

Abstract
Cholinergic afferents from the basal forebrain (BF) can influence cortical activity on rapid time scales, enabling sensory information processing and exploratory behavior. However, our understanding of how synaptically released acetylcholine (ACh) influences cellular targets in distinct cortical layers remains incomplete. Previous studies have shown that rapid changes in cortical dynamics induced by phasic BF activity can be mediated by the activation of nicotinic ACh receptors (nAChRs) expressed in distinct types of GABAergic interneurons. In contrast, muscarinic ACh receptors (mAChRs) are assumed to be involved in slower and more diffuse ACh signaling following sustained increases in afferent activity. Here, we examined the mechanisms underlying fast cholinergic control of cortical circuit dynamics by pairing optical stimulation of cholinergic afferents with evoked activity in somatosensory cortical slices of mice of either sex. ACh release evoked by single stimuli led to a rapid and persistent suppression of cortical activity, mediated by mAChRs expressed in layer 4 and to a lesser extent, by nAChRs in layers 1–3. In agreement, we found that cholinergic inputs to layer 4 evoked short-latency and long-lasting mAChR-dependent inhibition of the large majority of excitatory neurons, whereas inputs to layers 1–3 primarily evoked nAChR-dependent excitation of different classes of interneurons. Our results indicate that the rapid cholinergic control of cortical network dynamics is mediated by both nAChRs and mAChRs-dependent mechanisms, which are expressed in distinct cortical layers and cell types.
SIGNIFICANCE STATEMENT Acetylcholine (ACh) release from basal forebrain (BF) afferents to cortex influences a variety of cognitive functions including attention, sensory processing, and learning. Cholinergic control occurs on the time scale of seconds and is mediated by BF neurons that generate action potentials at low rates, indicating that ACh acts as a point-to-point neurotransmitter. Our findings highlight that even brief activation of cholinergic afferents can recruit both nicotinic and muscarinic ACh receptors expressed in several cell types, leading to modulation of cortical activity on distinct time scales. Furthermore, they indicate that the initial stages of cortical sensory processing are under direct cholinergic control.
Keywords: acetylcholine, interneuron, muscarinic, neocortex, nicotinic, synaptic
Introduction
Cholinergic afferents from the basal forebrain (BF) to neocortex play critical roles in a diverse set of cognitive functions such as attentional performance (Parikh et al., 2007; Herrero et al., 2008), learning (Letzkus et al., 2011), and sensory processing (Fu et al., 2014; Nelson and Mooney, 2016). Increases in cholinergic activity are strongly associated with changes in internal cortical state, defined by a switch from spontaneous, low-frequency, rhythmic activity to desynchronized activity. Such state changes can be global, slow and persistent, as during transitions from sleep to wakefulness (Brown et al., 2012). More recent studies have revealed much more rapid transitions in cortical dynamics within the awake state, allowing for the regulation of sensory processing and behavioral performance on the time scale of seconds (Crochet and Petersen, 2006; Reimer et al., 2014; McGinley et al., 2015a; Vinck et al., 2015). Mounting evidence suggests that BF cholinergic inputs trigger such rapid fluctuations of cortical state (Parikh et al., 2007; Eggermann et al., 2014; Reimer et al., 2016), predicting a high degree of specificity and precision in the underlying signaling mechanisms (Muñoz and Rudy, 2014).
BF cholinergic afferents target all cortical layers (Bloem et al., 2014; Wu et al., 2014) and the various effects of acetylcholine (ACh) are mediated by both nicotinic ACh receptors (nAChRs) and muscarinic ACh receptors (mAChRs) expressed in a layer- and cell-specific manner (Arroyo et al., 2012, 2014; Muñoz and Rudy, 2014; Hedrick and Waters, 2015; Hay et al., 2016; Verhoog et al., 2016; Obermayer et al., 2017). The signaling modes involved in the recruitment of nAChRs and mAChRs are assumed to be very distinct. Brief activation of cholinergic afferents can trigger fast-latency postsynaptic nAChR-mediated responses in both excitatory neurons (Hedrick and Waters, 2015; Hay et al., 2016; Nelson and Mooney, 2016) and different types of inhibitory interneurons (Letzkus et al., 2011; Arroyo et al., 2012; Fu et al., 2014). In turn, nAChR-mediated activation of interneurons can lead to either inhibition (Arroyo et al., 2012) or disinhibition (Letzkus et al., 2011; Fu et al., 2014) of pyramidal cell activity. The recruitment of mAChRs is thought to occur on much longer time scales. Although studies performed in vivo have shown that BF-mediated changes in cortical network properties are at least partly due to mAChR activation (Pinto et al., 2013; Eggermann et al., 2014; Kalmbach and Waters, 2014; Muñoz et al., 2017), the evidence for short-latency activation of mAChRs is sparse. Instead, mAChR recruitment appears to require sustained increases in cholinergic afferent activity, resulting in more gradual and diffuse changes in cortical dynamics (Descarries et al., 1997).
Here we determined the contributions of nAChRs and mAChRs to the fast cholinergic control of cortical circuits. Using an in vitro model of mouse barrel cortex and optogenetics, we temporally paired brief ACh increases with evoked neuronal population activity aimed to mimic slow and synchronous cortical activity patterns observed during quiet wakefulness (Crochet and Petersen, 2006; Eggermann et al., 2014). This paradigm allowed us to quantify the influence of ACh release on cortical responses with sufficient temporal precision while eliminating confounding influences present in vivo, such as changes in thalamic or other neuromodulatory inputs. We found that ACh release rapidly and reliably inhibited cortical network responses for several seconds. This inhibition was mediated in large part by the activation of mAChRs in layer 4, and to a lesser extent by the recruitment of nAChRs in the supragranular layers. In agreement, we found that synaptically released ACh produced long-lasting mAChR-mediated IPSCs in the majority of layer 4 excitatory neurons and nAChR EPSCs in superficial layer interneurons. Our findings reveal that mAChRs are critically involved in short-latency control of cortical network activity.
Materials and Methods
Animals.
We used bacterial artificial chromosome (BAC)-transgenic mice of either sex expressing ChR2 under the control of the choline acetyltransferase (ChAT) promoter (ChAT–ChR2-EYFP; Zhao et al., 2011). Animals were purchased from The Jackson Laboratory (https://www.jax.org/strain/014546) and maintained as hemizygous. This mouse line carries additional copies of the vesicular ACh transporter (VAChT) gene, potentially leading to enhanced release of ACh (Kolisnyk et al., 2013). Therefore, additional experiments were performed using ChAT-Cre/Ai32(ChR2-YFP) mice, generated by crossing ChAT-Cre animals (https://www.jax.org/strain/006410) with Cre-dependent reporter Ai32(ChR2-YFP) mice (https://www.jax.org/strain/012569), as described previously (Hedrick et al., 2016). Some experiments were performed using C57BL/6 wild-type mice. All animals used in this study were treated following procedures in accordance with National Institutes of Health guidelines and approved by the University of Texas Health Science Center at Houston (UTHealth) animal welfare committee.
Slice preparation.
Animals aged P12–P16 were anesthetized using isoflurane and then decapitated. The brains were rapidly removed and placed in ice-cold cutting solution saturated with 95% O2–5% CO2, which consisted of the following (in mm): 212 sucrose, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, 0.5 CaCl2, 26 NaHCO3, and 11 glucose. Thalamocortical slices (400 μm; Agmon and Connors, 1991) were cut using a vibratome (VT1200 S, Leica Biosystems) and immediately transferred to artificial CSF (ACSF; saturated with 95% O2–5% CO2), maintained at 35°C and consisting of the following (in mm): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose. Slices were incubated at 35°C for 20 min and then stored at room temperature until used for experiments.
Electrophysiology.
Electrophysiological recordings were performed in a recording chamber (RC-26GLP, Warner Instruments) perfused with ACSF saturated with 95% O2–5% CO2 and warmed to 31–34°C using an in-line heater connected to a temperature controller (TC-324B, Warner Instruments). Cells were visualized via infrared differential interference contrast using a fixed stage microscope (BX51WI, Olympus) equipped with an infrared camera (IR-1000, Dage-MTI). Recordings were acquired using an amplifier (MultiClamp 700B, Molecular Devices), filtered at 3–10 kHz, and digitized at 20 kHz with a 16-bit analog-to-digital converter (Digidata 1440A; Molecular Devices). For voltage-clamp recordings of glutamatergic or GABAergic activity in the absence of mAChR-dependent postsynaptic responses, glass pipettes (3–5 MΩ) were filled with a cesium-based internal solution consisting of the following (in mm): 120 CsMeSO3, 1 MgCl2, 1 CaCl2, 10 CsCl, 10 HEPES, 3 QX-314, 11 EGTA, 2 Mg-ATP, and 0.3 Na-GTP (adjusted to 295 mOsm, pH 7.3). For current-clamp recordings and voltage-clamp recordings of cholinergic postsynaptic responses, we used a potassium-based internal solution consisting of the following (in mm): 133 K-gluconate, 1 KCl, 2 MgCl2, 0.16 CaCl2, 10 HEPES, 0.5 EGTA, 2 Mg-ATP, and 0.4 Na-GTP (adjusted to 290 mOsm, pH 7.3). Where indicated, 5 mm BAPTA was included to block increases in intracellular calcium concentration.
Cortical activity was evoked using extracellular electrical stimuli (1–20 μA). Stimuli were generated using an isolated pulse stimulator (Model 2100, A-M Systems) and delivered via a glass electrode filled with ACSF. For some experiments, exogenous cholinergic agonists were applied using a Picospritzer (Parker Automation).
NBQX, DHβE, AF-DX 116, picrotoxin, CGP 55845, D-APV, and MLA were obtained from R&D Systems. All other chemicals were obtained from Sigma-Aldrich.
