

Wang et al. review the dual role of superoxide dismutases in controlling reactive oxygen species (ROS) damage and regulating ROS signaling across model systems as well as their involvement in human diseases.
Abstract
Superoxide dismutases (SODs) are universal enzymes of organisms that live in the presence of oxygen. They catalyze the conversion of superoxide into oxygen and hydrogen peroxide. Superoxide anions are the intended product of dedicated signaling enzymes as well as the byproduct of several metabolic processes including mitochondrial respiration. Through their activity, SOD enzymes control the levels of a variety of reactive oxygen species (ROS) and reactive nitrogen species, thus both limiting the potential toxicity of these molecules and controlling broad aspects of cellular life that are regulated by their signaling functions. All aerobic organisms have multiple SOD proteins targeted to different cellular and subcellular locations, reflecting the slow diffusion and multiple sources of their substrate superoxide. This compartmentalization also points to the need for fine local control of ROS signaling and to the possibility for ROS to signal between compartments. In this review, we discuss studies in model organisms and humans, which reveal the dual roles of SOD enzymes in controlling damage and regulating signaling.
The first superoxide dismutase (SOD) was discovered half a century ago (McCord and Fridovich, 1969). It was subsequently well established that SODs are the first line of defense against oxygen free radicals, and the vast majority of organisms that live in the presence of oxygen express at least one SOD. Three classes of SOD have evolved in various organisms possessing different catalytic metal ions: Cu/Zn SODs, Mn SOD/Fe SODs, and Ni SODs (Table S1; Levanon et al., 1985; Campbell et al., 1986; Chang et al., 1988; Wan et al., 1994; Jones et al., 1995; Strålin et al., 1995; Duttaroy et al., 1997; Folz et al., 1997; Antonyuk et al., 2009; Jung et al., 2011; Blackney et al., 2014). In addition to requirements for metal ion cofactors, SOD enzymes also have distinct subcellular localizations. Eukaryotes only express Cu/Zn SODs (in the cytoplasm and extracellularly) and Mn SODs (in the mitochondria; Miller, 2012).
Chemically, the dismutase activity of SODs accelerates the reaction of the superoxide anion (O2•−) with itself to form hydrogen peroxide (H2O2) and oxygen (2O2•−+2H+ → H2O2+O2; Fridovich, 1997). Superoxide is a negatively charged free radical formed through a single electron donation to oxygen (Hayyan et al., 2016). It is only moderately reactive by itself (Winterbourn, 2008), but it participates in several reactions yielding a variety of reactive oxygen species (ROS) and reactive nitrogen species (RNS) such as H2O2 and peroxynitrite (ONOO−), from which many additional secondary radical species can be generated (Fig. 1; Stamler et al., 1992; Beckman and Koppenol, 1996; Fridovich, 1997). By controlling O2•−, SODs also control the concentrations of these species. The SOD-catalyzed dismutation reaction is extremely efficient, occurring at the almost diffusion-limited rate of ∼2 × 109 M-1·s-1, which is ∼104 times the rate constant for spontaneous dismutation (Fridovich, 1975).
Figure 1.
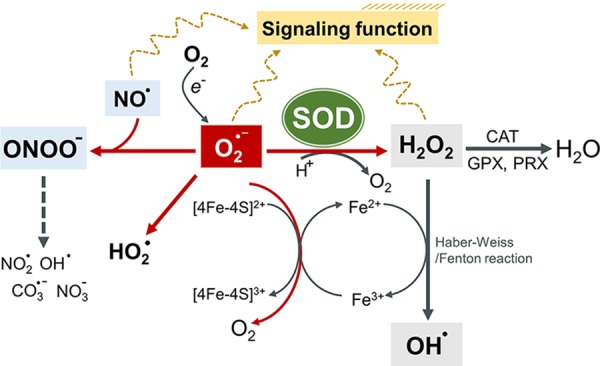
Reactions and transformations of the superoxide anion. SOD enzymes catalyze the dismutation of superoxide (O2•-), generating hydrogen peroxide (H2O2). The catalase (CAT), glutathione peroxidases (GPXs), and PRXs convert H2O2 into water. H2O2 can react with redox-active metals (e.g., iron) to generate the hydroxy radical (OH•) through the Fenton/Haber-Weiss reaction. The reaction between O2•- and nitric oxide (NO•) produces ONOO−, whose decomposition in turn gives rise to some highly oxidizing intermediates including NO2•, OH•, and CO3•- as well as, ultimately, stable NO3−. Therefore, raised O2•- levels can also decrease NO• bioavailability and generate ONOO− toxicity. O2•- by itself can reduce ferric iron (Fe3+) to ferrous iron (Fe2+) in iron–sulfur centers of proteins, leading to enzyme inactivation and concomitant loss of Fe2+ from the enzymes, which in turn fuels Fenton chemistry. The protonation of O2•- can form the more reactive hydroperoxyl radical (HO2•).
In respiring organisms, many spontaneous and enzymatically catalyzed reactions can give rise to O2•− (Fig. 2). These include the mitochondrial electron transport chain (ETC), the plasma membrane–associated NADPH oxidase complex (NOX), the cytosolic xanthine oxidase, and the cytochrome p450 monooxygenases, which are present mainly in the ER (Holmström and Finkel, 2014). Despite their potential toxicity, O2•− and some of its derivatives, especially H2O2, are also signaling molecules that mediate a variety of biological responses such as cell proliferation, differentiation, and migration (Holmström and Finkel, 2014). Furthermore, evidence is presented that burst production of ROS such as O2•− and/or H2O2 is an important component of the pathogen defense mechanism (Cross and Segal, 2004; Ha et al., 2005; Chávez et al., 2007). Superoxide does not readily pass though cell membranes and is relatively short lived; thus, it presumably acts where it is produced. In contrast, H2O2 is uncharged, more stable, and can traverse membranes freely, making it a more versatile signaling molecule (Fig. 2; Cardoso et al., 2012; Holmström and Finkel, 2014). The presence of specific SOD isoforms in distinct subcellular compartments highlights the need for a tight control of ROS homeostasis and suggests a role for ROS in signaling between compartments. For example, changes in SOD activity in a particular compartment could lead to production of an H2O2 concentration gradient, leading to an H2O2 flux and subsequently activation of particular redox-sensitive pathways. In this review, we will discuss the functions of SODs by focusing principally on findings arising from the study of SOD mutants in model organisms.
Figure 2.

SOD-dependent ROS signaling in mammalian cells. In aerobic organisms, many processes produce O2•-, including cytosolic xanthine oxidase (OX), the cytochrome P450-monooxygenases (CYP) in the ER, the mitochondrial ETC, and NADPH oxidase (NOX). NOX is a membrane-bound enzyme complex that can be found in the plasma membrane as well as within intracellular membrane structures or vesicles (Meitzler et al., 2014). O2•- produced by the plasma membrane–bound NOX (e.g., NOX2) can act both intra- and extracellularly. H2O2 produced by SOD3 outside the cell can transverse into the cell interior in part through aquaporin channels to initiate intracellular signaling, whereas O2•- could influx through the chloride channel-3 (Fisher, 2009). The intracellular NOX complexes produce ROS in the lumen of a vesicular compartment, where ROS acts locally or from which it enters the cytosol (Brown and Griendling, 2009). H2O2 has been implicated in ROS signaling through oxidative modification of critical redox-sensitive cysteines in signaling proteins. The relatively well-recognized targets of ROS signaling include protein phosphatases (PTPs), nonreceptor protein tyrosine kinases (PTKs), protein kinase C (PKC), mitogen-activated protein kinases (MAPKs), and transcriptional factors (TFs). The signaling function of O2•- is yet largely uncharacterized. In C. elegans, it was shown that mitochondrial ROS act by signaling in part through the intrinsic apoptotic pathway, likely via H2O2, triggering processes that promote longevity (Yee et al., 2014). Compartmentalization of different forms of SOD provides an important mechanism for fine spatial control of ROS homeostasis and signaling, whose exact significance remains to be understood. CAT, catalase; GPX, glutathione peroxidase.
Limiting simple chemical ROS toxicity
Although O2•− is generally not considered to be a strongly oxidizing agent, it is still potentially toxic. In particular, O2•− can oxidize iron–sulfur clusters in proteins (e.g., the Krebs cycle and cytosolic enzymes aconitase), resulting in cluster degradation and loss of the catalytic iron and hence enzymatic activity (Liochev and Fridovich, 1999; Imlay, 2003). Furthermore, the iron atom released in this process can directly reduce H2O2 to generate the highly toxic hydroxyl radical (OH•; Fig. 1; Liochev and Fridovich, 1999). In organisms ranging from bacteria to mice, loss of SOD activity is associated with increased levels of oxidative damage such as membrane lipid peroxidation, protein carbonylation, and DNA breakage (Dukan and Nyström, 1999; Van Remmen et al., 2003; Elchuri et al., 2005; Yang et al., 2007). This indicates that insufficient removal of endogenously generated superoxide is indeed deleterious. In addition to the toxicity of superoxide itself, this is likely because of the generation of ONOO− when O2•− reacts with nitric oxide (NO•; Pacher et al., 2007). Decomposition of ONOO− also results in the formation of some very reactive species such as OH•, NO2•, and CO3•- (Tharmalingam et al., 2017). The SODs are the only enzymes that can prevent ONOO− formation by removing O2•−. In fact, ONOO− accumulation resulting from inefficient O2•− removal in the mitochondria can significantly impair mitochondrial function and could thus contribute to the early lethality of Sod2 null mice (Brown and Borutaite, 2007) as will be discussed.
Cysteine-mediated redox signaling
Besides potentially evoking oxidative and nitrosative stress, the loss of SODs might also be toxic by perturbing ROS signaling by lowering H2O2 concentration. Indeed, the best-known mechanism of how ROS act as signaling molecules involves H2O2-mediated oxidation of key cysteines within proteins that possess regulatory functions (Finkel, 2001). The thiol (RSH) groups of cysteines, including those at active sites, can exist in different redox states (Paulsen and Carroll, 2010, 2013). Oxidation of the reactive cysteine residues can result in a wide range of posttranslational modifications (e.g., S-glutathiolation, sulfenic acid, and disulfide formation), which can produce structural changes that are sufficient to alter protein function, stability, and subcellular localization (Rhee, 2006). Also, importantly, the reactivity of any cysteine thiol group is greatly influenced by the protein context, meaning that some cysteine residues are more susceptible to oxidation than others, which provides a molecular basis for selectivity and specificity in redox signaling (Marino and Gladyshev, 2012; Cremers and Jakob, 2013; Marinho et al., 2014). Like cysteines, methionines also contain reactive sulfur atoms that are susceptible to reversible redox modifications and thus may also mediate redox signaling (Drazic and Winter, 2014).
One widely studied class of targets of redox regulation is the protein tyrosine phosphatase (PTPs) family. Acting in concert with receptor tyrosine kinases, PTPs play a regulatory role in many cellular functions such as responses to growth factor signaling (Trachootham et al., 2008). The active site of PTPs contains an essential cysteine residue that is in the thiolate anion form (Cys-S−) at neutral pH and is therefore highly susceptible to oxidation (Tonks, 2005; Rhee, 2006). Oxidation of the active-site cysteine leads to PTP inactivation, which is generally reversible and thus transient (den Hertog et al., 2005; Rhee, 2006).
H2O2 has also been shown to play a role in phosphorylation-dependent signaling via activating intracellular tyrosine kinases. In this case, specific oxidation of the cysteine sulfhydryl (SH) group induces conformational changes that lead to kinase activation. For example, the first discovered oncogene Src, which is such a nonreceptor tyrosine kinase, is well demonstrated to be redox sensitive (Giannoni et al., 2005; Corcoran and Cotter, 2013). Furthermore, serine/threonine-specific protein kinases MAPK and Akt, both important effectors of growth factor receptor signaling pathways, have been shown to undergo direct redox activation (Nakashima et al., 2002; Corcoran and Cotter, 2013). Finally, transcriptional factors constitute an important class of ROS-responsive proteins. Those include NF-κB, AP-1, H1F-1a, p53, and upstream stimulatory factor (USF), with active sites that contain low-pKa cysteine residues, whose oxidative modifications cause either their activation (e.g., p53, HIF-1, HSF1, and NRF2) or inactivation (e.g., USF; Fig. 2; Ahn and Thiele, 2003; Brandes et al., 2009).
Oxidation of cysteine thiols by H2O2 is a two-electron reaction that leads to the production of sulfenic acids, whereas one-electron oxidation by superoxide and the OH• leads to thiyl radicals (Trujillo et al., 2016). It is postulated that cysteine thiyl radicals can undergo intramolecular hydrogen transfer reactions with the surrounding amino acids, causing protein damage (Schöneich, 2008). They are also a potential source for S-nitrosylation, S-glutathiolation, and sulfenic acid production regulation. But little is known about the in vivo role of cysteine thiyl radicals in redox signaling (Schöneich, 2011).
