

Abstract
An 18-step synthesis of the antibiotic (+)-pleuromutilin is disclosed. The key steps of the synthesis include a highly stereoselective SmI2-mediated cyclization to establish the eight-membered ring, and a stereospecific transannular [1,5]-hydrogen atom transfer to set the C10 stereocenter. This strategy was also used to prepare (+)-12-epi-pleuromutilin. The chemistry described here will enable efforts to prepare new mutilin antibiotics.
Graphical Abstract
(+)-Pleuromutilin (1) is a diterpene natural product first isolated from the fungus Clitopilus passeckerianus in 1951 (Scheme 1).1 (+)-Pleuromutilin binds to the peptidyl transferase center of bacterial ribosomes, preventing protein synthesis.2 Semi-synthetic derivatives of 1 in which the C14 ester is modified have been identified as potent antibiotics; for example, retapamulin is an FDA-approved topical antibiotic.3 Recently, derivatives of 12-epi-mutilin have been developed as broad-spectrum antibiotics with efficacy against gram-negative pathogens.4 Given its promising antibacterial properties, four total syntheses of 1 have been reported to date, the most recent of which was disclosed by Herzon and coworkers in 2017.5,6,7 Here we report an approach that enables the preparation of (+)-pleuromutilin and (+)-12-epi-pleuromutilin in 18 steps from (+)-trans-dihydrocarvone.
Scheme 1.

Retrosynthetic analysis.
In considering a design plan for a synthesis of (+)-1, we targeted a modular approach in which a bifunctional hydrindane fragment (e.g. 5) would be annulated to form the eight-membered ring through two sequential C–C bond forming steps. In particular, the C5–C14 and C11–C12 bonds, which each link vicinal stereogenic centers, were identified as strategic points of disconnection.
Applying this general plan in a retrosynthesis, (+)-1 was simplified to 2, which was further disconnected through the C5–C14 bond to aldehyde 3 (Scheme 1). In the forward sense, we envisioned forming the eight-membered ring through a late-stage SmI2-mediated ketyl radical cyclization of 3.8 It is important to distinguish this approach from the SmI2-mediated cascade cyclization employed in Procter’s synthesis of 1,5c,d which formed the C3–C4 bond by a ketyl radical conjugate addition, and the C5–C14 bond—and 8-membered ring—by intramolecular aldol cyclization. To enable the cascade reaction, Proctor employed an ester as a precursor to the C15 methyl, and required several additional steps to adjust the oxidation states at C3 and C15. In contrast, a SmI2 cyclization of 3 was expected to provide 2 with C3, C14, and C15 in the correct oxidation states for advancement to 1.
Aldehyde 3 was anticipated to arise from enone 4, which, depending on the targeted stereochemistry at C12, could be prepared by crotylation of enal 5 with either Z- or E-boronic acid 6.9 Thus, through appropriate design of the crotylation reaction, either 1 or 12-epi-1 would be accessible through this route. Hydrindanone enal 5 was mapped back to enone 7 via sequential conjugate addition reactions and functional group interconversions.
The synthesis began with the preparation of enone 7 in one step from (+)-trans-dihydrocarvone.10 Conjugate addition of the cuprate derived from 8, followed by Pd-catalyzed desaturation furnished 9 (Scheme 2). A second conjugate addition furnished the C9-quaternary stereocenter; however, attempts to promote an intramolecular aldol condensation under Brønsted- or Lewis-acid catalysis resulted in the formation of undesired Prins-type products. Hypothesizing that electronic deactivation of the isopropenyl group would mitigate this non-productive reactivity, 10 was converted to the allylic chloride using trichloroisocyanuric acid (TCCA).11 Indeed, treatment of the ketal with HCl at 70 °C provided enone 11 as a 4.4:1 mixture of diastereomers at C6; the major diastereomer was isolated in 52% yield. 1,2-Addition of methylmagnesium chloride was achieved with the aid of CeCl3•2LiCl 12 and the diastereomeric mixture was submitted to pyridinium chlorochromate (PCC) to effect an oxidative transposition.13 Kornblum oxidation11 of 12 delivered enal 5 in 8 steps from 7.
Scheme 2.

Synthesis of a cyclization substrate.
With enal 5 in hand, the first of two key C–C bond constructions required to form the bridging eight-membered ring was investigated. Reaction of 5 with boronic acid Z-614 under the conditions developed by Szabó and coworkers provided a mixture of diastereomers 13a and 13b (Scheme 4).9b While the reaction proceeded with excellent selectivity for syn crotylation—consistent with a closed transition state—the catalyst did not discriminate between the diasterofaces of the aldehyde during the nucleophilic attack. Use of the S catalyst provides a 1:1.4 mixture of 13a and 13b.15 A brief investigation of alternative catalytic asymmetric crotylation conditions proved unfruitful. Separation of the diastereomers by column chromatography, followed by protection of 13a as the methoxymethyl (MOM) ether, cleavage of the trityl ether, and oxidation under the conditions developed by Stahl16 delivered aldehyde 14.