Optogenetics.
Cholinergic afferents were activated using 5 ms pulses of blue light using a LED light source (UHP-T-450-EP, Prizmatix) delivered through a 60×, 0.9 NA water-immersion objective (Olympus) with an effective illumination diameter of <250 μm. Light intensity was adjusted to ∼60 mW at the back aperture of the objective and kept constant throughout all experiments. During recordings, the objective was centered over the soma of the recorded neuron. For dual recordings of cells located in distinct cortical layers, postsynaptic responses for each cell were recorded sequentially.
Experimental design and statistical analyses.
In order to minimize response variability due to differences of ChR2 expression between animals, we first quantified cholinergic synaptic responses onto neurons in the thalamic reticular neurons (TRNs) for each animal (Sun et al., 2013). Neurons were recorded in voltage-clamp and cholinergic afferents were activated locally with individual pulses (0.5 ms) of constant intensity, as described above. If nicotinic EPSCs had amplitudes <50 pA, ChR2 expression was considered too low and slices were not used for experiments. A fraction of ChAT-Cre/Ai32(ChR2-YFP) animals show ChR2 expression in glutamatergic neurons (Hedrick et al., 2016). For TRN recordings, such ectopic expression resulted in light-evoked fast EPSCs and slices were not further considered for experiments.
Data were analyzed using custom macros written in IGOR Pro (WaveMetrics). Statistical tests were performed in Prism 5 (GraphPad). Evoked recurrent activity recorded in voltage-clamp was quantified as charge transferred to the recorded cell, by calculating the area under the PSC trace in a time window starting 90 ± 3 ms after the first electrical pulse, and ending when evoked activity returned to baseline. For a given cell, the same time window was used for paired and unpaired trials. To account for changes in response magnitudes in unpaired trials over the course of pharmacological experiments, responses recorded in paired trials in a given drug condition were normalized to responses recorded in unpaired trials in the same drug condition and time period. Decay time constants of postsynaptic cholinergic currents were determined by fitting single exponential functions to responses averaged over >10 trials. Unpaired comparisons were performed using the two-tailed unpaired t test. Paired comparisons were made using the Wilcoxon signed rank test or paired Student's t test. Differences were considered significant when p < 0.05. Data are shown as mean ± SEM.
Results
Synaptic release of ACh suppresses evoked cortical activity
We investigated the role of cholinergic synaptic signaling in regulating cortical activity by using optogenetic techniques in somatosensory (barrel) cortical slices of ChAT-ChR2-EYFP mice expressing ChR2 in cholinergic neurons (Zhao et al., 2011). Where indicated, experiments were performed in slices derived from ChAT-Cre/Ai32(ChR2-YFP) mice (Hedrick et al., 2016). Cortical activity was evoked by applying brief stimulus bursts (4 stimuli, 40 Hz) delivered through extracellular glass electrodes placed in layer 4. To monitor activity, we targeted layer 2/3 pyramidal cells in the same cortical column and performed voltage-clamp recordings using a Cs-based internal solution (Fig. 1A). Stimulus bursts generated postsynaptic responses consisting of short-latency monosynaptic EPSCs with little latency jitter, as well as long-latency polysynaptic activity (onset: 45.7 ± 6 ms, duration: 678.9 ± 50.9 ms, n = 19 cells), which displayed considerable jitter from trial-to-trial (Fig. 1B). Stimulus intensity was adjusted to reliably evoke polysynaptic activity for the majority of trials (90.3 ± 3%, n = 19 cells) in a given recording. Because polysynaptic responses are mediated by recurrent excitatory connections in local cortical networks, we will refer to these responses as recurrent activity (quantified as EPSC charge transfer; see Materials and Methods). To examine fast cholinergic modulation of recurrent activity, we paired extracellular stimulation in layer 4 with single light pulses (5 ms duration), to mimic the phasic discharge pattern of BF cholinergic neurons observed in vivo (Lee et al., 2005; Hangya et al., 2015). Light pulses were centered on the recorded neuron and applied 15 ms before the onset of stimulus bursts. This led to a reliable and repeatable suppression of recurrent activity (29.8 ± 0% compared with unpaired trials, n = 19 cells, p < 0.001, Wilcoxon signed rank test; Fig. 1B–E). Similar findings were obtained for ChAT-Cre/Ai32(ChR2-YFP) mice (17.7 ± 0% compared with unpaired trials, n = 2 cells). For neurons recorded in current-clamp, optical stimulation led to a reduction of spiking activity (36.4 ± 11% compared with unpaired trials, n = 6 cells, p = 0.01, Wilcoxon signed rank test; data not shown). In contrast, monosynaptic EPSCs evoked by the first two stimuli were unaffected by optical stimulation (100.9 ± 2% compared with unpaired trials, n = 19, p = 0.5, two-tailed paired t test; Fig. 1F). Next, we tested whether cholinergic signaling equally reduced activity in local inhibitory neuronal networks by simultaneously recording EPSCs and IPSCs in neighboring neurons, voltage-clamped at −70 and 0 mV, respectively (Fig. 1G,H). Across cell pairs, recurrent activity under paired conditions ranged from 71.1 to 1.2% compared with unpaired trials, with suppression of excitatory and inhibitory activity being virtually identical for a given cell pair (r2 = 0.98; Fig. 1H). Thus, cholinergic signaling did not alter the balance of synaptic excitation and inhibition in layer 2/3 during recurrent activity. Together, our data indicate that brief activation of cholinergic afferents reliably suppresses recurrent activity in cortical networks.
Figure 1.
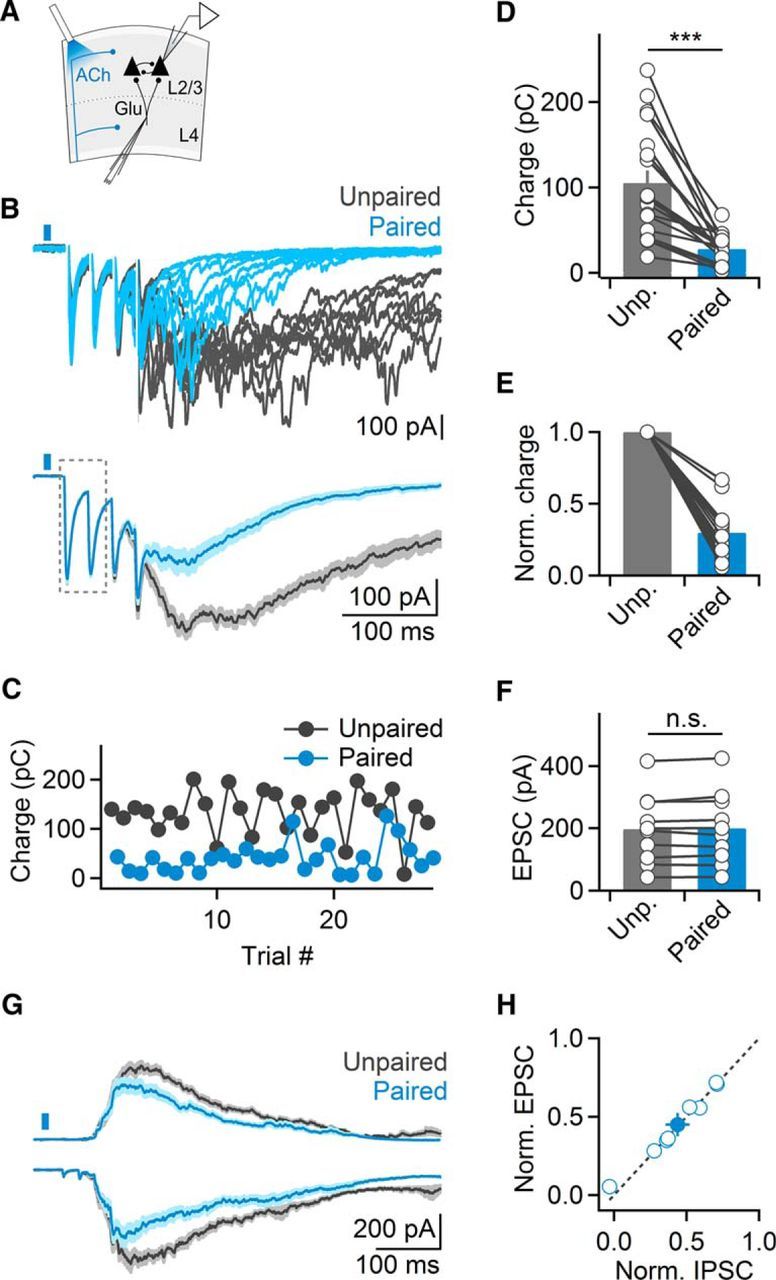
ACh release evoked by single light pulses suppresses evoked cortical recurrent activity. A, Schematic of experimental setup. Cortical recurrent activity was evoked using brief bursts of extracellular stimuli applied in layer 4 and was recorded in layer 2/3 neurons in voltage-clamp. Cholinergic afferents were activated using single light pulses (5 ms), 15 ms before electrical stimulation. B, Top, Representative recording showing multiple trials of recurrent activity, in the absence of (unpaired, black traces) or following optical stimulation (paired, blue traces). Bottom, EPSCs averaged across all unpaired and paired trials. Note lack of amplitude reduction of monosynaptic EPSCs (outlined). C, For the same cell shown in B, plot depicts recurrent activity (quantified as EPSC charge transfer), in paired trials (blue) alternated with unpaired trials (black). D, Summary data showing light-evoked suppression of recurrent activity in layer 2/3 neurons (n = 19 cells). ***p < 0.001. E, Same data as in D, normalized to unpaired responses. F, Summary data showing average amplitude of monosynaptic EPSC evoked by the first two stimuli (n = 10 cells) for unpaired and paired trials. G, Recurrent activity recorded as EPSCs and IPSCs (black, unpaired; blue, paired) from pairs of neighboring layer 2/3 cells, held at −70 and 0 mV, respectively. H, Summary data plotting normalized suppression of EPSCs and IPSCs, for all cell pairs (n = 8). Shaded areas and error bars denote SEM.
