

Romo1 regulates mitochondrial reactive oxygen species production and acts as an essential redox sensor in mitochondrial dynamics. Lee et al. demonstrate that Romo1 is a unique mitochondrial ion channel with viroporin-like characteristics that distinguish Romo1 from other known eukaryotic ion channels.
Abstract
Reactive oxygen species (ROS) modulator 1 (Romo1) is a nuclear-encoded mitochondrial inner membrane protein known to regulate mitochondrial ROS production and to act as an essential redox sensor in mitochondrial dynamics. Although its physiological roles have been studied for a decade, the biophysical mechanisms that explain these activities of Romo1 are unclear. In this study, we report that Romo1 is a unique mitochondrial ion channel that differs from currently identified eukaryotic ion channels. Romo1 is a highly conserved protein with structural features of class II viroporins, which are virus-encoded nonselective cation channels. Indeed, Romo1 forms a nonselective cation channel with its amphipathic helical transmembrane domain necessary for pore-forming activity. Notably, channel activity was specifically inhibited by Fe2+ ions, an essential transition metal ion in ROS metabolism. Using structural bioinformatics, we designed an experimental data–guided structural model of Romo1 with a rational hexameric structure. We propose that Romo1 establishes a new category of viroporin-like nonselective cation channel in eukaryotes.
Graphical Abstract

Introduction
Mitochondria are multifunctional cellular organelles present in most eukaryotes. As cellular power plants, mitochondria generate energy in the form of ATP, which is used for diverse cellular activities (Friedman and Nunnari, 2014). Unlike other cellular organelles with a single membrane, mitochondria are surrounded by outer and inner membranes (Palade, 1953). The outer membrane allows ions and small molecules <6.5 kD to freely diffuse through pore-forming channels; thus, the membrane potential across the outer membrane is negligibly small (Kühlbrandt, 2015; Hockenbery, 2016). In contrast, the inner mitochondrial membrane has low permeability, preserving the membrane potential (Δψm) generated by the mitochondrial respiratory chain. Transport of ions through the inner membrane is therefore tightly regulated by ion channels to prevent undesired energy dissipation (O’Rourke, 2007).
Δψm is known to be controlled in the narrow range of −136 to −139 mV for optimal ATP synthesis (Gerencser et al., 2012; Bagkos et al., 2014). Even subtle (±10%) changes beyond this range critically decrease the ATP synthesis rate and result in significant mitochondrial ROS production (Korshunov et al., 1997). For this reason, maintenance of membrane integrity and Δψm within the optimal range is essential to ensure a sufficient cellular ATP supply and for intracellular reactive oxygen species (ROS) management. Mitochondrial K+ channels in the inner membrane have been proposed to finely regulate Δψm and ROS production (Szewczyk et al., 2009). These channels transport K+ ions—the most abundant intracellular monovalent cations—through the inner membrane from the mitochondrial intermembrane space to the negatively charged mitochondrial matrix (Laskowski et al., 2016). This leads to a subtle depolarization (e.g., ≤2 mV by mitoKATP channels), which stimulates the respiratory chain to restore Δψm within the optimal range (Kowaltowski et al., 2001; Heinen et al., 2007; Leanza et al., 2015). Although mitochondrial K+ channels are primarily involved in this process, nonselective cation channels can also play a role. The most famous nonselective cation channel in the inner membrane is adenine nucleotide translocase (ANT), which switches roles from an ADP/ATP exchanger to a nonselective cation channel (PK/PCl = 4:1) in response to high Ca2+ concentration (Brustovetsky and Klingenberg, 1996). Because this channel type has a more powerful depolarizing effect than selective ion channels, opening of nonselective ion channels has been mainly studied in the context of pathological events such as mitochondrial swelling followed by cell death (Halestrap and Richardson, 2015). For example, ANT is known to be part of the permeability transition pore that facilitates diffusion of ions and small molecules inducing cell death in response to pathological signals. However, flickering of nonselective ion channels in low conductance mode has also been suggested to promote subtle mitochondrial depolarization spikes to maintain mitochondrial homeostasis (Brenner and Moulin, 2012).
Although it is clear that mitochondrial nonselective ion channels in the inner membrane have crucial roles in Δψm regulation, few novel mitochondrial ion channels have been identified because of the limitations of conventional homology-based studies. These limitations can be overcome using methods that evaluate local structural similarities (Marcotte, 2000; Sael et al., 2012). For example, it is noteworthy that many viruses encode nonconventional ion channels collectively termed viroporins. These are small hydrophobic proteins with one or two α-helical transmembrane domains (TMDs) that form homooligomeric channels through which protons, cations, and large molecules for viral production are transported (Nieva et al., 2012). Interestingly, most viroporins are known to be nonselective cation channels even though they do not exhibit interspecies homology (Delcour, 2015). Considering that their structural features confer an ion channel function, we posed the following question: do eukaryotes harbor novel nonselective cation channels with viroporin-like structural features that cannot be identified by conventional homology-based methods?
In 2006, the novel mitochondrial protein ROS modulator 1 (Romo1) was identified (Chung et al., 2006). This is a nuclear-encoded small hydrophobic protein composed of 79 amino acids and is expressed ubiquitously in many tissues (brain, heart, skeletal muscle, kidney, pancreas, liver, lung, and placenta; Zhao et al., 2009). For a decade, Romo1 has been studied in the context of mitochondrial ROS production (Chung et al., 2006, 2009; Na et al., 2008), cancer cell invasion (Chung et al., 2012, 2014; Lee et al., 2015), inflammation (Kim et al., 2010), replicative senescence (Chung et al., 2008), and mitochondrial dynamics (Norton et al., 2014). Although Romo1 is thought to be involved in mitochondrial superoxide production and functions as an essential redox sensor in mitochondrial dynamics, the mechanism by which Romo1 is involved in mitochondrial ROS generation is unknown. In this study, we performed biophysical characterization of Romo1 and demonstrated that it is a unique mitochondrial nonselective cation channel with viroporin-like characteristics.
Results
Romo1 is a highly conserved protein with an amphipathic helical TMD
The roles of Romo1 in mitochondria have been studied for a decade, but its biophysical functions have not been elucidated. Prediction of Romo1’s biophysical function using homology-based methods is not possible because Romo1 shows no detectable homology to well-known eukaryotic or prokaryotic protein families. Therefore, we attempted to deduce the biophysical functions of Romo1 using a nonhomology-based method that utilizes local structural similarities; this method was proposed as an attractive alternative to homology-based methods for predicting the functions of uncharacterized proteins (Marcotte, 2000; Sael et al., 2012). We found that Homo sapiens Romo1 has two TMDs, each consisting of an α helix, that are connected by a basic loop, whereas the N terminus is predicted to be a variable disordered region (Fig. 1 A). In contrast with TMD1, which contains a hydrophobic α helix, TMD2 contains polar amino acids (K58, T59, Q62, S63, T66, and T69). Interestingly, these amino acids are each separated by three to four amino acids, implying that they are clustered at a single surface in α helix 2, because there are 3.6 amino acids per α-helical turn. We examined the α-helical wheel of the N- and C-terminal α helices and confirmed that the C-terminal α helix in TMD2 forms an amphipathic helix as predicted (Fig. 1 B). To investigate whether these secondary structural features of Romo1 are conserved throughout eukaryotes, we constructed a phylogenetic tree using the publicly available Romo1 sequences of 460 species (247 animals, 120 fungi, 57 plants, and 36 protists; Fig. 1 C). Romo1 was sufficiently conserved to distinguish evolutionary relationships among the four kingdoms of Eucarya. Moreover, the amino acid sequences of Romo1 in 82 of 247 animal species were 100% identical with that of H. sapiens Romo1, indicating high sequence conservation.
Figure 1.

