

Abstract
(+)-Psiguadial B is a diformyl phloroglucinol meroterpenoid that exhibits anti-proliferative activity against the HepG2 human hepatoma cancer cell line. This full account details the evolution of a strategy that culminated in the first enantioselective total synthesis of (+)-psiguadial B. A key feature of the synthesis is the construction of the trans-cyclobutane motif by a Wolff rearrangement with in situ catalytic, asymmetric trapping of the ketene. An investigation of the substrate scope of this method to prepare enantioenriched 8-aminoquinolinamides is disclosed. Three routes toward (+)-psiguadial B were evaluated that featured the following key steps: 1) an ortho-quinone methide hetero–Diels–Alder cycloaddition to prepare the chroman framework; 2) a Prins cyclization to form the bridging bicyclo[4.3.1]decane system, and 3) a modified Norrish–Yang cyclization to generate the chroman. Ultimately, the successful strategy employed a ring-closing metathesis to form the seven-membered ring and an intramolecular O-arylation reaction to complete the polycyclic framework of the natural product.
INTRODUCTION
Plant extracts used in traditional folk medicine have long served as rich sources of structurally complex, bioactive compounds. For example, the bark, leaves, and fruit of the Psidium guajava plant are known for their medicinal properties, and have been used to treat ailments such as diabetes and hypertension.1 Efforts to isolate and characterize the bioactive constituents have identified a variety of diformyl phloroglucinol meroterpenoids with interesting structures,2 including 1 and 2 (Figure 1), which inhibit phosphodiesterase-4 (PDE4D2), a drug target for inflammatory and respiratory diseases.3 In 2010, Shao and coworkers reported the discovery of four new meroterpenoids, psiguadials A–D (3–6),4,5 which exhibit potent cytotoxicity against the HepG2 human hepatoma cancer cell line (IC50 = 46–128 nM). The most potent antiproliferative agent in this family, (+)-psiguadial B (3), is unique from a structural standpoint in that it possesses a strained bicyclo[4.3.1]decane terpene core, fused to a trans-cyclobutane ring.
Figure 1.
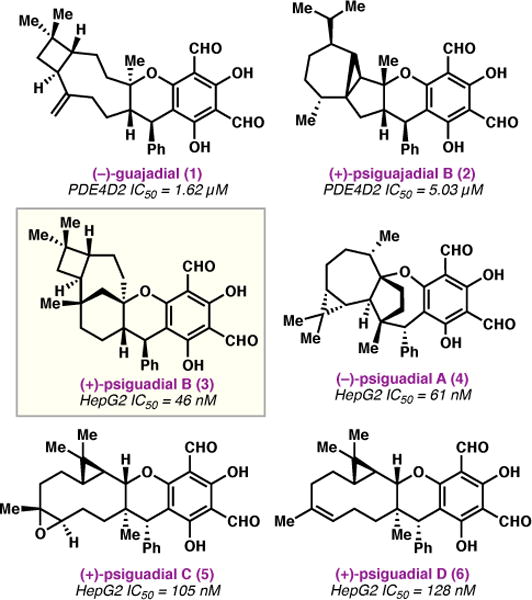
Bioactive diformyl phloroglucinol meroterpenoids isolated from Psidium guajava.
Biosynthetically, this motif is proposed to arise via a mixed terpene-polyketide pathway.5 Intramolecular cyclization of farnesyl pyrophosphate (7) generates humulyl cation 8,6 which undergoes stereoselective ring closure guided by caryophyllene synthase to produce β-caryophyllene (9, Scheme 1). Michael reaction of 9 with ortho-quinone methide (o-QM) 10—likely derived from the known P. guajava metabolite 3,5-dimethyl-2,4,6-trihydroxybenzophenone7—is proposed to afford tertiary carbocation 11,8 which can cyclize to give (−)-guajadial (1)1g and (+)-psidial A (12),9 isomeric natural products that have also been isolated from P. guajava. Alternatively, carbocation 11 can isomerize through proton transfer processes to form tertiary carbocation 13,5 which can undergo transannular ring closure to generate bridgehead cation 14. Finally, this species can be trapped by the pendant phenol to furnish (+)-psiguadial B (3). Cramer8,10 and Lee11 have validated this biosynthetic hypothesis by semi-syntheses of 3, 1, and 11, from β-caryophyllene (9).
Scheme 1.
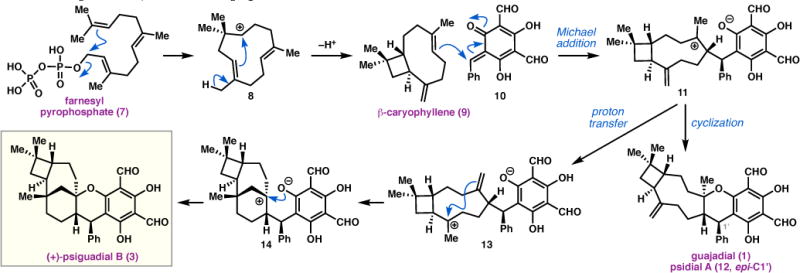
Proposed biosynthesis of (+)-psiguadial B.
While semi-synthetic approaches to phloroglucinol meroterpenoids provide direct access to β-caryophyllene-derived natural products, we viewed an abiotic approach to 3 as an opportunity to develop new chemistry and strategy concepts that could be applicable in broader synthetic contexts. Here, we describe a full account of our efforts to develop an enantioselective total synthesis of (+)-psiguadial B (3),12,13 which was enabled by an asymmetric Wolff-rearrangement to construct the trans-fused cyclobutane.
DESIGN PLAN: FIRST GENERATION STRATEGY
As disclosed in our prior communication,12 the construction of the central bicyclo[4.3.1]decane, which is trans-fused to a cyclobutane, was recognized as the primary synthetic challenge posed by 3. Closer analysis identified the C1−C2 bond (Figure 2), which links the A and C rings through vicinal stereogenic centers, as a strategic disconnection. On the basis of this analysis, we were interested in forming this bond by a Pd-catalyzed C(sp3)−H alkenylation reaction between cyclobutane 18 and vinyl iodide 19.
Figure 2.
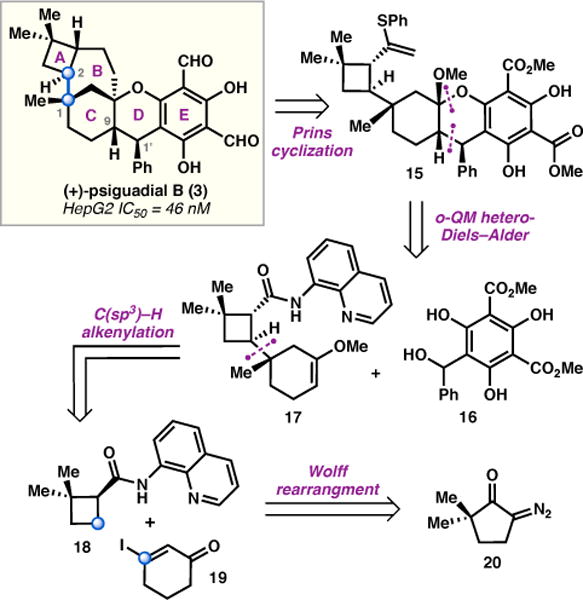
First generation retrosynthetic analysis.
Having identified a tactic to join the A and C rings, a retrosynthesis of 3 was conceived in which the 7-membered B ring would be generated via a late-stage intramolecular Prins cyclization, thus allowing simplification of 3 to 15. Although the ring closure to form this strained system was expected to be challenging, the Prins reaction has been previously used for the preparation of bridging polycycles.14 Tricycle 15 could be assembled through a bioinspired ortho-quinone methide hetero-Diels–Alder (o-QMHDA) reaction between enol ether 17 and an o-QM generated from 16.15 Although o-QMHDA reactions are widely used to construct chroman frameworks, simple acyclic enol ethers or styrenes are typically employed as dienophiles, and are used in excess to avoid o-QM dimerization.16 In contrast, the proposed strategy necessitates use of a functionalized cyclic enol ether, ideally as the limiting reagent. At the outset of these studies, we were unaware of any reported examples in which cyclohexanone-derived enol ethers were employed as dienophiles in o-QMHDA cycloadditions; thus, the proposed studies could potentially contribute a new substrate class for o-QMHDA reactions.17 Based on stereochemical analysis of reported o-QMHDA cycloadditions, we anticipated that the reaction would favor the desired anti-relationship between C1′ and C9, however, whether the stereochemistry of 17 would impart the desired facial selectivity in the approach of the heterodiene was less clear.15,18
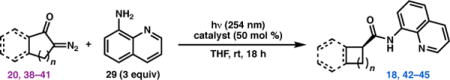
Enol ether 17 was envisioned to be accessible in short order from the product of the directed C(sp3)−H alkenylation reaction, joining fragments 18 and 19. Although the direct product of this reaction would be a cis-cyclobutane, the thermodynamically more stable trans-cyclobutane was anticipated to be accessible through an epimerization process. While C(sp3)−H functionalization using 8-aminoquinoline as a directing group was well established as a powerful strategy in the context of total synthesis,19,20 it was uncertain whether the proximal methyl C−H bonds might intervene unproductively. Finally, the required cyclobutane, 18, could be easily prepared from known diazoketone 20 via photochemical Wolff rearrangement.21
A key question presented by the proposed retrosynthesis was how best to synthesize cyclobutane 18 in enantioenriched form. Elegant studies by Fu and coworkers had demonstrated that N-acylpyrroles can be prepared with excellent enantioselectivity from the reaction between aryl ketenes (e.g. 21) and 2-cyanopyrrole (22) using chiral DMAP catalyst 23 (Scheme 2a).22 We hypothesized that a similar transformation could be used to prepare 18 directly from 20 by using 8-aminoquinoline (29) as a nucleophile in the presence of an appropriate catalyst. While there were no examples from Fu’s work in which the ketene was generated in situ photochemically, a single example from Lectka showed that a ketene could be generated in situ by a Wolff rearrangement, and engage in an enantioselective reaction (Scheme 2b).23
Scheme 2.
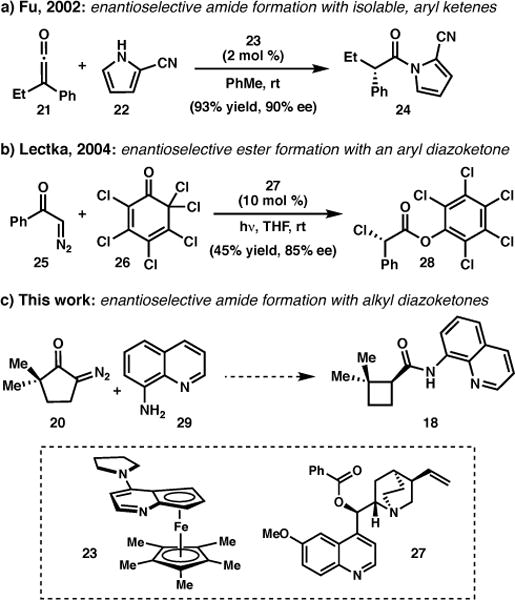
Enantioselective reactions with ketenes.
Following a survey of chiral nucleophilic catalysts known to engage with ketenes, 22–24 it was discovered that irradiation of a mixture of 20 and 3 equiv 2925 in the presence of 50 mol % (+)-cinchonine (30) produced 18 in 61% yield, and 79% ee (Table 1, entry 1). Investigation of various solvents revealed that THF provided the highest levels of enantioselectivity.26 More concentrated reaction mixtures led to lower yields, presumably due to poor light penetration as a result of the sparing solubility of 30 in THF. When scaling the reaction to quantities relevant for total synthesis (30 mmol), the catalyst loading of 30 could be reduced to 10 mol %, which provided 18 in 62% yield and 79% ee (see Scheme 3). Moreover, enantiomerically pure 18 was obtained after a single recrystallization by layer diffusion.
Table 1.
Optimization and exploration of substrate scope for tandem Wolff rearrangement/ketene addition.
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||
| catalyst |
entry |
yielda (%) |
eeb (%) |
entry |
yielda (%) |
eeb (%) |
entry |
yielda (%) |
eeb (%) |
entry |
yielda (%) |
eeb (%) |
entry |
yielda (%) |
eeb (%) |
|
| 30 | 1 | 61c | 79 | 9 | 64 | 58 | 17 | 49 | 64 | 25 | 48 | 9 | 33 | 54 | 35 |
|
| 31 | 2 | 50 | 24 | 10 | 52 | 64 | 18 | 60 | 7 | 26 | 43 | 7 | 34 | 65 | 42 | |
| 32 | 3 | 64 | −57 | 11 | 62 | −22 | 19 | 56 | −51 | 27 | 48 | −3 | 35 | 38 | −20 | |
| 33 | 4 | 67 | 64 | 12 | 69 | 50 | 20 | 59c | 71 | 28 | 48 | 25 | 36 | 42 | 23 | |
| 34 | 5 | 58 | 48 | 13 | 77c | 71 | 21 | 55 | −39 | 29 | 40 | 7 | 37 | 61 | 41 | |
| 35 | 6 | 65 | −54 | 14 | 66 | 0 | 22 | 54 | −34 | 30 | 48 | −26 | 38 | 42 | −23 | |
| 36 | 7 | 90 | −16 | 15 | 60 | −59 | 23 | 65 | −7 | 31 | 47 | −5 | 39 | 42c | −75 | |
| 37 | 8 | 50 | 53 | 16 | 49 | −23 | 24 | 50 | 37 | 32 | 31c | 34 | 40 | 35 | −1 | |
Reactions performed on 0.050 mmol scale and irradiated for 18 hours. Yield determined by 1H NMR analysis versus an added internal standard.
Determined by SFC using a chiral stationary phase.
Reactions performed on 0.200 mmol scale and irradiated for 48 hours, isolated yield reported.
Scheme 3.
C(sp3)–H alkenylation and quaternary center formation via conjugate addition.
Although our total synthesis efforts focused on the preparation of 18, we wondered if this tandem Wolff rearrangement/enantioselective addition reaction could be applied to other α-diazoketone substrates. Unfortunately, substantially lower levels of enantioinduction (9–64% ee) were observed using 30 as a catalyst with these substrates (Table 1, entries 9, 17, 25, and 33). Evaluation of alternative cinchona derivatives 31–37 revealed that synthetically useful levels of enantioselectivity could be achieved for each substrate, depending on the catalyst. For instance, while 31–37 produced 18 with lower enantioinduction (16–64% ee, entries 2–8), catalysts 34 and 33 proved optimal for the 6- and 7-membered analogs of 20, providing amides 42 and 43 each in 71% ee (entries 13 and 20). When these reactions were conducted on preparative scale, the catalyst loading could be dropped to 20 mol %, providing cyclopentyl amide 42 (n = 2) in 80% yield and 67% ee and cyclohexyl amide 43 (n = 3) in 67% yield and 67% ee.26 On the other hand, benzo-fused diazoketones, 40 and 41, performed best in the presence of dimeric cinchona catalysts 37 and 36 (entries 32 and 39). At present, a general catalyst for the tandem Wolff rearrangement/enantioselective addition of 8-aminoquinoline has not been identified, though further mechanistic investigations may inform future efforts to improve the generality of this reaction.
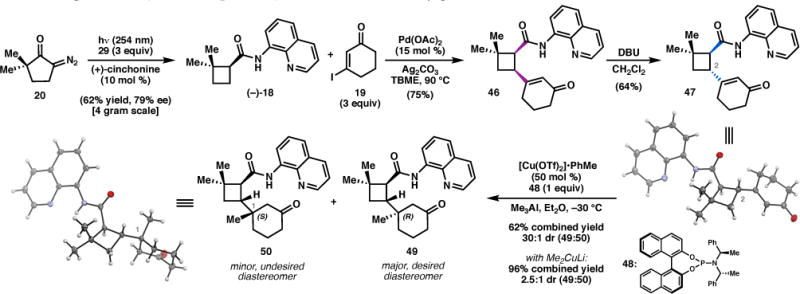
Having identified conditions to prepare multigram quantities of 18 in enantiopure form, we were pleased to find that treatment of 18 with Pd(OAc)2 (15 mol %), Ag2CO3, and 19 in TBME at 90 °C smoothly effected the C(sp3)–H alkenylation reaction to give 46 in 75% yield on gram scale (Scheme 3). Exposure of 46 to DBU furnished the requisite trans-cyclobutane via selective epimerization at C2, as determined by deuterium-labeling studies.26 It was at this stage that single crystals of trans-cyclobutane 47 suitable for X-ray diffraction were obtained. Unfortunately, 47 was found to be in the incorrect enantiomeric series for elaboration to natural 3. To our dismay, this problem could not be circumvented by simply employing (−)-cinchonidine (32) in the tandem Wolff rearrangement/asymmetric ketene addition, as this pseudoenantiomeric catalyst afforded (+)-18 in only 57% ee (Table 1, entry 3). Nevertheless, we elected to advance (–)-47 in the interest of validating the key reactions in our retrosynthetic analysis as soon as possible.
To this end, attention turned to formation of the C1 quaternary center (Scheme 3). Subjection of cis-cyclobutane 46 to a number of standard conjugate addition conditions provided only trace yields of the corresponding product (not shown), presumably due to steric encumbrance by the proximal large aminoquinoline group. On the other hand, treatment of trans-cyclobutane 47 with excess Gilman’s reagent smoothly furnished 49 and 50 in near quantitative yield as a 2.5:1 mixture of diastereomers, respectively. Separation of the diastereomers by HPLC allowed single crystals of 50 to be obtained, and X-ray analysis unambiguously confirmed that the major diastereomer (49) possessed the desired (R) configuration of the methyl group at the C1 quaternary center.
In an effort to improve the diastereoselectivity of this transformation, we turned to asymmetric catalysis. Fortunately, application of the conditions developed by Alexakis and coworkers for copper-catalyzed conjugate addition27 provided 49 in 62% yield and 30:1 dr, albeit using 50 mol % [Cu(OTf)2]•PhMe and a stoichiometric equivalent of phosphoramidite ligand 48. Presumably, the high catalyst loading is required due to the presence of the highly-coordinating 8-aminoquinolinamide, which can deactivate the catalyst or inhibit turnover.
With the quaternary center secured, ketone 49 was converted to the corresponding dimethyl ketal 51 (Scheme 4a), a precursor to the dienophile for the o-QMHDA reaction (vide infra). While phenolic aldol conditions28 failed to produce 16, this acid-labile o-QM precursor was prepared from phloroglucinol 5429 via the morpholine adduct (55, Scheme 4b).30 A control experiment determined that heating of 51 to 170 °C in toluene results in thermal extrusion of methanol to afford a 1:1 mixture of enol ethers 17 (Scheme 5).31 When a mixture of 51 and 16 was heated to 170 °C for 21 h, the cycloadduct was obtained in 68% yield, albeit as a complex mixture of diastereomers.
Scheme 4.
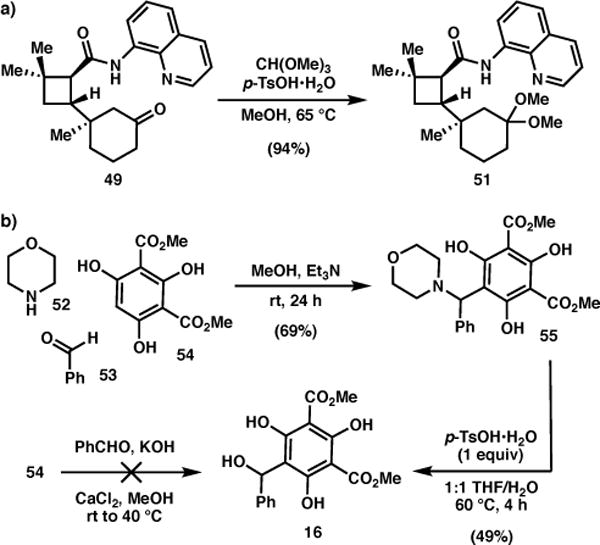
Synthesis of o-QMHDA cycloaddition reactants.
Scheme 5.
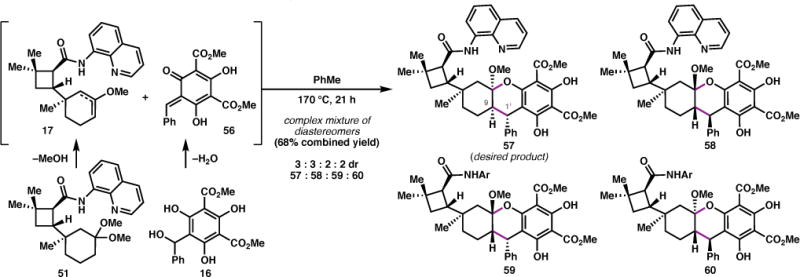
Evaluation of the thermal o-QMHDA cycloaddition.
