

Abstract
High-grade gliomas (HGG), including glioblastomas, are characterized by invasive growth, resistance to therapy, and high inter- and intra-tumoral heterogeneity. The key histological hallmarks of glioblastoma are pseudopalisading necrosis and microvascular proliferation, which allow pathologists to distinguish glioblastoma from lower-grade gliomas. In addition to being genetically and molecularly heterogeneous, HGG are also heterogeneous with respect to the composition of their microenvironment. The question of whether this microenvironmental heterogeneity is driven by the molecular identity of the tumor remains controversial. However, this question is of utmost importance since microenvironmental, non-neoplastic cells are key components of the most radiotherapy- and chemotherapy-resistant niches of the tumor. Our work demonstrates a versatile, reliable, and reproducible adult HGG mouse model with NF1-silencing as a driver mutation. This model shows significant differences in tumor microenvironment, expression of subtype-specific markers, and response to standard therapy when compared to our established PDGFB-overexpressing HGG mouse model. PDGFB-overexpressing and NF1-silenced murine tumors closely cluster with human proneural and mesenchymal subtypes, as well as PDGFRA-amplified and NF1-deleted/mutant human tumors, respectively, at both the RNA and protein expression levels. These models can be generated in fully immunocompetent mixed or C57BL/6 genetic background mice, and therefore can easily be incorporated into preclinical studies for cancer cell-specific or immune cell-targeting drug discovery studies.
Keywords: Glioma, microenvironment, temozolomide, GEMM of HGG
Introduction
High-grade gliomas (HGG) are the most common and aggressive primary brain tumors in adults, with the most common being grade IV- glioblastoma. The tendency of glioblastoma cells to infiltrate the normal brain tissue surrounding the tumor makes complete surgical resection impossible. In addition, these tumors are extremely resistant to radiation and chemotherapy. Therefore, recurrence is inevitable despite an advanced multimodal standard therapy that consists of maximal surgical resection followed by radiotherapy and concomitant and adjuvant temozolomide (TMZ/RT→TMZ). Despite this aggressive regimen, the median survival of glioblastoma patients is only ~15 months (Stupp et al., 2005). Our current understanding of the complex biology of HGG is largely derived from studies of genetic and molecular changes within cancer cells. The Cancer Genome Atlas (TCGA) initiative has driven characterization of the genome, epigenome, and transcriptome of glioblastoma and has provided a higher-resolution picture of frequent alterations, revealing different subtypes with distinct expression signatures (C. W. Brennan et al., 2013; Cancer Genome Atlas Research, 2008; Huse, Phillips, & Brennan, 2011; Verhaak et al., 2010). These studies have driven the characterization of robust gene expression-based subtypes of glioblastoma named Proneural (PN), Mesenchymal (MES), and Classical (CL) (Wang et al., 2016). Integration of multi-dimensional genomic data has also established subtype-specific patterns of somatic mutations and DNA copy numbers. This analysis associated aberrations in gene expression of epidermal growth factor receptor (EGFR), neurofibromatosis type I (NF1), and platelet-derived growth factor receptor alpha (PDGFRA) with the CL, MES, and PN subtypes, respectively. With the advances provided by TCGA, it is important to understand that these subtypes are determined based on dominant transcriptional patterns at the time of resection, so they are not mutually exclusive and, more importantly, are plastic. This point was reinforced by recent data revealing that multiple subtypes can co-exist within a single tumor at both regional and single cell levels, further underlining the substantial inter- and intra-tumoral heterogeneity of HGG (Patel et al., 2014; Sottoriva et al., 2013).
In addition to genetic heterogeneity within HGG, the tumor microenvironment in which tumor cells develop and grow displays heterogeneous phenotypes that contribute to the characteristic hallmarks of HGG. In fact, the HGG microenvironment contains a wide variety of non-neoplastic stromal cells including the vasculature, various infiltrating and resident immune cells, and other glial cell types (Charles, Holland, Gilbertson, Glass, & Kettenmann, 2012). The majority of the non-neoplastic cells in the microenvironment are tumor-associated macrophages (TAMs), either of hematopoietic origin or from brain intrinsic microglia (Szulzewsky et al., 2016), which create a supportive stroma for neoplastic cell expansion and invasion. Utilizing gene expression data from TCGA and the Gene Expression Omnibus databases, several studies have demonstrated an enrichment of immune response-related gene expression, especially of TAM genes, in the MES subtype of glioblastoma compared to the other subtypes (Engler et al., 2012), suggesting that TAMs may play a subtype-specific role in glioblastoma.
When designing our mouse models, we chose to focus on major genetic driver mutations within subtypes and employed RCAS/tv-a, a somatic cell-specific gene transfer system, as a delivery method. The replication-competent, ALV-splice acceptor, retroviral system (RCAS) allows for the insertion of particular genetic alterations within tightly regulated windows of differentiation in a cell type-specific manner (Fisher et al., 1999). This system utilizes the RCAS retroviral vector to infect mice genetically engineered to express the RCAS receptor (tv-a) in specific cell populations (Dai et al., 2001; Hambardzumyan, Amankulor, Helmy, Becher, & Holland, 2009). Our system allows for the transfer of selected oncogenes, or shRNA to known tumor suppressors, into Nestin-positive cells with transgenic Nestin-tv-a (Ntv-a) mice. To model human PN HGG with these mice, we delivered RCAS PDGFB to the right frontal striatum. For our model of human MES HGG, we chose to co-deliver RCAS shNF1, shp53, and Cre (to delete Pten) to the subventricular zone (SVZ). As we showed previously, PDGFB-overexpression results in adult HGG formation independent from the location of the injection. Here we found that silencing tumor suppressors NF1, p53, and Pten can generate tumors in adult mice only when injections were performed in the progenitor cell-rich SVZ. Using these mouse models, we interrogated various markers by qPCR and immunohistochemistry (IHC) to define subtype-specific signatures of PDGFB-overexpressing and NF1-silenced murine HGG (mHGG). We then correlated these signatures with human PN and MES as well as PDGFRA-amplified and NF1-deleted/mutant tumors to compare expression patterns between our mouse models and human HGG (hHGG). When examining the microenvironmental cell composition we show that, like MES hHGG, NF1-silenced mHGG exhibits a significantly higher number of tumor-associated macrophages (TAMs). While little difference was observed in total vessel area with IHC and functional vessel analysis, the PDGFB-overexpressing model showed increased average vessel size and vascular permeability. Using MRI, we detected no differences in the growth kinetics of the models, but IHC staining for phosphohistone H3 (pH3) showed that PDGFB-driven mHGG has more proliferating cells. When tumors were treated with TMZ and RT, no differences were observed in response to RT; however, PDGFB-overexpressing tumors were sensitive to TMZ, while NF1-silenced tumors were resistant. Overall, these models offer an attractive system for interrogations of subtype-specific phenomena in HGG. Additionally, they provide a platform for reliable preclinical trials for therapies targeting neoplastic cells, non-neoplastic cells, or a combination of the two.
