

Abstract
A microfluidic sample preparation multiplexer (SPM) and assay procedure is developed to improve amplification-free detection of Ebola virus RNA from blood. While a previous prototype successfully detected viral RNA following off-chip RNA extraction from infected cells, the new device and protocol can detect Ebola virus in raw blood with clinically relevant sensitivity. The Ebola RNA is hybridized with sequence specific capture and labeling DNA probes in solution and then the complex is pulled down onto capture beads for purification and concentration. After washing, the captured RNA target is released by irradiating the photocleavable DNA capture probe with ultraviolet (UV) light. The released, labeled, and purified RNA is detected by a sensitive and compact fluorometer. Exploiting these capabilities, a detection limit of 800 attomolar (aM) is achieved without target amplification. The new SPM can run up to 80 assays in parallel using a pneumatic multiplexing architecture. Importantly, our new protocol does not require time-consuming and problematic off-chip probe conjugation and washing. This improved SPM and labeling protocol is an important step toward a useful POC device and assay.
Graphical abstract
Point-of-care (POC) testing enables medical diagnosis to be performed immediately in the infected communities with simple instruments and processing.1,2 For example, the recent outbreak of Ebola in West Africa highlighted the need for rapid and quantitative POC detection.3,4 Nucleic acid-based detection techniques such as real-time polymerase chain reaction (qPCR)5,6 have been developed for pathogen detection as they show excellent sensitivity and specificity. However, qPCR requires expensive reagents, bulky instruments, and well trained personnel, all of which are in short supply in low resource settings. Isothermal amplification, on the other hand, provides a simple way for performing target amplification and shows excellent sensitivity.7,8 However, nucleic acid prepurification is required because organic and inorganic substances can inhibit the target amplification.9 In addition, multiplex detection is often limited when using isothermal amplification due to the primer–primer interactions.10 Thus, a rapid, sensitive, low cost, and fully integrated analysis process which can work with raw blood sample is needed to address POC viral testing needs.
Recently, amplification-free nucleic acid techniques such as surface-enhanced Raman scattering (SERS),11 electrochemical sensing,12 and fluorescence imaging13 have been introduced to address these diagnostic needs. These techniques do not require complicated target amplification and are intended for POC applications. As the Ebola viral loads are extremely low at the initial stages of infection, attomolar sensitivity is required to enable early clinical decisions.14,15 Although SERS and electrochemical sensing are label-free and amplification-free techniques, the sensitivity of these techniques is poor16,17 and does not reach the level required for clinical decision making. Microscopy provides highly sensitive single molecule sensing capability, but it is expensive and difficult to perform in field environments.18,19
State-of-the-art optofluidic and microfluidic technology enables an alternative approach for amplification-free detection of pathogens20,21 that is advantageous because it integrates microfluidic processing and optical sensing functions.22–27 We previously developed an efficient sample preparation multiplexer (SPM) and a sensitive liquid-core antiresonant reflecting optical waveguide (ARROW) biosensor chip for Ebola RNA sensing.25 Without target amplification, a limit of detection (LOD) of 800 aM was achieved with ∼2 h detection time.28 The automated SPM captured and concentrated Ebola RNA molecules on DNA probe labeled magnetic beads and the ARROW chip detected individual labeled RNA molecules in femtoliter excitation volumes. This optofluidic system demonstrated excellent sensitivity, specificity, and dynamic range for quantitative RNA molecule counting. However, several challenges still need to be addressed if optofluidic systems are to be used for POC applications. Most importantly, the sample processing procedure must be simplified and optimized to allow virus detection directly from body fluids without additional off-chip RNA purification steps. An on-chip filtering process could be used to isolate nucleic acid targets from blood,26,29 but our focus here is on simpler approaches with higher specificity and sensitivity. In addition, the processing throughput of the SPM needs to be improved to match high throughput techniques such as qPCR.30
Here we present a new SPM and analysis protocol that is based on a sequence specific barcode fluorescence reporter and a photocleavable capture probe. This method enables efficient extraction of Ebola RNA from raw blood using a SPM that is more automated and integrated as a result of the facile target release process. The sequence specific hybridization of the fluorescence reporter and the capture probe to the target RNA is simply and rapidly performed completely in solution phase before bead binding. The target RNA is efficiently released after solid-phase concentration and cleanup by activating the photocleavable linker with ultraviolet light. Exploiting a pneumatic multiplexing architecture, the SPM is capable of running 80 samples on a single chip. Combined with a simple, sensitive, and inexpensive fluorometer system, a detection limit of 800 aM for Ebola RNA in blood is achieved within 90 min total analysis time. This new method significantly simplifies the sample preparation and detection procedures for Ebola and other hemorrhagic fever viruses pointing the way to POC applications.