Analytically pure samples of the four highest abundance diastereomers (57–60) were obtained by HPLC purification. Spectroscopic analysis by 2D NMR led to the assignment of 57 and 58 as the two major diastereomers, which bear the expected relative anti relationship between C9 and C1′. The formation of these products in a ~1:1 ratio indicates that 17 does not exert significant facial selectivity in the o-QMHDA reaction. The trans-fused isomer, 60, presumably results from thermal equilibration of the ketal under the reaction conditions.
In considering how to improve the selectivity for desired diastereomer 57, we drew inspiration from Evans’ highly enantioselective inverse-demand hetero-Diels–Alder chemistry, which proceeds via bidentate coordination of heterodienes such as 61 to a chiral Cu(II)-BOX Lewis acid catalyst (Scheme 6a).32 We envisioned that chelation of the aminoquinoline in 17 to a Cu complex could engage 56 as depicted in Scheme 6b, thereby directing the o-QM to the top face of enol ether 17 (Scheme 6b). Formation of 56 could be induced by the equivalent of triflic acid generated via complexation of Cu(OTf)2 with aminoquinoline.17,33,33
Scheme 6.
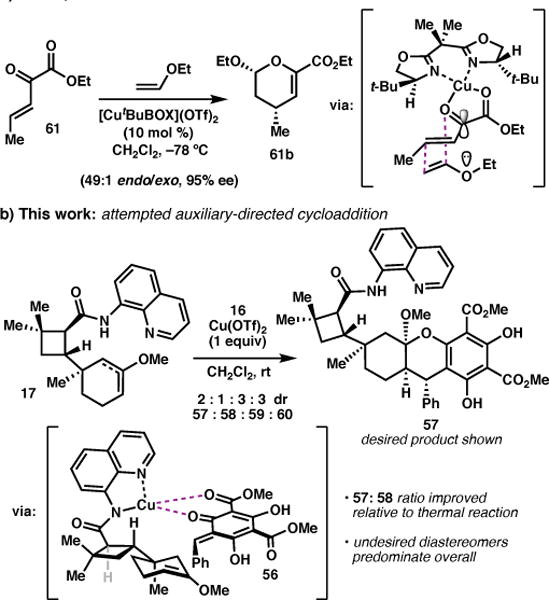
Attempted auxiliary-directed cycloaddition.
To test this hypothesis, enol ether 17 was prepared by heating in PhMe,34 and after exchanging the solvent for CH2Cl2, Cu(OTf)2 and 16 were added. Analysis of the crude reaction mixture by 1H NMR revealed that although the ratio of 57:58 had improved relative to the thermal reaction, significant quantities of the undesired isomers, 59 and 60, were still formed. Moreover, this reaction suffered from lower overall yields due to rapid hydrolysis of 17 and reversion of 16 to phloroglucinol 54. At this stage, it was clear that implementation of this strategy would require a significant investment in reaction optimization and we felt that such an effort would only be warranted if the proposed late-stage Prins reaction were proved feasible. Thus, attention turned to assessing this key reaction in a model system.
To this end, the aminoquinoline auxiliary in 51 was reductively cleaved by treatment with Schwartz’s reagent to furnish aldehyde 62, which was homologated to alkyne 63 using the Ohira–Bestmann reagent (Scheme 7). Nickel-catalyzed hydrothiolation35 proceeded with good regioselectivity to give vinyl sulfide 65 in low yield, mainly due to the facile conversion of this intermediate to a mixture of enol ethers 64 under the reaction conditions.
Scheme 7.
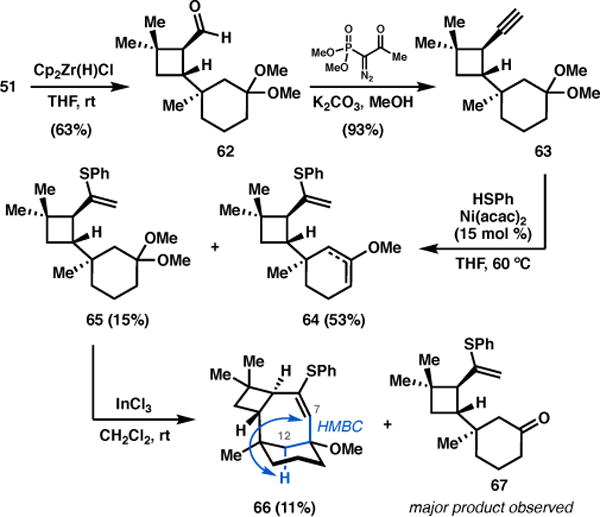
Model studies toward Prins cyclization.
Unfortunately, exposure of ketal 65 to a variety of Lewis acids led to hydrolysis, yielding ketone 67 in nearly all cases. The use of InCl3,36 however, delivered the desired Prins product 66 in 11% yield. Formation of the 7-membered ring was confirmed by a key HMBC correlation between the C12 axial proton and the distinct sp2 C7 signal at δ 140 ppm. Although the formation of the seven-membered ring through a Prins cyclization was promising, our excitement was tempered by the fact that 66 was obtained in poor yield and challenges were encountered with reproducibility. Taken together with the significant diastereoselectivity issues plaguing the o-QMHDA reaction, we revised our retrosynthetic analysis.
SECOND GENERATION STRATEGY
In our revised retrosynthesis, we envisioned that the chroman substructure could be constructed via a modified Norrish–Yang cyclization,37 revealing 68 as a key intermediate (Figure 3a). Benzophenones such as 68 are known to undergo photoexcitation upon irradiation with UV light38 to give triplet species (i.e. 68*) that can engage in Norrish type-II 1,5-hydrogen atom abstraction and subsequent radical recombination.37,39 In the absence of any available γ or δ-hydrogens, it was hypothesized that 68* could abstract a hydrogen atom from C9 to generate diradical 72.40 Recombination of the carbon-centered radicals would furnish the core of 3. We recognized that achieving the desired regioselectivity could prove challenging since the C7 and C12 methylenes in 68* were also within range for 1,7-H–atom abstraction (Figure 3b). Although the outcome of this transformation was uncertain, conformational analysis suggested that the product resulting from hydrogen atom abstraction at C9 would produce the least sterically encumbered chroman product. Moreover, this strategy was particularly appealing since it was expected that 68 could be assembled in an expedient and convergent fashion. Benzophenone 68 was envisioned to be accessible from tertiary alcohol 70 via an intermolecular O-arylation reaction with aryl bromide 69.41 We reasoned that the strained 7-membered ring in 70 could be formed by ring-closing metathesis,42 leading back to vinyl ketone 71, which could in turn be synthesized from known intermediates prepared during our studies of the C(sp3)–H alkenylation/asymmetric Wolff rearrangement.
Figure 3.
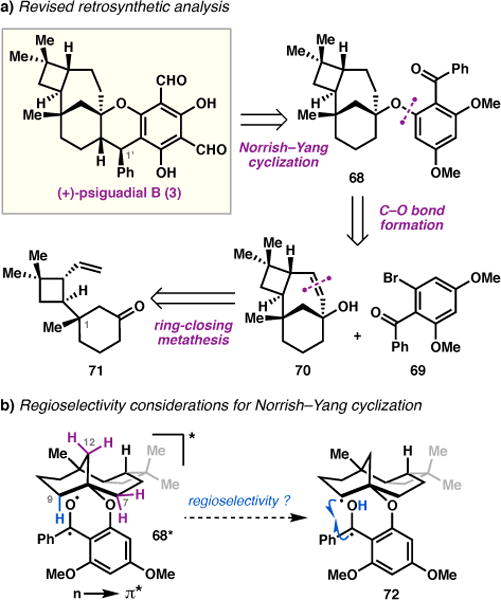
Second-generation retrosynthetic analysis.
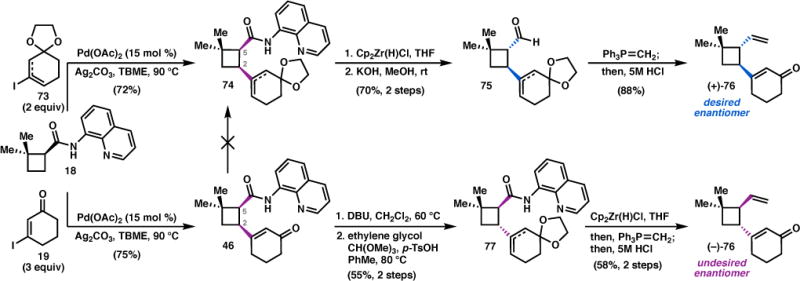
With this revised retrosynthetic plan, we set out to prepare vinyl ketone 71, and to also address two key challenges identified in the first generation approach: 1) to lower the catalyst loading in the conjugate addition reaction used to set the C1 quaternary center, and 2) to develop an epimerization sequence to prepare vinyl ketone 71 in the correct enantiomeric series from quinolinamide (–)-18. In terms of the latter challenge, we anticipated that the desired enantiomeric series could be accessed by epimerization of compounds derived from 18 (e.g. 46) at C5 instead of C2 (Scheme 8). A straightforward approach would involve disfavoring γ-deprotonation at C2 by masking the ketone of 46 in order to advance to a C5 epimerization substrate. Unfortunately, these efforts proved unfruitful, as ketalization of 46 under a variety of conditions always resulted in rapid epimerization at C2 to furnish trans-cyclobutane 77 in low yields.43
Scheme 8.
Development of enantiodivergent epimerization strategies.
Instead, it was recognized that 74 could be accessed directly by coupling 18 with vinyl iodide 73.44 To our delight, the Pd-catalyzed coupling with vinyl iodide 73 performed even better than its enone counterpart (19), requiring only 2 equiv of 73 to furnish 74 in 72% yield on a gram scale. Exposure of 74 to Schwartz’s reagent effected reduction to the corresponding cis-aldehyde, which was epimerized at C5 by treatment with KOH in methanol to give trans-aldehyde 75 in 70% yield over the two steps. Gratifyingly, Wittig methylenation and hydrolysis provided (+)-76, the required enantiomer for synthesis of natural psiguadial B (3). In addition, cross-coupling of 73 eliminated a linear protection step and substantially improved the material throughput.45
To demonstrate that either enantiomer of 76 can be prepared using a single enantiomer of organocatalyst, an alternative sequence was also developed. Epimerization of 46 to the trans-cyclobutane under the previously developed conditions, followed by ketalization provided 77. Reductive cleavage of the aminoquinoline auxiliary gave the corresponding aldehyde (ent-75), which was telescoped through a Wittig olefination and hydrolysis as before to afford vinyl enone (−)-76 in 58% yield over the two steps.
With the desired enantiomer of enone 76 in hand, attention turned to the installation of the C1 quaternary center using a catalytic asymmetric conjugate addition. In the absence of the amino-quinoline auxiliary, we were pleased to find that use of CuTC (15 mol %) in conjunction with ligand ent-48 (30 mol %) provided 71 in 94% yield and 19:1 dr (Scheme 9). Addition of vinyl Grignard to ketone 71 proceeded uneventfully, providing alcohol 78 in excellent yield and diastereoselectivity. Gratifyingly, exposure of 78 to second-generation Hoveyda–Grubbs catalyst at elevated temperature delivered bridged bicycle 70 in 93% yield. Subsequent hydrogenation under standard conditions led to tertiary alcohol 79. After some experimentation, we found that the combination of Pd(OAc)2 and dppf catalyzed the intermolecular O-arylation between 79 and aryl bromide 69, affording aryl ether 68 in 45% yield. Unfortunately, the reproducibility of this transformation proved capricious, and attempts to improve the yield through further optimization were unsuccessful. Nevertheless, a sufficient amount of 68 was obtained to evaluate the key Norrish–Yang cyclization.
Scheme 9.
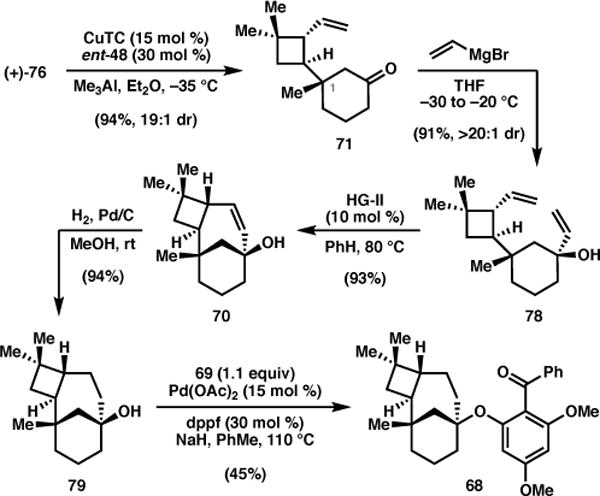
Elaboration to Norrish–Yang benzophenone.
In the event, irradiation of 68 with 254 nm light in rigorously deoxygenated dioxane led to complete consumption of starting material within 1 hour and produced a complex mixture of several new products.46 The formation of the undesired Norrish–Yang product 80 was confirmed by 2D NMR spectroscopy; a prominent HMBC correlation was apparent between C5 and the newly formed methine proton at C7, and several key NOE signals were consistent with the stereochemical assignment (Scheme 10). Thus, while the anticipated reactivity was observed, 80 results from the wrong regioselectivity, and was isolated in a mere 6.5% yield—a result that would likely be difficult to substantially improve through reaction optimization.
Scheme 10.
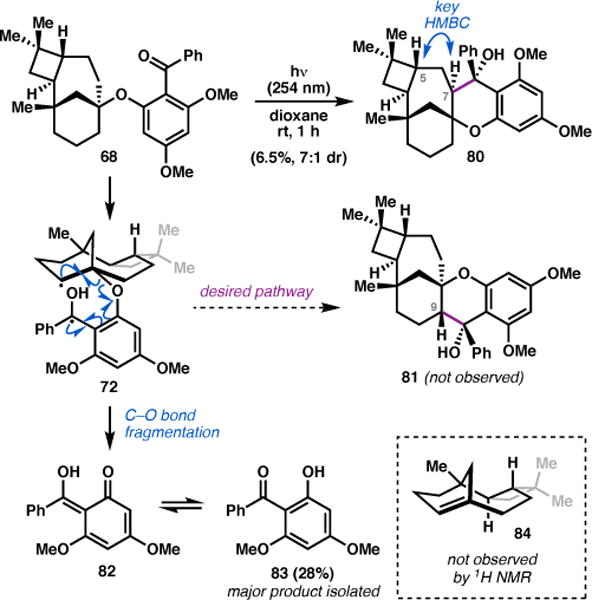
Evaluation of the Norrish–Yang cyclization.
Notably, the major compound isolated from this reaction is phenol 83,47 which was obtained in 28% yield. This side product presumably arises by fragmentation of diradical species 72 (or the diradical resulting from H-atom abstraction at C7), wherein C–O bond cleavage expels enol tautomer 82; the resulting terpene-based fragment likely undergoes further decomposition, as alkene 84 or related compounds were not isolated.48 In an effort to investigate whether this competing pathway could be suppressed, we examined a number of different solvents and irradiation wavelengths in a model system, but observed rapid formation of phenol 83 in all cases. Having determined that the late-stage Norrish–Yang cyclization was an untenable strategy to complete the chroman core of 3, an alternative synthetic route was devised.
THIRD GENERATION STRATEGY
In our final revision of the retrosynthesis, we simplified 3 to 85 and elected to construct the C9–C1′ bond at an earlier stage (Figure 4). Invoking a similar disconnection through the C–O aryl bond as in our second-generation route, it was anticipated that an intramolecular ring closure would prove more reliable than the challenging intermolecular arylation employed previously (see Scheme 9). This bond scission revealed aryl bromide 86, which could be accessed using the established ring-closing metathesis, while the arene functionality at C9 could be installed via aldol condensation with vinyl ketone 71.
Figure 4.
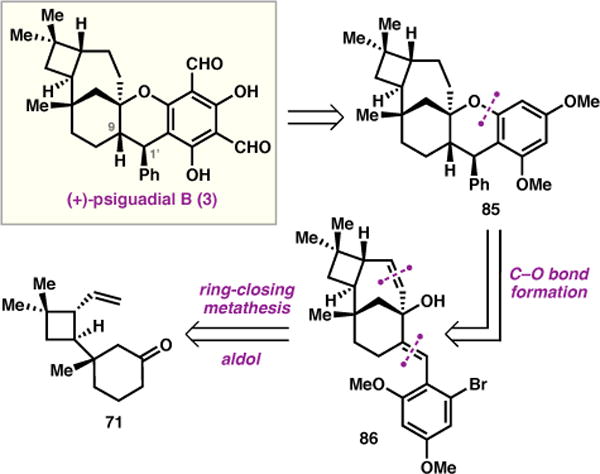
Third generation retrosynthetic analysis.
In the forward sense, a methanolic solution of vinyl ketone 71 and aldehyde 87 was treated with potassium hydroxide at elevated temperature to afford exo-enone 88 in 92% yield (Scheme 11). Attempts to incorporate the C1′ phenyl group at this stage via conjugate addition were met with limited success, yielding only trace amounts of 90 as an inseparable mixture of diastereomers at C9 and C1′. An alternative aldol condensation between 71 and benzophenone 69 (see Figure 3 for structure) failed to produce 89 under otherwise identical conditions or Mukaiyama aldol conditions. Given the inability to introduce the C1′ phenyl substituent at this point, we elected to advanced 88 and attempt to install this group at a later stage in the synthesis.
Scheme 11.
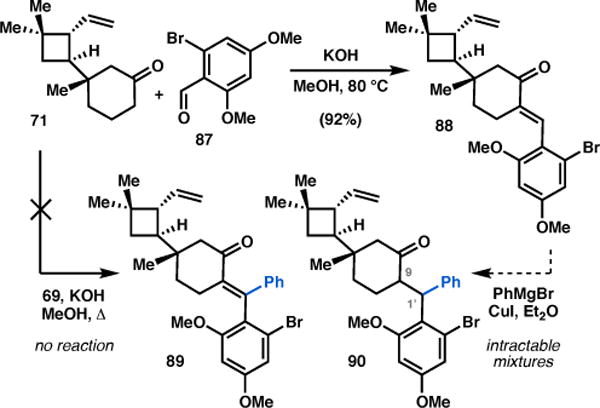
Installation of C9–C1′ bond via aldol reaction.
In contrast to the previous system lacking substitution at C9 (i.e. 71), 1,2-addition into this more sterically hindered ketone proved challenging. Treatment of 88 with vinyl magnesium bromide under the conditions used previously led to incomplete conversion—presumably due to competitive α-deprotonation— affording 91 (Scheme 12) in low yields and moderate diastereoselectivity.26 Attempts to improve conversion using Lewis acid activators gave higher yields of 91, but resulted in lower levels of diastereoselectivity (<2:1). Ultimately, the desired allylic alcohol was obtained in good yield with serviceable dr by employing vinyllithium in THF at −78 °C, allowing isolation of 91 as a single diastereomer in 54% yield. The ring-closing metathesis proceeded with equal efficiency on this new substrate to furnish 86 in 93% yield. With the strained sesquiterpene framework secured, both the di- and trisubstituted olefins in 86 were hydrogenated in the presence of Crabtree’s catalyst, which engaged in a hydroxyl-directed reduction49 to establish the C9 stereogenic center with 16:1 dr, providing 93 in excellent yield. The final ring of the psiguadial framework was formed by a Cu-catalyzed intramolecular O-arylation reaction, which furnished pentacycle 94 in 75% yield.50
Scheme 12.
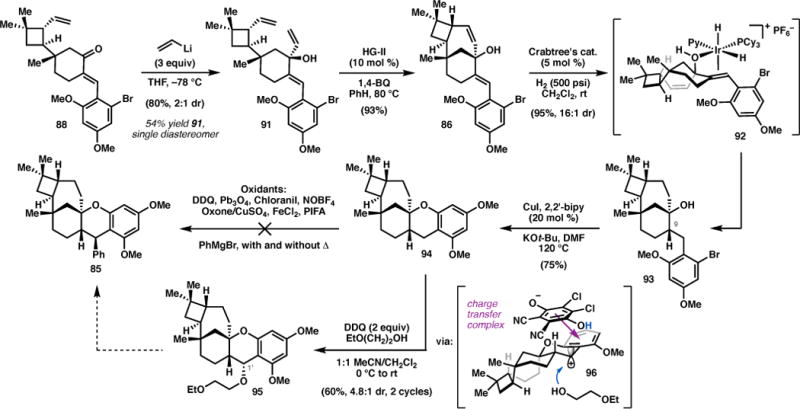
Synthesis of the core of (+)-psiguadial B.