Although the removal of H2O2 is highly regulated by numerous enzymatic systems (e.g., catalase, glutathione peroxidases, and peroxiredoxins [Prxs]), this might be for the purpose of regulating its signaling activity rather than its potential toxicity. This is suggested by the observation that there is little phenotypic effect of the loss of catalase in mice. Catalase is normally found at high concentrations in peroxisomes, where H2O2 is mainly produced by the β oxidation of fatty acids and might have no signaling role (Schrader and Fahimi, 2006).
Additional aspects of the diverse role of SODs in the regulation of redox signaling
Among ROS, superoxide and H2O2 have the most favorable properties to operate as signaling molecules. However, H2O2 has been given the most attention. First, it is relatively stable and can cross through cellular membranes, implicating the ability to deliver a redox signal to distant targets (Bienert et al., 2006). Second, its enzymatic production and removal potentially provides site and time specificity for its action (Forman et al., 2010; Finkel, 2011; Marinho et al., 2014). There are also some practical reasons for why H2O2 is best understood. For example, it is very difficult to target O2•− specifically (e.g., manipulations of O2•− levels will also affect H2O2), but many more tools are available to modify steady-state levels of H2O2, such as exposure of culture cells to H2O2 directly or treatment with the antioxidant N-acetyl cysteine, which impacts mainly H2O2. Note that superoxide production can also be tightly regulated, such as for example its production by NADPH oxidases (Bedard and Krause, 2007). If, as some research suggests, ROS signaling is largely elicited by a transient burst of ROS generation (part of the mechanisms conferring signal specificity; Terada, 2006), O2•− should be able to oxidize thiols on regulatory proteins in the immediate vicinity of its point of generation despite its relative instability and poor diffusibility (Cardoso et al., 2012). Still, relatively little is known about O2•− signaling (Forman et al., 2010). Interestingly, it has been shown that O2•− participates in major epigenetic processes such as DNA methylation, histone methylation, and histone acetylation (Afanas’ev, 2015). O2•− has also been shown to regulate autophagy (Chen et al., 2009). Cells lacking functional ETC showed reduced autophagy during starvation, indicating ROS of mitochondrial origin playing a major role (Li et al., 2013).
The study of SODs helps clarify the signaling roles of superoxide and H2O2. For example, the SOD1 inhibitor tetrathiomolybdate (ATN-224) protects PTPs from oxidation, and this, in turn, attenuates growth factor–mediated phosphorylation of ERK1/2 in endothelial and tumor cells (Juarez et al., 2008). The implication is that it is H2O2 rather than superoxide that mediates these effects (Juarez et al., 2008). Of note, however, is that ATN-224 was shown to increase O2•− but also elevates intracellular H2O2 levels, which was interpreted to suggest an unconventional function of SOD1, though this remains to be fully elucidated (Glasauer et al., 2014). Overexpression of human SOD1 in mouse NIH 3T3 fibroblasts enhances vascular endothelial growth factor expression, which can be suppressed by overexpression of human catalase, again pointing to a role of H2O2 in these phenomena (Marinho et al., 2014). Similarly, SOD2 overexpression in human fibrosarcoma mitochondria leads to phosphatase and tensin homolog (PTEN) inactivation and subsequent activation of PI3K-Akt signaling. This can be inhibited by overexpression of catalase in the cytosol or mitochondria (Connor et al., 2005).
How closely is ROS signaling controlled by SODs? There is consensus that ROS fulfills its signaling function at low or moderate levels. One mechanism that has been proposed for how ROS signaling specificity is achieved is confinement of the sources of ROS, such as SODs and NOXs, close to their intended targets (Mittler et al., 2011). Several lines of evidence further suggest that NOX ROS production may be localized proximal to signal proteins (e.g., PTPs), and such spatial restriction of ROS signal is required for effective oxidation of specific redox-sensitive proteins (Ushio-Fukai, 2006; Chen et al., 2008). Thus, the compartmentalization of SODs is likely critically important for spatial control of ROS-based signaling. Altered SOD expression or activity changes are observed in various pathological conditions such as, for example, cancer and amyotropic lateral sclerosis (ALS; Dhar and St Clair, 2012; Robberecht and Philips, 2013). These changes may not only perturb protection from potential ROS toxicity but also misregulate many redox-sensitive pathways that contribute to disease outcome.
Finally, it is noteworthy that at neutral pH, the rate constant for thiol oxidation by superoxide (<103 M−1 s−1) is far lower than the rate constant for SODs (>109 M−1s−1; Forman et al., 2010). Similarly, the redox-active thiols in some signaling proteins like the phosphatases Cdc25B and PTB1B were found to have a lower reactivity toward H2O2 (i.e., the rate of oxidation by H2O2) than H2O2 scavengers, especially Prxs (Winterbourn, 2008; Marinho et al., 2014). The kinetic disadvantage of protein thiol oxidation appears difficult to reconcile with them being a direct sensor and target of ROS signal. Explaining how poorly reactive targets outcompete ROS scavengers in reacting with ROS, there are currently two main schools of thought. The floodgate hypothesis posits that oxidation of redox-regulated target proteins requires the inactivation of Prxs, which allows for local accumulation of a sufficiently high concentration of H2O2, resulting in a temporal oxidation of target thiols of signaling pathways. The redox relay model proposes that thiol peroxidases, in particular Prxs, are the initial recipients of H2O2 and subsequently transfer redox equivalents to the end targets (Reczek and Chandel, 2015; Stöcker et al., 2018). Clarification of how each antioxidant system and their subcellular localization play into the mechanisms of ROS signaling is fundamental to better understanding ROS signaling and modulation of ROS-dependent signaling pathways.
Lastly, although this review focuses on animals, a study in yeast reveals how highly specifically SODs can act in signaling (Reddi and Culotta, 2013). It was shown that active SOD1 enzyme is required for glucose repression of respiration by binding to a C-terminal degron of Yck1p/Yck2p (two casein kinase 1-γ [CK1γ] homologues). This binding results in a very localized rise in H2O2 levels, which stabilizes the CK1γ kinases, whose activity ultimately results in respiratory repression (Reddi and Culotta, 2013).
Role of SODs in RNS control and signaling
Like ROS, RNS, including NO•, NO2, ONOO−, and others, are reactive molecules that are both functionally important and potentially dangerous. NO• is the main RNS and the primary source of all other RNS (Adams et al., 2015). Superoxide can directly react with NO• to form ONOO− (Fig. 1). At physiological pH, the direct biradical reaction of NO• with O2•− proceeds approximately three times faster than the decomposition of O2•− by SODs (Beckman and Koppenol, 1996), which might be one of the reasons why SODs (especially SOD1, which acts in the largest compartment, comprising cytoplasm and mitochondrial intermembrane space) are so abundant (Chang et al., 1988; Banci et al., 2013). Chemically, NO• is not intrinsically more reactive than oxygen, nor is it highly toxic by itself. However, ONOO− is both a strong oxidant and a nitrating agent toward a wide range of macromolecules. Moreover, it is considered to be relatively stable under physiological conditions and can diffuse across several cell diameters, making it far more toxic (Szabó et al., 2007).
NO• is an important signaling molecule modulating diverse physiological processes such as vasodilation, neurotransmission, and the immune response. Thus, by controlling the level of O2•− available to react with NO•, SODs also preserve the physiological functions of NO•. For example, in the blood vessels of mammals, NO• produced by endothelial cells is a vasodilator of the underlying smooth muscles (Zhao et al., 2015). It diffuses from the endothelium into the subjacent vascular smooth muscle cells, where it activates guanylate cyclase to increase synthesis of cyclic guanosine monophosphate. This in turn activates cyclic guanosine monophosphate–dependent protein kinases, leading to muscle relaxation. SOD3, the extracellular matrix SOD isoform, inhibits inactivation of vascular NO• by scavenging O2•−, and thus it plays an essential role in maintaining proper vascular tone and blood pressure (Fukai et al., 2002).
The biology of SOD in model animals
Several genetic tools have been created in model organisms that have allowed for the study of the roles of SOD function and ROS metabolism in vivo. In this section, we discuss the findings in three major model organisms: Caenorhabditis elegans, Drosophila melanogaster, and mouse.
C. elegans
In C. elegans, five genes encode SODs: sod-1 to sod-5. SOD-1, SOD-4, and SOD-5 are Cu/Zn SODs, whereas SOD-2 and SOD-3 are Mn SODs (Table S1; Jensen and Culotta, 2005; Yang et al., 2007; Doonan et al., 2008). SOD-1 itself contributes almost 80% of the total sod mRNA expression as well as 80% of the total SOD activity in C. elegans (Doonan et al., 2008). The fact that control of superoxide and peroxide levels is spread over five different SODs with different properties, subcellular distributions, and possibly tissue distributions makes C. elegans a particularly good model to explore the variety of biological roles of the SODs.
Cellular and subcellular patterns of expression of SODs in C. elegans
SOD-1 and SOD-5 are predicted to be intracellular cytoplasmic Cu/Zn SODs (Larsen, 1993; Jensen and Culotta, 2005). SOD-4 is predicted to be an extracellular membrane–bound Cu/Zn SOD, which exists in two different forms arising from alternative splicing, although neither form has been directly detected (Fujii et al., 1998). SOD-1 appears expressed in the cytoplasm of most cells of the worm (Doonan et al., 2008; Yanase et al., 2009). SOD-5 expression has been detected in the cytoplasm of a small subset of neurons (Doonan et al., 2008). However, given that sod-5 is an inducible sod (see below) this may not reflect the complete pattern of expression (Doonan et al., 2008).
SOD-2 and SOD-3 are very similar in amino acid sequence and are predicted to be mitochondrial Mn SODs (Hunter et al., 1997; Henderson et al., 2006; Doonan et al., 2008; Honda et al., 2008). Although the two proteins may be coexpressed in the same tissue, it is unclear whether they are colocalized to the same cells, and if so, to the same mitochondria (Henderson et al., 2006; Doonan et al., 2008). Furthermore, the interpretation of the expression of sod-3 is complicated by the fact that sod-3 is inducible. Interestingly, SOD-2 and SOD-3 have been reported to localize in mitochondria at the site of the supercomplex I:III:IV of the ETC. This raises the possibility that these SODs could participate in detoxifying O2•− right at the site where it is produced (Suthammarak et al., 2013).
Induction of sod genes in C. elegans
The expression of both of the two major sod genes, sod-1 and sod-2, has been found to be mildly up-regulated by oxidative stress (Tawe et al., 1998; Yang et al., 2007; Meng et al., 2017) and possibly by the loss of other sod genes (Van Raamsdonk and Hekimi, 2009; Yanase et al., 2009; Back et al., 2010). However, the findings about sod-1 and sod-2 up-regulation are conflicting, and the conditions under which the studies were done vary greatly. For example, to induce oxidative stress, the prooxidant paraquat (PQ) has been used at concentrations ranging from 0.1 mM to 100 mM. In WT C. elegans, there is little to no expression of sod-3 and sod-5 under normal conditions (Honda and Honda, 1999; Doonan et al., 2008). They are thus unusual SOD isoforms in that they are strictly inducible. Many environmental conditions, in particular stresses, and genetic mutations have been shown to induce sod-3 transcription (Feng et al., 2001; Honda and Honda, 2001; Yanase et al., 2002; Henderson et al., 2006; Wolf et al., 2008; Dingley et al., 2010; González-Cabo et al., 2011; Erkut et al., 2013; Suetomi et al., 2013; Suthammarak et al., 2013; Song et al., 2014; Oh et al., 2015; Rathor et al., 2015; Wu et al., 2016). However, the importance of sod-3 up-regulation for resistance to most of these stresses has not been established (see text box for discussion of aging). One notable exception is resistance to Enterococcus faecalis, which requires sod-3 (Chávez et al., 2007). sod-5, which is also induced by several conditions, has been studied less extensively (Doonan et al., 2008; Erkut et al., 2013; Suetomi et al., 2013; Song et al., 2014).
SODs and longevity in worms and flies
The Free Radical Theory of Aging, also known as the Oxidative Stress Theory, proposes that free radicals including ROS are toxic molecules that produce oxidative damage to cellular constituents (Harman, 1956; Sohal and Weindruch, 1996). Over time, the accumulation of this oxidative damage contributes to or may even cause aging. Given this proposed link between ROS and aging as well as the direct role SODs play in regulating ROS, it is not surprising that the phenotype that has been most studied for the sod genes in both C. elegans and Drosophila is aging. In C. elegans, most studies have focused on the effects of sod knockout mutants. Aging phenotypes have only been reported for sod-1 and sod-2, and only a subset of studies that examined their lifespan found any effect. sod-1 mutants were found to be slightly short lived (Doonan et al., 2008; Yanase et al., 2009), whereas surprisingly, sod-2 mutants have been found to be long lived (Van Raamsdonk and Hekimi, 2009; Dingley et al., 2010; Yang and Hekimi, 2010).
