mainly at the transcriptional level [82–84] by the transcription factor Upstream Stimulatory Factor (USF)-1/2 heterodimer [85–87]. Although it was generally believed that the feeding/fasting cycle is relayed to USF via the insulin signaling, which increases with feeding, the precise mechanism of how insulin signaling is relayed to USF and to these lipogenic gene promoters, was unknown until Wong et al. discovered that insulin activates USF and increases FAS expression through DNA-PK [88]. Insulin begins the cascade by activating protein phosphatase 1 (PP1), which then dephosphorylates and activates DNA-PK. Activation of DNA-PK also requires DNA DSBs, and indeed Wong et al. observed signs of DNA DSBs in the FAS promoter region after insulin treatment: DNA ends capable of being labeled by biotin-UTP and binding of topoisomerase IIβ, which can cleave DS DNA. Activated DNA-PK phosphorylates Ser262 in USF, which promotes recruitment of and acetylation by coactivator p300/CBP-associated factor. In Scid mice, USF-1 phosphorylation and acetylation in response to insulin is reduced, blunting transcriptional activation of FAS and de novo lipogenesis after carbohydrate feeding [88] (Fig. 2).
Fig. 2.
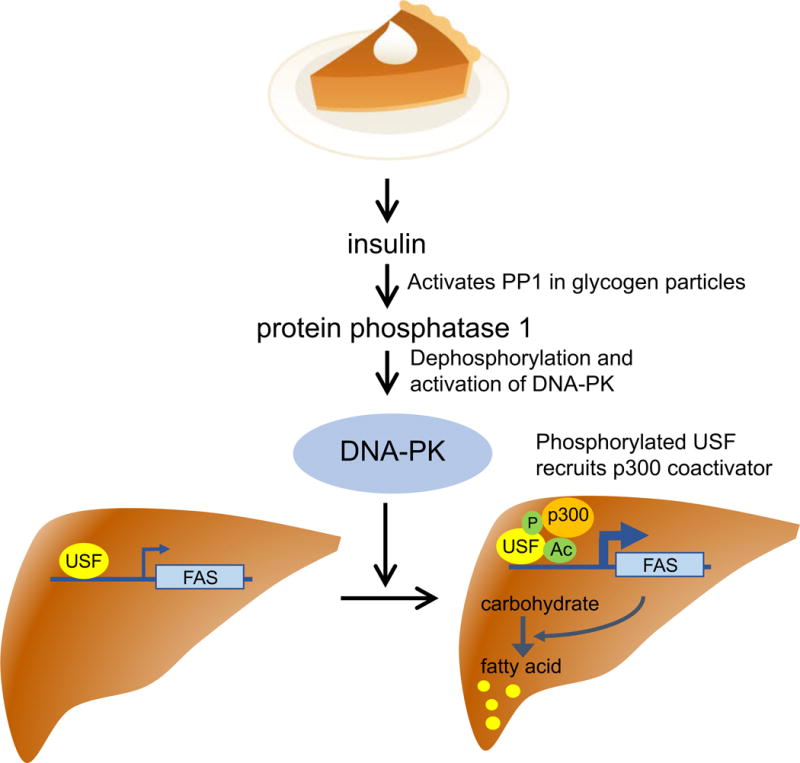
DNA-PK promotes fatty acid synthesis in the liver after feeding. Carbohydrate intake stimulates insulin secretion, which activates DNA-PK via PP1. DNA-PK phosphorylates Ser262 of transcription factor USF, which results in the recruitment of p300 acetyl transferase and acetylation-mediated activation of USF. Activated USF increases transcription of FAS and conversion of carbohydrate to fatty acid.
DNA-PK inhibits AMPK at older age
Aging is associated with many common diseases such as cardiovascular diseases, cancer, type 2 diabetes and neurodegenerative diseases. Obesity increases the risk for all of them. One typically gains approximately 30 pounds from his/her 20s to 50s, which translates to roughly one pound gain per year. In the US, over 70% of adults are either overweight or obese, and the percent of people who are obese is rising. This weight gain occurs even though food intake typically decreases with aging, indicating that the metabolic rate decreases with aging. One potential cause for the decline in metabolic rate with age may be the loss of mitochondria, which metabolizes nutrients to generate energy and heat [89–91]. Loss of mitochondria in skeletal muscle may also explain, at least in part, the decline in physical fitness during aging.
Why does aging lead to mitochondrial loss in skeletal muscle? Accumulating evidence indicates that the aging-associated decline in the activity of AMP-activated protein kinase (AMPK), a key regulator of mitochondrial function and energy balance [92–94], plays an important role in the aging-associated decrease in mitochondrial function and insulin response [95–98]. Activation of AMPK has healthful effects including stimulation of glucose uptake, fat oxidation, energy production and mitochondrial biogenesis [94, 99]. Metformin, the first line drug for type 2 diabetes, acts in part by activating AMPK [100]. Increased AMPK activity decreases visceral fat [101] and increases mitochondrial biogenesis [102] and energy production in skeletal muscle resulting in improved physical fitness. On the other hand, AMPK-deficiency in skeletal muscle leads to mitochondrial loss, impaired glucose uptake and exercise intolerance [103]. AMPK activity declines with age, and this appears to the mediated by elevated intra-mitochondrial ROS because reducing ROS in skeletal muscle by targeting the antioxidant catalase to mitochondria prevents loss of AMPK activity and mitochondrial function at older age [97]. However, the molecular mechanism by which aging and ROS decrease AMPK activity in skeletal muscle was poorly understood.
Like many kinases, AMPK [104] and its upstream activator kinase LKB1 [105] are folded in part by chaperone protein HSP90, which is unique among chaperone proteins in that it binds to and folds metastable proteins and only a small fraction of the total proteome. Most of the HSP90 client proteins are involved in signal transduction, including kinases and steroid hormone receptors [106]. HSP90 is composed of two isoforms: HSP90α, which is stress induced and is not essential for cellular survival, and HSP90β, which is constitutively expressed and is essential for cellular survival. Inhibiting HSP90 activity can result in misfolding of clients, which can lead to their degradation. Although HSP90α and HSP90β are 85% identical in protein sequence and have highly overlapping functions, they appear to have some distinct roles. It appears that HSP90α is particularly important for folding LKB1 and AMPK because knocking down HSP90α with siRNA decreases LKB1 and AMPK levels; whether HSP90β plays any role in folding LKB1 and/or AMPK is not known [107].