With the successful development of a scalable and high-yielding route to 94, the task of appending the C1′ phenyl group was now at hand. Ideally, the electron rich arene in 94 would be engaged directly in a benzylic arylation reaction; a possible mechanism would involve benzylic oxidation at C1′ followed by trapping with a phenyl nucleophile. Whereas a number of laboratories have shown that electron rich arenes can trap benzylic cations in simple systems,51 it was unclear whether an electronically neutral, unsubstituted phenyl group would be sufficiently reactive to engage as the nucleophile in this type of transformation. Nonetheless, we investigated this possibility with reagents commonly used in flavonoid chemistry (e.g. DDQ,51a,d,52 Chloranil, Pb3O4,53 Oxone/CuSO4,54 and NOBF455), followed by trapping with benzene, PhMgBr, or PhB(OH)2, all without success. Efforts to apply Shi’s FeCl2-catalyzed benzylic dehydrogenative arylation,56 or Muramatsu’s C(sp3)–H arylation using DDQ and PIFA57 were also unfruitful.
Having failed to achieve a direct arylation, a stepwise protocol was employed. Oxidation with DDQ in the presence of ethoxyethanol52 afforded 95—a relatively stable product—which could be isolated in modest yields. The remaining mass balance of the reaction consisted of side products suspected to result from over oxidation and elimination of the benzylic ether. A survey of reaction parameters revealed that adding acetonitrile as a co-solvent led to cleaner reaction profiles, albeit at the expense of conversion. Presumably, the acetonitrile solvent helps to stabilize the intermediate benzylic cation (i.e. 96), favoring more efficient trapping with ethoxyethanol over unproductive side reactions. Synthetically useful yields of 95 were obtained under these conditions by re-subjecting recovered 94 to the reaction conditions a second time.
With respect to the stereochemistry at C1′, 95 was isolated as a 4.8:1 mixture of diastereomers, favoring the α-disposed ether. Conformational analysis of 94 indicates that the 7-membered ring protrudes from the bottom face of the molecule, suggesting that C–O bond formation appears to proceed with contrasteric selectivity. The observed stereochemical outcome might result from an overall double inversion process that proceeds by initial association of DDQ from the less hindered top face of 94 to form a tightly bound charge-transfer complex (i.e. 96).58 If this complex remains closely associated, ethoxyethanol would then attack from the bottom face, thus leading to α-ether 95 as the major diastereomer.
With a functional handle installed at C1′, TMSOTf was initially investigated as a Lewis acid to activate the ethoxyethyl benzyl ether, however, no phenylated product was obtained using PhB(OH)2, or PhMgBr as nucleophiles (Table 2, entries 1 and 2).52b Simple heating59 or nickel-catalyzed Kumada coupling with PhMgBr in PhMe60 yielded only eliminated products and complex reaction profiles (entries 3–5). Likewise, Bode’s conditions for the addition of aryl trifluoroborates to oxonium ions, which use BF3•OEt2 as the Lewis acid, failed to produce 85 (entries 6 and 7).61 We were therefore delighted to obtain a near quantitative yield of 85 (in 1.7:1 dr) by treating a mixture of 95 and lithium diphenylcyanocuprate with BF3•OEt2 (entry 8).62 After some experimentation, it was found that the diastereoselectivity could be slightly improved to 2:1 by holding the reaction at –45 °C (entry 9). Although colder temperatures led to a further improvement in dr, this was accompanied by a lower yield (entry 10).
Table 2.
Investigation of the C1′ Phenylation.
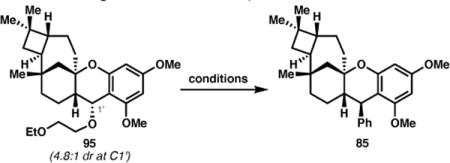
| |||||
|---|---|---|---|---|---|
| entry | Lewis acid or catalyst | nucleophile | solvent | T (°C) | yield (%)85 (dr)a |
| 1 | TMSOTf | PhB(OH)2 | THF | –78 to 0 | 0 |
| 2 | TMSOTf | PhMgBr | THF | −78 to 0 | 0 |
| 3 | none | PhMgBr | PhMe | 110 | 0 |
| 4 | Ni(dppe)Cl2 | PhMgBr | PhMe | 25 | 0 |
| 5 | Ni(dppe)Cl2/MgI2 | PhMgBr | PhMe | 25 | 0 |
| 6 | BF3•OEt2 | PhBF3K | CH2Cl2 | 0 to 25 | 0 |
| 7 | BF3•OEt2 | PhBF3K | MeCN | 0 to 25 | 0 |
| 8 | BF3•OEt2 | Ph2Cu(CN)Li | Et2O | −78 to 0 | 99 (1.7:1) |
| 9 | BF3•OEt2 | Ph2Cu(CN)Li | Et2O | −78 to –45 | 90 (2.0:1) |
| 10 | BF3•OEt2 | Ph2Cu(CN)Li | Et2O | −78 to –60 | 57 (3.3:1) |
Diastereomeric ratio obtained by 1H NMR analysis of crude reaction mixture, provided in parenthesis.
As the C1′ diastereomers of 85 were inseparable by silica gel chromatography, the mixture was subjected to pyridine hydrochloride at 200 °C, which afforded the corresponding demethylated products in 92% combined yield (Scheme 13). At this stage, the diastereomeric resorcinols were readily separable by column chromatography, providing 97 as a single diastereomer in 62% yield. Finally, the remaining two aryl aldehydes were simultaneously installed using Rieche formylation conditions,63 delivering (+)-psiguadial B (3) in 50% yield. Synthetic 3 was found to be spectroscopically identical in all respects to the natural sample reported by Shao et al.4
Scheme 13.
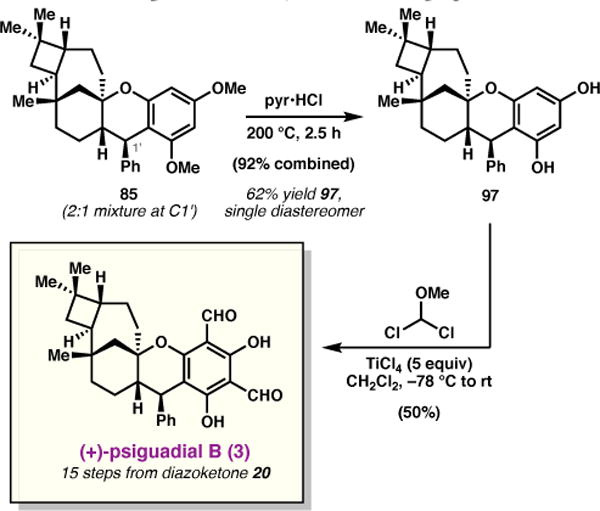
Completion of the synthesis of (+)-psiguadial B.
CONCLUSION
In summary, the first enantioselective total synthesis of the cytotoxic natural product, (+)-psiguadial B (3), was achieved in 15 steps from diazoketone 20. The successful synthetic strategy was enabled by the implementation of a tandem photochemical Wolff rearrangement/asymmetric ketene addition reaction. Having developed a novel protocol for the enantioselective preparation of quinolinamide 18, a variety of substrates were evaluated and conditions were identified to prepare the corresponding 5- and 6-membered ring products. De novo construction of the trans-fused cyclobutane ring in 3 was accomplished using a strategic Pd-catalyzed C(sp3)−H alkenylation reaction, followed by one of two distinct epimerization strategies, which permit access to both enantiomers of the natural product from a single enantiomer of organocatalyst.
In the course of this work, three different synthetic routes toward (+)-psiguadial B were investigated. These studies have led to the evaluation of several challenging transformations, including 1) an o-QMHDA cycloaddition between a highly functionalized enol ether and a phloroglucinol-derived o-QM; 2) a seven-membered ring-forming Prins cyclization; and 3) a modified Norrish–Yang cyclization. Ultimately, the strained sesquiterpene core was built using a remarkably efficient ring-closing metathesis, and elaborated through a short sequence to afford the natural product in 1.3% overall yield. We believe that the development of this route to 3 may enable the synthesis of unnatural analogs of 3, that would be difficult to access through semi-synthetic methods. Application of the key strategy concepts described herein to the synthesis of other trans-cyclobutane-containing natural products are currently ongoing in our laboratory.
EXPERIMENTAL
General Procedures
Unless otherwise stated, reactions were performed under a nitrogen atmosphere using freshly dried solvents. Tetrahydrofuran (THF), methylene chloride (CH2Cl2), acetonitrile (MeCN), benzene (PhH), 1,4-dioxane, and toluene (PhMe) were dried by passing through activated alumina columns. Triethylamine (Et3N) and methanol (MeOH) were distilled over calcium hydride prior to use. Unless otherwise stated, chemicals and reagents were used as received. All reactions were monitored by thin-layer chromatography (TLC) using EMD/Merck silica gel 60 F254 pre-coated plates (0.25 mm) and were visualized by UV, p-anisaldehyde, or 2,4-dinitrophenylhydrazine staining. Flash column chromatography was performed either as described by Still et al.64 using silica gel (particle size 0.032-0.063) purchased from Silicycle or using prepackaged RediSep®Rf columns on a CombiFlash Rf system (Teledyne ISCO Inc.). Optical rotations were measured on a Jasco P-2000 polarimeter using a 100 mm path-length cell at 589 nm. 1H and 13C NMR spectra were recorded on a Bruker Avance III HD with Prodigy cryoprobe (at 400 MHz and 101 MHz respectively), a Varian 400 MR (at 400 MHz and 101 MHz, respectively), a Varian Inova 500 (at 500 MHz and 126 MHz, respectively), or a Varian Inova 600 (at 600 MHz and 150 MHz, respectively), and are reported relative to internal CHCl3 (1H, δ = 7.26) and CDCl3 (13C, δ = 77.1), C6H5 (1H, δ = 7.16) and C6D6 (13C, δ = 128), or d8-THF (1H, δ = 3.58) and (13C, δ = 67.6). Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). Multiplicity and qualifier abbreviations are as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, app = apparent. IR spectra were recorded on a Perkin Elmer Paragon 1000 spectrometer and are reported in frequency of absorption (cm–1). HRMS were acquired using an Agilent 6200 Series TOF with an Agilent G1978A Multimode source in electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI), or mixed (MM) ionization mode. Analytical SFC was performed with a Mettler SFC supercritical CO2 analytical chromatography system with a Chiralcel AD-H column (4.6 mm × 25 cm).
Procedures and characterization data for compounds 3, 18, 20, 46, 47, 71, 73, 74, 75, 76, 77, 85, 86, 88, 91, 93, 94, 95, 97 were reported previously.12
Preparation of diazoketone 20:21,65
To each of two flame-dried 1 L round-bottom flasks was added NaH (60% dispersion in mineral oil, 3.17 g, 79.2 mmol, 1.20 equiv) and the atmosphere was exchanged for N2 one time. Dry Et2O (30.0 mL) was then added via syringe and the suspension cooled to 0 °C. Ethyl formate (12.4 mL, 152 mmol, 2.30 equiv) was then added, followed by 2,2-dimethylcyclopentanone (7.40 g, 66.0 mmol) either neat, or as a 3.0 M solution in Et2O. A catalytic amount of wet methanol (~100 μL) was then added and the reaction left to stir at 0 °C.66 Upon completion, the reaction solidifies to a chunky, white solid that dissolved readily upon the addition of DI H2O. At this point, both reaction mixtures were combined for workup: after dilution with Et2O, the layers were separated and the aqueous layer was washed with Et2O 3x to remove organic impurities and a small amount of unreacted starting material. The aqueous layer was then cooled to 0 °C and acidified to pH = 3 using 5 M HCl. Et2O was then added and the acidified aqueous layer was extracted 6x. The combined organics were then dried over Mg2SO4, filtered, and concentrated in vacuo into a 500 mL round-bottom flask.
The crude α-formyl ketone was taken up in CH2Cl2 (132 mL) and the solution cooled to –10 °C. Triethylamine (55.2 mL, 396 mmol, 5.00 equiv) was added, followed by solid p-ABSA67 (31.8 g, 132 mmol, 1.00 equiv) in three portions. The reaction was stirred for 3 hours and allowed to gradually reach 10 °C, at which point an aqueous solution of KOH (55.0 mL, 4 M) was added. Additional CH2Cl2 and H2O were added, the layers were separated and the aqueous layer extracted with CH2Cl2 until no product remains by TLC. The combined organics were dried over Mg2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (20–30% Et2O/pentane) to afford 20 (17.4 g, 95% yield) as a bright yellow oil. 1H NMR (400 MHz, CDCl3) δ 2.88 (t, J = 7.0 Hz, 2H), 1.77 (t, J = 7.2 Hz, 2H), 1.04 (d, J = 1.0 Hz, 6H). 1H NMR (400 MHz, d8-THF) δ 2.94 (t, J = 7.0 Hz, 2H), 1.79 (t, J = 7.2 Hz, 2H), 1.04 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 204.8, 56.6, 46.3, 35.7, 24.1, 21.2. 13C NMR (101 MHz, d8-THF) δ 203.6, 56.1, 46.9, 36.6, 24.5, 21.9. FTIR (NaCl, thin film) 3754, 3414, 3332, 2962, 2934, 2892, 2869, 2672, 2642, 2578, 2510, 2080, 1981, 1673, 1581, 1471, 1460, 1382, 1362, 1339, 1309, 1267, 1245, 1204, 1133, 1110, 1058, 1030, 994, 977, 948, 919, 893, 780, 726, 697 cm.−1
Diazoketones 38–41 were prepared according to the procedure developed for 20. Spectroscopic data for 40 and 41 are consistent with that reported in the literature.68
(38)
Yellow Oil, (1.76 g, 36% yield over 2 steps). 1H NMR (400 MHz, CDCl3) δ 2.71 (t, J = 6.5 Hz, 2H), 1.82 – 1.73 (m, 2H), 1.68 –1.61 (m, 2H), 1.15 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 200.1, 62.6, 42.0, 37.5, 26.7, 22.9, 18.5. FTIR (NaCl, thin film) 2943, 2864, 2082, 1626, 1472, 1449, 1381, 1342, 1317, 1275, 1261, 1220, 1201, 1162, 1122, 1044, 1011, 910, 853, 738, 658 cm.−1 HRMS (EI) calc’d for C8H12N2O [M]+ 152.0950, found 152.0956.
(39)
Yellow Oil, (400.0 mg, 26% yield over 2 steps) 1H NMR (400 MHz, CDCl3) δ 2.55 (ddt, J = 7.0, 4.8, 2.3 Hz, 2H), 1.75 (dt, J = 4.4, 2.8 Hz, 4H), 1.57 (ddt, J = 6.3, 3.4, 1.7 Hz, 2H), 1.17 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 202.2, 68.3, 47.0, 37.9, 29.5, 25.8, 25.7, 25.6. FTIR (NaCl, thin film) 2981, 2966, 2927, 2858, 2083, 1704, 1617, 1474. 1448, 1387. 1364, 1350, 1324, 1272, 1251, 1231, 1203, 1147, 1113, 1057, 1020, 980, 953, 871, 845, 736, 656 cm.−1 HRMS (EI) calc’d for C9H14N2O [M]+ 166.1106, found 166.1095.
Small-scale screening protocol for enantioenriched amides 18, 42–45
Oven-dried quartz tubes were each charged with aminoquinoline (21.6 mg, 0.150 mmol, 3.00 equiv) and catalyst (50 mol %). Inside a N2-filled glovebox, diazoketones 20, 38–40 (0.05 mmol) were then added to each as a solution in 0.500 mL THF (excluding diazoketone 41, which was added as a solid outside of the glovebox). The reactions were then sealed with a 19/38 rubber septum around the outside of each tube and sealed with electrical tape. The reactions were then brought out of the glovebox and placed in a bottomless test tube rack in front of a Honeywell 254 nm lamp. The reactions were irradiated with stirring at room temperature for 18 hours. The reactions were then concentrated in vacuo, and the crude reaction mixtures were analyzed by 1H NMR with an added internal standard to determine % yield. The crude residues were purified by silica gel preparative TLC (2% Et2O/CH2Cl2) to provide 18, 42–45 in varying yields and enantiopurities.
Large-scale protocol for enantioenriched amide 18:65
To a flame-dried, 1 L quartz flask was added 8-aminoquinoline (29) (12.9 g, 89.5 mmol, 3.00 equiv) and (+)-cinchonine (30) (879 mg, 2.99 mmol, 0.100 equiv). The flask was evacuated and backfilled with N2 three times and dry THF (600 mL) was then added via cannula. Diazoketone 20 (4.12 g, 29.8 mmol, 1.00 equiv) was added last via syringe and the reaction was irradiated with stirring using a Honeywell 254 nm lamp at room temperature. Reaction progress was monitored by TLC (72-168 hours are typically required for complete conversion on this scale, and rotation of the flask every day provided faster conversion).69 Upon completion, the reaction mixture was concentrated in vacuo, the solids were taken up in CH2Cl2, and the suspension filtered. The filter cake was washed with CH2Cl2 three times and the filtrate was concentrated in vacuo to give a crude residue that was purified by silica gel flash chromatography (isocratic: 6% EtOAc/hexane) to provide 18 (4.69 g, 62%) as a pale-yellow solid. The enantiomeric excess was determined to be 79% by chiral SFC analysis (AD-H, 2.5 mL/min, 20% IPA in CO2, λ = 254 nm): tR (major) = 4.23 min, tR (minor) = 5.64 min. (c = 0.560, CHCl3). Enantioenriched cyclobutane 18 was dissolved in a minimal amount of CH2Cl2 in a 100 mL round-bottom flask. An equal amount of hexanes was carefully layered on top of the CH2Cl2 to form a biphasic mixture. The layers were allowed to diffuse overnight to provide 18 as white, crystalline needles (mp: 66–68 °C). The supernatant was concentrated under reduced pressure and this process was repeated again to provide additional 18 (3.50 g total, 83% recovery of theoretical total of the desired enantiomer, 46% overall from 20): (c = 0.720, CHCl3). 1H NMR (400 MHz, CDCl3) δ 9.68 (s, 1H), 8.80 (t, J = 1.8 Hz, 1H), 8.79 (dd, J = 13.6, 1.6 Hz, 1H), 8.15 (dd, J = 8.3, 1.7 Hz, 1H), 7.52 (q, J = 8.2, 7.5 Hz, 1H), 7.48 (dd, J = 8.3, 1.6 Hz, 1H), 7.45 (dd, J = 8.3, 4.2 Hz, 1H), 3.07 (ddd, J = 9.1, 8.2, 0.9 Hz, 1H), 2.48 (dq, J = 11.4, 9.4 Hz, 1H), 2.06 (dtd, J = 11.6, 8.6, 3.3 Hz, 1H), 1.85 (dt, J = 10.8, 9.1 Hz, 1H), 1.74 (dddd, J = 10.7, 9.5, 3.3, 0.9 Hz, 1H), 1.39 (s, 3H), 1.14 (s, 3H). 13C NMR δ 171.8, 148.3, 138.6, 136.4, 134.7, 128.1, 127.6, 121.7, 121.3, 116.4, 51.0, 40.4, 32.3, 30.9, 23.4, 17.4. FTIR (NaCl, thin film) 3353, 3047, 2952, 2861, 1685, 1595, 1577, 1526, 1485, 1460, 1424, 1385, 1324, 1261, 1239, 1187, 1169, 1153, 825, 791,756 cm.−1 HRMS (MM) calc’d for C16H19N2O [M+H]+ 255.1492, found 255.1501.
(42)
0.2 mmol scale: An oven-dried quartz tube was charged with aminoquinoline (29) (86.5 mg, 0.600 mmol, 3.00 equiv) and (34) (32.5 mg, 0.100 mmol, 0.500 equiv) and brought into a N2 filled glovebox. Diazoketone (38) (33.2 mg, 0.200 mmol, 1.00 equiv) was added as a solution in 2.00 mL THF and the tube was sealed with a 19/38 rubber septum and secured with electrical tape. The reaction was removed from the glovebox and placed in a bottomless test tube rack in front of a Honeywell 254 nm lamp for 48 hours. The reaction mixture was then concentrated in vacuo. The crude residue was purified via silica gel flash chromatography (6% EtOAc/hexanes) to afford 42 (37.5 mg, 77% yield) as a brown oil. The enantiomeric excess was determined to be 71% by chiral SFC analysis (AD-H, 2.5 mL/min, 20% IPA in CO2, λ = 254 nm): tR (major) = 4.28 min, tR (minor) = 5.41 min.