Several groups have also combined sod mutations with other long-lived mutants and examined the effects on lifespan. The increase in sod levels reported for many long-lived mutants has been shown repeatedly not to be necessary for their longevity (Yang et al., 2007). This is especially striking for the case of sod-3 and daf-2. Indeed, sod-3 is strongly up-regulated in daf-2 mutants (Honda and Honda, 1999), but knocking down sod-3 does not suppress daf-2 longevity (Henderson et al., 2006; Doonan et al., 2008); in fact, it can actually lengthen it (Honda et al., 2008).
Another key finding is that increased sensitivity to ROS does not shorten lifespan. RNAi knockdown of sod-2 leads to a measurable increase in ROS sensitivity but increases lifespan (Yang et al., 2007). Overexpression of sod-1, against expectation, can also lead to ROS hypersensitivity and an extended lifespan (Doonan et al., 2008). Most strikingly, the quintuple sod knockout, which is hypersensitive to ROS as well as to other stressors, has a normal lifespan (Van Raamsdonk and Hekimi, 2012). A lack of correlation has also been observed by combining sod mutations with other mutations that affect lifespan (for examples, see Schaar et al. [2015] and Dues et al. [2017]).
In Drosophila, mutation of any of the Sods leads to ROS sensitivity and a shortened lifespan, so there has been a major effort by several laboratories to determine whether overexpression of SODs could be beneficial for lifespan. Indeed, lifespan-lengthening effects were observed in several overexpression models. For example, using P-element–mediated introduction of DNA, transgenic flies overexpressing both Sod1 and catalase were observed to have an increased lifespan and reduced oxidative damage, whereas overexpression of either enzyme alone had no effect (Orr and Sohal, 1994). Using a different transgenic system that used FLP recombinase to mediate overexpression of transgenes, it was observed that overexpression of either Sod1 or catalase conferred protection to H2O2, but only Sod1 overexpression could increase lifespan in some lines (Sun and Tower, 1999). Another study used the GAL4-UAS system to overexpress human SOD1 specifically in motor neurons and observed both an increased protection from oxidative stress and an extension of lifespan (Parkes et al., 1998). Overexpression of Sod2 has also been found to increase lifespan (Sun et al., 2002). However, for every study that found an effect on lifespan, there appears to be an equal number of studies that found no effect, even in cases where an increase in oxidative stress resistance was observed (for examples, see (Seto et al. [1990], Orr et al. [2003], Bayne et al. [2005], and Mockett et al. [2010]). Thus, it appears that increasing SOD activity can increase stress resistance and lifespan in some experimental contexts, but it is not sufficient. Other experimental and/or genetic factors must also be necessary to achieve lifespan extension.
Collectively, studies of the SODs in both C. elegans and Drosophila do not lend support to the oxidative stress theory of aging. Indeed, measurable increases in ROS do not necessarily decrease lifespan and can even lengthen it. In fact, in C. elegans, conditions of increased ROS generation such as ETC dysfunction, loss of sod-2, PQ treatment, and reduced glucose metabolism were shown to promote longevity (Schulz et al., 2007; Durieux et al., 2011; Wang and Hekimi, 2015). The elements that were demonstrated to be necessary for the longevity phenotypes include the intrinsic apoptosis signaling pathway, which is stimulated by ROS, and the mitochondrial unfolded protein response, which is required for the survival of worms with damaged mitochondria (Houtkooper et al., 2013; Yee et al., 2014). Whether some of the effects on aging could be attributed to phenomena akin to mitohormesis (Ristow, 2014) could not be discussed because of space constraints.
ROS sensitivity of C. elegans sod mutants
Several studies have examined the ROS sensitivity of sod mutants. The consensus seems to be that loss of the major sod genes sod-1 or sod-2 leads to hypersensitivity to oxidative stress. For example, sod-1 mutants are hypersensitive to 0.1 M PQ (Yanase et al., 2009), RNAi knockdown of either sod-1 or sod-2 leads to hypersensitivity to 4 mM PQ (Yang et al., 2007), and knockouts of either sod-1 or sod-2 (but not other sod genes) are sensitive to 40 mM PQ (Doonan et al., 2008). Similarly, knockout of either sod-1 or sod-2 leads to sensitivity to both 4 mM PQ and 240 µM juglone (Van Raamsdonk and Hekimi, 2009). Finally, quintuple knockout mutant animals lacking all the sods are extremely hypersensitive to PQ and juglone as well as to other stresses such as osmotic stress, extreme heat, and cold (Van Raamsdonk and Hekimi, 2012). In contrast with sod-1 and sod-2, loss of sod-3 leads only to very mild hypersensitivity to PQ, and loss of sod-4 or -5 do not lead to hypersensitivity to either PQ or juglone (Van Raamsdonk and Hekimi, 2009). Given the link between ROS damage and aging, several studies have examined how the sod mutations affect lifespan (see text box).
Roles of SOD isoforms in ROS signaling in C. elegans
Although a role for ROS in signaling has been well established, it has not yet been studied extensively in C. elegans. However, there are some observations from studies of sod genes that are consistent with ROS-related roles in signaling. As described above, sod-3 expression is induced by a variety of stresses. However, the four following considerations suggest that this is not because of an increased need for O2•− detoxification but is more likely part of a stress response signaling pathway: (1) It is unclear that all the conditions that up-regulate sod-3 described above also increase oxidative stress. (2) There is very little evidence that the SOD-3 protein accumulates in amounts that are consistent with a role in detoxification. For example, sod-3 transcripts but not SOD-3 protein levels are up-regulated in daf-2 mutants (Doonan et al., 2008; Dingley et al., 2010). Similarly, the massive up-regulation of sod-3 that is observed in dauer larvae contributes very little or nothing to overall SOD activity (Doonan et al., 2008). (3) Loss of SOD-3 increases the lifespan of the insulin-signaling mutant daf-2 and suppresses the shortened lifespan of daf-2; sod-2 double mutants (Honda et al., 2008). It was also demonstrated that, at least in daf-2 mutants, sod-2 and sod-3 are expressed in fully distinct subsets of cells. They therefore concluded that SOD-3 and SOD-2 are involved in a ROS signal that functions in intercellular communication and that regulates longevity. (4) Despite the fact that neither decreasing (Doonan et al., 2008; Honda et al., 2008; Van Raamsdonk and Hekimi, 2009) nor increasing (Henderson et al., 2006) sod-3 expression has any effect on lifespan, the expression of a sod-3::gfp reporter is reported as being the best predictor of remaining lifespan from a collection of genes whose expression is age dependent (Sánchez-Blanco and Kim, 2011). Possibly, this means that sod-3 is coregulated with other genes that affect lifespan or that it is the combined effect of these coregulated genes that can affect lifespan.
There is also a very interesting study on cell-specific up-regulation of sod-1 (Horspool and Chang, 2017). Horspool and Chang (2017) show that sod-1 expression is specifically induced in the gustatory neuron ASER in response to pathogenic bacteria and that this up-regulation is required to mediate pathogen avoidance. Horspool and Chang (2017) interpret these findings to suggest that pathogen-induced ROS activate a sod-1–dependent pathway to mediate the avoidance behavior. SODs have also been shown to be required in a developmental context: the development of the vulva, which is regulated by the RAS signaling pathway. RAS signaling has been shown to be regulated by ROS in a variety of organisms. In C. elegans, RNAi knockdown of sod-1 or sod-4 down-regulates RAS signaling in a Ras gain-of-function mutant, suggesting that the RAS pathway is down-regulated by O2•− or the absence of H2O2 in the context of vulval development (Shibata et al., 2003).
Drosophila
SOD enzymes in Drosophila and their expression
In Drosophila, there are three genes that encode SOD (Table S1; Landis and Tower, 2005). Sod1 and Sod3 encode Cu/Zn SODs, and Sod2 encodes an Mn SOD. It has been shown experimentally that SOD1 and SOD2 confer protection from oxidative stress specifically in their respective compartments, i.e., the cytosol and the mitochondria (Missirlis et al., 2003). Sod3 is extracellular (Jung et al., 2011). Like for the C. elegans extracellular SOD (SOD-4), there are two isoforms of Sod3 in Drosophila, so that in addition to the extracellular form, there is also predicted to be a form that is membrane associated (Blackney et al., 2014). The Drosophila genome also contains an insect-specific SOD-related gene Sodesque (Sodq), which is related to the extracellular Cu/Zn SOD family but has lost critical residues required for SOD enzymatic activity, as well as a member of another conserved family of proteins related to Cu/Zn SOD, Related to Sod (Rsod), whose function is unknown (Landis and Tower, 2005).
Induction of sod genes in Drosophila
Although there are not strictly inducible Sod genes in Drosophila, up-regulation of SOD activity or expression has been reported in a few contexts. As in C. elegans, SOD activity is up-regulated in long-lived insulin/IGF receptor (INR) mutants as well as in mutants of the insulin receptor substrate CHICO. However, in these cases, it is Cu/Zn SOD activity that is elevated, rather than up-regulation of an Mn SOD (Clancy et al., 2001; Tatar et al., 2001; Kabil et al., 2007). However, the up-regulation in activity does not appear to correlate with an increase in resistance to oxidative stress. It has also been shown that the Cu/Zn SOD Sod3 is up-regulated in a fly model of Aβ-toxicity (Favrin et al., 2013). Surprisingly, knocking down Sod3 by RNAi improved both behavioral phenotypes and survival, suggesting that it may not act simply to reduce oxidative damage, as will be discussed.
There appears to be an interesting interaction between the two Cu/Zn SODs. Half of Cu/Zn SOD activity is lost in homozygous Sod3 mutants, suggesting it contributes about half the Cu/Zn SOD activity. However, all Cu/Zn SOD activity in lost in homozygous Sod1 mutants, suggesting some dependence of Sod3 on Sod1 (Phillips et al., 1989; Blackney et al., 2014). Moreover, in the Sod3KG06029 hypomorph mutant, when Sod3 expression is restored as a result of excision of the KG p-element upstream of the Sod3 coding sequence, higher mRNA expression was observed for all types of Sod, especially Sod1, suggesting that their levels may be coregulated, although it is unclear how or why (Blackney et al., 2014).
ROS sensitivity of Drosophila sod mutants
Sod1 mutants harboring a point mutation show a complete lack of Cu/Zn activity and display hypersensitivity to oxidative stress induced by PQ, increased rates of DNA damage, and a shortened lifespan (Phillips et al., 1989; Rogina and Helfand, 2000; Woodruff et al., 2004). However, a deletion allele is homozygous lethal (Phillips et al., 1995; Blackney et al., 2014). Similarly, RNAi-mediated knockdown of Sod2 leads to increased sensitivity to oxidative stress induced by PQ and a dramatic increase in adult-onset mortality (Kirby et al., 2002). Sod2 heterozygotes are viable and also display hypersensitivity to PQ treatment (Duttaroy et al., 2003). However, mutants homozygous for a null allele of Sod2 suffer from neonatal lethality. Collectively, these results suggest that both Sod1 and Sod2 offer protection from oxidative stress. Similarly, mutation of Sod3 and RNAi knockdown of Sod3 leads to increased sensitivity to PQ and shortened lifespan (Jung et al., 2011). In contrast, Sod3 hypomorphic mutants have normal survival but are less sensitive to H2O2 and PQ. This suggests that at least in some contexts, loss of Sod3 can somehow confer protection (Blackney et al., 2014). Given that mutation of any of the Sods can increase ROS sensitivity and shorten lifespan, several studies have examined whether overexpression of Sod genes could be beneficial for stress resistance and longevity (see text box).
Roles for SOD in Ros signaling in Drosophila
Similar to the situation in C. elegans, there are few studies that have directly addressed the roles of ROS in signaling in vivo in Drosophila. However, some of the phenotypes of the Sod mutants are suggestive of ROS functions in signaling. For example, mutants carrying a particular reduction-of-function missense mutation of Sod2 exhibit increased mitochondrial ROS and sensitivity to hyperoxia as well as behavioral defects, defects in axonal targeting, and neurodegeneration (Celotto et al., 2012). Although some of these phenotypes could be caused by ROS toxicity, it is likely that specific neural phenotypes like impairments of axon targeting arise from aberrant intracellular signaling caused by increased mitochondrial O2•−. Similarly, it is difficult to imagine how loss of the extracellular Sod3 can be protective both in an Aβ toxicity model (Favrin et al., 2013) and to H2O2 and ROS generators like PQ (Blackney et al., 2014) unless it also serves to regulate some cellular functions in response to ROS levels rather than protect from superoxide damage.
Mouse
Mammalian SOD enzymes
There are three known SOD isoforms in mammals (Table S1). The Cu/Zn SOD SOD1 is the cytoplasmic isoform but has also been identified in the nucleus, lysosome, peroxisome, and the mitochondrial intermembrane space (Chang et al., 1988; Crapo et al., 1992; Okado-Matsumoto and Fridovich, 2001). The Mn SOD SOD2 localizes to the mitochondrial matrix (Slot et al., 1986). Although complex I superoxide is released nearly exclusively to the mitochondrial matrix, complex III generates superoxide not only to the matrix but also outward to the inner membrane space, where it becomes a substrate for SOD1 (Murphy, 2009; Holmström and Finkel, 2014). The Cu/Zn SOD SOD3 is secreted into the extracellular space (Fukai et al., 2002). SOD1 is most abundant in most tissues, with a surprisingly high cellular concentration (estimated to be 10–40 µM; Chang et al., 1988; Banci et al., 2013). SOD1 and SOD2 are expressed in practically all cells. High expression of Sod3 was identified in selected tissues, particularly in blood vessels, lung, kidney, and heart (Marklund, 1984; Folz and Crapo, 1994; Strålin et al., 1995; Fattman et al., 2003). SOD3 can also be inducible in other tissues, which we will discuss.
