

Abstract
Communication between cells is necessary for the functioning of a multicellular organism. Cells process a large amount of information through G-protein-coupled receptors, and activation of this receptor class has been implicated in neuronal differentiation. In this study, we used a method based on PCR with degenerated primers to identify G-protein-coupled receptors regulated by retinoic acid-induced differentiation of the human teratocarci-noma cell line NTera-2/D1. Subtracted cDNA libraries and control cDNA served as templates in half-sided PCR with a forward degenerate primer based on a conserved sequence from human serotonergic, adrenergic, and dopaminergic receptors and reverse primers on adaptors with long terminal repeats commonly employed in subtractive suppression hybridization. We developed conditions to amplify G-protein-coupled receptors from adaptor-ligated cDNA and found the β2-adrenergic receptor to be upregulated fourfold. This seems to be physiologically relevant, as it could also be shown in rat primary cortical cultures maturing in vitro. The method presented here makes use of the otherwise unused control cDNA from subtractive suppression hybridization experiments and could be easily adapted to other gene families.
Keywords: NT2 cells, Neuronal differentiation, β2-Adrenergic receptor, Subtractive suppression hybridization, Degenerate PCR
COMMUNICATION between individual cells is an essential prerequisite for the coordinated functioning of a multicellular organism. Cells process a large amount of extracellular information through the activation of receptors coupled to heterotrimeric guanine nucleotide binding proteins. These G-protein-coupled receptors (GPCRs) are integral membrane proteins characterized by amino acid sequences, which contain seven hydrophobic domains, predicted to represent the transmembrane spanning regions of these proteins. They respond to a diverse range of agents, including small peptides, lipid analogues, amino acid derivatives, and special stimuli such as light, taste, and odor (1). The vast majority of known receptors belong to the rhodopsin/β2-adrenergic receptor family of GPCRs (22). As certain amino acid residues of this receptor family are well conserved, approaches like PCR with degenerate primers have been used to clone new GPCRs (20).
We were interested in the isolation of GPCRs differentially regulated by neuronal differentiation, as this opens up the possibility to study the physiological role of these receptors by pharmacology. Neurons and glia are derived from pluripotent precursor cells and differentiate in response to environmental and intrinsic factors, which include the regulation of GPCRs (15,16,18). The process of neuronal differentiation can be studied in vitro by the maturation of primary neuronal cultures or by retinoic acid (RA)-induced differentiation of the human teratocarcinoma cell line NTera-2/D1 (NT2). NT2 cells remain as undifferentiated mitotic neuronal precursor cells, when maintained under defined cell culture conditions. Upon exposure to RA and the use of differential adhesion matrices and mitotic inhibitors, the cells develop the morphological and cytoskeletal characteristics of post-mitotic central nervous system (CNS) neurons (21). This seems to model the in vivo situation, as high concentrations of RA have been detected in the embryonic CNS and the developing spinal cord (14,27). RA binding proteins and receptors are present in the developing nervous system, suggesting a role in neurogenesis (19,26,28). We recently identified genes upregulated by RA-induced differentiation of NT2 cells using subtractive suppression hybridization (SSH) (17). SSH uses PCR with primers located on long inverted terminal repeats attached to DNA fragments in combination with hybridization procedures to enrich differentially regulated transcripts. Although SSH is supposed to combine a high subtraction efficiency with an equalized representation of low and highly abundant differentially expressed sequences (8), we could not detect differentially regulated GPCRs in a cDNA library of differentially regulated transcripts generated using this method. This could be due to the rare abundance of most GPCR transcripts even after presumed amplification by SSH. To overcome this problem we tried to develop a PCR-based technique to screen SSH-generated libraries for differentially regulated GPCRs in our paradigm of neuronal differentiation.
MATERIALS AND METHODS
Cell Culture
NT2 cells were cultured and differentiated as previously described (4,17). Briefly, cells were maintained in OptiMEM™ (Life Technologies) supplemented with 5% fetal calf serum (FCS, Linaris), 500 IU/ml penicillin, and 500 μg/ml streptomycin (P/S). For aggregation, cells were trypsinized and 106 cells/ml plated into 140-mm bacteriological grade petri dishes in 50 ml Dulbecco’s minimal Eagle medium DMEM-HG (high glucose, Life Technologies) with 10% FCS and P/S. After overnight incubation at 37°C in a 4% CO2 incubator, the aggregates were treated with 1 μM RA. Medium and culture dish were replaced every 3 days. After 21 days, the aggregates were plated onto tissue culture grade petri dishes or coverslips precoated with 10 μg/ml poly-D-lysine (Sigma) and 10 μg/ml mouse laminin (Sigma) in DMEM-HG with 10% FCS and P/S supplemented with 10 μg/ml cytosin-D-arabinofuranosid (Sigma) and 1 μg/ml uridine (Sigma) for 7 days. Medium was replaced once after 4 days. Cells were harvested after an additional day without cytosin-D-arabinofuranosid and uridine.
Primary cortical neurons were prepared from whole cerebral cortices of fetal Wistar rats (E16-18) as described previously (3). After removing the me-ninges, the tissue was minced and digested with tryp-sin (0.005%; 0.002% EDTA; 15 min, 37°C) followed by mechanical dissociation. Culture dishes were pre-coated with 10 |ig/ml poly-L-lysine (Biochrom, Germany) and 10 |ig/ml collagen G (Biochrom, Germany) and 2 × 106 cells seeded in 35-mm culture dishes in plating medium consisting of neurobasal medium with B-27 serum-free supplement (Gibco, Germany), 100 U/ml penicillin and 100 μg/ml streptomycin, 25 μM glutamate, and 2 mM L-glutamine. After 4 days, the medium was changed to neurobasal medium supplemented with B-27 only. Experiments were performed at days in vitro (DIV) 7 and 14. Medium was changed at DIV 8 and 12.