The connection between DNA-PK and the functions of HSP90α clients such as LKB1 and AMPK was made when it was discovered that DNA-PK phosphorylates Thr5,7 (T5,7) in HSP90α both in vitro [17] and in vivo [107–109]. Thr5,7 phosphorylation decreases HSP90α-client interaction and presumably the folding of these clients [107]. Consistent with the increase in genetic breaks [107, 110, 111] and DNA-PK activity [107] at older age, T5,7 phosphorylation increased with age in skeletal muscle of both rhesus monkeys and mice. (Fig. 3). In line with this concept, AMPK activity is decreased in skeletal muscle of older WT mice but not of older Scid mice compared to young mice. Muscle-specific knockout of DNA-PKcs also decreased T5,7 phosphorylation and prevented aging-associated loss of AMPK activity in skeletal muscle, indicating that the effect of DNA-PK on AMPK activity is cell autonomous and is unrelated to its immune function [107]. Interestingly, in tissue culture cells, AMPK activation by glucose deprivation is decreased in the absence of DNA-PK [112], suggesting that the role of DNA-PK in AMPK regulation is complicated.
Fig. 3.
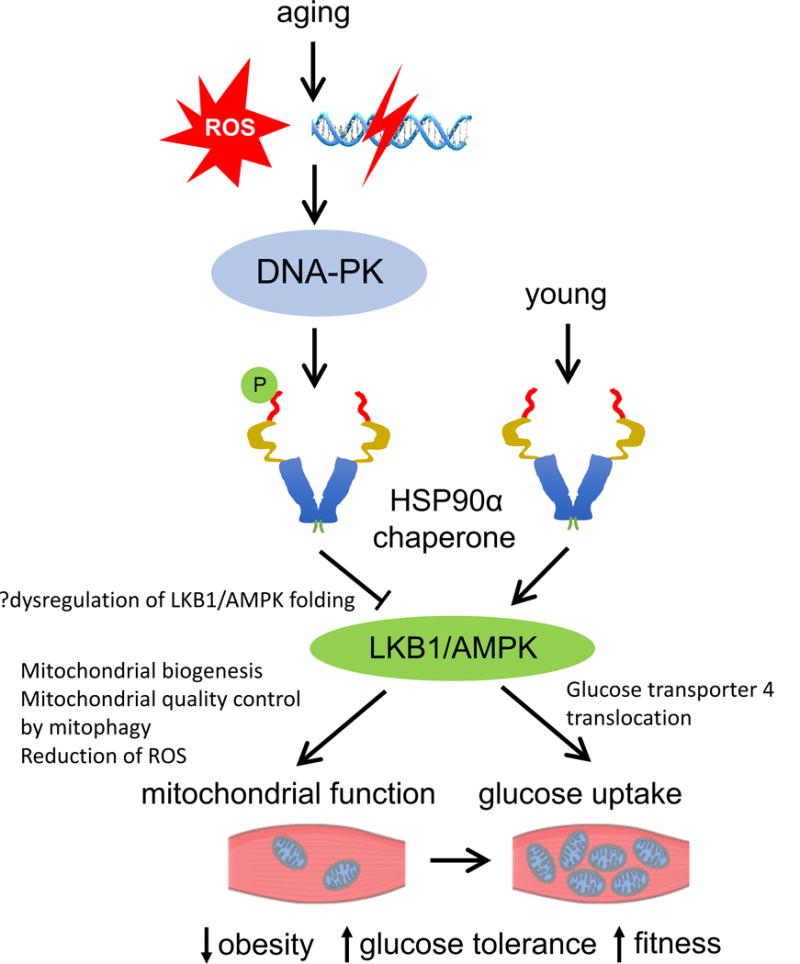
Aging promotes DNA-PK-mediated suppression of AMPK in skeletal muscle. DSBs and/or ROS, both of which increase with age, likely activate DNA-PK. Activated DNA-PK phosphorylates Thr5,7 in HSP90α which decreases the activity (folding) of its client proteins LKB1/AMPK. AMPK activity increases mitochondrial biogenesis, quality control and function (e.g. fat oxidation) and glucose uptake and decreases ROS, protecting against obesity and improving glucose tolerance and physical fitness.
DNA-PK drives mitochondrial loss at older age
As AMPK plays a critical role in mitochondrial biogenesis, increasing DNA-PK activity at older age may promote mitochondrial loss in skeletal muscle. Indeed, there is a strong inverse correlation between mitochondrial content and DNA-PK activity in skeletal muscle of middle-aged rhesus monkeys [107], and mitochondrial loss does not occur in older SCID mice. In skeletal muscle, activation of AMPK promotes a switch from glycolytic fibers to oxidative fibers [101, 113, 114]. Consistent with DNA-PK being a negative regulator of AMPK, older SCID muscle had more oxidative fibers than WT muscle as did the muscle from the mice treated with DNA-PK inhibitor NU7441. Glycolytic fibers generate lactic acid, a major source of muscle fatigue [115, 116]; and switching to oxidative fibers confers fatigue-resistance [101, 117, 118]. Consistent with this, older SCID mice have lower serum lactic acid levels and have reduced aging-associated decline in exercise capacity on treadmill running compared to WT mice.
Higher percentage of oxidative fibers in SCID muscle is consistent with higher AMPK activity in SCID muscle, but it conflicts with the observation that it is the glycolytic fiber, not the oxidative fiber, that is preferentially lost in the elderly in a process called sarcopenia. This seeming contradiction most likely reflects the complex nature of sarcopenia, a process marked by overall declines in size and in number of glycolytic skeletal muscle fibers and an infiltration of fibrous and adipose tissue into the skeletal muscle [119]. In addition, satellite cells, the skeletal muscle precursor cells that are activated and trigger skeletal muscle repair and regeneration in response to the stress of heavy muscle use or injury [120], also undergo aging-related changes, including reduction of satellite cell content, particularly in the glycolytic skeletal muscle fibers [121].
Could a small molecule inhibitor of DNA-PK be an effective fitness enhancer? Indeed, feeding obese and middle-aged WT mice NU7441 increased the exercise capacity of mice by approximately 60% and 40%, respectively [107]. The metabolic effects of NU7441 were mediated by AMPK as NU7441 did not increase mitochondrial biogenesis in skeletal muscle of or exercise capacity in AMPKα2 KO mice, indicating that the fitness-enhancing effect of NU7441 requires AMPK.