Secondary structure analysis of Romo1 in eukaryotes. (A) Secondary structure prediction of H. sapiens Romo1. The Romo1 sequence was described using the WebLogo 3 server. The indicated secondary structures were predicted using JPRED (α-helix), HMMTOP (TMDs), and DISOPRED (disordered region). (B) α-Helical wheel of H. sapiens Romo1. Illustrations were recreated based on results from the Helical Wheel Projections server. Blue, nonpolar amino acids; red, polar amino acids. (C) Phylogenetic relationships of Romo1. A phylogenetic tree was built from 460 Romo1 amino acid sequences using the maximum likelihood method based on the JTT matrix–based model. Evolutionary analyses were performed using MEGA7 software. Blue, fungi; green, plants; red, animals; purple, protists. (D–F) Analysis of position-specific polarity conservation. Multiple sequence alignments of nonidentical Romo1 sequences from 104 animal species to determine position-specific polarity conservation (D). Numbers below sequences represent the amino acid position of H. sapiens Romo1 based on the results of multiple sequence alignment. Position-specific polarity was calculated as the ratio of conservation of polar amino acids at the indicated position, and bars represent the conservation percentage of polar amino acids. The phylogenetic tree was built with 104 nonidentical sequences using the maximum likelihood method based on the JTT matrix–based model (E). The 104 nonidentical sequences were classified as originating from vertebrates or invertebrates to determine position-specific polarity conservation (F). (G) α-Helical wheel of position-specific polarity conservation in animal and fungal species. Numbers around the α-helical wheel represent the amino acid position of H. sapiens Romo1.
We examined whether the position-specific polarity observed in H. sapiens Romo1 was conserved across Eucarya. To minimize sequence redundancy, we selected 104 nonidentical sequences from the 247 species of animals. Amino acid positions corresponding with positions 58, 62, and 66 of H. sapiens Romo1 showed the same polarity in all animal species investigated, whereas amino acids in positions 54, 55, 59, and 63 did not show consistent polarity across the phylogeny (Fig. 1 D). We found that the Romo1 sequences of 104 animals could be divided into two groups: vertebrates and invertebrates (Fig. 1 E). Interestingly, amino acids in positions 54, 55, 59, and 63 showed opposite polarity between vertebrates and invertebrates (Figs. 1 F and S1). In vertebrates, the polar surface of the amphipathic helix is formed by amino acids at positions 58, 59, 62, 63, 66, and 69; in invertebrates, loss of polarity at positions 59 and 63 appears to be compensated for by acquisition of polar amino acids at positions 54 and 55. This translates into differences in the charge of the polar surface of the amphipathic helix between vertebrates and invertebrates, but amphipathic helical features were still present in both vertebrates and invertebrates (Fig. 1 G). Similarly, the polar surface of the amphipathic helix was conserved in 120 species of fungi, but the conserved polar surface in fungi was relatively wider than that in animals. These results indicate that Romo1 is a highly conserved protein with amphipathic helical features.
Romo1 forms homooligomers in a membrane environment
To deduce the biophysical function of Romo1 from its secondary structural features, we attempted to find a protein family consisting of small α-helical transmembrane proteins with one amphipathic helical TMD. One family that met this description was class II viroporins, a family of virus-encoded nonselective ion channels. These proteins harbor two TMDs with at least one amphipathic helical TMD, and an amphipathic helical TMD is required for these proteins to form homooligomeric nonselective ion channels. For example, the first TMD of hepatitis C virus (HCV) p7 and the second TMD of picornavirus 2B each consist of an amphipathic α helix that is responsible for ion channel activity (Patargias et al., 2006; Sánchez-Martínez et al., 2012). These led us to hypothesize that Romo1 is a viroporin-like eukaryotic ion channel. To evaluate this hypothesis, we first examined Romo1 oligomerization, which is a prerequisite for ion channel formation. We lysed isolated mitochondria from HEK293 cells with two detergents: digitonin, which is generally used to preserve protein complex interactions, and octyl-β-d-glucoside (ODG). We found that Romo1 migrated as three bands in digitonin lysates, whereas it migrated as only one band at ∼30–50 kD in ODG lysates in blue native (BN)/SDS-PAGE (Fig. 2 A). We added DTT to the ODG lysates to exclude the possibility of disulfide bonds and found that DTT treatment did not affect Romo1 oligomerization (Fig. 2 B). This indicates that Romo1 oligomerization is not dependent on disulfide bonds.
Figure 2.
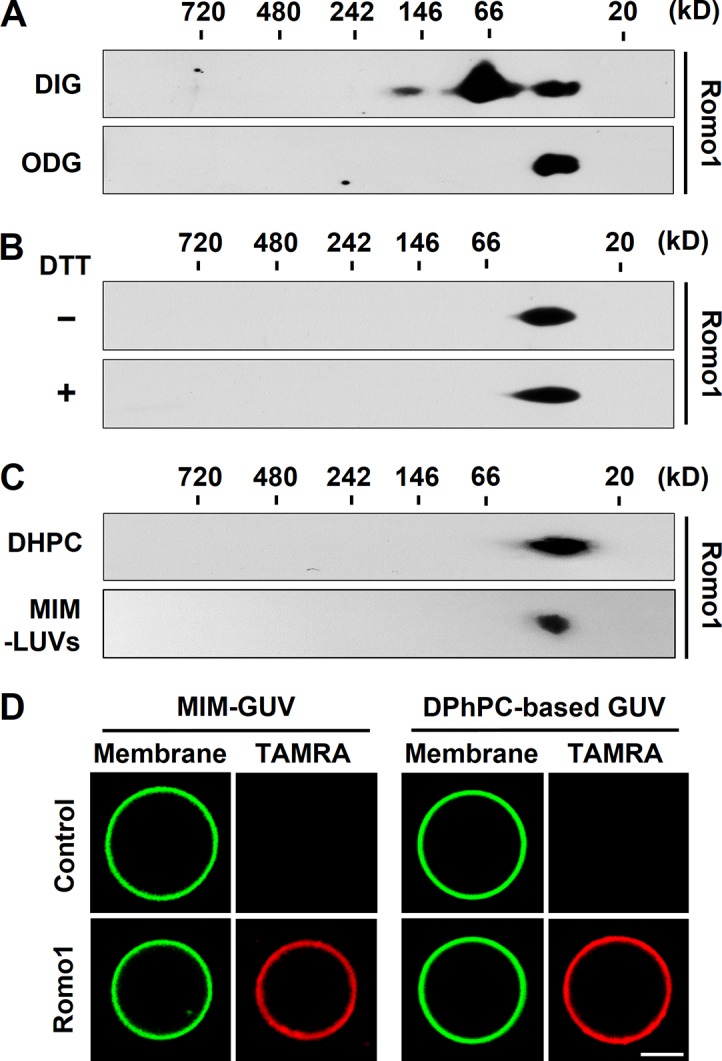
Romo1 oligomerization in mitochondria and artificial membrane environments. (A and B) Romo1 oligomerization in mitochondria. Isolated mitochondria from HEK293 cells were lysed with 0.5% digitonin (DIG) or ODG. The resultant lysates were analyzed by BN/SDS-PAGE (A). Mitochondrial lysates with 1% ODG were incubated with DTT for 30 min and then analyzed by BN/SDS-PAGE (B). (C) Synthesized Romo1 was incubated in 300 mM sucrose and 10 mM Hepes/Tris, pH 7, with 50 mM DHPC or added to MIM-mimicking LUVs (MIM-LUVs) in 300 mM sucrose and 10 mM Hepes/Tris, pH 7, for 2 h at room temperature. The resultant protein products were analyzed by BN/SDS-PAGE. (D) MIM-mimicking GUVs (MIM-GUVs) and 8:1:1 (molar ratio) DPhPC/DPhPG/cholesterol (DPhPC-based GUVs) with 0.2% TopFluor-cholesterol were prepared by GUV electroformation. Synthesized TAMRA-labeled Romo1 was added to GUVs for 5 min followed by confocal microscopy analysis. Bar, 10 µm.
Although we showed that Romo1 forms oligomers in mitochondria, we could not exclude the possibility of detergent-resistant heterooligomers. To clarify whether Romo1 forms homo- or heterooligomers, we chemically synthesized Romo1 and reconstituted it with 1,2-diheptanoyl-sn-glycero-3-phosphocholine (DHPC), a detergent widely used for viroporin reconstitution. We found that DHPC-reconstituted Romo1 homooligomerized as tetramers to hexamers (8.2 kD/monomer; Fig. 2 C); these oligomers were identical to the Romo1 oligomers detected in mitochondria isolated from HEK293 cells (Fig. 2 A). We also reconstituted Romo1 in large unilamellar vesicles (LUVs) with a previously reported rat liver mitochondrial inner membrane (MIM) composition (Daum and Vance, 1997) to mimic the membrane environment. We confirmed that Romo1 forms homooligomers in a membrane environment similar to DHPC-reconstituted Romo1 (Fig. 2 C). To determine whether Romo1 targets liposomes, we visualized Romo1 targeting using 5-carboxy-tetramethylrhodamine (TAMRA)-labeled Romo1 and giant unilamellar vesicles (GUVs). As expected, TAMRA-Romo1 was clearly targeted to MIM-GUVs and 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC)-based GUVs (Fig. 2 D). We also confirmed the oligomerization of MIM-GUV–reconstituted TAMRA-Romo1 (Fig. S2 A). These results indicate that Romo1 forms stable homooligomers of 30–50 kD in mitochondria and an artificial membrane environment.