Materials and Methods
Mice
Ntv-a/Ink4a-Arf−/−/Pten fl/fl and Ntv-a/Pten fl/fl mice in the age range of 4–8 weeks were used for all experiments. Ntv-a/Ink4a-Arf−/−/Pten fl/fl are in a mixed genetic background (Dai et al., 2001; Hu et al., 2005; Schaper & van de Heyning, 1976; Trotman et al., 2003). Ntv-a/Pten fl/fl are in C57BL/6 background and were generated by 10 generations of back-crossesing.
Cell cultures and transfections
DF-1 cells were purchased from ATCC. Cells were grown at 39°C according to ATCC instructions, used in early passages, and tested for Mycoplasma. Transfections with RCAS PDGFB-HA, RCAS Cre, RCAS shNF1, and RCAS sh53-RFP were performed using a Fugene 6 transfection kit (Roche # 11814443001) according to the manufacturer’s protocol.
Intracranial injections
Injections were performed using a stereotactic fixation device (Stoelting, Wood Dale, IL). Mice used for these experiments were adults ranging from 4 to 8 weeks old as previously utilized (Alcantara Llaguno et al., 2009; Fox JG, 2002). Mice were anaesthetized with intraperitoneal (ip) injections of ketamine (0.1 mg/g) and xylazine (0.01 mg/g). For injections, one microliter of 4×104 transfected DF-1 cells in suspension was delivered using a 30-gauge needle attached to Hamilton syringe. A 1:1:1 mixture was used for co-injections. Locations were determined according to the coordinates in the mouse brain atlas (Keith B. J. Franklin, 1997). Coordinates for SVZ injections were: AP-0mm from bregma, Lat-0.5mm (right of midline), and depth-1.5mm from the dural surface. Right frontal striatum coordinates were: AP-1.7mm from bregma, Lat-0.5mm (left or right), and depth-2mm from the dural surface. Mice were monitored carefully and sacrificed when they displayed endpoint symptoms.
Tissue processing
Animals used for histological analysis were sacrificed and brains were removed and fixed in 10% neutral-buffered formalin for 72 h. Fixed tissues were then embedded in paraffin. Formalin-fixed, paraffin-embedded (FFPE) specimens were serially sectioned and slide mounted. The sections were deparaffinized in histo-clear (Richard-Allan Scientific) and were passed through graded alcohols before staining with hematoxylin and eosin (H&E) reagent.
Treatment of tumor-bearing mice
For long-term survival studies with RT, mice were sedated with ketamine (0.1 mg/g) and xylazine (0.01 mg/g) and irradiation of the head was performed using an X-RAD 320 from Precision X-Ray at 115 cGy/min (the rest of the mouse was shielded with a lead cover in a homemade apparatus). RT was performed with a daily 2 Gy single dose for 5 consecutive days, two days off, and then another cycle of 2 Gy per day for 5 consecutive days. TMZ treatment was performed by oral gavage as monotherapy at a dose of 25mg/kg/day (10% DMSO in saline) once daily for a total of 10 doses (5 days on/2 days off for 2 cycles). Vehicle was delivered to control mice on the same schedule. Mice were monitored carefully and were sacrificed when they displayed endpoint symptoms.
Immunohistochemistry
The FFPE specimens were cut into 5 μm sections and stained on the DISCOVERY XT platform (Ventana Medical Systems, Inc., Tucson, USA). Anti-CD31 antibodies were obtained from BD Pharmingen (558736, 1:50), anti-CD44 from BD Pharmingen (550538, 1:1000), anti-pH3 (Ser10) from Millipore (06-570, 1:400), anti-GFAP from Dako (Z0334, 1:8000), anti-Olig2 from Millipore (AB9610, 1:400), and anti-Iba1 from Wako (019-19741, 1:250) at the indicated dilutions in 2% bovine serum albumin (BSA) in phosphate-buffered saline (PBS).
Magnetic Resonance Imaging (MRI)
All MRI data were acquired with a 9.4 T/20 cm horizontal bore Bruker magnet, interfaced to an Avance console (Bruker, Billerica, MA, USA) and equipped with an actively shielded gradient set (inner diameter, 11.0 cm; maximum gradient strength, 400 mT/m; rise time, 110 ms). A two-coil actively decoupled imaging set-up was used (a 2 cm diameter surface coil for reception and a 7.2 cm diameter volume coil for transmission) to achieve maximal signal-to-noise ratio over the cortical and subcortical areas of interest. All animals were imaged with a custom-made ID 32 mm quadrature birdcage body resonator (Stark Contrast MRI Research, Erlangen, Germany) while anesthetized using 1.5% isoflurane (TerrellTM, MINRAD Inc., Bethlehem, PA). All images were acquired on a Bruker USR 9.4T scanner (Bruker Biospin MRI Inc., Billerica, MA). The mouse head was imaged in the coronal orientation using a T2-weighted rapid acquisition with relaxation enhancement (RARE) sequence with TR= 3.5 s, TE= 60 ms, RARE factor of 8, NE= 4, FOV= 40 × 40 mm2, slice thickness = 0.7 mm, with an in-plane resolution of 0.156 × 0.156 mm2. Animal breathing was monitored using an animal physiological monitor system (SA instruments, Stony Brook, NY) (Koutcher et al., 2002). Tumor volumes were determined by outlining the tumor region apparent in each MRI section and multiplying it by the slice thickness. The sum of the tumor volume from each slice was used as the total tumor volume. The growth curves generated with this analysis were fitted with an exponential growth equation of the form y = y0 + ekg*t. The mean value for the growth rate constant (kg) was compared between the two tumor subtypes.
Image acquisition and quantification of cell populations
The Nanozoomer 2.0HT (Hamamatsu Photonic K.K., Hamamatsu, Japan) whole slide scanner was utilized to convert the stained tissue sections into digital files. Subsequent image analysis was carried out using Fiji (Schindelin et al., 2012). For Iba1, CD44, CD31, and GFAP, the percentage of stained area per 2560 x 1417 pixel area (pixel width: 454 nm) field at 20x magnification was analyzed, whereas positive nuclei per field were quantified for pH3. Olig2-positive nuclei number was estimated using Aperio’s Image Analysis Toolbox (Leica Biosystems, Wetzlar Germany). The number of representative images obtained from each tissue section analyzed with Fiji was standardized based on total tumor size. To account for heterogeneity within individual samples, each tissue section was subdivided into different regions. Each region was subsequently attributed a percentage of the total number of images per section based on its relative size. The investigators were blinded as to the tumor type of the HGG samples during the quantification process. Necrotic and peri-tumoral areas were not included. Final values were standardized to an area of one mm2.