(42)
1 mmol scale: An oven-dried quartz tube was charged with aminoquinoline (29) (433 mg, 3.00 mmol, 3.00 equiv) and (34) (64.9 mg, 0.200 mmol, 0.200 equiv) and brought into a N2 filled glovebox. Diazoketone (38) (166 mg, 1.00 mmol, 1.00 equiv) was added as a solution in 10.0 mL THF and the tube was sealed with a 19/38 rubber septum and secured with electrical tape. The reaction was removed from the glovebox and placed in a bottomless test tube rack in front of a Honeywell 254 nm lamp for 48 hours. The reaction mixture was then concentrated in vacuo. The crude residue was purified via silica gel flash chromatography (6% EtOAc/hexanes) to afford 42 (215 mg, 80% yield) as a brown oil. The enantiomeric excess was determined to be 67% by chiral SFC analysis (AD-H, 2.5 mL/min, 20% IPA in CO2, λ = 254 nm): tR (major) = 4.28 min, tR (minor) = 5.41 min. (c = 2.075, CHCl3). 1H NMR (400 MHz, CDCl3) δ 9.80 (s, 1H), 8.81 (d, J = 1.7 Hz, 1H), 8.80 (dd, J = 3.0, 1.7 Hz, 1H), 8.16 (dd, J = 8.3, 1.7 Hz, 1H), 7.57 – 7.47 (m, 2H), 7.45 (dd, J = 8.3, 4.2 Hz, 1H), 2.61 (t, J = 8.4 Hz, 1H), 2.38 – 2.22 (m, 1H), 2.02 (dtd, J = 13.2, 8.5, 4.4 Hz, 1H), 1.95 – 1.82 (m, 1H), 1.79 – 1.65 (m, 2H), 1.63 – 1.57 (m, 1H), 1.31 (s, 3H), 1.01 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 173.1, 148.3, 138.6, 136.5, 134.8, 128.1, 127.6, 121.7, 121.3, 116.4, 58.1, 43.2, 42.1, 29.7, 27.9, 24.0, 22.5. FTIR (NaCl, thin film) 3362, 2957, 2924, 2854, 1729, 1690, 1525, 1486, 1464, 1424, 1381, 1325, 1262, 1164, 1145, 1132, 1072, 825, 791, 720 cm.−1 HRMS (MM) calc’d for C17H21N2O [M+H]+ 269.1648, found 269.1645.
(43)
0.2 mmol scale: An oven-dried quartz tube was charged with 8-aminoquinoline (29) (86.5 mg, 0.600 mmol, 3.00 equiv) and (33) (32.5 mg, 0.100 mmol, 0.500 equiv) and brought into a N2 filled glovebox. Diazoketone (39) (31.0 mg, 0.200 mmol, 1.00 equiv) was added as a solution in 2.00 mL THF and the tube was sealed with a 19/38 rubber septum and secured with electrical tape. The reaction was removed from the glovebox and placed in a bottomless test tube rack in front of a Honeywell 254 nm lamp for 48 hours. The reaction mixture was then concentrated in vacuo. The crude residue was purified via silica gel flash chromatography (6% EtOAc/hexanes) to afford 43 (33.3 mg, 59% yield) as a brown oil. The enantiomeric excess was determined to be 71% by chiral SFC analysis (AD-H, 2.5 mL/min, 12% IPA in CO2, λ = 254 nm): tR (major) = 9.67 min, tR (minor) = 10.34 min.
(43)
1 mmol scale: An oven-dried quartz tube was charged with 8-aminoquinoline (29) (433 mg, 3.00 mmol, 3.00 equiv) and (33) (64.9 mg, 0.200 mmol, 0.200 equiv) and brought into a N2 filled glovebox. Diazoketone (39) (152 mg, 1.00 mmol, 1.00 equiv) was added as a solution in 10.0 mL THF and the tube was sealed with a 19/38 rubber septum and secured with electrical tape. The reaction was removed from the glovebox and placed in a bottomless test tube rack in front of a Honeywell 254 nm lamp for 48 hours. The reaction mixture was then concentrated in vacuo. The crude residue was purified via silica gel flash chromatography (6% EtOAc/hexanes) to afford 43 (189 mg, 67% yield) as a brown oil. The enantiomeric excess was determined to be 67% by chiral SFC analysis (AD-H, 2.5 mL/min, 12% IPA in CO2, λ = 254 nm): tR (major) = 9.67 min, tR (minor) = 10.34 min. (c = 1.68, CHCl3). 1H NMR (400 MHz, CDCl3) δ 9.79 (s, 1H), 8.82 (d, J = 1.7 Hz, 1H), 8.80 (dd, J = 2.7, 1.7 Hz, 1H), 8.16 (dd, J = 8.3, 1.7 Hz, 1H), 7.57 – 7.47 (m, 2H), 7.45 (dd, J = 8.3, 4.2 Hz, 1H), 2.30 (dd, J = 11.8, 3.5 Hz, 1H), 1.99 – 1.78 (m, 3H), 1.55 – 1.47 (m, 2H), 1.39 – 1.27 (m, 3H), 1.13 (s, 3H), 1.10 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 173.6, 148.3, 138.6, 136.5, 134.7, 128.1, 127.6, 121.7, 121.3, 116.5, 56.5, 41.6, 33.4, 31.5, 25.7, 25.7, 22.1, 21.2. FTIR (NaCl, thin film) 3364, 2956, 2923, 2852, 1729, 1691, 1523, 1486, 1462, 1424, 1378, 1326, 1273, 1129, 1072, 825, 790 cm.−1 HRMS (MM) calc’d for C18H23N2O [M+H]+ 283.1805, found 283.1796.
(44)
An oven-dried quartz tube was charged with aminoquinoline (29) (86.5 mg, 0.600 mmol, 3.00 equiv) and (37) (85.7 mg, 0.100 mmol, 0.500 equiv) and brought into a N2 filled glovebox. Diazoketone (40) (31.6 mg, 0.200 mmol, 1.00 equiv) was added as a solution in 2.00 mL THF and the tube was sealed with a 19/38 rubber septum and secured with electrical tape. The reaction was removed from the glovebox and placed in a bottomless test tube rack in front of a Honeywell 254 nm lamp for 48 hours. The reaction mixture was then concentrated in vacuo. The crude residue was purified via silica gel flash chromatography (5–50 % EtOAc/hexanes followed by 0–1% Et2O/CH2Cl2) to afford 44 (16.8 mg, 31% yield). The enantiomeric excess was determined to be 34% by chiral SFC analysis (AD-H, 2.5 mL/min, 30% IPA in CO2, λ = 254 nm): tR (major) = 5.06 min, tR (minor) = 6.89 min. (c = 0.565, CHCl3). 1H NMR (400 MHz, CDCl3) δ 10.21 (s, 1H), 8.79 (dd, J = 11.5, 1.7 Hz, 1H), δ 8.78 (d, J = 1.7 Hz, 1H), 8.15 (dd, J = 8.3, 1.7 Hz, 1H), 7.54 (dd, J = 8.3, 7.2 Hz, 1H), 7.50 (dd, J = 8.3, 1.8 Hz, 1H), 7.46 – 7.41 (m, 2H), 7.38 – 7.29 (m, 2H), 7.22 – 7.16 (m, 1H), 4.56 (ddt, J = 5.8, 2.9, 0.8 Hz, 1H), 3.69 (ddd, J = 14.2, 5.7, 0.7 Hz, 1H), 3.60 (ddd, J = 14.2, 2.9, 0.8 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 170.6, 148.4, 144.7, 142.9, 138.7, 136.4, 134.5, 128.6, 128.0, 127.8, 127.5, 123.5, 122.7, 121.7, 121.7, 116.5, 49.3, 35.2. FTIR (NaCl, thin film) 3347, 3066, 2928, 2851, 1680, 1596, 1578, 1526, 1485, 1458, 1424, 1386, 1328, 1262, 1240, 1202, 1162, 1132, 869, 826, 791, 759, 734, 707, 679 cm.−1 HRMS (MM) calc’d for C18H15N2O [M+H]+ 275.1179, found 275.1178.
(45)
An oven-dried quartz tube was charged with diazoketone (41) (34.4 mg, 0.200 mmol, 1.00 equiv), aminoquinoline (29) (86.5 mg, 0.600 mmol, 3.00 equiv), and diazoketone (36) (88.1 mg, 0.100 mmol, 0.500 equiv) and brought into a N2 filled glovebox. The mixture was suspended in 2.00 mL THF and the tube was sealed with a 19/38 rubber septum and secured with electrical tape. The reaction was removed from the glovebox and placed in a bottomless test tube rack in front of a Honeywell 254 nm lamp for 48 hours. The reaction mixture was then concentrated in vacuo. The crude residue was purified via silica gel flash chromatography (5–10% EtOAc/hexanes) to afford 45 (24.1 mg, 42% yield) as a brown oil. The enantiomeric excess was determined to be 75% by chiral SFC analysis (AD-H, 2.5 mL/min, 30% IPA in CO2, λ = 254 nm): tR (major) = 5.73 min, tR (minor) = 4.86 min. (c = 0.91, CHCl3). 1H NMR (400 MHz, CDCl3) δ 10.06 (s, 1H), 8.79 (dd, J = 7.1, 1.9 Hz, 1H), 8.75 (dd, J = 4.2, 1.7 Hz, 1H), 8.15 (dd, J = 8.3, 1.7 Hz, 1H), 7.56 – 7.46 (m, 3H), 7.44 (dd, J = 8.3, 4.2 Hz, 1H), 7.33 (d, J = 7.2 Hz, 1H), 7.31 – 7.18 (m, 2H), 4.27 (dd, J = 8.4, 6.1 Hz, 1H), 3.23 (dt, J = 15.2, 7.4 Hz, 1H), 3.09 – 2.95 (m, 1H), 2.69 – 2.48 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 172.7, 148.4, 144.8, 141.5, 138.7, 136.4, 134.7, 128.0, 127.9, 127.53, 126.9, 125.1, 125.0, 121.7, 121.7, 116.6, 54.0, 32.1, 30.4. FTIR (NaCl, thin film) 3347, 2957, 2923, 2852, 1728, 1689, 1524, 1484, 1461, 1424, 1380, 1325, 1272, 1163, 1132, 1072, 826, 791, 743 cm.−1 HRMS (MM) calc’d for C19H17N2O [M+H]+ 289.1335, found 289.1334.
(50)
To a flame-dried 100 mL flask was added copper (I) iodide (1.48 g, 7.75 mmol, 5.00 equiv) and Et2O (15.5 mL). The resulting suspension was cooled to –40 °C and methyllithium (1.6 M in Et2O; 9.68 mL, 15.5 mmol, 10 equiv) was added dropwise. The reaction mixture was stirred at −40 °C for 2 hours before 47 (540 mg, 1.55 mmol) was added dropwise as a solution in 5:2 CH2Cl2/Et2O. The reaction mixture was gradually warmed to 0 °C over 4 hours, then quenched with saturated aqueous NH4Cl (10 mL) and diluted with EtOAc. NH4OH was added until all of the solid copper salts were sequestered and two homogenous layers remained. The aqueous layer was extracted with EtOAc (3 × 20 mL) and the combined organics dried over MgSO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (isocratic: 20% EtOAc/Hexane) to afford a 2.5:1 mixture of 49 and 50 (543 mg, 96% yield), respectively as a white amorphous solid. Subsequent purification by reverse-phase HPLC using two Agilent Eclipse XDB-C8 5um 9.4 × 250 mm columns connected in series (gradient: 77–85%MeCN/H2O) afforded analytically pure samples of each diastereomer, from which 50 was crystallized for X-Ray analysis26 (mp: 80–83 °C). Data for minor diastereomer 50: (c = 1.50, CHCl3). 1H NMR (500 MHz, CDCl3) δ 9.64 (s, 1H), 8.82 (dd, J = 4.2, 1.7 Hz, 1H), 8.75 (dd, J = 7.4, 1.6 Hz, 1H), 8.17 (dd, J = 8.3, 1.7 Hz, 1H), 7.56 – 7.48 (m, 2H), 7.46 (dd, J = 8.2, 4.2 Hz, 1H), 2.89 – 2.77 (m, 2H), 2.35 – 2.26 (m, 2H), 2.24 (d, J = 13.3 Hz, 1H), 2.09 (d, J = 13.4 Hz, 1H), 2.07 – 1.99 (m, 1H), 1.88 – 1.77 (m, 1H), 1.72 – 1.61 (m, 3H), 1.55 – 1.48 (m, 1H), 1.35 (s, 3H), 1.13 (s, 3H), 0.92 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 212.4, 170.6, 148.4, 138.5, 136.5, 134.6, 128.1, 127.6, 121.7, 121.4, 116.4, 51.5, 50.8, 41.2, 39.8, 39.6, 35.2, 34.1, 33.0, 30.8, 23.7, 22.2, 21.3. FTIR (NaCl, thin film) 3349, 3044, 2952, 2863, 1706, 1687, 1595, 1577, 1523, 1484, 1460, 1424, 1383, 1325, 1238, 1228, 1163, 827, 792 cm.−1 HRMS (MM) calc’d for C23H29N2O2 [M+H]+ 365.2224, found 365.2261. XRCD: A suitable crystal of C23H28N2O2 (50) was selected for analysis. Low-temperature diffraction data (φ- and ω-scans) were collected on a Bruker AXS D8 VENTURE KAPPA diffractometer coupled to a PHOTON 100 CMOS detector with Cu-Kα radiation (λ = 1.54178 Å) from a IμS HB micro-focus sealed X-ray tube. All diffractometer manipulations, including data collection, integration, and scaling were carried out using the Bruker APEXII software.70
(49)
Inside a N2-filled glovebox, [Cu(OTf)]2•PhMe (72.4 mg, 0.140 mmol, 0.25 equiv) and (S,R,R) ligand 4871 (302 mg, 0.560 mmol, 1.00 equiv) were added to a 25 mL flask. The reagents were suspended in Et2O (5.60 mL) and stirred at room temperature for 30 mins before trans-cyclobutane 47 (195 mg, 0.560 mmol) was added as a solid, in one portion. The reaction was sealed under N2, removed from the glovebox and cooled to –30 °C under argon using a cryocool unit to control the temperature. Me3Al (2.0 M in heptane; 560 μL, 1.12 mmol, 2.00 equiv) was then added dropwise, taking care to avoid an exotherm and the reaction mixture stirred vigorously at –30 °C for 16 hours. MeOH (1.00 mL) was then added to quench excess Me3Al and then the reaction was warmed to room temperature. The mixture was diluted with EtOAc and H2O, then the organic layer was separated. The aqueous layer was extracted with EtOAc (3 × 5 mL) and the combined organics dried over MgSO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (2% Et2O/CH2Cl2 until ligand/impurities elute, then 4% Et2O/CH2Cl2) to afford a 30:1 mixture of 49 and 50 (126 mg, 62% yield), respectively as a white solid: (c = 0.600, CHCl3). 1H NMR (400 MHz, CDCl3) δ 9.64 (s, 1H), 8.81 (dd, J = 4.2, 1.7 Hz, 1H), 8.75 (dd, J = 7.2, 1.8 Hz, 1H), 8.16 (dd, J = 8.3, 1.7 Hz, 1H), 7.56 – 7.47 (m, 2H), 7.45 (dd, J = 8.3, 4.2 Hz, 1H), 2.89 – 2.76 (m, 2H), 2.36 – 2.28 (m, 2H), 2.25 (ddd, J = 12.5, 6.6, 1.1 Hz, 1H), 2.04 (dt, J = 13.4, 2.0 Hz, 1H), 1.96 (ddq, J = 13.7, 7.0, 3.6 Hz, 1H), 1.81 (dtt, J = 13.7, 12.0, 5.0 Hz, 1H), 1.68 – 1.62 (m, 2H), 1.62 – 1.51 (m, 2H), 1.35 (s, 3H), 1.13 (s, 3H), 0.89 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 212.4, 170.6, 148.3, 138.5, 136.5, 134.6, 128.1, 127.5, 121.7, 121.4, 116.4, 51.5, 50.4, 41.3, 40.9, 39.5, 35.2, 33.8, 32.6, 30.8, 23.7, 22.1, 20.8. FTIR (NaCl, thin film) 3351, 3047, 2954, 2870, 1708, 1688, 1524, 1485, 1460, 1424, 1384, 1325, 1281, 1259, 1240, 1228, 1163, 919, 827, 792, 757, 732 cm.−1 HRMS (MM) calc’d for C23H29N2O2 [M+H]+ 365.2224, found 365.2228.
(51)
To a flame-dried 15 mL flask was added ketone 49 (100 mg, 0.274 mmol) and dissolved in freshly distilled MeOH (2.7 mL). Trimethylorthoformate (150 μL, 1.37 mmol, 5.00 equiv) was then added, followed by p-toluenesulfonic acid monohydrate (2.60 mg, 0.014 mmol, 0.05 equiv). The reaction was topped with a reflux condenser and heated to 65 °C for 1 hour, then quenched with saturated aqueous NaHCO3. The aqueous layer was extracted with EtOAc (3 × 5 mL), and the combined organics were dried over MgSO4, filtered, and concentrated in vacuo. The crude residue was purified by Florisil® flash chromatography (isocratic: 10% EtOAc/Hexane) to afford 51 (106 mg, 94% yield) as a white, foamy solid: (c = 1.60, CHCl3). 1H NMR (400 MHz, CDCl3) δ 9.66 (s, 1H), 8.81 (dd, J = 4.3, 1.7 Hz, 1H), 8.78 (dd, J = 7.4, 1.6 Hz, 1H), 8.14 (dd, J = 8.3, 1.7 Hz, 1H), 7.56 – 7.46 (m, 2H), 7.44 (dd, J = 8.3, 4.2 Hz, 1H), 3.16 (s, 3H), 3.13 (s, 3H), 2.80 (d, J = 10.0 Hz, 1H), 2.69 (q, J = 9.7 Hz, 1H), 2.01 (ddd, J = 13.2, 3.5, 1.6 Hz, 1H), 1.74 (dt, J = 14.0, 2.4 Hz, 1H), 1.70 – 1.50 (m, 4H), 1.31 (s, 3H), 1.28 – 1.13 (m, 4H), 1.11 (s, 3H), 1.01 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.0, 148.3, 138.5, 136.4, 134.7, 128.0, 127.6, 121.6, 121.2, 116.4, 100.8, 51.3, 47.9, 47.3, 42.3, 38.6, 34.8, 34.7, 34.0, 33.3, 32.5, 30.7, 23.9, 21.4, 18.8. FTIR (NaCl, thin film) 3356, 3048, 2950, 2867, 2828, 1690, 1525, 1485, 1460, 1424, 1384, 1368, 1325, 1288, 1276, 1261, 1242, 1155, 1108, 1096, 1048, 946, 927, 826, 792, 756, 690, 666 cm.−1 HRMS (MM) calc’d for C24H31N2O2 [M–OCH3]+ 379.2380, found 379.2376.
(17)
To a 15 mL thick-walled, screw top pressure vessel were added dimethyl ketal 51 (59.8 mg, 0.146 mmol) and PhMe (5.0 mL). The tube sealed under a stream of N2. The reaction was heated to 170 °C in a preheated oil bath for 3.5 hours. The reaction was then cooled to room temperature and concentrated in vacuo to afford 17 (55.1 mg, quantitative yield), an inseparable ~1:1 mixture of enol ether isomers, as a foamy colorless gum: (c = 1.25, CHCl3). 1H NMR (400 MHz, CDCl3) δ 9.70 (s, 1H), 8.90 – 8.72 (m, 2H), 8.15 (dd, J = 8.2, 1.5 Hz, 1H), 7.57 – 7.40 (m, 3H), 4.48 (s, 1H), 3.48 (s, 3H), 2.87 – 2.74 (m, 2H), 2.12 – 1.93 (m, 2H), 1.74 – 1.57 (m, 4H), 1.48 – 1.36 (m, 1H), 1.33 (s, 3H), 1.31 – 1.27 (m, 1H), 1.12 (s, 3H), 0.97 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.6, 171.0, 156.3, 154.3, 148.3, 148.3, 138.6, 138.5, 136.4, 136.4, 134.8, 134.7, 128.5, 128.1, 128.1, 127.6, 126.9, 126.8, 121.7, 121.6, 121.2, 121.2, 116.4, 116.3, 99.4, 92.1, 54.1, 53.9, 52.6, 51.5, 40.9, 40.1, 36.4, 35.4, 35.2, 34.0, 33.5, 33.0, 32.6, 30.9, 30.8, 30.7, 29.9, 28.2, 26.1, 25.1, 24.1, 23.9, 21.2, 20.7, 19.5. FTIR (NaCl, thin film) 3354, 3051, 2949, 2930, 2862, 1690, 1668, 1524, 1484, 1461, 1424, 1384, 1368, 1326, 1238, 1215, 1162, 1147, 1026, 826, 791, 756, 694 cm.−1 HRMS (MM) calc’d for C24H31N2O2 [M+H]+ 379.2380, found 379.2395.