Lethality of Sod2 knockout mice
As valuable tools to study ROS metabolism, an extensive collection of Sod mutant mice has been generated and characterized (Table S2). Loss of a single copy of Sod2 (Sod2+/−) does not produce any overt pathology except for increased incidence of cancer in old age (Li et al., 1995; Lebovitz et al., 1996; Van Remmen et al., 2003). Close examination revealed that Sod2+/− mutants display higher levels of oxidative damage, susceptibility to oxidative stress, inhibition of mitochondrial function, and premature initiation of apoptosis, yet exhibit close-to-normal lifespans (Li et al., 1995; Lebovitz et al., 1996; Kokoszka et al., 2001; Van Remmen et al., 2001, 2003). Along the same lines, several studies showed that increased SOD2 expression is associated with a reduction of mitochondrial O2•− levels, attenuated age-dependent increase of oxidative damage, and better preservation of mitochondrial function especially in old age (Silva et al., 2005; Hu et al., 2007; Lee et al., 2009). Despite this, increased SOD2 expression had little or no effect on lifespan (Hu et al., 2007; Jang et al., 2009).
To study the requirement for SOD2 in vivo, several conditional knockout lines have already been created (Table S2). Among those, conditional knockout of Sod2 in the highly aerobic tissues (e.g., cardiac and skeletal muscles) appears to be most damaging (Nojiri et al., 2006; Izuo et al., 2015). Especially, in contrast with other tissue-specific knockouts of Sod2, loss of Sod2 in the brain is lethal within 3–4 wk after birth, suggesting a particularly high sensitivity of neurons to elevated mitochondrial O2•− (Izuo et al., 2015).
The exact pathophysiological effects and the resulting animal lethality of loss of mitochondrial SOD are not entirely clear, as superoxide is not a particularly reactive species and its toxicity has been most clearly linked to its transformation into other more toxic species rather than by direct oxidative damage by itself (Benov, 2001). Another important and direct target of superoxide is iron–sulfur cluster proteins, which might be particularly relevant for the condition of Sod2 knockout as many of the mitochondrial respiratory chain enzymes (e.g., succinate dehydrogenase) and TCA enzymes (e.g., aconitase) contain iron–sulfur clusters in their active sites. For example, aconitase has long been recognized as a sensitive index of steady-state superoxide, and loss of mitochondrial aconitase activity has been observed in many conditions where excessive superoxide is expected to be present, including Sod2 mutants (Nojiri et al., 2006; Lapointe and Hekimi, 2008; Kuwahara et al., 2010). Finally, reduced NO• bioavailability, in addition to ONOO- accumulation resulting from inefficient superoxide removal in the mitochondria, could significantly impair mitochondrial function (Brown and Borutaite, 2007), contributing at least in part to the early lethality of Sod2 null mice.
Phenotypes associated with loss of Sod1 and Sod3 in mice
In contrast with Sod2−/− mice, homozygous mutations in the Sod1 and Sod3 genes are not lethal but rather appear superficially normal (Pouyet and Carrier, 2010). Sod1−/− mice, as expected, showed extensive oxidative damage in the cytoplasm as well as to a lesser extent in the nucleus and mitochondria (Elchuri et al., 2005). In addition to ∼30% reduction in lifespan, many abnormalities have been reported for Sod1−/− mice including acceleration of age-dependent skeletal muscle atrophy, hearing loss, delayed wound healing, increase in cellular senescence, locomotor defects, denervation, motor axonopathy, impaired motivational behavior, accelerated age-related macular degeneration, impaired olfactory sexual signaling in the male, and lower female fertility (Table S2; Matzuk et al., 1998; Flood et al., 1999; Elchuri et al., 2005; Imamura et al., 2006; Muller et al., 2006; Iuchi et al., 2010; Fischer et al., 2012; Zhang et al., 2013, 2017; Garratt et al., 2014; Shi et al., 2014; Yoshihara et al., 2016). Sod1−/− mice also have increased incidence of hepatocellular carcinoma in old age (Elchuri et al., 2005).
Sod3−/− mice under normal conditions develop normally and remain healthy for the majority of their adult lives (Carlsson et al., 1995). However, under high oxygen pressure (>99%), they show a significantly shorter survival time than the WT and develop an earlier onset of lung edema (Carlsson et al., 1995). The lack of SOD3 is also shown to exacerbate angiotensin-induced hypertension, which is likely attributed to a decrease in endothelial NO• bioavailability (see above; Jung et al., 2003). As lung and blood vessels have high amounts of SOD3 expression (Fukai et al., 2002), it is not surprising that they are most affected by the loss of SOD3. In contrast with the germline Sod3 knockout, acute deletion of Sod3 in adult mice results in severe acute lung injury and death within 7 d of induction of the gene knockout (Table S2; Gongora et al., 2008). The striking phenotypic difference between the germline and induced adult-onset knockouts points to the presence of powerful compensatory mechanisms that acts in the conventional Sod3 knockout model.
Interestingly, in cell culture, the effects of the loss of different Sod genes appear to be exactly opposite to those observed in mice. That is, the mouse embryonic fibroblasts isolated from Sod1−/− mice die a few days after isolation (Tsunoda et al., 2013), whereas Sod2−/− mouse embryonic fibroblasts and equivalent human cells can grow and proliferate under normal conditions despite compromised mitochondrial function (Zhang et al., 2010; Cramer-Morales et al., 2015). This phenomenon is not yet understood.
Another type of SOD signaling in mammals
In mammals, Cu/Zn SOD SOD3 (also extracellular SOD [EcSOD]) is found in the extracellular matrix of tissues as well as extracellular fluids including the blood (Karlsson and Marklund, 1988). It is a glycoprotein, and because of its affinity for heparan-sulfate polysaccharides, a portion of SOD3 binds to the glycocalyx of cell surfaces, where it acts on O2•− released by the membrane-bound NADPH oxidase (Fukai et al., 2002). O2•− diffuses poorly across membranes but can penetrate the plasma membrane through anion channels. Thus, both O2•− and H2O2 produced in the extracellular space potentially can travel back into the cells, where they could initiate signaling by interacting with nearby proteins (Fisher, 2009). It is not yet known what regulatory role SOD3 could have in the cross-membrane signaling.
Because SOD3 is secreted, it potentially can be redistributed via circulation and have an action at a distance. Indeed, a recent study shows that overexpression of SOD3 in skeletal muscles led to significantly elevated SOD3 levels both in the serum and in most of the peripheral tissues, which protected against organ damage and improved animal survival under lipopolysaccharide-induced endotoxemia. Similar beneficial effects were also seen in the parabiotic mice who shared blood with ones that overexpress SOD3 in muscle (Call et al., 2017). The observed increase of SOD3 protein in peripheral organs could be mainly caused by its accumulation on cell surface; however, internalization by endothelial cells is also possible (Chu et al., 2006). Interestingly, exercise was reported to sufficiently enhance expression of SOD3 in muscle tissues, though the effect appears to depend on exercise mode and intensity (Hitomi et al., 2008; Zhao et al., 2013). Lastly, as mentioned above, altered SOD3 levels could affect NO• availability and endothelial function. Therefore, one possible effect of increased circulating SOD3 that has not been studied is how vascular tone and tissue blood supply are affected, the result of which could significantly impact tissue oxygenation and ultimately viability.
The roles of SODs in human diseases
As SOD enzymes are key player in antioxidant defense, there has long been an interest in their roles in disease and the potential of increasing SOD levels for therapeutic use. In fact, alterations in the expression level or catalytic activity of SODs have been found in a variety of pathological conditions, including some of the most common age-dependent diseases such as cardiovascular diseases, neurodegenerative diseases, and cancer (Marcus et al., 2006; Robbins and Zhao, 2014). We will briefly discuss a few diseases for which the role of SOD has been studied.
Mutation of SOD1 in ALS
The most widely studied connection between SOD and human diseases is ALS, a late-onset fatal neurodegenerative disorder that primarily affects motor neurons (Robberecht and Philips, 2013). Mutations in the SOD1 gene were first identified as a cause of familial ALS in 1993 (Rosen et al., 1993). Since then, >170 different mutations spread throughout the SOD1 gene have been described to result in ALS, and a general estimate is that SOD1 mutations account for ∼14–20% of familiar ALS and 1–3% of sporadic cases (Andersen and Al-Chalabi, 2011; Taylor et al., 2016). Although it has been generally accepted that acquisition of a toxic property by mutant SOD1 forms rather than loss or gain of its superoxide scavenging activity is responsible for the manifestation of ALS phenotype, how exactly SOD1 mutations lead to the selective motor neuron death is not yet entirely clear.
SODs in cancer redox signals
Oxidative stress has been heavily implicated in many aspects of cancer biology. First, higher ROS level could promote DNA mutations, thus playing a potential causal role in tumorigenesis. After transformation and during tumor maintenance, cancer cells must cope with toxic effects of high oxidative stress and potentially alter their redox homeostasis to support fast growth (Panieri and Santoro, 2016). Tumor cells often display enhanced intracellular ROS levels (Schumacker, 2006; Sabharwal and Schumacker, 2014). A more recent view is that their increasing ROS production is an adaptive process that tumor cells use to induce changes in signaling pathways beneficial for their normal function. For example, activation of the PI3K/AKT signaling transduction pathway, one of the major targets of ROS, can promote proliferation, metabolic adaption, and cell mobility (Cantley, 2002; Leslie et al., 2003; Pelicano et al., 2006; Sullivan and Chandel, 2014). ROS stabilize HIF1α and activate NRF2, and their aberrant activation can facilitate adaptations to hypoxia, stimulate angiogenesis, and enhance cell proliferation (Chandel et al., 2000; Semenza, 2003; Mitsuishi et al., 2012). Furthermore, several oncogenes like KRAS and MYC have been linked to increased mitochondrial ROS production (Sabharwal and Schumacker, 2014).
It has been shown that many tumor cells also display changes in antioxidant enzyme profiles. For example, alterations in SOD2 gene expression and/or protein levels have been reported in various types of cancer cells, but some with conflicting results (Hempel et al., 2011). This is perhaps not surprising given that the studies dealt with different cancer types, samples at varying stages of metastatic progression, and different detection techniques. It remains to be fully understood how SOD2 contributes to aberrant protumorigenic signaling; however, one point of view particularly noteworthy is that SOD2 activity may change with cell transformation and malignant progression in accordance with the different needs of tumor cells at different stages to maintain their viability and growth advantage (Hempel et al., 2011). That is, in the initial onset/proliferative stage of tumor initiation, SOD2 level is low, which may help promote malignant transformation, yet once the tumor progresses to a more aggressive phenotype, SOD2 level/activity is increased, contributing to a mitochondrial H2O2 increase that, through activation of oncogenic and angiogenic pathways, helps the cell to acquire invasive properties (Hempel et al., 2011; Miriyala et al., 2012). In fact, using transgenic mice expressing a luciferase reporter gene under the control of human SOD2 promoter and a chemically induced skin tumorigenesis model, a shift in SOD2 expression from low at early stage to high at advanced squamous cell carcinoma was identified (Dhar et al., 2011). Furthermore, catalase levels are found to be decreased in some tumor cells, and more notably, catalase overexpression, which is expected to accelerate H2O2 destruction, was shown to inhibit invasive and migratory phenotype (Connor et al., 2007; Hempel et al., 2011). In vitro induction of SOD2 overexpression was shown to slow cancer cell proliferation (Hurt et al., 2007), which presumably is mediated by redox-dependent signaling events, though how is not completely clear.
SOD3 in cardiovascular diseases
Because of the key role of SOD3 in modulating O2•- levels in the vasculature, this SOD has been linked to pathological conditions that involve vascular dysfunction such as diabetes, hypertension, and atherosclerosis (Fukai et al., 2002). Oxidative stress, especially production of O2•-, is believed to induce endothelial dysfunction, a crucial early step in atherosclerosis (Münzel et al., 2010). Furthermore, as previously mentioned, excess O2•- itself can directly antagonize NO• by a direct chemical interaction. The endothelium-derived NO• controls the extent of vascular smooth muscle relaxation, inhibits platelet aggregation, and attenuates neutrophil adherence to the endothelium (Tousoulis et al., 2012). SOD3, by opposing inactivation of vascular NO•, might therefore be crucial for the maintenance of blood vessel tone and protection against plaque formation. In support of this, decreased SOD3 activity was observed in human atherosclerotic lesions (Fukai and Ushio-Fukai, 2011). In patients with coronary artery disease, the activity of SOD3 was also found to be reduced, whereas SOD1 and SOD2 remain unaffected (Fukai and Ushio-Fukai, 2011).