Cholinergic suppression is largely mediated by mAChRs
Both nAChRs and mAChRs are expressed in different types of neocortical neurons (Muñoz and Rudy, 2014), but how these receptors are activated by endogenous ACh to mediate cholinergic control of cortical circuits is not well understood. We found that bath application of atropine to block mAChRs significantly reduced cholinergic suppression (paired: 36.7 ± 5% compared with unpaired trials, atropine: 77.9 ± 4% compared with unpaired trials in atropine, n = 10, p < 0.01, Wilcoxon signed rank test; Fig. 2A,B), indicating that ACh increases evoked by single light pulses can recruit mAChRs. Atropine application led to a small increase in recurrent activity in unpaired trials (118.7 ± 9% compared with control, n = 15, p = 0.03, Wilcoxon signed rank test; Fig. 2B), suggesting that cortical activity is also controlled via persistent activation of mAChRs. To determine whether this reduction was due to enhanced levels of ambient ACh in our transgenic mouse model (Kolisnyk et al., 2013), we repeated these experiments in slices derived from wild-type animals. Bath application of atropine still led to an increase in recurrent activity, although this effect did not reach statistical significance (120 ± 11% compared with control, n = 10, p = 0.07, Wilcoxon signed rank test). This suggests that persistent activation of mAChRs might not be limited to ChAT-ChR2-EYFP mice.
Figure 2.
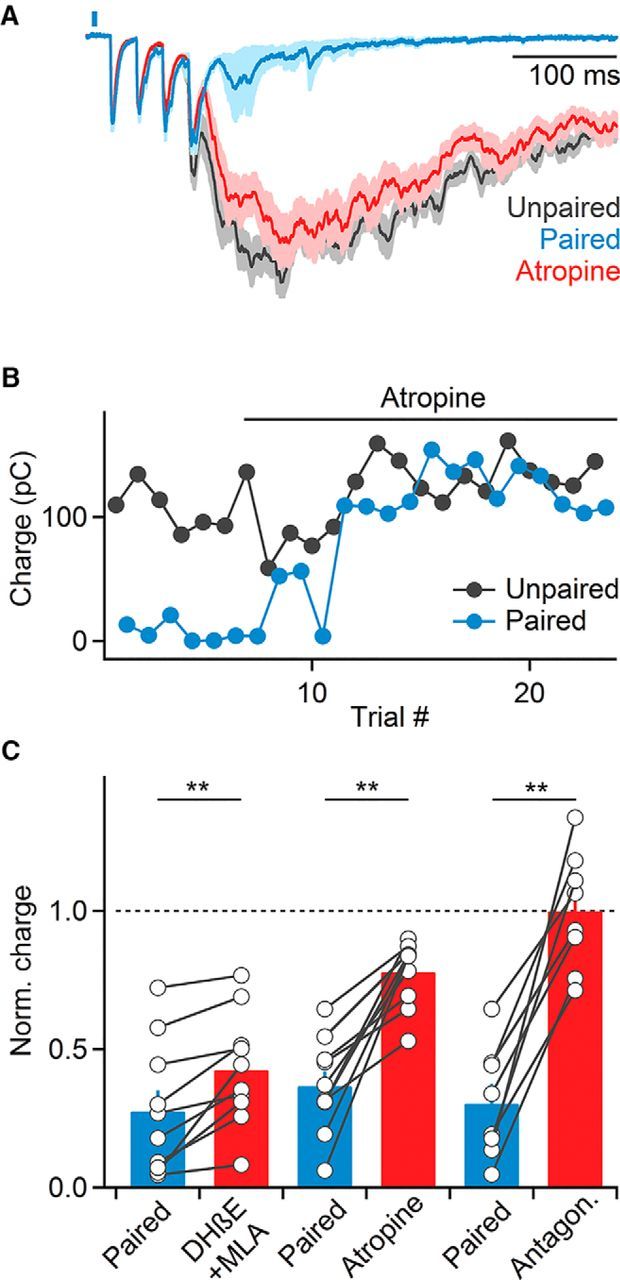
Cholinergic suppression of recurrent activity is mediated by both nAChRs and mAChRs. A, Voltage-clamp recordings from a representative layer 2/3 cell showing that bath application of the mAChR antagonist atropine (10 μm) largely blocks cholinergic suppression of recurrent activity (blue, normalized EPSCs in paired control trials; red, normalized average EPSCs in paired trials following atropine application; gray, EPSCs in unpaired trials). B, Magnitude of recurrent activity for the same cell across unpaired (black) and paired (blue) trials during atropine application. C, Summary data of recurrent activity (normalized to activity in unpaired trials over the same time period) before and after bath application of nAChR antagonists (500 nm DHβE + 5 nm MLA, n = 10 cells), atropine (10 μm Atr, n = 10 cells), or both (n = 8 cells). **p < 0.01. Shaded areas and error bars denote SEM.
Compared with the effects of blocking mAChRs, wash-in of MLA and DHβE to block α7 and non-α7 nAChRs, respectively, led to a smaller but significant reduction of cholinergic suppression (paired: 27.5 ± 7% compared with unpaired trials; MLA and DHβE: 42.5 ± 6%, n = 10, p < 0.01, Wilcoxon signed rank test; Fig. 2C). Furthermore, MLA and DHβE application did not lead to an increase in recurrent activity in unpaired trials (96.4 ± 7% compared with control, n = 7, p = 0.25, Wilcoxon signed rank test) suggesting that tonic activation of nAChRs is not prominent.
In addition to evoking ACh release, optical stimuli might lead to the liberation of GABA from BF afferents (Saunders et al., 2015) or from neocortical ChAT-positive GABAergic neurons (von Engelhardt et al., 2007), which express ChR2 in our transgenic mouse lines. However, we found that the combined application of both mAChR and nAChR antagonists completely eliminated suppression of recurrent activity (control: 30.4 ± 7% compared with unpaired trials; antagonists: 99.9 ± 8%, n = 8, p < 0.01, Wilcoxon signed rank test; Fig. 2C), suggesting that light-evoked effects on recurrent activity were exclusively mediated by ACh.
Transient ACh increases lead to prolonged suppression of recurrent activity
The crucial role of mAChRs in the suppression of recurrent activity predicts that BF-evoked suppression should be long-lasting. To examine this possibility, we progressively increased the delay between optical activation of cholinergic afferents and extracellular stimulation to evoke recurrent activity. Suppression of recurrent activity was maximal for delays of 1 and 2 s and remained robust even at 5 s delays, with delays of 8 s no longer yielding significant reductions in activity (Fig. 3A,B).
Figure 3.

Synaptic recruitment of mAChRs mediates sustained suppression of recurrent activity. A, Representative recording showing normalized EPSCs during unpaired (black) and paired (blue) trials, for a range of temporal delays between optical and electrical stimulation (15–8000 ms). B, Summary data quantifying light-evoked suppression of recurrent activity (normalized to responses in unpaired trials) as a function of temporal delay between and electrical stimulation (n = 5 cells). Summary data were fit by a third order polynomial (χ2 = 0.013). C, Representative recording showing that for 5 s delays between optical and electrical stimulation, suppression of recurrent activity (blue) was entirely reversed by bath application of atropine. D, Summary data showing elimination of light-evoked suppression of recurrent activation following bath application of either atropine or 10 μm AF-DX 116 (circles: atropine, n = 7 cells; triangles: AF-DX 116, n = 4 cells), for experiments as shown in C. **p < 0.01. Shaded areas and error bars denote SEM.
A strong reduction of recurrent activity several seconds after the release of ACh does not appear to be compatible with a role for nAChRs. Indeed, for experiments with delays of 5 s between optical and electrical stimulation, bath application of atropine or the M2/M4 mAChR antagonist AF-DX 116 completely eliminated cholinergic suppression (control: 46.1 ± 8% suppression, atropine/AF: 99.2 ± 5% suppression, n = 11, p < 0.01, Wilcoxon signed rank test; Fig. 3C,D). Thus, nAChRs and mAChRs mediate cholinergic suppression of recurrent activity on distinct timescales, with nAChRs mediating transient reduction and mAChRs being responsible for long-lasting reduction of cortical activity.
Cholinergic suppression via mAChRs is prominent in layer 4
Next, we tested whether the contributions of nAChRs and mAChRs to cholinergic suppression could be localized to distinct cortical layers. To address this question, we surgically removed layers 1–3 by performing cuts parallel to the pial surface just above layer 4, and performed recordings from layer 4 neurons deemed excitatory (Fig. 4A). Extracellular stimulation applied to the same barrel still led to recurrent activity, but with reduced magnitude (uncut slice: 105 ± 15 pC, n = 19, layer 4–6 slice: 54.9 ± 8 pC, n = 15). Furthermore, we still observed robust light-evoked suppression of recurrent activity (38.5 ± 5% compared with unpaired trials, n = 15, p < 0.001, Wilcoxon signed rank test; Fig. 4B,C). However, in contrast to our findings in intact slices, atropine almost completely reversed cholinergic suppression (control: 35.4 ± 7% compared with unpaired trials, atropine: 92.8 ± 4%, n = 6, p = 0.01, Wilcoxon signed rank test; Fig. 4B,C), whereas application of MLA and DHβE to block nAChRs no longer reduced cholinergic suppression (control: 35.1 ± 6% compared with unpaired trials, MLA and DHβE: 30.2 ± 5%, n = 6, p = 0.23, Wilcoxon signed rank test; Fig. 4D,E). Furthermore, increasing the delay between optical and extracellular stimuli to 5 s still led to atropine-sensitive suppression of recurrent activity (control: 46.4 ± 8% compared with unpaired trials, atropine: 124.6 ± 27%, n = 5, p = 0.02, Wilcoxon signed rank test; Fig. 4F,G). These data indicate that cholinergic inputs to layers 4–6 can mediate robust and long-lasting mAChR-mediated suppression of cortical activity. Furthermore, they suggest that the nAChR-dependent suppression of network activity primarily occurs in more superficial layers. However, it is possible that severing dendrites and translaminar projections eliminated the contributions of nAChR activation in deeper cortical layers.