MATERIALS AND METHODS
Microfluidic Chip Fabrication
The sample preparation multiplexer (SPM) is designed to run 80 on-chip solid-phase extraction assays in parallel (Figure 1a). The chip consists of 16 identical units and each unit controls five assays (inset of Figure 1a). For the pneumatic layer (green), each inlet controls the opening and closing of 4 microvalves in the array driven by programmable solenoid valves. The dimensions of the pneumatic channels are 90 μm wide and 40 μm deep. For the fluidic layer (red), each unit consists of one fluidic inlet and one fluidic outlet. Reagents are transported automatically in the fluidic layer by controlling the opening and closing of the microvalves (a video is available in the Supporting Information). A photograph of the device is presented in Figure 1b. For the fluidic layer, two 100 μm wide, 25 μm deep channels are connected to each 3 mm diameter, 1.4 cm deep incubation reservoir. The normally closed lifting-gate microvalve-based microfluidic SPM fabricated and assembled from two polydimethylsiloxane (PDMS) layers31,32 is advantageous for our applications compared to normally open microvalve systems.33,34 To form the pneumatic layer, SU-8 micropatterns (thickness ∼ 40 μm) were created on a 15 cm diameter silicon wafer by using photolithography. PDMS was poured on the SU-8 micropatterns and cured to a final thickness of ∼7 mm. After peeling off the pneumatic layer from the SU-8 micropatterns, holes for the pneumatic control layer were punched. To form the fluidic layer, SU-8 micropatterns (thickness ∼ 25 μm) were created on a 12 cm diameter silicon wafer by using photolithography followed by spin coating a 300 μm PDMS membrane. Then, the oxygen plasma activated pneumatic layer and UV-Ozone activated fluidic layer were bonded. After bonding, the fused layers were peeled off the silicon wafer and transferred to a 12 cm diameter glass substrate. The diameter of the microvalves is ∼2 mm and the diameter of the incubation reservoirs is ∼3 mm (capacity of ∼50 μL). To increase the volume of the incubation reservoirs, another 7 mm thick PDMS layer was bonded on top of the pneumatic layer with ∼6 mm diameter reservoirs (capacity ∼ 200 μL).
Figure 1.
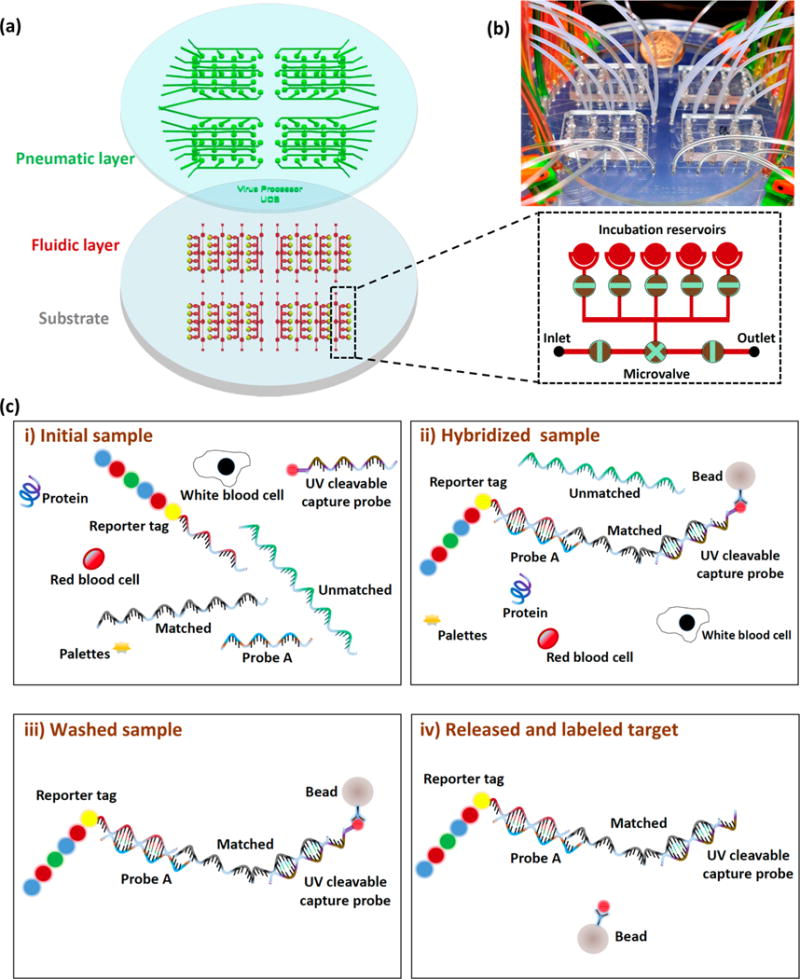
(a) Design of the sample preparation multiplexer (SPM) for on-chip solid-phase extraction with 80 incubation reservoirs. Each inlet/outlet line controls five incubation reservoirs (black dashed box). (b) Photograph of the SPM. (c) Schematic of the on-chip solid phase extraction experiment: (i) Reporter tag and UV cleavable capture probe mixed with Ebola RNA spiked blood samples. (ii) Hybridization of reporter tag, probe A, Ebola RNA, UV cleavable capture probe, and the streptavidin bead. (iii) Incubation reservoirs washed with buffer solution to remove unbound materials, excess reagents, and contamination. (iv) UV cleavable linker dissociated with UV light from the streptavidin bead to release the labeled target.
Sample Preparation
Digital color-coded barcode fluorescence reporter (nCounter Elements TagSet-12) was purchased from NanoString Technologies, Inc. Synthetic nucleic acid probe A (84-mer) and capture probe modified with photocleavable spacer/biotin (58-mer) were obtained from Integrated DNA Technologies, Inc. (IDT). They were designed to complement part of the Zaire Ebola Virus (AY354458.1:12638) and the barcode fluorescence reporter (for sequences see Table S1). Purified Zaire Ebola Virus RNA (AY354458.1) was extracted in the Biosafety Level 4 Laboratory at Texas Biomedical Research Institute using Trizol LS reagents (Thermo Fisher Scientific) as reported previously.28,35,36 Purified Ebola RNA was spiked in mouse whole blood samples containing sodium heparin to avoid coagulation (Biochemed Services) for on-chip microfluidic experiments. MyOne streptavidin T1 beads (10 mg/mL) were purchased from Thermo Fisher Scientific and washed 4 times with RNase-free water (Takara Bio Inc.) for on-chip experiments.