RNA extraction and Real-Time Quantitative PCR
Tumors and naïve brains from tumor-bearing or control animals were removed after perfusion with ice-cold Ringer’s solution, snap-frozen, and stored at −80°C until RNA extraction. RNA was extracted using an RNeasy Lipid Tissue Mini (QIAGEN #74804) kit according to the manufacturer’s instructions. RNA concentrations were measured with a NanoDrop (Thermo Scientific) and samples were stored at −80°C. cDNA was synthesized from total RNA using the SuperScript III First-Strand Synthesis System (Invitrogen). Real-time, quantitative PCR (qPCR) was performed using the FastStart SYBR Green Master mix (Roche Applied Science) according to the manufacturer’s instructions. Amplification was performed in the Applied Biosystems 7500 Fast Real-Time PCR System (Life Technologies). Optimized PCR primers were utilized (BioRad). β-actin was used as an internal control and the comparative CT method was used to calculate changes in expression. mRNA expression results were transformed to z-scores and utilized to generate heat-maps and to perform principal component analysis (PCA) in MatLab (MathWorks).
Hoechst leakage assay for vessel integrity
Ten minutes prior to euthanasia of mice, 50 μl of DyLight488-conjugated lectin (Vector, #DL-1174, 2 mg/ml) was injected intravenously (iv) via the retro-orbital sinus. Five minutes later, 50 μl of Hoechst 33342 (Sigma, #H6024) was injected iv through the other sinus. Animals were then perfused with 4% PFA and their brains were dissected. The brains were cut into 30 μm sections on a cryostat (Leica). Sections were stained with PI for 20 min at room temperature and washed with PBS before mounting with 70% glycerol. Images were taken on an Olympus confocal microscope and analyzed with Fiji.
TCGA analysis
Patient survival data, mRNA expression, gene mutation and copy number values for each gene of interest were obtained from the MSKCC computational biology cancer genomics data server using the R package cgdsr (https://github.com/cBioPortal/cgdsr) (Cerami et al., 2012; Gao et al., 2013). A total of 273 samples were included for analysis based on availability of DNA copy number and mutational data. Expression subclasses were assigned based on annotation as previously described (Brennan et al., 2013), and G-CIMP-positive tumors were excluded. EGFR- and PDGFRA-altered tumors were defined by gene amplification (GISTIC score = 2). NF1-altered tumors were identified as the union of cases with gene loss (GISTIC score = −1) and silencing mutations (nonsense or frameshift). Analysis was performed on two datasets; 1) a dataset where patients with two or more hits (co-amplifications of EGFR, PDGFRA and deletions of NF1) are included and 2) a dataset where patients with two or more hits are excluded.
Histopathological analysis
Hematoxylin and eosin-stained tumor samples were analyzed in a blinded fashion by a certified neuropathologist (PJC). Grading was performed according to the guidelines presented in the 2016 World Health Organization Classification of Tumors of the Central Nervous System (Louis D. N., Ohgaki H., Wiestler O.D., & Cavanee W.K., 2016).
Statistical analysis
GraphPad Prism 6 (GraphPad Software, San Diego, CA) was used for statistical analysis. The details of specific statistical tests are included in the legends of each figure. Data are presented as mean±SD. Significant values were those with P<0.05.
Results
The RCAS/tv-a system produces adult NF1-silenced mHGG from cells in the SVZ
The RCAS/tv-a system has been a useful and productive tool for studying adult PDGFB-overexpressing HGG. We have previously shown that RCAS PDGFB injection into the right and left hemispheres and the SVZ of Ntv-a/Ink4a-Arf−/− mice results in 100% formation of grade IV glioblastoma with equal median survivals (Fig. S1) (Hambardzumyan et al., 2009). These tumors displayed microvascular proliferation and pseudopalisading necrosis, which are hallmark histologic characteristics of human HGG (Hambardzumyan et al., 2009; Hambardzumyan, Squatrito, Carbajal, & Holland, 2008). The expression profile of PDGFB-driven mHGG has been shown to be similar to the human PN subtype when generated with various retroviral-based model systems, including RCAS/tv-a (Halliday et al., 2014; Lei et al., 2011). To model MES hHGG using the RCAS/tv-a system, we used mixed strain Ntv-a/Ink4a-Arf−/−/Pten fl/fl mice and C57BL/6-background Ntv-a/Pten fl/fl mice and co-injected RCAS shNF1, RCAS shp53, and RCAS Cre in the striatum and SVZ. NF1-silenced tumors only formed when injected into the SVZ of adult mice (Fig. 1A/B, data not shown), in contrast to what we previously showed with PDGFB-overexpressing tumors (Hambardzumyan et al., 2009). These results suggest that cells at various locations can serve as the cell-of-origin for transformation by PDGFB-overexpression, but only cells in the SVZ can be the cell-of-origin of NF1-silenced mHGG. This provides additional support to previous publications suggesting that the cell-of-origin for NF1-silenced HGG is either SVZ stem and progenitor cells (Alcantara Llaguno et al., 2009) or only progenitors (Liu et al., 2011). Moreover, the unique, stem cell-promoting microenvironment of the SVZ may contribute to the ability of NF1-silenced tumors to form only in this location (Lim & Alvarez-Buylla, 1999).
Fig. 1. PDGFB-overexpressing and NF1-silenced mHGG display significantly different median survivals and tumor initiation times, but similar tumor growth kinetics.

A) Kaplan-Meier survival curve showing overall survival of PDGFB-overexpressing and NF1-silenced mHGG in mixed strain Ntv-a/Ink4a-Arf−/−/Pten fl/fl mice and C57BL/6 background Ntv-a/Pten fl/fl mice. NF1-silenced mHGG was generated by SVZ RCAS injection, while PDGFB-overexpressing mHGG was generated by injections in the frontal striatum. P-values were calculated using a log-rank (Mantel-Cox) test. B) Representative MR images of PDGFB-overexpressing and NF1-silenced mHGG showing the injection site as well as small and large tumors. Tumor growth curves were fitted with an exponential regression. Comparison of the mean growth rate constant (kg) for each group indicated no significant differences in growth kinetics. The x-axis (days) indicates the number of days after initial MRI when the scan was taken. C) Tumor volumes as measured by T2-weighted MRI scans taken 30 days following RCAS injection for both PDGFB-overexpressing (N=31) and NF1-silenced (N=5) mHGG as well as 60 days following RCAS injection for the NF1-silenced (N=5) model. Percent tumor formation (TF) is indicated above each group. D) Representative images of immunohistochemistry for pH3 staining of PDGFB-overexpressing (N=5) and NF1-silenced (N=4) mHGG in Ntv-a/Ink4a-Arf−/−/Pten fl/fl mice. pH3 quantification shows a significantly increased number of cells in the M-phase of the cell cycle in PDGFB-driven tumors compared to NF1-silenced tumors. Analysis was performed with unpaired, two-tailed Student’s t-tests. Scale bars= 100 μm. *=P<0.05, ****=P<0.0001.