Figure 4.
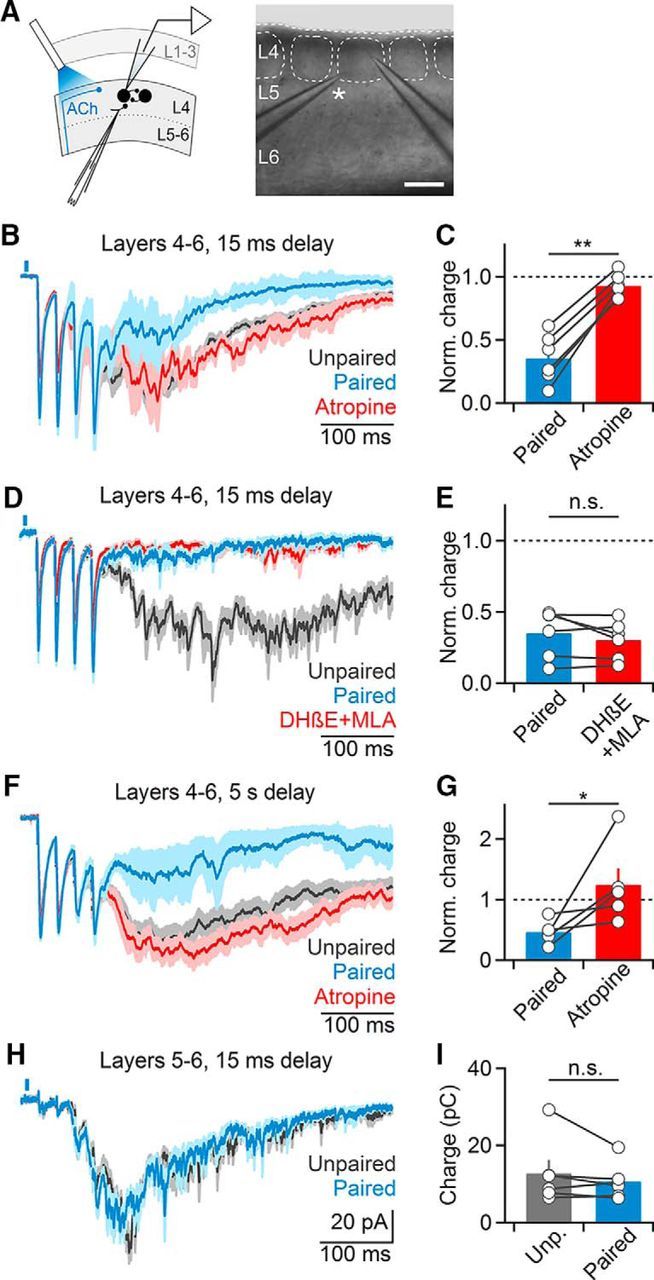
Cholinergic suppression of recurrent activity is layer-specific. A–G, Recordings were performed in slices following surgical removal of layers 1–3. A, Left, Schematic indicating recording arrangement. Right, Bright-field image of slice preparation. Asterisk denotes stimulating electrode. Scale bar, 150 μm. B, Representative recording of layer 4 neuron showing that cholinergic suppression of recurrent activity is entirely mAChR-mediated. C, Summary data (n = 6 cells) showing complete reversal of cholinergic suppression following atropine application. **p < 0.01. D, Representative recording showing that nAChR antagonist application no longer reduces cholinergic suppression (n = 4 cells, DHβE alone: n = 2, DHβE+MLA: n = 2). E, Summary data (n = 5 cells) for experiments as shown in D. F, Cholinergic suppression of recurrent activity is maintained for long delays (5 s) between optical and electrical stimuli and mediated by mAChRs. G, Summary data (n = 5 cells) for experiments as shown in F. *p = 0.02. H, I, Surgical removal of layers 1–4 eliminates cholinergic suppression. Recordings were performed from layer 5 neurons and activity was evoked in the white matter below the same column. H, Representative recording showing EPSCs averaged across paired and unpaired trials. I, Summary data (n = 6 cells) for recordings as shown in H. All shaded areas and error bars denote SEM.
To further constrain the location of mAChR-mediated suppression, we performed recordings from layer 5 pyramidal neurons in slices with layers 1–4 surgically removed, and evoked recurrent activity using electrodes placed in the white matter. The magnitude of recurrent activity was further reduced under these conditions (uncut slice: 105 ± 15 pC, n = 19, layer 5–6 slice: 12.8 ± 3 pC, n = 6). Importantly, optical stimulation no longer reduced recurrent activity (90.9 ± 8% compared with unpaired trials, n = 6, p = 0.12, Wilcoxon signed rank test; Fig. 4H,I), suggesting that fast synaptic ACh release in the infragranular layers is not involved in the control of cortical activity, at least under our experimental conditions.
Together, our findings show that the contributions of nAChRs and mAChRs to the suppression of network activity are not uniform across cortical layers. Instead, they indicate that nAChR-dependent suppression is primarily mediated by layers 1–3, whereas mAChR-dependent suppression is particularly prominent in layer 4.
Cholinergic postsynaptic responses are cell-type and layer-specific
Our results described so far are consistent with the activation of nAChRs expressed in layer 2/3 GABAergic interneurons, leading to transient suppression of cortical activity. In addition, they suggest a strong recruitment of mAChRs in layer 4, resulting in a long-lasting depolarization of GABAergic interneurons, a long-lasting inhibition of excitatory neurons, or both. Next, we performed recordings from neurons in layers 1–4 using a K+-based recording solution and determined the nature and frequency of light-evoked postsynaptic responses in different cell types. Neurons were classified as either regular-spiking (RS) cells considered excitatory, or as fast-spiking (FS) or non-fast-spiking (non-FS) cells considered inhibitory, based on their intrinsic firing properties (Beierlein et al., 2003; Fig. 5A). In agreement with previous findings (Arroyo et al., 2012), neurons in layer 1 showed nAChR-mediated EPSCs (nEPSCs; 11/12 neurons) that were fully blocked by a combination of MLA and DHβE (data not shown). In layer 2/3, a large percentage of inhibitory interneurons displayed nEPSCs that were blocked by DHβE (FS: 39%, non-FS: 77%; Fig. 5B,C), although a minority of neurons displayed long-lasting mAChR-dependent currents (FS: 23%, non-FS: 5%; Fig. 5B,C). To confirm the existence of functional mAChRs in non-FS neurons as shown previously (Chen et al., 2015), we used a Picospritzer to apply brief puffs of muscarine. For all neurons examined (n = 9), which showed a light-evoked nEPSP only, muscarine application led to a robust depolarization, which was blocked by atropine (Fig. 6). These data indicate that although mAChRs are expressed in layer 2/3 non-FS neurons, they do not appear to be recruited by brief activation of cholinergic afferents.
Figure 5.
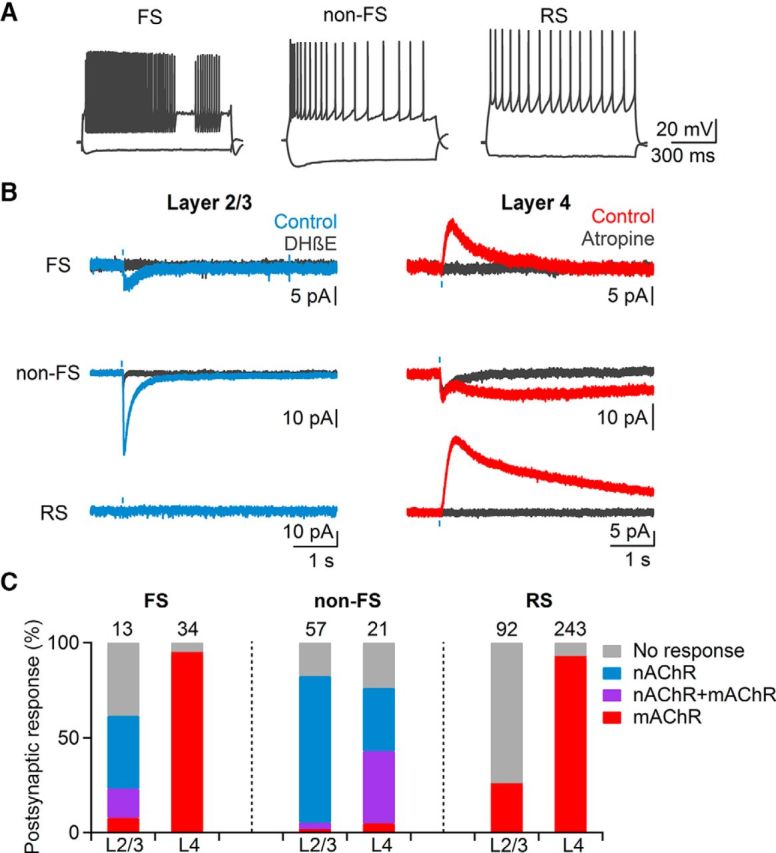
Cholinergic postsynaptic responses are layer-specific. A, Cells in layer 2/3 and layer 4 were classified as either inhibitory FS, or non-FS cells or excitatory RS cells based on their intrinsic firing characteristics. B, Example voltage-clamp recordings carried out in the presence of NBQX (10 μm), D-APV (25 μm), picrotoxin (50 μm), and CGP 55845 (5 μm) showing typical light-evoked responses. Most cells in layer 2/3 (left column) showed either no response or fast EPSCs (blue traces) blocked by DHβE (black traces). In layer 4 (right column) the majority of neurons displayed slow postsynaptic responses (red traces) that were blocked by atropine (black traces). C, Summary data showing likelihood of nAChRs and mAChR-mediated responses for each cell type. Numbers above bars indicate total number of cells recorded.