(55)
To a flame-dried 100 mL round-bottom flask were added phloroglucinol 5472 (1.00g, 4.13 mmol), followed by freshly distilled MeOH (41.0 mL). Benzaldehyde (421 μL, 4.13 mmol, 1.00 equiv), morpholine (361 μL, 4.13 mmol, 1.00 equiv), and triethylamine (576 μL, 4.13 mmol, 1.00 equiv) were then added successively via syringe and the reaction stirred at room temperature for 24 hours. The precipitate thus formed was collected by vacuum filtration and washed with MeOH (20 mL) and dried under high vacuum to afford analytically pure 55 (1.19 g, 69% yield) as a white powder. 1H NMR (400 MHz, CDCl3) δ 15.34 (s, 1H), 13.16 (s, 1H), 12.53 (s, 1H), 7.45 (d, J = 7.2 Hz, 2H), 7.34 – 7.20 (m, 3H), 4.88 (s, 1H), 3.99 (s, 3H), 3.91 (s, 3H), 3.90 – 3.40 (br m, 4H), 3.08 (br s, 1H), 2.46 (ddd, J = 11.9, 6.2, 3.1 Hz, 2H), 2.18 (br s, 1H). 13C NMR (101 MHz, CDCl3) δ 171.7, 166.2, 165.6, 165.1, 138.2, 128.9, 128.4, 103.8, 96.5, 94.2, 69.0, 66.6, 52.7, 52.6. FTIR (NaCl, thin film) 3404 (br), 3062, 3030, 2955, 2894, 2854, 2716, 2562 (br), 2252, 1953 (br), 1731, 1654, 1603, 1494, 1454, 1431, 1403, 1326, 1290, 1250, 1205, 1169, 1121, 1080, 1029, 1006, 986, 942, 915, 878, 843, 825, 808, 761, 732, 700, 648 cm.−1HRMS (MM) calc’d for C21H24NO8 [M+H]+418.1496, found 418.1515.
(16)
To a 50 mL round-bottom flask was added benzhydryl morpholine 55 (200 mg, 0.479 mmol), followed by a 1:1 mixture of THF/H2O (9.6 mL). p-Toluenesulfonic acid monohydrate (91.1 mg, 0.479 mmol, 1.00 equiv) was then added in one portion and the reaction was heated to 60 °C for 4 hours. Note: it is best to monitor this reaction closely by TLC to mitigate degradation of the product to 54, presumably via acid-mediated retro aldol. Upon completion, the reaction was cooled to room temperature and quenched with saturated aqueous NaHCO3. The reaction was diluted with EtOAc and the organic layer separated. The aqueous layer was extracted with EtOAc (2 × 5 mL) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (isocratic: 5% EtOAc/CH2Cl2 + 0.5% AcOH, necessary to avoid streaking on the column). Fractions containing pure product were combined, washed with saturated aqueous NaHCO3, dried over MgSO4, filtered, and concentrated in vacuo to afford 16 (82.0 mg, 49% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 11.89 (s, 2H), 11.70 (s, 1H), 7.46 – 7.39 (m, 2H), 7.31 (t, J = 7.4 Hz, 2H), 7.26 – 7.19 (m, 1H), 6.38 – 6.23 (m, 1H), 4.09 (d, J = 11.6 Hz, 1H), 4.02 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 170.9, 165.0, 164.7, 143.9, 128.2, 127.0, 125.6, 110.2, 94.5, 68.1, 53.2. FTIR (NaCl, thin film) 3563 (br), 3357 (br), 3085, 3058, 3028, 3006, 2956, 2851, 2749 (br), 1727, 1655, 1623, 1599, 1492, 1434, 1333, 1318, 1245, 1201, 1170, 1129, 1039, 1026, 972, 909, 836, 816, 733, 698, 622 cm.−1 HRMS (MM) calc’d for C17H15O7 [M–OH]+331.0812, found 331.0825.
(57–60, thermal reaction)
To a 15 mL thick-walled, screw top pressure vessel were added dimethyl ketal 51 (105 mg, 0.256 mmol) and o-QM precursor 16 (98.0 mg, 0.281 mmol, 1.10 equiv). PhMe (4.3 mL) was then added and the tube sealed under a stream of argon. The reaction was heated to 170 °C in a preheated oil bath for 21 hours. The reaction was then cooled to room temperature and concentrated in vacuo. The crude residue was first purified by silica gel flash chromatography to remove separable impurities (4% EtOAc/CH2Cl2 + 0.5% AcOH) to afford a complex mixture of diastereomers, including 57–60 (109 mg, 68% yield). Analytically pure samples of the four diastereomers produced in greatest abundance (i.e. 57–60) were obtained by subsequent reverse-phase HPLC purification using an Agilent XDB-C18 5 μm 30 × 250 mm column (gradient: 83–100% MeCN/H2O).
Data for 57 (peak 2)
(c = 0.360, CHCl3) White Solid. 1H NMR (400 MHz, CDCl3) δ 12.81 (s, 1H), 12.08 (s, 1H), 9.65 (s, 1H), 8.78 – 8.74 (m, 2H), 8.15 (dd, J = 8.3, 1.6 Hz, 1H), 7.53 – 7.46 (m, 2H), 7.43 (dd, J = 8.3, 4.2 Hz, 1H), 7.22 (d, J = 7.5 Hz, 2H), 7.14 – 7.07 (m, 3H), 3.93 (s, 3H), 3.93 (s, 3H), 3.91 (d, J = 7.8 Hz, 1H), 3.39 (s, 3H), 2.82 – 2.76 (m, 2H), 2.12 (s, 1H), 1.86 – 1.73 (m, 2H), 1.69 – 1.49 (m, 5H), 1.33 (s, 3H), 1.25 (d, J = 9.6 Hz, 1H), 1.10 (s, 3H), 1.05 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.4, 170.8, 169.9, 166.0, 164.7, 158.9, 148.3, 145.9, 138.5, 136.5, 134.7, 128.1, 128.1, 127.8, 127.6, 126.0, 121.7, 121.3, 116.3, 104.2, 104.1, 97.1, 95.7, 52.7, 52.7, 52.2, 49.0, 44.2, 41.7, 39.9, 37.7, 35.1, 35.1, 33.9, 30.8, 28.9, 24.0, 23.5, 22.8. FTIR (NaCl, thin film) 3412 (br), 3354 (br), 3059, 3022, 3006, 2951, 2928, 2864, 1731, 1686, 1654, 1648, 1643, 1594, 1524, 1484, 1459, 1426, 1384, 1338, 1325, 1249, 1222, 1201, 1157, 1122, 1081, 1092, 1028, 976, 945, 936, 847, 826, 792, 755, 700, 667 cm.−1 HRMS (MM) calc’d for C41H45N2O9 [M+H]+ 709.3120, found 709.3141.
Data for 58 (peak 1)
(c = 0.420, CHCl3) White Solid. 1H NMR (400 MHz, CDCl3) δ 12.22 (s, 1H), 11.68 (s, 1H), 9.65 (s, 1H), 8.81 (dd, J = 4.2, 1.7 Hz, 1H), 8.77 (dd, J = 7.3, 1.7 Hz, 1H), 8.16 (dd, J = 8.3, 1.7 Hz, 1H), 7.56 – 7.48 (m, 2H), 7.46 (dd, J = 8.3, 4.2 Hz, 1H), 7.24 – 7.16 (m, 2H), 7.16 – 7.09 (m, 1H), 7.09 – 7.02 (m, 2H), 3.96 (s, 3H), 3.93 (s, 3H), 3.91 (s, 1H), 3.05 (s, 3H), 2.82 – 2.67 (m, 2H), 2.16 (dd, J = 12.4, 3.5 Hz, 1H), 1.98 (d, J = 13.8 Hz, 1H), 1.76 (dd, J = 13.3, 4.1 Hz, 1H), 1.70 – 1.39 (m, 6H), 1.32 (s, 3H), 1.12 (s, 3H), 0.96 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.0, 170.8, 169.9, 165.0, 163.1, 157.2, 148.3, 145.6, 138.5, 136.5, 134.7, 128.1, 127.8, 127.6, 127.3, 125.6, 121.7, 121.3, 116.4, 102.4, 102.1, 99.0, 95.3, 52.8, 52.5, 51.3, 47.8, 45.3, 41.9, 41.4, 40.1, 34.7, 34.6, 32.7, 32.6, 30.8, 27.4, 23.8, 22.3. FTIR (NaCl, thin film) 3410 (br), 3355 (br), 3055, 3021, 3000, 2950, 2864, 1734, 1686, 1654, 1643, 1599, 1524, 1484, 1460, 1426, 1384, 1336, 1326, 1279, 1247, 1225, 1163, 1142, 1093, 1063, 988, 973, 949, 841, 826, 791, 754, 698, 667 cm.−1 HRMS (MM) calc’d for C41H45N2O9 [M+H]+ 709.3120, found 709.3119.
Data for 59 (peak 3)
(c = 0.206, CHCl3) White Solid. 1H NMR (400 MHz, CDCl3) δ 12.11 (s, 1H), 11.61 (s, 1H), 9.64 (s, 1H), 8.82 (dd, J = 4.3, 1.7 Hz, 1H), 8.76 (dd, J = 7.3, 1.7 Hz, 1H), 8.16 (dd, J = 8.2, 1.7 Hz, 1H), 7.57 – 7.43 (m, 3H), 7.30 (dd, J = 8.6, 5.1 Hz, 2H), 7.17 (s, 2H), 6.81 (s, 1H), 4.54 (d, J = 7.3 Hz, 1H), 3.94 (s, 3H), 3.92 (s, 3H), 3.21 (s, 3H), 2.74 (q, J = 9.8 Hz, 2H), 2.10 (d, J = 13.7 Hz, 1H), 1.97 – 1.83 (m, 1H), 1.62 (d, J = 8.9 Hz, 2H), 1.45 (d, J = 13.8 Hz, 1H), 1.32 (s, 3H), 1.29 – 1.24 (m, 1H), 1.18 (d, J = 13.2 Hz, 1H), 1.11 (s, 3H), 1.10 – 1.06 (m, 1H), 1.04 (s, 3H), 0.76 (dd, J = 13.1, 3.9 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 171.0, 170.9, 169.7, 164.9, 162.7, 158.0, 148.3, 142.2, 138.5, 136.5, 134.7, 128.5, 128.1, 127.7, 127.6, 125.9, 121.7, 121.3, 116.4, 104.2, 102.2, 99.3, 95.5, 52.8, 52.5, 51.3, 49.0, 43.7, 42.2, 40.3, 38.5, 34.8, 34.4, 33.0, 32.6, 30.8, 23.8, 22.1, 21.7. FTIR (NaCl, thin film) 3408 (br), 3354 (br), 3059, 3022, 3009, 2952, 2868, 1738, 1732, 1682, 1658, 1652, 1645, 1599, 1525, 1485, 1462, 1455, 1426, 1385, 1327, 1281, 1251, 1225, 1165, 1133, 1090, 1077, 1031, 991, 946, 872, 826, 792, 755, 703 cm.−1 HRMS (MM) calc’d for C41H45N2O9 [M+H]+ 709.3120, found 709.3133.
Data for 60 (peak 4)
(c = 0.226, CHCl3) White Solid. 1H NMR (400 MHz, CDCl3) δ 11.95 (s, 1H), 11.23 (s, 1H), 9.66 (s, 1H), 8.81 (dd, J = 4.2, 1.7 Hz, 1H), 8.76 (dd, J = 7.3, 1.7 Hz, 1H), 8.16 (dd, J = 8.3, 1.7 Hz, 1H), 7.55 – 7.42 (m, 3H), 7.22 (dd, J = 7.9, 6.5 Hz, 2H), 7.18 – 7.13 (m, 1H), 7.10 (d, J = 7.4 Hz, 2H), 3.90 (s, 3H), 3.87 (s, 3H), 3.67 (d, J = 11.0 Hz, 1H), 3.16 (s, 3H), 2.81 – 2.66 (m, 2H), 2.10 (dd, J = 14.2, 1.6 Hz, 1H), 1.71 (td, J = 10.8, 5.3 Hz, 1H), 1.64 (dd, J = 9.2, 2.6 Hz, 2H), 1.56 – 1.48 (m, 2H), 1.45 (d, J = 14.3 Hz, 1H), 1.38 – 1.32 (m, 1H), 1.31 (s, 3H), 1.21 – 1.14 (m, 1H), 1.12 (s, 3H), 1.08 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 170.8, 170.8, 169.5, 164.0, 162.4, 157.4, 148.3, 145.4, 138.5, 136.5, 134.7, 128.1, 128.1, 127.6, 125.9, 121.7, 121.3, 116.4, 108.0, 101.7, 99.6, 95.5, 52.7, 52.5, 51.2, 49.4, 49.0, 42.0, 41.1, 36.2, 35.3, 34.8, 33.3, 32.6, 30.8, 23.8, 22.5, 21.1. FTIR (NaCl, thin film) 3412 (br), 3354 (br), 3055, 3023, 3003, 2950, 2866, 1732, 1688, 1656, 1598, 1524, 1484, 1453, 1426, 1384, 1327, 1277, 1248, 1225, 1165, 1062, 993, 954, 925, 826, 792, 755, 702 cm.−1 HRMS (MM) calc’d for C41H45N2O9 [M+H]+ 709.3120, found 709.3139.
(57–60, Cu-mediated reaction)
Inside a N2-filled glovebox, methyl enol ether 17 (17.0 mg, 0.045 mmol) and o-QM precursor 16 (16.4 mg, 0.047 mmol, 1.05 equiv) were added to a 1 dram vial and dissolved in CH2Cl2 (400 μL). Cu(OTf)2 was then added as a solid in one portion and the reaction immediately turns a light green color, then yellow-brown within the first 5 minutes. The reaction was stirred at room temperature for 1 hour, then quenched with saturated aqueous NaHCO3 and diluted with CHCl3. The reaction mixture was extracted with CHCl3 (3 × 1 mL) and the organics filtered through a plug of Na2SO4 and concentrated in vacuo. The crude residue was analyzed by 1H NMR and determined to contain 57, 58, 59, and 60 in an approximate ratio of 2:1:3:3, respectively.
(62)
Inside a N2-filled glovebox, Schwartz’s reagent (119 mg, 0.462 mmol, 2.00 equiv) was added to a 10 mL flask and sealed under N2. The flask was removed from the glovebox and THF (1.2 mL) was added via syringe. To the milky-white suspension was added ketal 51 (94.8 mg, 0.231mmol) as a solution in THF (1.2 mL) in a quick drip. The reaction immediately beings to turn yellow, eventually becoming a darker orange color over 1 hour, at which time the reaction was quenched by the addition of saturated aqueous NaHCO3. The reaction was diluted with EtOAc and the organic layer separated. The aqueous layer was extracted with EtOAc (2 × 5 mL) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (isocratic: 5% EtOAc/hexane + 1% Et3N) to afford 62 (36.9 mg, 63% yield) as a pale yellow oil: (c = 0.500, CHCl3). 1H NMR (400 MHz, CDCl3) δ 9.70 (d, J = 3.0 Hz, 1H), 3.14 (s, 3H), 3.09 (s, 3H), 2.64 (td, J = 9.7, 8.5 Hz, 1H), 2.58 (dd, J = 9.9, 3.0 Hz, 1H), 1.86 (ddt, J = 13.6, 4.2, 2.6 Hz, 1H), 1.61 (t, J = 10.3 Hz, 1H), 1.57 – 1.44 (m, 4H), 1.34 – 1.18 (m, 2H), 1.16 (s, 3H), 1.14 (s, 3H), 1.13 – 1.05 (m, 2H), 0.89 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 204.7, 100.5, 55.2, 47.6, 47.1, 39.8, 39.3, 35.7, 34.4, 33.9, 33.0, 32.7, 31.1, 24.3, 21.7, 18.6. FTIR (NaCl, thin film) 2952, 2868, 2828, 2705, 1713, 1461, 1383, 1368, 1341, 1288, 1262, 1246, 1180, 1166, 1110, 1098, 1048, 1009, 945, 924, 823, 828 cm.−1 HRMS (FAB) calc’d for C15H25O2 [M–OCH3]+ 237.1849, found 237.1855.
(63)
To a 10 mL round bottom flask were added aldehyde 62 (36.0 mg, 0.134 mmol) and K2CO3 (37.0 mg, 0.268 mmol, 2.00 equiv). The flask was fitted with a septum and the atmosphere exchanged 2x for N2. Freshly distilled MeOH (1.5 mL) was then added via syringe and the solution cooled to 0 °C. Dimethyl-1-diazo-2-oxopropylphosphonate73 (38.6 mg, 0.201 mmol, 1.50 equiv) was weighed into a tared syringe and added dropwise to the reaction, neat. The reaction was allowed to gradually warm to room temperature and stirred for 12 hours. The reaction was then diluted with Et2O, saturated aqueous NaHCO3 was added, and the organic layer separated. The aqueous layer was extracted with Et2O (3 × 5 mL) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude residue was purified by Florisil® flash chromatography (isocratic: 5% Et2O/pentane) to afford 63 (32.9 mg, 93% yield) as a pale yellow oil: (c = 0.355, CHCl3). 1H NMR (400 MHz, CDCl3) δ 3.17 (s, 3H), 3.13 (s, 3H), 2.43 (dd, J = 10.1, 2.4 Hz, 1H), 2.16 – 2.05 (m, 2H), 2.04 – 1.93 (m, 1H), 1.69 (ddd, J = 13.9, 2.8, 1.8 Hz, 1H), 1.59 – 1.50 (m, 2H), 1.48 (d, J = 9.6 Hz, 2H), 1.29 – 1.17 (m, 5H), 1.16 (d, J = 2.7 Hz, 4H), 1.03 (s, 3H), 0.97 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 100.7, 85.8, 70.5, 49.1, 47.9, 47.3, 39.1, 35.2, 35.1, 34.0, 33.5, 33.2, 33.2, 29.9, 24.8, 21.1, 18.8. FTIR (NaCl, thin film) 3310, 3263, 2953, 2866, 2828, 1459, 1383, 1364, 1342, 1323, 1288, 1266, 1243, 1180, 1157, 1106, 1094, 1048, 945, 926, 858, 830, 655, 621 cm.−1 HRMS (MM) calc’d for C16H25O2 [M–OCH3]+233.1900, found 233.1887.
(64–65)
Inside a N2-filled glovebox, THF (400 μL) was added to a 1 dram vial containing alkyne 63 (12.4 mg, 0.047 mmol), followed by Ni(acac)2 as a stock solution in THF (0.10 M, 70 μL, 0.007 mmol, 0.15 equiv). The reaction was stirred for 10 minutes at room temperature before thiophenol (10 μL, 0.094 mmol, 2.00 equiv) was added neat. The reaction was sealed with a Teflon cap and heated to 60 ° C in a preheated aluminum block inside the glovebox. After 3 hours, the reaction was cooled to room temperature and diluted with CH2Cl2. The reaction mixture was filtered over a small pad of celite, washed with CH2Cl2 until the filtrate runs colorless, and concentrated in vacuo. The crude residue was taken up in EtOAc and shaken with 5M NaOH (to remove excess thiophenol). The organic layer was then filtered through a plug of Na2SO4, concentrated, and purified by silica gel preparative TLC (5% EtOAc/hexane + 1% Et3N) to afford 64 (8.50 mg, 53% yield) and 65 (2.7 mg, 15% yield) each as colorless oils.
Data for 65
(c = 0.115, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.42 (dd, J = 8.1, 1.6 Hz, 2H), 7.36 – 7.28 (m, 3H), 5.17 (d, J = 1.3 Hz, 1H), 4.96 (s, 1H), 3.17 (s, 3H), 3.13 (s, 3H), 2.51 (d, J = 10.2 Hz, 1H), 2.26 (q, J = 9.7 Hz, 1H), 2.04 – 1.92 (m, 1H), 1.65 (ddd, J = 13.8, 2.8, 1.6 Hz, 1H), 1.50 (ddd, J = 9.6, 7.0, 3.7 Hz, 2H), 1.45 – 1.39 (m, 2H), 1.21 – 1.12 (m, 4H), 1.11 (s, 3H), 0.98 (s, 3H), 0.91 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 145.9, 133.8, 133.4, 129.2, 127.9, 111.6, 100.8, 49.4, 47.9, 47.3, 45.1, 39.7, 35.6, 34.8, 34.6, 33.2, 32.4, 30.6, 23.2, 21.7, 18.9. FTIR (NaCl, thin film) 2950, 2863, 2827, 1610, 1583, 1476, 1459, 1439, 1379, 1364, 1322, 1274, 1260, 1247, 1178, 1145, 1130, 1100, 1083, 1049, 1024, 946, 926, 856, 831, 822, 747, 691 cm.−1 HRMS (MM) calc’d for C22H31OS [M–OCH3]+343.2090, found 343.2073.
Data for 64
(c = 0.982, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.46 – 7.36 (m, 2H), 7.36 – 7.27 (m, 3H), 5.23 – 4.84 (m, 2H), 4.63 – 4.29 (m, 1H), 3.40 (s, 3H), 2.58 – 2.50 (m, 1H), 2.40 (dq, J = 34.9, 9.5 Hz, 1H), 2.11 – 1.91 (m, 3H), 1.64 (ddd, J = 15.0, 5.9, 2.4 Hz, 2H), 1.48 – 1.40 (m, 2H), 1.39 – 1.30 (m, 1H), 1.11 (d, J = 9.9 Hz, 3H), 1.00 (d, J = 2.4 Hz, 3H), 0.83 (d, J = 21.0 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 155.3, 154.4, 146.0, 145.9, 133.8, 133.5, 133.4, 133.3, 129.2, 129.1, 127.9, 127.7, 112.3, 111.1, 100.6, 92.0, 54.1, 53.9, 50.2, 49.5, 43.5, 40.9, 37.7, 36.1, 35.1, 35.1, 34.5, 34.1, 32.9, 32.6, 31.6, 30.6, 30.5, 28.2, 25.8, 23.3, 23.2, 22.0, 20.8, 19.4. FTIR (NaCl, thin film) 3061, 2991, 2950, 2930, 2862, 2843, 1667, 1609, 1583, 1476, 1460, 1453, 1440, 1380, 1366, 1251, 1215, 1148, 1066, 1024, 940, 817, 747, 691 cm.−1 HRMS (MM) calc’d for C22H31OS [M+H]+ 343.2090, found 343.2087.
