

Quality control (QC) pathways for misfolded proteins depend on E3 ubiquitin ligases and associated chaperones. Prasad et al. show that Hsp40/70/110 chaperones traffic and manage misfolded proteins in the nucleus, extending the nuclear protein QC pathway to include cytosolic clients.
Abstract
Misfolded cytosolic proteins are degraded by the ubiquitin proteasome system through quality control (QC) pathways defined by E3 ubiquitin ligases and associated chaperones. Although they work together as a comprehensive system to monitor cytosolic protein folding, their respective contributions remain unclear. To bridge existing gaps, the pathways mediated by the San1 and Ubr1 E3 ligases were studied coordinately. We show that pathways share the same complement of chaperones needed for substrate trafficking, ubiquitination, and degradation. The significance became clear when Ubr1, like San1, was localized primarily to the nucleus. Appending nuclear localization signals to cytosolic substrates revealed that Ydj1 and Sse1 are needed for substrate nuclear import, whereas Ssa1/Ssa2 is needed both outside and inside the nucleus. Sis1 is required to process all substrates inside the nucleus, but its role in trafficking is substrate specific. Together, these data show that using chaperones to traffic misfolded cytosolic proteins into the nucleus extends the nuclear protein QC pathway to include cytosolic clients.
Introduction
Proteins are involved in all cellular functions. Accordingly, intricate intracellular pathways have evolved that govern the synthesis, quantity, delivery, activity, and lifetime of their protein constituents. As they work in concert, these mechanisms are collectively termed the protein homeostasis (or proteostasis) network (Balchin et al., 2016). Their importance is underscored by the prevalence and diversity of human disease when they become compromised (Labbadia and Morimoto, 2015). Although specialized mechanisms heighten the fidelity of information flow from genes to proteins, rare errors emanating from DNA replication, transcription, and translation can result in aberrant protein products. In addition, stochastic deviations from protein folding and maturation processes contribute to the total load. Because misfolded proteins can be cytotoxic through interference with cellular functions, quality control (QC) pathways are positioned throughout to monitor the folding state of nascent and preexisting polypeptides. Molecules beyond repair are targeted for degradation via the ubiquitin–proteasome system (UPS) or in lysosomes (Amm et al., 2014; Brandman and Hegde, 2016; Casson et al., 2016; Voos et al., 2016; Preston and Brodsky, 2017).
Aberrant proteins exist in two basic forms: aggregate and soluble. Protein aggregates, both ordered and amorphous, are associated with various human diseases including Alzheimer’s, Huntington’s, and Parkinson’s (Currais et al., 2017; Dubnikov et al., 2017; Pearce and Kopito, 2018). Although it remains controversial whether large aggregates are harmful or protective, cellular mechanisms exist to counter them. They can undergo targeted degradation through autophagy or the UPS (Gamerdinger et al., 2009; Scior et al., 2016). Alternatively, they can be found sequestered as ubiquitin-modified and/or chaperone-associated macromolecular structures (Kopito and Sitia, 2000; Kaganovich et al., 2008). Intracellular segregation presumably reduces their toxicity by limiting inappropriate interactions. Although many misfolded proteins have the tendency to aggregate on their own, they can be maintained in a soluble state through direct chaperone interactions (Balchin et al., 2016). Substrate solubility is particularly important for protein QC pathways that rely on the UPS (Amm et al., 2016; Comyn et al., 2016). In the better-understood ER-associated degradation (ERAD) pathways, luminal substrates must translocate across the ER membrane, and integral membrane proteins must be extracted for ubiquitination and degradation by the 26S proteasome (McCaffrey and Braakman, 2016; Preston and Brodsky, 2017). ER proteins forming large aggregates cannot use ERAD and are instead degraded in lysosomes through an autophagic mechanism (Kruse et al., 2006; Jackson and Hewitt, 2016).
Currently, the best understood protein QC mechanism is glycan-directed ERAD (Caramelo and Parodi, 2015; Cherepanova et al., 2016; Neubert and Strahl, 2016; Roth and Zuber, 2017). In this study, a series of glycosidases sequentially trim N-linked glycans to generate a degradation signal recognized by the Yos9 (OS9 and XTP3-B in mammals) lectin receptor located at the ER membrane. The sum of the steps is believed to set a timer for folding, culminating in degradation should it fail at the end of the sequence. Because N glycosylation doesn’t occur anywhere else in the cell, this mechanism is not generally applicable.
The QC of cytoplasmic proteins (CytoQCs) also relies on the UPS, but this is where the similarity to ERAD ends. In budding yeast, CytoQC is surprisingly complex, with at least five E3 ubiquitin ligases that function in substrate recognition and ubiquitination identified so far (Park et al., 2007; Eisele and Wolf, 2008; Kohlmann et al., 2008; Lewis and Pelham, 2009; Heck et al., 2010; Prasad et al., 2010; Fang et al., 2011, 2014; Stolz et al., 2013; Summers et al., 2013; Maurer et al., 2016). Interestingly, each already had established roles in other pathways. San1 ubiquitinates damaged nuclear proteins for degradation, where it resides (Gardner et al., 2005); Ubr1 is best known for its role in recognizing N-terminal degrons (N-end rule) of folded proteins (Bartel et al., 1990; Varshavsky, 2011); Doa10, located in the ER and inner nuclear envelope, mediates membrane protein ERAD (Swanson et al., 2001; Huyer et al., 2004; Vashist and Ng, 2004); Ltn1 targets to ribosomes to ubiquitinate stalled translation products (Bengtson and Joazeiro, 2010; Brandman et al., 2012); and Rsp5 is found throughout the cell and has a broad array of targets (Rotin and Kumar, 2009; Lauwers et al., 2010; Domanska and Kaminska, 2015). In addition, the E4 ubiquitin ligase Hul5 is required for the degradation of heat-damaged proteins, particularly those that display low solubility (Kohlmann et al., 2008; Fang et al., 2011). None of these enzymes are functionally redundant, so it remains enigmatic how they are organized within the CytoQC system.
Among CytoQC pathways, the San1 and Ubr1 pathways share several similarities. Although the pathways have some distinct substrates, there is substantial substrate overlap whereby both genes must be disabled to fully disrupt degradation (Lewis and Pelham, 2009; Heck et al., 2010; Khosrow-Khavar et al., 2012; Prasad et al., 2012; Guerriero et al., 2013; Amm and Wolf, 2016). Both pathways use the Hsp70 proteins Ssa1 and Ssa2 and the Hsp40 proteins Ydj1 and Sis1 (Park et al., 2007, 2013; Heck et al., 2010; Prasad et al., 2010; Stolz et al., 2013; Summers et al., 2013; Amm et al., 2016). As San1 is a nuclear protein, misfolded cytosolic proteins must traffic into the nucleus for ubiquitination and degradation (Gardner et al., 2005). The localization of Ubr1 has not been unequivocally determined, partly because of its low levels, but its substrates can be found in both the nucleus and cytosol, so it should be active in both. Ssa1 and Ssa2 are required for substrate transport, but this could be a consequence of protein aggregation in their absence, a state incompatible with nuclear import should aggregates exceed size constraints of nuclear pores (Kaganovich et al., 2008; Amm et al., 2016). Importantly, Ssa1 facilitates San1–substrate interactions so that it sequentially facilitates nuclear transport and targeting to the E3 ligase (Guerriero et al., 2013). Accordingly, the Hsp70 nucleotide exchange factor Fes1 is required for CytoQC. Cells lacking FES1 fail to ubiquitinate and degrade misfolded proteins (Gowda et al., 2013). Sis1 facilitates nuclear import likely through substrate handoff from Hsp70 in the cytosol, but the role of Ydj1 remains unclear (Park et al., 2013; Gowda et al., 2016).
To analyze the functional relationship between the San1 and Ubr1 pathways, we measured the behavior of pathway-specific substrates in well-characterized mutant strains. Importantly, Ubr1, whose localization was unclear, is shown to reside primarily in the nucleus. Although the Hsp70 chaperones Ssa1 and Ssa2 are functionally redundant in the San1 and Ubr1 pathways, their cochaperones Ydj1 and Sis1 are equally critical but act sequentially. Ydj1 functions specifically to traffic substrates into the nucleus, whereas Sis1 cooperates with Ydj1 and also plays a role in the nucleus to facilitate ubiquitination and degradation. The Hsp110 protein Sse1, however, is entirely dispensable for nuclear QC and functions primarily to traffic misfolded cytosolic substrates into the nucleus. These data show that the Hsp40/Hsp70/Hsp110 chaperone system is recruited by CytoQC to adapt the nuclear protein QC system to degrade misfolded cytosolic proteins.