On-Chip Solid-Phase Extraction
Ebola spiked blood samples were pumped into the incubation reservoirs through the microvalve system. For 1× protocol, 15 μL blood was used and for 10× protocol, 150 μL blood was used with the same Ebola RNA concentration as the 1× protocol. Then, Probe A (5 nM, 1 μL), UV cleavable probe (15 nM, 1 μL), barcode fluorescence reporter (5 μL) and 10 μL nCounter Sprint hybridization buffer (Nanostring Technologies, Inc.) were mixed off-chip and pumped into the incubation reservoirs for the hybridization with Ebola RNA at 50 °C on a hot plate (Figure S1a). A glass slide was used to cover the incubation reservoirs during probe hybridization to avoid evaporation. After probe hybridization (2–40 min), 3 μL of magnetic beads were manually added to each incubation reservoir and metered air bubbles (0.7 μL/s) were introduced to each incubation reservoir for 30 s to improve mixing (Figure S1b). After incubation for 15–120 min, a magnet was placed under each incubation reservoir to isolate the beads so that the supernatant could be automatically evacuated. Then, the magnet was removed and RNase-free water was pumped into each incubation reservoir followed by 30 s of air bubble mixing to suspend the beads. The automated washing cycle was repeated three times followed by pumping 15 μL of RNase-free water into each incubation reservoir. After that, the SPM was placed under a 311 nm ultraviolet (UV) lamp (Kernel-4003B) to release captured nucleic acid targets from the beads (Figure S1c). Finally, the beads were isolated by a magnet and the supernatant was removed manually from each incubation reservoir for the fluorescence measurements (Figure S1d).
Fluorescence Quantitation
The supernatant was excited by a continuous wave laser at 488 nm (Sapphire 488 LP, Coherent Inc.), and the power was reduced to ∼10 mW by a variable neutral-density filter (Thorlabs, Inc.) to avoid photo damage. The laser was focused onto the sample to a ∼1 mm diameter spot size and the illumination volume was ∼1.5 μL. Fluorescence was collected by an off-axis parabolic (OAP) mirror with a centered hole for passing the excitation beam (2″ diameter, 50 mm focal length, 3 mm center hole diameter, Thorlabs, Inc.). Unwanted scattered light was eliminated by a 488 nm notch filter (NF488-15, ∼6 optical density, fwhm 15 nm, Thorlabs, Inc.). The fluorescence was focused into an optical fiber (M93L01, Thorlabs, Inc.) on a XYZ translation stage (Newport Corporation), connected to a mini USB spectrometer (USB 2000+, Ocean Optics, Inc.). Data measurement and analysis were carried out by using Spectra Suite (Ocean Optics, Inc.) and Origin Pro (OriginLab Corporation) software. To characterize the limit of detection (LOD) of the fluorometer, fluorescence reporters were serially diluted with RNase-free water and measured by the fluorometer. The emission curve of the reporter (centered at ∼520 nm) versus various concentrations (200 aM–8 pM) is presented in Figure 2b. The fluorescence signals were integrated from 510 to 540 nm and presented in Figure 2c. The integrated counts show a linear dependence on concentration for the reporters and the measured LOD is ∼200 aM.
Figure 2.
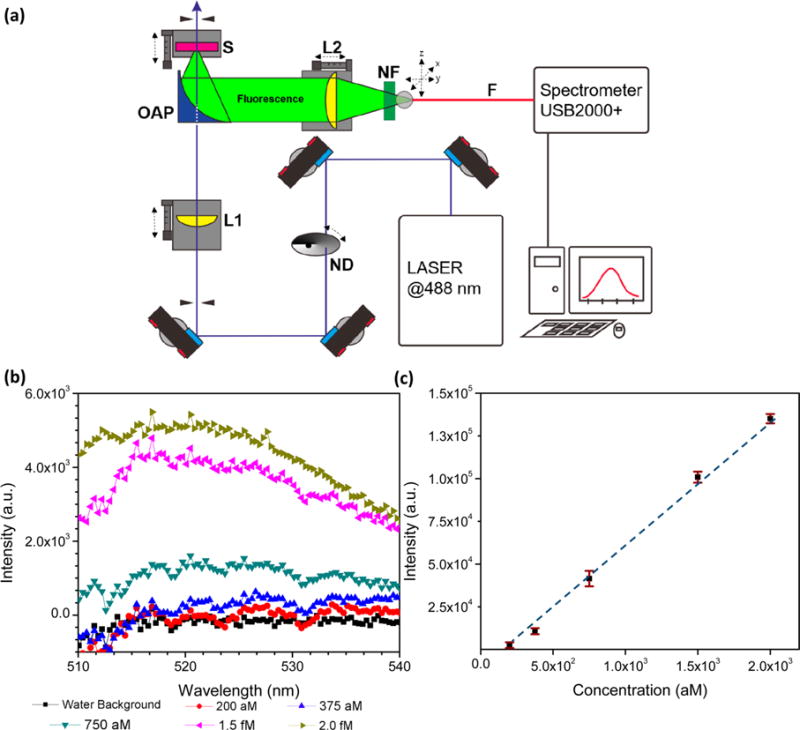
(a) Schematic of the optical-fiber-based fluorometer. Fluorescence signal is collected by the parabolic mirror and detected by a USB 2000 spectrometer. Apparatus: ND, Neutral-density filter; L1 and L2, lenses; OAP, off-axis parabolic mirror; S, sample; F, optical fiber; and NF, notch filter. (b) Uncorrected emission curve of barcode reporter vs various concentrations. The peak is centered at ∼520 nm. (c) Fluorescence intensity vs barcode reporter concentrations shows linear dependence at the attomolar level. The limit of detection of using pure fluorescence probe for the system is ∼200 aM. Error bars indicate standard error of the mean.