Hypertension has also been linked to increased vascular oxidative stress. Sod3−/− mice were shown to display augmented hypertension in response to angiotensin II or development of higher renovascular hypertension after renal artery clipping (Jung et al., 2003; Gongora et al., 2006). These findings suggest a role for SOD3 and superoxide in the genesis of hypertension. Interestingly, more recent studies demonstrated that deletion of SOD3 in the circumventricular organs raised basal blood pressure and augmented the hypertension caused by angiotensin II, whereas removal of SOD3 specifically in the vascular smooth muscle showed no effect on blood pressure either at baseline or in response to angiotensin II despite observable effects on vascular O2•- and NO• (Lob et al., 2010, 2011).
Conclusions
It has been well established that ROS and RNS function as important diffusible signaling molecules. Because the SODs are the only enzymes that interact with superoxide specifically and thus control the levels of ROS and RNS, they also serve as key regulators of signaling. Of course, in addition to acting as signaling molecules, at high levels, these reactive species can become toxic. In fact, most attention has focused on their toxic properties because when their levels are dramatically altered, damaging effects become a more obvious phenomenon than alteration in signaling, although the deleteriousness of the loss of SODs might involve both aspects. This is clearly shown by some of the studies in model organisms discussed in this review, where the loss of a particular SOD leads to specific developmental or physiological changes without impairing viability.
Online supplemental material
Table S1 show nomenclature and characteristics of SOD enzymes across various species. Table S2 shows Sod gene knockout mouse models and their phenotypes.
Supplementary Material
Acknowledgments
Research in laboratory of S. Hekimi is funded by grants from the Canadian Institutes of Health Research (MOP-142774, MOP-123295, and MOP-97869) as well as by McGill University. S. Hekimi is Campbell Chair of Developmental Biology at McGill University.
The authors declare no competing financial interests.
References
- Adams L., Franco M.C., and Estevez A.G.. 2015. Reactive nitrogen species in cellular signaling. Exp. Biol. Med. (Maywood). 240:711–717. 10.1177/1535370215581314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afanas’ev I. 2015. Mechanisms of superoxide signaling in epigenetic processes: relation to aging and cancer. Aging Dis. 6:216–227. 10.14336/AD.2014.0924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S.G., and Thiele D.J.. 2003. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 17:516–528. 10.1101/gad.1044503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P.M., and Al-Chalabi A.. 2011. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7:603–615. 10.1038/nrneurol.2011.150 [DOI] [PubMed] [Google Scholar]
- Antonyuk S.V., Strange R.W., Marklund S.L., and Hasnain S.S.. 2009. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 A resolution: insights into heparin and collagen binding. J. Mol. Biol. 388:310–326. 10.1016/j.jmb.2009.03.026 [DOI] [PubMed] [Google Scholar]
- Back P., Matthijssens F., Vlaeminck C., Braeckman B.P., and Vanfleteren J.R.. 2010. Effects of sod gene overexpression and deletion mutation on the expression profiles of reporter genes of major detoxification pathways in Caenorhabditis elegans. Exp. Gerontol. 45:603–610. 10.1016/j.exger.2010.01.014 [DOI] [PubMed] [Google Scholar]
- Banci L., Barbieri L., Bertini I., Luchinat E., Secci E., Zhao Y., and Aricescu A.R.. 2013. Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat. Chem. Biol. 9:297–299. 10.1038/nchembio.1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne A.C., Mockett R.J., Orr W.C., and Sohal R.S.. 2005. Enhanced catabolism of mitochondrial superoxide/hydrogen peroxide and aging in transgenic Drosophila. Biochem. J. 391:277–284. 10.1042/BJ20041872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman J.S., and Koppenol W.H.. 1996. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. 271:C1424–C1437. 10.1152/ajpcell.1996.271.5.C1424 [DOI] [PubMed] [Google Scholar]
- Bedard K., and Krause K.H.. 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87:245–313. 10.1152/physrev.00044.2005 [DOI] [PubMed] [Google Scholar]
- Benov L. 2001. How superoxide radical damages the cell. Protoplasma. 217:33–36. 10.1007/BF01289410 [DOI] [PubMed] [Google Scholar]
- Bienert G.P., Schjoerring J.K., and Jahn T.P.. 2006. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta. 1758:994–1003. 10.1016/j.bbamem.2006.02.015 [DOI] [PubMed] [Google Scholar]
- Blackney M.J., Cox R., Shepherd D., and Parker J.D.. 2014. Cloning and expression analysis of Drosophila extracellular Cu Zn superoxide dismutase. Biosci. Rep. 34:e00164 10.1042/BSR20140133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes N., Schmitt S., and Jakob U.. 2009. Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal. 11:997–1014. 10.1089/ars.2008.2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.I., and Griendling K.K.. 2009. Nox proteins in signal transduction. Free Radic. Biol. Med. 47:1239–1253. 10.1016/j.freeradbiomed.2009.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G.C., and Borutaite V.. 2007. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc. Res. 75:283–290. 10.1016/j.cardiores.2007.03.022 [DOI] [PubMed] [Google Scholar]
- Call J.A., Donet J., Martin K.S., Sharma A.K., Chen X., Zhang J., Cai J., Galarreta C.A., Okutsu M., Du Z., et al. 2017. Muscle-derived extracellular superoxide dismutase inhibits endothelial activation and protects against multiple organ dysfunction syndrome in mice. Free Radic. Biol. Med. 113:212–223. 10.1016/j.freeradbiomed.2017.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S.D., Hilliker A.J., and Phillips J.P.. 1986. Cytogenetic analysis of the cSOD microregion in Drosophila melanogaster. Genetics. 112:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley L.C. 2002. The phosphoinositide 3-kinase pathway. Science. 296:1655–1657. 10.1126/science.296.5573.1655 [DOI] [PubMed] [Google Scholar]
- Cardoso A.R., Chausse B., da Cunha F.M., Luévano-Martínez L.A., Marazzi T.B., Pessoa P.S., Queliconi B.B., and Kowaltowski A.J.. 2012. Mitochondrial compartmentalization of redox processes. Free Radic. Biol. Med. 52:2201–2208. 10.1016/j.freeradbiomed.2012.03.008 [DOI] [PubMed] [Google Scholar]
- Carlsson L.M., Jonsson J., Edlund T., and Marklund S.L.. 1995. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc. Natl. Acad. Sci. USA. 92:6264–6268. 10.1073/pnas.92.14.6264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celotto A.M., Liu Z., Vandemark A.P., and Palladino M.J.. 2012. A novel Drosophila SOD2 mutant demonstrates a role for mitochondrial ROS in neurodevelopment and disease. Brain Behav. 2:424–434. 10.1002/brb3.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel N.S., McClintock D.S., Feliciano C.E., Wood T.M., Melendez J.A., Rodriguez A.M., and Schumacker P.T.. 2000. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 275:25130–25138. 10.1074/jbc.M001914200 [DOI] [PubMed] [Google Scholar]
- Chang L.Y., Slot J.W., Geuze H.J., and Crapo J.D.. 1988. Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J. Cell Biol. 107:2169–2179. 10.1083/jcb.107.6.2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez V., Mohri-Shiomi A., Maadani A., Vega L.A., and Garsin D.A.. 2007. Oxidative stress enzymes are required for DAF-16-mediated immunity due to generation of reactive oxygen species by Caenorhabditis elegans. Genetics. 176:1567–1577. 10.1534/genetics.107.072587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Kirber M.T., Xiao H., Yang Y., and Keaney J.F. Jr. 2008. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 181:1129–1139. 10.1083/jcb.200709049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Azad M.B., and Gibson S.B.. 2009. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 16:1040–1052. 10.1038/cdd.2009.49 [DOI] [PubMed] [Google Scholar]
- Chu Y., Piper R., Richardson S., Watanabe Y., Patel P., and Heistad D.D.. 2006. Endocytosis of extracellular superoxide dismutase into endothelial cells: role of the heparin-binding domain. Arterioscler. Thromb. Vasc. Biol. 26:1985–1990. 10.1161/01.ATV.0000234921.88489.5c [DOI] [PubMed] [Google Scholar]
- Clancy D.J., Gems D., Harshman L.G., Oldham S., Stocker H., Hafen E., Leevers S.J., and Partridge L.. 2001. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 292:104–106. 10.1126/science.1057991 [DOI] [PubMed] [Google Scholar]
- Connor K.M., Subbaram S., Regan K.J., Nelson K.K., Mazurkiewicz J.E., Bartholomew P.J., Aplin A.E., Tai Y.T., Aguirre-Ghiso J., Flores S.C., and Melendez J.A.. 2005. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 280:16916–16924. 10.1074/jbc.M410690200 [DOI] [PubMed] [Google Scholar]
- Connor K.M., Hempel N., Nelson K.K., Dabiri G., Gamarra A., Belarmino J., Van De Water L., Mian B.M., and Melendez J.A.. 2007. Manganese superoxide dismutase enhances the invasive and migratory activity of tumor cells. Cancer Res. 67:10260–10267. 10.1158/0008-5472.CAN-07-1204 [DOI] [PubMed] [Google Scholar]
- Corcoran A., and Cotter T.G.. 2013. Redox regulation of protein kinases. FEBS J. 280:1944–1965. 10.1111/febs.12224 [DOI] [PubMed] [Google Scholar]
- Cramer-Morales K., Heer C.D., Mapuskar K.A., and Domann F.E.. 2015. SOD2 targeted gene editing by CRISPR/Cas9 yields Human cells devoid of MnSOD. Free Radic. Biol. Med. 89:379–386. 10.1016/j.freeradbiomed.2015.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapo J.D., Oury T., Rabouille C., Slot J.W., and Chang L.Y.. 1992. Copper,zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proc. Natl. Acad. Sci. USA. 89:10405–10409. 10.1073/pnas.89.21.10405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremers C.M., and Jakob U.. 2013. Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 288:26489–26496. 10.1074/jbc.R113.462929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross A.R., and Segal A.W.. 2004. The NADPH oxidase of professional phagocytes--prototype of the NOX electron transport chain systems. Biochim. Biophys. Acta. 1657:1–22. 10.1016/j.bbabio.2004.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hertog J., Groen A., and van der Wijk T.. 2005. Redox regulation of protein-tyrosine phosphatases. Arch. Biochem. Biophys. 434:11–15. 10.1016/j.abb.2004.05.024 [DOI] [PubMed] [Google Scholar]
- Dhar S.K., and St Clair D.K.. 2012. Manganese superoxide dismutase regulation and cancer. Free Radic. Biol. Med. 52:2209–2222. 10.1016/j.freeradbiomed.2012.03.009 [DOI] [PubMed] [Google Scholar]
- Dhar S.K., Tangpong J., Chaiswing L., Oberley T.D., and St Clair D.K.. 2011. Manganese superoxide dismutase is a p53-regulated gene that switches cancers between early and advanced stages. Cancer Res. 71:6684–6695. 10.1158/0008-5472.CAN-11-1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingley S., Polyak E., Lightfoot R., Ostrovsky J., Rao M., Greco T., Ischiropoulos H., and Falk M.J.. 2010. Mitochondrial respiratory chain dysfunction variably increases oxidant stress in Caenorhabditis elegans. Mitochondrion. 10:125–136. 10.1016/j.mito.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doonan R., McElwee J.J., Matthijssens F., Walker G.A., Houthoofd K., Back P., Matscheski A., Vanfleteren J.R., and Gems D.. 2008. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 22:3236–3241. 10.1101/gad.504808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drazic A., and Winter J.. 2014. The physiological role of reversible methionine oxidation. Biochim. Biophys. Acta. 1844:1367–1382. 10.1016/j.bbapap.2014.01.001 [DOI] [PubMed] [Google Scholar]
- Dues D.J., Schaar C.E., Johnson B.K., Bowman M.J., Winn M.E., Senchuk M.M., and Van Raamsdonk J.M.. 2017. Uncoupling of oxidative stress resistance and lifespan in long-lived isp-1 mitochondrial mutants in Caenorhabditis elegans. Free Radic. Biol. Med. 108:362–373. 10.1016/j.freeradbiomed.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukan S., and Nyström T.. 1999. Oxidative stress defense and deterioration of growth-arrested Escherichia coli cells. J. Biol. Chem. 274:26027–26032. 10.1074/jbc.274.37.26027 [DOI] [PubMed] [Google Scholar]
- Durieux J., Wolff S., and Dillin A.. 2011. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 144:79–91. 10.1016/j.cell.2010.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttaroy A., Parkes T., Emtage P., Kirby K., Boulianne G.L., Wang X., Hilliker A.J., and Phillips J.P.. 1997. The manganese superoxide dismutase gene of Drosophila: structure, expression, and evidence for regulation by MAP kinase. DNA Cell Biol. 16:391–399. 10.1089/dna.1997.16.391 [DOI] [PubMed] [Google Scholar]