Results
Ydj1 and Sse1 chaperones mediate trafficking of misfolded cytosolic proteins to the nucleus
The cytosolic/ER membrane J class chaperone Ydj1 is required for CytoQC, but its role in the pathway is unclear (Park et al., 2007; Fang et al., 2014; Maurer et al., 2016). To understand its contribution, we analyzed three model substrates in conditional and null mutants of YDJ1 (Caplan et al., 1992). Δ2GFP and ΔssPrA are San1-dependent substrates, and Ste6*C is a Ubr1-dependent substrate (Prasad et al., 2010, 2012). The conditional ydj1-151 mutant strongly stabilized each substrate at the restrictive temperature as expected (Fig. 1). At the permissive temperature, substrate stabilization remained strong in mutant cells. This suggests that the 37°C growth restriction reflects a loss of function other than San1/Ubr1-dependent CytoQC, which is nonessential (Park et al., 2007; Eisele and Wolf, 2008; Heck et al., 2010; Prasad et al., 2010; Maurer et al., 2016). We also observed that the higher incubation temperature also reduced substrate degradation rates in the WT cells (Fig. 1, A, C, and E). This likely reflects competition from endogenous substrates that increase under stress conditions or the formation of aggregates that are more difficult to degrade (Fang et al., 2014). In all cases, stabilization was accompanied by a proportional reduction in polyubiquitination, indicating that Ydj1 functions at or upstream of this step (Fig. 1, B, D, and F).
Figure 1.

Ydj1 is required for both San1 and Ubr1 pathways. (A, C, and E) All constructs were appended with a C-terminal HA epitope tag. The turnover of Δ2GFP, ΔssPrA, or Ste6*C in WT and ydj1-151 cells was determined by metabolic pulse chase. Cells were grown to log phase at indicated temperatures before labeling. Each strain was pulse labeled for 10 min and chased for the times indicated. After immunoprecipitation (IP), proteins were resolved by SDS-PAGE and quantified by phosphorimager analysis. Representative phosphor screen scans are shown. All data plotted were processed using Excel, reflecting three independent experiments with the means and SD indicated. (B, D, and F) Δ2GFP, ΔssPrA, or Ste6*C expressed in WT and ydj1-151 cells was immunoprecipitated, resolved by SDS-PAGE, and analyzed by immunoblotting (IB) to detect the ubiquitinated proteins. Protein amounts used for immunoprecipitation were normalized using an Odyssey infrared imaging system.
Because CytoQC is a dynamic process, we examined whether Ydj1 plays a role in substrate trafficking. For this question, we first performed indirect immunofluorescence to localize substrates in WT and mutant cells. In WT cells, Δ2GFP and ΔssPrA were enriched in the nuclei as expected (Fig. 2, A and B). Ste6*C displayed a similar pattern, which was interesting because it was unclear where Ubr1-dependent substrates are degraded (Fig. 2 C). Intriguingly, all three substrates displayed reduced nuclear localization in ydj1-151 and Δydj1 mutant cells (Figs. 2 and S1 A). The effect is specific and not a general consequence of substrate stabilization because Δ2GFP and ΔssPrA accumulate strongly in the nuclei of Δsan1 cells (Prasad et al., 2010). The import defect is specific for misfolded proteins because folded nuclear proteins are unaffected (Fig. S1 D). Together, these data suggest a role for Ydj1 in substrate trafficking into the nucleus. To test the hypothesis, the superfolder (sf) GFP variant was appended to Δ2GFP to analyze substrate dynamics in live cells. sf-Δ2GFP behaves indistinguishably from Δ2GFP except that it more readily forms large intracellular foci at 37°C, reminiscent of structures termed insoluble protein deposits (IPODs) and juxtanuclear QC compartments (JUNQs; Figs. S1, E–G; Kaganovich et al., 2008). Live WT, ydj1-151, and Δsse1 cells expressing sf-Δ2GFP were given a high-intensity laser pulse to bleach fluorescent proteins in the nucleus. Our analysis was extended to cells lacking the Hsp110 factor Sse1 because these cells suggested a defect in substrate trafficking from immunofluorescence images (Prasad et al., 2010). The experiment was performed at 25°C to avoid the formation of the intracellular foci. Fluorescence intensities over photobleached areas were then measured at short intervals over a 100-s span and plotted (Fig. 3 A). Nuclear sf-Δ2GFP fluorescence recovered within a minute, revealing a significant lag between import and degradation. In both mutant strains, import was strongly impaired (Fig. 3, B and C). These data show that the import of misfolded cytosolic proteins into the nucleus is rapid and that the Ydj1 and Sse1 chaperones are required for this step.
Figure 2.

Localization of misfolded cytosolic proteins is defective in ydj1-151 cells. (A–C) WT and ydj1-151 cells expressing ΔssPrA, Δ2GFP, and Ste6*C were grown to log phase at room temperature and incubated at 37°C for 30 or 60 min. Cells were prepared for indirect immunofluorescence as described in Materials and methods. Substrates were detected using anti-HA antibodies in the green channel. ER and nuclear envelope were visualized in the red channel using anti-Kar2 antiserum. Nuclei were localized using DAPI staining. Cell imaging and acquisition were performed by confocal microscopy. Quantification of fluorescence intensity was done as described in Materials and methods. One-way ANOVA was used to test for significance (35 < n < 50; ****, P < 0.0001). The results shown are representative of two independent experiments. Bars, 2 µm.
Figure 3.

Misfolded protein nuclear trafficking is defective in YDJ1 and SSE1 mutants. (A) Nuclear import of sf-Δ2GFP in WT, ydj1-151, and Δsse1 cells was analyzed by FRAP. Nuclei were photobleached (100% 488-nm laser transmission; 15–20 iterations) after three images, and recovery was monitored by acquiring images immediately after bleaching (postbleach). Images show cells before bleaching (prebleach), immediately after bleaching (0 s), and 9, 30, 60, and 90 s after the initial bleach (postbleach). Dashed circles indicate positions of bleached nuclei. Bars, 2 µm. (B) The recovery of nuclear localized sf-Δ2GFP was plotted over the time. The fluorescence intensity (F. I.) of sf-Δ2GFP was quantified using ImageJ. Unpaired t test: *, P < 0.033; **, P < 0.002; ***, P < 0.0002; ****, P < 0.0001. n = 10. (C) Import rates were calculated using a pseudo–first order association kinetics curve using the model equation Y = Y0 + (Yindef − Y0) × [1 − e(−K × t)]. n = 10. The result shown is representative of three independent experiments. Error bars indicate means ± SEM.
Ubr1 is a nuclear E3 ubiquitin ligase
The dependence of both substrate classes on Ydj1 suggests a shared mechanism in the cytosol, which deviates only after nuclear import (Fig. 1). Although San1 is an established nuclear enzyme, Ubr1’s location was unclear because of its low abundance, but it is often described as a cytosolic protein in yeast (Amm et al., 2014). However, some fraction of Ubr1 must be in the nucleus because that is the location of some of its folded substrates. Applying two independent algorithms, Ubr1 is predicted to be a nuclear protein containing a strong nuclear localization signal (NLS; Fig. 4 A; Brameier et al., 2007; Kosugi et al., 2009). To test the prediction, we applied complementary experimental approaches. First, functional FLAG-tagged Ubr1 was expressed in WT cells using the strong TDH3 promoter to enhance detection (McAlister and Holland, 1985). For known nuclear proteins, expression from this promoter does not cause mislocalization (Gardner et al., 2005). Using indirect immunofluorescence, Ubr1 localizes entirely within the nucleus, discerned by the ER/nuclear envelope marker Kar2 and DAPI (Fig. 4 B). In addition, Ubr1 localization is identical to San1, indicating that they function within the same compartment (Fig. 4 B). In the second experiment, a Ubr1-specific polyclonal antiserum was generated to detect endogenous Ubr1 protein (Fig. S2 B). Initially, cell fractionation experiments using standard protocols were unsatisfactory because the integrity of the nucleus was partially compromised (Fig. S2 C). For this reason, we applied a gentler method to maintain the integrity of nuclei. The proprietary Y-PER detergent solution is designed to extract cytosolic proteins from yeast cells (Thermo Fisher Scientific). Because Y-PER is also used as a first step to remove excess proteins before nuclear DNA isolation, we surmised that it could serve this role only if nuclear factors are excluded (yeast DNA extraction kit; Thermo Fisher Scientific). After treatment of whole cells, the cytosolic protein Pgk1 was extracted efficiently in the cytosolic fraction (C), whereas the nuclear protein San1 is recovered exclusively in the post-cytosolic fraction (Fig. 4 C, San1p post-cytosolic fraction [PCF] lane). Indeed, we observed the expected patterns for GFP, which is mostly cytosolic in yeast, and for nuclear GFP-NLS (Fig. 4 C). As the GFP pair is well below the molecular weight cutoff for passive diffusion through nuclear pores, the assay proves effective for distinguishing nuclear and cytosolic proteins. When probed for endogenous Ubr1, it too was found almost exclusively in the pellet fraction (Fig. 4 C). Although it is necessary that some Ubr1 remain in the cytosol for cytosolic substrates (Shemorry et al., 2013; Stolz et al., 2013; Amm and Wolf, 2016), these data show that the bulk is localized in the nucleus under unstressed conditions. Collectively, these data show that like San1, Ubr1 is primarily a nuclear enzyme in Saccharomyces cerevisiae and provide an explanation for why some misfolded cytosolic proteins traffic to the nucleus for degradation.