Scheme 4.
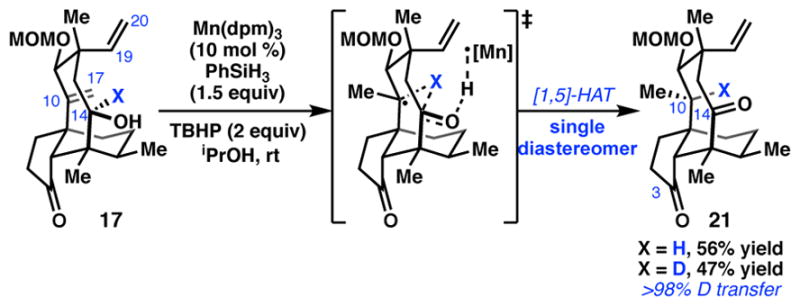
Redox relay by transannular [1,5]-HAT.
At this stage, attention turned to the second key C–C bond construction: a SmI2-mediated cyclization to form the eight-membered ring (Scheme 3). When 14 was treated with a freshly-prepared solution of SmI2 in THF at 0 °C, then quenched with aqueous ammonium chloride, carboxylic acid 16 was obtained as a single diastereomer. Presumably 16 arises from exposure of SmIII-enolate 15 to oxygen, resulting in formation of an α-peroxyketone and subsequent oxidative ring scission.17 Although the ring scission was deleterious, it nonetheless confirmed that C–C bond formation occurred with high diastereoselectivity.
Scheme 3.

Completion of the synthesis of (+)-pleuromutilin (1).
In an effort to prevent the unwanted formation of 16, a variety of conditions were evaluated. After substantial optimization, it was found that dropwise addition of SmI2 (3 equiv) to 14 and 6 equiv H2O as a solution in THF at 0 °C, under rigorously anaerobic conditions, followed by quenching first with trimethylsilyl chloride (TMSCl), then aqueous workup delivered tricycle 17 in 93% yield as a separable 23:1 mixture of diastereomers (Scheme 3). The addition of H2O was found to be critical to minimize undesired side-product formation and achieve high diastereoselectivity; reactions conducted in the absence of H2O afforded 17 with 1:1 dr at C14.
To complete the synthesis of (+)-1, chemoselective reduction of the C10–C17 exocyclic olefin and installation of the glycolate ester were required. Unfortunately, standard hydrogenation conditions employing cationic transition metal complexes gave rapid and exclusive reduction of the more sterically-accessible C19–C20 vinyl group. Instead, we turned to hydrogen-atom transfer (HAT) reactions, seeking to leverage the thermodynamic preference for formation of a 3°carbon-centered radical.18 Indeed, use of tris(2,2,6,6-tetramethyl-3,5-heptanedionato)manganese (III) (Mn(dpm)3) in the presence of phenylsilane and tert-butyl hydroperoxide (TBHP) in degassed, anhydrous isopropanol resulted in highly diastereoselective reduction of the C10–C17 olefin (Scheme 4).19 We were surprised to discover, however, that alkene reduction was accompanied by oxidation of the C14 alcohol. This redox relay process delivered diketone 21 in 55% yield as a single diastereomer. Only trace products arising from competing C19-C20 vinyl reduction were observed. Substrates in which the C14 alcohol is protected gave only 6–10% conversion after 6 h, and the resulting C10 stereocenter was formed as a mixture of diastereomers. To test if this reaction proceeds by a transannular [1,5]-HAT process,20 deuterium-labeled substrate 17-d was prepared and exposed to the optimized reaction conditions (Scheme 4). Tricycle 21-d was formed as a single diastereomer with complete transfer of the deuterium label. The observation that substrates in which the C14 alcohol is protected perform poorly under the HAT conditions suggests that cleavage of the O–H bond to form the C14 ketone serves as a driving force for this transformation.21
Having solved the problem of chemoselective alkene reduction, the selective reduction the C14 ketone in the presence of the C3 ketone was now required. Ultimately, selective reduction of diketone 21 proved untenable. Instead, triisopropylsilyl (TIPS) enol ether 18 was prepared and submitted to radical reduction to obtain ketone 19 as a single diastereomer (Scheme 3). To complete the total synthesis, 19 was submitted to excess lithium in ammonia, which furnished alcohol 20 as a separable 14:1 mixture of diastereomers. Subsequent one-pot acylation with 2-(2,2,2-trifluoroacetoxy)acetic acid followed by trifluoroacetate methanolysis, then acidic hydrolysis effected global deprotection to deliver (+)-1.