Although both our PDGFB-overexpressing and NF1-silenced mHGG models demonstrate reliable tumor formation, there was a significant difference in their median survivals (Fig. 1A). Since median survival can be affected by tumor initiation and/or growth kinetics, we sought to determine to what extent the difference in median survival is driven by growth kinetics. To do so, we used T2-weighted MRI imaging to determine the tumor volume once per week following initiation, thereby allowing us to determine the rate of tumor growth over time. Our data (Fig. 1B) show that the differences seen in the survival curves cannot be attributed to significantly altered growth kinetics. Moreover, MRI scans at 30 days post-RCAS injection in both models, and 60 days post-injection in the NF1-silenced model, demonstrate that PDGFB-overexpressing tumors form within 30 days of injection, while NF1-silenced tumors take around 60 days to initiate (Fig. 1C). To support the point that these tumors grow at a similar rate following initiation, we assessed proliferation in our two models using IHC for pH3, which labels replicating cells in the M phase of the cell cycle. The quantification of pH3-positive nuclei showed a significantly higher number of pH3-positive nuclei in PDGFB-overexpressing mHGG, despite the apparent heterogeneity in both groups (Fig. 1D). These results suggest that the main difference seen between survival curves of PDGFB-overexpressing and NF1-silenced mHGG can be attributed to differences in their latency following RCAS injection. To corroborate this phenomenon in humans, we evaluated whether there is difference in survival of PN vs. MES and well as PDGFRA-amplified vs. NF1-deleted/mutant human HGG using TCGA database and observed no significant differences (Fig. S2).
Genetic driver mutations determine tumor cell expression patterns for various markers as well as histological properties
Since we have previously shown that enrichment of EGFR, PDGFRA, and NF1 genomic alterations associate with overall transcriptome patterns, we asked whether genetic alterations can directly influence the transcription of factors that are known to be associated with the defined HGG subtypes (C. Brennan et al., 2009; Verhaak et al., 2010). Our TCGA analysis demonstrates that amplifications in PDGFRA are most prevalent in PN hHGG, while NF1 loss is most frequently observed in MES hHGG (Table S1). Moreover, the fact that PDGFB-overexpression in murine models can drive a transcriptome pattern like that of the human PN subtype is direct evidence that genetic mutations can drive defined transcriptomic patterns (Halliday et al., 2014; Lei et al., 2011). It was also recently shown that the loss of NF1 can directly drive a MES expression profile in pediatric mouse glioblastoma models that utilized the RCAS/tv-a system to silence NF1 in the brain of newborn mice (Ozawa et al., 2014). Here, we evaluated a panel of 18 differentially-expressed genes among HGG subtypes using qPCR. We assembled this panel by mining TCGA and literature that examined subtype-specific gene expression signatures. We have also incorporated a subset of TCGA samples in our analysis, which had either PDGFRA-amplification or NF1-deletion/mutation to serve as a comparator between our mouse models and the human tumors with mutations that are enriched PN and MES subtypes (Fig. 2A). Heat maps demonstrating z-scores of mRNA levels indicate that there is a consistent expression pattern of these 18 genes between human PN and MES, human PDGFRA-amplified and NF1-deleted/mutant, and murine PDGFB-overexpressing and NF1-silenced HGG, respectively (Fig. 2A, Fig. S3, S4, S5). Principal component analysis shows that PDGFB-overexpressing mHGG clusters together with PDGFRA-amplified hHGG and PN hHGG, while NF1-silenced mHGG clusters with NF1-deleted/mutant hHGG and MES hHGG (Fig. 2B). Histologically, the PDGFB-overexpressing tumors were almost universally grade IV glioblastomas, while the NF1-silenced tumors were a mixture of grade III anaplastic astrocytomas and grade IV glioblastomas in both mixed strain Ntv-a/Ink4a-Arf−/−/Pten fl/fl mice and C57BL/6-background Ntv-a/Pten fl/fl mice (Fig. 2C/D).
Fig. 2. Genetic driver mutations determine expression patterns of various markers.
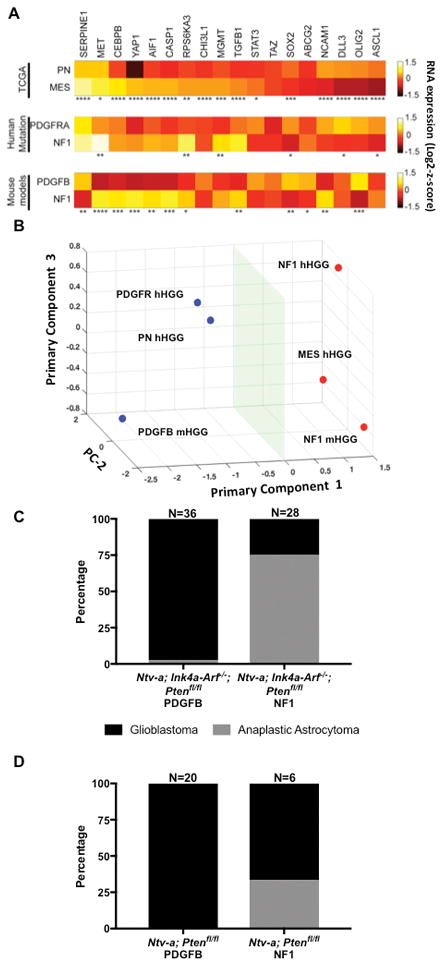
A) Heat maps demonstrating 18 selected genes that are differentially expressed in PN (N=69) and MES (N=106) hHGG, PDGFRA-amplified (N=18) and NF1-deleted/mutant (N=26) hHGG, and PDGFB-overexpressing (N=13) and NF1-silenced (N=13) mHGG. B) Principal component analysis showing that PN hHGG, PDGFRA-amplified hHGG, and PDGFB-overexpressing mHGG cluster, as do MES hHGG, NF1-deleted/mutant hHGG, and NF1-silenced mHGG. C) Histological grading of PDGFB-overexpressing (N=36) and NF1-silenced (N=28) mHGG in Ntv-a/Ink4a-Arf−/−/Pten fl/fl mice. D) Histological grading of PDGFB-overexpressing (N=20) and NF1-silenced (N=6) mHGG in Ntv-a/Pten fl/flmice. Analysis was performed with unpaired, two-tailed Student’s t-tests. Welch’s correction was performed for samples with unequal variance as determined by an F-test. *=P<0.05, **=P<0.01, ***=P<0.001, ****=P<0.0001.