Figure 4.

The San1 and Ubr1 E3 ubiquitin ligases are nuclear enzymes. (A) San1 and Ubr1 NLSs were predicted using cNLS Mapper (Kosugi et al., 2009) and NucPred (Brameier et al., 2007), respectively. (B) Localization of overexpressed Ubr1 and San1 was revealed by immunostaining. ER membranes were visualized using Kar2 antibodies and nuclei by DAPI staining. Arrowheads indicate the positions of nuclei. Bars, 2 µm. (C) Cells were incubated with Y-PER reagent and centrifuged to separate cytosolic (C) and post-cytosolic fractions (PCFs). Each fraction including the total extract (T) was analyzed by immunoblotting with antibodies against Ubr1, San1 (nuclear protein), Pkg1 (cytosolic protein), 20S-proteasome (proteasomal protein), and GFP (nuclear GFP and cytosolic GFP).
Hsp40/Hsp70/Hsp110 chaperones adapt nuclear QC for cytosolic proteins
We next sought to better understand how misfolded proteins selectively traffic into the nucleus. For this, monopartite NLSs were appended to Δ2GFP and ΔssPrA to generate Δ2GFP-NLS and ΔssPrA-NLS, respectively (Fig. S2 D). As shown in Figs. 5 D and S2 E, Δ2GFP-NLS and ΔssPrA-NLS are exclusively nuclear localized in WT and mutant cells. Because the appended substrates no longer depend on CytoQC for import, their analyses can reveal factors specifically required for the import and/or degradation of misfolded cytosolic proteins. In pulse–chase experiments, NLS substrates require San1, Ubr1, and Ssa1/Ssa2, like their cytosolic counterparts (Figs. 5 A and S3 A). In contrast, the Sse1 chaperone, required for Δ2GFP and ΔssPrA (Prasad et al., 2010), is entirely dispensable for their NLS counterparts (Figs. 5 A and S3 A). This shows that the function of Sse1 in Δ2GFP and ΔssPrA CytoQC is exclusively for their nuclear import. Similarly, Δ2GFP-NLS bypasses the degradation block of Δ2GFP in ydj1-151 cells (Figs. 5 B and S3 B). As ubiquitination and degradation of Δ2GFP-NLS is only slightly impaired in ydj1-151 cells, the data indicate that like Sse1, the primary role of Ydj1 in CytoQC is in the nuclear import step (Fig. 5, B and C; and Fig. S3, B and C).
Figure 5.

Appending an NLS to Δ2GFP bypasses requirement for Ydj1 and Sse1. (A and B) Turnover of Δ2GFP-NLS in WT, Δsan1, Δubr1, Δsan1Δubr1, Δssa1Δssa2, Δsse1, and ydj1-151 cells was determined by pulse chase at temperatures indicated. All data plotted were processed using Excel, reflecting three independent experiments with the means and SD indicated. (C) Ubiquitination of Δ2GFP-NLS was determined as described in Fig. 1 B. IB, immunoblot; IP, immunoprecipitation. (D) Cells expressing Δ2GFP-NLS were grown to log phase at room temperature and shifted to 37°C for 30 or 60 min. After fixation, Δ2GFP-NLS and Kar2 were visualized by immunostaining with respective primary and secondary antibodies. Nuclei were marked by DAPI staining. Arrowheads indicate positions of nuclei. Bars, 2 µm.
The efficient degradation of NLS-marked substrates in ydj1-151 background suggested a role for alternative Hsp40-like proteins. Of the 22 J domain proteins in budding yeast, we examined mutants of those localized in the nucleus or cytosol (Kampinga and Craig, 2010). Other than YDJ1, only SIS1 displayed a role in cytosolic/nuclear protein QC. Its role in CytoQC was already demonstrated previously in mediating the nuclear transport of the model protein CG* (ΔssCPY*-GFP fusion protein). It is also the target of polyglutamine aggregates, which helped explain the toxicity of such disease-related proteins (Park et al., 2013). Sis1 is also involved in localizing aberrant proteins to stress-inducible foci comprised of chaperones and misfolded proteins (Malinovska et al., 2012). In this study, analysis was performed in cells with SIS1 expression controlled by the tetracycline-responsive promoter (Tet-Off). Sis1 levels in Tet-Off SIS1 cells were 10–15% the level of WT even in the absence of doxycycline (Fig. 6, A and B; Summers et al., 2013). When misfolded proteins were expressed, these strains grew like WT (Fig. S5 D). At the same time, nuclear localization of San1 and Ubr1 was completely intact in these strains (Fig. S5, A and B). Thus, in this strain, reduced Sis1 impairs degradation of both cytosolic and NLS-targeted substrates, with the strongest effects in the presence of doxycycline (Fig. 6, A and B; and Fig. S4, A and B). Sis1 is required for the ubiquitination of QC substrates carrying the DegAB degron (Shiber et al., 2013). To test whether Sis1 is required for ubiquitination, Δ2GFP, ΔssPrA, and their NLS-modified variants were immunoprecipitated, resolved by SDS-PAGE, and immunoblotted. Membranes were probed with anti-Myc antibodies to detect ubiquitinated substrates. PolyUb-substrate conjugates were recovered from WT cells, whereas Tet-Off Sis1 cells produced diminished levels for all substrates (Fig. 6, C and D; and Fig. S4, C and D). This was expected for the CytoQC substrates because Sis1 was reported to play a role in substrate import so it would act upstream of ubiquitination (Malinovska et al., 2012; Park et al., 2013). It was more surprising that the NLS-tagged substrates were similarly affected. Thus, we wondered whether Sis1 also is required for their import. To address this question, the same strains were processed for indirect immunofluorescence to localize substrates. In the absence of doxycycline, under which substrate degradation is moderately disrupted, Δ2GFP and ΔssPrA can be found in both cytosol and nucleus, whereas Δ2GFP-NLS and ΔssPrA-NLS are primarily in the nucleus (Fig. 6, E and F; and Fig. S4, E and F). The complete depletion of Sis1 did not change the pattern, indicating that the degradation defect cannot be attributed to a defect in nuclear import alone (Fig. 6, E and F; and Fig. S4, E and F, +DOX). Collectively, these data show that Sis1 is required for ubiquitination and degradation of both substrate classes, independent of its role in substrate nuclear import.
Figure 6.

Sis1 is required for CytoQC and nuclear QC but dispensable for Δ2GFP nuclear import. (A and B) Cycloheximide decay experiments were performed in WT cells (R1158) and Tet-Off SIS1 cells expressing Δ2GFP or Δ2GFP-NLS in the absence and presence of doxycycline (DOX; 10 µg/ml, 20 h treatment before the experiment). Total protein extract was prepared as described above, and immunoblotting (IB) was performed to quantify the protein level of substrates. Sis1 and Pgk1 were used as loading controls. All data plotted were processed using Excel, reflecting three independent experiments with means and SD indicated. (C and D) The ubiquitination of Δ2GFP or Δ2GFP-NLS was determined in cells with or without SIS1 expression as described in Fig. 1 B. IP, immunoprecipitation. (E and F) The cellular localization of Δ2GFP or Δ2GFP-NLS in cells with or without SIS1 expression was examined by indirect immunostaining as described in Fig. 2. Arrowheads indicate positions of nuclei. Asterisks indicate the position of a nonspecific band. Bars, 2 µm.