RESULTS
The protocol for on-chip sample preparation is outlined in Figure 1c. (i) First Ebola RNA spiked blood samples are pumped into the incubation reservoirs and mixed with fluorescence reporters, Probe A oligonucleotides, Ebola RNA targets, and photocleavable probes. (ii) After hybridization is complete, streptavidin beads are added into the incubation reservoirs to capture the biotin modified photocleavable probes. Through the process, fluorescence reporters, Probe A oligonucleotides, Ebola RNA targets, and photocleavable capture probes are hybridized and linked with magnetic beads. (iii) Magnet beads are isolated and washed to remove unbound targets, excess reagents, and contamination. (iv) Captured RNA targets are released from the magnetic beads for analysis by dissociating the photocleavable spacer in the capture probe.
To characterize the release efficiency of the photocleavable capture probes from the magnetic beads, an experiment was carried out mixing 0.85 picomoles photocleavable probes with 1 μL magnetic beads. After 2 h, the excess probes were removed and UV exposure was used to release the probes from the beads. Then, the supernatant was removed and diluted with 280 μL of RNase-free water. SYBR Gold dye (1×) was added to the supernatant for fluorescence detection. As shown in Figure 3a, distances of the UV lamp from the samples ranging from 0 to 10 cm and release times ranging from 0 to 20 min were examined. The release efficiency is very sensitive to the release time. The release efficiency increases dramatically from 5 to 10 min but plateaued after that. A NanoDrop Micro volume Fluorometer (Thermo Fisher Scientific) was also used to confirm that all the linked capture probes were released into the supernatant with the 20 min UV exposure.
Figure 3.
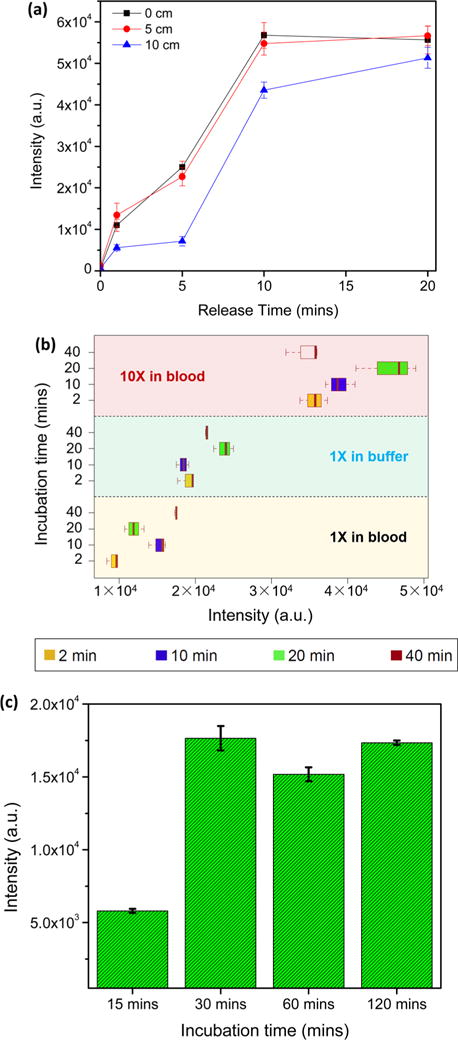
Optimization of solid-phase extraction process and assay. (a) Release efficiency of UV-cleavable probes vs release time with UV power ranging from ∼0.8 mW/cm2 (10 cm) to ∼1.6 mW/cm2 (0 cm). Error bars are standard error of the mean. (b) Capture efficiency results for 30 fM Ebola RNA (1× in buffer) as a function of incubation time (yellow box: 2 min; purple box: 10 min; green box: 20 min; red box: 40 min) compared with (1× in blood) and (10× in blood) target concentration procedure (1×: input 15 μL, release 15 μL; 10×: input 150 μL, release 15 μL). Error bars are confidence intervals. (c) Magnetic bead capture efficiency results as a function of incubation time for Ebola RNA-probe A-reporter tag-UV cleavable probe in blood. Error bars are standard error of the mean.
The probe hybridization conditions for Ebola RNA, probe A oligonucleotide, fluorescence reporter, and photocleavable probe were optimized by adjusting the incubation time as well as an on-chip concentration procedure. For all the experiments, 1 h bead incubation and 15 min UV release were employed after the probe hybridization at 50 °C. As presented in Figure 3b, incubation times of probes ranging from 2 to 40 min were explored (yellow box: 2 min; purple box: 10 min; green box: 20 min; red box: 40 min). The target capture efficiency improves somewhat by increasing the incubation time from 2 to 40 min. Magnetic bead adsorption on the PDMS walls and the glass substrate was observed in the 10× blood experiment, which likely explains the decrease of capture efficiency for 40 min incubation.28 A roughly 2-fold increase of the fluorescence signal was detected with the 1× buffer process (15 μL Ebola sample in RNase-free water input, 15 μL release) over the 1× blood process (15 μL of Ebola-spiked blood sample input, 15 μL release), regardless of the incubation time (blue and black label). The 10× blood protocol (red label) capture efficiency is ∼4× higher than the 1× blood protocol (black label) and ∼2× higher than the 1× buffer protocol (blue label); this represents capture of ∼20–25% of the total input Ebola RNA molecules.