Figure 6.

Synaptic release of ACh primarily recruits nAChRs in layer 2/3 non-FS neurons. A, Responses to hyperpolarizing and depolarizing current steps in a representative layer 2/3 non-FS neuron. B, Average response in current-clamp from layer 2/3 non-FS cells (n = 4) to 5 ms optical activation (blue bar) followed by a 200 ms puff of muscarine chloride (1 mm; red bar). C, Summary data (n = 4 cells), showing that responses evoked by muscarine were completely blocked by atropine. Shaded areas and error bars denote SEM.
In contrast to interneurons, most RS cells in layer 2/3 did not show light-evoked postsynaptic responses (75%; Fig. 5B,C), with the remaining neurons displaying small-amplitude mAChR-dependent IPSCs (mIPSCs; 25%). Together, these findings indicate that the synaptic release of ACh in superficial layers can control cortical activity via the recruitment of nAChRs in distinct types of interneurons.
Recordings in layer 4 yielded dramatically different results. Almost all FS cells displayed atropine-sensitive mIPSCs (94%; Fig. 5B,C), and never showed nAChR-dependent responses. Non-FS interneurons responding to ACh release displayed either isolated nEPSCs (33%), or biphasic responses consisting of nEPSCs and mAChR EPSCs (mEPSCs; 38%; Fig. 5B,C). Furthermore, the large majority of RS cells showed mIPSCs (92%) that were blocked by atropine and AF-DX 116. mAChR-mediated responses were also detected in slices derived from ChAT-Cre/Ai32(ChR2-YFP) mice (FS: n = 2 cells, non-FS: n = 1 cell, RS: n = 29 cells). Postsynaptic mAChR-dependent responses displayed large cell-type-specific differences in their kinetics. Whereas mIPSCs in FS had relatively fast kinetics (rise time: 165.9 ± 10 ms, decay time constant: 844.2 ± 78 ms, n = 20), mIPSCs in RS cells were considerably slower (rise time: 328.3 ± 23 ms, decay time constant: 3281.7 ± 157 ms, n = 21). For non-FS cells, mEPSCs displayed extremely slow kinetics (rise time: 1248.3 ± 125 ms, decay time constant: 23.7 ± 5.4 s, n = 7). Thus, the recruitment of mAChRs via brief ACh transients can control postsynaptic activity on dramatically different time scales, depending on the cell type.
Our data suggest that excitatory neurons in layer 4 are much more likely to receive cholinergic inputs compared with excitatory neurons in layer 2/3. Next, we compared the strength of cholinergic postsynaptic responses in excitatory neurons located in distinct layers. To account for postsynaptic response variability due to differences of ChR2 expression between slices and animals, we performed dual recordings from RS neurons in layer 4 and layer 2/3 or layer 4 and layer 5 in the same cortical column. For almost all pairs examined, mIPSC amplitudes in layer 4 were larger compared with responses in either layer 2/3 or layer 5 (layer 2/3: 21.5 ± 10% compared with layer 4, n = 11 pairs, p < 0.001; layer 5: 16.0 ± 5% compared with layer 4, n = 14 pairs, p < 0.0001, two-tailed paired t test; Fig. 7A,B). Finally, mIPSC amplitudes in layer 4 RS cells were indistinguishable between the two transgenic mouse lines (Cre/Ai32(ChR2-YFP): 9.0 ± 1.0 pA, n = 8 cells; ChAT-ChR2-EYFP: 11.0 ± 1.0 pA, n = 23 cells, p = 0.38, two-tailed unpaired t test) suggesting that VAChT overexpression does not lead to a significant enhancement of response amplitudes.
Figure 7.

mIPSCs in RS cell are strongest in layer 4 and are mediated by GIRK conductances. Recordings were obtained in the presence of antagonists for GABAergic and glutamatergic synaptic transmission. A, Left, Average mIPSCs for simultaneously recorded pairs of layer 2/3 and layer 4 neurons (n = 11 pairs). Right, Summary data (11 layer 2/3 and 4 pairs). ***p < 0.001. B, Left, Average mIPSCs for simultaneously recorded pairs of layer 4 and 5 neurons (n = 14 pairs). Right, Summary data (14 layer 4 and 5 pairs). ***p < 0.001. C, Left, Light-evoked mIPSC in a layer 4 RS cell was blocked following Ba2+ (200 μm) application. Right, Summary data quantifying mIPSC reduction following Ba2+ application (n = 4 cells). *p = 0.05. D, Left, Application of the SK channel antagonist apamin (10–100 nm) has no effect on mIPSCs, as shown for layer 4 RS neuron. Right, Summary data quantifying mIPSC responses before and following apamin wash-in (circles: 10 nm, n = 3 cells; triangles: 100 nm, n = 3 cells). All shaded areas and error bars denote SEM.
Next, we probed the mechanisms mediating mIPSCs in layer 4 RS neurons. Synaptic currents had onset latencies of 30.6 ± 1 ms (n = 19 cells), reversed at ∼−96 mV, displayed strong inward rectification and could be blocked by bath application of barium (15.11 ± 2% of control, n = 4 cells, p = 0.05, two-tailed paired t test; Fig. 7C), indicating that the mIPSCs were mediated by G-protein-coupled inwardly-rectifying potassium (GIRK) conductances. By contrast, bath application of the small conductance calcium-activated potassium (SK) channel antagonist apamin had little effect on mIPSC amplitudes (93.0 ± 4% of control, n = 6 cells, p = 0.09, two-tailed paired t test; Fig. 7D) and recordings using an internal solution containing 5 mm BAPTA did not attenuate mIPSCs (n = 4 cells; data not shown), suggesting that SK channel activation is not involved in mediating mIPSCs in layer 4 neurons.
Together, our data show that the synaptic release of ACh in layer 4 leads to the recruitment of mAChRs in all major cell types including the large majority of RS cells, suggesting that the monosynaptic inhibition of excitatory neurons contributes to the prolonged suppression of recurrent activity.
Synaptic ACh reduces neuronal firing in layer 4 RS cells via hyperpolarizing inhibition
Next, we determined the impact of light-evoked mAChR IPSPs (mIPSPs) on postsynaptic action potential activity in RS cells, in the absence of recurrent activity. For RS neurons held at −70 mV, mIPSPs had amplitudes of 2.9 ± 0 mV and decay time constants of 5140.5 ± 427 ms (n = 25). When mIPSPs were paired with action potential firing evoked by depolarizing current steps, firing frequencies were rapidly (<100 ms) and persistently reduced compared with unpaired trials (n = 11; Fig. 8A–C). To examine cholinergic control under more physiological conditions, we paired optical stimulation with action potential activity (Δt = 1 s) evoked by extracellular stimulation of glutamatergic afferents (4 stimuli at 40 Hz). For these experiments, we added the NMDAR antagonist APV (25 μm) to block recurrent activity and to isolate fast monosynaptic responses (Beierlein et al., 2002). Light-evoked mIPSPs reduced synaptically-evoked action potential activity (57.9 ± 5% compared with unpaired trials, n = 10 cells, p < 0.01, Wilcoxon signed rank test; Fig. 9A–C). To test whether these effects were mediated in part by a reduction of glutamate release, we paired glutamatergic and cholinergic inputs but performed voltage-clamp recordings using a Cs-based internal solution to block postsynaptic mIPSCs. Glutamatergic EPSCs were slightly but not significantly reduced (91.5 ± 2% compared with unpaired trails, n = 8 cells, p = 0.13, two-tailed paired t test). These data suggest that cholinergic inputs trigger a rapid and long-lasting reduction of layer 4 RS cell activity by activating postsynaptic mAChRs.
Figure 8.