We additionally tested a panel of four IHC markers, which have been shown to exhibit polarized expression patterns in human PN vs. MES HGG (Fig. 3, 4). These known markers included the PN markers Olig2 and DCX (C. Brennan et al., 2009) and the MES markers GFAP (Ozawa et al., 2014) and CD44 (Bhat et al., 2013). As expected, our results show that PDGFB-overexpressing mHGG has a significantly higher percentage of DCX- and Olig2-positive cells (Fig. 3A, 4A). Conversely, quantification of CD44- and GFAP-positive areas showed significantly increased levels in NF1-silenced mHGG (Fig. 3B, 4B).
Fig. 3. Inverse correlation of Dcx and CD44 expression in PDGFB-overexpressing and NF1-silenced mHGG.

Representative images of immunohistochemistry for A) Dcx (N=5 for PDGFB-overexpressing and N=4 for NF1-silenced) and B) CD44 (N=5 for both groups) staining of PDGFB-overexpressing and NF1-silenced mHGG. Sections were counterstained with hematoxylin. A) Quantification of Dcx showed a significant increase in positive nuclei in PDGFB-overexpressing compared to NF1-silenced mHGG. B) Quantification of CD44, showed a significant increase in positive area in NF1-silenced mHGG. In the dentate gyrus (DG), new neurons showed positive staining for Dcx and were used as a positive control for staining. Analysis was performed with unpaired, two-tailed Student’s t-tests. Welch’s correction was performed for samples with unequal variance as determined by an F-test. Scale bars = 100 μm. *=P<0.05.
Fig. 4. Inverse correlation of Olig2 and GFAP expression in PDGFB-overexpressing and NF1-silenced mHGG.
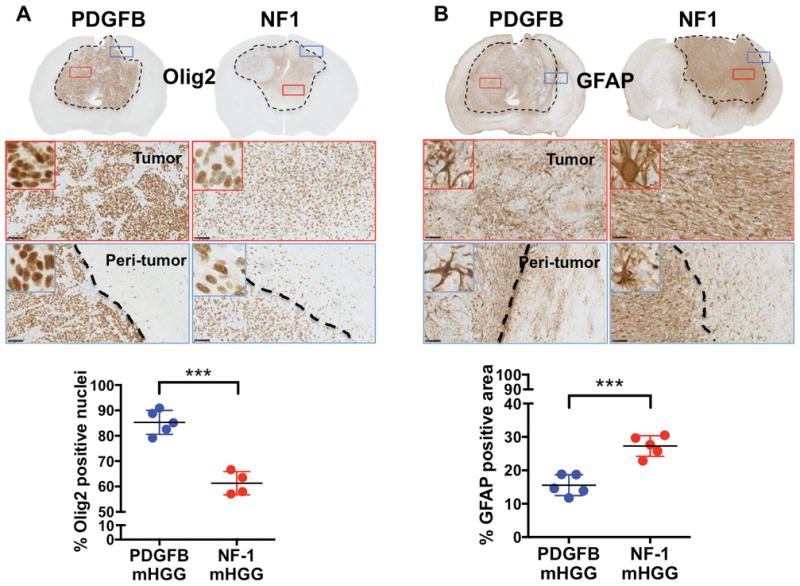
Representative images of immunohistochemistry for A) Olig2 (N=5 for PDGFB-overexpressing and N=4 for NF1-silenced) and B) GFAP (N=5 for both groups) staining of PDGFB-overexpressing and NF1-silenced mHGG. Sections were counterstained with hematoxylin. A) Quantification of Olig2 showed significantly increased Olig2-positive nuclei in PDGFB-overexpressing mHGG. B) In contrast, NF1-silenced mHGG showed a higher percentage of GFAP-positive area. This suggests more oligodendroglioma character in PDGFB-overexpressing mHGG and more astrocytoma character in NF1-silenced mHGG. Analysis was performed with unpaired, two-tailed Student’s t-tests. Scale bars = 100 μm. ***=P<0.001.
Genetic driver mutations determine the microenvironmental composition of mHGG
Next, we investigated whether different genetic-drivers influence the microenvironmental composition of HGG tumors. Non-neoplastic cell types that form the tumor microenvironment include various infiltrating and resident immune cells, the vasculature, and other glial cell types (Hambardzumyan & Bergers, 2015). The most abundant non-neoplastic population, which can be up to 30% of tumor mass, is tumor associated-macrophages (TAMs), which are comprised of both resident brain microglia and bone marrow-derived macrophages (Chen et al., 2017; Hambardzumyan & Bergers, 2015; Hambardzumyan, Gutmann, & Kettenmann, 2016). To assess this population, we used immunohistochemistry for Iba1, which labels TAMs independent of their origin. Quantification of Iba1-positive area showed a significantly higher percentage in NF1-silenced mHGG (Fig. 5A). These results are in line with correlative expression data from the TCGA and the Gene Expression Omnibus databases, which have demonstrated an enrichment of TAM genes in the MES subtype (Engler et al., 2012), suggesting that TAMs may play a subtype-specific role. Paired with our results, this implies that specific genetic alterations in tumor cells can define the immune cell composition within the tumor. To examine whether there are differences in vessel density and size between PDGFB-overexpressing and NF1-silenced mHGG, we stained tumors for the endothelial cell-specific marker CD31. Quantification of average vessel size demonstrated a significantly increased average vessel size in PDGFB-overexpressing mHGG (Fig. 5B). The vessels in NF1-silenced mHGG were both smaller and more uniformly distributed. These data suggest that major genetic driver mutations can also impact the architecture of the tumor vasculature. Since we observed significantly increased vessel size in PDGFB-driven mHGG, we next evaluated the permeability of the vessels with a Hoechst dye leakage assay and functional vessel labeling with FITC-lectin (Fig. 6). Consistent with the CD31 staining (Fig. 5B), vasculature in PDGFB-overexpressing mHGG was shown to be disorganized and enlarged. There was no statistically significant difference in total vessel area observed in this assay. The area occupied by Hoechst dye, indicative of vessel leakage, was significantly higher in PDGFB-overexpressing mHGG, indicating increased blood-brain barrier permeability (Fig. 6A). The Hoechst dye was mostly restricted to the vessel walls in NF1-silenced tumors, as shown by the line profile, but freely diffused from the vessels into tumor parenchyma in PDGFB-driven tumors (Fig. 6B). In brain tissues that are distant from the tumors, the blood vessels appeared to be normal and no Hoechst leakage was noticed in either mHGG model (Fig. S6).
Fig. 5. NF1-silenced mHGG exhibits increased tumor-associated macrophage infiltration and reduced vessel size compared to PDGFB-overexpressing mHGG.