Inhibiting DNA-PK improves glucose metabolism
Incidences of disease associated with dysregulation of metabolism increase with aging. For example, among adults aged 18-44, 4% have diabetes (diagnosed and undiagnosed) whereas among adults 65 years or older, 25% have diabetes [122]. One of the characteristics of both type 2 diabetic and obese insulin-resistant nondiabetic individuals is decreased fat oxidation in skeletal muscle [123], which is consistent with impaired mitochondrial oxidative capacity in muscle [124, 125]. Even insulin-resistant offspring of type 2 diabetic parents have impaired mitochondrial activity [126–128]. The decline in mitochondrial content and function also occurs with aging in healthy humans and rats and may contribute to insulin resistance at older age [89–91]. These findings have led some to conclude that the primary defect is muscle mitochondrial dysfunction, resulting in elevated intramyocellular fatty acid metabolites and insulin resistance [129, 130]. However, small increases in palmitoyl carnitine can decrease ATP synthesis in mitochondria [131], which suggests that increased muscle lipid content resulting from increased fatty acid delivery could also be the primary defect that results in mitochondrial dysfunction and insulin resistance. Regardless of the nature of the primary defect, inhibiting DNA-PK may improve insulin sensitivity and protect against type 2 diabetes by activating AMPK and thereby increasing mitochondrial function. Indeed, in mice fed with a high fat diet, the inclusion of NU7441 in the food significantly decreased weight gain [107]. Moreover, NU7441 increased both insulin sensitivity and glucose tolerance primarily by increasing glucose uptake in skeletal muscle and adipose tissue. Therefore, increased DNA-PK activity at older age may contribute to the development of insulin resistance and ultimately, type 2 diabetes (Fig. 3).
Calorie restriction and aerobic fitness are associated with decreased DNA-PK activity
A calorie restricted diet can activate AMPK, increase mitochondrial biogenesis, protect against a diverse array of diseases and disease risk factors such as insulin resistance and metabolic syndrome [132, 133] and extend life span compared to an ad libitum diet [132, 133]. Similarly, rats selectively bred for high intrinsic exercise capacity [134] have higher mitochondrial biogenesis and are protected from a number of diseases and have a longer lifespan compared to rats bred for low intrinsic exercise capacity [135]. Interestingly, both calorie-restricted middle age rhesus monkeys and rats with high intrinsic exercise capacity have decreased DNA-PK activity and HSP90α phosphorylation in muscle. These findings suggest that decreased DNA-PK activity may contribute to the metabolic benefits produced by both calorie restriction and intrinsic aerobic fitness and may warrant further studies.
DISCUSSION
DNA-PK is largely known for its function in NHEJ [11, 48], but more recent evidence indicates that DNA-PK has functions far beyond NHEJ. It is this complex repertoire of functions that can result in seemingly paradoxical observations. Given its function in telomere maintenance and genetic stability, it was not at all surprising that DNA-PK−/− mice undergo accelerated aging [68, 69]. Therefore, it was unexpected that the SCID mutation or treatment with a DNA-PK inhibitor protected against aging-associated decline in metabolism and physical fitness in mice. The most likely explanation for this paradox is that neither the SCID mutation nor the DNA-PK inhibitor inhibit DNA-PK completely, but incomplete loss of DNA-PK activity may be sufficient to decrease its metabolic function while at the same time providing some protective function against the level of DNA DSBs generated naturally. It is also possible that the results may depend on the cell type in DNA-PK−/− or SCID mice.
The study of DNA-PK in non-NHEJ functions such as aging and metabolism is in its early stages and a number of unanswered questions remain. First, what other proteins does DNA-PK phosphorylate in the aging-related pathway and what do they do? One such candidate is Ser473 of AKT, which is required for AKT activation. This site can be phosphorylated by mTORC2 [136] in response to growth signals or by DNA-PK [137–143] in response to DNA breaks. Both insulin and insulin-like growth factor 1 pathways promote aging, and they do so in part by activating AKT [144]. Decreasing the activity of mTOR, which is activated by AKT, increases longevity [145–148]. Aging is also associated with inflammation [149], which is a common denominator of most chronic diseases, including metabolic syndrome [150] and type 2 diabetes [151]. NF-κB, the master transcription factor for inflammation [152], can be activated by AKT [153]. DNA-PK has also been reported to phosphorylate and activate NF-κB [154], but evidence contradicting this has also been reported [155]. Therefore, the DNA-PK-AKT pathway, may contribute to the aging phenotype, not only by increasing mTOR activity, but also by increasing inflammation as evidenced by the role of DNA-PK in inflammation-driven diseases such as asthma [156, 157]. Second, why would nature use a DNA break sensor such as DNA-PK for non-NHEJ functions? This raises the question as to whether DNA-PK is really sensing chromosomal breaks for the aging-related pathways. Although both chromosomal breaks and DNA-PK activity increase with aging [107, 111], we cannot conclude that the increase in DNA-PK activity is mediated solely by chromosomal breaks as DNA-PK is also activated by ROS independently of chromosomal breaks [158]. This may be relevant because ROS also increases with aging [97] and mitochondria, which are regulated by DNA-PK (via AMPK), are the main source of ROS production [159]. This relationship may hint at some type of feedback mechanism, but more work is needed to demonstrate this feedback relationship. The published study of the role of DNA-PK in metabolism and aging has been largely limited to the liver and skeletal muscle, and it is not known whether the role of DNA-PK in metabolism and aging extends to other tissues as well.
HSP90 is considered to be a capacitor of phenotypic variation [160, 161]: by folding metastable proteins and therefore acting as a buffer, HSP90 allows mutations or polymorphisms (inherited or acquired) to accumulate unseen, unaffected by the pressures of natural selection. In this context, HSP90α T5,7 phosphorylation might decrease the ability of HSP90α to provide the necessary buffering at older age. As a consequence, HSP90 chaperone function may be decreased and mutations or polymorphisms may be more exposed at older age. In this scenario, aging and aging-associated diseases may partly be a manifestation of the exposure of these mutations or polymorphisms.
Finally, could a DNA-PK inhibitor be useful for delaying aging and/or ameliorating the risks for aging-associated diseases? Even though a DNA-PK inhibitor may inhibit DNA-PK incompletely, the function of DNA-PK in NHEJ and telomere maintenance may be affected by the inhibitor, potentially increasing the risk for cancer. Therefore, a safer approach may be to decrease the generation of the trigger that activates DNA-PK at older age, but this requires further investigation into the nature of the in vivo trigger(s) of DNA-PK.
Acknowledgments
This work was supported by the Intramural Research Program, National Heart Lung and Blood Institute, in the National Institutes of Health. I thank Dr. Alexandra Brown and Stephanie Mao for their help with the manuscript.