Immunocytochemistry
Antibodies used were a monoclonal rabbit anti-human neurofilament H (NF-H) IgG antibody (Serotec, 1:1000), a monoclonal rabbit anti-human glial fibrillary acidic protein (GFAP) IgG antibody (Boehringer, 1:500), and a monoclonal mouse anti-human NF-M IgG antibody (Zymed, 1:200). Secondary antibodies were a polyclonal alkaline phosphatase (AP)-conjugated goat anti-mouse IgG (Sigma, 1:700) and a polyclonal AP-conjugated goat anti-rabbit IgG (Promega, 1:700). Cultures were plated on coverslips and fixed with a solution containing 4% formaldehyde, 7% acetic acid, and 7% glycerol in H2O for 1 h and washed twice for 10 min with 0.05% Triton X-100 in phosphate-buffered saline (PBS). The cover-slips were then incubated with the primary antibodies followed by AP-conjugated secondary antibodies and the AP substrates 133 μg/ml 5-bromo-4-chloro-3-indolylphosphat and 266 μg/ml nitrobluetetrazolium. The reaction was stopped with 25 mM Tris-HCl, pH 8.0, and 10 mM EDTA, pH 8.0. After drying, the coverslips were mounted in glycerol.
cDNA Synthesis and Adaptor Ligation
Approximately 2 × 108 RA-induced and untreated cells were used for total RNA extraction using the TRIzol™ reagent (Life Technologies) and poly(A)+ RNA purified with two rounds of DynaBeads (Dynal). Approximately 2 μg poly(A)+ RNA was reverse transcribed with RNAse H− mutant Moloney murine leukemia virus reverse transcriptase (Promega) and oligo-(dT)15 primers. The second strand was synthesized immediately afterwards by the Gubler and Hofman procedure using E. coli DNA Polymerase I, RNAse H and DNA ligase at 16°C for 2 h. Double-stranded cDNA was blunted by T4 DNA polymerase for 30 min at 16°C and digested with 15 U RsaI (Pro-mega) or TaqI (New England Biolabs) at 37°C for 1.5 h to obtain short cDNA fragments. After heat in-activation, phenol/chloroform extraction, and ethanol precipitation, approximately 7% of both cDNAs were ligated with 10 pmol of adaptor 1 and the ligation efficiency was tested by a PCR-based assay according to the manual (PCR Select, Clontech). The SSH libraries tested were those previously described (17).
Northern Blotting
Five micrograms total RNA of RA-induced and untreated NT2 cells was separated by denaturing 1.2% gel electrophoresis and transferred to a nylon membrane. A specific glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probe was generated by linear PCR in the presence of 50 μCi [α-32P]dATP (Amersham) using the GAPDH reverse primer and 25 ng GAPDH template generated by PCR. Cycle parameters were 94°C 2 min, 65°C 2 min, and 72°C 5 min for 30 cycles. Specific activity was >107 cpm/μg DNA. The blots were hybridized overnight in UltraHyb (Ambion) solution at 42°C and washed under high-stringency conditions. Autoradiographs were exposed for 4 h and analyzed using a phosphoimag-ing system (Fujix).
PCR Amplification and Dot Blot Analysis
We used adaptor-ligated cDNA from RA-induced (I) and untreated (U) NT2 cells, as well as subtracted libraries (U-I) and (I-U) as templates in a primary PCR with the degenerate primer DP and the adaptor-specific primer P1 (Table 1). DP is based on the highly conserved L C W L P F F sequence located in the sixth transmembrane region of human serotonergic, adrenergic, and dopaminergic receptors and was constructed using the codehop algorithm (http://block-s.fhcrc.org/blocks/codehop.html) (25). The subtracted libraries were generated by subtractive hybridization and suppressive PCR using primer P1. The PCR conditions were 94°C 10 s, 66°C 30 s, and 72°C 90 s for 27 cycles. The PCR product was diluted 10-fold and 1 μl used as template for a second PCR using primers DP and NP1 for 30, 35, and 40 cycles using annealing temperatures from 56°C, 62°C, to 68°C. For PCR amplification of unsubtracted libraries, the RsaI- or TaqI-digested libraries ligated to adaptor 1 were diluted 1:200 and 1 μl used as template for the first half-degenerate PCR using P1 and DP for 20 cycles at 50°C, 58°C, or 65°C. The amplified product was diluted 1:50 and 1 μl used as template in a secondary half-nested PCR with the primers NP1 and DP. Conditions were 30, 35, or 40 cycles at 68°C annealing temperature with the remaining conditions unchanged to the primary PCR. One microliter of each secondary PCR was dotted onto a nylon membrane (Pora-blot, Machery & Nagel) and hybridized to the end-labeled degenerate oligonucleotide DC located downstream from DP to identify transcripts belonging to the GPCR family. The blot was washed under low-stringency conditions and exposed to a phosphoimaging plate for 30 min. A PCR fragment of the 5-HT1B receptor containing the sequence complementary to DP served as positive control. Furthermore, PCR products were size fractionated by agarose gel electrophoresis, blotted, and hybridized with DC. Aliquots of the two PCR products using I as template that revealed the most robust hybridization signal by dot blotting and bands of differential hybridization intensity compared to U in the Southern blots were cloned into the pCR2.1-TOPO vector (Invitrogen) and transformed into DH5α competent cells. Inserts of 27 clones of each PCR product were PCR amplified with NP1 and DP, dot blotted onto a nylon membrane (Porablot, Machery & Nagel), and hybridized to the end-labeled oligonucleotide DC to identify specific products. The blot was washed under low-stringency conditions and exposed for 30 min. Ten hybridizing clones from the RsaI-digested and four of the TaqI-digested libraries were sequenced with an Applied Biosystems automated DNA sequencer. The same PCR conditions mentioned above were also applied to the subtracted libraries digested with RsaI. Dot blots were hybridized with a specific probe representing the cloned insert of the β2-adrenergic receptor (β2-adrenoceptor) radiolabeled by random hexameric primers (MegaPrime) and washed under high-stringency conditions. PCR products obtained from amplification of gpr19, 5-HT7, and β2-adrenoceptor dotted in different dilutions were used to control specificity.
TABLE 1.