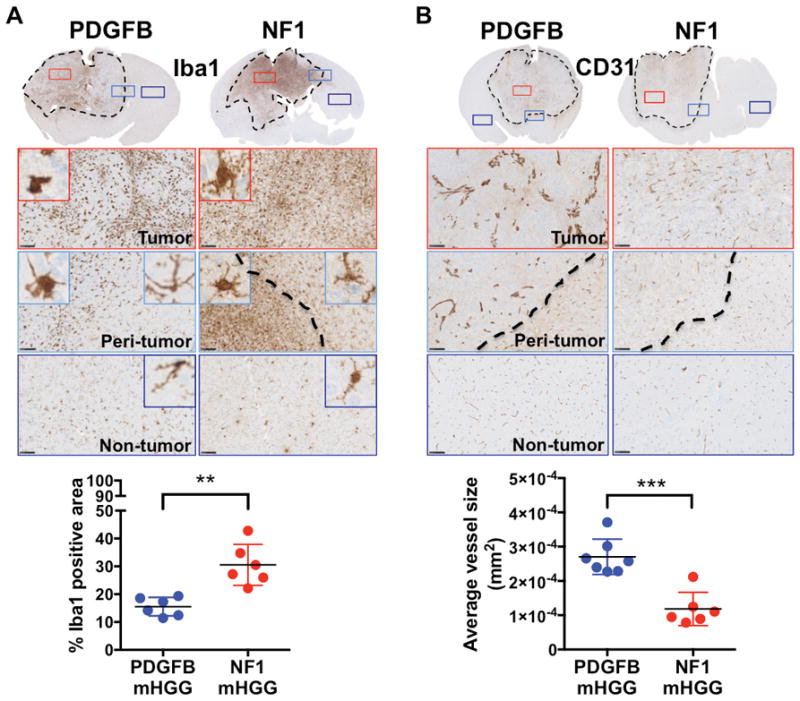
Representative images of immunohistochemistry for A) Iba1 (N=6 for both groups) and B) CD31 (N=7 for PDGFB-overexpressing and N=6 for NF1-silenced) staining of PDGFB-overexpressing and NF1-silenced mHGG. Sections were counterstained with hematoxylin. A) Quantification of Iba1 showed significantly increased positive area in NF1-silenced mHGG. B) CD31 staining showed smaller vessels in NF1-silenced mHGG compared to PDGFB-driven mHGG. Analysis was performed with unpaired, two-tailed Student’s t-tests. Welch’s correction was performed for samples with unequal variance as determined by an F-test. Scale bars = 100 μm. **=P<0.01, ***=P<0.001.
Fig. 6. Blood vessels in PDGFB-overexpressing mHGG are larger and more permeable than those in NF1-silenced mHGG.

A) Representative images of vessel functionality and permeability assessments with FITC-conjugated lectin and Hoechst dye injections in PDGFB-overexpressing and NF1-silenced mHGG. The corresponding quantification of vessel area and Hoechst-positive area are shown. Scale bar = 50 μm. B) Arbitrary fluorescent intensities of either Hoechst or FITC-lectin were measured along defined lines and plotted against the length of the lines. Scale bar = 25 μm. Analysis was performed with unpaired, two-tailed Student’s t-tests. **=P<0.01.
Genetic driver mutations determine the response to TMZ
Next, we evaluated the sensitivity of PDGFB-overexpressing and NF1-silenced mHGG to radiation therapy (RT) and temozolomide (TMZ) treatment. Using a calculation for dose translation from human to mouse, we determined that the standard human dose of 75 mg/m2 of TMZ corresponds to 25 mg/kg in mice and we used radiation at the dose of 20 Gy. Since various doses have been used in the literature to treat xenografts and other murine models, we first compared whether escalating doses of TMZ would increase the effect on median survival of PDGFB-overexpressing mHGG. Based on T2-weighted MRI scans, we distributed size-matched tumor-bearing mice in 25 mg/kg versus 100 mg/kg groups with endpoint survival time as the primary readout. As shown (Fig. S7), there was no difference in median survival time between the groups. Since PDGFB-overexpressing mHGG develops rapidly and within a defined period, we used T2-MRI images to distribute equal volume tumors into vehicle, TMZ, and RT treatment groups according to the indicated dosing regimen (Fig. 7A/B). TMZ treatment significantly prolonged the median survival time of PDGFB-overexpressing mHGG (Fig. 7C). RT provided a survival advantage compared to both the TMZ and vehicle groups (Fig. 7C). Next, we used the same treatment paradigm (Fig. 7A) to determine treatment response in mice with NF1-silenced tumors. This time, mice were randomly distributed into treatment groups due to variance in tumor initiation in NF1-silenced tumors and MRI facility constraints. In contrast to PDGFB-overexpressing mHGG, our data showed no difference in median survival between the vehicle and TMZ groups in NF1-silenced mHGG, while significant differences were shown in the RT group compared to both the TMZ and vehicle groups (Fig. 7D). To further explore the difference in response to TMZ of PDGFB-driven versus to NF1-silenced mHGG, we next evaluated levels of the DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) since the response to TMZ in human HGG patients has been associated with expression levels of MGMT (Hegi et al., 2005). We first looked at MGMT expression levels in TCGA database comparing PN and MES hHGG as well as PDGFRA-amplified and NF1-deleted/mutant hHGG and saw significantly higher expression of MGMT in MES and NF1-deleted/mutant hHGG compared to PN and PDGFRA-amplified hHGG, respectively (Fig. 2A, S3, S4). qPCR results from mouse tumors demonstrated a trend towards higher expression of MGMT in NF1-silenced compared to PDGFB-overexpressing mHGG (Fig. 2A, S5); however, this trend is not sufficient to explain the observed differences in response to TMZ. Following treatment, the recurrent NF1 tumors displayed a trend towards increased malignancy (Fig. S7).
Fig. 7. RT treatment provides a survival advantage to mice with either PDGFB-overexpressing or NF1-silenced mHGG, while TMZ provides a survival advantage only to mice with PDGFB-overexpressing tumors.