Discussion
Of the broad classes of protein QC pathways, CytoQC is emerging as the most complex. In budding yeast, the numbers of E2, E3, and E4 enzymes of the UPS and chaperones involved in CytoQC are numerous and continue to grow (Park et al., 2007; Eisele and Wolf, 2008; Kohlmann et al., 2008; Lewis and Pelham, 2009; Heck et al., 2010; Prasad et al., 2010; Fang et al., 2011, 2014; Stolz et al., 2013; Summers et al., 2013; Ibarra et al., 2016; Maurer et al., 2016). Unlike ER mechanisms, distinct CytoQC pathways are deployed for normal and stress conditions (Kaganovich et al., 2008; Fang et al., 2011, 2014, 2016; Khosrow-Khavar et al., 2012). Some proteins traffic to the nucleus for processing, whereas others are degraded in the cytosol (Prasad et al., 2010; Park et al., 2013; Fang et al., 2014; Amm and Wolf, 2016). The reasons for the complexity remain unclear. The rationale to the complexity of CytoQC can only emerge through the coordinate analyses of the various pathways and their clients.
The discovery of San1 as a key CytoQC ubiquitin ligase upended the nascent field because San1 is a nuclear enzyme active in nuclear protein QC (Lewis and Pelham, 2009; Heck et al., 2010; Prasad et al., 2010). These studies led to the demonstration that misfolded cytosolic proteins traffic into the nucleus, where they are ubiquitinated and degraded (Heck et al., 2010; Prasad et al., 2010; Park et al., 2013). Such behavior is consistent with proteasomes being distributed mainly in the nucleus in dividing yeast cells (Russell et al., 1999). A nuclear import mechanism was confirmed and extended with additional San1-dependent substrates. The CG* substrate (ΔssCPY*-GFP fusion) traffics into the nucleus for degradation in a Sis1-dependent manner (Park et al., 2013). Importantly, this mechanism is disrupted in the presence of polyglutamine protein aggregates, providing a biochemical basis for the toxicity of polyQ disease proteins. This study further demonstrates a nuclear import–based CytoQC mechanism in mammalian cells, indicating that the basic pathway is conserved.
Although previously thought to be unique, we now know that the cytosol-to-nucleus mechanism is comprised of multiple pathways. This view was borne out of the expansion of model CytoQC substrates. Indeed, an early hint came from the discovery that Ura3 models (Ura3-2/3 and Ura3-CL1) use the Doa10 E3 ubiquitin ligase (Metzger et al., 2008; Lewis and Pelham, 2009). This was notable because Doa10 also mediates the degradation of some integral membrane proteins by ERAD (Swanson et al., 2001; Huyer et al., 2004; Vashist and Ng, 2004). Although it is not yet determined where the Ura3 substrates are ubiquitinated and degraded, Doa10 is found in the inner nuclear envelope and the ER (Deng and Hochstrasser, 2006). Doa10 certainly functions in the nucleus for QC because a misfolded variant of the kinetochore protein Ndc10 (Ndc10-2) requires Doa10 for its degradation (Furth et al., 2011). Thus, Doa10 is involved in ERAD, CytoQC, and nuclear protein QC, making it the most versatile E3 in protein homeostasis. Whether its CytoQC mode also requires the trafficking of substrates into the nucleus remains to be determined.
Ubr1 is yet another CytoQC ubiquitin ligase that straddles pathways. The RING-type enzyme is best known as the E3 ligase of the well-characterized and conserved N-end rule degradation pathway (Bartel et al., 1990; Varshavsky, 2011). Here, the recognition mechanism is through substrate N-terminal sequences that directly bind pockets within Ubr1 (N degrons; Choi et al., 2010; Matta-Camacho et al., 2010). Ubr1 works with HECT-type Ufd4 E3 enzyme, and together, the complex plays a role in QC (Hwang et al., 2010; Nillegoda et al., 2010). Although this recognition was first characterized for folded proteins, Ubr1’s QC mode also uses substrate N-terminal sequences for recognition (Kim et al., 2014). However, why it incorporates the N-end rule for the recognition of misfolded proteins was enigmatic. More recently, it was shown that some proteins bearing Ubr1 N degrons are normally stable (Shemorry et al., 2013). Their stability is caused by “shielding” of N degrons by interacting partners or ligands. Should such partners be limiting, the N degron is exposed and recognized by Ubr1, thus underlying a mechanism of QC for protein complexes.
Because the location of Ubr1 was previously unclear, misfolded Ubr1 substrates were thought to be ubiquitinated and degraded in the cytosol (Heck et al., 2010; Stolz et al., 2013). The data in this study support the view that Ubr1 is mainly a nuclear enzyme, and some of its substrates—misfolded proteins in particular—traffic into the nucleus for ubiquitination and degradation (Fig. 7). This explains the substrate overlap between San1 and Ubr1 (Lewis and Pelham, 2009; Heck et al., 2010; Khosrow-Khavar et al., 2012; Prasad et al., 2012; Guerriero et al., 2013; Amm and Wolf, 2016). Despite its preponderance as a nuclear enzyme, a fraction of active Ubr1 functions in the cytosol as some substrates are ubiquitinated there (Heck et al., 2010; Stolz et al., 2013). Perhaps diagnostic to identify such substrates is their exclusive dependence on Ubr1 because San1 has the intrinsic ability to recognize any unfolded protein in the nucleus (Rosenbaum et al., 2011).
Figure 7.
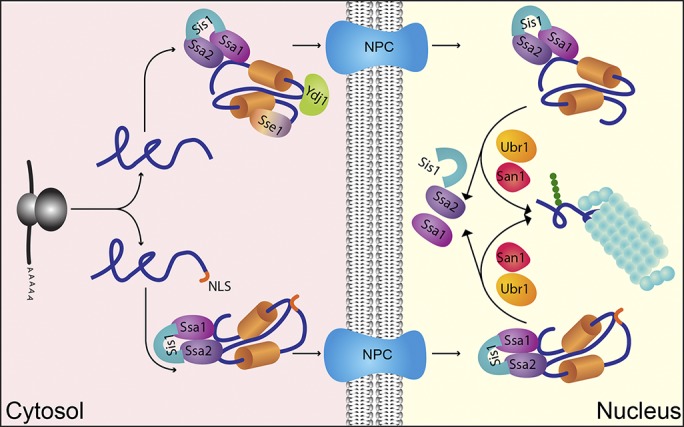
Proposed mechanism of nuclear-based CytoQC. The cell uses the Ssa1/2, Sis1, Ydj1, and Sse1 chaperone system to traffic misfolded cytosolic proteins to the nucleus for degradation. In the cytosol, Ssa1/2 and Sis1 are generally required for recognition as well as nuclear trafficking of misfolded cytosolic and nuclear proteins. If the misfold protein does not contain NLS, additional factors such as Sse1 and Ydj1 are needed for the trafficking step. Once inside the nucleus, these two factors are no longer essential. Misfolded proteins are chaperoned by Ssa1/2 and Sis1 and presented to the nuclear E3 ligases Ubr1 and San1 for degradation. Thus, by adapting Hsp40/70/110 chaperones, the nuclear protein control system can be exploited to serve cytosolic clients.
Although the San1, Ubr1, and Doa10 pathways provide surveillance during normal conditions, stress conditions that increase protein misfolding require additional pathways. The Rsp5 and Hul5 ubiquitin ligases play specialized roles in targeting heat-damaged proteins for degradation (Fang et al., 2011, 2014). Although Rsp5 is involved in diverse cellular processes, during heat stress, it partners with the Ubp2 and Ubp3 deubiquitinases to efficiently degrade misfolded proteins (Fang et al., 2016). Interestingly, Rsp5 is also involved in the clearance of damaged membrane proteins that have already trafficked out of the ER (Haynes et al., 2002; Wang et al., 2011). Thus, it follows the theme of CytoQC ubiquitin ligases that participate in multiple pathways. Some damaged proteins localize to large intracellular complexes called JUNQs and IPODs under heat stress (Kaganovich et al., 2008), so it would be interesting to learn whether Rsp5 and Hul5 are constituents of these sites. San1, Ubr1, and Doa10 are less likely to function at cytosolic JUNQs and IPODs because they are localized primarily in the nucleus. However, it is possible they play a role in the processing nuclear inclusions (Miller et al., 2015). San1 and Ubr1 also play significant roles during heat stress (Khosrow-Khavar et al., 2012). Thus, the nuclear route is active under broad conditions, whereas the Rsp5/Hul5 ligases may be better suited to process more aggregate-prone proteins.