- Duttaroy A., Paul A., Kundu M., and Belton A.. 2003. A Sod2 null mutation confers severely reduced adult life span in Drosophila. Genetics. 165:2295–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elchuri S., Oberley T.D., Qi W., Eisenstein R.S., Jackson Roberts L., Van Remmen H., Epstein C.J., and Huang T.T.. 2005. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 24:367–380. 10.1038/sj.onc.1208207 [DOI] [PubMed] [Google Scholar]
- Erkut C., Vasilj A., Boland S., Habermann B., Shevchenko A., and Kurzchalia T.V.. 2013. Molecular strategies of the Caenorhabditis elegans dauer larva to survive extreme desiccation. PLoS One. 8:e82473 10.1371/journal.pone.0082473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattman C.L., Schaefer L.M., and Oury T.D.. 2003. Extracellular superoxide dismutase in biology and medicine. Free Radic. Biol. Med. 35:236–256. 10.1016/S0891-5849(03)00275-2 [DOI] [PubMed] [Google Scholar]
- Favrin G., Bean D.M., Bilsland E., Boyer H., Fischer B.E., Russell S., Crowther D.C., Baylis H.A., Oliver S.G., and Giannakou M.E.. 2013. Identification of novel modifiers of Aβ toxicity by transcriptomic analysis in the fruitfly. Sci. Rep. 3:3512 10.1038/srep03512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J., Bussière F., and Hekimi S.. 2001. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell. 1:633–644. 10.1016/S1534-5807(01)00071-5 [DOI] [PubMed] [Google Scholar]
- Finkel T. 2001. Reactive oxygen species and signal transduction. IUBMB Life. 52:3–6. 10.1080/15216540252774694 [DOI] [PubMed] [Google Scholar]
- Finkel T. 2011. Signal transduction by reactive oxygen species. J. Cell Biol. 194:7–15. 10.1083/jcb.201102095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer L.R., Li Y., Asress S.A., Jones D.P., and Glass J.D.. 2012. Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp. Neurol. 233:163–171. 10.1016/j.expneurol.2011.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher A.B. 2009. Redox signaling across cell membranes. Antioxid. Redox Signal. 11:1349–1356. 10.1089/ars.2008.2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood D.G., Reaume A.G., Gruner J.A., Hoffman E.K., Hirsch J.D., Lin Y.G., Dorfman K.S., and Scott R.W.. 1999. Hindlimb motor neurons require Cu/Zn superoxide dismutase for maintenance of neuromuscular junctions. Am. J. Pathol. 155:663–672. 10.1016/S0002-9440(10)65162-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folz R.J., and Crapo J.D.. 1994. Extracellular superoxide dismutase (SOD3): tissue-specific expression, genomic characterization, and computer-assisted sequence analysis of the human EC SOD gene. Genomics. 22:162–171. 10.1006/geno.1994.1357 [DOI] [PubMed] [Google Scholar]
- Folz R.J., Guan J., Seldin M.F., Oury T.D., Enghild J.J., and Crapo J.D.. 1997. Mouse extracellular superoxide dismutase: primary structure, tissue-specific gene expression, chromosomal localization, and lung in situ hybridization. Am. J. Respir. Cell Mol. Biol. 17:393–403. 10.1165/ajrcmb.17.4.2826 [DOI] [PubMed] [Google Scholar]
- Forman H.J., Maiorino M., and Ursini F.. 2010. Signaling functions of reactive oxygen species. Biochemistry. 49:835–842. 10.1021/bi9020378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridovich I. 1975. Superoxide dismutases. Annu. Rev. Biochem. 44:147–159. 10.1146/annurev.bi.44.070175.001051 [DOI] [PubMed] [Google Scholar]
- Fridovich I. 1997. Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J. Biol. Chem. 272:18515–18517. 10.1074/jbc.272.30.18515 [DOI] [PubMed] [Google Scholar]
- Fujii M., Ishii N., Joguchi A., Yasuda K., and Ayusawa D.. 1998. A novel superoxide dismutase gene encoding membrane-bound and extracellular isoforms by alternative splicing in Caenorhabditis elegans. DNA Res. 5:25–30. 10.1093/dnares/5.1.25 [DOI] [PubMed] [Google Scholar]
- Fukai T., and Ushio-Fukai M.. 2011. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 15:1583–1606. 10.1089/ars.2011.3999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukai T., Folz R.J., Landmesser U., and Harrison D.G.. 2002. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc. Res. 55:239–249. 10.1016/S0008-6363(02)00328-0 [DOI] [PubMed] [Google Scholar]
- Garratt M., Pichaud N., Glaros E.N., Kee A.J., and Brooks R.C.. 2014. Superoxide dismutase deficiency impairs olfactory sexual signaling and alters bioenergetic function in mice. Proc. Natl. Acad. Sci. USA. 111:8119–8124. 10.1073/pnas.1322282111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoni E., Buricchi F., Raugei G., Ramponi G., and Chiarugi P.. 2005. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 25:6391–6403. 10.1128/MCB.25.15.6391-6403.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasauer A., Sena L.A., Diebold L.P., Mazar A.P., and Chandel N.S.. 2014. Targeting SOD1 reduces experimental non–small-cell lung cancer. J. Clin. Invest. 124:117–128. 10.1172/JCI71714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gongora M.C., Qin Z., Laude K., Kim H.W., McCann L., Folz J.R., Dikalov S., Fukai T., and Harrison D.G.. 2006. Role of extracellular superoxide dismutase in hypertension. Hypertension. 48:473–481. 10.1161/01.HYP.0000235682.47673.ab [DOI] [PubMed] [Google Scholar]
- Gongora M.C., Lob H.E., Landmesser U., Guzik T.J., Martin W.D., Ozumi K., Wall S.M., Wilson D.S., Murthy N., Gravanis M., et al. 2008. Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: a potential mechanism underlying adult respiratory distress syndrome. Am. J. Pathol. 173:915–926. 10.2353/ajpath.2008.080119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Cabo P., Bolinches-Amorós A., Cabello J., Ros S., Moreno S., Baylis H.A., Palau F., and Vázquez-Manrique R.P.. 2011. Disruption of the ATP-binding cassette B7 (ABTM-1/ABCB7) induces oxidative stress and premature cell death in Caenorhabditis elegans. J. Biol. Chem. 286:21304–21314. 10.1074/jbc.M110.211201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha E.M., Oh C.T., Bae Y.S., and Lee W.J.. 2005. A direct role for dual oxidase in Drosophila gut immunity. Science. 310:847–850. 10.1126/science.1117311 [DOI] [PubMed] [Google Scholar]
- Harman D. 1956. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11:298–300. 10.1093/geronj/11.3.298 [DOI] [PubMed] [Google Scholar]
- Hayyan M., Hashim M.A., and AlNashef I.M.. 2016. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 116:3029–3085. 10.1021/acs.chemrev.5b00407 [DOI] [PubMed] [Google Scholar]
- Hempel N., Carrico P.M., and Melendez J.A.. 2011. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anticancer. Agents Med. Chem. 11:191–201. 10.2174/187152011795255911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S.T., Bonafè M., and Johnson T.E.. 2006. daf-16 protects the nematode Caenorhabditis elegans during food deprivation. J. Gerontol. A Biol. Sci. Med. Sci. 61:444–460. 10.1093/gerona/61.5.444 [DOI] [PubMed] [Google Scholar]
- Hitomi Y., Watanabe S., Kizaki T., Sakurai T., Takemasa T., Haga S., Ookawara T., Suzuki K., and Ohno H.. 2008. Acute exercise increases expression of extracellular superoxide dismutase in skeletal muscle and the aorta. Redox Rep. 13:213–216. 10.1179/135100008X308894 [DOI] [PubMed] [Google Scholar]
- Holmström K.M., and Finkel T.. 2014. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15:411–421. 10.1038/nrm3801 [DOI] [PubMed] [Google Scholar]
- Honda Y., and Honda S.. 1999. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 13:1385–1393. 10.1096/fasebj.13.11.1385 [DOI] [PubMed] [Google Scholar]
- Honda Y., and Honda S.. 2001. Life span extensions associated with upregulation of gene expression of antioxidant enzymes in Caenorhabdms elegans; studies of mutation in the AGE-1, PI3 kinase homologue and short-term exposure to hyperoxia. J. Am. Aging Assoc. 24:179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda Y., Tanaka M., and Honda S.. 2008. Modulation of longevity and diapause by redox regulation mechanisms under the insulin-like signaling control in Caenorhabditis elegans. Exp. Gerontol. 43:520–529. 10.1016/j.exger.2008.02.009 [DOI] [PubMed] [Google Scholar]
- Horspool A.M., and Chang H.C.. 2017. Superoxide dismutase SOD-1 modulates C. elegans pathogen avoidance behavior. Sci. Rep. 7:45128 10.1038/srep45128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper R.H., Mouchiroud L., Ryu D., Moullan N., Katsyuba E., Knott G., Williams R.W., and Auwerx J.. 2013. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 497:451–457. 10.1038/nature12188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D., Cao P., Thiels E., Chu C.T., Wu G.Y., Oury T.D., and Klann E.. 2007. Hippocampal long-term potentiation, memory, and longevity in mice that overexpress mitochondrial superoxide dismutase. Neurobiol. Learn. Mem. 87:372–384. 10.1016/j.nlm.2006.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T., Bannister W.H., and Hunter G.J.. 1997. Cloning, expression, and characterization of two manganese superoxide dismutases from Caenorhabditis elegans. J. Biol. Chem. 272:28652–28659. 10.1074/jbc.272.45.28652 [DOI] [PubMed] [Google Scholar]
- Hurt E.M., Thomas S.B., Peng B., and Farrar W.L.. 2007. Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines. Br. J. Cancer. 97:1116–1123. 10.1038/sj.bjc.6604000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura Y., Noda S., Hashizume K., Shinoda K., Yamaguchi M., Uchiyama S., Shimizu T., Mizushima Y., Shirasawa T., and Tsubota K.. 2006. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA. 103:11282–11287. 10.1073/pnas.0602131103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay J.A. 2003. Pathways of oxidative damage. Annu. Rev. Microbiol. 57:395–418. 10.1146/annurev.micro.57.030502.090938 [DOI] [PubMed] [Google Scholar]
- Iuchi Y., Roy D., Okada F., Kibe N., Tsunoda S., Suzuki S., Takahashi M., Yokoyama H., Yoshitake J., Kondo S., and Fujii J.. 2010. Spontaneous skin damage and delayed wound healing in SOD1-deficient mice. Mol. Cell. Biochem. 341:181–194. 10.1007/s11010-010-0449-y [DOI] [PubMed] [Google Scholar]
- Izuo N., Nojiri H., Uchiyama S., Noda Y., Kawakami S., Kojima S., Sasaki T., Shirasawa T., and Shimizu T.. 2015. Brain-Specific Superoxide Dismutase 2 Deficiency Causes Perinatal Death with Spongiform Encephalopathy in Mice. Oxid. Med. Cell. Longev. 2015:238914 10.1155/2015/238914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y.C., Pérez V.I., Song W., Lustgarten M.S., Salmon A.B., Mele J., Qi W., Liu Y., Liang H., Chaudhuri A., et al. 2009. Overexpression of Mn superoxide dismutase does not increase life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 64:1114–1125. 10.1093/gerona/glp100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen L.T., and Culotta V.C.. 2005. Activation of CuZn superoxide dismutases from Caenorhabditis elegans does not require the copper chaperone CCS. J. Biol. Chem. 280:41373–41379. 10.1074/jbc.M509142200 [DOI] [PubMed] [Google Scholar]
- Jones P.L., Kucera G., Gordon H., and Boss J.M.. 1995. Cloning and characterization of the murine manganous superoxide dismutase-encoding gene. Gene. 153:155–161. 10.1016/0378-1119(94)00666-G [DOI] [PubMed] [Google Scholar]
- Juarez J.C., Manuia M., Burnett M.E., Betancourt O., Boivin B., Shaw D.E., Tonks N.K., Mazar A.P., and Doñate F.. 2008. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. USA. 105:7147–7152. 10.1073/pnas.0709451105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung I., Kim T.Y., and Kim-Ha J.. 2011. Identification of Drosophila SOD3 and its protective role against phototoxic damage to cells. FEBS Lett. 585:1973–1978. 10.1016/j.febslet.2011.05.033 [DOI] [PubMed] [Google Scholar]
- Jung O., Marklund S.L., Geiger H., Pedrazzini T., Busse R., and Brandes R.P.. 2003. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. Circ. Res. 93:622–629. 10.1161/01.RES.0000092140.81594.A8 [DOI] [PubMed] [Google Scholar]
- Kabil H., Partridge L., and Harshman L.G.. 2007. Superoxide dismutase activities in long-lived Drosophila melanogaster females: chico1 genotypes and dietary dilution. Biogerontology. 8:201–208. 10.1007/s10522-006-9065-3 [DOI] [PubMed] [Google Scholar]
- Karlsson K., and Marklund S.L.. 1988. Extracellular superoxide dismutase in the vascular system of mammals. Biochem. J. 255:223–228. [PMC free article] [PubMed] [Google Scholar]