A key design aspect of our strategy was the ability to easily vary the stereochemistry of the cyclization substrates at C11 and C12. In particular, given the recent interest in derivatives of C12-epi-mutilin as broad-spectrum antibiotics,4 we sought to demonstrate that the 12-epi-mutilin framework could be prepared. To this end, enal 5 was subjected to crotylation with E-6 under the previously developed conditions, to deliver 13c and 13d as a 2:1 mixture in 85% combined yield (Scheme 5, a). Elaboration of 13c to 12-epi-14 proceeded without difficulty.15 Exposure of 12-epi-14 to the optimal conditions for the SmI2 cyclization furnished 12-epi-17 in 77% yield and 17:1 dr. 12-epi-17 was smoothly advanced four steps to complete the synthesis of 12-epi-1.15
Scheme 5.

Reactivity of diastereomeric cyclization substrates.
In contrast, attempts to cyclize aldehyde 22, prepared from crotylation product 13b, revealed that the C11 stereochemistry exerts a pronounced effect on reactivity (Scheme 5, b). Subjection of 22 to the SmI2-mediated cyclization conditions provided tricycle 26 as the major product in 20% yield.22 It is proposed that conformational gearing to minimize A1,2 strain at C11 reverses the regioselectivity of the Sm-ketyl addition to the enone, producing radical 23. Subsequent Dowd-Beckwith rearrangement proceeding through cyclopropane 24 delivers the product bearing a bridgehead olefin.
In summary, the total syntheses of (+)-pleuromutilin and (+)-12-epi-pleuromutilin were each completed in 18 steps (longest linear sequence) from (+)-trans-dihydrocarvone. These syntheses were enabled by a modular approach, which employed a highly diastereoselective SmI2-mediated radical cyclization to form the eight-membered ring. In addition, we uncovered a transannular [1,5]-HAT that effects a stereospecific redox relay to set the C10 stereocenter. The brevity and modularity of the route will enable the design and synthesis of new fully synthetic variants of mutilin antibiotics.
Supplementary Material
Acknowledgments
We thank Dr. Michael Takase and Larry Henling for X-ray data collection, Ms. Julie Hofstra for X-ray data refinement, Dr. David VanderVelde for assistance with NMR structure determination, Dr. Scott Virgil for assistance with crystallization of 26, and the Caltech 3CS for access to analytical equipment. Fellowship support was provided by the NIH (E.P.F., Grant 1F32GM117764) and NSF (S.S.F., DGE-1144469). Financial support from the Heritage Medical Research Institute is gratefully acknowledged.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: xx.xxxx/jacs.xxxxxxxxx.
Crystallographic data for 16, 17, and 26 (CIF)
Experimental procedures and characterization and spectral data for all compounds (PDF)
References
- 1.(a) Kavanagh F, Hervey A, Robbins WJ. Proc Natl Acad Sci USA. 1951;37:570. doi: 10.1073/pnas.37.9.570. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Birch AJ, Holzapfel CW, Rickards RW. Tetrahedron. 1966;(Suppl 8)(Part II):359. [Google Scholar]
- 2.Poulsen SM, Karlsson M, Johansson LB, Vester B. Mol Microbiol. 2001;41:1091–1099. doi: 10.1046/j.1365-2958.2001.02595.x. [DOI] [PubMed] [Google Scholar]
- 3.(a) Rittenhouse S, Biswas S, Broskey J, McCloskey L, Moore T, Vasey S, West J, Zalacain M, Zonis R, Payne D. Antimicrob Agents Chemother. 2006;50:3882. doi: 10.1128/AAC.00178-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fazakerley NJ, Procter DJ. Tetrahedron. 2014;70:6911. [Google Scholar]
- 4.Thirring K, Heilmayer W, Riedl R, Kollmann H, Ivezic-Schoenfeld Wicha W, Paukner S, Strickmann D. WO2015110481A1. 2015 Jul 30;
- 5.(a) Gibbons EG. J Am Chem Soc. 1982;104:1767. [Google Scholar]; (b) Boeckman RK, Jr, Springer DM, Alessi TR. J Am Chem Soc. 1989;111:8284. [Google Scholar]; (c) Helm MD, Da Silva M, Sucunza D, Findley TJK, Procter DJ. Angew Chem Int Ed. 2009;48:9315. doi: 10.1002/anie.200905490. [DOI] [PubMed] [Google Scholar]; (d) Fazakerley NJ, Helm MD, Procter DJ. Chem Eur J. 2013;19:6718. doi: 10.1002/chem.201300968. [DOI] [PubMed] [Google Scholar]; (e) Murphy SK, Zeng M, Herzon SB. Science. 2017;356:956. doi: 10.1126/science.aan0003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zeng M, Murphy SK, Herzon SB. J Am Chem Soc. 2017;139:16377. doi: 10.1021/jacs.7b09869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Synthetic studies toward pleuromutilin: Paquette LA, Wiedeman PE, Bulman-Page PC. J Org Chem. 1988;53:1441.Bacque E, Pautrat F, Zard S. Org Lett. 2003;5:325. doi: 10.1021/ol027312m.Loresta SD, Liu J, Yates EV, Krieger I, Sacchettini JC, Freundlich JS, Sorensen EJ. Chem Sci. 2011;2:1258. doi: 10.1039/C1SC00116G.