Romo1 induces membrane permeabilization
The ability of viroporins to induce membrane permeabilization has been studied extensively (Nieva et al., 2012); we therefore examined whether Romo1 possesses viroporin-like pore-forming activity in a membrane environment. To evaluate the pore-forming activity of Romo1, we used a liposome permeabilization assay frequently used in viroporin research (Nieva et al., 2012). We generated MIM-LUVs filled with carboxyfluorescein (CF), a fluorescent dye quenched at high concentrations, and found that Romo1 induced liposome permeabilization in a concentration- and time-dependent manner (Fig. 3, A and B). Moreover, the activity of Romo1 was more potent than that of the well-known pore-forming antimicrobial peptide alamethicin. Because our analysis implied that TMD2 forms a membrane-active pore-forming domain, we synthesized the 1–43 and 44–79 regions of Romo1 (Fig. 3 C) and found that 44–79 induced liposome permeabilization, whereas 1–43 did not (Fig. 3 D). We also synthesized the sequential deletion mutants from the C terminus of 44–79 (44–75, 44–70, and 44–65), but its sequential deletion mutants did not induce liposome permeabilization (Fig. 3 E).
Figure 3.

Romo1-induced membrane permeabilization. (A–E) Romo1-induced liposome permeabilization. CF-LUVs were incubated with the indicated concentrations of Romo1, and liposome permeabilization was monitored by a spectrophotometric assay (A). (B) CF release was quantified at 10 min after Romo1 treatment. (C) Illustration of deletion mutants of Romo1. (D and E) CF-LUVs were incubated with Romo1 or the indicated deletion mutants (1 µM) for 10 min, after which liposome permeabilization was analyzed. Alamethicin (Ala.) was used as a positive control. (F–K) Romo1-induced mitochondrial depolarization. (F) The purity of isolated mouse liver mitochondria was examined by flow cytometry using the mitochondrial-specific marker MitoTracker green FM. (G and H) Mitochondrial targeting of Romo1 was analyzed by flow cytometry with 0.2 µM TAMRA-Romo1 (G) and confirmed by confocal microscopy (H). Bar, 10 µm. (I and J) Isolated liver mitochondria were treated with the indicated concentrations of Romo1 for 5 min and then stained with the Δψm indicator JC-1, after which they were analyzed by flow cytometry (I) and spectrophotometric assay (J). (K) Romo1 or its deletion mutants (1 µM) were added to isolated liver mitochondria for 5 min followed by staining with JC-1, after which Δψm was quantified by spectrophotometric assay. Carbonyl cyanide m-chlorophenyl hydrazine (CCCP) was used as a positive control. (L–N) CF-LUVs were incubated with the indicated deletion or point mutants of Romo1 (1 µM) for 10 min, and liposome permeabilization was quantified by a spectrophotometric assay. Data represent means ± SD of three independent experiments. ***, P ≤ 0.001 by two-way ANOVA. Cont., control.
We next investigated the effects of Romo1-induced membrane permeabilization on mitochondria. Because MIM permeabilization can be assessed by measuring the decrease in Δψm, we examined whether Romo1 depolarizes mitochondria. Although Romo1 expression has been shown to decrease Δψm in cells (Chung et al., 2008; Kim et al., 2010), it is difficult to determine whether this mitochondrial depolarization is caused by the direct effect of Romo1 on pore-forming activity or an indirect effect. We therefore designed an experimental workflow for directly targeting synthesized Romo1 to isolated liver mitochondria to minimize the effects of cellular factors. We first confirmed the purity (>90%) of isolated liver mitochondria by flow cytometry using MitoTracker green FM (Fig. 3 F). Next, we evaluated the targeting of Romo1 by adding TAMRA-Romo1 to isolated liver mitochondria and found that Romo1 was efficiently targeted to the isolated mitochondria within 5 min (Fig. 3, G and H). To evaluate Romo1-induced mitochondrial depolarization, Romo1-treated mitochondria were stained with the Δψm indicator JC-1. We found that Romo1 induced mitochondrial depolarization in a concentration-dependent manner (Fig. 3, I and J). The deletion mutant 44–79 also induced mitochondrial depolarization, but the sequential deletion mutants 44–75, 44–70, and 44–65 had negligible effects (Figs. 3 K and S3). Because it is still unclear whether the length of TMD2 or C-terminal amino acids is essential for the formation of pores, we synthesized additional deletion mutants (44–78, 44–77, and 44–76) based on 44–79. Surprisingly, deletion of C79 affected the Romo1 activity significantly (Fig. 3 L). Moreover, the C79A mutant showed decreased activity compared with 44–79 (Fig. 3 M). These results are consistent with results of Norton et al. (2014) showing that C15 and C79 are essential for redox sensor function of Romo1 in mitochondrial dynamics, and C-terminally tagged Romo1 showed a dominant-negative effect on mitochondrial fusion. We also examined the F67A + F70A mutant (FFAA), which has been demonstrated as a dominant-negative mutant (Norton et al., 2014). We found that it forms a stable oligomer in mitochondria regardless of DTT treatment (Fig. S2 B). We synthesized the FFAA mutant (based on 44–79) to examine whether it affects Romo1 channel activity. Surprisingly, the FFAA mutant showed no effect on liposome permeabilization (Fig. 3 N). Therefore, the dominant-negative effect of Romo1 FFAA mutant on mitochondrial dynamics may be caused by loss of channel activity. Collectively, these results indicate that the homooligomeric form of Romo1 can induce membrane permeabilization and that the C-terminal amphipathic helix TMD2 is essential for pore-forming activity.
Romo1 shows ion channel activities in bilayer patch clamp experiments, and its activity is inhibited by a viroporin inhibitor
To determine whether Romo1 functions as an ion channel, we performed planar bilayer patch clamp assays using synthesized Romo1. Because Romo1 readily aggregated when salts were used in the targeting buffer, we designed a novel targeting method applicable to our bilayer patch clamp system to maximize targeting efficiency (Fig. 4 A). Because 1 M sorbitol has a greater density than the recording buffer used (150 mM KCl and Hepes/Tris, pH 7), we speculated that Romo1 diluted with sorbitol solution could be overlaid on the preformed planar bilayer, thus effectively targeting Romo1 to the bilayer. To ensure that the method we designed functioned as planned, we added GUVs to form a planar bilayer on a glass chamber filled with 150 mM KCl and 10 mM Hepes/Tris, pH 7, and then overlaid the Romo1-containing sorbitol solution. As expected, Romo1 (red) exhibited satisfactory targeting to the preformed bilayer (green) within 5 min (Fig. 4 B). We applied this method to a planar bilayer patch clamp system. A symmetrical buffer containing 150 mM KCl and 10 mM Hepes/Tris, pH 7, was used as both the external and internal buffer (Fig. 4 C). After Romo1-containing sorbitol solution was added to the preformed bilayer, the ion channel activity of Romo1 was monitored after application of −120 mV. We found that Romo1 showed single-channel activity (Fig. 4 D) with low conductance (21 pS; Fig. 4 E). Because Romo1 currents increased continuously throughout Romo1 targeting, the single-channel current was observed for only a limited time and then changed to viroporin-like macroscopic currents (Fig. 4 F). The macroscopic currents of Romo1 showed a double-rectification current–voltage (I–V) relationship (Fig. 4 G). We also tested the 44–79 region that was predicted to be a pore-forming domain; as expected, this region showed ion channel activity with low conductance (17 pS; Fig. 4 H).
Figure 4.