Abbreviations
- DNA-PK
DNA-dependent protein kinase
- DNA-PKcs
DNA-PK catalytic subunit
- dsDNA
double-strand DNA
- DSB
double-strand break
- ATM
ataxia-telangiectasia-mutated
- ATR
ATM- and Rad3-Related
- NHEJ
non-homologous end joining
- SCID
severe combined immune deficiency
- AMPK
AMP-activated protein kinase
- LKB1
liver kinase B1
- MEFs
mouse embryonic fibroblasts
- HGPS
Hutchison-Gilford progeria syndrome
- FAS
fatty acid synthase
- ROS
reactive oxygen species
- USF1
upstream stimulatory factor
- PP1
protein phosphatase 1
References
- 1.Christensen K, Doblhammer G, Rau R, Vaupel JW. Ageing populations: the challenges ahead. Lancet. 2009;374:1196–208. doi: 10.1016/S0140-6736(09)61460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bulterijs S, Hull RS, Bjork VC, Roy AG. It is time to classify biological aging as a disease. Front Genet. 2015;6:205. doi: 10.3389/fgene.2015.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finkel T. The metabolic regulation of aging. Nat Med. 2015;21:1416–23. doi: 10.1038/nm.3998. [DOI] [PubMed] [Google Scholar]
- 5.Soares JP, Cortinhas A, Bento T, Leitao JC, Collins AR, Gaivao I, Mota MP. Aging and DNA damage in humans: a meta-analysis study. Aging (Albany NY) 2014;6:432–9. doi: 10.18632/aging.100667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vermeij WP, Hoeijmakers JH, Pothof J. Aging: not all DNA damage is equal. Curr Opin Genet Dev. 2014;26:124–30. doi: 10.1016/j.gde.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 7.d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 8.Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol. 2012;13:579–90. doi: 10.1038/nrm3420. [DOI] [PubMed] [Google Scholar]
- 9.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 12.Jette N, Lees-Miller SP. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog Biophys Mol Biol. 2015;117:194–205. doi: 10.1016/j.pbiomolbio.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohtsuki K, Yamada E, Nakamura M, Ishida N. Mouse spleen cell nuclear protein kinases and the stimulating effect of dsDNA on NHP phosphorylation by cyclic AMP-independent protein kinase in vitro. J Biochem. 1980;87:35–45. doi: 10.1093/oxfordjournals.jbchem.a132745. [DOI] [PubMed] [Google Scholar]
- 14.Walker AI, Hunt T, Jackson RJ, Anderson CW. Double-stranded DNA induces the phosphorylation of several proteins including the 90,000 mol. wt. heat-shock protein in animal cell extracts. EMBO J. 1985;4:139–45. doi: 10.1002/j.1460-2075.1985.tb02328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter TH, Kopman CR, James CB. DNA-stimulated protein phosphorylation in HeLa whole cell and nuclear extracts. Biochem Biophys Res Commun. 1988;157:535–40. doi: 10.1016/s0006-291x(88)80282-1. [DOI] [PubMed] [Google Scholar]
- 16.Jackson SP, MacDonald JJ, Lees-Miller S, Tjian R. GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell. 1990;63:155–65. doi: 10.1016/0092-8674(90)90296-q. [DOI] [PubMed] [Google Scholar]
- 17.Lees-Miller SP, Anderson CW. The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues. J Biol Chem. 1989;264:17275–80. [PubMed] [Google Scholar]
- 18.Lees-Miller SP, Chen YR, Anderson CW. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53 and the human Ku autoantigen. Mol Cell Biol. 1990;10:6472–81. doi: 10.1128/mcb.10.12.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dvir A, Peterson SR, Knuth MW, Lu H, Dynan WS. Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc Natl Acad Sci U S A. 1992;89:11920–4. doi: 10.1073/pnas.89.24.11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–42. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 21.Hartley KO, Gell D, Smith GC, Zhang H, Divecha N, Connelly MA, Admon A, Lees-Miller SP, Anderson CW, Jackson SP. DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell. 1995;82:849–56. doi: 10.1016/0092-8674(95)90482-4. [DOI] [PubMed] [Google Scholar]
- 22.Poltoratsky VP, Shi X, York JD, Lieber MR, Carter TH. Human DNA-activated protein kinase (DNA-PK) is homologous to phosphatidylinositol kinases. J Immunol. 1995;155:4529–33. [PubMed] [Google Scholar]
- 23.Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–6. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 24.Smith GC, Divecha N, Lakin ND, Jackson SP. DNA-dependent protein kinase and related proteins. Biochem Soc Symp. 1999;64:91–104. [PubMed] [Google Scholar]
- 25.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 26.Savitsky K, Sfez S, Tagle DA, Ziv Y, Sartiel A, Collins FS, Shiloh Y, Rotman G. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum Mol Genet. 1995;4:2025–32. doi: 10.1093/hmg/4.11.2025. [DOI] [PubMed] [Google Scholar]
- 27.Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD. TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell. 1995;82:823–9. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- 28.Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell. 1995;82:831–40. doi: 10.1016/0092-8674(95)90480-8. [DOI] [PubMed] [Google Scholar]
- 29.Bentley NJ, Holtzman DA, Flaggs G, Keegan KS, DeMaggio A, Ford JC, Hoekstra M, Carr AM. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J. 1996;15:6641–51. [PMC free article] [PubMed] [Google Scholar]
- 30.Cimprich KA, Shin TB, Keith CT, Schreiber SL. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci U S A. 1996;93:2850–5. doi: 10.1073/pnas.93.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Araki R, Fujimori A, Hamatani K, Mita K, Saito T, Mori M, Fukumura R, Morimyo M, Muto M, Itoh M, Tatsumi K, Abe M. Nonsense mutation at Tyr-4046 in the DNA-dependent protein kinase catalytic subunit of severe combined immune deficiency mice. Proc Natl Acad Sci U S A. 1997;94:2438–43. doi: 10.1073/pnas.94.6.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Labhart P. mRNA encoding the catalytic subunit of DNA-dependent protein kinase is widely expressed in Xenopus cells. Gene. 1997;203:235–40. doi: 10.1016/s0378-1119(97)00498-8. [DOI] [PubMed] [Google Scholar]
- 33.Shin EK, Perryman LE, Meek K. A kinase-negative mutation of DNA-PK(CS) in equine SCID results in defective coding and signal joint formation. J Immunol. 1997;158:3565–9. [PubMed] [Google Scholar]