OLIGONUCLEOTIDES USED FOR PCR AND HYBRIDIZATION EXPERIMENTS
| Gene | Forward Primer | Reverse Primer | Hybridization Probes |
|---|---|---|---|
| GAPDH | 5′-ACCACAGTCCATGCCATCAC-3′ | 5′-TCCACCACCCTGTTGCTGTA-3′ | 5′-GGATGACCTTGCCCACAGC-3′ |
| P1 | 5′-CTAATACGACTCACTATACGGC-3′ | ||
| NP1 | 5′-TCGAGCGGCCGCCCGGGCAGGT-3′ | ||
| 5-HT7 receptor | 5′-GGCTGCCATTTTTCCTCCTCT-3′ | 5′-CCGGTGGCCTCTTTTCTGGTCT-3′ | |
| Gpr19 | 5′-GGCCGAACGGTGAGGAGGACAA-3′ | 5′-CATGAAAAGGCAGCCAGGAGAGC-3′ | |
| Human β2-adrenoceptor | 5′-TGCCATTGCCTCTTCCATCGTGTC-3′ | 5′-AAGGGCAGCCAGCAGAGGGTGAAA-3′ | 5′-TGCCCCGTCCGCCCATCCT-3′ |
| Rat β2-adrenoceptor | 5′-CTGTGCCTTCGCCGGTCTTCTT-3′ | 5′-TGTCAGGGAGGGGCCGTTCTTA-3′ | 5′-CCTGCCCCAGCTGATATGCGC-3′ |
| Oligo-dT primer | 5′-TTTTGTACAACTT30NN-3′ |
Quantitative (RT)-PCR
RT-PCR was used to quantitate the transcript amount in cDNA from RA-induced and untreated NT2 cells, and DIV 7 and 14 rat primary cortical cultures. The expression was normalized for GAPDH expression. In order to determine the exponential phase of amplification, aliquots of the PCR reaction were removed after 15, 20, 25, 30, 35, and 40 cycles and checked on ethidium bromide-stained agarose gels. PCR reactions were performed with either GAPDH or specific primer pairs alone or together in a multiplex reaction for both templates. All results were reproduced twice. Aliquots were removed every other round of amplification starting at the estimated start of linear amplification, run on a 2% agarose gel (SeaKem LE Agarose, FMC BioProducts), and blotted onto a nylon membrane (Porablot, Machery & Nagel). A specific β2-adrenoceptor or GAPDH oligonucleotide was end-labeled and hybridized with the membranes overnight at 42°C. Specific activity was >107 cpm/pmol oligonucleotide. Membranes were washed under high-stringency conditions and exposed to phosphoimaging plates. Autoradiographies were analyzed using Tina Version 2.10h (Raytest). A density profile (not shown) was created for each lane in the autoradiography (e.g., Fig. 5A), the baseline estimated and subtracted from all values. The area under the curve for each band was determined and plotted on a semilogarithmic scale against the cycle number for each reaction. All values were presumed to be valid when compared curves were parallel at a given cycle number, indicating that both reactions were still in the exponential phase of amplification. Valid data were normalized to the corresponding GAPDH photostimulated luminescence. The mean, standard deviation, and standard error of ratios of normalized photostimulated luminescence of amplification products from RA-induced compared to untreated templates were calculated and Student’s ratio paired t-test performed to determine the significance.
Figure 5.
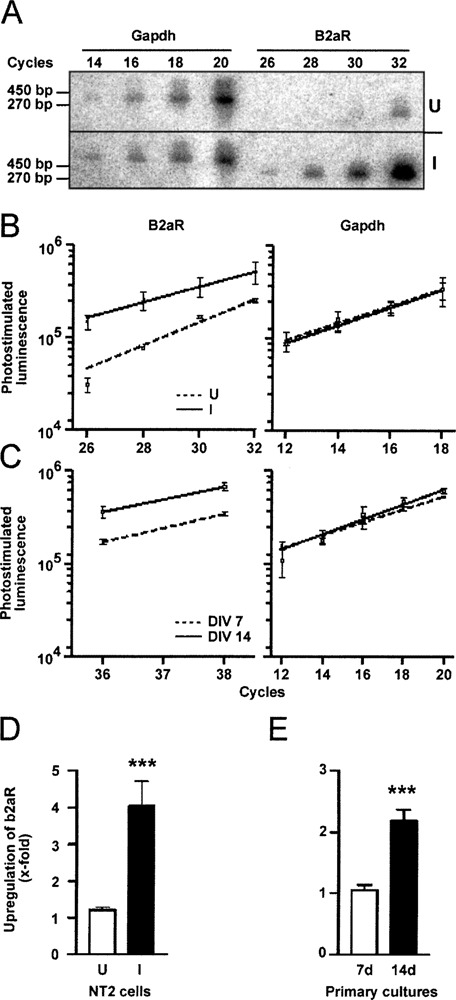
Regulation of β2-adrenoceptor by neuronal differentiation. (A) Southern blot of GAPDH and β2-adrenoceptor (B2aR) PCR products from RA-induced (I) and untreated (U) NT2 cDNA. Aliquots were removed at the indicated cycle numbers, blotted after electrophoresis, and hybridized with specific end-labeled oligonucleotides. Sizes of PCR products are indicated. (B) Hybridization intensities of β2-adrenoceptor and GAPDH PCR products plotted against the cycle number. (□, dashed line) cDNA from U; (▴, solid line) cDNA from I. (C) Similar experiment with cDNA from rat primary neuronal cultures at DIV 7 (▪, dashed line) and 14 (□, solid line). (D) Statistical analysis of quantitative β2-adrenoceptor PCR experiments. Three values from three independent quantitative PCR experiments were used and normalized to relative GAPDH expression. Onefold upregulation corresponds to equal expression. Bar graphs represent the mean ± SE. Significance was calculated by Student’s ratio paired t-test (***p < 0.001). (E) Similar analysis showing upregulation of β2-adrenergic receptor in cDNA from rat primary cultures prepared at the indicated time points.
RESULTS
We differentiated NT2 cells by combining cell aggregation, treatment with RA, and mitotic inhibition (4). These cultures consisted of cell aggregates and postmitotic CNS-like neurons that express late neuronal markers like phosphorylated NF-H (Fig. 1). An antibody against GFAP did not stain the RA-induced NT2 cultures, suggesting the absence or at least underrepresentation of glial-like cells (not shown).
Figure 1.