Measurement of the ion channel activity of Romo1. (A) Illustration of planar bilayer formation and Romo1 targeting. (B) Romo1 targeting to the planar bilayer. GUVs (8:1:1 DPhPC/DPhPG/cholesterol molar ratio) were used to form a planar bilayer in a glass chamber filled with 150 mM KCl and 10 mM Hepes/Tris, pH 7. TAMRA-Romo1 (in 1 M sorbitol) was overlaid onto the planar bilayer for 5 min, after which the bilayer was washed with 150 mM KCl and 10 mM Hepes/Tris, pH 7. Images were captured using a fluorescence microscope. Bar, 40 µm. (C) Illustration of the NPC-1 chip used in the Port-a-Patch system. External and internal buffers were the same, i.e., 150 mM KCl and 10 mM Hepes/Tris, pH 7. Gray, NPC-1 chip; green, planar bilayer, pale blue; internal and external buffers. (D–F) Bilayer patch clamp assay of Romo1. Romo1 (in 1 M sorbitol) was added to a preformed planar bilayer, and a voltage of −120 mV was applied. Activities were then monitored. D shows the single channel current at −120 mV and E shows histogram of single channel conductance of Romo1. (F) Macroscopic currents using a step protocol from −150 to 150 mV were recorded. (G) I–V relationship of macroscopic currents. Currents from a ramp protocol from −150 to 150 mV (2-s duration) were averaged from five sweeps and plotted on the I–V curve. (H) Ion channel activity of the 44–79 region of Romo1. Control data were acquired before treatment with 44–79.
Because we suggested that Romo1 is a viroporin-like ion channel, we tested viroporin inhibitors on Romo1 channel activity. Interestingly, channel activity of Romo1 was inhibited by hexamethylene amiloride (HMA) but not by amantadine, rimantadine, or N-nonyldeoxynojirimycin (Fig. S4 A). Because knockdown of Romo1 expression was shown to increase the Δψm (Kim et al., 2010), we applied these inhibitors to HEK293 cells and found that HMA increased the Δψm, but no other viroporin inhibitor had this effect (Fig. S4, B and C). Next, we examined the inhibitory effects of HMA on the previously reported Romo1-induced mitochondrial depolarization (Kim et al., 2010). We found that 1 µM HMA was sufficient to inhibit Romo1-induced mitochondrial depolarization (Fig. S4 D) and that Flag-Romo1 expression was not altered by HMA treatment (Fig. S4 E). Because Romo1-induced mitochondrial depolarization was shown to be directly linked with TNF-α/cycloheximide (CHX)–induced mitochondrial depolarization (Kim et al., 2010), we tested the effect of HMA on TNF-α/CHX–induced mitochondrial depolarization. As expected, only HMA blocked TNF-α/CHX–induced mitochondrial depolarization (Fig. S4 F). These results demonstrate that Romo1 functions as an viroporin-like ion channel in artificial bilayer and that TMD2 is sufficient to induce ion channel activity.
Romo1 is a nonselective monovalent cation channel
We hypothesized that Romo1 is a nonselective cation channel like class II viroporins. To examine the selectivity of the channel for K+ versus Cl− ions, the external buffer was exchanged to a buffer containing 1.5 M KCl and 10 mM Hepes/Tris, pH 7. The reversal potential was measured to be 42.3 ± 1.4 mV (means ± SD), indicating that Romo1 exhibits ∼10 times more selectivity for K+ over Cl− ions (Fig. 5 A). Because Romo1 lacks a conserved potassium filter such as the TVGYG sequence, we assumed that Romo1 would not display specific potassium ion selectivity. As expected, Romo1 did not exhibit specific selectivity among monovalent cations (Fig. 5 A) but did show Stokes radius-dependent cation selectivity (Fig. 5 B), implying that cations diffuse through the channel without dehydration. Interestingly, when a buffer containing 100 mM CaCl2 and 10 mM Hepes/Tris, pH 7, was used to measure the ion selectivity of monovalent over divalent cations, CaCl2 inhibited the Romo1 current, and no reversal potential was measured (Fig. 5 C). This phenomenon was also observed in liposome permeabilization assays with CaCl2 or MgCl2 (Fig. 5 D). We also examined the effect of Gd3+, a nonselective cation channel blocker. As expected, Romo1-induced liposome permeabilization was efficiently blocked by Gd3+ ions (Fig. 5 E). These results indicate that Romo1 is a nonselective monovalent cation channel.
Figure 5.

Determination of ion selectivity of Romo1. (A and B) Ion selectivity of Romo1. The external buffer was changed to 1.5 M KCl and 10 mM Hepes/Tris, pH 7, to determine selectivity for K+ versus Cl−. To determine selectivity for K+ versus cations, the external buffer was changed to 150 mM XCl and 10 mM Hepes/Tris, pH 7, and the reversal potential was calculated where X represents the indicated cation. Reversal potential was corrected by the liquid junction potential. (C and D) Effects of divalent cation on the channel activity of Romo1. (C) The external buffer was changed to 100 mM CaCl2, and the current was recorded using a step protocol from −150 to 150 mV. (D) CF-LUVs were incubated with the indicated concentrations of CaCl2 or MgCl2, and Romo1 (50 nM) was added for 10 min, after which liposome permeabilization was analyzed. (E) Effects of GdCl3 on Romo1-induced liposome permeabilization. CF-LUVs were incubated with the indicated concentration of GdCl3, and Romo1 (50 nM) was added for 10 min, after which liposome permeabilization was analyzed. Data represent means ± SD of three independent experiments. ***, P ≤ 0.001 by two-way ANOVA.
Romo1 activity is inhibited by Fe2+ ions
We showed that Romo1 induces membrane permeabilization (Fig. 3) and functions as a nonselective monovalent cation channel (Fig. 5). However, Romo1 appears to open without any activators under our artificial experimental conditions, i.e., liposome permeabilization and bilayer patch clamp assays. Because constitutively activated Romo1 in mitochondria could induce the undesired depolarization of Δψm, we expected that certain cellular factors would keep Romo1 in the closed state. Because Romo1 has been studied mainly in the context of mitochondrial ROS production, we first examined the effect of H2O2—the most important ROS in intracellular signaling—on Romo1-induced liposome permeabilization, but it showed no inhibitory effect (Fig. 6 A). We next explored factors known to be involved in mitochondrial ROS metabolism. Transition metal ions (Cu2+, Zn2+, and Fe2+) are essential for mitochondrial function and are closely related to intracellular ROS management (Rines and Ardehali, 2013); we therefore tested whether they inhibited Romo1 activity. Surprisingly, we found that Fe2+ specifically inhibited Romo1-induced liposome permeabilization, whereas Cu2+ and Zn2+ did not (Fig. 6 B).
Figure 6.

Inhibitory effect of Fe2+ ions on Romo1 activity. (A) Inhibitory effect of H2O2 on Romo1-induced liposome permeabilization. CF-LUVs were incubated with the indicated concentrations of H2O2, and Romo1 (50 nM) was added for 10 min. (B–D) Determination of the inhibitory effects of transition metal ions on Romo1 activity. CF-LUVs were incubated with the indicated ions (10 µM), and Romo1 (50 nM) was added for 10 min. Alamethicin (Ala.; 0.5 µM) was used as a negative control (B). The IC50 of Fe2+ was quantified by liposome permeabilization assay (C). CF-LUVs were incubated with the indicated ions (1 µM), and Romo1 (50 nM) was added for 10 min (D). Data represent means ± SD of three independent experiments. ***, P ≤ 0.001 by two-way ANOVA. Cont., control; D.W., distilled water.
It is noteworthy that iron is indispensable for mitochondrial enzymatic reactions and thus crucial for mitochondrial respiration. Iron ions can readily cycle between oxidized and reduced states in response to changes in the intracellular ROS environment. Because redox-active cytosolic free iron ions (also known as the cytosolic labile iron pool) are highly toxic in the cellular environment, most intracellular iron is stored in the nontoxic form of ferritin. Therefore, cytosolic free iron (0.2–1.5 µM, variable according to cell type) accounts for <5% of the total iron concentration and mainly comprises Fe2+ because of the reductive nature of the intracellular environment (Kakhlon and Cabantchik, 2002; Urrutia et al., 2014). We therefore investigated whether Romo1 function is inhibited by physiological concentrations of Fe2+. The half-maximal inhibition (IC50) value of Fe2+ was determined to be 0.4 µM (Fig. 6 C). Moreover, Romo1-induced liposome permeabilization was not inhibited by Fe3+, an oxidized form of Fe2+ (Fig. 6 D). Considering that the intracellular free iron concentration ranges from 0.2–1.5 µM, our results indicate that Romo1 could be regulated in response to fluctuation in free iron concentration and redox state of iron.