- Kirby K., Hu J., Hilliker A.J., and Phillips J.P.. 2002. RNA interference-mediated silencing of Sod2 in Drosophila leads to early adult-onset mortality and elevated endogenous oxidative stress. Proc. Natl. Acad. Sci. USA. 99:16162–16167. 10.1073/pnas.252342899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka J.E., Coskun P., Esposito L.A., and Wallace D.C.. 2001. Increased mitochondrial oxidative stress in the Sod2 (+/-) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. USA. 98:2278–2283. 10.1073/pnas.051627098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara H., Horie T., Ishikawa S., Tsuda C., Kawakami S., Noda Y., Kaneko T., Tahara S., Tachibana T., Okabe M., et al. 2010. Oxidative stress in skeletal muscle causes severe disturbance of exercise activity without muscle atrophy. Free Radic. Biol. Med. 48:1252–1262. 10.1016/j.freeradbiomed.2010.02.011 [DOI] [PubMed] [Google Scholar]
- Landis G.N., and Tower J.. 2005. Superoxide dismutase evolution and life span regulation. Mech. Ageing Dev. 126:365–379. 10.1016/j.mad.2004.08.012 [DOI] [PubMed] [Google Scholar]
- Lapointe J., and Hekimi S.. 2008. Early mitochondrial dysfunction in long-lived Mclk1+/- mice. J. Biol. Chem. 283:26217–26227. 10.1074/jbc.M803287200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen P.L. 1993. Aging and resistance to oxidative damage in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 90:8905–8909. 10.1073/pnas.90.19.8905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz R.M., Zhang H., Vogel H., Cartwright J. Jr., Dionne L., Lu N., Huang S., and Matzuk M.M.. 1996. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA. 93:9782–9787. 10.1073/pnas.93.18.9782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Van Remmen H., and Csete M.. 2009. Sod2 overexpression preserves myoblast mitochondrial mass and function, but not muscle mass with aging. Aging Cell. 8:296–310. 10.1111/j.1474-9726.2009.00477.x [DOI] [PubMed] [Google Scholar]
- Leslie N.R., Bennett D., Lindsay Y.E., Stewart H., Gray A., and Downes C.P.. 2003. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 22:5501–5510. 10.1093/emboj/cdg513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon D., Lieman-Hurwitz J., Dafni N., Wigderson M., Sherman L., Bernstein Y., Laver-Rudich Z., Danciger E., Stein O., and Groner Y.. 1985. Architecture and anatomy of the chromosomal locus in human chromosome 21 encoding the Cu/Zn superoxide dismutase. EMBO J. 4:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Chen Y., and Gibson S.B.. 2013. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell. Signal. 25:50–65. 10.1016/j.cellsig.2012.09.020 [DOI] [PubMed] [Google Scholar]
- Li Y., Huang T.T., Carlson E.J., Melov S., Ursell P.C., Olson J.L., Noble L.J., Yoshimura M.P., Berger C., Chan P.H., et al. 1995. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 11:376–381. 10.1038/ng1295-376 [DOI] [PubMed] [Google Scholar]
- Liochev S.I., and Fridovich I.. 1999. Superoxide and iron: partners in crime. IUBMB Life. 48:157–161. 10.1080/713803492 [DOI] [PubMed] [Google Scholar]
- Lob H.E., Marvar P.J., Guzik T.J., Sharma S., McCann L.A., Weyand C., Gordon F.J., and Harrison D.G.. 2010. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 55:277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lob H.E., Vinh A., Li L., Blinder Y., Offermanns S., and Harrison D.G.. 2011. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension. 58:232–239. 10.1161/HYPERTENSIONAHA.111.172718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus D.L., Strafaci J.A., and Freedman M.L.. 2006. Differential neuronal expression of manganese superoxide dismutase in Alzheimer’s disease. Med. Sci. Monit. 12:BR8–BR14. [PubMed] [Google Scholar]
- Marinho H.S., Real C., Cyrne L., Soares H., and Antunes F.. 2014. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2:535–562. 10.1016/j.redox.2014.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S.M., and Gladyshev V.N.. 2012. Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 287:4419–4425. 10.1074/jbc.R111.275578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund S.L. 1984. Extracellular superoxide dismutase in human tissues and human cell lines. J. Clin. Invest. 74:1398–1403. 10.1172/JCI111550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk M.M., Dionne L., Guo Q., Kumar T.R., and Lebovitz R.M.. 1998. Ovarian function in superoxide dismutase 1 and 2 knockout mice. Endocrinology. 139:4008–4011. 10.1210/endo.139.9.6289 [DOI] [PubMed] [Google Scholar]
- McCord J.M., and Fridovich I.. 1969. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244:6049–6055. [PubMed] [Google Scholar]
- Meitzler J.L., Antony S., Wu Y., Juhasz A., Liu H., Jiang G., Lu J., Roy K., and Doroshow J.H.. 2014. NADPH oxidases: a perspective on reactive oxygen species production in tumor biology. Antioxid. Redox Signal. 20:2873–2889. 10.1089/ars.2013.5603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J., Lv Z., Qiao X., Li X., Li Y., Zhang Y., and Chen C.. 2017. The decay of Redox-stress Response Capacity is a substantive characteristic of aging: Revising the redox theory of aging. Redox Biol. 11:365–374. 10.1016/j.redox.2016.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A.F. 2012. Superoxide dismutases: ancient enzymes and new insights. FEBS Lett. 586:585–595. 10.1016/j.febslet.2011.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miriyala S., Spasojevic I., Tovmasyan A., Salvemini D., Vujaskovic Z., St Clair D., and Batinic-Haberle I.. 2012. Manganese superoxide dismutase, MnSOD and its mimics. Biochim. Biophys. Acta. 1822:794–814. 10.1016/j.bbadis.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missirlis F., Hu J., Kirby K., Hilliker A.J., Rouault T.A., and Phillips J.P.. 2003. Compartment-specific protection of iron-sulfur proteins by superoxide dismutase. J. Biol. Chem. 278:47365–47369. 10.1074/jbc.M307700200 [DOI] [PubMed] [Google Scholar]
- Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., and Motohashi H.. 2012. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 22:66–79. 10.1016/j.ccr.2012.05.016 [DOI] [PubMed] [Google Scholar]
- Mittler R., Vanderauwera S., Suzuki N., Miller G., Tognetti V.B., Vandepoele K., Gollery M., Shulaev V., and Van Breusegem F.. 2011. ROS signaling: the new wave? Trends Plant Sci. 16:300–309. 10.1016/j.tplants.2011.03.007 [DOI] [PubMed] [Google Scholar]
- Mockett R.J., Sohal B.H., and Sohal R.S.. 2010. Expression of multiple copies of mitochondrially targeted catalase or genomic Mn superoxide dismutase transgenes does not extend the life span of Drosophila melanogaster. Free Radic. Biol. Med. 49:2028–2031. 10.1016/j.freeradbiomed.2010.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F.L., Song W., Liu Y., Chaudhuri A., Pieke-Dahl S., Strong R., Huang T.T., Epstein C.J., Roberts L.J. II, Csete M., et al. 2006. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 40:1993–2004. 10.1016/j.freeradbiomed.2006.01.036 [DOI] [PubMed] [Google Scholar]
- Münzel T., Gori T., Bruno R.M., and Taddei S.. 2010. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 31:2741–2748. 10.1093/eurheartj/ehq396 [DOI] [PubMed] [Google Scholar]
- Murphy M.P. 2009. How mitochondria produce reactive oxygen species. Biochem. J. 417:1–13. 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima I., Kato M., Akhand A.A., Suzuki H., Takeda K., Hossain K., and Kawamoto Y.. 2002. Redox-linked signal transduction pathways for protein tyrosine kinase activation. Antioxid. Redox Signal. 4:517–531. 10.1089/15230860260196326 [DOI] [PubMed] [Google Scholar]
- Nojiri H., Shimizu T., Funakoshi M., Yamaguchi O., Zhou H., Kawakami S., Ohta Y., Sami M., Tachibana T., Ishikawa H., et al. 2006. Oxidative stress causes heart failure with impaired mitochondrial respiration. J. Biol. Chem. 281:33789–33801. 10.1074/jbc.M602118200 [DOI] [PubMed] [Google Scholar]
- Oh S.I., Park J.K., and Park S.K.. 2015. Lifespan extension and increased resistance to environmental stressors by N-acetyl-l-cysteine in Caenorhabditis elegans. Clinics (São Paulo). 70:380–386. 10.6061/clinics/2015(05)13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okado-Matsumoto A., and Fridovich I.. 2001. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu/Zn-SOD in mitochondria. J. Biol. Chem. 276:38388–38393. 10.1074/jbc.M105395200 [DOI] [PubMed] [Google Scholar]
- Orr W.C., and Sohal R.S.. 1994. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science. 263:1128–1130. 10.1126/science.8108730 [DOI] [PubMed] [Google Scholar]
- Orr W.C., Mockett R.J., Benes J.J., and Sohal R.S.. 2003. Effects of overexpression of copper-zinc and manganese superoxide dismutases, catalase, and thioredoxin reductase genes on longevity in Drosophila melanogaster. J. Biol. Chem. 278:26418–26422. 10.1074/jbc.M303095200 [DOI] [PubMed] [Google Scholar]
- Pacher P., Beckman J.S., and Liaudet L.. 2007. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87:315–424. 10.1152/physrev.00029.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panieri E., and Santoro M.M.. 2016. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 7:e2253 10.1038/cddis.2016.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes T.L., Elia A.J., Dickinson D., Hilliker A.J., Phillips J.P., and Boulianne G.L.. 1998. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat. Genet. 19:171–174. 10.1038/534 [DOI] [PubMed] [Google Scholar]
- Paulsen C.E., and Carroll K.S.. 2010. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol. 5:47–62. 10.1021/cb900258z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen C.E., and Carroll K.S.. 2013. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem. Rev. 113:4633–4679. 10.1021/cr300163e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicano H., Xu R.H., Du M., Feng L., Sasaki R., Carew J.S., Hu Y., Ramdas L., Hu L., Keating M.J., et al. 2006. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 175:913–923. 10.1083/jcb.200512100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J.P., Campbell S.D., Michaud D., Charbonneau M., and Hilliker A.J.. 1989. Null mutation of copper/zinc superoxide dismutase in Drosophila confers hypersensitivity to paraquat and reduced longevity. Proc. Natl. Acad. Sci. USA. 86:2761–2765. 10.1073/pnas.86.8.2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J.P., Tainer J.A., Getzoff E.D., Boulianne G.L., Kirby K., and Hilliker A.J.. 1995. Subunit-destabilizing mutations in Drosophila copper/zinc superoxide dismutase: neuropathology and a model of dimer dysequilibrium. Proc. Natl. Acad. Sci. USA. 92:8574–8578. 10.1073/pnas.92.19.8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouyet L., and Carrier A.. 2010. Mutant mouse models of oxidative stress. Transgenic Res. 19:155–164. 10.1007/s11248-009-9308-6 [DOI] [PubMed] [Google Scholar]
- Rathor L., Akhoon B.A., Pandey S., Srivastava S., and Pandey R.. 2015. Folic acid supplementation at lower doses increases oxidative stress resistance and longevity in Caenorhabditis elegans. Age (Dordr.). 37:113 10.1007/s11357-015-9850-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczek C.R., and Chandel N.S.. 2015. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 33:8–13. 10.1016/j.ceb.2014.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddi A.R., and Culotta V.C.. 2013. SOD1 integrates signals from oxygen and glucose to repress respiration. Cell. 152:224–235. 10.1016/j.cell.2012.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee S.G. 2006. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 312:1882–1883. 10.1126/science.1130481 [DOI] [PubMed] [Google Scholar]
- Ristow M. 2014. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat. Med. 20:709–711. 10.1038/nm.3624 [DOI] [PubMed] [Google Scholar]
- Robberecht W., and Philips T.. 2013. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14:248–264. 10.1038/nrn3430 [DOI] [PubMed] [Google Scholar]
- Robbins D., and Zhao Y.. 2014. Manganese superoxide dismutase in cancer prevention. Antioxid. Redox Signal. 20:1628–1645. 10.1089/ars.2013.5297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogina B., and Helfand S.L.. 2000. Cu, Zn superoxide dismutase deficiency accelerates the time course of an age-related marker in Drosophila melanogaster. Biogerontology. 1:163–169. 10.1023/A:1010039813107 [DOI] [PubMed] [Google Scholar]
- Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O’Regan J.P., Deng H.X., et al. 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 362:59–62. 10.1038/362059a0 [DOI] [PubMed] [Google Scholar]