There is broad agreement that cytosolic and nuclear chaperones are important for CytoQC in budding yeast (McClellan et al., 2005; Park et al., 2007, 2013; Metzger et al., 2008; Lewis and Pelham, 2009; Heck et al., 2010; Stolz et al., 2013; Comyn et al., 2016). Although it is known that the Hsp70 Ssa1/Ssa2 and the Hsp40 Sis1 proteins traffic some misfolded cytosolic proteins to the nucleus, how other chaperones act was less clear (Prasad et al., 2010; Park et al., 2013). A simple modification of established substrates with NLSs to bypass the CytoQC import step provided some clarity. These experiments showed that Ssa1/Ssa2 and Sis1 are also required for degradation after nuclear import (Figs. 5 A and 7). The Hsp110 protein Sse1, however, is only required for the import step (Figs. 5 A and 7). Similarly, Ydj1 is required for the import step, but its role in the nucleus is less clear (Fig. 7). Although the degradation of Δ2GFP-NLS is efficient in ydj1-151 cells, it is delayed somewhat compared with WT (Fig. 5 B). Because ydj1 mutant cells grow poorly compared with WT, it’s possible that the small difference is attributable to one of Ydj1’s critical housekeeping functions.
Collectively, these data clarify the functional relationship between San1 and Ubr1, which were previously linked mostly through substrate overlaps. Their shared localization in the nucleus helps to explain much of the previous observations. In this study, we show that chaperones serve to traffic their cytosolic substrates to the nuclear protein QC system for processing (Fig. 7). This critical step suggests that chaperones make the first decision for which proteins should be degraded. However, the established mechanisms of substrate recognition by San1 and Ubr1 suggest that they comprise an additional step that improves stringency. San1 functioning through direct recognition of misfolded structures while Ubr1’s binding of N-terminal degrons (Rosenbaum et al., 2011 Kim et al., 2014) might also broaden the scope of potential substrates. San1 recognizes grossly misfolded proteins of all types, whereas Ubr1 recognizes orphan subunits whose N termini are normally shielded by partners or ligands (Shemorry et al., 2013). Thus, the N degron mechanism of Ubr1 would be particularly important for orphan subunits that do not grossly misfold. The purpose of segregating QC E3 enzymes to the nucleus could be as simple as to prevent promiscuous targeting of newly synthesized folding polypeptides, activities that occur in the cytosol. Thus, the complexity of the multistep process quickly can be seen as elegant and efficient if it is used also to protect newly synthesized proteins in their most vulnerable state. To accomplish this by making use of a standalone QC system for nuclear proteins makes the system even more efficient.
Materials and methods
Plasmids used in this study
Plasmids were constructed using standard cloning protocols (Sambrook et al., 1989). All genes encoding expression constructs were verified by DNA sequencing. All substrates contained an engineered single HA epitope tag at their C termini. Unless otherwise noted, expression plasmids were constructed by placing coding sequences under the control of the constitutive TDH3 promoter in yeast centromeric vectors. Plasmid descriptions and oligonucleotide primers used in plasmid construction are listed in Tables S2 and S3, respectively.
pRP86, pRP88, and pRP120: Δ2GFP-HA, ΔssPrA-HA, and Ste6*C-HA fragments were released from pRP44, pRP42, and pRP22 (Prasad et al., 2012), respectively, by digesting the plasmids with BamHI and XbaI. Digested fragments were placed under the control of the TDH3 promoter in pRS316 vector to generate pRP86 and pRP88. pRP90: the SV40 NLS sequence SPKKKRKVEASGS was added to C terminus of Δ2GFP-HA by site-directed mutagenesis (Sawano and Miyawaki, 2000) using primers RP163 and pRP44. pRP91: an SV40 NLS sequence was added to the C terminus of ΔssPrA-HA by site-directed mutagenesis using primers RP164 and pRP42. pRP92 and pRP93: Δ2GFP-HA-NLS and ΔssPrA-HA-NLS fragments were released from pRP90 and pRP91, respectively, by digesting the plasmids with BamHI and XbaI. Digested fragments were ligated into pRP86 to generate pRP92 and pRP93, respectively. pRP96: an sf-Δ2GFP-HA fragment was released from pSK172 (Ng Laboratory plasmid collection) by digesting the plasmid with BamHI and XbaI. The digested fragment was ligated into pRP44 to generate pRP96. pRP97: an sf-Δ2GFP-HA fragment along with ACT1 terminator was released from pRP96 by digesting the plasmid with BamHI and SphI. The digested fragment was placed under the control of the TDH3 promoter in pRS314 vector to generate pRP97.
Strains and antibodies
S. cerevisiae strains used in this study are described in Table S1. Anti-HA monoclonal antibody (HA.11, raised in mouse) and anti-myc monoclonal antibody (9E10 c-myc, raised in mouse) were purchased from Covance. Anti-Kar2 and anti-Sec61 antibodies (rabbit) were provided by P. Walter (University of California, San Francisco, San Francisco, CA). Anti-Sis1 antiserum (rabbit) was a gift from D. Cyr (University of North Carolina, Chapel Hill, NC). Monoclonal antiproteasome 20S α (mouse) and polyclonal anti–histone H3 (rabbit) were purchased from Abcam. Monoclonal anti–3-phosphoglycerate kinase antibody (mouse) and monoclonal anti-V5 antibody (mouse) were purchased from Invitrogen, monoclonal anti-Ydj1 antibody (mouse) was from StressMarq, monoclonal anti-GFP antibody (mouse) was purchased from Roche, and monoclonal anti-FLAG (mouse) was purchased from Sigma-Aldrich. Secondary antibodies labeled with Alexa Fluor 488 (anti–mouse) or Alexa Fluor 596 (anti–rabbit) were purchased from Molecular Probes. Anti–rabbit IRDye 680 and anti–mouse IRDye 800 secondary antibodies were purchased from LI-COR Biosciences. Anti-Ubr1 antiserum was raised in rabbit against a recombinant protein containing the 100 N-terminal amino acids of Ubr1. Anti-San1 antiserum was raised in rabbit against a protein containing the 100 C-terminal amino acids of San1.
Metabolic pulse–chase assay
Three OD600 units of log-phase cells were labeled with 82.5 µCi of [35S]methionine/cysteine (EasyTag EXPRESS [35S]; PerkinElmer) and chased with excess cold amino acids (final concentration, 2 mM methionine/cysteine) at the times indicated. Protein extract preparation, immunoprecipitation, and SDS-PAGE analyses were carried as described previously (Prasad et al., 2010). In brief, 100 µl of 100% TCA was directly added to 900 µl cell culture to terminate the metabolic labeling at each time point (e.g., 0, 30, and 60 min). Cells were disrupted with zirconium beads. The whole-cell lysate was transferred to a new tube and subjected to 10 min centrifugation at 16,000 g. The resulting pellet was resuspended in TCA resuspension buffer (100 mM Tris, pH 11.0, 3% SDS, and 1 mM PMSF; 50 µl/OD) and boiled at 100°C for 10 min. The insoluble fraction was removed after 10 min spin at 16,000 g. The volume of the soluble cell extract used for the following immunoprecipitation analysis was normalized based on the scintillation counting of total isotopically labeled proteins. Typically, we added ≤50 µl of total cell extract into solution containing 700 µl of IPS II buffer (13.3 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, and 0.02% sodium azide), 6 µl of 100 mM PMSF, 1 µl of protease inhibitor cocktail (1:200; Sigma-Aldrich), and 1 µl of specific antibody. After 1 h incubation at 4°C, the diluted extract was subject to 10 min centrifugation at 16,000 g. The soluble fraction was transferred a new tube, mixed with IgG agarose beads, and incubated at 4°C for an additional 2 h on a nutator plate. The precipitants were washed three times with IPS I buffer (0.2% SDS, 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, and 0.02% sodium azide) and once with PBS. Lastly, the precipitants were eluted with protein loading buffer and analyzed by SDS-PAGE. The resulting gels were vacuum dried and exposed to phosphor screens for 24–48 h and then scanned and quantified using a Typhoon phosphorimager and ImageQuant TL software (GE Healthcare). All data plotted reflect three independent experiments with means and SD indicated. They were processed in Excel (Microsoft) with the AVERAGE and STDEV functions.
Cycloheximide chase assay and immunoblotting
Cells were grown to mid-log phase in synthetic media. Cessation of protein synthesis was initiated by adding cycloheximide to 200 µg/ml to begin the chase. Aliquots of cells were collected at the times indicated, and total protein extract was prepared by TCA precipitation as described above. A portion of total protein extract was separated on a 4–15% gradient gel by SDS-PAGE and transferred to nitrocellulose membranes. After blocking, the membranes were incubated with appropriate primary antibodies and followed by fluorescence dye–labeled secondary antibodies (anti–mouse IRDye 800 and/or anti–rabbit IRDye 680). The protein level was quantified by the Odyssey infrared imaging system (LI-COR Biosciences). All data plotted reflect three independent experiments with means and SD indicated. They were processed in Excel with the AVERAGE and STDEV functions.