Computational modeling of Romo1 reveals a hexameric ion channel model
In this study, we showed that Romo1 is a small α-helical nonselective cation channel with characteristics of class II viroporins. These structural and functional features, which differ from those of currently identified eukaryotic ion channels, motivated us to determine the tertiary structure of the Romo1 oligomer. Because Romo1 is difficult to characterize by x-ray crystallography or nuclear magnetic resonance imaging because of its small size and hydrophobic nature, we designed an experimental data–guided structure prediction workflow extending from sequence to oligomer models by applying structural bioinformatics (Fig. 7 A). As the first step, we deleted the flexible M1–S14 region predicted to be a disordered domain (Fig. 1 A) because it hindered oligomeric structural prediction. The C15–C79 region was used for tertiary structure prediction using Robetta (Kim et al., 2004) and LOMETS (Wu and Zhang, 2007), leading to the prediction of 15 monomer models. We selected monomer models based on the α-helical structure of Romo1 predicted in Fig. 1 A and the reported membrane topology of Romo1 (the N and C termini are in the same direction; Ieva et al., 2013). This led to the selection of five potential monomer models from among the 15 monomer models (Fig. 7 B). Next, we docked the Romo1 monomers from tetramers to hexamers using M-ZDOCK (Pierce et al., 2005). This oligomer choice was in the range of predicted oligomers shown in Fig. 2 and generated 150 oligomer models. Because we already demonstrated that the TMD2 of Romo1 possesses pore-forming activity (Fig. 3 D), we selected TMD2-based pore-forming models (47 oligomers) and excluded highly tilted or TMD1-based pore-forming models. Among the 47 oligomer models, we selected 11 top oligomer models (3 tetramers, 4 pentamers, and 4 hexamers), each of which showed a different structural shape based on the M-ZDOCK criteria with visual inspection. Next, we performed structure refinement using GalaxyRefine-Complex (Heo et al., 2013) to improve the structural quality of the 11 oligomer models. As a last step, we evaluated the 11 oligomer models that had pores that were sufficiently large to allow passage of hydrated Li+ ions, consistent with our bilayer patch clamp assay results (Fig. 5 A), using PoreWalker (Pellegrini-Calace et al., 2009) and Chexvis (Fig. S5; Masood et al., 2015). Finally, one hexamer model remained (Fig. 7 C). In this hexamer model, the polar surface of the amphipathic helix faced the inside of the pore (Fig. 7 D). The bottleneck diameter of the Romo1 channel was predicted to be 4.79 Å in PoreWalker and 4.94 Å in Chexvis. K58, T59, Q62, T66, and T69 were predicted to be pore-lining residues by both PoreWalker and Chexvis (Fig. 7 E). These amino acids were those already predicted to be polar surface residues of the amphipathic helix, as shown in Fig. 1 B.
Figure 7.

Experimental data–guided Romo1 structural modeling. (A) Computational modeling workflow designed to predict Romo1 structure. (B–E) The Robetta and LOMETS servers predicted 15 monomers from the H. sapiens Romo1 sequence, of which five were selected (B). After application of the selection criteria, 1 hexamer remained (C and D). Pore-lining residues that were predicted in both PoreWalker and Chexvis server were visualized (E). (F–H) Romo1 point mutants from 44–79 were synthesized based on the predicted hexamer structure. The following mutants were synthesized: E51A, K58A, Q62A, and R78A. Mitochondrial depolarization (F) and liposome permeabilization (G and H) induced by the four Romo1 point mutants (1 µM) were quantified by spectrophotometric assay. Data represent means ± SD of three independent experiments. ***, P ≤ 0.001 by two-way ANOVA. Cont., control.
Interestingly, the positively charged amino acid K58 is located inside the pore (Fig. 7 E) in this model. This finding is somewhat counterintuitive because a positively charged lysine conflicts with the cation selectivity of Romo1. However, we found a similar example in a recent viroporin study: the nonselective cation channel HCV p7 harbors a positively charged amino acid, arginine (R35), at the pore entrance (OuYang et al., 2013). This residue was proposed to bind to anions and obstruct their diffusion while allowing diffusion of cations through the pore. We investigated whether this concept applied to Romo1. We synthesized a K58A mutant in addition to mutants of two charged residues: the pore-lining residue E51 and the residue R78 (outside the pore) in the 44–79 region. Surprisingly, the K58A mutant showed decreased induction of mitochondrial depolarization (Fig. 7 F) and liposome permeabilization (Fig. 7 G), whereas these effects were not observed in the E51A or R78A mutant; rather, the E51A mutant showed increased activity. To examine whether the K58A mutant actually forms an oligomer, we overexpressed the Flag–Romo1-K58A in HEK293 cells and found that it forms a stable oligomer (Fig. S2 C). We also predicted that the bottleneck of the Romo1 channel is located near the Q62 region (Fig. 7 E). We expected that a widened bottleneck would increase the activity compared with that of 44–79. Therefore, we synthesized the Q62A mutant based on 44–79 and found that it showed increased activity compared with 44–79 (Fig. 7 H). Collectively, our results imply that Romo1 can function as a hexameric nonselective cation channel in a manner analogous to viroporins.
Discussion
Romo1 was first identified in 2006 and has been reported to have a role in mitochondrial ROS production and related signaling pathways (Chung et al., 2006). Subsequent experiments demonstrated that Romo1 induces mitochondrial ROS production in response to various cellular stresses, e.g., inflammation, senescence, and serum deprivation. In the case of inflammation, for example, internalized TNF-α complex II binds to Romo1, inducing mitochondrial depolarization within 15 min (Kim et al., 2010). Notably, TNF-α–mediated mitochondrial depolarization and ROS production were efficiently inhibited by knockdown of Romo1 expression. Considering the channel activity that we demonstrated, Romo1 is likely to be closed to minimize undesired energy dissipation and specifically activated in certain stress environments. This assumption is also consistent with the expected function of a mitochondrial cation channel: it should be tightly regulated for optimal mitochondrial respiration. For example, mitoKATP channels are generally accepted to be mainly closed at high concentrations of intracellular ATP, but they can be activated in response to a decrease in ATP/ADP ratio (Laskowski et al., 2016). In this study, we found that the channel activity of Romo1 was inhibited by free Fe2+ ions and that the IC50 for Fe2+ (0.4 µM) was within the cytosolic free iron concentration range (0.2–1.5 µM; Fig. 6). Considering this, the channel activity of Romo1 could be regulated by the cytosolic free iron concentration, and channel activity is likely maintained at the lowest acceptable level by cellular factors such as Fe2+ to minimize undesired loss of Δψm.
This raises the questions of how and why Romo1 regulates mitochondrial ROS production. Romo1-induced mitochondrial superoxide production originates from complex III of the respiratory chain (Chung et al., 2008). Interestingly, knockdown of Romo1 expression has been reported to reduce (Norton et al., 2014) or increase (Chung et al., 2008) mitochondrial superoxide production depending on experimental conditions. Similar to these studies, even using the same isolated mitochondria models, mitochondrial K+ channels have been reported to increase or decrease ROS production (Szewczyk et al., 2009). These findings can be explained in the context of the fine tuning of Δψm. Because Δψm is tightly regulated in a narrow range for optimal respiration, a value even 10% out of this range could increase the production of ROS significantly. Romo1 and mitochondrial cation channel–induced subtle depolarization could fit the optimal range or lower than this range depending on experimental conditions. For example, if Δψm is slightly higher than the optimal range, activation of these channels could fine tune the Δψm, in turn reducing ROS production. In contrast, if Δψm is already in the optimal range, further depolarization could induce ROS production. Collectively, these results suggest that Romo1 fine tunes Δψm to regulate mitochondrial ROS production in response to cellular factors such as Fe2+. In future studies, we intend to focus on potential correlations between Romo1-induced ROS production and free intracellular iron fluctuations including variations in the redox state of irons.
Romo1 might interact with other mitochondrial protein complexes to maintain their optimal activities. We observed homooligomers with two additional bands in digitonin-lysed mitochondria. These bands could represent Romo1 homooligomers, but this might be a result of interaction with other protein complexes (Fig. 2 A). Indeed, Romo1 has been reported to interact with protein complexes involved in mitochondrial dynamics and protein transport. In the context of mitochondrial dynamics, Romo1 has been shown to bind to OPA1 and the MIM organizing system (MINOS) complex, both of which are required to form cristae junctions for maximum respiration potential (Norton et al., 2014). Moreover, knockdown of Romo1 was shown to decrease the number of cristae junctions and the mitochondrial fusion rate. This can be explained by plasma membrane–resident viroporins, which have been shown to decrease the electrostatic repulsion of contact membranes for viral budding (Nieva et al., 2012). Binding of Romo1 to the MINOS complex transiently decreases the local membrane potential between the inner membranes to reduce electrostatic repulsion, after which membrane contact is formed and cristae junctions are created. This hypothesis can also be applied to the role of Romo1 in mitochondrial protein transport. Mgr2 (Romo1 in Saccharomyces cerevisiae) was shown to bind to the translocase of the inner membrane 23 (TIM23) complex, which is essential for protein translocation into the MIM and matrix (Gebert et al., 2012). Interestingly, TIM and translocase of the outer membrane (TOM) complexes did not interact at elevated temperatures in Romo1-knockout S. cerevisiae. Multiple studies have shown that the TIM and TOM complexes are coupled at contact sites of the two membranes (Schleyer and Neupert, 1985; Jascur et al., 1992; Endo et al., 2003); therefore, the distance between the inner and outer membranes must be sufficiently small to enable contact between the TIM and TOM complexes. One possibility is that Romo1 decreases local electrostatic repulsion below the threshold to maintain contact between the TIM and TOM complexes as we suggested above for cristae junction formation. However, there are not any currently available techniques by which local membrane potential decreases in mitochondria can be detected; such techniques would need to be developed to demonstrate the function of Romo1 in reducing electrostatic repulsion between membranes.