After optimization of the probe hybridization, the capture efficiency was further examined by tuning the incubation time of hybridized probes with streptavidin beads at room temperature. Ebola RNA spiked blood, probe A oligonucleotide, fluorescence reporter, and photocleavable probe were mixed in the incubation reservoir at 50 °C for 20 min and then cooled down to room temperature. A total of 3 μL of streptavidin beads were added to the incubation reservoir and air bubbles were applied every 10 min. After incubation, the incubation reservoir was washed 3 times to eliminate all the impurities from blood followed by 15 min target release with UV exposure. As shown in Figure 3c, the bead-biotin capture efficiency is very sensitive to the incubation time. A 3-fold increase of the fluorescence signal was detected by increasing the incubation time from 15 to 30 min. After 30 min, the capture efficiency does not change significantly with incubation time which indicates that the biotin–streptavidin reaction is complete.
With the optimized solid-phase extraction conditions in hand, we determined the LOD and linearity for the 10× blood protocol. Ebola RNA spiked blood (150 μL), probe A oligonucleotide (1 μL), fluorescence reporter (5 μL), and photocleavable probe (1 μL) were mixed on-chip for 20 min at 50 °C followed by the bead incubation for 30 min at room temperature. During the incubation, 30 s air bubble mixing was applied every 10 min. The captured targets were UV released into 15 μL of RNase-free water. The measurements are presented in Figure 4. Due to the high specificity of the fluorescence reporter and photocleavable probe, no fluorescence peak counts were higher than RNase-free water background for the negative control sample (red). On the other hand, fluorescence peak counts show a linear dependence on concentration for positive Ebola samples over 3 orders of magnitude (black). We achieved a LOD of ∼800 aM combining the 10× on-chip concentration protocol and optical-fiber based fluorometer. The total processing and detection time was less than 90 min (20 min probe hybridization, 30 min bead incubation, 15 min automated washes, 15 min target release, and 5 min fluorescence detection).
Figure 4.
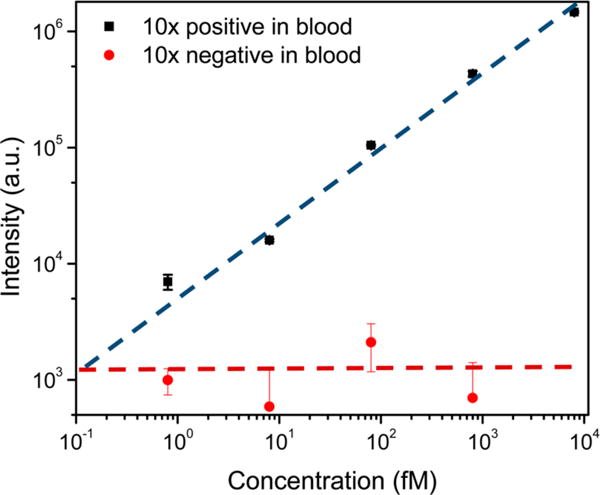
Detection of Ebola virus with optical-fiber based fluorometer. Calibration curve showing the relationship between the target concentrations and fluorescence intensity on a logarithmic scale. RNase-free water background signal was subtracted from both of the positive and negative signals. The limit of detection of the Ebola virus in blood with the 10× on-chip concentration protocol is ∼800 aM determined with a positive-to-negative signal ratio of ∼3:1. Error bars are standard error of the mean.
DISCUSSION
The SPM and analysis protocol presented here are designed to efficiently extract and sensitively detect viral RNA from blood all of which are necessary steps toward POC applications. Compared to our previous studies, the current approach has a number of advantages including higher throughput sample processing, higher specificity capture with two sequence specific probes binding to the RNA target, simpler target release, and faster solution phase hybridization.
In our previous work, a proprietary unsymmetrical cyanine dye was used to stain the Ebola RNA target for fluorescence detection.28,35,37 Although this dye shows a high fluorescence sensitivity, it binds nonspecifically to target RNA and to off-target RNA and DNA. When working with blood samples, a high concentration of contaminating RNA and DNA is often present. Also capture probes can bleed off from the magnetic beads in the thermal processing steps. Any irrelevant sequence in the assay leads to elevated background and degrades the LOD. In this paper, a sequence specific barcode fluorescence reporter is used to provide target sequence specific fluorescence labeling to enhance specificity and reduce background.