(66–67)
Inside a N2-filled glovebox, CH2Cl2 was added to a 1 dram vial containing 65 (9.30 mg, 0.025 mmol), followed by InCl3 (5.49 mg, 0.025 mmol, 1.00 equiv). The reaction was stirred at room temperature for 2 hours, then quenched with saturated aqueous NaHCO3 and diluted with CH2Cl2. The reaction was extracted with CH2Cl2 (3 × 500 μL), the combined organics filtered a plug of Na2SO4, and concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (40–60% CH2Cl2/hexane) to afford 66 (0.900 mg, 11% yield) as a colorless oil, with the remaining mass balance accounted for by ketone 67, as determined by crude 1H NMR.
Data for 66
(c = 0.053, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.40 – 7.36 (m, 2H), 7.30 (ddd, J = 8.3, 7.1, 0.8 Hz, 2H), 7.24 – 7.18 (m, 1H), 5.52 (dd, J = 2.8, 1.7 Hz, 1H), 3.18 (s, 3H), 2.91 – 2.81 (m, 1H), 2.00 – 1.72 (m, 5H), 1.66 (ddd, J = 11.6, 6.8, 3.5 Hz, 1H), 1.44 – 1.37 (m, 2H), 1.35 (dd, J = 12.3, 1.7 Hz, 1H), 1.29 (m, 1H), 1.28 (s, 3H), 1.19 (dd, J = 14.1, 7.0 Hz, 1H), 1.00 (s, 3H), 0.80 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 139.7, 139.6, 135.0, 131.7, 129.2, 127.2, 80.6, 52.4, 50.2, 49.2, 48.6, 40.1, 38.3, 36.2, 33.7, 31.3, 31.2, 28.9, 22.4, 21.2. FTIR (NaCl, thin film) 3062, 2945, 2927, 2860, 2820, 1734, 1718, 1701, 1654, 1583, 1560, 1476, 1458, 1438, 1370, 1294, 1254, 1232, 1151, 1086, 1066, 1024, 950, 870, 840, 800, 743, 690 cm.−1 HRMS (MM) calc’d for C22H31OS [M+H]+343.2090, found 343.2077.
Data for 67
(c = 0.100, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.46 (dd, J = 8.1, 1.6 Hz, 2H), 7.40 – 7.32 (m, 3H), 5.13 (d, J = 1.3 Hz, 1H), 4.98 (s, 1H), 2.53 (d, J = 10.1 Hz, 1H), 2.38 (td, J = 10.0, 8.9 Hz, 1H), 2.32 – 2.23 (m, 2H), 2.17 (d, J = 13.6 Hz, 1H), 2.02 (dt, J = 13.4, 1.9 Hz, 1H), 1.93 – 1.90 (m, 1H), 1.82 (dddd, J = 9.7, 8.1, 3.9, 2.3 Hz, 1H), 1.57 (q, J = 4.4 Hz, 1H), 1.49 – 1.44 (m, 1H), 1.44 – 1.33 (m, 2H), 1.16 (s, 3H), 1.03 (s, 3H), 0.82 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 212.6, 145.5, 134.1, 133.0, 129.3, 128.2, 111.4, 51.1, 49.8, 43.1, 41.3, 40.3, 35.1, 34.2, 32.5, 30.6, 23.1, 22.1, 21.8. FTIR (NaCl, thin film) 3059, 2953, 2927, 2860, 1711, 1680, 1611, 1583, 1476, 1461, 1440, 1381, 1364, 1347, 1311, 1283, 1253, 1228, 1151, 1087, 1067, 1024, 890, 855, 749, 692 cm.−1 HRMS (MM) calc’d for C21H29OS [M+H]+ 329.1934, found 329.1943.
(78)
To a 15 mL round-bottom flask was added vinyl ketone 71 (91.0 mg, 0.413 mmol) and the atmosphere was exchanged 3x for N2. Dry THF (4.10 mL) was then added via syringe and the reaction cooled to –30 °C using a closely monitored acetone/CO2 bath. Vinylmagnesium bromide (2.06 mL, 1.0 M in THF, 2.06 mmol, 5.00 equiv) was then added dropwise. The reaction was maintained at –30 °C for 30 minutes, then quenched at that temperature with saturated aqueous NaH2PO4. The reaction mixture was diluted with Et2O and the layers separated. The aqueous layer was extracted with Et2O (2 × 5 mL) and the combined organics were dried over Mg2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (10% EtOAc/hexane) to afford 78 (92.7 mg, 91% yield) as a colorless oil: (c = 1.75, CHCl3). 1H NMR (400 MHz, CDCl3) δ 5.88 (dd, J = 17.3, 10.6 Hz, 1H), 5.75 (ddd, J = 17.1, 10.2, 8.7 Hz, 1H), 5.18 (dd, J = 17.3, 1.3 Hz, 1H), 5.01 – 4.85 (m, 3H), 2.32 (t, J = 9.3 Hz, 1H), 1.92 (q, J = 9.6 Hz, 1H), 1.82 (qt, J = 13.5, 3.4 Hz, 1H), 1.55 (dddd, J = 14.0, 5.3, 3.5, 1.9 Hz, 1H), 1.48 (dq, J = 13.8, 3.5 Hz, 1H), 1.45 – 1.39 (m, 2H), 1.35 (dd, J = 13.5, 4.0 Hz, 1H), 1.31 – 1.22 (m, 3H), 1.16 – 1.11 (m, 1H), 1.11 (s, 1H), 1.06 (s, 3H), 0.97 (s, 3H), 0.97 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 148.1, 140.6, 114.6, 110.5, 73.1, 49.0, 48.0, 45.0, 37.6, 34.6, 34.3, 33.9, 32.8, 30.1, 23.8, 22.7, 17.8. FTIR (NaCl, thin film) 3601, 3452 (br), 3077, 2996, 2950, 2932, 2865, 1635, 1459, 1441, 1413, 1380, 1367, 1343, 1291, 1275, 1250, 1200, 1170, 1081, 1058, 994, 974, 909, 858, 846, 666 cm.−1 HRMS (ESI) calc’d for C17H27 [M–OH]+ 231.2107, found 231.2101.
(70)
A 50 mL round-bottom flask containing divinyl alcohol 78 (88.0 mg, 0.355 mmol) was pumped into a N2-filled glovebox where Hoveyda–Grubbs second-generation catalyst (22.2 mg, 0.035 mmol, 0.100 equiv) was added. The flask was sealed under nitrogen, removed from the glovebox and dry PhH (17.7 mL) was added via syringe. The green reaction mixture was heated to 80 °C for 3.5 hours, then cooled to room temperature. Ethyl vinyl ether was added to inactivate the catalyst and stirred for 15 minutes before the reaction mixture was concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (isocratic: 30% Et2O/hexane) to afford allylic alcohol 70 (72.5 mg, 93% yield) as a pale yellow oil and a single diastereomer at C1: (c = 2.67, CHCl3). 1H NMR (400 MHz, CDCl3) δ 5.84 (dd, J = 10.9, 2.5 Hz, 1H), 5.15 (ddd, J = 11.0, 2.9, 2.2 Hz, 1H), 2.41 (dt, J = 11.6, 2.7 Hz, 1H), 2.09 (td, J = 11.5, 10.7, 7.9 Hz, 2H), 1.69 (ddd, J = 13.0, 3.2, 1.1 Hz, 1H), 1.64 (s, 1H), 1.63 – 1.56 (m, 2H), 1.54 – 1.41 (m, 2H), 1.34 – 1.25 (m, 2H), 1.15 (dd, J = 12.8, 2.2 Hz, 1H), 1.12 – 1.06 (m, 1H), 1.05 (s, 3H), 1.03 (s, 3H), 0.87 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 134.1, 132.5, 75.1, 49.9, 45.3, 43.9, 39.0, 38.1, 37.8, 35.0, 32.6, 30.9, 26.9, 21.3, 20.2. FTIR (NaCl, thin film) 3350 (br), 3004, 2948, 2930, 2866, 1460, 1443, 1369, 1380, 1366, 1329, 1270, 1256, 1238, 1175, 1106, 1044, 1030, 999, 973, 958, 925, 875, 864, 766, 723 cm.−1 HRMS (MM) calc’d for C15H23 [M–OH]+ 203.1794, found 203.1790.
(79)
To a 100 mL round-bottom flask were added allylic alcohol 70 (107 mg, 0.486 mmol) and Pd/C (103 mg, 10% by weight, 0.097 mmol, 0.200 equiv). The flask was fitted with a septum and the atmosphere exchanged 1x for N2. MeOH (9.7 mL) was then added via syringe and the reaction placed under a balloon atmosphere of H2 (purged through a needle for 30 seconds). The reaction was stirred vigorously at room temperature for 2.5 hours, at which time the atmosphere was purged with argon. The reaction mixture was filtered over celite, washed thoroughly with Et2O, and the filtrate concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (isocratic: 40% Et2O/pentane) to afford 79 (101 mg, 94% yield) as a colorless oil: (c = 0.800, CHCl3). 1H NMR (400 MHz, CDCl3) δ 1.97 (ddd, J = 11.8, 10.7, 7.9 Hz, 1H), 1.86 – 1.78 (m, 1H), 1.78 – 1.68 (m, 3H), 1.67 (d, J = 0.8 Hz, 3H), 1.51 – 1.39 (m, 2H), 1.34 (dt, J = 3.5, 2.0 Hz, 1H), 1.33 – 1.22 (m, 4H), 1.15 – 1.04 (m, 1H), 1.02 (d, J = 12.8 Hz, 1H), 0.97 (s, 3H), 0.96 (s, 3H), 0.80 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 74.0, 50.2, 46.3, 40.3, 40.1, 39.7, 38.2, 36.4, 34.6, 32.8, 30.7, 27.1, 22.7, 20.9, 20.7. FTIR (NaCl, thin film) 3368 (br), 2948, 2927, 2863, 1460, 1443, 1384, 1364, 1332, 1288, 1249, 1217, 1183, 1124, 1102, 1050, 1022, 993, 976, 936, 918, 873, 862 cm.−1 HRMS (ESI) calc’d for C15H25 [M–OH]+ 205.1951, found 205.1951.
(68)
Inside a N2-filled glovebox, to a 1 dram vial containing tertiary alcohol 79 (14.4 mg, 0.065 mmol) were added Pd(OAc)2 (4.36 mg, 0.019 mmol, 0.300 equiv), dppf (21.6 mg, 0.039 mmol, 0.600 equiv), and NaH (95%, 3.11 mg, 0.130 mmol, 2.00 equiv). PhMe (650 μL) was then added and the orange reaction mixture stirred at room temperature for 5 minutes before aryl bromide 6974 (22.8 mg, 0.071 mmol, 1.10 equiv) was added as a solid in one portion. The reaction was sealed with a Teflon cap and heated to 110 °C in a preheated aluminum block inside the glovebox. After 13.5 hours, the reaction was cooled to room temperature, diluted with EtOAc and saturated aqueous Na2HPO4 was added. The layers were separated and the aqueous layer was extracted with EtOAc until the organic layer was colorless. The combined organics were filtered over a plug of celite and Na2SO4. The filtrate was concentrated in vacuo and the crude residue purified by silica gel flash chromatography (isocratic: 30% hexane/CH2Cl2 + 1% EtOAc) to afford 68 (13.4 mg, 45% yield) as a milky white gum: (c = 0.345, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.86 – 7.76 (m, 2H), 7.51 (tt, J = 7.5, 2.7 Hz, 1H), 7.44 – 7.35 (m, 2H), 6.29 (d, J = 2.1 Hz, 1H), 6.23 (d, J = 2.1 Hz, 1H), 3.83 (s, 3H), 3.70 (s, 3H), 1.90 (ddd, J = 12.0, 10.7, 7.9 Hz, 1H), 1.78 (d, J = 2.3 Hz, 1H), 1.73 (t, J = 6.5 Hz, 2H), 1.68 (dt, J = 13.0, 2.3 Hz, 1H), 1.65 – 1.57 (m, 1H), 1.55 – 1.45 (m, 2H), 1.45 – 1.35 (m, 3H), 1.27 – 1.23 (m, 2H), 1.22 – 1.11 (m, 2H), 1.04 (d, J = 12.9 Hz, 1H), 0.92 (s, 3H), 0.91 (s, 3H), 0.69 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 196.0, 161.3, 158.7, 155.2, 138.8, 132.8, 129.6, 128.3, 116.8, 100.3, 92.5, 86.9, 55.9, 55.6, 47.6, 45.6, 39.7, 37.5, 36.8, 36.2, 36.1, 34.6, 32.7, 30.7, 27.1, 22.5, 20.9, 20.5. FTIR (NaCl, thin film) 3059, 2948, 2930, 2861, 1671, 1601, 1582, 1458, 1451, 1438, 1420, 1364, 1335, 1312, 1266, 1216, 1199, 1157, 1138, 1107, 1052, 1015, 998, 948, 917, 843, 819, 802, 721, 702, 689 cm.−1 HRMS (MM) calc’d for C30H38NaO4 [M+Na]+ 485.2662, found 485.2672.
(80)
To a 13 × 100 quartz test tube was added benzophenone 68 (15.5 mg, 0.034 mmol). The tube was fitted with a 19/38 rubber septum and the atmosphere was exchanged 3 × for N2. Rigorously degassed dioxane (4.70 mL, freeze-pump-thawed 3x) was then added via syringe and the tube was sealed with electrical tape. The reaction was then placed in a bottomless test tube rack in front of a Honeywell 254 nm lamp and irradiated for 1 hour at room temperature. The reaction mixture was transferred to a cone-bottom flask and concentrated in vacuo. The crude residue was purified by silica gel preparative TLC (30% hexane/CH2Cl2 + 1% EtOAc) to afford 8375 (2.4 mg, 28% yield) as a white solid and 80 (1.00 mg, 6.5% yield) as a colorless oil: (c = 0.050, CHCl3). Note: an additional ~18% yield of a complex mixture of products is also isolated as a single band. Although this mixture generally appears similar to 80 by 1H NMR, definitive characterization was not achieved. 1H NMR (400 MHz, CDCl3) δ 7.34 – 7.26 (m, 1H), 7.24 – 7.09 (m, 4H), 6.11 (dd, J = 2.5, 1.1 Hz, 1H), 6.01 (dd, J = 2.4, 1.2 Hz, 1H), 3.95 (d, J = 1.1 Hz, 1H), 3.78 (d, J = 1.2 Hz, 3H), 3.35 (d, J = 1.1 Hz, 3H), 2.65 (dd, J = 12.7, 3.5 Hz, 1H), 2.62 – 2.52 (m, 1H), 2.40 (t, J = 14.4 Hz, 1H), 2.26 (q, J = 10.4 Hz, 1H), 2.11 – 1.90 (m, 1H), 1.86 (d, J = 13.0 Hz, 1H), 1.83 – 1.72 (m, 1H), 1.69 – 1.57 (m, 1H), 1.43 – 1.34 (m, 2H), 1.30 – 1.09 (m, 4H), 0.80 (s, 3H), 0.78 (s, 3H), 0.75 (s, 3H), 0.50 (dt, J = 14.5, 4.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 160.5, 158.8, 154.3, 149.3, 127.4, 126.1, 125.8, 111.4, 94.2, 93.5, 80.6, 74.6, 55.6, 55.4, 48.5, 48.1, 44.0, 37.3, 36.8, 35.7, 35.5, 34.6, 33.2, 30.5, 26.4, 25.0, 20.6, 20.4. FTIR (NaCl, thin film) 3542 (br), 3312, 3187 (br), 2960, 2924, 2854, 1738, 1726, 1710, 1666, 1614, 1592, 1492, 1462, 1453, 1445, 1423, 1376, 1366, 1351, 1332, 1261, 1215, 1203, 1150, 1112, 1045, 1020, 865, 800, 736, 702, 664 cm.−1 HRMS (MM) calc’d for C30H37O3 [M–OH]+ 445.2737, found 445.2729.
(+)-psiguadial B (3):76
To a 2-dram vial was added resorcinol 97 (15.4 mg, 0.037 mmol) and the atmosphere exchanged three times for N2. CH2Cl2 (1.30 mL) was then added via syringe, followed by dichloromethyl methyl ether (0.083 mL, 0.920 mmol, 25.0 equiv). The solution was cooled to –78 °C and a freshly prepared stock solution of TiCl4 (0.190 mL, 0.912 M in CH2Cl2, 0.173 mmol, 4.68 equiv) was added dropwise. The reaction immediately turns dark red. The reaction was stirred at –78 °C for 5 minutes, then warmed to room temperature and stirred for an additional 3 hours and 40 minutes. DI H2O (2.00 mL) was then added via syringe and the reaction stirred vigorously for 15 minutes before the layers were separated. The aqueous layer was extracted five times with CH2Cl2 and the combined organic layers were filtered over a plug of Na2SO4 and concentrated in vacuo. The crude residue was purified by silica gel flash chromatography (isocratic: 2% EtOAc/hexane + 1% AcOH) to afford (+)-psiguadial B (3) (8.7 mg, 50%) as an ivory solid. Note: 3 is streaky on SiO2 and after an initial concentrated band elutes, approximately 12% of the product is contained in the following very dilute fractions. The natural product is weakly UV active, but can also be visualized by TLC using 2,4-dinitrophenylhydrazine stain. (c = 0.265, CHCl3). 1H NMR (400 MHz, CDCl3) δ 13.51 (s, 1H), 13.04 (s, 1H), 10.07 (s, 2H), 7.26 (dd, J = 14.6, 1.5 Hz, 2H), 7.23 – 7.17 (m, 1H), 7.10 (br s, 2H), 3.49 (d, J = 11.5 Hz, 1H), 2.20 – 2.12 (m, 1H), 2.09 (dd, J = 12.7, 2.4 Hz, 1H), 1.92 (ddd, J = 14.9, 12.8, 4.2 Hz, 1H), 1.82 (ddd, J = 12.3, 8.8, 5.6 Hz, 1H), 1.73 – 1.59 (m, 3H), 1.53 – 1.44 (m, 1H), 1.49 (ddd, J = 11.6, 8.1, 2.9 Hz, 2H), 1.44 – 1.29 (m, 4H), 1.05 (dd, J = 7.6, 5.8 Hz, 1H), 1.02 (s, 3H), 1.00 (s, 3H), 0.85 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 192.3, 191.5, 169.6, 168.5, 163.5, 143.4, 128.2, 126.2, 105.7, 104.6, 104.1, 84.1, 50.0, 47.4, 44.0, 40.4, 37.6, 36.9, 35.4, 35.1, 33.4, 30.6, 29.3, 26.1, 23.9, 20.7, 20.1. FTIR (NaCl, thin film) 3026, 2945, 2926, 2864, 2720, 1633, 1603, 1493, 1437, 1382, 1363, 1300, 1270, 1251, 1231, 1184, 1154, 1143, 1031, 1006, 976, 926, 917, 875, 851, 840, 824, 768, 701, 636, 618, 606, 564 cm.−1 HRMS (MM) calc’d for C30H35O5 [M+H]+ 475.2479, found 475.2487.
Supplementary Material
Figure 5.

Setup for small scale photochemical reaction screening.
Acknowledgments
Funding Sources
Prof. Greg Fu is gratefully acknowledged for insightful discussions. We thank Dr. Allen Oliver, Dr. Nathan Schley, and Ms. Julie Hofstra for X-ray crystallographic structure determination and Dr. David VanderVelde for assistance with NMR structure determination. We thank Dr. Scott Virgil and the Caltech Center for Catalysis and Chemical Synthesis for access to analytical equipment, and Materia, Inc. for a donation of HG-II catalyst. Fellowship support was provided by the NSF (L. M. C., C. M. L., Grant No. DGE-1144469), NIH Training Grant (J. C. B., Grant No. 5T32GM007616-39) and SNF (L. W., Grant No. PBZHP2-147311). S.E.R. is an American Cancer Society Research Scholar and a Heritage Medical Research Foundation Investigator. Financial support from the NSF (CAREER-1057143), the American Cancer Society, the Research Corporation Cottrell Scholars program, and DuPont is gratefully acknowledged.