It is noteworthy that viroporins possess an unusual feature referred to as channel-pore dualism (Fischer and Krüger, 2009). This phenomenon indicates that viroporins can function as an ion channel or large pore depending on the experimental conditions. For example, HCV p7 conducts H+, Na+, K+, and Ca2+ ions and even allows passage of large molecules like CF and 10-kD dextran (Premkumar et al., 2004; StGelais et al., 2007). Interestingly, HCV p7 has been shown to adopt two different structures for proton versus fluorescent dye release, which provides some support for the channel-pore dualism phenomenon (Gan et al., 2014). Because Romo1 also induces release of CF from liposomes (Fig. 3 A), it is also possible that Romo1 adopts other structures based on the channel-pore dualism theory.
In conclusion, we described a novel mitochondrial nonselective cation channel with viroporin-like characteristics. To our knowledge, this is the first demonstration of a viroporin-like nonselective cation channel in eukaryotes. We provided key evidence of the core function of Romo1 in the context of mitochondrial ROS production and also suggested that some membrane proteins that were not previously considered to be ion channels could potentially be classified as viroporin-like nonselective cation channels. We expect that identification of additional viroporin-like nonselective cation channels will shed light on ion channel function in eukaryotes.
Materials and methods
Chemicals
Egg l-α-phosphatidylcholine (PC), egg l-α-phosphatidylethanolamine (PE), liver l-α-phosphatidylinositol (PI), brain l-α-phosphatidylserine (PS), bovine heart cardiolipin (CL), DPhPC, 1,2-diphytanoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (DPhPG), and TopFluor-cholesterol were purchased from Avanti Polar Lipids. All other chemicals were purchased from Sigma-Aldrich. Anti-Romo1 antibody (TA505580) was purchased from Origene. H. sapiens Romo1 (MPVAVGPYGQ10 SQPSCFDRVK20 MGFVMGCAVG30 MAAGALFGTF40 SCLRIGMRGR50 ELMGGIGKTM60 MQSGGTFGTF70 MAIGMGIRC79) was chemically synthesized with or without TAMRA by GL Biochem and purified by HPLC.
Liposome permeabilization assay
For LUV preparation, 100 µl of a MIM-mimic lipid mixture (egg PC/egg PE/liver PI/brain PS/heart CL = 40:34:5:3:18 [weight ratio] dissolved in 4:1 chloroform/methanol [10 mg/ml]) was dried under a stream of nitrogen and rehydrated with 500 µl of 50 mM CF, 100 mM sucrose, and 5 mM Hepes/KOH, pH 8. Multilamellar liposomal suspensions were extruded through a 0.1-µm polycarbonate membrane using a Mini Extruder (Avanti Polar Lipids) and purified on a PD-10 desalting column (GE Healthcare). The standard external buffer was 150 mM KCl and 10 mM Hepes/Tris, pH 7. The osmotic pressures of all experimental buffers were adjusted to ∼300 mOsm with sucrose using a VAPRO osmometer (Wescor Inc.). For CF-LUVs incubated with 10 mM CaCl2 or MgCl2, 135 mM KCl were used to adjust the osmotic pressure. LUVs treated with 90% trifluoroethanol (TFE) were used as a control. The final TFE concentration was restricted to 0.9%, a percentage at which no significant CF release was observed. CF leakage was analyzed at λ 488-nm excitation and 517-nm emission using a SpectraMax 384 microplate reader (Molecular Devices) and calculated using the formula CF leakage (%) = 100 × (F − F0)/(Fmax − F0), where F is the measured fluorescence intensity, F0 is the fluorescence intensity of intact LUVs, and Fmax is the fluorescence intensity of LUVs treated with 0.1% Triton X-100.
Romo1 oligomerization assay
Mitochondria were isolated from HEK293 cells using a mitochondrial isolation kit (Thermo Fisher Scientific). To assess Romo1 oligomerization, mitochondria isolated from HEK293 cells were lysed with 0.5% digitonin or 1% ODG for 1 h on ice. Lysates were analyzed by BN-PAGE using NativePAGE Novex 4–16% Bis-Tris gels (Invitrogen). A BN-PAGE gel strip was used for secondary SDS-PAGE. For the homooligomerization assay, synthesized Romo1 was incubated with 50 mM DHPC, 300 mM sucrose, and 10 mM Hepes/Tris, pH 7, or added to MIM-LUVs in 300 mM sucrose and 10 mM Hepes/Tris, pH 7, for 2 h at room temperature. For MIM-LUV–reconstituted Romo1, samples were lysed with 1% ODG before BN/SDS-PAGE.
GUV preparation
An 8:1:1 DPhPC/DPhPG/cholesterol (molar ratio) mixture dissolved in chloroform (5 mM) or a MIM-mimic lipid mixture was used to produce GUVs using a Vesicle Prep Pro (Nanion Technologies). GUVs were electroformed following the manufacturer’s instructions. In brief, the lipid mixture was dried on indium tin oxide–coated glass, and the chamber was filled with 300 mM sucrose. GUVs were produced by application of 3 V peak-to-peak and 5 Hz for 2 h at 36°C. For confocal microscopy, each cover glass was spin coated with 3% polyvinyl alcohol to ensure GUV stability. Data were acquired using an LSM 700 microscope (ZEISS).
Bilayer patch clamp assay
Bilayer patch clamp assays were performed with a Port-a-Patch system (Nanion Technologies) using a 3–5-Mohm NPC-1 chip and amplified with an EPC-10 amplifier (HEKA Electronics). The same buffer was used in the external and internal chambers (150 mM KCl and 10 mM Hepes/Tris, pH 7). DPhPC-based GUVs were dropped onto the chip, and a pressure of −15 to −25 mbar was applied. After forming the correct seal over 5 Gohm, the bilayer was tested using a −150 to 150 mV ramp protocol to ensure bilayer stability. The baseline value was subtracted before Romo1 targeting to generate the I–V curve. For Romo1 targeting, Romo1 (in 90% TFE) was diluted with 1 M sorbitol and rapidly overlaid on the chip. Next, −120 mV was applied. The external buffer was quickly exchanged with the indicated buffer after the Romo1 signal was detected. For ion-selectivity calculations, the reversal potential of the zero mean current was recorded and corrected by the liquid junction potential calculated using JPCalcWin software (Barry, 1994). The Goldman-Hodgkin-Katz equation was used to obtain PX/PK (Eq. 1) and PK/PCl (Eq. 2). RT/zF was 25.6 at 25°C. All data were Gaussian low pass–filtered at 1 kHz.
Erev = (RT/zF)ln([X]exPX/[K]inPK) (Eq. 1)
Erev= (RT/zF)ln({[K]ex + [Cl]inPCl/PK}/{[K]in + [Cl]exPCl/PK}) (Eq. 2)
Measurement of Δψm
All experiments were approved by the Institutional Animal Care and Use Committee of the Korea University College of Medicine (KOREA-2016-0174). Male C57BL/6N mice (6 wk old) were sacrificed for liver mitochondrial isolation. Isolated liver mitochondria were incubated in mitochondrial incubation buffer composed of 250 mM mannitol, 1 mM KH2PO4, 10 µM EGTA, 2 µM rotenone, and 5 mM Hepes/KOH, pH 7.4, for flow cytometry and spectrophotometric assays. Romo1 was added directly to the isolated liver mitochondria at the indicated concentrations for 5 min, after which the mitochondria were stained with JC-1 (excitation at 488 nm and emission at 525 nm for monomers or 594 nm for aggregates). Isolated mitochondria treated with 90% TFE were used as control. The final TFE concentration was restricted to 0.18%, a percentage at which no significant decrease in Δψm was observed.