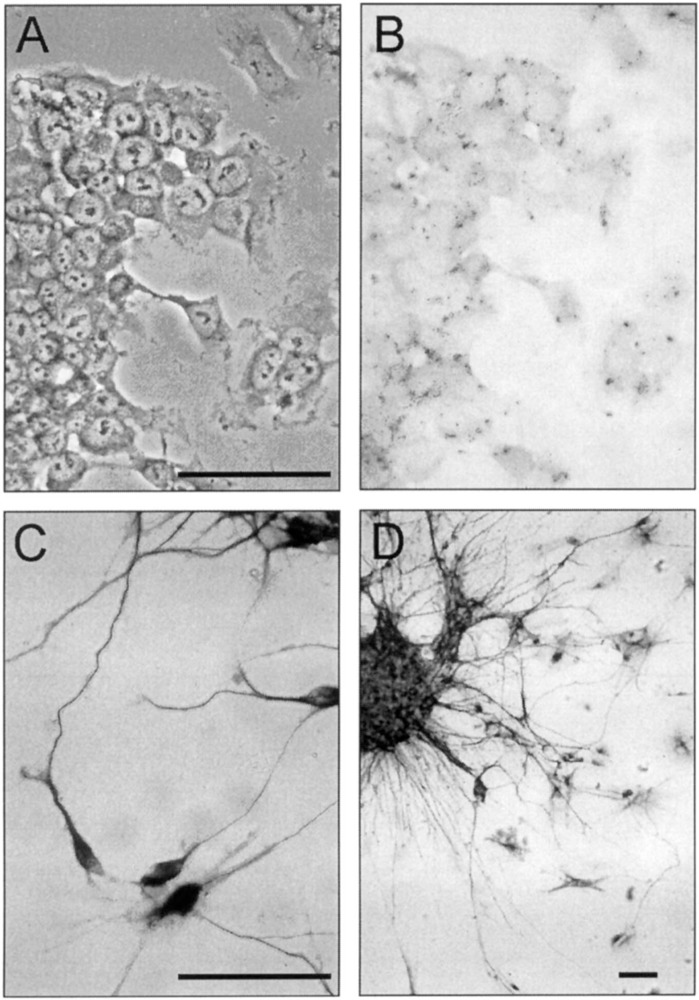
Retinoic acid induces morphological and cytoskeletal changes in NT2 cells. (A) Untreated NT2 cells have large nuclei with prominent nucleoli and granular cytoplasm. (B) Untreated NT2 cells show no visible staining for the late neuronal marker NF-H. (C) RA-induced cells possess a conical cell body with long and thin interconnected extensions. The cell bodies and most extensions of RA-induced cells are stained with antibodies against NF-H. (D) Aggregates of RA-induced cells and axons protruding from them are heavily stained with antibodies against NF-H. (A) Phase-contrast photomicrography, (B–D) bright-field photomicrographies. Scale bar: 100 μm.
We previously prepared mRNA and double-stranded cDNA from these cultures and ligated SSH adaptors to the cDNA. We then used SSH to screen for transcripts regulated by neuronal differentiation (17). The SSH protocol consists of two hybridization steps that enrich differentially regulated transcripts. These transcripts have different adaptors on either side, which allows efficient amplification by PCR (see diagram in the upper part of Fig. 2). SSH adaptors have long inverted repeats and the suppression PCR effect inhibits amplification of unregulated cDNA molecules flanked by identical adaptors, when a primer corresponding to the outer part of the repeat is used (8,9). We found differentially regulated transcripts in this screen and assumed that our SSH cDNA libraries contained differentially regulated transcripts. We hypothesized that a degenerate primer based on a sequence conserved in GPCRs could be used in combination with a primer on the SSH adaptor to amplify differentially regulated GPCRs from this template. We first investigated whether one GAPDH primer alone used in combination with an SSH adaptor primer amplifies GAPDH as efficiently as two specific primers. GAPDH can serve as a suitable internal standard, because RA-induced and untreated NT2 cells expressed similar amounts of RNA as shown by Northern blotting (Fig.3A) and quantitative PCR with two specific GAPDH primers (Fig. 3B). A specific GAPDH primer used with primer P1, corresponding to the outer part of the adaptor, resulted in a very inefficient amplification (not shown), but the nested primer NP1 downstream of the inverted repeat amplified GAPDH as well as specific primers (Fig. 4A). We designed the degenerate primer DP based on the highly conserved L C W L P F F sequence located in the sixth transmembrane region of human serotonergic, adrenergic, and dopaminergic receptors (see Table 2) and used it in a half-sided PCR approach with the SSH primer NP1 similar to the control experiment. Templates were SSH-subtracted libraries after the first round of P1 amplification. A second degenerate oligonucleotide DC based on a sequence approximately 60 bp downstream of DP was hybridized under low-stringency conditions to dot blots of various PCR reactions to optimize the conditions for GPCR amplification (see diagram in the lower part of Fig. 2 for the relative position of the primers and probes used). The libraries used were derived from a digest of two different restriction endo-nucleases (TaqI and RsaI) to minimize the chance that the restriction enzyme digest removed the DC binding site and to obtain fragments of an ideal size for PCR. PCR with DP and NP1 under various conditions resulted in some DC hybridizing dot blots, but shotgun cloning and sequencing of hybridizing clones did not yield GPCRs (Fig. 4B).
Figure 2.
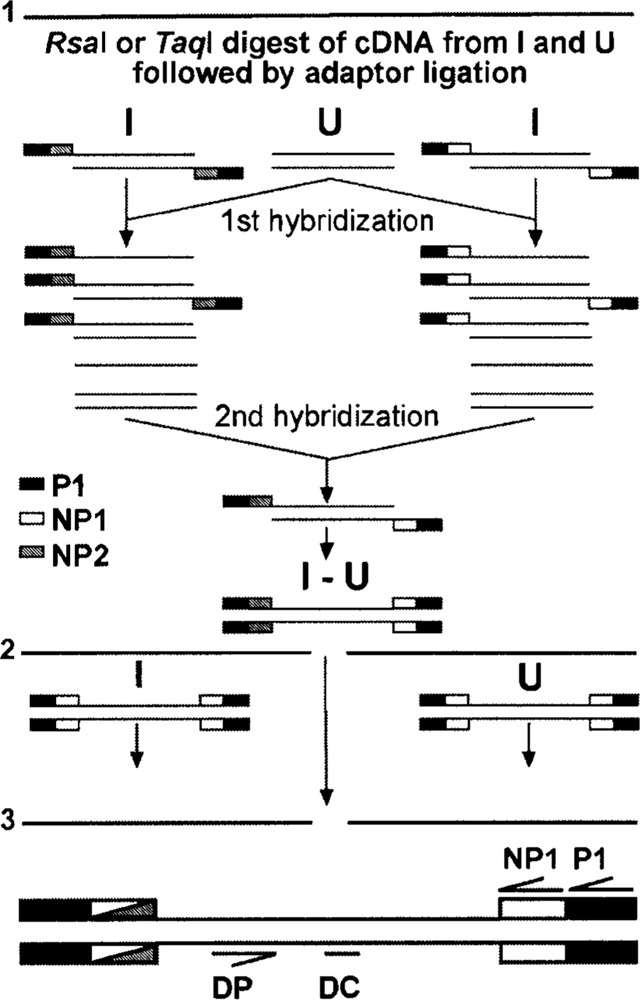
Generation of subtracted and adaptor-ligated cDNA. 1: Generation of SSH libraries. 2: Templates for PCR amplification were the adaptor ligated cDNA from RA-induced (I) and untreated NT2 cells (U), as well as the amplified subtracted library (I-U). 3: Binding sites of primers used in this study.
Figure 3.