- 34.van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, Mari PO, Tezcan I, Chen DJ, Zdzienicka MZ, van Dongen JJ, van Gent DC. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009;119:91–8. doi: 10.1172/JCI37141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blunt T, Finnie NJ, Taccioli GE, Smith GC, Demengeot J, Gottlieb TM, Mizuta R, Varghese AJ, Alt FW, Jeggo PA, et al. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell. 1995;80:813–23. doi: 10.1016/0092-8674(95)90360-7. [DOI] [PubMed] [Google Scholar]
- 36.Finnie NJ, Gottlieb TM, Blunt T, Jeggo PA, Jackson SP. DNA-dependent protein kinase activity is absent in xrs-6 cells: implications for site-specific recombination and DNA double-strand break repair. Proc Natl Acad Sci U S A. 1995;92:320–4. doi: 10.1073/pnas.92.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Getts RC, Stamato TD. Absence of a Ku-like DNA end binding activity in the xrs double-strand DNA repair-deficient mutant. J Biol Chem. 1994;269:15981–4. [PubMed] [Google Scholar]
- 38.Lees-Miller SP, Godbout R, Chan DW, Weinfeld M, Day RS, 3rd, Barron GM, Allalunis-Turner J. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 1995;267:1183–5. doi: 10.1126/science.7855602. [DOI] [PubMed] [Google Scholar]
- 39.Peterson SR, Kurimasa A, Oshimura M, Dynan WS, Bradbury EM, Chen DJ. Loss of the catalytic subunit of the DNA-dependent protein kinase in DNA double-strand-break-repair mutant mammalian cells. Proc Natl Acad Sci U S A. 1995;92:3171–4. doi: 10.1073/pnas.92.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rathmell WK, Chu G. Involvement of the Ku autoantigen in the cellular response to DNA double-strand breaks. Proc Natl Acad Sci U S A. 1994;91:7623–7. doi: 10.1073/pnas.91.16.7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smider V, Rathmell WK, Lieber MR, Chu G. Restoration of X-ray resistance and V(D)J recombination in mutant cells by Ku cDNA. Science. 1994;266:288–91. doi: 10.1126/science.7939667. [DOI] [PubMed] [Google Scholar]
- 42.Taccioli GE, Gottlieb TM, Blunt T, Priestley A, Demengeot J, Mizuta R, Lehmann AR, Alt FW, Jackson SP, Jeggo PA. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 1994;265:1442–5. doi: 10.1126/science.8073286. [DOI] [PubMed] [Google Scholar]
- 43.Ashley AK, Shrivastav M, Nie J, Amerin C, Troksa K, Glanzer JG, Liu S, Opiyo SO, Dimitrova DD, Le P, Sishc B, Bailey SM, Oakley GG, Nickoloff JA. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair (Amst) 2014;21:131–9. doi: 10.1016/j.dnarep.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ying S, Chen Z, Medhurst AL, Neal JA, Bao Z, Mortusewicz O, McGouran J, Song X, Shen H, Hamdy FC, Kessler BM, Meek K, Helleday T. DNA-PKcs and PARP1 Bind to Unresected Stalled DNA Replication Forks Where They Recruit XRCC1 to Mediate Repair. Cancer Res. 2016;76:1078–88. doi: 10.1158/0008-5472.CAN-15-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saintigny Y, Delacote F, Vares G, Petitot F, Lambert S, Averbeck D, Lopez BS. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–70. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu S, Opiyo SO, Manthey K, Glanzer JG, Ashley AK, Amerin C, Troksa K, Shrivastav M, Nickoloff JA, Oakley GG. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012;40:10780–94. doi: 10.1093/nar/gks849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 2007;6:923–35. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18:495–506. doi: 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blunt T, Gell D, Fox M, Taccioli GE, Lehmann AR, Jackson SP, Jeggo PA. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci U S A. 1996;93:10285–90. doi: 10.1073/pnas.93.19.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527–30. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 51.Kirchgessner CU, Patil CK, Evans JW, Cuomo CA, Fried LM, Carter T, Oettinger MA, Brown JM. DNA-dependent kinase (p350) as a candidate gene for the murine SCID defect. Science. 1995;267:1178–83. doi: 10.1126/science.7855601. [DOI] [PubMed] [Google Scholar]
- 52.Meek K, Kienker L, Dallas C, Wang W, Dark MJ, Venta PJ, Huie ML, Hirschhorn R, Bell T. SCID in Jack Russell terriers: a new animal model of DNA-PKcs deficiency. J Immunol. 2001;167:2142–50. doi: 10.4049/jimmunol.167.4.2142. [DOI] [PubMed] [Google Scholar]
- 53.Anderson CW, Carter TH. The DNA-activated protein kinase -- DNA-PK. Curr Top Microbiol Immunol. 1996;217:91–111. doi: 10.1007/978-3-642-50140-1_7. [DOI] [PubMed] [Google Scholar]
- 54.Sibanda BL, Chirgadze DY, Ascher DB, Blundell TL. DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science. 2017;355:520–524. doi: 10.1126/science.aak9654. [DOI] [PubMed] [Google Scholar]
- 55.Huston E, Lynch MJ, Mohamed A, Collins DM, Hill EV, MacLeod R, Krause E, Baillie GS, Houslay MD. EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc Natl Acad Sci U S A. 2008;105:12791–6. doi: 10.1073/pnas.0805167105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–62. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 57.Coppede F, Migliore L. DNA repair in premature aging disorders and neurodegeneration. Curr Aging Sci. 2010;3:3–19. doi: 10.2174/1874609811003010003. [DOI] [PubMed] [Google Scholar]
- 58.Neveling K, Bechtold A, Hoehn H. Genetic instability syndromes with progeroid features. Z Gerontol Geriatr. 2007;40:339–48. doi: 10.1007/s00391-007-0483-x. [DOI] [PubMed] [Google Scholar]
- 59.Maccormick RE. Possible acceleration of aging by adjuvant chemotherapy: a cause of early onset frailty? Med Hypotheses. 2006;67:212–5. doi: 10.1016/j.mehy.2006.01.045. [DOI] [PubMed] [Google Scholar]
- 60.Bianchi A, de Lange T. Ku binds telomeric DNA in vitro. J Biol Chem. 1999;274:21223–7. doi: 10.1074/jbc.274.30.21223. [DOI] [PubMed] [Google Scholar]