Indirect immunofluorescence microscopy
Indirect immunofluorescence was performed as described previously (Prasad et al., 2010) with minor modifications. In brief, log-phase cells were fixed with 3.7% formaldehyde at 30°C for 90 min. For temperature-sensitive mutants, cells were shifted to 37°C for 30 or 60 min and fixed at 37°C for 90 min by rotating in the incubator. Cells were spheroplasted by zymolyase digestion (1 mg/ml zymolase 20T [US Biological], 0.1 M potassium phosphate, pH 7.5, and 1.4 M sorbitol). Spheroplasts were applied to a poly-l-lysine–coated slide. Slides were sequentially immersed in methanol for 6 min and in acetone for 30 s at −20°C. After blocking with 5% BSA, cells were incubated with primary antibodies followed by secondary antibodies. HA.11 mAb (Covance) and polyclonal anti-Kar2 were diluted to 1:200 and 1:500, respectively. Secondary antibodies Alexa Fluor 488 goat anti–mouse and Alexa Fluor 596 goat anti–rabbit were diluted to 1:500. Nuclei were visualized by DAPI staining. Samples were examined at room temperature with a three-photomultiplier-tube detector using a 100× 1.4 NA oil Plan Apochromat objective in an upright confocal microscope (EXCITER or LSM510; ZEISS) controlled by LSM5 program software (ZEISS). Images were archived by LSM Image Browser and Photoshop (Adobe). For Fig. 2, an upright confocal LSM880 microscope using a 63× 1.4 NA oil Plan Apochromat (controlled by Zen 2012; ZEISS) with a tunable chameleon multiphoton laser, two conventional PMTs, and a high-sensitivity GaAsP detector (also with an airyscan detector) was used. For the analysis of images, background-subtracted images were quantified by drawing two regions of interest (ROIs) in the cytoplasm and one ROI in nucleus of each cell, and the ROIs’ mean fluorescence intensities were calculated using ImageJ (National Institutes of Health). Later for cytoplasm, means of two ROIs intensities were calculated. The reported relative nuclear enrichment was calculated as the ratio between mean nuclear and cytoplasmic fluorescence intensities. For drawing ROIs, the vacuole was avoided. Data of replicates were pooled before significance testing. One-way ANOVA was used to test for significance.
Substrate ubiquitination assay
The experiments were performed using mid-log-phase cells overexpressing ubiquitin. For temperature-sensitive mutants, cells were shifted to 37°C for 1 h before lysate preparation. 10 OD600 units of cells were collected, and total protein extract was prepared by TCA precipitation as described previously (Vashist and Ng, 2004). Protein levels were normalized before immunoprecipitation, and lysates were then mixed with IPS II (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 1% Triton X-100) supplemented with protease inhibitor cocktail (Roche), 100 mM PMSF, anti-HA affinity matrix, and 10 mM N-ethylmaleimide (Sigma-Aldrich). Immunoprecipitated samples were resolved by SDS-PAGE and transferred onto nitrocellulose. Total substrates and ubiquitinated substrates were detected by using anti-HA antibody and anti-myc antibody, respectively.
GFP fluorescence microscopy
Cells expressing sf-Δ2GFP were grown at room temperature and shifted to 37°C for 30 min. Cells were examined at room temperature using a Meta 510 confocal microscope under a 100× 1.4 NA oil-immersion objective lens (ZEISS). Images were archived by LSM Image Browser and Photoshop.
FRAP
For all FRAP experiments, fresh cells expressing sf-Δ2GFP were grown at room temperature, resuspended in fresh Synthetic Complete medium, and immobilized on a 2% agar pad containing Synthetic Complete medium. An LSM780 microscope (controlled by Zen 2012; ZEISS) with a multiarray 32PMT GaAsP detector and an Apochromat 63× 1.4 NA oil differential interference contrast Plan Apochromat objective was used for acquiring FRAP. Each nucleus was bleached with 15–20 iterations with 100% laser power of 488-nm light at 30°C. The FRAP protocol involved five prebleach images followed by bleaching and postbleach recording at 3-s intervals for 100 s. Imaging was typically performed by using a 488-nm argon laser at 3% laser intensity at 30°C. FRAP quantification was performed using ImageJ. The mean fluorescence recovery signal was quantified in the bleached nucleus. As a control, fluorescence of nonbleached nucleus of three neighboring cells was measured. After background subtraction, the fluorescence signals of nucleus were normalized to the means of the three control nuclei and set to 100% at beginning of the experiment. 10 different FRAP experiments were pooled and transferred to Prism 6 (GraphPad Software) and plotted on an exponential FRAP curve. Nuclear import rates were calculated using nonlinear curve fitting of Prism software to a pseudo–first order association kinetics curve using the model equation Y = Y0 + (Yindef − Y0) × [1 − e(−K × t)] to determine the apparent transport rate constant K for each strain. Images were also archived by ImageJ and Photoshop. All the error bars indicate ±SEM.
Statistics
A two-tailed unpaired Student’s test was used for significance of FRAP analysis. For Fig. 2, one-way ANOVA was used to test for significance. Unless otherwise indicated, all the analyzed mutants were always compared with WT.
Cytosol/membrane fractionation
Cells were grown to log phase (OD600 = 0.7–1) at 30°C. Cells (10 OD600 equivalent) were resuspended in lysis buffer (20 mM Hepes-KOH, pH 6.8, 250 mM sorbitol, 150 mM KOAc, and 5 mM MgOAc containing 1 mM PMSF and protease inhibitor cocktail). Glass beads (300 µl) were added, and cells were disrupted by agitation on a Vortex mixer (at full speed) 10 times for 30 s with 30-s intervals on ice between each cycle. The homogenate was collected and pooled after rinsing the beads with buffer 88 (20 mM Hepes, pH 7.4, 150 mM KOAc, 250 mM sorbitol, and 5 mM MgOAc). Unbroken cells were removed by centrifugation at 300 g for 5 min at 4°C twice. Subsequently, the supernatant was centrifuged at 18,000 g for 30 min at 4°C. The membrane pellet was resuspended in buffer 88, pH 7.4. The resulting supernatant (S18K) and the pellet (P18K) fractions were further processed for Western blotting.
Separation of cytosol and post-cytosolic fraction
10 OD600 units of log-phase cells were collected and resuspended in 1 ml Y-PER reagent (Thermo Fisher Scientific) containing 1 mM PMSF and protease inhibitor cocktail (Sigma-Aldrich) and then were incubated at room temperature for 30 min. Cells were centrifuged at 4°C for 30 min at 18,000 g to separate the cytosolic and post-cytosolic fractions. Each fraction was precipitated in 10% TCA and analyzed on a 4–15% gradient gel by SDS-PAGE, followed by immunoblotting.
Online supplemental material
The Supplemental material consists of three tables (Tables S1, S2, and S3) and five figures (Figs. S1, S2, S3, S4, and S5). Supplementary figures show extended control experiments and data of additional examples to those shown in main figures. Tables list strains, plasmids, and oligonucleotide primers used in this study as well as their pertinent information.
Supplementary Material
Acknowledgments
We thank P. Walter and D. Cyr for gifts of antibodies. We thank N. Bedford and Y. Liu for technical assistance. We are indebted to the immense generosity of Yves Barral (ETH Zürich), who provided access to equipment and reagents in experiments performed by R. Prasad for the revised version of this manuscript.
This work was supported by funds from the Temasek Trust. R. Prasad and C. Xu were recipients of the National University of Singapore Graduate Research Scholarship.
The authors declare no competing financial interests.
Author contributions: R. Prasad performed molecular, genetic, biochemical, and imaging analyses. R. Prasad and C. Xu performed quantitative analyses. D.T.W. Ng conceived and supervised the project. R. Prasad, C. Xu, and D.T.W. Ng wrote the paper.