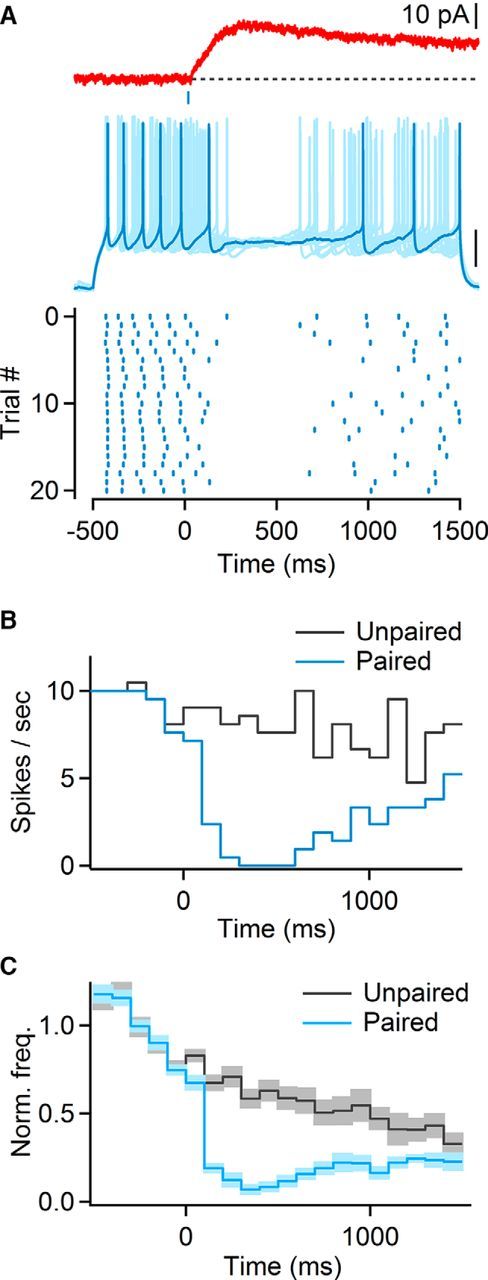
Cholinergic synaptic inputs to layer 4 reduce neuronal firing in RS cells. A, Top, mIPSC in layer 4 RS cell. Middle, Multiple overlaid trials showing neuronal activity evoked by 2 s depolarizing current step paired with light stimulation applied with a 500 ms delay (indicated by blue bar). Scale bar, 20 mV. Bottom, Raster plot showing timing of action potentials over multiple trials, for the same neuron. B, For the same cell as in A, peristimulus time histogram (PSTH) for paired (blue) and unpaired (gray) current steps. C, Average PSTH (n = 11 RS cells), normalized to the average firing frequency over a 500 ms window before light stimulation. Shaded areas denote SEM.
Figure 9.
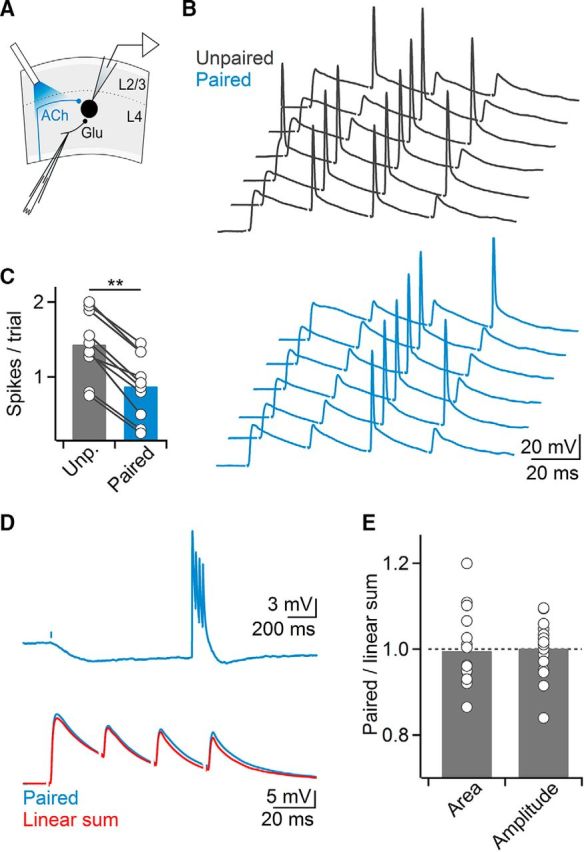
Cholinergic afferents to layer 4 mediate hyperpolarizing inhibition. A, Schematic of experimental setup. Glutamatergic EPSPs in layer 4 neuron were paired with single optical stimulus (5 ms), applied 1 s before electrical stimulation. Recordings were performed in the presence of APV (25 μm) to block recurrent activity. B, Glutamatergic-evoked spikes are significantly suppressed or delayed, as shown for several trials in control (black) or with paired optical stimulation (blue). C, Summary data showing cholinergic-mediated suppression of spiking suppression (n = 10 cells). **p < 0.01. D, mAChR-mIPSPs do not cause shunting of glutamatergic EPSPs. Top, Light-evoked mIPSP was paired with a train (40 Hz) of electrically evoked glutamatergic EPSPs (delay: 1 s). Bottom, Close-up of glutamatergic EPSPs in top trace showing that paired response (blue trace) is identical to linear sum of mIPSP and EPSPs evoked separately (red trace). E, Summary data quantifying both area under the paired EPSPs and amplitude of the first paired EPSP, normalized to their respective unpaired controls (n = 16 cells).
How does postsynaptic mAChR signaling influence the processing of subthreshold synaptic inputs? Activation of mAChRs and the opening of GIRK conductances will lead to a hyperpolarization of membrane potential and in addition, to an increase in membrane conductance generating a potential “shunt” (Eggermann and Feldmeyer, 2009). Shunting inhibition is thought to be a critical mechanism underlying the spatiotemporal summation of excitatory and inhibitory synaptic inputs in neocortex and other brain areas (Koch, 1999). When probed with brief (300 ms) hyperpolarizing current steps, mIPSPs led to a significant reduction in input resistance of the postsynaptic RS cell (86.2 ± 3% compared with control, n = 14 cells, p < 0.001, Wilcoxon signed rank test). Next, we examined whether subthreshold glutamatergic EPSPs are controlled by mAChR-mediated shunting inhibition, by activating glutamatergic afferents (4 stimuli at 40 Hz) during light-evoked mIPSPs (Δt = 1 s). Surprisingly, we found that both glutamatergic EPSP amplitude and area of the paired postsynaptic response were on average nearly identical to the linear sum of the EPSP and the mIPSP evoked separately (EPSP amplitude: 100.1 ± 2% compared with linear sum, n = 16, p = 0.16, EPSP area: 99.6 ± 2%, n = 16, p = 0.34, Wilcoxon signed rank test; Fig. 9D,E). Together, our data suggest that cholinergic inputs to layer 4 excitatory cells control synaptic integration primarily via hyperpolarizing inhibition.
Discussion
Recent in vivo and in vitro work has shown that the fast activation of GABAergic interneurons by nAChRs can mediate cholinergic control of cortical activity. However, the conditions leading to the recruitment of mAChRs are far less understood. Here we have demonstrated that the release of ACh evoked by brief afferent activity can reliably activate postsynaptic mAChRs in the large majority of excitatory neurons of layer 4, leading to a long-lasting inhibition of neuronal firing. Our results highlight a critical role of mAChRs in the rapid and flexible modulation of cortical circuit dynamics.
Mechanisms underlying cholinergic synaptic signaling
We found that activation of cholinergic afferents led to mAChR-dependent IPSCs in excitatory neurons of layer 4. IPSCs were blocked by the M2/M4-specific antagonist AF-DX 116, showed strong inward rectification and were blocked by barium, consistent with the involvement of M2/M4 mAChRs leading to GIRK opening by a membrane delimited pathway, as was observed previously for cholinergic synapses targeting thalamic neurons (Sun et al., 2013). This extends previous findings in layer 4 of rat barrel cortex showing GIRK activation following exogenous agonist application (Eggermann and Feldmeyer, 2009). In contrast, we did not find evidence for an involvement of SK channels in generating mAChR-dependent inhibition, as observed using exogenous agonists in other cortical areas and layers (Gulledge and Stuart, 2005; Gulledge et al., 2007).
Surprisingly, mAChR-dependent postsynaptic responses in layer 4 RS cells did not generate shunting inhibition of glutamatergic postsynaptic responses. In addition to being strongly distance-dependent, shunting inhibition is most effective for inhibitory synapses located “on-path”, i.e., between excitatory synapses and the spike initiation zone in the axon (Vu and Krasne, 1992). Because our extracellular stimuli likely recruited both intracortical and thalamocortical glutamatergic synapses targeting the entire dendritic arbor, cholinergic synapses are predicted to be located in more distal dendritic regions or on branches devoid of glutamatergic synapses. Alternatively, cholinergic transmission might trigger multiple signaling cascades with opposing effects, resulting in little overall change in the PSP waveform. How cholinergic synaptic signals dynamically regulate postsynaptic integration in dendritic trees will require further investigation.
What is the signaling mode responsible for the synaptic recruitment of mAChRs in layer 4? It remains unclear how widespread ultrastructurally defined cholinergic synapses are in neocortex, with studies reporting either classic synapses (Turrini et al., 2001), small varicosities (Umbriaco et al., 1994), or a complete absence of distinct release sites. Furthermore, specific mAChR subtypes do not appear to be preferentially expressed near suspected sites of ACh release (Yamasaki et al., 2010), suggesting that their activation requires sustained increases in BF afferent activity that lead to widespread increases in ambient ACh. However, such a scenario is difficult to reconcile with the phasic discharge pattern of BF cholinergic neurons during the awake state (Lee et al., 2005; Hangya et al., 2015). Our demonstration of short-latency (∼30 ms) mAChR-mediated postsynaptic responses in the large majority of layer 4 neurons suggests that mAChRs can participate in point-to-point transmission, via either conventional synapses or unique forms of nonsynaptic transmission, as has been proposed for certain types of nAChR-mediated responses (Bennett et al., 2012).
In this study we focused exclusively on postsynaptic mechanisms of cholinergic signaling. We did not examine whether fast release of ACh leads to the recruitment presynaptic nAChRs or mAChRs and it remains possible that presynaptic modulation of glutamatergic (Gil et al., 1997; Eggermann and Feldmeyer, 2009; Urban-Ciecko et al., 2018) or GABAergic (Kruglikov and Rudy, 2008) responses is at least partly responsible for the suppression of network activity we observed. Finally, our results also indicate the presence of functional mAChRs in layer 2/3 non-FS neurons, which were not readily recruited by brief ACh transients (Fig. 6) suggesting the existence of more diffuse forms of ACh signaling that are involved in the recruitment of extrasynaptic receptors.