The use of a UV photocleavable capture probe enables a simpler and more efficient capture procedure. In our previous work, 4-formyl benzamine functionalized (4FB) beads were used for target capture and release.28 Although the covalent bond between 4FB and Hynic linker is more stable at the temperature (70 °C) used for target release, several prewashes are still required to reduce the capture probe loss. There were two problems with this previous method: The captured RNA targets were released from the magnetic beads by denaturing the RNA–DNA hybrid strands at 80 °C. Thus, a microheater and a micro temperature controller had to be added to monitor the temperature in the incubation reservoir for on-chip experiments. Furthermore, the 80 °C release still resulted in some capture probe loss and elevated background because the fluorescence labeling is not sequence specific. Our new protocol adds a photocleavable linker between the biotinylated linker and the capture probe that can be dissociated with a simple UV exposure. The results show that 15 min UV exposure is sufficient to cleave all the capture probe-Ebola RNA-fluorescence reporter hybridized strands from the magnetic beads. Thus, the new protocol requires less processing time and complexity. Because the barcode fluorescence reporter is on a separate hybridized probe, the presence of the other irrelevant probe sequences in the assay released by UV exposure does not increase the fluorescence background. In addition, the fluorescence reporter and the photocleavable capture probe each bind to a unique sequence in the 100-mer of the Ebola RNA target, thus our sample preparation protocol is more stringent, should work with degraded or short nucleic acid targets and does not require an ultra clean working environment. All these advantages will be helpful when working in low resource settings.
A second advantage of the new protocol is the performance of an initial solution phase hybridization. In our previous work, the hybridization of complementary strands was performed in solid phase with the capture probes immobilized on magnetic bead. The reaction rate was significantly decreased by the slow diffusion of targets to the beads. Additional devices such as an acoustic mixer,38 motor controlled rotational magnets,39 on-chip centrifuge,40 and metered air bubbles28 are utilized to improve the transport properties when dealing with beads. On the other hand, solution phase reactions are much faster.41 In this work, the hybridization of probes and Ebola RNA targets is in the liquid phase reducing diffusion barriers and the annealing process is rapid and specific even when the target concentration is very low. This solution phase hybridization step significantly improves the sample preparation efficiency.42
The SPM can analyze 80 unique samples in parallel with a pneumatic multiplexing architecture. This architecture enables independent control of 128 microvalves and 80 incubation reservoirs with only 16 solenoid valves. Nanoliter to microliter scale reagents are automatically transferred and mixed in the SPM by controlling the opening and close of the microvalves without using syringe pumps or pipettes. Furthermore, we demonstrated that metered air bubbles can be injected into the incubation reservoirs from the microvalves to enhance convective mixing without adding complicated instruments.28 This automated, programmable, and versatile system is ideal for POC analysis.
We also developed a sensitive fluorometer based on an off-axis parabolic (OAP) mirror and a mini USB spectrometer. The OAP mirror (numerical aperture ∼ 0.5, see Supporting Information) provides high fluorescence collection efficiency with a simple optical system. Combined with the high sensitivity of the barcode fluorescence reporter, a detection limit of ∼200 aM is achieved. The detection time is less than ∼3 min from 25 μL of 200 aM samples. The OAP-based fluorometer system is simple and robust and should be readily miniatured and integrated for POC applications.43
The barcode fluorescence reporter is designed to anneal fluorescent dyes with different colors on one M13 single-strand DNA and thus it can be excited with different wavelengths for multiplexing detection.44 The highest signal-to-noise (S/N) ratio was achieved by using 488 nm excitation but it is comparable with 590 and 633 nm excitations. Thus, the developed protocol can be further extended for quantitatively multiplexing measurements of various viruses and microorganisms in blood samples by digital color-coding.45,46
Combining all these advantages, our system can process multiple assays in parallel with improved sample process ease and sensitivity. Although polymerase chain reaction (PCR) shows similar throughput, it often requires an off-chip plasma separation47 and an RNA isolation step48 before target amplification. In addition, PCR requires precise temperature control and expensive and sensitive enzymes.49,50 Our technique only requires microliters of raw blood and does not need additional sample amplification steps. Using the 10× on-chip concentration protocol, our technique shows a detection limit of 800 aM for Ebola RNA targets, which is comparable with PCR analysis.51 This high sensitivity can provide crucial information for early detection by confirming or clearing suspected cases.52 Thus, our technique is compatible with the fingerprick test and is easy to carry out in resource-limited settings.53 With the optimized sample preparation conditions, the sample preparation and detection time is less than 1.5 h which is comparable with typical PCR testing.5 We also demonstrate a wide dynamic range from attomolar to picomolar scale which can be used to precisely monitor the seriousness of the infection. Exploiting these advances, we achieved the same detection limit as our previous work but with a faster detection time (30 min less) and the current approach can directly deal with raw blood samples. Although isothermal amplification techniques show higher sensitivity and is a useful diagnostic method for POC,7,8,54 we can further extend our detection limit to match isothermal amplification by exploiting 80× on-chip concentration as we have done previously.28 In addition, our amplification-free protocol can be extended for multiplexing detection which is challenging when performing isothermal amplification due to the interactions of the large number of primers in the assay.10
In the future, it should be possible to reengineer and integrate the current system to provide rapid and sensitive detection at POC without using expensive instruments. Since the SPM, fluorometer, and UV lamp are small in size and low cost, they can be integrated in a small instrument enabling real-time detection of viruses in blood. Because the sample processing and detection processes presented here are simple, once the reengineering is complete it should be facile to diagnostic suspected patients in a POC environment.55
Supplementary Material
Acknowledgments
We would like to thank D. Wartmann, H. Li, H. Cai, and A. Hawkins for valuable discussions. This research was supported by the NIH under Grants 4R33AI100229 and 1R21AI100229 and the NSF under Grant CBET-1159453 (H.S.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.7b03527.
Figure S1, Procedures of Ebola RNA purification and detection; Figure S2, Design of the OAP; Figure S3a,b, UVB power vs the distance to the SPM and schematic of the characterization procedure for UV release; Figure S4, Characterization of fluorescence reporter with spectrofluorometer (PDF).