Footnotes
Supporting Information
Synthetic schemes, additional reaction optimization tables, spectral data for all compounds, and crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.(a) Arima H, Danno GI. Isolation of Antimicrobial Compounds from Guava (Psidium guajava L.) and their Structural Elucidation. Biosci Biotechnol Biochem. 2002;66:1727–1730. doi: 10.1271/bbb.66.1727. [DOI] [PubMed] [Google Scholar]; (b) Begum S, Hassan SI, Siddiqui BS, Shaheen F, Ghayur MN, Gilani AH. Triterpenoids from the Leaves of Psidium guajava. Phytochemistry. 2002;61:399–403. doi: 10.1016/s0031-9422(02)00190-5. [DOI] [PubMed] [Google Scholar]; (c) Mukhtar HM, Ansari SH, Bhat ZA, Naved T, Singh P. Antidiabetic Activity of an Ethanol Extract Obtained from the Stem Bark of Psidium guajava (Myrtaceae) Pharmazie. 2006;61:725–727. [PubMed] [Google Scholar]; (d) Oh WK, Lee CH, Lee MS, Bae EY, Sohn CB, Oh H, Kim BY, Ahn JS. Antidiabetic Effects of Extracts from Psidium guajava. J Ethnopharmacol. 2005;96:411–415. doi: 10.1016/j.jep.2004.09.041. [DOI] [PubMed] [Google Scholar]; (e) Mukhtar HM, Ansari SH, Ali M, Naved T, Bhat ZA. Effect of Water Extract of Psidium guajava Leaves on Alloxan-Induced Diabetic Rats. Pharmazie. 2004;59:734–735. [PubMed] [Google Scholar]; (f) Ojewole JA. Hypoglycemic and Hypotensive Effects of Psidium guajava Linn. (Myrtaceae) Leaf Aqueous Extract. Methods Find Exp Clin Pharmacol. 2005;27:689–695. doi: 10.1358/mf.2005.27.10.948917. [DOI] [PubMed] [Google Scholar]; For Isolation of Guajadial (1) and Additional Citations Describing Biological Studies of Psidium guajava, see:; (g) Yang XL, Hsieh KL, Liu JK. Guajadial: An Unusual Meroterpenoid from Guava Leaves. Psidium guajava Org Lett. 2007;9:5135–5138. doi: 10.1021/ol702537q. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 2.For a review of phloroglucinols derived from natural origins, see:; (a) Singh IP, Sidana J, Bharate SB, Foley WJ. Phloroglucinol Compounds of Natural Origin: Synthetic aspects. Nat Prod Rep. 2010;27:393–416. doi: 10.1039/b914364p. [DOI] [PubMed] [Google Scholar]; For new meroterpenoids isolated recently, see:; (b) Qin XJ, Yan H, Ni W, Yu MY, Khan A, Liu H, Zhang HX, He L, Hao XJ, Di YT, Liu HY. Cytotoxic Meroterpenoids with Rare Skeletons from Psidium guajava Cultivated in Temperate Zone. Sci Rep. 2016;6:32748. doi: 10.1038/srep32748. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shang ZC, Yang MH, Liu RH, Wang XB, Kong LY. New Formyl Phloroglucinol Meroterpenoids from the Leaves of Eucalyptus robusta. Sci Rep. 2016;6:39815. doi: 10.1038/srep39815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang GH, Dong Z, Guo YQ, Cheng ZB, Zhou CJ, Yin S. Sci Rep. doi: 10.1038/s41598-017-01028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; Psiguajadials A-K. Unusual Psidium Meroterpenoids as Phosphodiesterase-4 Inhibitors from the Leaves of Psidium guajava. 2017;7:1952. doi: 10.1038/s41598-017-01028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Psiguadial AB, Shao M, Wang Y, Liu Z, Zhang D-M, Cao H-H, Jiang R-W, Fan C-L, Zhang X-Q, Chen H-R, Yao X-S, Ye W-C. Psiguadials A and B, Two Novel Meroterpenoids with Unusual Skeletons from the Leaves of Psidium guajava. Org Lett. 2010;12:5040–5043. doi: 10.1021/ol102179u. [DOI] [PubMed] [Google Scholar]
- 5.Psiguadial C, D and proposed biosynthesis:; Shao M, Wang Y, Jian Y-Q, Huang X-J, Zhang D-M, Tang Q-F, Jiang R-W, Sun X-G, Lv Z-P, Zhang X-Q, Ye W-C. Guadial A and Psiguadials C and D, Three Unusual Meroterpenoids from. Psidium guajava Org Lett. 2012;14:5262–5265. doi: 10.1021/ol302423b. [DOI] [PubMed] [Google Scholar]
- 6.(a) Dehal SS, Croteau R. Partial Purification and Characterization of two Sesquiterpene Cyclases from Sage (Salvia officinalis) which Catalyze the Respective Conversion of Farnesyl Pyrophosphate to Humulene and Caryophyllene. Arch Biochem Biophys. 1988;261:346–356. doi: 10.1016/0003-9861(88)90350-5. [DOI] [PubMed] [Google Scholar]; (b) Cai Y, Jia JW, Crock J, Lin ZX, Chen XY, Croteau R. A cDNA Clone for β-Caryophyllene Aynthase from. Artemisia annua Phytochemistry. 2002;61:523–529. doi: 10.1016/s0031-9422(02)00265-0. [DOI] [PubMed] [Google Scholar]
- 7.Moussa GE. Phenol Dehydrogenations. 12. Oxidative Coupling of 3,5-Dimethyl-2,4,6-trihydroxybenzophenone. Acta Chem Scand. 1968;22:3329–3330. [Google Scholar]
- 8.Newton CG, Tran DN, Wodrich MD, Cramer N. One-Step Multigram-Scale Biomimetic Synthesis of Psiguadial B. Angew Chem Int Ed. 2017;56:13776–13780. doi: 10.1002/anie.201708333. [DOI] [PubMed] [Google Scholar]
- 9.Fu H-Z, Luo Y-M, Li C-J, Yang J-Z, Zhang D-M, Psidials A-C. Three Unusual Meroterpenoids from the Leaves of Psidium guajava L. Org Lett. 2010;12:656–659. doi: 10.1021/ol9024869. [DOI] [PubMed] [Google Scholar]
- 10.Tran DN, Cramer N. Biomimetic Synthesis of (+)-Ledene, (+)-Viridiflorol, (−)-Palustrol, (+)-Spathulenol, and Psiguadial A, C, and D via the Platform Terpene (+)-Bicyclogermacrene. Chem Eur J. 2014;20:10654–10660. doi: 10.1002/chem.201403082. [DOI] [PubMed] [Google Scholar]
- 11.Lawrence AL, Adlington RM, Baldwin JE, Lee V, Kershaw JA, Thompson AL. A Short Biomimetic Synthesis of the Meroterpenoids Guajadial and Psidial A. Org Lett. 2010;12:1676–1679. doi: 10.1021/ol100138k. [DOI] [PubMed] [Google Scholar]
- 12.Chapman LM, Beck JC, Wu L, Reisman SE. Enantioselective Total Synthesis of (+)-Psiguadial B. J Am Chem Soc. 2016;138:9803–9806. doi: 10.1021/jacs.6b07229. [DOI] [PubMed] [Google Scholar]
- 13.Tanino and coworkers recently disclosed an approach to (±)-3, see:; Kinebuchi M, Uematsu R, Tanino K. Synthetic Studies on Psiguadial B: Construction of Bicyclo[4.3.1]decane Skeleton via Double Cyclization Reaction of Alkyne Dicobalt Complex. Tetrahedron Lett. 2017;58:1382–1386. [Google Scholar]
- 14.(a) Sasmal PK, Maier ME. Formation of Bicyclic Ethers from Lewis Acid Promoted Cyclizations of Cyclic Oxonium Ions. Org Lett. 2002;4:1271–1274. doi: 10.1021/ol025570d. [DOI] [PubMed] [Google Scholar]; (b) López F, Castedo L, Mascareñas JL. Practical Asymmetric Approach to Medium-Sized Carbocycles Based on the Combination of Two Ru-Catalyzed Transformations and a Lewis Acid-Induced Cyclization. Org Lett. 2005;7:287–290. doi: 10.1021/ol0477125. [DOI] [PubMed] [Google Scholar]; For related examples, see:; (c) Blumenkopf TA, Bratz M, Castaneda A, Look GC, Overman LE, Rodriguez D, Thompson AS. Preparation of Eight-Membered Cyclic Ethers by Lewis Acid Promoted Acetal-Alkene Cyclizations. J Am Chem Soc. 1990;112:4386–4399. [Google Scholar]
- 15.Arduini A, Bosi A, Pochini A, Ungaro R. o-Quinone Methides 2. Stereoselectivity in Cycloaddition Reactions of o-Quinone Methides with Vinyl Ethers. Tetrahedron. 1985;41:3095–3103. [Google Scholar]
- 16.For reviews, see:; (a) Willis NJ, Bray CD. o-Quinone Methides in Natural Product Synthesis. Chem Eur J. 2012;18:9160–9173. doi: 10.1002/chem.201200619. [DOI] [PubMed] [Google Scholar]; (b) Van De Water RW, Pettus TR. o-Quinone Methides: Intermediates Underdeveloped and Underutilized in Organic Synthesis. Tetrahedron. 2002;58:5367–5405. [Google Scholar]; (c) Ferreira SB, da Silva F, de C, Pinto AC, Gonzaga DTG, Ferreira VF. Syntheses of Chromenes and Chromanes via o-Quinone Methide Intermediates. J Heterocyclic Chem. 2009;46:1080–1097. [Google Scholar]
- 17.Since 2012, Schneider and others have reported o-QMHDA reactions with cyclic enamides. For relevant examples, see:; (a) Saha S, Schneider C. Brønsted Acid-Catalyzed, Highly Enantioselective Addition of Enamides to In Situ-Generated o-Quinone Methides: A Domino Approach to Complex Acetamidotetrahydroxanthenes. Chem Eur J. 2015;21:2348–2352. doi: 10.1002/chem.201406044. [DOI] [PubMed] [Google Scholar]; (b) El-Sepelgy O, Haseloff S, Alamsetti SK, Schneider C. Brønsted acid Catalyzed, Conjugate Addition of β-Dicarbonyls to In Situ Generated o-Quinone Methides–Enantioselective Synthesis of 4-Aryl-4H-chromenes. Angew Chem, Int Ed. 2014;53:7923–7927. doi: 10.1002/anie.201403573. [DOI] [PubMed] [Google Scholar]; (c) Saha S, Schneider C. Directing Group Assisted Nucleophilic Substitution of Propargylic Alcohols via o-Quinone Methide Intermediates: Brønsted Acid Catalyzed, Highly Enantio- and Diastereoselective Synthesis of 7-Alkynyl-12a-acetamido-Substituted Benzoxanthenes. Org Lett. 2015;17:648–651. doi: 10.1021/ol503662g. [DOI] [PubMed] [Google Scholar]; (d) Zhao JJ, Zhang YC, Xu MM, Tang M, Shi F. Catalytic Chemo-, E/Z-, and Enantioselective Cyclizations of o-Hydroxybenzyl Alcohols with Dimedone-Derived Enaminones. J Org Chem. 2015;80:10016–10024. doi: 10.1021/acs.joc.5b01613. [DOI] [PubMed] [Google Scholar]
- 18.For examples of chiral enol ethers used in o-QMHDA reactions, see:; (a) Selenski C, Mejorado LH, Pettus TR. Diastereoselective [4+2] Reactions of o-Quinone Methides with a Chiral Enol Ether: Asymmetric Synthesis of (+)-R-Mimosifoliol. Synlett. 2004;6:1101–1103. [Google Scholar]; (b) Selenski C, Pettus TRR. Enantioselective [4 + 2] Cycloadditions of o-Quinone Methides: Total Synthesis of (+)-Mimosifoliol and Formal Synthesis of (+)-Tolterodine. J Org Chem. 2004;69:9196–9203. doi: 10.1021/jo048703c. [DOI] [PubMed] [Google Scholar]; (c) Wenderski TA, Marsini MA, Pettus TRR. A Diastereoselective Formal Synthesis of Berkelic Acid. Org Lett. 2011;13:118–121. doi: 10.1021/ol102652t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Marsini MA, Huang Y, Lindsey CC, Wu KL, Pettus TRR. Diastereoselective Syntheses of Chroman Spiroketals via [4 + 2] Cycloaddition of Enol Ethers and o-Quinone Methides. Org Lett. 2008;10:1477–1480. doi: 10.1021/ol8003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For seminal studies, see:; (a) Shabashov D, Daugulis O. Auxiliary-Assisted Palladium-Catalyzed Arylation and Alkylation of sp2 and sp3 Carbon−Hydrogen Bonds. J Am Chem Soc. 2010;132:3965–3972. doi: 10.1021/ja910900p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zaitsev VG, Shabashov D, Daugulis O. Highly Regioselective Arylation of sp3 C−H Bonds Catalyzed by Palladium Acetate. J Am Chem Soc. 2005;127:13154–13155. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]; (c) Reddy BVS, Reddy LR, Corey E. J Novel Acetoxylation and C−C Coupling Reactions at Unactivated Positions in α-Amino Acid Derivatives. Org Lett. 2006;8:3391–3394. doi: 10.1021/ol061389j. [DOI] [PubMed] [Google Scholar]; For reviews, see:; (d) Corbet M, De Campo F. 8-Aminoquinoline: a Powerful Directing Group in Metal-Catalyzed Direct Functionalization of C-H Bonds. Angew Chem Int Ed. 2013;52:9896–9898. doi: 10.1002/anie.201303556. [DOI] [PubMed] [Google Scholar]; For a review, see:; (e) Yamaguchi J, Itami K, Yamaguchi AD. C-H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew Chem, Int Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]
- 20.(a) Gutekunst WR, Baran PS. Total Synthesis and Structural Revision of the Piperarborenines via Sequential Cyclobutane C–H Arylation. J Am Chem Soc. 2011;133:19076–19079. doi: 10.1021/ja209205x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gutekunst WR, Gianatassio R, Baran PS. Sequential C(sp3)-H Arylation and Olefination: Total Synthesis of the Proposed Structure of Pipercyclobutanamide. A. Angew Chem, Int Ed. 2012;51:7507–7510. doi: 10.1002/anie.201203897. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Feng Y, Chen G. Total Synthesis of Celogentin C by Stereoselective C-H Activation. Angew Chem, Int Ed. 2010;49:958–961. doi: 10.1002/anie.200905134. [DOI] [PubMed] [Google Scholar]; For reports disclosed after our studies commenced, see:; (d) Gutekunst WR, Baran PS. Applications of C–H Functionalization Logic to Cyclobutane Synthesis. J Org Chem. 2014;79:2430–2452. doi: 10.1021/jo4027148. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Panish RA, Chintala SR, Fox JM. A Mixed-Ligand Chiral Rhodium(II) Catalyst Enables the Enantioselective Total Synthesis of Piperarborenine B. Angew Chem, Int Ed. 2016;55:4983–4987. doi: 10.1002/anie.201600766. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ting CP, Maimone TJ. C-H Bond Arylation in the Synthesis of Aryltetralin Lignans: a Short Total Synthesis of Podophyllotoxin. Angew Chem, Int Ed. 2014;53:3115–3119. doi: 10.1002/anie.201311112. [DOI] [PubMed] [Google Scholar]
- 21.(a) Ghosh A, Banerjee UK, Venkateswaran RV. Photolysis of α-diazocyclopentanones. Ring Contraction to Functionalised Cyclobutanes and Synthesis of Junionone, Grandisol and Planococcyl Acetate. Tetrahedron. 1990;46:3077–3088. [Google Scholar]; (b) Banerjes UK, Venkateswaran RV. PhoTolysis of α-Diazocyclopentanones: Ring Contraction to Functionalised Cyclobutanes and Synthesis of Precursors to Grandisol and Fragranol. Tetrahedron Lett. 1983;24:423–424. [Google Scholar]
- 22.(a) Hodous BL, Fu GC. Enantioselective Addition of Amines to Ketenes Catalyzed by a Planar-Chiral Derivative of PPY: Possible Intervention of Chiral Brønsted-Acid Catalysis. J Am Chem Soc. 2002;124:10006–10007. doi: 10.1021/ja027466x. [DOI] [PubMed] [Google Scholar]; (b) Wiskur SL, Fu GC. Catalytic Asymmetric Synthesis of Esters from Ketenes. J Am Chem Soc. 2005;127:6176–6177. doi: 10.1021/ja0506152. [DOI] [PubMed] [Google Scholar]
- 23.France S, Wack H, Taggi AE, Hafez AM, Wagerle TR, Shah MH, Dusich CL, Lectka T. Catalytic Asymmetric α-Chlorination of Acid Halides. J Am Chem Soc. 2004;126:4245–4255. doi: 10.1021/ja039046t. [DOI] [PubMed] [Google Scholar]
- 24.(a) Pracejus H. Organische Katalysatoren, LXI. Asymmetrische Synthesen mit Ketenen, I. Alkaloid-Katalysierte Asymmetrische Synthesen von α-Phenyl-Propionsäureestern. Justus Liebigs Ann Chem. 1960;634:9–22. [Google Scholar]; (b) Zhang Y-R, He L, Wu X, Shao P-L, Ye S. Chiral N-Heterocyclic Carbene Catalyzed Staudinger Reaction of Ketenes with Imines: Highly Enantioselective Synthesis of N-Boc β-Lactams. Org Lett. 2008;10:277–280. doi: 10.1021/ol702759b. [DOI] [PubMed] [Google Scholar]; For a review on catalytic, asymmetric additions to ketenes, see:; (c) Paull DH, Weatherwax A, Lectka T. Catalytic, Asymmetric Reactions of Ketenes and Ketene Enolates. Tetrahedron. 2009;65:6771–6803. doi: 10.1016/j.tet.2009.05.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.As discussed in reference 12, it was determined that 3 equiv 29 was necessary to mitigate a competing photodecarbonylation process, see:; Tidwell TT. Ketenes. 2nd. John Wiley & Sons; Hoboken, New Jersey: 2006. pp. 443–447. [Google Scholar]
- 26.See Supporting Information for full details.
- 27.(a) d’Augustin M, Palais LT, Alexakis A. Enantioselective Copper-Catalyzed Conjugate Addition to Trisubstituted Cyclohexenones: Construction of Stereogenic Quaternary Centers. Angew Chem, Int Ed. 2005;44:1376–1378. doi: 10.1002/anie.200462137. [DOI] [PubMed] [Google Scholar]; (b) Vuagnoux-d’Augustin M, Alexakis A. Copper-Catalyzed Asymmetric Conjugate Addition of Trialkylaluminium Reagents to Trisubstituted Enones: Construction of Chiral Quaternary Centers. Chem Eur J. 2007;13:9647–9662. doi: 10.1002/chem.200701001. [DOI] [PubMed] [Google Scholar]
- 28.Saimoto H, Yoshida K, Murakami T, Morimoto M, Sashiwa H, Shigemasa Y. Effect of Calcium Reagents on Aldol Reactions of Phenolic Enolates with Aldehydes in Alcohol. J Org Chem. 1996;61:6768–6769. doi: 10.1021/jo961352k. [DOI] [PubMed] [Google Scholar]
- 29.Prepared directly in <10% yield according to:; (a) Leuchs H, Theodorescu G. Chem Ber. 1910;43:1243. [Google Scholar]; The corresponding ethyl ester can also be prepared in higher yield (~20%) according to:; (b) Zhang Q, Botting NP, Kay C. A Gram Scale Synthesis of a Multi- 13C-Labelled Anthocyanin, [6,8,10,3′,5′-13C5]Cyanidin-3-glucoside, for Use in Oral Tracer Studies in Humans. Chem Commun. 2011;47:10596–10598. doi: 10.1039/c1cc14323a. [DOI] [PubMed] [Google Scholar]
- 30.Attempts to employ 55 directly as an o-QM precursor led to complex reaction profiles for the cycloaddition: The liberated equivalent of morpholine (52) displaced the methoxy ketal in the initially formed cycloaddition products (i.e. 57–60) via thermal oxonium ion formation. Efforts to mitigate this problem by activating 55 via N-methylation were unsuccessful. For relevant examples, see:; (a) Wilson PD, Pettigrew JD, Bexrud JA, Freeman RP. Total Synthesis of (±)-Xyloketal D and Model Studies towards the Total Synthesis of (−)-Xyloketal A. Heterocycles. 2004;62:445–452. [Google Scholar]; (b) Pettigrew JD, Freeman RP, Wilson PD. Total Synthesis of (−)-Xyloketal D and its Enantiomer – Confirmation of Absolute Stereochemistry. Can J Chem. 2004;82:1640–1648. [Google Scholar]
- 31.We do not observe products resulting from cycloaddition with the isomeric enol ether (indicated with a dashed line); presumably, only one isomer reacts due to steric hindrance.