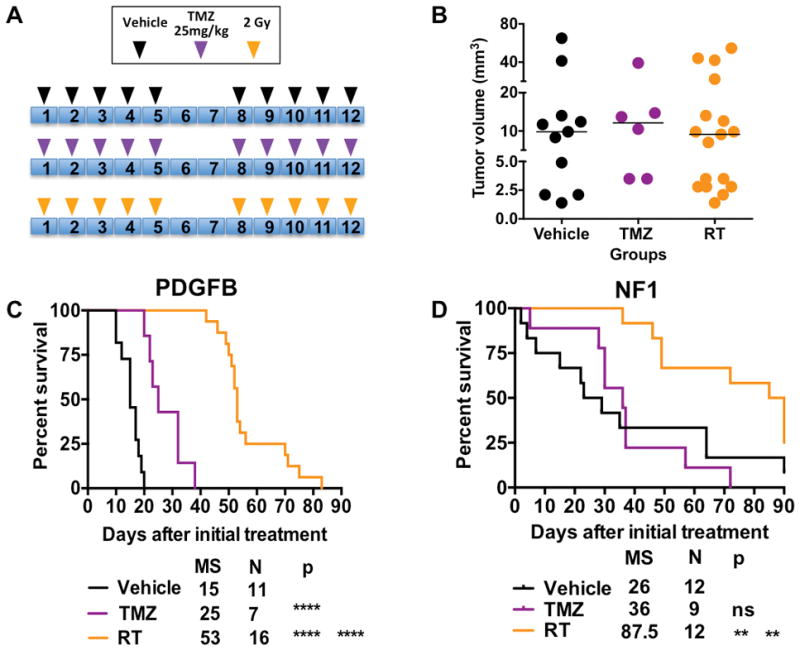
A) Schematic illustrations of treatment paradigms and groups. B) Four weeks post-injection, mice with PDGFB-overexpressing mHGG were separated into treatment groups with matched sex, age, and average tumor size as measured by T2-weighted MRI. Similarly, mice with NF1-silenced mHGG were randomized into vehicle, TMZ, and RT treatment cohorts 8 weeks post-injection with age and sex equally distributed. C) Kaplan-Meier survival curves for the different treatment groups for PDGFB-overexpressing mHGG. D) Kaplan-Meier survival curves for the different treatment groups for NF1-silenced mHGG. Analysis was performed with a log-rank (Mantel-Cox) test. **=P<0.01, ****=P<0.0001.
Discussion
Due to increased awareness of the genetic heterogeneity of HGG resulting from extensive studies of TCGA initiative and single cell-RNA sequencing data, intriguing questions have been raised regarding conversions between subtypes and the ability of subtypes to co-exist within a single tumor. This has increased the demand for models that allow for investigation of the genetic drivers associated with HGG expression signatures. We therefore decided to generate models based on genetic driver mutations that will allow the field to directly address these questions. Our PDGFB-overexpressing and NF1-silenced mHGG models will now allow investigators to scrutinize the role that these mutations play in determining the response to therapy, thereby improving our ability to develop and utilize precision therapeutics.
Single cell RNA sequencing has suggested that the clinical outcome of PN HGG is influenced by the presence of cancer cells of alternate subtypes within the tumor (Patel et al., 2014). Whether the PN to MES transition seen in response to radiation or anti-angiogenic therapy (Halliday et al., 2014; Lu et al., 2012) is because the predominant, treatment-sensitive population of PN cells dies and treatment-resistant cells of the MES subtype repopulate the tumor, or that tumor cells themselves change their expression profile from PN to MES in response to treatment, is debatable (Halliday et al., 2014). Additionally, it is unknown whether PN HGG acquires NF1 mutation during the transition to MES HGG. To enable the brain tumor community to answer these questions with direct methods, we developed an adult, NF1-silenced murine model of HGG, which can be used together with our PDGFB-overexpressing model to interrogate the role that these genetic driver mutations play in determining tumor phenotype and expression of tumor- and stroma-specific markers.
Our study also analyzed the influence of location on the tumorigenic potential of two known genetic driver mutations. We demonstrated that RCAS PDGFB injected into the right frontal striatum induced tumor formation in that area and spread through the frontal lobe and ultimately into the olfactory bulb, a common location for human gliomas (Larjavaara et al., 2007). We have previously demonstrated that tumor incidence, latency, and mortality do not differ when tumors are generated in different locations in the cerebral hemispheres, including the SVZ, when we use PDGFB-overexpression. By contrast, NF1-silencing resulted in tumor formation only when the SVZ was targeted and occurred with similar incidence and progression, but longer latency. In total, these data imply that the cell-of-origin may differ between these models and that the microenvironment of the SVZ may be a requirement for NF1-silenced tumor generation. We additionally demonstrated that tumors generated by overexpression of PDGFB exhibit more features or oligodendrogliomas with high expression of Olig2 and DCX, compared to NF1-silenced mHGG, which have more astrocytoma features including high expression of CD44 and GFAP. Moreover, our data demonstrate that genetic driver mutations influence the stromal composition, including TAM infiltration and the vasculature in HGG. Recently, it has been demonstrated that a different cell-of-origin, even (Ozawa et al., 2014) when the same genetic driver mutation is used, will directly influence the expression subtype and drug sensitivity of the resulting HGG (Jiang et al., 2017). These data together with our results demonstrate that genetic driver mutations and the cell-of-origin are major contributors to the inter- and intra-tumoral heterogeneity of HGG.
Finally, we turned our investigation towards how these two models respond to standard HGG therapy. TMZ has been shown to provide benefits in some patients, but not others. Initial studies showed that in nearly half of HGG patients, expression of the DNA repair enzyme MGMT is silenced by promoter methylation. These patients were shown to benefit from TMZ, whereas those without MGMT promoter methylation did not show such a benefit (Hegi et al., 2005). Later, studies documented that high MGMT expression levels were associated with low TMZ response in non-MGMT promoter methylated, patient-derived cell lines and xenografts (Kitange et al., 2009). We have previously shown the existence of intra-tumoral heterogeneity in response to TMZ. In PDGFB-overexpressing mHGG, stem-like populations showed higher MGMT expression compared to non-stem populations and were more resistant to TMZ (Bleau et al., 2009). It should be noted, however, that MGMT promoter methylation is not the only factor that predicts TMZ response. Recently, it was shown that mutations in the DNA-repair enzyme MSH6 can also predict TMZ response (Nguyen et al., 2014). Here, we demonstrate TMZ as efficacious in prolonging median survival only in PDGFB-overexpressing mHGG. The complete mechanism underlying this phenomenon is still unclear; however, it is logical to postulate that the microenvironmental changes we observed, with respect to both the immune cell profile and vasculature, play a substantial role. Moreover, intrinsic properties of the tumor cells within these subtypes may contribute to the observed differences in treatment response. It has been shown that oligodendroglioma cells are more responsive to TMZ than astrocytic glioma cells (Schindelin et al., 2012; Schmid et al., 2016). Since our Olig2 staining and histological analysis demonstrated that the PDGFB-overexpressing model has oligodendroglial features, and our GFAP staining and histological analysis showed that the NF1-silenced model has astrocytoma features, this point may account for some of the differences in TMZ response seen in our models. In both mouse models, radiation therapy resulted in significant extension of median survival.