ORCID
Ke Du: 0000-0003-2686-650X
Notes
The authors declare the following competing financial interest(s): Holger Schmidt has financial interest in Fluxus Inc. that is developing optofluidic devices.
References
- 1.Coarsey CT, Esiobu N, Narayanan R, Pavlovic M, Shafiee H, Asghar W. Crit Rev Microbiol. 2017;43:1–20. doi: 10.1080/1040841X.2017.1313814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zarei M. TrAC, Trends Anal Chem. 2017;91:26–41. [Google Scholar]
- 3.Shuaib F, Gunnala R, Musa EO, Mahoney FJ, Oguntimehin O, Nguku PM, Nyanti SB, Knight N, Gwarzo NS, Idigbe O. MMWR Morb Mortal Wkly Rep. 2014;63:867–872. [PMC free article] [PubMed] [Google Scholar]
- 4.Woolhouse ME, Rambaut A, Kellam P. Sci Transl Med. 2015;7:307rv305–307rv305. doi: 10.1126/scitranslmed.aab0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherpillod P, Schibler M, Vieille G, Cordey S, Mamin A, Vetter P, Kaiser L. J Clin Virol. 2016;77:9–14. doi: 10.1016/j.jcv.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 6.Drosten C, Göttig S, Schilling S, Asper M, Panning M, Schmitz H, Günther S. J Clin Microbiol. 2002;40:2323–2330. doi: 10.1128/JCM.40.7.2323-2330.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song J, Mauk MG, Hackett BA, Cherry S, Bau HH, Liu C. Anal Chem. 2016;88:7289–7294. doi: 10.1021/acs.analchem.6b01632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao S-C, Peng J, Mauk MG, Awasthi S, Song J, Friedman H, Bau HH, Liu C. Sens Actuators, B. 2016;229:232–238. doi: 10.1016/j.snb.2016.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deguo W, Guicheng H, Fugui W, Yonggang L, Daxi R. Afr J Food Sci. 2008;1:083–086. [Google Scholar]
- 10.Fischbach J, Frohme M, Glökler J. Sci Rep. 2017;7:7683. doi: 10.1038/s41598-017-08067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Granger JH, Schlotter NE, Crawford AC, Porter MD. Chem Soc Rev. 2016;45:3865–3882. doi: 10.1039/c5cs00828j. [DOI] [PubMed] [Google Scholar]
- 12.Henihan G, Schulze H, Corrigan DK, Giraud G, Terry JG, Hardie A, Campbell CJ, Walton AJ, Crain J, Pethig R. Biosens Bioelectron. 2016;81:487–494. doi: 10.1016/j.bios.2016.03.037. [DOI] [PubMed] [Google Scholar]
- 13.Culley S, Towers GJ, Selwood DL, Henriques R, Grove J. Viruses. 2016;8:201. doi: 10.3390/v8070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Towner JS, Rollin PE, Bausch DG, Sanchez A, Crary SM, Vincent M, Lee WF, Spiropoulou CF, Ksiazek TG, Lukwiya M. J Virol. 2004;78:4330–4341. doi: 10.1128/JVI.78.8.4330-4341.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de La Vega M-A, Caleo G, Audet J, Qiu X, Kozak RA, Brooks JI, Kern S, Wolz A, Sprecher A, Greig J. J Clin Invest. 2015;125:4421–4428. doi: 10.1172/JCI83162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahangar LE, Mehrgardi MA. Bioelectrochemistry. 2017;117:83–88. doi: 10.1016/j.bioelechem.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Tian S, Neumann O, McClain MJ, Yang X, Zhou L, Zhang C, Nordlander P, Halas NJ. Nano Lett. 2016;16:1478–1484. doi: 10.1021/acs.nanolett.5b05149. [DOI] [PubMed] [Google Scholar]
- 18.Ji X, Xiao C, Lau W-F, Li J, Fu J. Biosens Bioelectron. 2016;82:240–247. doi: 10.1016/j.bios.2016.04.022. [DOI] [PubMed] [Google Scholar]
- 19.Qin P, Parlak M, Kuscu C, Bandaria J, Mir M, Szlachta K, Singh R, Darzacq X, Yildiz A, Adli M. Nat Commun. 2017;8:14725. doi: 10.1038/ncomms14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altug H. Nano-Optics: Principles Enabling Basic Research and Applications. Springer; 2017. pp. 275–282. [Google Scholar]
- 21.Testa G, Persichetti G, Bernini R. Micromachines. 2016;7:47. doi: 10.3390/mi7030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song C, Tan SH. Micromachines. 2017;8:152. [Google Scholar]
- 23.Parks J, Schmidt H. Photonics Conference (IPC), 2016 IEEE. IEEE; 2016. pp. 370–371. [Google Scholar]
- 24.Lu X. Sensing Techniques for Food Safety and Quality Control: Sensing Techniques for Food Safety and Quality Control. Vol. 2 Royal Society of Chemistry; 2017. [Google Scholar]