- 32.Evans DA, Johnson JS, Olhava EJ. Enantioselective Synthesis of Dihydropyrans. Catalysis of Hetero Diels−Alder Reactions by Bis(oxazoline) Copper(II) Complexes. J Am Chem Soc. 2000;122:1635–1649. [Google Scholar]
- 33.For select examples of o-QMs generated using Brønsted acids, see:; (a) Hsiao CC, Raja S, Liao HH, Atodiresei I, Rueping M. O-Quinone Methides as Reactive Intermediates in Asymmetric Brønsted Acid Catalyzed Cycloadditions with Unactivated Alkenes by Exclusive Activation of the Electrophile. Angew Chem Int Ed. 2015;54:5762–5765. doi: 10.1002/anie.201409850. and references cited therein. [DOI] [PubMed] [Google Scholar]; (b) Gharpure SJ, Sathiyanarayanan AM, Vuram PK. Hetero Diels–Alder Reaction of Olefin with o-Quinone Methides Generated using (±)-Binolphosphoric acid for the Stereoselective Synthesis of 2,4-Diarylbenzopyrans: Application to the Formal Synthesis of Myristinin B/C. RSC Adv. 2013;3:18279–18282. [Google Scholar]
- 34.Notably, this method provides a 1:1 mixture of enol ethers that do not equilibrate at room temperature. Thus, only half of the starting material employed in the Cu(OTf)2-mediated o-QMHDA reaction can provide the desired cycloaddition product.
- 35.Ananikov VP, Orlov NV, Beletskaya IP. Efficient and Convenient Synthesis of β-Vinyl Sulfides in Nickel-Catalyzed Regioselective Addition of Thiols to Terminal Alkynes under Solvent-Free Conditions. Organometallics. 2006;25:1970–1977. [Google Scholar]
- 36.Cho YS, Kim HY, Cha JH, Pae AN, Koh HY, Choi JH, Chang MH. Indium Trichloride Mediated Intramolecular Prins-Type Cyclization. Org Lett. 2002;4:2025–2028. doi: 10.1021/ol025856i. [DOI] [PubMed] [Google Scholar]
- 37.For leading references on the Norrish–Yang cyclization, see:; (a) Yang NC, Yang DDH. Photochemical Reactions of Ketones in Solution. J Am Chem Soc. 1958;80:2913–2914. [Google Scholar]; (b) Chen C. The Past, Present, and Future of the Yang Reaction. Org Biomol Chem. 2016;14:8641–8647. doi: 10.1039/c6ob01214k. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an example in total synthesis, see:; Paquette LA, Sugimura T. Enantiospecific Total Synthesis and Absolute Configurational Assignment of (−)-Punctatin A (antibiotic M95464) J Am Chem Soc. 1986;108:3841–3842. [Google Scholar]
- 38.Schwinden MD. The Norrish Type II Reaction in Organic Synthesis (1990) (Paper 9890).Retrospective Theses and Dissertations. [Google Scholar]
- 39.(a) Bach T, Aechtner T, Neumüller B. Enantioselective Norrish–Yang Cyclization Reactions of N-(ω-Oxo- ω-phenylalkyl)-Substituted Imidazolidinones in Solution and in the Solid State. Chem Eur J. 2002;8:2464–2475. doi: 10.1002/1521-3765(20020603)8:11<2464::AID-CHEM2464>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]; (b) Singhal N, Koner AL, Mal P, Venugopalan P, Nau WM, Moorthy JN. Diastereomer-Differentiating Photochemistry of β-Arylbutyrophenones: Yang Cyclization versus Type II Elimination. J Am Chem Soc. 2005;127:14375–14372. doi: 10.1021/ja0523643. [DOI] [PubMed] [Google Scholar]; (c) Fleming I, Kemp-Jones AV, Long WE, Thomas EJ. Cyclobutanol: Fragmentation Ratios for the Singlet and Triplet Excited States in the Type II Photochemistry of some α-Alkylated Cyclohexanones. J Chem Soc, Perkin Trans 2. 1976;0:7–14. [Google Scholar]
- 40.(a) Winnik MA, Breslow R. Remote Oxidation of Unactivated Methylene Groups. J Am Chem Soc. 1969;91:3083–3984. [Google Scholar]; (b) Breslow R, Rothbard J, Herman F, Rodriguez ML. Remote Functionalization Reactions as Conformational Probes for Flexible Alkyl Chains. J Am Chem Soc. 1978;100:1213–1218. [Google Scholar]; (c) Andreu I, Palumbo F, Tilocca F, Morera IM, Boscá F, Miranda MA. Solvent Effects in Hydrogen Abstraction from Cholesterol by Benzophenone Triplet Excited State. Org Lett. 2011;13:4096–4099. doi: 10.1021/ol2016059. [DOI] [PubMed] [Google Scholar]
- 41.(a) Palucki M, Wolfe JP, Buchwald SL. Synthesis of Oxygen Heterocycles via a Palladium-Catalyzed C−O Bond-Forming Reaction. J Am Chem Soc. 1996;118:10333–10334. [Google Scholar]; (b) Palucki M, Wolfe JP, Buchwald SL. Palladium-Catalyzed Intermolecular Carbon−Oxygen Bond Formation: A New Synthesis of Aryl Ethers. J Am Chem Soc. 1997;119:3395–3396. [Google Scholar]; (c) Parrish CA, Buchwald SL. Palladium-Catalyzed Formation of Aryl tert-Butyl Ethers from Unactivated Aryl Halides. J Org Chem. 2001;66:2498–2500. doi: 10.1021/jo001426z. [DOI] [PubMed] [Google Scholar]; (d) Vorogushin AV, Huang X, Buchwald SL. Use of Tunable Ligands Allows for Intermolecular Pd-Catalyzed C−O Bond Formation. J Am Chem Soc. 2005;127:8146–8149. doi: 10.1021/ja050471r. [DOI] [PubMed] [Google Scholar]
- 42.We were aware of unsuccessful efforts to prepare β-caryophyllene by ring-closing metathesis. See:; Dowling MS, Vanderwal CD. Ring-Closing Metathesis of Allylsilanes As a Flexible Strategy toward Cyclic Terpenes. Short Syntheses of Teucladiol, Isoteucladiol, Poitediol, and Dactylol and an Attempted Synthesis of Caryophyllene. J Org Chem. 2010;75:6908–6922. doi: 10.1021/jo101439h. [DOI] [PubMed] [Google Scholar]
- 43.Attempts to apply Noyori’s aprotic ketalization protocol caused rapid decomposition of 46:; Tsunoda T, Suzuki M, Noyori R. A Facile Procedure for Acetalization under Aprotic Conditions. Tetrahedron Lett. 1980;21:1357–1358. [Google Scholar]
- 44.Iodide 73 was prepared as an inconsequential 8:1 mixture of olefin isomers.
- 45.The major side product in the epimerization of 46 to 47 is spirocyclic lactam S1 (see Supporting Information). Formation of S1 is precluded using this alternative sequence, since aza-Michael addition cannot occur when the enone is protected as the corresponding dioxolane.
- 46.We were unable to separate all the components in this complex reaction mixture with sufficient purity for definitive characterization.
- 47.Spectroscopic data for 83 matches that reported in the literature:; Lee H, Yi CS. Synthesis of 2-Acylphenol and Flavene Derivatives from the Ruthenium-Catalyzed Oxidative C–H Acylation of Phenols with Aldehydes. Eur J Org Chem. 2015;2015:1899–1904. doi: 10.1002/ejoc.201403518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.(a) Khomenko TM, Korchagina DV, Gatilov YV, Bagryanskaya IY, Tkachev AV, Vyalkov AI, Kun OB, Salenko VL, Dubovenko ZV, Barkash VA. Synthesis of Some Dienes with a Caryophyllane Skeleton and their Reactions in Acid Media. Zh Org Khim. 1990;26:2129–2145. [Google Scholar]; Khomenko TM, Korchagina DV, Gatilov YV, Bagryanskaya IY, Tkachev AV, Vyalkov AI, Kun OB, Salenko VL, Dubovenko ZV, Barkash VA. Synthesis of Some Dienes with a Caryophyllane Skeleton and their Reactions in Acid Media. Russ J Org Chem (English Translation) 1990;26:1839–1852. [Google Scholar]; See also:; (b) Khomenko TM, Bagryanskaya IY, Gatilov YV, Korchagina DV, Gatilova VP, Dubovenko ZV, Barkhash VA. Molecular Rearrangements of Isocaryophyllene in a Super Acid. Zh Org Khim. 1985;21:677–678. [Google Scholar]; Khomenko TM, Bagryanskaya IY, Gatilov YV, Korchagina DV, Gatilova VP, Dubovenko ZV, Barkhash VA. Molecular Rearrangements of Isocaryophyllene in a Super Acid. Russ J Org Chem (English Translation) 1985;21:614–615. [Google Scholar]
- 49.For an example of a tertiary alcohol-directed hydrogenation using Crabtree’s catalyst, see:; (a) Hong AY, Stoltz BM. Biosynthesis and Chemical Synthesis of Presilphiperfolanol Natural Products. Angew Chem Int Ed. 2014;53:5248–5260. doi: 10.1002/anie.201309494. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review, see:; (b) Hoveyda AH, Evans DA, Fu GC. Substrate-Directable Chemical Reactions. Chem Rev. 1993;93:1307–1370. [Google Scholar]
- 50.Suchand B, Krishna J, Mritunjoy K, Satyanarayana G. Lewis acid Promoted C–C and Copper-Catalyzed C–O Bond Formation: Synthesis of Neoflavans. RSC Adv. 2014;4:13941–13945. [Google Scholar]
- 51.For select examples, see:; (a) Selenski C, Pettus TRR. (±)-Diinsininone: Made Nature’s Way. Tetrahedron. 2006;62:5298–5307. doi: 10.1016/j.tet.2006.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Achilonu MC, Bonnet SL, van der Westhuizen JH. Synthesis of Proanthocyanidins. Part 1. The First Oxidative Formation of the Interflavanyl Bond in Procyanidins. Org Lett. 2008;10:3865–3868. doi: 10.1021/ol801353u. [DOI] [PubMed] [Google Scholar]; (c) Bezuidenhoudt BCB, Brandt EV, Roux DG. Synthesis of lsoflavanoid Oligomers Using a Pterocarpan as Inceptive Electrophile. J Chem Soc, Perkin Trans 1. 1984:2767–2778. [Google Scholar]; (d) Hayes CJ, Whittaker BP, Watson SA, Grabowska AM. Synthesis and Preliminary Anticancer Activity Studies of C4 and C8-Modified Derivatives of Catechin Gallate (CG) and Epicatechin Gallate (ECG) J Org Chem. 2006;71:9701–9712. doi: 10.1021/jo061740e. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 52.(a) Saito A, Nakajima N, Tanaka A, Ubukata M. Synthetic Studies of Proanthocyanidins. Part 2: Stereoselective Gram-Scale Synthesis of Procyanidin-B3. Tetrahedron. 2002;58:7829–7837. doi: 10.1271/bbb.66.1764. [DOI] [PubMed] [Google Scholar]; (b) Dennis EG, Jeffery DW, Johnston MR, Perkins MV, Smith PA. Procyanidin Oligomers. A New Method for 4→8 Interflavan Bond Formation using C8-Boronic Acids and Iterative Oligomer Aynthesis through a Boron-Protection Strategy. Tetrahedron. 2012;68:340–348. [Google Scholar]
- 53.Feng ZG, Bai WJ, Pettus TRR. Unified Total Syntheses of (−)-Medicarpin, (−)-Sophoracarpan A, and (±)-Kushecarpin A with Some Structural Revisions. Angew Chem Int Ed. 2015;54:1864–1867. doi: 10.1002/anie.201408910. [DOI] [PubMed] [Google Scholar]
- 54.Hendrik C, Mouton L, Steenkamp JA, Young DA, Bezuidenhoudt BCB, Ferreira D. Regio- and Stereoselective Oxygenation of Flavan-S-Ol-, 4-Arylflavan- 3-Ol-, and Biflavanoid-Derivatives with Potassium Persulphate. Tetrahedron. 1990;46:6885–6894. [Google Scholar]
- 55.(a) Day JJ, McFadden RM, Virgil SC, Kolding H, Alleva JL, Stoltz BM. The Catalytic Enantioselective Total Synthesis of (+)-Liphagal. Angew Chem Int Ed. 2011;50:6814–6818. doi: 10.1002/anie.201101842. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Olah GA, Salem G, Staral JS, Ho T-L. Preparative Carbocation Chemistry. 13. Preparation of Carbocations from Hydrocarbons Via Hydrogen Abstraction with Nitrosonium Hexafluorophosphate and Sodium Nitrite-Trifluoromethanesulfonic Acid. J Org Chem. 1978;43:173–175. [Google Scholar]
- 56.Li YZ, Li BJ, Lu XY, Lin S, Shi ZJ. Cross Dehydrogenative Arylation (CDA) of a Benzylic C-H Bond with Arenes by Iron Catalysis. Angew Chem Int Ed. 2009;48:3817–3820. doi: 10.1002/anie.200900341. [DOI] [PubMed] [Google Scholar]
- 57.Muramatsu W, Nakano K. Organocatalytic Approach for C(sp3)–H Bond Arylation, Alkylation, and Amidation of Isochromans under Facile Conditions. Org Lett. 2014;16:2042–2045. doi: 10.1021/ol5006399. [DOI] [PubMed] [Google Scholar]
- 58.Steenkamp JA, Mouton C, Ferreira D. Regio- and Stereoselective Oxidation of Flavan-3-Ol-4-Arylflavan-3-Ol- and Biflavanoid Derivatives with 2,3-Dichloro-56-Dicyano-1,4-Benzoquinone (DDQ) Tetrahedron. 1991;47:6705–6716. [Google Scholar]
- 59.Willson TM, Amburgey J, Denmark SE. Synthesis of α- and β-Branched Ethers from Alcohols by Reaction of Acetals with Grignard Reagents: Synthesis of Isopropyl and Isobutyl Ethers of (1S*,2R*S*,4R*)-6-methylenebicyclo[2.2.2]octan-2-ol. J Chem Soc, Perkin Trans 1. 1991;12:2899–2906. [Google Scholar]
- 60.(a) Greene MA, Yonova IM, Williams FJ, Jarvo ER. Traceless Directing Group for Stereospecific Nickel-Catalyzed Alkyl−Alkyl Cross-Coupling Reactions. Org Lett. 2012;14:4293–4296. doi: 10.1021/ol300891k. [DOI] [PubMed] [Google Scholar]; (b) Yonova IM, Johnson AG, Osborne CA, Moore CE, Morrissette NS, Jarvo ER. Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Alkyl Grignard Reagents and Identification of Selective Anti-Breast-Cancer Agents0. Angew Chem Int Ed. 2014;53:2422–2427. doi: 10.1002/anie.201308666. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dawson DD, Jarvo ER. Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Benzylic Ethers with Isotopically-Labeled Grignard Reagents. Org Process Res Dev. 2015;19:1356–1359. doi: 10.1021/acs.oprd.5b00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.(a) Mitchell TA, Bode JW. Synthesis of Dialkyl Ethers from Organotrifluoroborates and Acetals. J Am Chem Soc. 2009;131:18057–18059. doi: 10.1021/ja906514s. [DOI] [PubMed] [Google Scholar]; (b) Vo C-VT, Mitchell TA, Bode JW. Expanded Substrate Scope and Improved Reactivity of Ether-Forming Cross-Coupling Reactions of Organotrifluoroborates and Acetals. J Am Chem Soc. 2011;133:14082–14089. doi: 10.1021/ja205174c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.(a) Lipshutz BH, Wilhelm RS, Kozlowski JA. Conjugate Addition Reactions of α,β,-Unsaturated ketones with Higher Order, Mixed Organocuprate Reagents, R2Cu(CN)Li2. J Org Chem. 1984;49:3938–3942. [Google Scholar]; (b) Lipshutz BH, Parker DA, Kozlowski JA, Nguyen SM. Effects of Lewis Acids on Higher Order, Mixed Cuprate Couplings. Tetrahedron Lett. 1984;25:5959–5962. [Google Scholar]
- 63.(a) Rieche A, Gross H, Höft E. Aromatic aldehydes Mesitaldehyde. Org Synth. 1967;47:1. [Google Scholar]; (b) Aukrust IR, Skattebol L. The Syntehsis of (–)-Robustadial A and Some Analogues. Acta Chem Scand. 1996;50:132–140. [Google Scholar]; (c) Kraus GA, Mengwasser J, Maury W, Oh C. Synthesis of Chroman Aldehydes that Inhibit HIV. Bioorg Med Chem Lett. 2011;21:1399–1401. doi: 10.1016/j.bmcl.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Still WC, Kahn M, Mitra A. Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution. J Org Chem. 1978;43:2923–2925. [Google Scholar]
- 65.Characterization for this compound was reported previously (reference 12). It is re-presented here for the convenience of the readers.
- 66.We have found that concentrated reaction mixtures (e.g. ≥2.2 M) react to full conversion at 0 °C, whereas more dilute reactions (i.e. 1.5 M, as reported in ref 21a) often require warming to room temperature in order to initiate. This can be dangerous on large scale, as the reaction proceeds quickly and produces H2.
- 67.For safety reasons, p-4-acetamidobenzenesulfonyl azide (p-ABSA) was used as an alternative diazo transfer reagent in place of tosyl azide.
- 68.(a) Vuluga D, Legros J, Crousse B, Bonnet-Delpon D. Synthesis of Pyrazoles Through Catalyst-Free Cycloaddition of Diazo Compounds to Alkynes. Green Chem. 2009;11:156–159. [Google Scholar]; (b) Sato Y, Fujisawa H, Mukaiyama T. Bull Chem Soc Jpn. 2006;79:1275. [Google Scholar]; (c) Rosenfeld MJ, Shankar BKR, Shechter H. Rhodium(II) Acetate-Catalyzed Reactions of 2-Diazo-1,3-Indandione and 2-Diazo-1-Indanone with Various Substrates. J Org Chem. 1998;53:2699–2705. [Google Scholar]; For preparation of the 6- and 7-membered ketones used to access 38 and 39, see:; (d) Cernijenko A, Risgaard R, Baran PS. 11-Step Total Synthesis of (−)-Maoecrystal V. J Am Chem Soc. 2016;138:9425–9428. doi: 10.1021/jacs.6b06623. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu H, Drizin I, Koenig JR, Cowart MD, Wakefield BD, Altenbach RJ, Black LA, Zhao C. Macrocyclic pyrimidine derivatives WO200912. 2009;3967 [Google Scholar]
- 69.Reaction time varies with the age of the lamp. A UV-opaque film slowly develops on the inside surface of the flask facing the lamp upon prolonged irradiation. This film can be removed by soaking the flask in an alkali base bath (KOH, 4:1 i-PrOH/H2O).
- 70.APEX2, Version 2 User Manual, M86-E0 1078 Bruker Analytical X-ray Systems, Madison, WI, June 2006.
- 71.Prepared according to the ligand protocol described in:; Bao H, Qi X, Tambar UK. Catalytic Enantioselective [2,3]-Rearrangements of Amine N-Oxides. J Am Chem Soc. 2011;133:1206–1208. doi: 10.1021/ja110500m. We found that the use of CH2Cl2 as a reaction solvent provided higher and more reproducible yields, compared with THF. [DOI] [PubMed] [Google Scholar]
- 72.Prepared directly in <10% yield according to:; Leuchs H, Theodorescu G. Chem Ber. 1910;43:1243. [Google Scholar]; The corresponding ethyl ester can also be prepared in higher yield (~20%) according to:; (b) Zhang Q, Botting NP, Kay C. A Gram Scale Synthesis of a Multi-13C-Labelledanthocyanin, [6,8,10,3′,5′-13C5]Cyanidin-3-Glucoside, for Use in Oral Tracer Studies in Humans. Chem Commun. 2011;47:10596–10598. doi: 10.1039/c1cc14323a. [DOI] [PubMed] [Google Scholar]
- 73.Ohira–Bestmann reagent prepared according to:; Pietruszka J, Witt A. Synthesis of the Bestmann-Ohira Reagent. Synthesis. 2006;2006:4266–4268. [Google Scholar]
- 74.Prepared according to:; Gambacorti-Passerini C, Mologni L, Scapozza L, Bisson W, Ahmed S, Goekjian P, Tardy S, Orsato A, Gueyrard D, Benoit J. Alpha-carbolines for the treatment of cancer WO 201316. 2013;7730 [Google Scholar]
- 75.Spectroscopic data for 83 matches that reported in the literature:; Lee H, Yi CS. Synthesis of 2-Acylphenol and Flavene Derivatives from the Ruthenium-Catalyzed Oxidative C-H Acylation of Phenols with Aldehydes. Eur J Org Chem. 2015;2015:1899–1904. doi: 10.1002/ejoc.201403518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.The characterization data were fully consistent with the isolation data reported in ref 4a. See Supporting Information for NMR comparison tables.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.