The role of interneurons in cholinergic control of cortical activity
Many questions remain regarding the role of interneurons in mediating cholinergic control of cortical processing. Elegant in vivo studies have shown that cholinergic afferents can rapidly engage layer 1 interneurons (Letzkus et al., 2011) as well as vasoactive intestinal peptide (VIP)-expressing interneurons (Fu et al., 2014) by activating postsynaptic nAChRs. As both types of neurons primarily target other interneurons (Letzkus et al., 2011, 2015; Lee et al., 2013; Pfeffer et al., 2013; Pi et al., 2013), which in turn project onto pyramidal neurons, cholinergic inputs in superficial cortical layers can increase activity in cortical networks by disinhibition (Letzkus et al., 2015). Here we find that nAChR-mediated cholinergic signaling in fact moderately suppresses cortical activity. This suggests that under our experimental conditions, cholinergic afferent inputs recruit interneurons, which generate feedforward inhibition of excitatory neurons, including somatostatin-expressing (SOM) non-FS cells as proposed previously (Chen et al., 2015) or FS cells. Further activation of SOM cells might be generated by an increased excitatory drive mediated by presynaptically expressed nAChRs (Urban-Ciecko et al., 2018). The balance between BF-evoked inhibition and disinhibition is likely to be highly dependent on the activity levels of distinct types of interneurons associated with different cortical activity patterns (Moore et al., 2010; Kuchibhotla et al., 2017).
Contrary to our findings for layer 2/3 neurons, a high percentage of layer 4 non-FS neurons displayed mAChR-mediated depolarizations, consistent with recent in vivo results (Muñoz et al., 2017). Therefore, mAChR-mediated excitation of layer 4 non-FS interneurons might be partly responsible for cholinergic suppression. However, because mAChR-mediated responses in these cells show extremely slow rise times, they are unlikely to contribute to cortical suppression at short latencies. Furthermore, the majority of non-FS cells we recorded from in layer 4 are likely to be SOM neurons (Rudy et al., 2011) and activation of these cells is predicted to generate disinhibition of layer 4 cortical activity, via their preferential inhibition of FS cells (Xu et al., 2013). In conclusion, the rapid suppression of cortical activity in layer 4 is at least partly mediated by a direct mAChR-dependent inhibition of excitatory neurons.
Finally, whether cortical interneurons not only form an important target for BF cholinergic afferents, but also act as a source of ACh remains unclear (Granger et al., 2017). Some reports suggest that ChAT-expressing GABAergic neurons which constitute a subgroup of VIP cells can liberate ACh acting on presynaptic or postsynaptic nAChRs (von Engelhardt et al., 2007; Karnani et al., 2016). Because these neurons express ChR2 in the mouse lines we used it is possible that their activation is at least partly responsible for the fast cholinergic control of cortical circuits examined here.
Cholinergic control of cortical state
Information processing in cortical circuits is strongly modulated by the internal cortical state, as defined by the degree of low-frequency synchronous activity in the local network (Harris and Thiele, 2011). Transitions between cortical states can occur on a range of time scales, from very slow and sustained typical for sleep–wake transitions, to very rapid and highly transient, as observed for sub-states within the awake state (McGinley et al., 2015b). Fast state transitions from quiet wakefulness to a medium arousal state or to locomotor behavior are characterized by a suppression of low-frequency rhythmic activity, thereby enabling increased sensory responses and improved behavioral performance. The mechanisms underlying fast cortical state transitions remain poorly understood but likely involve a number of highly coordinated processes, including several distinct neuromodulatory systems (Aston-Jones and Cohen, 2005; McGinley et al., 2015b) and changes in thalamocortical and corticothalamic activity patterns (Zagha and McCormick, 2014). BF cholinergic afferent activity is likely critical for mediating moment-to-moment changes in brain state, for example during whisking (Eggermann et al., 2014) or locomotor activity (Nelson and Mooney, 2016; Reimer et al., 2016).
Our findings are consistent with previous in vitro studies that have shown a reduction of spontaneous slow oscillatory cortical activity following exogenous ACh application (Favero et al., 2012; Wester and Contreras, 2013; Castro-Alamancos and Gulati, 2014). In addition, we have identified a physiologically plausible mechanism for the precise spatiotemporal control of cortical activity by synaptically released ACh. In our hands, even brief ACh transients are sufficient to cause a prolonged suppression of cortical activity, via the rapid activation of mAChRs in layer 4. As an example, this might explain how transient BF activity at the onset of whisking suppresses low-frequency synchronous activity typical for quiet wakefulness (Eggermann et al., 2014).
More generally, our findings suggest that layer 4 excitatory neurons are a critical target of cholinergic control, with BF afferent activity tracking increases and decreases in thalamic afferent activity associated with different behavioral states. Such ongoing adjustments in layer 4 gain might underlie low-noise cortical computations during periods of heightened arousal. In addition, long-lasting gain control in layer 4 might allow for more rapid nAChR-mediated computations involving both inhibition and disinhibition in local circuits in superficial layers (Letzkus et al., 2015).
Footnotes
This work was supported by a Zilkha Family Discovery Fellowship in Neuroengineering to R.D. and a National Institute of Neurological Disorders and Stroke Grant R01 NS077989 to M.B. We thank John O'Malley for comments on the paper.
The authors declare no competing financial interests.
References
- Agmon A, Connors BW (1991) Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41:365–379. 10.1016/0306-4522(91)90333-J [DOI] [PubMed] [Google Scholar]
- Arroyo S, Bennett C, Aziz D, Brown SP, Hestrin S (2012) Prolonged disynaptic inhibition in the cortex mediated by slow, non-α7 nicotinic excitation of a specific subset of cortical interneurons. J Neurosci 32:3859–3864. 10.1523/JNEUROSCI.0115-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo S, Bennett C, Hestrin S (2014) Nicotinic modulation of cortical circuits. Front Neural Circuits 8:30. 10.3389/fncir.2014.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Cohen JD (2005) An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci 28:403–450. 10.1146/annurev.neuro.28.061604.135709 [DOI] [PubMed] [Google Scholar]
- Beierlein M, Fall CP, Rinzel J, Yuste R (2002) Thalamocortical bursts trigger recurrent activity in neocortical networks: layer 4 as a frequency-dependent gate. J Neurosci 22:9885–9894. 10.1523/JNEUROSCI.22-22-09885.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Gibson JR, Connors BW (2003) Two dynamically distinct inhibitory networks in layer 4 of the neocortex. J Neurophysiol 90:2987–3000. 10.1152/jn.00283.2003 [DOI] [PubMed] [Google Scholar]
- Bennett C, Arroyo S, Berns D, Hestrin S (2012) Mechanisms generating dual-component nicotinic EPSCs in cortical interneurons. J Neurosci 32:17287–17296. 10.1523/JNEUROSCI.3565-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloem B, Schoppink L, Rotaru DC, Faiz A, Hendriks P, Mansvelder HD, van de Berg WD, Wouterlood FG (2014) Topographic mapping between basal forebrain cholinergic neurons and the medial prefrontal cortex in mice. J Neurosci 34:16234–16246. 10.1523/JNEUROSCI.3011-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW (2012) Control of sleep and wakefulness. Physiol Rev 92:1087–1187. 10.1152/physrev.00032.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Gulati T (2014) Neuromodulators produce distinct activated states in neocortex. J Neurosci 34:12353–12367. 10.1523/JNEUROSCI.1858-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Sugihara H, Sur M (2015) An acetylcholine-activated microcircuit drives temporal dynamics of cortical activity. Nat Neurosci 18:892–902. 10.1038/nn.4002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crochet S, Petersen CC (2006) Correlating whisker behavior with membrane potential in barrel cortex of awake mice. Nat Neurosci 9:608–610. 10.1038/nn1690 [DOI] [PubMed] [Google Scholar]
- Descarries L, Gisiger V, Steriade M (1997) Diffuse transmission by acetylcholine in the CNS. Prog Neurobiol 53:603–625. 10.1016/S0301-0082(97)00050-6 [DOI] [PubMed] [Google Scholar]
- Eggermann E, Feldmeyer D (2009) Cholinergic filtering in the recurrent excitatory microcircuit of cortical layer 4. Proc Natl Acad Sci U S A 106:11753–11758. 10.1073/pnas.0810062106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Kremer Y, Crochet S, Petersen CCH (2014) Cholinergic signals in mouse barrel cortex during active whisker sensing. Cell Rep 9:1654–1660. 10.1016/j.celrep.2014.11.005 [DOI] [PubMed] [Google Scholar]
- Favero M, Varghese G, Castro-Alamancos MA (2012) The state of somatosensory cortex during neuromodulation. J Neurophysiol 108:1010–1024. 10.1152/jn.00256.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Tucciarone JM, Espinosa JS, Sheng N, Darcy DP, Nicoll RA, Huang ZJ, Stryker MP (2014) A cortical circuit for gain control by behavioral state. Cell 156:1139–1152. 10.1016/j.cell.2014.01.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y (1997) Differential regulation of neocortical synapses by neuromodulators and activity. Neuron 19:679–686. 10.1016/S0896-6273(00)80380-3 [DOI] [PubMed] [Google Scholar]
- Granger AJ, Wallace ML, Sabatini BL (2017) Multi-transmitter neurons in the mammalian central nervous system. Curr Opin Neurobiol 45:85–91. 10.1016/j.conb.2017.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ (2005) Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci 25:10308–10320. 10.1523/JNEUROSCI.2697-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Park SB, Kawaguchi Y, Stuart GJ (2007) Heterogeneity of phasic cholinergic signaling in neocortical neurons. J Neurophysiol 97:2215–2229. 10.1152/jn.00493.2006 [DOI] [PubMed] [Google Scholar]