- 25.Schmidt H, Hawkins AR. Bioanalysis. 2016;8:867–870. doi: 10.4155/bio-2016-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai H, Stott MA, Ozcelik D, Parks JW, Hawkins AR, Schmidt H. Biomicrofluidics. 2016;10:064116. doi: 10.1063/1.4968033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu K, Liu Q, Jackson KR, Landers JP. Sci Rep. 2016;6:22246. doi: 10.1038/srep22246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du K, Cai H, Park M, Wall T, Stott M, Alfson K, Griffiths A, Carrion R, Patterson J, Hawkins A. Biosens Bioelectron. 2017;91:489–496. doi: 10.1016/j.bios.2016.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim B, Oh S, You D, Choi S. Anal Chem. 2017;89:1439–1444. doi: 10.1021/acs.analchem.6b04587. [DOI] [PubMed] [Google Scholar]
- 30.Maan S, Maan NS, Belaganahalli MN, Potgieter AC, Kumar V, Batra K, Wright IM, Kirkland PD, Mertens PP. PLoS One. 2016;11:e0163014. doi: 10.1371/journal.pone.0163014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J, Stockton AM, Jensen EC, Mathies RA. Lab Chip. 2016;16:812–819. doi: 10.1039/c5lc01397f. [DOI] [PubMed] [Google Scholar]
- 32.Kim J, Kang M, Jensen EC, Mathies RA. Anal Chem. 2012;84:2067–2071. doi: 10.1021/ac202934x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcus JS, Anderson WF, Quake SR. Anal Chem. 2006;78:956–958. doi: 10.1021/ac0513865. [DOI] [PubMed] [Google Scholar]
- 34.Marcus JS, Anderson WF, Quake SR. Anal Chem. 2006;78:3084–3089. doi: 10.1021/ac0519460. [DOI] [PubMed] [Google Scholar]
- 35.Cai H, Parks JW, Wall TA, Stott MA, Stambaugh A, Alfson K, Griffiths A, Mathies RA, Carrion R, Patterson JL, Hawkins AR, Schmidt H. Sci Rep. 2015;5:14494. doi: 10.1038/srep14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ro YT, Ticer A, Carrion R, Patterson JL. Microbiol Immunol. 2017;61:130–137. doi: 10.1111/1348-0421.12475. [DOI] [PubMed] [Google Scholar]
- 37.Cai H, Stott M, Ozcelik D, Hawkins A, Schmidt H. Lasers and Electro-Optics (CLEO), 2016 Conference. IEEE; 2016. pp. 1–2. [Google Scholar]
- 38.Frommelt T, Kostur M, Wenzel-Schafer M, Talkner P, Hanggi P, Wixforth A. Phys Rev Lett. 2008;100:034502. doi: 10.1103/PhysRevLett.100.034502. [DOI] [PubMed] [Google Scholar]
- 39.Jackson K, Borba J, Meija M, Mills D, Haverstick D, Olson K, Aranda R, Garner G, Carrilho E, Landers J. Anal Chim Acta. 2016;937:1–10. doi: 10.1016/j.aca.2016.06.036. [DOI] [PubMed] [Google Scholar]
- 40.Jeon H, Kim D, Kim M, Nguyen X, Park D, Go J. J Micromech Microeng. 2015;25:114001. [Google Scholar]
- 41.Tijssen P. Lab Tech Biochem Mol Biol. 1993;24:375–436. [Google Scholar]
- 42.Söderlund H. Annales de Biologie Clinique. 1990:489–491. [PubMed] [Google Scholar]
- 43.Amselem E, Marklund E, Kipper K, Johansson M, Deindl S, Elf J. Biophys J. 2017;112:295a. [Google Scholar]
- 44.Tsang H-F, Xue VW, Koh S-P, Chiu Y-M, Ng LP-W, Wong S-CC. Expert Rev Mol Diagn. 2017;17:95–103. doi: 10.1080/14737159.2017.1268533. [DOI] [PubMed] [Google Scholar]
- 45.Fortina P, Surrey S. Nat Biotechnol. 2008;26:293–295. doi: 10.1038/nbt0308-293. [DOI] [PubMed] [Google Scholar]
- 46.Lin C, Jungmann R, Leifer AM, Li C, Levner D, Church GM, Shih WM, Yin P. Nat Chem. 2012;4:832–839. doi: 10.1038/nchem.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu C, Liao S-C, Song J, Mauk MG, Li X, Wu G, Ge D, Greenberg RM, Yang S, Bau HH. Lab Chip. 2016;16:553–560. doi: 10.1039/c5lc01235j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan SC, Yiap BC. BioMed Res Int. 2009;2009:574398. doi: 10.1155/2009/574398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marx V. Nat Methods. 2016;13:475–479. doi: 10.1038/nmeth.3868. [DOI] [PubMed] [Google Scholar]
- 50.Duan R, Zuo X, Wang S, Quan X, Chen D, Chen Z, Jiang L, Fan C, Xia F. Nat Protoc. 2014;9:597–607. doi: 10.1038/nprot.2014.036. [DOI] [PubMed] [Google Scholar]
- 51.Benzine JW, Brown KM, Agans KN, Godiska R, Mire CE, Gowda K, Converse B, Geisbert TW, Mead DA, Chander Y. J Infect Dis. 2016;214:S234–S242. doi: 10.1093/infdis/jiw330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Broadhurst MJ, Brooks TJ, Pollock NR. Clin Microbiol Rev. 2016;29:773–793. doi: 10.1128/CMR.00003-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson T, SU X-Z, Bockarie M, Lagog M, Day K. Parasitology. 1999;119:113–125. doi: 10.1017/s0031182099004552. [DOI] [PubMed] [Google Scholar]
- 54.Mori Y, Notomi T. J Infect Chemother. 2009;15:62–69. doi: 10.1007/s10156-009-0669-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gostin LO, Lucey D, Phelan A. Jama. 2014;312:1095–1096. doi: 10.1001/jama.2014.11176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.