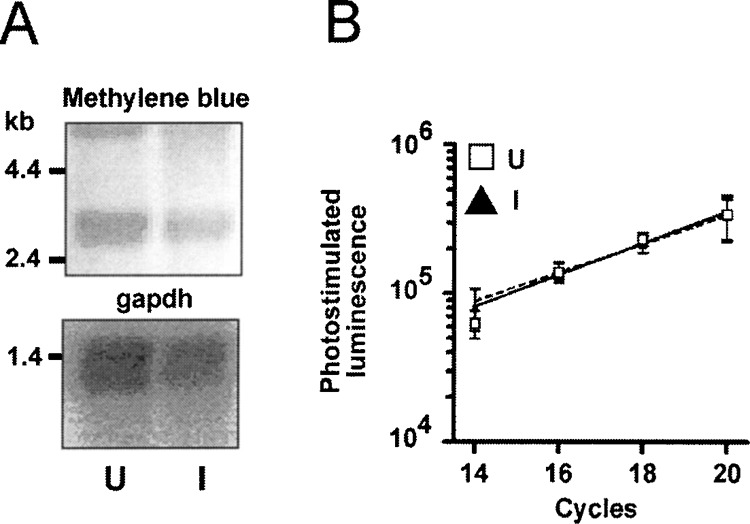
GAPDH is a suitable control transcript. (A) Five micrograms total RNA of untreated (U) and RA-induced (I) NT2 cells was stained in the upper panel with methylene blue and in the lower panel hybridized with a 450 bp GAPDH probe. (B) Quantitative PCR with specific primers comparing GAPDH expression in U and I. (□, dashed line) cDNA from U; (▴, solid line) cDNA from I. Relative hybridization intensities of aliquots removed at the indicated cycle number and hybridized with a specific end-labeled probe are blotted against the cycle number. Error bars show SEM of three independent experiments.
Figure 4.
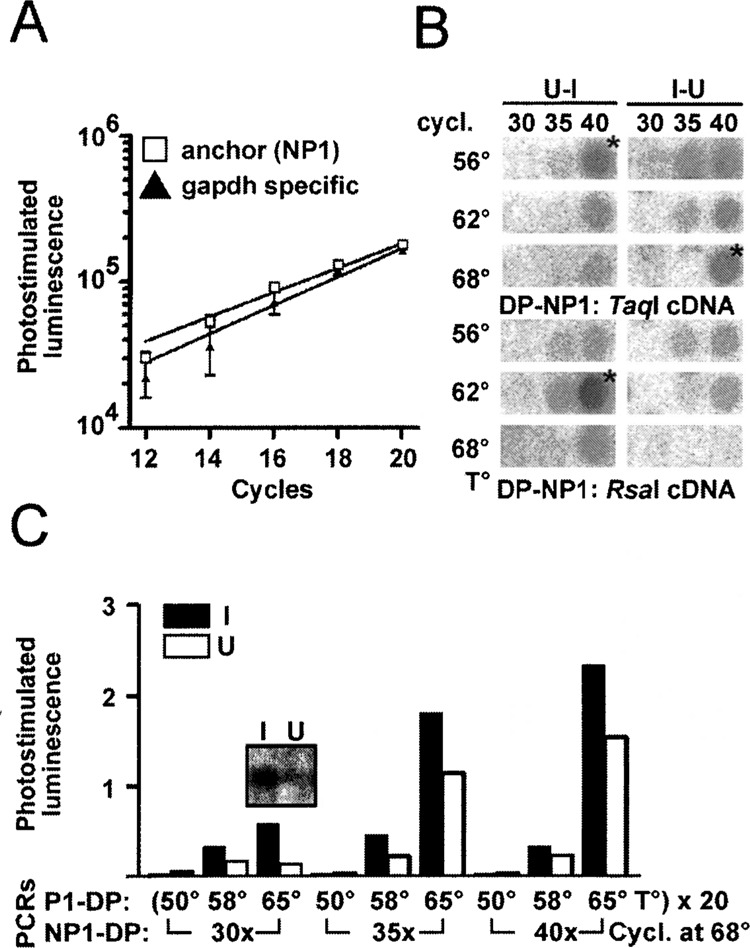
Degenerate suppression PCR identifies regulated GPCRs. (A) Comparison of specific GAPDH primers (▴) and the anchored primer NP1 with one specific primer (□) on cDNA from RA-induced NT2 cells (I). (B) Dot blot of PCR products generated with the indicated primer pairs hybridized with end-labeled DC. Templates were subtracted libraries I-U and U-I from RsaI- or TaqI-digested cDNA. Products labeled with an asterisk were cloned and sequenced. (C) Densitometric analyses of dot blots from nested PCR products hybridized with end-labeled DC. Templates were RsaI-digested adaptor-ligated I and U cDNA. First PCR with degenerate primer DP and adaptor-specific primer P1 for 20 cycles at the temperatures indicated. Second half nested PCR with primers DP and NP1 using a 1:50 dilution of the first PCR at 68°C annealing temperature for the cycle number indicated. The inset contains a Southern blot of the PCR reaction with the most pronounced difference between I and U.
TABLE 2.