Computational modeling
Romo1 sequences were acquired from publicly available sequences in the NCBI RefSeq database with its name “Romo1” on 06/23/2016; partial sequences were excluded. Each group (animals, plants, fungi, and protists) was extracted as a fasta format file. The human Romo1 sequence was described using the WebLogo 3 server (Crooks et al., 2004). The secondary structures were predicted using JPRED (Drozdetskiy et al., 2015), HMMTOP (Tusnády and Simon, 2001), and DISOPRED (Ward et al., 2004). α-Helical wheel illustrations were recreated based on results from the Helical Wheel Projections server (http://rzlab.ucr.edu/scripts/wheel/wheel.cgi).
Multiple sequence alignments of Romo1 were obtained using the ClustalW program in MEGA7. To obtain nonredundant sequences of Romo1, we manually deleted the redundant sequences of animals in MEGA7 software using the Find function. If the first species in the Romo1 sequence list was found in other species, these reductant species were deleted. We repeated this protocol to obtain 104 nonredundant sequences. For tertiary structure prediction, the C15–C79 region of the H. sapiens Romo1 sequence was used as input for the Robetta and LOMETS servers. The M-ZDOCK server was used to oligomerize Romo1 from tetramers to hexamers without the residue selection option. Structural refinement was performed with the GalaxyRefine-Complex server using symmetric refinement for homooligomers. Oligomers with the lowest MolProbity scores were selected. The pore diameters and pore-lining residues were determined using both PoreWalker and Chexvis. All predicted Romo1 structures were visualized with Jmol software (http://www.jmol.org/).
Online supplemental material
Fig. S1 shows numerical results of position-specific polarity conservation of Romo1 sequences in 104 animals and 120 fungi. Fig. S2 shows Romo1 oligomerization of chemically synthesized TAMRA-Romo1 or Flag-Romo1 (WT and point mutants) in BN/SDS-PAGE. Fig. S3 shows mitochondrial depolarization induced by Romo1 deletion mutants. Fig. S4 shows the effects of viroporin inhibitors on the channel activity of Romo1. Fig. S5 shows the Romo1 oligomers predicted by experimental data–guided Romo1 structural modeling and their predicted pore diameters.
Supplementary Material
Acknowledgments
This study was supported by National Research Foundation of Korea grants funded by the Korean government (NRF-2017M2A2A7A01070813, NRF-2013R1A1A2063171, and NRF-2017R1D1A1B03032322).
The authors declare no competing financial interests.
Author contributions: Y.D. Yoo and G.Y. Lee conceived the project. G.Y. Lee designed all experiments. C.J. Lee and S.W. Hwang guided the patch clamp experiments. G.Y. Lee carried out almost all experiments, and D.-G. You carried out flow cytometric assays. G.Y. Lee, D.-G. You, H.-R. Lee, and Y.D. Yoo analyzed the results. G.Y. Lee wrote the manuscript. Y.D. Yoo and C.J. Lee edited the manuscript. All authors reviewed the manuscript.
References
- Bagkos G., Koufopoulos K., and Piperi C.. 2014. A new model for mitochondrial membrane potential production and storage. Med. Hypotheses. 83:175–181. 10.1016/j.mehy.2014.05.001 [DOI] [PubMed] [Google Scholar]
- Barry P. 1994. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J. Neurosci. Methods. 51:107–116. [DOI] [PubMed] [Google Scholar]
- Brenner C., and Moulin M.. 2012. Physiological roles of the permeability transition pore. Circ. Res. 111:1237–1247. 10.1161/CIRCRESAHA.112.265942 [DOI] [PubMed] [Google Scholar]
- Brustovetsky N., and Klingenberg M.. 1996. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry. 35:8483–8488. 10.1021/bi960833v [DOI] [PubMed] [Google Scholar]
- Chung J.S., Lee S.B., Park S.H., Kang S.T., Na A.R., Chang T.S., Kim H.J., and Yoo Y.D.. 2009. Mitochondrial reactive oxygen species originating from Romo1 exert an important role in normal cell cycle progression by regulating p27(Kip1) expression. Free Radic. Res. 43:729–737. 10.1080/10715760903038432 [DOI] [PubMed] [Google Scholar]
- Chung J.S., Park S., Park S.H., Park E.R., Cha P.H., Kim B.Y., Chung Y.M., Woo S.R., Han C.J., Kim S.B., et al. 2012. Overexpression of Romo1 promotes production of reactive oxygen species and invasiveness of hepatic tumor cells. Gastroenterology. 143:1084–1094. 10.1053/j.gastro.2012.06.038 [DOI] [PubMed] [Google Scholar]
- Chung J.S., Lee S., and Yoo Y.D.. 2014. Constitutive NF-κB activation and tumor-growth promotion by Romo1-mediated reactive oxygen species production. Biochem. Biophys. Res. Commun. 450:1656–1661. 10.1016/j.bbrc.2014.07.059 [DOI] [PubMed] [Google Scholar]
- Chung Y.M., Kim J.S., and Yoo Y.D.. 2006. A novel protein, Romo1, induces ROS production in the mitochondria. Biochem. Biophys. Res. Commun. 347:649–655. 10.1016/j.bbrc.2006.06.140 [DOI] [PubMed] [Google Scholar]
- Chung Y.M., Lee S.B., Kim H.J., Park S.H., Kim J.J., Chung J.S., and Yoo Y.D.. 2008. Replicative senescence induced by Romo1-derived reactive oxygen species. J. Biol. Chem. 283:33763–33771. 10.1074/jbc.M805334200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G.E., Hon G., Chandonia J.M., and Brenner S.E.. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G., and Vance J.E.. 1997. Import of lipids into mitochondria. Prog. Lipid Res. 36:103–130. 10.1016/S0163-7827(97)00006-4 [DOI] [PubMed] [Google Scholar]
- Delcour A.H. 2015. Electrophysiology of Unconventional Channels and Pores. Springer; New York. 10.1007/978-3-319-20149-8 [DOI] [Google Scholar]
- Drozdetskiy A., Cole C., Procter J., and Barton G.J.. 2015. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43(W1):W389–W394. 10.1093/nar/gkv332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo T., Yamamoto H., and Esaki M.. 2003. Functional cooperation and separation of translocators in protein import into mitochondria, the double-membrane bounded organelles. J. Cell Sci. 116:3259–3267. 10.1242/jcs.00667 [DOI] [PubMed] [Google Scholar]
- Fischer W.B., and Krüger J.. 2009. Viral channel-forming proteins. Int. Rev. Cell Mol. Biol. 275:35–63. 10.1016/S1937-6448(09)75002-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J.R., and Nunnari J.. 2014. Mitochondrial form and function. Nature. 505:335–343. 10.1038/nature12985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan S.W., Surya W., Vararattanavech A., and Torres J.. 2014. Two different conformations in hepatitis C virus p7 protein account for proton transport and dye release. PLoS One. 9:e78494 10.1371/journal.pone.0078494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebert M., Schrempp S.G., Mehnert C.S., Heißwolf A.K., Oeljeklaus S., Ieva R., Bohnert M., von der Malsburg K., Wiese S., Kleinschroth T., et al. 2012. Mgr2 promotes coupling of the mitochondrial presequence translocase to partner complexes. J. Cell Biol. 197:595–604. 10.1083/jcb.201110047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser A.A., Chinopoulos C., Birket M.J., Jastroch M., Vitelli C., Nicholls D.G., and Brand M.D.. 2012. Quantitative measurement of mitochondrial membrane potential in cultured cells: calcium-induced de- and hyperpolarization of neuronal mitochondria. J. Physiol. 590:2845–2871. 10.1113/jphysiol.2012.228387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap A.P., and Richardson A.P.. 2015. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 78:129–141. 10.1016/j.yjmcc.2014.08.018 [DOI] [PubMed] [Google Scholar]
- Heinen A., Camara A.K.S., Aldakkak M., Rhodes S.S., Riess M.L., and Stowe D.F.. 2007. Mitochondrial Ca2+-induced K+ influx increases respiration and enhances ROS production while maintaining membrane potential. Am. J. Physiol. Cell Physiol. 292:C148–C156. 10.1152/ajpcell.00215.2006 [DOI] [PubMed] [Google Scholar]
