

Abstract
SR31747A is a sigma ligand that exhibits a potent antitumoral activity on various human tumor cell lines both in vitro and in vivo. To understand its mode of action, we used DNA microarray technology combined with a new bioinformatic approach to identify genes that are modulated by SR31747A in different human breast or prostate cancer cell lines. The SR31747A transcriptional signature was also compared with that of seven different representative anticancer drugs commonly used in the clinic. To this aim, we performed a two-dimensional hierarchical clustering analysis of drugs and genes which showed that 1) standard molecules with similar mechanism of action clustered together and 2) SR31747A does not belong to any previously characterized class of standard anticancer drugs. Moreover, we showed that 3) SR31747A mainly exerted its antiproliferative effect by inhibiting the expression of genes playing a key role in DNA replication and cell cycle progression. Finally, contrasting with other drugs, we obtained evidence that 4) SR31747A strongly inhibited the expression of three key enzymes of the nucleotide synthesis pathway (i.e., dihydrofolate reductase, thymidylate synthase, and thymidine kinase) with the latter shown both at the mRNA and protein levels. These results, obtained through a novel molecular approach to characterize and compare anticancer agents, showed that SR31747A exhibits an original mechanism of action, very likely through unexpected targets whose modulations may account for its antitumoral effect.
Keywords: SR31747A, Anticancer drugs, Transcriptional signature, DNA chip, Thymidine kinase
IDENTIFIED as a sigma receptor ligand, SR31747A exhibits potent antitumoral properties (50). Low concentrations of the drug were shown to inhibit proliferation in yeast and in several human breast and prostate cancer cell lines both in vitro and in vivo, suggesting that it has cancer therapy potential (2,10,28). To date, four specific high-affinity SR31747A binding sites have been identified in humans, three of which have been molecularly characterized: SR31747A binding protein 1 (SRBP-1), which corresponds to the sigma-1 receptor (25,26); the human sterol isomerase (HSI) (19,61), also called the emopamil binding protein, which belongs to the sterol biosynthesis pathway; and SRBP-2 (67), which is homologous to HSI but whose function is not known. The fourth receptor is sigma-2, which has not yet been cloned (2,19,33). SR31747A binding on HSI is known to block cholesterol synthesis, but this does not fully account for the proliferation arrest induced by the drug, and additional pathways may be involved (2). The mechanisms by which SR31747A stops cell cycling and induces apoptosis have not yet been clearly defined. This work is aimed at understanding the SR31747A antitumoral properties through a comparison with conventional anticancer drugs. Our strategy was to apply the DNA chip approach to analyze global gene expression in cells treated with either SR31747A or seven different standard anticancer molecules commonly used in the clinic and to compare SR31747A-induced gene modulations with those induced by the standard molecules.
Antitumoral drugs used in cancer therapy exert their antiproliferative and proapoptotic effects by targeting different process such as DNA synthesis or mitosis in tumor cells. They have been classified according their mechanism of action on the basis of biochemical and cellular biology studies: topoisomerase inhibitors and alkylating agents have been shown to induce DNA strand breaks; spindle poisons bind to microtubules, inhibiting their functions; and antimetabolites disrupt nucleotide pools, thereby inhibiting DNA synthesis. Considering a class, subgroups can be distinguished. For example, although they activate common cellular responses leading to cell cycle arrest and DNA repair and/or apoptosis, DNA-damaging agents can be distinguished by the physical form of the DNA damages they induce or the kinetics of their antiproliferative effects (76). Until recently, these classifications were based on biochemical criteria. With the emergence of global gene expression measurements, such a classification can now be considered according to gene modulations. Many reports on gene expression-based classification have been published. For example, Scherf et al. demonstrated that statistical correlations between cellular transcriptional profiling and drug sensitivity of various cancer cell lines can be used to classify anticancer drugs (58). Dan et al. used a similar approach to demonstrate that different sets of genes can be considered as predictive markers for chemosensitivity to drugs exhibiting similar mechanisms of action (12). However, no drug classifications based on the gene modulations they induced have been reported to date.
Here, we investigated the antitumoral properties of SR31747A. The aim of our study was twofold: 1) to explore the feasibility of drug classification based on their transcriptional signatures, and 2) to characterize the effect of SR31747A in human cancer cells at the gene expression level. Using DNA microarrays combined with a novel bioinformatic approach, we identified gene expression changes following treatment with different classes of standard anticancer drugs or SR31747A in different breast or prostate cancer cell lines. We exploited these signatures to compare the molecules and to identify drug specific biomarkers. This approach made it possible to analyze the impact of SR31747A on tumor cells at the genomic level, to highlight original properties of the molecule, and, as a result, brought new insights into the understanding of its antitumoral activity.