- 61.Hsu HL, Gilley D, Blackburn EH, Chen DJ. Ku is associated with the telomere in mammals. Proc Natl Acad Sci U S A. 1999;96:12454–8. doi: 10.1073/pnas.96.22.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bailey SM, Meyne J, Chen DJ, Kurimasa A, Li GC, Lehnert BE, Goodwin EH. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc Natl Acad Sci U S A. 1999;96:14899–904. doi: 10.1073/pnas.96.26.14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gilley D, Tanaka H, Hande MP, Kurimasa A, Li GC, Oshimura M, Chen DJ. DNA-PKcs is critical for telomere capping. Proc Natl Acad Sci U S A. 2001;98:15084–8. doi: 10.1073/pnas.261574698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goytisolo FA, Samper E, Edmonson S, Taccioli GE, Blasco MA. The absence of the dna-dependent protein kinase catalytic subunit in mice results in anaphase bridges and in increased telomeric fusions with normal telomere length and G-strand overhang. Mol Cell Biol. 2001;21:3642–51. doi: 10.1128/MCB.21.11.3642-3651.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hsu HL, Gilley D, Galande SA, Hande MP, Allen B, Kim SH, Li GC, Campisi J, Kohwi-Shigematsu T, Chen DJ. Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev. 2000;14:2807–12. doi: 10.1101/gad.844000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Samper E, Goytisolo FA, Slijepcevic P, van Buul PP, Blasco MA. Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep. 2000;1:244–52. doi: 10.1093/embo-reports/kvd051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruis BL, Fattah KR, Hendrickson EA. The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol Cell Biol. 2008;28:6182–95. doi: 10.1128/MCB.00355-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Espejel S, Klatt P, Menissier-de Murcia J, Martin-Caballero J, Flores JM, Taccioli G, de Murcia G, Blasco MA. Impact of telomerase ablation on organismal viability, aging, and tumorigenesis in mice lacking the DNA repair proteins PARP-1, Ku86, or DNA-PKcs. J Cell Biol. 2004;167:627–38. doi: 10.1083/jcb.200407178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Espejel S, Martin M, Klatt P, Martin-Caballero J, Flores JM, Blasco MA. Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice. EMBO Rep. 2004;5:503–9. doi: 10.1038/sj.embor.7400127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Espejel S, Franco S, Sgura A, Gae D, Bailey SM, Taccioli GE, Blasco MA. Functional interaction between DNA-PKcs and telomerase in telomere length maintenance. EMBO J. 2002;21:6275–87. doi: 10.1093/emboj/cdf593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H, Vogel H, Holcomb VB, Gu Y, Hasty P. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol Cell Biol. 2007;27:8205–14. doi: 10.1128/MCB.00785-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nussenzweig A, Chen C, da Costa Soares V, Sanchez M, Sokol K, Nussenzweig MC, Li GC. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature. 1996;382:551–5. doi: 10.1038/382551a0. [DOI] [PubMed] [Google Scholar]
- 73.Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P. Deletion of Ku86 causes early onset of senescence in mice. Proc Natl Acad Sci U S A. 1999;96:10770–5. doi: 10.1073/pnas.96.19.10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hande P, Slijepcevic P, Silver A, Bouffler S, van Buul P, Bryant P, Lansdorp P. Elongated telomeres in scid mice. Genomics. 1999;56:221–3. doi: 10.1006/geno.1998.5668. [DOI] [PubMed] [Google Scholar]
- 75.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–8. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature. 2003;423:298–301. doi: 10.1038/nature01631. [DOI] [PubMed] [Google Scholar]
- 77.Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, Thompson J, Boue S, Fung HL, Sancho-Martinez I, Zhang K, Yates J, 3rd, Izpisua Belmonte JC. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–5. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Paulauskis JD, Sul HS. Cloning and expression of mouse fatty acid synthase and other specific mRNAs. Developmental and hormonal regulation in 3T3-L1 cells. J Biol Chem. 1988;263:7049–54. [PubMed] [Google Scholar]
- 79.Paulauskis JD, Sul HS. Hormonal regulation of mouse fatty acid synthase gene transcription in liver. J Biol Chem. 1989;264:574–7. [PubMed] [Google Scholar]
- 80.Dircks LK, Sul HS. Mammalian mitochondrial glycerol-3-phosphate acyltransferase. Biochim Biophys Acta. 1997;1348:17–26. doi: 10.1016/s0005-2760(97)00106-9. [DOI] [PubMed] [Google Scholar]
- 81.Sul HS, Wang D. Nutritional and hormonal regulation of enzymes in fat synthesis: studies of fatty acid synthase and mitochondrial glycerol-3-phosphate acyltransferase gene transcription. Annu Rev Nutr. 1998;18:331–51. doi: 10.1146/annurev.nutr.18.1.331. [DOI] [PubMed] [Google Scholar]
- 82.Moustaid N, Beyer RS, Sul HS. Identification of an insulin response element in the fatty acid synthase promoter. J Biol Chem. 1994;269:5629–34. [PubMed] [Google Scholar]
- 83.Moustaid N, Sakamoto K, Clarke S, Beyer RS, Sul HS. Regulation of fatty acid synthase gene transcription. Sequences that confer a positive insulin effect and differentiation-dependent expression in 3T3-L1 preadipocytes are present in the 332 bp promoter. Biochem J. 1993;292(Pt 3):767–72. doi: 10.1042/bj2920767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moustaid N, Sul HS. Regulation of expression of the fatty acid synthase gene in 3T3-L1 cells by differentiation and triiodothyronine. J Biol Chem. 1991;266:18550–4. [PubMed] [Google Scholar]
- 85.Sawadogo M, Roeder RG. Interaction of a gene-specific transcription factor with the adenovirus major late promoter upstream of the TATA box region. Cell. 1985;43:165–75. doi: 10.1016/0092-8674(85)90021-2. [DOI] [PubMed] [Google Scholar]
- 86.Wang D, Sul HS. Upstream stimulatory factors bind to insulin response sequence of the fatty acid synthase promoter. USF1 is regulated. J Biol Chem. 1995;270:28716–22. doi: 10.1074/jbc.270.48.28716. [DOI] [PubMed] [Google Scholar]
- 87.Wang D, Sul HS. Upstream stimulatory factor binding to the E-box at -65 is required for insulin regulation of the fatty acid synthase promoter. J Biol Chem. 1997;272:26367–74. doi: 10.1074/jbc.272.42.26367. [DOI] [PubMed] [Google Scholar]
- 88.Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell. 2009;136:1056–72. doi: 10.1016/j.cell.2008.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barazzoni R, Short KR, Nair KS. Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. J Biol Chem. 2000;275:3343–7. doi: 10.1074/jbc.275.5.3343. [DOI] [PubMed] [Google Scholar]
- 90.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–2. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–23. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nature reviews Molecular cell biology. 2007;8:774–85. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 93.Garcia D, Shaw RJ. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol Cell. 2017;66:789–800. doi: 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Carling D. AMPK signalling in health and disease. Curr Opin Cell Biol. 2017;45:31–37. doi: 10.1016/j.ceb.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 95.Qiang W, Weiqiang K, Qing Z, Pengju Z, Yi L. Aging impairs insulin-stimulated glucose uptake in rat skeletal muscle via suppressing AMPKalpha. Exp Mol Med. 2007;39:535–43. doi: 10.1038/emm.2007.59. [DOI] [PubMed] [Google Scholar]