References
- Amm I., and Wolf D.H.. 2016. Molecular mass as a determinant for nuclear San1-dependent targeting of misfolded cytosolic proteins to proteasomal degradation. FEBS Lett. 590:1765–1775. 10.1002/1873-3468.12213 [DOI] [PubMed] [Google Scholar]
- Amm I., Sommer T., and Wolf D.H.. 2014. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim. Biophys. Acta. 1843:182–196. 10.1016/j.bbamcr.2013.06.031 [DOI] [PubMed] [Google Scholar]
- Amm I., Kawan M., and Wolf D.H.. 2016. Characterization of protein quality control components via dual reporter-containing misfolded cytosolic model substrates. Anal. Biochem. 515:14–21. 10.1016/j.ab.2016.09.012 [DOI] [PubMed] [Google Scholar]
- Balchin D., Hayer-Hartl M., and Hartl F.U.. 2016. In vivo aspects of protein folding and quality control. Science. 353:aac4354 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- Bartel B., Wünning I., and Varshavsky A.. 1990. The recognition component of the N-end rule pathway. EMBO J. 9:3179–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson M.H., and Joazeiro C.A.. 2010. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 467:470–473. 10.1038/nature09371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brameier M., Krings A., and MacCallum R.M.. 2007. NucPred--predicting nuclear localization of proteins. Bioinformatics. 23:1159–1160. 10.1093/bioinformatics/btm066 [DOI] [PubMed] [Google Scholar]
- Brandman O., and Hegde R.S.. 2016. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 23:7–15. 10.1038/nsmb.3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O., Stewart-Ornstein J., Wong D., Larson A., Williams C.C., Li G.W., Zhou S., King D., Shen P.S., Weibezahn J., et al. 2012. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 151:1042–1054. 10.1016/j.cell.2012.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan A.J., Cyr D.M., and Douglas M.G.. 1992. YDJ1p facilitates polypeptide translocation across different intracellular membranes by a conserved mechanism. Cell. 71:1143–1155. 10.1016/S0092-8674(05)80063-7 [DOI] [PubMed] [Google Scholar]
- Caramelo J.J., and Parodi A.J.. 2015. A sweet code for glycoprotein folding. FEBS Lett. 589:3379–3387. 10.1016/j.febslet.2015.07.021 [DOI] [PubMed] [Google Scholar]
- Casson J., McKenna M., and High S.. 2016. On the road to nowhere: cross-talk between post-translational protein targeting and cytosolic quality control. Biochem. Soc. Trans. 44:796–801. 10.1042/BST20160045 [DOI] [PubMed] [Google Scholar]
- Cherepanova N., Shrimal S., and Gilmore R.. 2016. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 41:57–65. 10.1016/j.ceb.2016.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W.S., Jeong B.C., Joo Y.J., Lee M.R., Kim J., Eck M.J., and Song H.K.. 2010. Structural basis for the recognition of N-end rule substrates by the UBR box of ubiquitin ligases. Nat. Struct. Mol. Biol. 17:1175–1181. 10.1038/nsmb.1907 [DOI] [PubMed] [Google Scholar]
- Comyn S.A., Young B.P., Loewen C.J., and Mayor T.. 2016. Prefoldin Promotes Proteasomal Degradation of Cytosolic Proteins with Missense Mutations by Maintaining Substrate Solubility. PLoS Genet. 12:e1006184 10.1371/journal.pgen.1006184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currais A., Fischer W., Maher P., and Schubert D.. 2017. Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J. 31:5–10. 10.1096/fj.201601184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M., and Hochstrasser M.. 2006. Spatially regulated ubiquitin ligation by an ER/nuclear membrane ligase. Nature. 443:827–831. 10.1038/nature05170 [DOI] [PubMed] [Google Scholar]
- Domanska A., and Kaminska J.. 2015. Role of Rsp5 ubiquitin ligase in biogenesis of rRNA, mRNA and tRNA in yeast. RNA Biol. 12:1265–1274. 10.1080/15476286.2015.1094604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnikov T., Ben-Gedalya T., and Cohen E.. 2017. Protein Quality Control in Health and Disease. Cold Spring Harb. Perspect. Biol. 9:a023523 10.1101/cshperspect.a023523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele F., and Wolf D.H.. 2008. Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett. 582:4143–4146. 10.1016/j.febslet.2008.11.015 [DOI] [PubMed] [Google Scholar]
- Fang N.N., Ng A.H., Measday V., and Mayor T.. 2011. Hul5 HECT ubiquitin ligase plays a major role in the ubiquitylation and turnover of cytosolic misfolded proteins. Nat. Cell Biol. 13:1344–1352. 10.1038/ncb2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang N.N., Chan G.T., Zhu M., Comyn S.A., Persaud A., Deshaies R.J., Rotin D., Gsponer J., and Mayor T.. 2014. Rsp5/Nedd4 is the main ubiquitin ligase that targets cytosolic misfolded proteins following heat stress. Nat. Cell Biol. 16:1227–1237. 10.1038/ncb3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang N.N., Zhu M., Rose A., Wu K.P., and Mayor T.. 2016. Deubiquitinase activity is required for the proteasomal degradation of misfolded cytosolic proteins upon heat-stress. Nat. Commun. 7:12907 10.1038/ncomms12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furth N., Gertman O., Shiber A., Alfassy O.S., Cohen I., Rosenberg M.M., Doron N.K., Friedler A., and Ravid T.. 2011. Exposure of bipartite hydrophobic signal triggers nuclear quality control of Ndc10 at the endoplasmic reticulum/nuclear envelope. Mol. Biol. Cell. 22:4726–4739. 10.1091/mbc.E11-05-0463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamerdinger M., Hajieva P., Kaya A.M., Wolfrum U., Hartl F.U., and Behl C.. 2009. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 28:889–901. 10.1038/emboj.2009.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R.G., Nelson Z.W., and Gottschling D.E.. 2005. Degradation-mediated protein quality control in the nucleus. Cell. 120:803–815. 10.1016/j.cell.2005.01.016 [DOI] [PubMed] [Google Scholar]
- Gowda N.K., Kandasamy G., Froehlich M.S., Dohmen R.J., and Andréasson C.. 2013. Hsp70 nucleotide exchange factor Fes1 is essential for ubiquitin-dependent degradation of misfolded cytosolic proteins. Proc. Natl. Acad. Sci. USA. 110:5975–5980. 10.1073/pnas.1216778110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda N.K., Kaimal J.M., Masser A.E., Kang W., Friedländer M.R., and Andréasson C.. 2016. Cytosolic splice isoform of Hsp70 nucleotide exchange factor Fes1 is required for the degradation of misfolded proteins in yeast. Mol. Biol. Cell. 27:1210–1219. 10.1091/mbc.E15-10-0697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero C.J., Weiberth K.F., and Brodsky J.L.. 2013. Hsp70 targets a cytoplasmic quality control substrate to the San1p ubiquitin ligase. J. Biol. Chem. 288:18506–18520. 10.1074/jbc.M113.475905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C.M., Caldwell S., and Cooper A.A.. 2002. An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. J. Cell Biol. 158:91–101. 10.1083/jcb.200201053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck J.W., Cheung S.K., and Hampton R.Y.. 2010. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. USA. 107:1106–1111. 10.1073/pnas.0910591107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyer G., Piluek W.F., Fansler Z., Kreft S.G., Hochstrasser M., Brodsky J.L., and Michaelis S.. 2004. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J. Biol. Chem. 279:38369–38378. 10.1074/jbc.M402468200 [DOI] [PubMed] [Google Scholar]
- Hwang C.S., Shemorry A., Auerbach D., and Varshavsky A.. 2010. The N-end rule pathway is mediated by a complex of the RING-type Ubr1 and HECT-type Ufd4 ubiquitin ligases. Nat. Cell Biol. 12:1177–1185. 10.1038/ncb2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra R., Sandoval D., Fredrickson E.K., Gardner R.G., and Kleiger G.. 2016. The San1 Ubiquitin Ligase Functions Preferentially with Ubiquitin-conjugating Enzyme Ubc1 during Protein Quality Control. J. Biol. Chem. 291:18778–18790. 10.1074/jbc.M116.737619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M.P., and Hewitt E.W.. 2016. Cellular proteostasis: degradation of misfolded proteins by lysosomes. Essays Biochem. 60:173–180. 10.1042/EBC20160005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaganovich D., Kopito R., and Frydman J.. 2008. Misfolded proteins partition between two distinct quality control compartments. Nature. 454:1088–1095. 10.1038/nature07195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga H.H., and Craig E.A.. 2010. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11:579–592. 10.1038/nrm2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosrow-Khavar F., Fang N.N., Ng A.H., Winget J.M., Comyn S.A., and Mayor T.. 2012. The yeast ubr1 ubiquitin ligase participates in a prominent pathway that targets cytosolic thermosensitive mutants for degradation. G3 (Bethesda). 2:619–628. 10.1534/g3.111.001933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.K., Kim R.R., Oh J.H., Cho H., Varshavsky A., and Hwang C.S.. 2014. The N-terminal methionine of cellular proteins as a degradation signal. Cell. 156:158–169. 10.1016/j.cell.2013.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmann S., Schäfer A., and Wolf D.H.. 2008. Ubiquitin ligase Hul5 is required for fragment-specific substrate degradation in endoplasmic reticulum-associated degradation. J. Biol. Chem. 283:16374–16383. 10.1074/jbc.M801702200 [DOI] [PubMed] [Google Scholar]