GENE SYMBOLS AND SWISSPROT ACCESSION NUMBERS OF THE GPCRS USED TO DESIGN THE DEGENERATE OLIGONUCLEOTIDES
| Gene Symbol | Accession | RsaI | TaqI | DP (5′-3′) Homology | DC (5′-3′) Homology |
|---|---|---|---|---|---|
| β1-adrenoceptor | P35368 | 764 | 770* | ATCTTGTGCTGGCTACCCTTCTTCAT | TGGCTGGGCTACTTCAAC |
| α1D-adrenoceptor | P25100 | 1060 | 413 | GTGCTCTGCTGGTTCCCTTTCTTCTT | TGGCTCGGCTACTTCAAC |
| α2A-adrenoceptor | P08913 | 527 | 281 | GTGGTGTGCTGGTTCCCCTTCTTCTT | TGGTTCGGCTACTGCAAC |
| α2B-adrenoceptor | P18089 | 1490 | 955 | GTGCTCTGCTGGTTCCCCTTCTTCTT | TGGATCGGCTACTGCAAC |
| α2C-adrenoceptor | P18825 | 62† | 829* | GTGCTCTGCTGGTTCCCCTTCTTCTT | TGGATCGGCTACTGCAAC |
| β1-adrenoceptor | P08588 | 696 | 405 | ACGCTCTGCTGGCTGCCCTTCTTCCT | TGGCTGGGCTACGCCAAC |
| β2-adrenoceptor | P07550 | 353 | 1015* | ACCCTCTGCTGGCTGCCCTTCTTCAT | TGGATAGGCTATGTCAAT |
| β3-adrenoceptor | P13945 | 1602 | 319 | ACTCTCTGCTGGTTGCCCTTCTTTCT | TGGCTAGGTTATGCCAAT |
| Dopamine D1 | P21728 | 603 | 1287* | GTGTGCTGTTGGCTACCTTTCTTCAT | TGGTTTGGGTGGGCTAAT |
| Dopamine D2 | P14416 | 98† | 1381* | ATCATCTGCTGGCTGCCCTTCTTCAT | TGGCTGGGCTATGTCAAC |
| Dopamine D3 | P35462 | 253 | 172 | ATTGTCTGCTGGCTGCCCTTCTTCTT | TGGCTGGGCTACGTGAAT |
| Dopamine D4 | P21917 | 292 | 359* | CTGCTGTGCTGGACGCCCTTCTTCGT | TGGCTGGGCTACGTCAAC |
| Dopamine D5 | P21918 | 745 | 130 | GTGTGTTGCTGGCTGCCCTTCTTCAT | TGGTTCGGCTGGGCTAAC |
| Gprl9 | Q15760 | 590 | 330 | TTGCTCTCCTGGCTGCCTTTTCATGT | TGGGAAGGCACTGCCTAC |
| 5-HT1A | P08908 | 265 | 271* | ATCCTCTGCTGGCTGCCCTTCTTCAT | TGGCTGGGCTACTCCAAC |
| 5-HT1B | P28222 | 262 | 270* | ATTGTGTGTTGGCTACCCTTCTTCAT | TGGCTGGGCTATCTCAAC |
| 5-HT1D | P28221 | 1168 | 672 | ATCATCTGCTGGCTGCCCTTCTTCGT | TGGCTAGGCTATTTAAAC |
| 5-HT1E | P28566 | 503 | 508* | ATTTTATCCTGGCTGCCATTTTTCAT | TGGCTCGGTTATGTGAAT |
| 5-HT1F | P30939 | 253 | 207 | GTAATATGTTGGCTTCCTTTTTTTGT | TGGCTTGGGTATCTCAAT |
| 5-HT5A | P47898 | 250 | 256* | GTGCTCTGCTGGATCCCCTTCTTTCT | TGGCTTGGCTACTCCAAC |
| 5-HT6 | P50406 | 744 | 97† | TTTGTGACCTGGTTGCCCTTCTTTGT | TGGCTGGGTTACTGTAAC |
| 5-HT 7 | P34969 | 221 | 54 † | ACCGTGTGCTGGCTGCCATTTTTCCT | TGGCTAGGCTATGCAAAC |
| 5-HT 7 pseudogene | XR-000I03 | 219 | 809 | ACCATGTGCTGGCCGCCCTTTTTCCT | TGGCTGGGCTATGCAAAC |
| GTGCTCTGCTGGCTGCCHTTYTTYNT | TGGMTVGGYTAYBBMAAY |
Restriction fragment length from DP to the next downstream RsaI or TaqI site is indicated. Receptors shown in italic were amplified from the respective underlined cDNA. Mismatches between the putative binding site and the degenerate oligonucleotides are shown in bold.
No TaqI site in the 3′ sequence.
Signifies lacking DC binding site.
We therefore used unamplified and unsubtracted cDNAs from RA-induced and untreated NT2 cells digested with RsaI or TaqI and ligated to the respective SSH adaptor as an alternative template. PCR with DP and NP1 alone resulted in no DC-hybridizing products (not shown), but a half-nested PCR with 20 cycles DP and P1 and 30–40 cycles DP and NP1 gave reproducible and strong signals already after 30 cycles of the second PCR. Annealing at 50°C and even 58°C in the primary PCR resulted in significantly lower signals. The most prominent difference in hybridization intensity between amplified RA-induced and untreated NT2 cDNA was observed with an annealing temperature of 65°C in the primary PCR and 30 cycles secondary PCR using the RsaI-digested template (Fig. 4C). Separation of the product from this PCR reaction by gel electrophoresis resulted in an undistinguishable smear and the DC oligonucleotide hybridized only to one prominent band of approximately 350 bp on the corresponding Southern blot, which was remarkably more intense in I (Fig. 4C, inset). Dotted PCR products of the TaqI-digested DNA showed a similar pattern with less hybridization intensity and an additional, very faintly hybridizing band at approximately 800 bp not detected in DNA from uninduced cells (not shown).
Shotgun cloning of the reaction products from RA-induced cDNA that showed a hybridizing band, and analysis of 27 randomly picked clones from each cDNA by dot blotting identified 10 clones hybridizing with the DC oligonucleotide obtained from RsaI-digested and four from TaqI-digested templates. Two of the 10 clones obtained from the RsaI-digested template corresponded to the β2-adrenergic receptor (β2-adrenoceptor) and one to the 5-HT7 receptor. TaqI-digested and amplified cDNA yielded one clone corresponding to the orphan receptor gpr19 and one to the 5-HT7 receptor pseudogene. Only β2-adrenoceptor and the 5-HT7 pseudogene corresponded in size (Table 2) with the presumed regulated transcripts hybridizing with the control oligonucleotide using RsaI- and TaqI-digested templates. The β2-adrenoceptor was found to be upregulated 4.05-fold (SEM 0.66, p < 0.01, n = 74) by quantitative PCR with specific primers in cDNA from RA-induced NT2 cells (Fig. 5A, B, D). Gpr19 and 5-HT7 receptors showed no difference in ethidium bromide-stained gels and were not further analyzed. The 5-HT7 pseudogene was not analyzed further due to the lack of functional significance. Thus, the conditions chosen allowed the amplification of four different receptors and identified one differentially expressed GPCR. We next applied the successful conditions to the substracted libraries and analyzed the amplification of the β2-adrenoceptor by hybridizing dotted PCR products with a specific probe. No hybridization was observed in the amplificates of the subtracted libraries.
We then evaluated the proposed upregulation of β2-adrenoceptor in rat primary neuronal cultures as a model closer to the in vivo situation. At DIV 14, these cultures are mature as judged by morphology and the expression of neurite and synaptic markers (7). DIV 14 cultures contained 2.3-fold more β2-adrenoceptor mRNA than DIV 7 cultures, as shown by quantitative PCR (Fig. 5C, E).