In total, PDGFB-overexpressing and NF1-silenced HGG mouse models offer excellent tools for the evaluation of subtype-specific phenomena and for pharmaceutical assessment in preclinical trials. MRI imaging techniques further complement these models, allowing for non-invasive monitoring of therapeutic response, tumor initiation, and tumor progression in a clinically relevant manner. These models will additionally allow investigators to interrogate the roles of tumor location, genetic driver mutations, stromal composition, and the mechanism of the PN to MES shift. We hope that data obtained from preclinical studies in these mouse models will contribute to the development of individualized, targeted therapies and accelerate their progression into clinical trials. Since we have also demonstrated the ability to generate PDGFB-overexpressing and NF1-silenced tumors in mice of pure genetic backgrounds, these models will also offer invaluable tools for researchers investigating the burgeoning field of immunotherapy.
Supplementary Material
Main Points.
Genetic driver mutations create defined expression signatures in glioma.
Driver mutations define microenvironment composition and response to temozolomide.
Acknowledgments
We thank Drs. Eric Holland and Tatsuya Ozawa for providing reagents and plasmids (Fred Hutchinson Cancer Research Center and the University of Washington, Seattle). We additionally thank Neil Anthony of the Integrated Cellular Imaging Core at Emory University and the Pathology Core Laboratory and MRI facility of the Winship Cancer Institute.
Funding: This work was supported by NIH/NCI (U01 CA160882), NIH/NINDS R01 NS096956 and from the Aflac Cancer and Blood Disorders Center for DH and PSTP Training Grant (4T32GM008602-20) for CH.
References
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, … Parada LF. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15(1):45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, … Aldape K. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24(3):331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, Holland E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One. 2009;4(11):e7752. doi: 10.1371/journal.pone.0007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, … Network TR. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, … Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2012;60(3):502–514. doi: 10.1002/glia.21264. [DOI] [PubMed] [Google Scholar]
- Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, … Hambardzumyan D. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017;77(9):2266–2278. doi: 10.1158/0008-5472.CAN-16-2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15(15):1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, … Phillips JJ. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS One. 2012;7(8):e43339. doi: 10.1371/journal.pone.0043339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher GH, Orsulic S, Holland E, Hively WP, Li Y, Lewis BC, … Varmus HE. Development of a flexible and specific gene delivery system for production of murine tumor models. Oncogene. 1999;18(38):5253–5260. doi: 10.1038/sj.onc.1203087. [DOI] [PubMed] [Google Scholar]
- Fox JG, AL, Loew FM, Quimby FW. Labaratory Animal Medicine. 2. 2002. 43-Table VII. [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, … Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday J, Helmy K, Pattwell SS, Pitter KL, LaPlant Q, Ozawa T, Holland EC. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc Natl Acad Sci U S A. 2014;111(14):5248–5253. doi: 10.1073/pnas.1321014111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling Adult Gliomas Using RCAS/t-va Technology. Transl Oncol. 2009;2(2):89–95. doi: 10.1593/tlo.09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Bergers G. Glioblastoma: Defining Tumor Niches. Trends Cancer. 2015;1(4):252–265. doi: 10.1016/j.trecan.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–27. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Squatrito M, Carbajal E, Holland EC. Glioma formation, cancer stem cells, and akt signaling. Stem Cell Rev. 2008;4(3):203–210. doi: 10.1007/s12015-008-9021-5. [DOI] [PubMed] [Google Scholar]
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, … Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7(4):356–368. doi: 10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse JT, Phillips HS, Brennan CW. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia. 2011;59(8):1190–1199. doi: 10.1002/glia.21165. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Marinescu VD, Xie Y, Jarvius M, Maturi NP, Haglund C, … Uhrbom L. Glioblastoma Cell Malignancy and Drug Sensitivity Are Affected by the Cell of Origin. Cell Rep. 2017;18(4):977–990. doi: 10.1016/j.celrep.2017.01.003. [DOI] [PubMed] [Google Scholar]
- Keith BJ, Franklin GP. The Mouse Brain in Steriotaxic Coordinates 1997 [Google Scholar]
- Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, … Sarkaria JN. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. 2009;11(3):281–291. doi: 10.1215/15228517-2008-090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutcher JA, Hu X, Xu S, Gade TP, Leeds N, Zhou XJ, … Holland EC. MRI of mouse models for gliomas shows similarities to humans and can be used to identify mice for preclinical trials. Neoplasia. 2002;4(6):480–485. doi: 10.1038/sj.neo.7900269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larjavaara S, Mantyla R, Salminen T, Haapasalo H, Raitanen J, Jaaskelainen J, Auvinen A. Incidence of gliomas by anatomic location. Neuro Oncol. 2007;9(3):319–325. doi: 10.1215/15228517-2007-016. 15228517-2007-016 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, … Canoll P. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS One. 2011;6(5):e20041. doi: 10.1371/journal.pone.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DA, Alvarez-Buylla A. Interaction between astrocytes and adult subventricular zone precursors stimulates neurogenesis. Proc Natl Acad Sci U S A. 1999;96(13):7526–7531. doi: 10.1073/pnas.96.13.7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, … Zong H. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146(2):209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavanee WK. World Health Organization Histological Classification of Tumours of the Central Nervous System. 4. International Agency for Research on Cancer; France: 2016. [Google Scholar]
- Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, … Bergers G. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22(1):21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen SA, Stechishin OD, Luchman HA, Lun XQ, Senger DL, Robbins SM, … Weiss S. Novel MSH6 mutations in treatment-naive glioblastoma and anaplastic oligodendroglioma contribute to temozolomide resistance independently of MGMT promoter methylation. Clin Cancer Res. 2014;20(18):4894–4903. doi: 10.1158/1078-0432.CCR-13-1856. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, … Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26(2):288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, … Bernstein BE. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper J, van de Heyning J. Cholesteatoma of the middle ear in human patients. An ultrastructural study. Arch Otolaryngol. 1976;102(11):663–668. doi: 10.1001/archotol.1976.00780160059004. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, … Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid RS, Simon JM, Vitucci M, McNeill RS, Bash RE, Werneke AM, … Miller CR. Core pathway mutations induce de-differentiation of murine astrocytes into glioblastoma stem cells that are sensitive to radiation but resistant to temozolomide. Neuro Oncol. 2016;18(7):962–973. doi: 10.1093/neuonc/nov321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, … Tavare S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013;110(10):4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ … National Cancer Institute of Canada Clinical Trials, G. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Szulzewsky F, Arora S, de Witte L, Ulas T, Markovic D, Schultze JL, … Kettenmann H. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia. 2016;64(8):1416–1436. doi: 10.1002/glia.23014. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, … Pandolfi PP. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1(3):E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD … Cancer Genome Atlas Research, N. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Hu X, Muller F, Kim H, Squatrito M, Millelsen T, … Verhaak RGW. Tumor evolution of glioma intrinsic gene expression subtype associates with immunological changes in the microenvironment. bioRxiv. 2016 doi: 10.1101/052076. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.