- 96.Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, Befroy D, Pypaert M, Hardie DG, Young LH, Shulman GI. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell metabolism. 2007;5:151–6. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, Petersen KF, Samuel VT, Shulman GI. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell metabolism. 2010;12:668–74. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koonen DP, Sung MM, Kao CK, Dolinsky VW, Koves TR, Ilkayeva O, Jacobs RL, Vance DE, Light PE, Muoio DM, Febbraio M, Dyck JR. Alterations in skeletal muscle fatty acid handling predisposes middle-aged mice to diet-induced insulin resistance. Diabetes. 2010;59:1366–75. doi: 10.2337/db09-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123:2764–72. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ, Evans RM. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–15. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–7. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, Kemp BE, Richter EA, Steinberg GR. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. 2011;108:16092–7. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nony P, Gaude H, Rossel M, Fournier L, Rouault JP, Billaud M. Stability of the Peutz-Jeghers syndrome kinase LKB1 requires its binding to the molecular chaperones Hsp90/Cdc37. Oncogene. 2003;22:9165–75. doi: 10.1038/sj.onc.1207179. [DOI] [PubMed] [Google Scholar]
- 106.Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017;18:345–360. doi: 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 107.Park SJ, Gavrilova O, Brown AL, Soto JE, Bremner S, Kim J, Xu X, Yang S, Um JH, Koch LG, Britton SL, Lieber RL, Philp A, Baar K, Kohama SG, Abel ED, Kim MK, Chung JH. DNA-PK Promotes the Mitochondrial, Metabolic, and Physical Decline that Occurs During Aging. Cell Metab. 2017;25:1135–1146.e7. doi: 10.1016/j.cmet.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Quanz M, Herbette A, Sayarath M, de Koning L, Dubois T, Sun JS, Dutreix M. Heat shock protein 90alpha (Hsp90alpha) is phosphorylated in response to DNA damage and accumulates in repair foci. The Journal of biological chemistry. 2012;287:8803–15. doi: 10.1074/jbc.M111.320887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Solier S, Kohn KW, Scroggins B, Xu W, Trepel J, Neckers L, Pommier Y. Feature Article: Heat shock protein 90alpha (HSP90alpha), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proceedings of the National Academy of Sciences of the United States of America. 2012 doi: 10.1073/pnas.1203617109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sedelnikova OA, Horikawa I, Redon C, Nakamura A, Zimonjic DB, Popescu NC, Bonner WM. Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging cell. 2008;7:89–100. doi: 10.1111/j.1474-9726.2007.00354.x. [DOI] [PubMed] [Google Scholar]
- 111.Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol. 2004;6:168–70. doi: 10.1038/ncb1095. [DOI] [PubMed] [Google Scholar]
- 112.Amatya PN, Kim HB, Park SJ, Youn CK, Hyun JW, Chang IY, Lee JH, You HJ. A role of DNA-dependent protein kinase for the activation of AMP-activated protein kinase in response to glucose deprivation. Biochim Biophys Acta. 2012;1823:2099–108. doi: 10.1016/j.bbamcr.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 113.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 114.Rockl KS, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes. 2007;56:2062–9. doi: 10.2337/db07-0255. [DOI] [PubMed] [Google Scholar]
- 115.Hanson RW, Hakimi P. Born to run; the story of the PEPCK-Cmus mouse. Biochimie. 2008;90:838–42. doi: 10.1016/j.biochi.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mason SD, Howlett RA, Kim MJ, Olfert IM, Hogan MC, McNulty W, Hickey RP, Wagner PD, Kahn CR, Giordano FJ, Johnson RS. Loss of skeletal muscle HIF-1alpha results in altered exercise endurance. PLoS Biol. 2004;2:e288. doi: 10.1371/journal.pbio.0020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Suwa M, Nakano H, Kumagai S. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3, and PGC-1 in rat muscles. J Appl Physiol. 2003;95:960–8. doi: 10.1152/japplphysiol.00349.2003. [DOI] [PubMed] [Google Scholar]
- 118.Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci. 1995;50(Spec No):11–6. doi: 10.1093/gerona/50a.special_issue.11. [DOI] [PubMed] [Google Scholar]
- 120.Snijders T, Verdijk LB, van Loon LJ. The impact of sarcopenia and exercise training on skeletal muscle satellite cells. Ageing Res Rev. 2009;8:328–38. doi: 10.1016/j.arr.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 121.Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab. 2007;292:E151–7. doi: 10.1152/ajpendo.00278.2006. [DOI] [PubMed] [Google Scholar]
- 122.Kirkman MS, Briscoe VJ, Clark N, Florez H, Haas LB, Halter JB, Huang ES, Korytkowski MT, Munshi MN, Odegard PS, Pratley RE, Swift CS. Diabetes in older adults. Diabetes Care. 2012;35:2650–64. doi: 10.2337/dc12-1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kelley DE. Skeletal muscle fat oxidation: timing and flexibility are everything. J Clin Invest. 2005;115:1699–702. doi: 10.1172/JCI25758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 125.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–71. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M, Shulman GI. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005;115:3587–93. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Short KR, Nair KS, Stump CS. Impaired mitochondrial activity and insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:2419–21. doi: 10.1056/NEJM200406033502320. author reply 2419-21. [DOI] [PubMed] [Google Scholar]
- 129.Morino K, Neschen S, Bilz S, Sono S, Tsirigotis D, Reznick RM, Moore I, Nagai Y, Samuel V, Sebastian D, White M, Philbrick W, Shulman GI. Muscle-specific IRS-1 Ser->Ala transgenic mice are protected from fat-induced insulin resistance in skeletal muscle. Diabetes. 2008;57:2644–51. doi: 10.2337/db06-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Petersen KF, Dufour S, Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Abdul-Ghani MA, Muller FL, Liu Y, Chavez AO, Balas B, Zuo P, Chang Z, Tripathy D, Jani R, Molina-Carrion M, Monroy A, Folli F, Van Remmen H, DeFronzo RA. Deleterious action of FA metabolites on ATP synthesis: possible link between lipotoxicity, mitochondrial dysfunction, and insulin resistance. Am J Physiol Endocrinol Metab. 2008;295:E678–85. doi: 10.1152/ajpendo.90287.2008. [DOI] [PubMed] [Google Scholar]