DISCUSSION
We recently identified genes upregulated by retinoic acid differentiation of human NT2 cells (17) by subtractive suppression hybridization. We were, however, unable to detect regulated GPCRs in this study, although the process of neuronal differentiation is supposed to include the activation of GPCRs (15,16, 18). We assumed at the time that this was due to the rare abundance of most GPCR transcripts, even though SSH is supposed to normalize and enrich differentially expressed low-abundance transcripts (13).
To overcome the problem, we used these cDNA libraries as templates in a half-sided PCR using a GPCR-specific degenerate primer and a primer on the SSH adaptor. A second degenerate oligonucleotide was hybridized under low-stringency conditions to the amplification products to identify true positives. The cDNA was digested with two different restriction enzymes to minimize the possibility that the primer or probe binding site might be destroyed by the digest. We hoped to identify known GPCRs by their restriction fragment length and to find new differentially regulated human receptors. We were, however, not able to amplify GPCRs from the subtracted libraries. Therefore, we amplified unsubtracted adaptor-ligated cDNAs from RA-induced and untreated NT2 cells as an alternative approach. This allowed us to identify four different GPCRs from RA-induced NT2 cells. The β2-adrenoceptor and the 5-HT7 receptor were amplified from RsaI-digested cDNA and the 5-HT7 pseudogene and the orphan receptor gpr19 from TaqI-digested cDNA. PCR was only efficient at 65°C, probably due to the self-annealing properties of the long inverted repeats at lower temperatures. Theoretically, only the best fitting primers anneal at these high temperatures. The β2-adrenoceptor is, indeed, one of the best fitting receptors with only three mismatches at the 5′ end (Table 2). The 5-HT7 receptor, however, has five mismatches and gpr19 had four mismatches. The mismatches in gpr19 are even very close to the 3′ end, which makes it the least fitting receptor of all. This probably means that there were no better fitting receptors present in the template in the amount necessary for PCR amplification. Possibly, we could not detect the β2-adrenoceptor from the TaqI library due to the fact that the expected β2-adrenoceptor restriction fragment length in this library is 1015 bp as opposed to 353 bp in the RsaI library, leading to less efficient amplification. We could not amplify any GCPRs from the subtracted libraries, although these are supposed to consist of molecules with different adaptors on each side that do not self-anneal at low temperatures. We think that this failure might be due to the reduced complexity of these libraries. The number of molecules amplifiable by suppressive PCR after subtractive hybridization is estimated to be in the thousands and the restriction digestion step even leads to more than one amplifiable molecule per differentially expressed transcript (23), which further reduces the complexity. Thus, the subtracted libraries most probably do not represent all upregulated transcripts. Although this problem could be ameliorated by pooling numerous first PCR reactions, it is difficult to predict how many reactions are sufficient to achieve a representational library. It might be safer to use the unsubtracted adaptor ligated libraries as presented here.
We next used quantitative PCR to verify the presumed differential expression. The β2-adrenoceptor was found to be upregulated fourfold in cDNA from RA-induced NT2 cells, and we assume that the differentially amplified product of approximately 350 bp in cDNA from induced cells (Fig. 4C, inset) corresponds to the β2-adrenoceptor. To further elucidate the role of β2-adrenoceptor upregulation in neuronal differentiation, we chose primary neuronal cultures kept under serum-free conditions (2) as an in vitro model close to the in vivo situation with a mixed glial and neuronal environment. The in vitro differentiation of neurons in serum-free cortical cultures closely resembles the development of cortical neurons in the intact tissue (10). At DIV 14, these cultures are mature as judged by morphology and the expression of neurite and synaptic markers (7). DIV 14 cultures contained 2.3-fold more β2-adrenoceptor mRNA than DIV 7 cultures, as shown by quantitative PCR. This upregulation was probably due to neuronal maturation in vitro, as our cultures consisted mainly of neurons, though a glial component could not be completely ruled out. The upregulation observed is in accordance with previous work, where the levels of β2-adrenoceptor mRNA and binding sites in rat brain were low at postnatal day 1 and increased steadily to adult levels by days 16–25 (11). Neuronally restricted overexpression of β2-adrenoceptor in RA-treated NT2 cells was shown previously to increase the length of cell processes and expression of MAP2 (12), possibly by increasing nerve growth factor biosynthesis (6). However, there seems to be no overt neurological or developmental phenotype associated with the targeted disruption of the β2-adrenoceptor gene (5) or with the β1- and β2-adrenoceptor double knockout mice (24).
In summary, we successfully combined suppression PCR with PCR degenerate primers to identify a G-protein-coupled receptor upregulated by RA-induced differentiation of human NT2 cells. The inexpensive method presented here makes use of the otherwise unused control cDNA from SSH experiments and could be easily adapted to other gene families.
ACKNOWLEDGMENTS
We are most grateful for the continuous support and the laboratory space provided by Prof. Dr. Chica Schaller. This work was funded by the Deutsche Forschungsgemeinschaft Graduiertenkolleg 255 and the Dr. Kurt and Irmgard Meister-Stiftung.