- 7.For a review of the role of synthesis in antibacterial drug discovery: Wright PM, Seiple IB, Myers AG. Angew Chem Int Ed. 2014;53:8840. doi: 10.1002/anie.201310843.
- 8.For examples of medium size ring construction using SmI2, see: Molander GA, McKie J. J Org Chem. 1994;59:3186.Enholm EJ, Satici H, Trivellas A. J Org Chem. 1989;54:5841.Matsuda F, Sakai T, Okada N, Miyashita M. Tetrahedron Lett. 1998;39:863.Molander GA, George KM, Monovich LG. J Org Chem. 2003;68:9533. doi: 10.1021/jo0347361.Blot V, Reibig H-U. Eur J Org Chem. 2006:4989.For a review: Edmonds DJ, Johnston D, Procter DJ. Chem Rev. 2004;104:3371. doi: 10.1021/cr030017a.
- 9.(a) Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2006;128:12660. doi: 10.1021/ja0651308. [DOI] [PubMed] [Google Scholar]; (b) Alam R, Vollgraff T, Eriksson L, Szabó KL. J Am Chem Soc. 2015;137:11262. doi: 10.1021/jacs.5b07498. [DOI] [PubMed] [Google Scholar]
- 10.White JD, Grether UM, Lee C-S. Org Synth. 2005;82:108.. Commercially available (+)-dihydrocarvone is supplied as 4:1 mixture of trans and cis isomers, which can be chromatographically separated to give pure trans.
- 11.Singh D, McPhee D, Paddon CJ, Cherry J, Maurya G, Mahale G, Patel Y, Kumar N, Singh S, Sharma B, Kushwaha L, Singh S, Kumar A. Org Process Res Dev. 2017;21:551. [Google Scholar]
- 12.Krasovskiy A, Kopp F, Knochel P. Angew Chem Int Ed. 2006;45:497. doi: 10.1002/anie.200502485. [DOI] [PubMed] [Google Scholar]
- 13.Dauben WG, Michno DM. J Org Chem. 1977;42:682. [Google Scholar]
- 14.See the Supporting Information for the synthesis of Z-6 and E-6.
- 15.See Supporting Information.
- 16.Steves JE, Stahl SS. J Am Chem Soc. 2013;135:15742. doi: 10.1021/ja409241h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.This type of oxidative ring scission has previously been observed: Spring DM, Bunker A, Luh BY, Sorenson ME, Goodrich JT, Bronson JJ, DenBleyker K, Dougherty TJ, Fung-Tomc J. Eur J Med Chem. 2007;42:109. doi: 10.1016/j.ejmech.2006.07.018.
- 18.(a) Ma X, Herzon S. Chem Sci. 2015;6:6250. doi: 10.1039/c5sc02476e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crossely SWM, Obradors C, Martinez RM, Shenvi RA. Chem Rev. 2016;116:8912. doi: 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conditions were adapted from: Iwasaki K, Wan KK, Oppedisano A, Crossley SWM, Shenvi RA. J Am Chem Soc. 2014;136:1300. doi: 10.1021/ja412342g.
- 20.For examples of transannular [1,5]-HAT on a medium-sized ring, see: Winkler JD, Sridar V, Rubo L, Hey JP, Haddad N. J Org Chem. 1989;54:3004.Boivin J, da Silva E, Ourisson G, Zard SZ. Tetrahedron Lett. 1990;31:2501.
- 21.Intermolecular hydrogen-bonding with an acceptor molecule, such as isopropanol solvent, may be responsible for polarizing the O–H bond, leading to weakening of the C14 carbinol hydrogen atom. See: Gawlita E, Lantz M, Paneth P, Bell AF, Tonge PJ, Anderson VE. J Am Chem Soc. 2000;122:11660.Jeffrey JL, Terrett JA, MacMillan DWC. Science. 2015;349:1532. doi: 10.1126/science.aac8555.
- 22.The aldehyde derived from 13d underwent the analogous cyclization.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.