- 132.Lopez-Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P, de Cabo R. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006;103:1768–73. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Canto C, Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiology (Bethesda) 2011;26:214–24. doi: 10.1152/physiol.00010.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Koch LG, Britton SL, Wisloff U. A rat model system to study complex disease risks, fitness, aging, and longevity. Trends Cardiovasc Med. 2012;22:29–34. doi: 10.1016/j.tcm.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Garton FC, North KN, Koch LG, Britton SL, Nogales-Gadea G, Lucia A. Rodent models for resolving extremes of exercise and health. Physiol Genomics. 2016;48:82–92. doi: 10.1152/physiolgenomics.00077.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 137.Boehme KA, Kulikov R, Blattner C. p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc Natl Acad Sci U S A. 2008;105:7785–90. doi: 10.1073/pnas.0703423105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30:203–13. doi: 10.1016/j.molcel.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 139.Dragoi AM, Fu X, Ivanov S, Zhang P, Sheng L, Wu D, Li GC, Chu WM. DNA-PKcs, but not TLR9, is required for activation of Akt by CpG-DNA. Embo J. 2005;24:779–89. doi: 10.1038/sj.emboj.7600539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem. 2004;279:41189–96. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 141.Lu D, Huang J, Basu A. Protein kinase Cepsilon activates protein kinase B/Akt via DNA-PK to protect against tumor necrosis factor-alpha-induced cell death. J Biol Chem. 2006;281:22799–807. doi: 10.1074/jbc.M603390200. [DOI] [PubMed] [Google Scholar]
- 142.Park J, Feng J, Li Y, Hammarsten O, Brazil DP, Hemmings BA. DNA-dependent protein kinase-mediated phosphorylation of protein kinase B requires a specific recognition sequence in the C-terminal hydrophobic motif. J Biol Chem. 2009;284:6169–74. doi: 10.1074/jbc.C800210200. [DOI] [PubMed] [Google Scholar]
- 143.Surucu B, Bozulic L, Hynx D, Parcellier A, Hemmings BA. In vivo analysis of protein kinase B (PKB)/Akt regulation in DNA-PKcs-null mice reveals a role for PKB/Akt in DNA damage response and tumorigenesis. J Biol Chem. 2008;283:30025–33. doi: 10.1074/jbc.M803053200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.van Heemst D. Insulin, IGF-1 and longevity. Aging Dis. 2010;1:147–57. [PMC free article] [PubMed] [Google Scholar]
- 145.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–45. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schroder S, Adler T, Afonso LC, Aguilar-Pimentel JA, Becker L, Garrett L, Hans W, Hettich MM, Holtmeier R, Holter SM, Moreth K, Prehn C, Puk O, Racz I, Rathkolb B, Rozman J, Naton B, Ordemann R, Adamski J, Beckers J, Bekeredjian R, Busch DH, Ehninger G, Graw J, Hofler H, Klingenspor M, Klopstock T, Ollert M, Stypmann J, Wolf E, Wurst W, Zimmer A, Fuchs H, Gailus-Durner V, Hrabe de Angelis M, Ehninger D. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. 2013;123:3272–91. doi: 10.1172/JCI67674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wu JJ, Liu J, Chen EB, Wang JJ, Cao L, Narayan N, Fergusson MM, Rovira II, Allen M, Springer DA, Lago CU, Zhang S, DuBois W, Ward T, deCabo R, Gavrilova O, Mock B, Finkel T. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013;4:913–20. doi: 10.1016/j.celrep.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 150.Catrysse L, van Loo G. Inflammation and the Metabolic Syndrome: The Tissue-Specific Functions of NF-kappaB. Trends Cell Biol. 2017;27:417–429. doi: 10.1016/j.tcb.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 151.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–7. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–34. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Salminen A, Kaarniranta K. Insulin/IGF-1 paradox of aging: regulation via AKT/IKK/NF-kappaB signaling. Cell Signal. 2010;22:573–7. doi: 10.1016/j.cellsig.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 154.Ju J, Naura AS, Errami Y, Zerfaoui M, Kim H, Kim JG, Abd Elmageed ZY, Abdel-Mageed AB, Giardina C, Beg AA, Smulson ME, Boulares AH. Phosphorylation of p50 NF-kappaB at a single serine residue by DNA-dependent protein kinase is critical for VCAM-1 expression upon TNF treatment. J Biol Chem. 2010;285:41152–60. doi: 10.1074/jbc.M110.158352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu L, Kwak YT, Bex F, Garcia-Martinez LF, Li XH, Meek K, Lane WS, Gaynor RB. DNA-dependent protein kinase phosphorylation of IkappaB alpha and IkappaB beta regulates NF-kappaB DNA binding properties. Mol Cell Biol. 1998;18:4221–34. doi: 10.1128/mcb.18.7.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mishra A, Brown AL, Yao X, Yang S, Park SJ, Liu C, Dagur PK, McCoy JP, Keeran KJ, Nugent GZ, Jeffries KR, Qu X, Yu ZX, Levine SJ, Chung JH. Dendritic cells induce Th2-mediated airway inflammatory responses to house dust mite via DNA-dependent protein kinase. Nat Commun. 2015;6:6224. doi: 10.1038/ncomms7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ghonim MA, Pyakurel K, Ju J, Rodriguez PC, Lammi MR, Davis C, Abughazleh MQ, Mansy MS, Naura AS, Boulares AH. DNA-dependent protein kinase inhibition blocks asthma in mice and modulates human endothelial and CD4(+) T-cell function without causing severe combined immunodeficiency. J Allergy Clin Immunol. 2015;135:425–40. doi: 10.1016/j.jaci.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li M, Lin YF, Palchik GA, Matsunaga S, Wang D, Chen BP. The catalytic subunit of DNA-dependent protein kinase is required for cellular resistance to oxidative stress independent of DNA double-strand break repair. Free Radic Biol Med. 2014;76:278–85. doi: 10.1016/j.freeradbiomed.2014.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 160.Queitsch C, Sangster TA, Lindquist S. Hsp90 as a capacitor of phenotypic variation. Nature. 2002;417:618–24. doi: 10.1038/nature749. [DOI] [PubMed] [Google Scholar]
- 161.Specchia V, Piacentini L, Tritto P, Fanti L, D’Alessandro R, Palumbo G, Pimpinelli S, Bozzetti MP. Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature. 2010;463:662–5. doi: 10.1038/nature08739. [DOI] [PubMed] [Google Scholar]