REFERENCES
- 1. Baldwin J. M. Structure and function of receptors coupled to G proteins. Curr. Opin. Cell Biol. 6:180–190; 1994. [DOI] [PubMed] [Google Scholar]
- 2. Brewer G. J.; Torricelli J. R.; Evege E. K.; Price P. J. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 35:567–576; 1993. [DOI] [PubMed] [Google Scholar]
- 3. Bruer U.; Weih M. K.; Isaev N. K.; Meisel A.; Ruscher K.; Bergk A.; Trendelenburg G.; Wiegand F.; Victorov I. V.; Dirnagl U. Induction of tolerance in rat cortical neurons: Hypoxic preconditioning. FEBS Lett. 414:117–121; 1997. [DOI] [PubMed] [Google Scholar]
- 4. Cheung W. M.; Fu W. Y.; Hui W. S.; Ip N. Y. Production of human CNS neurons from embryonal carcinoma cells using a cell aggregation method. Biotechniques 26:946–948, 950–952, 954; 1999. [DOI] [PubMed] [Google Scholar]
- 5. Chruscinski A. J.; Rohrer D. K.; Schauble E.; Desai K. H.; Bernstein D.; Kobilka B. K. Targeted disruption of the beta2 adrenergic receptor gene. J. Biol. Chem. 274:16694–16700; 1999. [DOI] [PubMed] [Google Scholar]
- 6. Colangelo A. M.; Johnson P. F.; Mocchetti I. Beta-adrenergic receptor-induced activation of nerve growth factor gene transcription in rat cerebral cortex involves CCAAT/enhancer-binding protein delta. Proc. Natl. Acad. Sci. USA 95:10920–10925; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Lima A. D.; Merten M. D.; Voigt T. Neuritic differentiation and synaptogenesis in serum-free neuronal cultures of the rat cerebral cortex. J. Comp. Neurol. 382:230–246; 1997. [DOI] [PubMed] [Google Scholar]
- 8. Diatchenko L.; Lau Y. F.; Campbell A. P.; Chenchik A.; Moqadam F.; Huang B.; Lukyanov S.; Lukyanov K.; Gurskaya N.; Sverdlov E. D.; Siebert P. D. Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc. Natl. Acad. Sci. USA 93:6025–6030; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diatchenko L.; Lukyanov S.; Lau Y. F.; Siebert P. D. Suppression subtractive hybridization: A versatile method for identifying differentially expressed genes. Methods Enzymol. 303:349–380; 1999. [DOI] [PubMed] [Google Scholar]
- 10. Dotti C. G.; Banker G. A.; Binder L. I. The expression and distribution of the microtubule-associated proteins tau and microtubule-associated protein 2 in hippocampal neurons in the rat in situ and in cell culture. Neuroscience 23:121–130; 1987. [DOI] [PubMed] [Google Scholar]
- 11. Duman R. S.; Saito N.; Tallman J. F. Development of beta-adrenergic receptor and G protein messenger RNA in rat brain. Brain Res. Mol. Brain Res. 5:289–296; 1989. [DOI] [PubMed] [Google Scholar]
- 12. Fennell M.; Khawaja X. Z.; Cockett M. I.; Wood A. Enhanced neuronal differentiation of NTera-2 cells expressing neuronally restricted beta2 adrenergic receptor. Brain Res. 799:243–249; 1998. [DOI] [PubMed] [Google Scholar]
- 13. Gurskaya N. G.; Diatchenko L.; Chenchik A.; Siebert P. D.; Khaspekov G. L.; Lukyanov K. A.; Vagner L. L.; Ermolaeva O. D.; Lukyanov S. A.; Sverdlov E. D. Equalizing cDNA subtraction based on selective suppression of polymerase chain reaction: Cloning of Jurkat cell transcripts induced by phytohemaglutinin and phorbol 12-myristate 13-acetate. Anal. Biochem. 240:90–97; 1996. [DOI] [PubMed] [Google Scholar]
- 14. Horton C.; Maden M. Endogenous distribution of retinoids during normal development and teratogenesis in the mouse embryo. Dev. Dyn. 202:312–323; 1995. [DOI] [PubMed] [Google Scholar]
- 15. Jalink K.; Eichholtz T.; Postma F. R.; van Corven E. J.; Moolenaar W. H. Lysophosphatidic acid induces neuronal shape changes via a novel, receptor-mediated signaling pathway: Similarity to thrombin action. Cell Growth Differ. 4:247–255; 1993. [PubMed] [Google Scholar]
- 16. Kaiser E.; Forster R.; Wolf I.; Ebensperger C.; Kuehl W. M.; Lipp M. The G protein-coupled receptor BLR1 is involved in murine B cell differentiation and is also expressed in neuronal tissues. Eur. J. Immunol. 23:2532–2539; 1993. [DOI] [PubMed] [Google Scholar]
- 17. Leypoldt F.; Lewerenz J.; Methner A. Identification of genes upregulated by retinoic acid induced differentiation of the human neuronal precursor cell line Ntera 2cl.D/1. J. Neurochem. 76:806–814; 2001. [DOI] [PubMed] [Google Scholar]
- 18. MacLennan A. J.; Devlin B. K.; Marks L.; Gaskin A. A.; Neitzel K. L.; Lee N. Antisense studies in PC12 cells suggest a role for H218, a sphingosine 1-phosphate receptor, in growth-factor-induced cell-cell interaction and neurite outgrowth. Dev. Neurosci. 22:283–295; 2000. [DOI] [PubMed] [Google Scholar]
- 19. Maden M.; Ong D. E.; Chytil F. Retinoid-binding protein distribution in the developing mammalian nervous system. Development 109:75–80; 1990. [DOI] [PubMed] [Google Scholar]
- 20. Methner A.; Hermey G.; Schinke B.; Hermans-Borgmeyer I. A novel G protein-coupled receptor with homology to neuropeptide and chemoattractant receptors expressed during bone development. Biochem. Biophys. Res. Commun. 232(2):336–342; 1997. [DOI] [PubMed] [Google Scholar]
- 21. Pleasure S. J.; Lee V. M. NTera 2 cells: A human cell line which displays characteristics expected of a human committed neuronal progenitor cell. J. Neurosci. Res. 35:585–602; 1993. [DOI] [PubMed] [Google Scholar]
- 22. Probst W. C.; Snyder L. A.; Schuster D. I.; Brosius J.; Sealfon S. C. Sequence alignment of the G-protein coupled receptor superfamily. DNA Cell Biol. 11:1–20; 1992. [DOI] [PubMed] [Google Scholar]
- 23. Rebrikov D. V.; Britanova O. V.; Gurskaya N. G.; Lukyanov K. A.; Tarabykin V. S.; Lukyanov S. A. Mirror orientation selection (MOS): A method for eliminating false positive clones from libraries generated by suppression subtractive hybridization. Nucleic Acids Res. 28:E90; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rohrer D. K.; Bernstein D.; Chruscinski A.; Desai K. H.; Schauble E.; Kobilka B. K. The developmental and physiological consequences of disrupting genes encoding beta 1 and beta 2 adrenoceptors. Adv. Pharmacol. 42:499–501; 1998. [DOI] [PubMed] [Google Scholar]
- 25. Rose T. M.; Schultz E. R.; Henikoff J. G.; Pietrokovski S.; McCallum C. M.; Henikoff S. Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res. 26:1628–1635; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ruberte E.; Friederich V.; Chambon P.; Morriss-Kay G. Retinoic acid receptors and cellular retinoid binding proteins. III. Their differential transcript distribution during mouse nervous system development. Development 118:267–282; 1993. [DOI] [PubMed] [Google Scholar]
- 27. Wagner M.; Han B.; Jessell T. M. Regional differences in retinoid release from embryonic neural tissue detected by an in vitro reporter assay. Development 116:55–66; 1992. [DOI] [PubMed] [Google Scholar]
- 28. Zetterstrom R. H.; Lindqvist E.; de Urquiza A. M.; Tomac A.; Eriksson U.; Perlmann T.; Olson L. Role of retinoids in the CNS: Differential expression of retinoid binding proteins and receptors and evidence for presence of retinoic acid. Eur. J. Neurosci. 11:407–416; 1999. [DOI] [PubMed] [Google Scholar]