- Sabharwal S.S., and Schumacker P.T.. 2014. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer. 14:709–721. 10.1038/nrc3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Blanco A., and Kim S.K.. 2011. Variable pathogenicity determines individual lifespan in Caenorhabditis elegans. PLoS Genet. 7:e1002047 10.1371/journal.pgen.1002047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaar C.E., Dues D.J., Spielbauer K.K., Machiela E., Cooper J.F., Senchuk M., Hekimi S., and Van Raamsdonk J.M.. 2015. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 11:e1004972 10.1371/journal.pgen.1004972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöneich C. 2008. Mechanisms of protein damage induced by cysteine thiyl radical formation. Chem. Res. Toxicol. 21:1175–1179. 10.1021/tx800005u [DOI] [PubMed] [Google Scholar]
- Schöneich C. 2011. Cysteine residues as catalysts for covalent peptide and protein modification: a role for thiyl radicals? Biochem. Soc. Trans. 39:1254–1259. 10.1042/BST0391254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader M., and Fahimi H.D.. 2006. Peroxisomes and oxidative stress. Biochim. Biophys. Acta. 1763:1755–1766. 10.1016/j.bbamcr.2006.09.006 [DOI] [PubMed] [Google Scholar]
- Schulz T.J., Zarse K., Voigt A., Urban N., Birringer M., and Ristow M.. 2007. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6:280–293. 10.1016/j.cmet.2007.08.011 [DOI] [PubMed] [Google Scholar]
- Schumacker P.T. 2006. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 10:175–176. 10.1016/j.ccr.2006.08.015 [DOI] [PubMed] [Google Scholar]
- Semenza G.L. 2003. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer. 3:721–732. 10.1038/nrc1187 [DOI] [PubMed] [Google Scholar]
- Seto N.O., Hayashi S., and Tener G.M.. 1990. Overexpression of Cu-Zn superoxide dismutase in Drosophila does not affect life-span. Proc. Natl. Acad. Sci. USA. 87:4270–4274. 10.1073/pnas.87.11.4270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Ivannikov M.V., Walsh M.E., Liu Y., Zhang Y., Jaramillo C.A., Macleod G.T., and Van Remmen H.. 2014. The lack of CuZnSOD leads to impaired neurotransmitter release, neuromuscular junction destabilization and reduced muscle strength in mice. PLoS One. 9:e100834 10.1371/journal.pone.0100834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y., Branicky R., Landaverde I.O., and Hekimi S.. 2003. Redox regulation of germline and vulval development in Caenorhabditis elegans. Science. 302:1779–1782. 10.1126/science.1087167 [DOI] [PubMed] [Google Scholar]
- Silva J.P., Shabalina I.G., Dufour E., Petrovic N., Backlund E.C., Hultenby K., Wibom R., Nedergaard J., Cannon B., and Larsson N.G.. 2005. SOD2 overexpression: enhanced mitochondrial tolerance but absence of effect on UCP activity. EMBO J. 24:4061–4070. 10.1038/sj.emboj.7600866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot J.W., Geuze H.J., Freeman B.A., and Crapo J.D.. 1986. Intracellular localization of the copper-zinc and manganese superoxide dismutases in rat liver parenchymal cells. Lab. Invest. 55:363–371. [PubMed] [Google Scholar]
- Sohal R.S., and Weindruch R.. 1996. Oxidative stress, caloric restriction, and aging. Science. 273:59–63. 10.1126/science.273.5271.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S., Zhang X., Wu H., Han Y., Zhang J., Ma E., and Guo Y.. 2014. Molecular basis for antioxidant enzymes in mediating copper detoxification in the nematode Caenorhabditis elegans. PLoS One. 9:e107685 10.1371/journal.pone.0107685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler J.S., Singel D.J., and Loscalzo J.. 1992. Biochemistry of nitric oxide and its redox-activated forms. Science. 258:1898–1902. 10.1126/science.1281928 [DOI] [PubMed] [Google Scholar]
- Stöcker S., Van Laer K., Mijuskovic A., and Dick T.P.. 2018. The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs. Antioxid. Redox Signal. 28:558–573. 10.1089/ars.2017.7162 [DOI] [PubMed] [Google Scholar]
- Strålin P., Karlsson K., Johansson B.O., and Marklund S.L.. 1995. The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arterioscler. Thromb. Vasc. Biol. 15:2032–2036. 10.1161/01.ATV.15.11.2032 [DOI] [PubMed] [Google Scholar]
- Suetomi K., Mereiter S., Mori C., Takanami T., and Higashitani A.. 2013. Caenorhabditis elegans ATR checkpoint kinase ATL-1 influences life span through mitochondrial maintenance. Mitochondrion. 13:729–735. 10.1016/j.mito.2013.02.004 [DOI] [PubMed] [Google Scholar]
- Sullivan L.B., and Chandel N.S.. 2014. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2:17 10.1186/2049-3002-2-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., and Tower J.. 1999. FLP recombinase-mediated induction of Cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Mol. Cell. Biol. 19:216–228. 10.1128/MCB.19.1.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Folk D., Bradley T.J., and Tower J.. 2002. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics. 161:661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthammarak W., Somerlot B.H., Opheim E., Sedensky M., and Morgan P.G.. 2013. Novel interactions between mitochondrial superoxide dismutases and the electron transport chain. Aging Cell. 12:1132–1140. 10.1111/acel.12144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó C., Ischiropoulos H., and Radi R.. 2007. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 6:662–680. 10.1038/nrd2222 [DOI] [PubMed] [Google Scholar]
- Tatar M., Kopelman A., Epstein D., Tu M.P., Yin C.M., and Garofalo R.S.. 2001. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 292:107–110. 10.1126/science.1057987 [DOI] [PubMed] [Google Scholar]
- Tawe W.N., Eschbach M.L., Walter R.D., and Henkle-Dührsen K.. 1998. Identification of stress-responsive genes in Caenorhabditis elegans using RT-PCR differential display. Nucleic Acids Res. 26:1621–1627. 10.1093/nar/26.7.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J.P., Brown R.H. Jr., and Cleveland D.W.. 2016. Decoding ALS: from genes to mechanism. Nature. 539:197–206. 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada L.S. 2006. Specificity in reactive oxidant signaling: think globally, act locally. J. Cell Biol. 174:615–623. 10.1083/jcb.200605036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharmalingam S., Alhasawi A., Appanna V.P., Lemire J., and Appanna V.D.. 2017. Reactive nitrogen species (RNS)-resistant microbes: adaptation and medical implications. Biol. Chem. 398:1193–1208. 10.1515/hsz-2017-0152 [DOI] [PubMed] [Google Scholar]
- Tonks N.K. 2005. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 121:667–670. 10.1016/j.cell.2005.05.016 [DOI] [PubMed] [Google Scholar]
- Tousoulis D., Kampoli A.M., Tentolouris C., Papageorgiou N., and Stefanadis C.. 2012. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 10:4–18. 10.2174/157016112798829760 [DOI] [PubMed] [Google Scholar]
- Trachootham D., Lu W., Ogasawara M.A., Nilsa R.D., and Huang P.. 2008. Redox regulation of cell survival. Antioxid. Redox Signal. 10:1343–1374. 10.1089/ars.2007.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo M., Alvarez B., and Radi R.. 2016. One- and two-electron oxidation of thiols: mechanisms, kinetics and biological fates. Free Radic. Res. 50:150–171. 10.3109/10715762.2015.1089988 [DOI] [PubMed] [Google Scholar]
- Tsunoda S., Kibe N., Kurahashi T., and Fujii J.. 2013. Differential responses of SOD1-deficient mouse embryonic fibroblasts to oxygen concentrations. Arch. Biochem. Biophys. 537:5–11. 10.1016/j.abb.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M. 2006. Localizing NADPH oxidase-derived ROS. Sci. STKE. 2006:re8. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk J.M., and Hekimi S.. 2009. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 5:e1000361 10.1371/journal.pgen.1000361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk J.M., and Hekimi S.. 2012. Superoxide dismutase is dispensable for normal animal lifespan. Proc. Natl. Acad. Sci. USA. 109:5785–5790. 10.1073/pnas.1116158109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Remmen H., Williams M.D., Guo Z., Estlack L., Yang H., Carlson E.J., Epstein C.J., Huang T.T., and Richardson A.. 2001. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am. J. Physiol. Heart Circ. Physiol. 281:H1422–H1432. 10.1152/ajpheart.2001.281.3.H1422 [DOI] [PubMed] [Google Scholar]
- Van Remmen H., Ikeno Y., Hamilton M., Pahlavani M., Wolf N., Thorpe S.R., Alderson N.L., Baynes J.W., Epstein C.J., Huang T.T., et al. 2003. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics. 16:29–37. 10.1152/physiolgenomics.00122.2003 [DOI] [PubMed] [Google Scholar]
- Wan X.S., Devalaraja M.N., and St Clair D.K.. 1994. Molecular structure and organization of the human manganese superoxide dismutase gene. DNA Cell Biol. 13:1127–1136. 10.1089/dna.1994.13.1127 [DOI] [PubMed] [Google Scholar]
- Wang Y., and Hekimi S.. 2015. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science. 350:1204–1207. 10.1126/science.aac4357 [DOI] [PubMed] [Google Scholar]
- Winterbourn C.C. 2008. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4:278–286. 10.1038/nchembio.85 [DOI] [PubMed] [Google Scholar]
- Wolf M., Nunes F., Henkel A., Heinick A., and Paul R.J.. 2008. The MAP kinase JNK-1 of Caenorhabditis elegans: location, activation, and influences over temperature-dependent insulin-like signaling, stress responses, and fitness. J. Cell. Physiol. 214:721–729. 10.1002/jcp.21269 [DOI] [PubMed] [Google Scholar]
- Woodruff R.C., Phillips J.P., and Hilliker A.J.. 2004. Increased spontaneous DNA damage in Cu/Zn superoxide dismutase (SOD1) deficient Drosophila. Genome. 47:1029–1035. 10.1139/g04-083 [DOI] [PubMed] [Google Scholar]
- Wu J.Z., Huang J.H., Khanabdali R., Kalionis B., Xia S.J., and Cai W.J.. 2016. Pyrroloquinoline quinone enhances the resistance to oxidative stress and extends lifespan upon DAF-16 and SKN-1 activities in C. elegans. Exp. Gerontol. 80:43–50. 10.1016/j.exger.2016.04.008 [DOI] [PubMed] [Google Scholar]
- Yanase S., Yasuda K., and Ishii N.. 2002. Adaptive responses to oxidative damage in three mutants of Caenorhabditis elegans (age-1, mev-1 and daf-16) that affect life span. Mech. Ageing Dev. 123:1579–1587. 10.1016/S0047-6374(02)00093-3 [DOI] [PubMed] [Google Scholar]
- Yanase S., Onodera A., Tedesco P., Johnson T.E., and Ishii N.. 2009. SOD-1 deletions in Caenorhabditis elegans alter the localization of intracellular reactive oxygen species and show molecular compensation. J. Gerontol. A Biol. Sci. Med. Sci. 64:530–539. 10.1093/gerona/glp020 [DOI] [PubMed] [Google Scholar]
- Yang W., and Hekimi S.. 2010. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 8:e1000556 10.1371/journal.pbio.1000556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Li J., and Hekimi S.. 2007. A Measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics. 177:2063–2074. 10.1534/genetics.107.080788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C., Yang W., and Hekimi S.. 2014. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell. 157:897–909. 10.1016/j.cell.2014.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara D., Fujiwara N., Kitanaka N., Kitanaka J., Sakiyama H., Eguchi H., Takemura M., and Suzuki K.. 2016. The absence of the SOD1 gene causes abnormal monoaminergic neurotransmission and motivational impairment-like behavior in mice. Free Radic. Res. 50:1245–1256. 10.1080/10715762.2016.1234048 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Zhang H.M., Shi Y., Lustgarten M., Li Y., Qi W., Zhang B.X., and Van Remmen H.. 2010. Loss of manganese superoxide dismutase leads to abnormal growth and signal transduction in mouse embryonic fibroblasts. Free Radic. Biol. Med. 49:1255–1262. 10.1016/j.freeradbiomed.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Ikeno Y., Bokov A., Gelfond J., Jaramillo C., Zhang H.M., Liu Y., Qi W., Hubbard G., Richardson A., and Van Remmen H.. 2013. Dietary restriction attenuates the accelerated aging phenotype of Sod1(-/-) mice. Free Radic. Biol. Med. 60:300–306. 10.1016/j.freeradbiomed.2013.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Unnikrishnan A., Deepa S.S., Liu Y., Li Y., Ikeno Y., Sosnowska D., Van Remmen H., and Richardson A.. 2017. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1-/- mice is correlated to increased cellular senescence. Redox Biol. 11:30–37. 10.1016/j.redox.2016.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Liu J., Pan S., Sun Y., Li Q., Li F., Ma L., and Guo Q.. 2013. SOD mRNA and MDA expression in rectus femoris muscle of rats with different eccentric exercise programs and time points. PLoS One. 8:e73634 10.1371/journal.pone.0073634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Vanhoutte P.M., and Leung S.W.. 2015. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 129:83–94. 10.1016/j.jphs.2015.09.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.