- Heo L., Park H., and Seok C.. 2013. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 41(W1):W384–W388. 10.1093/nar/gkt458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenbery D.M. 2016. Mitochondria and Cell Death. Springer; New York. 10.1007/978-1-4939-3612-0 [DOI] [Google Scholar]
- Ieva R., Heißwolf A.K., Gebert M., Vögtle F.N., Wollweber F., Mehnert C.S., Oeljeklaus S., Warscheid B., Meisinger C., van der Laan M., and Pfanner N.. 2013. Mitochondrial inner membrane protease promotes assembly of presequence translocase by removing a carboxy-terminal targeting sequence. Nat. Commun. 4:2853 10.1038/ncomms3853 [DOI] [PubMed] [Google Scholar]
- Jascur T., Goldenberg D.P., Vestweber D., and Schatz G.. 1992. Sequential translocation of an artificial precursor protein across the two mitochondrial membranes. J. Biol. Chem. 267:13636–13641. [PubMed] [Google Scholar]
- Kakhlon O., and Cabantchik Z.I.. 2002. The labile iron pool: characterization, measurement, and participation in cellular processes(1). Free Radic. Biol. Med. 33:1037–1046. 10.1016/S0891-5849(02)01006-7 [DOI] [PubMed] [Google Scholar]
- Kim D.E., Chivian D., and Baker D.. 2004. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32(Web Server):W526–W531. 10.1093/nar/gkh468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.J., Lee S.B., Park J.K., and Yoo Y.D.. 2010. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death Differ. 17:1420–1434. 10.1038/cdd.2010.19 [DOI] [PubMed] [Google Scholar]
- Korshunov S.S., Skulachev V.P., and Starkov A.A.. 1997. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 416:15–18. 10.1016/S0014-5793(97)01159-9 [DOI] [PubMed] [Google Scholar]
- Kowaltowski A.J., Seetharaman S., Paucek P., and Garlid K.D.. 2001. Bioenergetic consequences of opening the ATP-sensitive K(+) channel of heart mitochondria. Am. J. Physiol. Heart Circ. Physiol. 280:H649–H657. 10.1152/ajpheart.2001.280.2.H649 [DOI] [PubMed] [Google Scholar]
- Kühlbrandt W. 2015. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 13:89 10.1186/s12915-015-0201-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski M., Augustynek B., Kulawiak B., Koprowski P., Bednarczyk P., Jarmuszkiewicz W., and Szewczyk A.. 2016. What do we not know about mitochondrial potassium channels? Biochim. Biophys. Acta. 1857:1247–1257. 10.1016/j.bbabio.2016.03.007 [DOI] [PubMed] [Google Scholar]
- Leanza L., Venturini E., Kadow S., Carpinteiro A., Gulbins E., and Becker K.A.. 2015. Targeting a mitochondrial potassium channel to fight cancer. Cell Calcium. 58:131–138. 10.1016/j.ceca.2014.09.006 [DOI] [PubMed] [Google Scholar]
- Lee S., Park Y.H., Chung J.S., and Yoo Y.D.. 2015. Romo1 and the NF-κB pathway are involved in oxidative stress-induced tumor cell invasion. Int. J. Oncol. 46:2021–2028. 10.3892/ijo.2015.2889 [DOI] [PubMed] [Google Scholar]
- Marcotte E.M. 2000. Computational genetics: finding protein function by nonhomology methods. Curr. Opin. Struct. Biol. 10:359–365. 10.1016/S0959-440X(00)00097-X [DOI] [PubMed] [Google Scholar]
- Masood T.B., Sandhya S., Chandra N., and Natarajan V.. 2015. CHEXVIS: a tool for molecular channel extraction and visualization. BMC Bioinformatics. 16:119 10.1186/s12859-015-0545-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na A.R., Chung Y.M., Lee S.B., Park S.H., Lee M.S., and Yoo Y.D.. 2008. A critical role for Romo1-derived ROS in cell proliferation. Biochem. Biophys. Res. Commun. 369:672–678. 10.1016/j.bbrc.2008.02.098 [DOI] [PubMed] [Google Scholar]
- Nieva J.L., Madan V., and Carrasco L.. 2012. Viroporins: structure and biological functions. Nat. Rev. Microbiol. 10:563–574. 10.1038/nrmicro2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton M., Ng A.C., Baird S., Dumoulin A., Shutt T., Mah N., Andrade-Navarro M.A., McBride H.M., and Screaton R.A.. 2014. ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci. Signal. 7:ra10 10.1126/scisignal.2004374 [DOI] [PubMed] [Google Scholar]
- O’Rourke B. 2007. Mitochondrial ion channels. Annu. Rev. Physiol. 69:19–49. 10.1146/annurev.physiol.69.031905.163804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- OuYang B., Xie S., Berardi M.J., Zhao X., Dev J., Yu W., Sun B., and Chou J.J.. 2013. Unusual architecture of the p7 channel from hepatitis C virus. Nature. 498:521–525. 10.1038/nature12283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palade G.E. 1953. An electron microscope study of the mitochondrial structure. J. Histochem. Cytochem. 1:188–211. 10.1177/1.4.188 [DOI] [PubMed] [Google Scholar]
- Patargias G., Zitzmann N., Dwek R., and Fischer W.B.. 2006. Protein-protein interactions: modeling the hepatitis C virus ion channel p7. J. Med. Chem. 49:648–655. 10.1021/jm050721e [DOI] [PubMed] [Google Scholar]
- Pellegrini-Calace M., Maiwald T., and Thornton J.M.. 2009. PoreWalker: a novel tool for the identification and characterization of channels in transmembrane proteins from their three-dimensional structure. PLOS Comput. Biol. 5:e1000440 10.1371/journal.pcbi.1000440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce B., Tong W., and Weng Z.. 2005. M-ZDOCK: a grid-based approach for Cn symmetric multimer docking. Bioinformatics. 21:1472–1478. 10.1093/bioinformatics/bti229 [DOI] [PubMed] [Google Scholar]
- Premkumar A., Wilson L., Ewart G.D., and Gage P.W.. 2004. Cation-selective ion channels formed by p7 of hepatitis C virus are blocked by hexamethylene amiloride. FEBS Lett. 557:99–103. 10.1016/S0014-5793(03)01453-4 [DOI] [PubMed] [Google Scholar]
- Rines A.K., and Ardehali H.. 2013. Transition metals and mitochondrial metabolism in the heart. J. Mol. Cell. Cardiol. 55:50–57. 10.1016/j.yjmcc.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sael L., Chitale M., and Kihara D.. 2012. Structure- and sequence-based function prediction for non-homologous proteins. J. Struct. Funct. Genomics. 13:111–123. 10.1007/s10969-012-9126-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Martínez S., Madan V., Carrasco L., and Nieva J.L.. 2012. Membrane-active peptides derived from picornavirus 2B viroporin. Curr. Protein Pept. Sci. 13:632–643. 10.2174/138920312804142165 [DOI] [PubMed] [Google Scholar]
- Schleyer M., and Neupert W.. 1985. Transport of proteins into mitochondria: translocational intermediates spanning contact sites between outer and inner membranes. Cell. 43:339–350. 10.1016/0092-8674(85)90039-X [DOI] [PubMed] [Google Scholar]
- StGelais C., Tuthill T.J., Clarke D.S., Rowlands D.J., Harris M., and Griffin S.. 2007. Inhibition of hepatitis C virus p7 membrane channels in a liposome-based assay system. Antiviral Res. 76:48–58. 10.1016/j.antiviral.2007.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk A., Jarmuszkiewicz W., and Kunz W.S.. 2009. Mitochondrial potassium channels. IUBMB Life. 61:134–143. 10.1002/iub.155 [DOI] [PubMed] [Google Scholar]
- Tusnády G.E., and Simon I.. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics. 17:849–850. 10.1093/bioinformatics/17.9.849 [DOI] [PubMed] [Google Scholar]
- Urrutia P.J., Mena N.P., and Núñez M.T.. 2014. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 5:38 10.3389/fphar.2014.00038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J.J., McGuffin L.J., Bryson K., Buxton B.F., and Jones D.T.. 2004. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 20:2138–2139. 10.1093/bioinformatics/bth195 [DOI] [PubMed] [Google Scholar]
- Wu S., and Zhang Y.. 2007. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35:3375–3382. 10.1093/nar/gkm251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Liu T., Jin S.B., Tomilin N., Castro J., Shupliakov O., Lendahl U., and Nistér M.. 2009. The novel conserved mitochondrial inner-membrane protein MTGM regulates mitochondrial morphology and cell proliferation. J. Cell Sci. 122:2252–2262. 10.1242/jcs.038513 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.