- Kopito R.R., and Sitia R.. 2000. Aggresomes and Russell bodies: Symptoms of cellular indigestion? EMBO Rep. 1:225–231. 10.1093/embo-reports/kvd052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S., Hasebe M., Tomita M., and Yanagawa H.. 2009. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. USA. 106:10171–10176. 10.1073/pnas.0900604106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse K.B., Brodsky J.L., and McCracken A.A.. 2006. Autophagy: an ER protein quality control process. Autophagy. 2:135–137. 10.4161/auto.2.2.2388 [DOI] [PubMed] [Google Scholar]
- Labbadia J., and Morimoto R.I.. 2015. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84:435–464. 10.1146/annurev-biochem-060614-033955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauwers E., Erpapazoglou Z., Haguenauer-Tsapis R., and André B.. 2010. The ubiquitin code of yeast permease trafficking. Trends Cell Biol. 20:196–204. 10.1016/j.tcb.2010.01.004 [DOI] [PubMed] [Google Scholar]
- Lewis M.J., and Pelham H.R.. 2009. Inefficient quality control of thermosensitive proteins on the plasma membrane. PLoS One. 4:e5038 10.1371/journal.pone.0005038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinovska L., Kroschwald S., Munder M.C., Richter D., and Alberti S.. 2012. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spatiotemporal distribution of protein aggregates. Mol. Biol. Cell. 23:3041–3056. 10.1091/mbc.E12-03-0194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta-Camacho E., Kozlov G., Li F.F., and Gehring K.. 2010. Structural basis of substrate recognition and specificity in the N-end rule pathway. Nat. Struct. Mol. Biol. 17:1182–1187. 10.1038/nsmb.1894 [DOI] [PubMed] [Google Scholar]
- Maurer M.J., Spear E.D., Yu A.T., Lee E.J., Shahzad S., and Michaelis S.. 2016. Degradation Signals for Ubiquitin-Proteasome Dependent Cytosolic Protein Quality Control (CytoQC) in Yeast. G3 (Bethesda). 6:1853–1866. 10.1534/g3.116.027953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister L., and Holland M.J.. 1985. Differential expression of the three yeast glyceraldehyde-3-phosphate dehydrogenase genes. J. Biol. Chem. 260:15019–15027. [PubMed] [Google Scholar]
- McCaffrey K., and Braakman I.. 2016. Protein quality control at the endoplasmic reticulum. Essays Biochem. 60:227–235. 10.1042/EBC20160003 [DOI] [PubMed] [Google Scholar]
- McClellan A.J., Scott M.D., and Frydman J.. 2005. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell. 121:739–748. 10.1016/j.cell.2005.03.024 [DOI] [PubMed] [Google Scholar]
- Metzger M.B., Maurer M.J., Dancy B.M., and Michaelis S.. 2008. Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J. Biol. Chem. 283:32302–32316. 10.1074/jbc.M806424200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S.B., Ho C.T., Winkler J., Khokhrina M., Neuner A., Mohamed M.Y., Guilbride D.L., Richter K., Lisby M., Schiebel E., et al. 2015. Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 34:778–797. 10.15252/embj.201489524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert P., and Strahl S.. 2016. Protein O-mannosylation in the early secretory pathway. Curr. Opin. Cell Biol. 41:100–108. 10.1016/j.ceb.2016.04.010 [DOI] [PubMed] [Google Scholar]
- Nillegoda N.B., Theodoraki M.A., Mandal A.K., Mayo K.J., Ren H.Y., Sultana R., Wu K., Johnson J., Cyr D.M., and Caplan A.J.. 2010. Ubr1 and Ubr2 function in a quality control pathway for degradation of unfolded cytosolic proteins. Mol. Biol. Cell. 21:2102–2116. 10.1091/mbc.E10-02-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.H., Bolender N., Eisele F., Kostova Z., Takeuchi J., Coffino P., and Wolf D.H.. 2007. The cytoplasmic Hsp70 chaperone machinery subjects misfolded and endoplasmic reticulum import-incompetent proteins to degradation via the ubiquitin-proteasome system. Mol. Biol. Cell. 18:153–165. 10.1091/mbc.E06-04-0338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.H., Kukushkin Y., Gupta R., Chen T., Konagai A., Hipp M.S., Hayer-Hartl M., and Hartl F.U.. 2013. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 154:134–145. 10.1016/j.cell.2013.06.003 [DOI] [PubMed] [Google Scholar]
- Pearce M.M., and Kopito R.R.. 2018. Prion-Like Characteristics of Polyglutamine-Containing Proteins. Cold Spring Harb. Perspect. Med. 8:a024257 10.1101/cshperspect.a024257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R., Kawaguchi S., and Ng D.T.. 2010. A nucleus-based quality control mechanism for cytosolic proteins. Mol. Biol. Cell. 21:2117–2127. 10.1091/mbc.E10-02-0111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R., Kawaguchi S., and Ng D.T.. 2012. Biosynthetic mode can determine the mechanism of protein quality control. Biochem. Biophys. Res. Commun. 425:689–695. 10.1016/j.bbrc.2012.07.080 [DOI] [PubMed] [Google Scholar]
- Preston G.M., and Brodsky J.L.. 2017. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 474:445–469. 10.1042/BCJ20160582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum J.C., Fredrickson E.K., Oeser M.L., Garrett-Engele C.M., Locke M.N., Richardson L.A., Nelson Z.W., Hetrick E.D., Milac T.I., Gottschling D.E., and Gardner R.G.. 2011. Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol. Cell. 41:93–106. 10.1016/j.molcel.2010.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J., and Zuber C.. 2017. Quality control of glycoprotein folding and ERAD: the role of N-glycan handling, EDEM1 and OS-9. Histochem. Cell Biol. 147:269–284. 10.1007/s00418-016-1513-9 [DOI] [PubMed] [Google Scholar]
- Rotin D., and Kumar S.. 2009. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10:398–409. 10.1038/nrm2690 [DOI] [PubMed] [Google Scholar]
- Russell S.J., Steger K.A., and Johnston S.A.. 1999. Subcellular localization, stoichiometry, and protein levels of 26 S proteasome subunits in yeast. J. Biol. Chem. 274:21943–21952. 10.1074/jbc.274.31.21943 [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.M., and Maniatis T.. 1989. Molecular Cloning: a laboratory manual. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY. 1626 pp. [Google Scholar]
- Sawano A., and Miyawaki A.. 2000. Directed evolution of green fluorescent protein by a new versatile PCR strategy for site-directed and semi-random mutagenesis. Nucleic Acids Res. 28:e78 10.1093/nar/28.16.e78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scior A., Juenemann K., and Kirstein J.. 2016. Cellular strategies to cope with protein aggregation. Essays Biochem. 60:153–161. 10.1042/EBC20160002 [DOI] [PubMed] [Google Scholar]
- Shemorry A., Hwang C.S., and Varshavsky A.. 2013. Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol. Cell. 50:540–551. 10.1016/j.molcel.2013.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiber A., Breuer W., Brandeis M., and Ravid T.. 2013. Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when Hsp70 chaperone levels are limiting. Mol. Biol. Cell. 24:2076–2087. 10.1091/mbc.E13-01-0010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A., Besser S., Hottmann H., and Wolf D.H.. 2013. Previously unknown role for the ubiquitin ligase Ubr1 in endoplasmic reticulum-associated protein degradation. Proc. Natl. Acad. Sci. USA. 110:15271–15276. 10.1073/pnas.1304928110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers D.W., Wolfe K.J., Ren H.Y., and Cyr D.M.. 2013. The Type II Hsp40 Sis1 cooperates with Hsp70 and the E3 ligase Ubr1 to promote degradation of terminally misfolded cytosolic protein. PLoS One. 8:e52099 10.1371/journal.pone.0052099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson R., Locher M., and Hochstrasser M.. 2001. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 15:2660–2674. 10.1101/gad.933301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. 2011. The N-end rule pathway and regulation by proteolysis. Protein Sci. 20:1298–1345. 10.1002/pro.666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S., and Ng D.T.. 2004. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 165:41–52. 10.1083/jcb.200309132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voos W., Jaworek W., Wilkening A., and Bruderek M.. 2016. Protein quality control at the mitochondrion. Essays Biochem. 60:213–225. 10.1042/EBC20160009 [DOI] [PubMed] [Google Scholar]
- Wang S., Thibault G., and Ng D.T.. 2011. Routing misfolded proteins through the multivesicular body (MVB) pathway protects against proteotoxicity. J. Biol. Chem. 286:29376–29387. 10.1074/jbc.M111.233346 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.