- Hangya B, Ranade SP, Lorenc M, Kepecs A (2015) Central cholinergic neurons are rapidly recruited by reinforcement feedback. Cell 162:1155–1168. 10.1016/j.cell.2015.07.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Thiele A (2011) Cortical state and attention. Nat Rev Neurosci 12:509–523. 10.1038/nrn3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay YA, Lambolez B, Tricoire L (2016) Nicotinic transmission onto layer 6 cortical neurons relies on synaptic activation of non-α7 receptors. Cereb Cortex 26:2549–2562. 10.1093/cercor/bhv085 [DOI] [PubMed] [Google Scholar]
- Hedrick T, Waters J (2015) Acetylcholine excites neocortical pyramidal neurons via nicotinic receptors. J Neurophysiol 113:2195–2209. 10.1152/jn.00716.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick T, Danskin B, Larsen RS, Ollerenshaw D, Groblewski P, Valley M, Olsen S, Waters J (2016) Characterization of channelrhodopsin and archaerhodopsin in cholinergic neurons of Cre-lox transgenic mice. PLoS One 11:e0156596. 10.1371/journal.pone.0156596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero JL, Roberts MJ, Delicato LS, Gieselmann MA, Dayan P, Thiele A (2008) Acetylcholine contributes through muscarinic receptors to attentional modulation in V1. Nature 454:1110–1114. 10.1038/nature07141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach A, Waters J (2014) Modulation of high- and low-frequency components of the cortical local field potential via nicotinic and muscarinic acetylcholine receptors in anesthetized mice. J Neurophysiol 111:258–272. 10.1152/jn.00244.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnani MM, Jackson J, Ayzenshtat I, Tucciarone J, Manoocheri K, Snider WG, Yuste R (2016) Cooperative subnetworks of molecularly similar interneurons in mouse neocortex. Neuron 90:86–100. 10.1016/j.neuron.2016.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch C. (1999) Biophysics of computation: information processing in single neurons. New York, NY: Oxford UP. [Google Scholar]
- Kolisnyk B, Guzman MS, Raulic S, Fan J, Magalhães AC, Feng G, Gros R, Prado VF, Prado MA (2013) ChAT-ChR2-EYFP mice have enhanced motor endurance but show deficits in attention and several additional cognitive domains. J Neurosci 33:10427–10438. 10.1523/JNEUROSCI.0395-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglikov I, Rudy B (2008) Perisomatic GABA release and thalamocortical integration onto neocortical excitatory cells are regulated by neuromodulators. Neuron 58:911–924. 10.1016/j.neuron.2008.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Gill JV, Lindsay GW, Papadoyannis ES, Field RE, Sten TA, Miller KD, Froemke RC (2017) Parallel processing by cortical inhibition enables context-dependent behavior. Nat Neurosci 20:62–71. 10.1038/nn.4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Alonso A, Jones BE (2005) Cholinergic basal forebrain neurons burst with theta during waking and paradoxical sleep. J Neurosci 25:4365–4369. 10.1523/JNEUROSCI.0178-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kruglikov I, Huang ZJ, Fishell G, Rudy B (2013) A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat Neurosci 16:1662–1670. 10.1038/nn.3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Wolff SB, Meyer EM, Tovote P, Courtin J, Herry C, Lüthi A (2011) A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature 480:331–335. 10.1038/nature10674 [DOI] [PubMed] [Google Scholar]
- Letzkus JJ, Wolff SB, Lüthi A (2015) Disinhibition, a circuit mechanism for associative learning and memory. Neuron 88:264–276. 10.1016/j.neuron.2015.09.024 [DOI] [PubMed] [Google Scholar]
- McGinley MJ, David SV, McCormick DA (2015a) Cortical membrane potential signature of optimal states for sensory signal detection. Neuron 87:179–192. 10.1016/j.neuron.2015.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinley MJ, Vinck M, Reimer J, Batista-Brito R, Zagha E, Cadwell CR, Tolias AS, Cardin JA, McCormick DA (2015b) Waking state: rapid variations modulate neural and behavioral responses. Neuron 87:1143–1161. 10.1016/j.neuron.2015.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CI, Carlen M, Knoblich U, Cardin JA (2010) Neocortical interneurons: from diversity, strength. Cell 142:189–193. 10.1016/j.cell.2010.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz W, Rudy B (2014) Spatiotemporal specificity in cholinergic control of neocortical function. Curr Opin Neurobiol 26:149–160. 10.1016/j.conb.2014.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz W, Tremblay R, Levenstein D, Rudy B (2017) Layer-specific modulation of neocortical dendritic inhibition during active wakefulness. Science 355:954–959. 10.1126/science.aag2599 [DOI] [PubMed] [Google Scholar]
- Nelson A, Mooney R (2016) The basal forebrain and motor cortex provide convergent yet distinct movement-related inputs to the auditory cortex. Neuron 90:635–648. 10.1016/j.neuron.2016.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermayer J, Verhoog MB, Luchicchi A, Mansvelder HD (2017) Cholinergic modulation of cortical microcircuits is layer-specific: evidence from rodent, monkey and human brain. Front Neural Circuits 11:100. 10.3389/fncir.2017.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Kozak R, Martinez V, Sarter M (2007) Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron 56:141–154. 10.1016/j.neuron.2007.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M (2013) Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat Neurosci 16:1068–1076. 10.1038/nn.3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi HJ, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, Kepecs A (2013) Cortical interneurons that specialize in disinhibitory control. Nature 503:521–524. 10.1038/nature12676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L, Goard MJ, Estandian D, Xu M, Kwan AC, Lee SH, Harrison TC, Feng G, Dan Y (2013) Fast modulation of visual perception by basal forebrain cholinergic neurons. Nat Neurosci 16:1857–1863. 10.1038/nn.3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer J, Froudarakis E, Cadwell CR, Yatsenko D, Denfield GH, Tolias AS (2014) Pupil fluctuations track fast switching of cortical states during quiet wakefulness. Neuron 84:355–362. 10.1016/j.neuron.2014.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer J, McGinley MJ, Liu Y, Rodenkirch C, Wang Q, McCormick DA, Tolias AS (2016) Pupil fluctuations track rapid changes in adrenergic and cholinergic activity in cortex. Nat Commun 7:13289. 10.1038/ncomms13289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S, Hjerling-Leffler J (2011) Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 71:45–61. 10.1002/dneu.20853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Granger AJ, Sabatini BL (2015) Corelease of acetylcholine and GABA from cholinergic forebrain neurons. eLife 4:e06412. 10.7554/eLife.06412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YG, Pita-Almenar JD, Wu CS, Renger JJ, Uebele VN, Lu HC, Beierlein M (2013) Biphasic cholinergic synaptic transmission controls action potential activity in thalamic reticular nucleus neurons. J Neurosci 33:2048–2059. 10.1523/JNEUROSCI.3177-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrini P, Casu MA, Wong TP, De Koninck Y, Ribeiro-da-Silva A, Cuello AC (2001) Cholinergic nerve terminals establish classical synapses in the rat cerebral cortex: synaptic pattern and age-related atrophy. Neuroscience 105:277–285. 10.1016/S0306-4522(01)00172-5 [DOI] [PubMed] [Google Scholar]
- Umbriaco D, Watkins KC, Descarries L, Cozzari C, Hartman BK (1994) Ultrastructural and morphometric features of the acetylcholine innervation in adult rat parietal cortex: an electron microscopic study in serial sections. J Comp Neurol 348:351–373. 10.1002/cne.903480304 [DOI] [PubMed] [Google Scholar]
- Urban-Ciecko J, Jouhanneau JS, Myal SE, Poulet JFA, Barth AL (2018) Precisely timed nicotinic activation drives SST inhibition in neocortical circuits. Neuron 97:611–625.e5. 10.1016/j.neuron.2018.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoog MB, Obermayer J, Kortleven CA, Wilbers R, Wester J, Baayen JC, De Kock CP, Meredith RM, Mansvelder HD (2016) Layer-specific cholinergic control of human and mouse cortical synaptic plasticity. Nat Commun 7:12826. 10.1038/ncomms12826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinck M, Batista-Brito R, Knoblich U, Cardin JA (2015) Arousal and locomotion make distinct contributions to cortical activity patterns and visual encoding. Neuron 86:740–754. 10.1016/j.neuron.2015.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Eliava M, Meyer AH, Rozov A, Monyer H (2007) Functional characterization of intrinsic cholinergic interneurons in the cortex. J Neurosci 27:5633–5642. 10.1523/JNEUROSCI.4647-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu ET, Krasne FB (1992) Evidence for a computational distinction between proximal and distal neuronal inhibition. Science 255:1710–1712. 10.1126/science.1553559 [DOI] [PubMed] [Google Scholar]
- Wester JC, Contreras D (2013) Differential modulation of spontaneous and evoked thalamocortical network activity by acetylcholine level in vitro. J Neurosci 33:17951–17966. 10.1523/JNEUROSCI.1644-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Williams J, Nathans J (2014) Complete morphologies of basal forebrain cholinergic neurons in the mouse. eLife 3:e02444. 10.7554/eLife.02444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Jeong HY, Tremblay R, Rudy B (2013) Neocortical somatostatin-expressing GABAergic interneurons disinhibit the thalamorecipient layer 4. Neuron 77:155–167. 10.1016/j.neuron.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Matsui M, Watanabe M (2010) Preferential localization of muscarinic M1 receptor on dendritic shaft and spine of cortical pyramidal cells and its anatomical evidence for volume transmission. J Neurosci 30:4408–4418. 10.1523/JNEUROSCI.5719-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagha E, McCormick DA (2014) Neural control of brain state. Curr Opin Neurobiol 29:178–186. 10.1016/j.conb.2014.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, Augustine GJ, Deisseroth K, Luo M, Graybiel AM, Feng G (2011) Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods 8:745–752. 10.1038/nmeth.1668 [DOI] [PMC free article] [PubMed] [Google Scholar]