MATERIALS AND METHODS
Reagents
SR31747A was synthesized at the chemistry department of Sanofi-Synthelabo Recherche (Montpellier, France). The stock solution (10 mM) was prepared in 100% ethanol and stored at −20°C. Taxol (paclitaxel), vincristine, 5-fluorouracile (5-FU), methotrexate, doxorubicin, etoposide (VP-16), and melphalan were purchased from Sigma Chemical Co. (St. Louis, MO). A polyclonal antibody was produced against the human thymidine kinase (TK1, referred as TK throughout the manuscript). Briefly, 2 mg TK C-terminal peptide conjugated to BSA (Neosystem, Strasbourg, France) was injected subcutaneously in 250 μl water and 250 μl complete Freund’s adjuvant. Animals were boosted monthly under the same conditions. Sera were collected 10 days after the second and subsequent injections.
Cell Culture and In Vitro Cell Proliferation Assays
The human androgen-independent PC-3 and DU-145 prostate cancer cell lines (American Type Culture Collection, ATCC, Rockville, MD) were maintained in RPMI-1640 medium (Gibco-BRL, MD, USA) while the human breast cancer cell line, MDA-MB-231 (ATCC), was cultured in a mix of DMEM/ Ham’s F12 medium (1:1, v/v Gibco-BRL). Media were supplemented with 10% heat-inactivated fetal bovine serum, 2.5 mM sodium pyruvate, 2 mM l-glutamine, and 20 μg/ml gentamicin (Gibco-BRL). Cells were grown in a humidified 5% CO2/95% air atmosphere at 37°C.
For proliferation assays, DU-145, PC-3, and MDA-MB-231 cells were seeded onto 96-well plates at 4000, 7500, and 10,000 cells/well, respectively. Twenty-four hours later, cells were incubated with or without different drugs for further 24, 48, and 72 h. Cell proliferation and viability were evaluated using the CellTiter 96 Aqueous cell proliferation assay kit (Promega, Madison, WI) as described previously (74). Each measurement was performed in triplicate and the percentage of cell survival was calculated as (DO experiment/DO control) × 100, where “control” referred to untreated cells.
RNA Preparation and Northern Blot Analysis
For transcriptional profiling, cells at 40–80% confluence were treated for 6, 24, or 48 h with different anticancer drugs. Total RNA was isolated from cells using the guanidium isothiocyanate method and purified by ultracentrifugation on cesium chloride gradient. Total RNA (10 μg) was subjected to Northern blot analysis. The RNA quality was assessed by analyzing 18S and 28S ribosomal RNA by electrophoresis through agarose gels containing ethidium bromide or using 2100 Bioanalyzer (Agilent Technologies). Poly(A)+ RNA was isolated using the Fast Track 2.0 Kit (Invitrogen NV, Leek, Netherlands).
The coding region of human TK was amplified by RT-PCR from HeLa 3 cells and subcloned in the NotI site of pGEX 6P-3 (Pharmacia Biotech). Labeling of TK probe was performed with the RTS RadPrime DNA Labeling System according the manufacturer’s recommendations (Gibco BRL).
Biotinylated Probes and Hybridization on Microarrays
Affymetrix human cancer HC-G110 arrays (Santa Clara, CA, USA) containing 1700 cancer-associated genes were used for mRNA expression profiling. Double-stranded cDNA was prepared from 3 μg poly(A)+ RNA using the Life Technologies superscript choice system and an oligo(dT)24 anchored T7 primer. Biotinylated RNA was synthesized using the T7 megascript system (Ambion, Inc., TX, USA) with biotin-11-CTP and biotin-16-UTP for 5 h at 37°C. Following purification, labeled cRNAs were fragmented to 50–200 bases in length at 94°C for 35 min in a buffer containing 200 mM Tris acetate, 500 mM potassium acetate, 150 mM magnesium acetate, pH 8.1. Duplicate arrays were then hybridized with biotinylated cRNA products (10 μg/chip) for 16 h at 45°C using the manufacturer’s hybridization buffer, which contains internal standard RNAs. After hybridization, arrays were washed, stained on the Affymetrix fluidic station 400, and then scanned using a specific scanner (Affymetrix, Hewlett-Packard), as described previously (10).
Statistical Data Analysis
On the Affymetrix human cancer HC-G110 micro-array, a gene is represented by 16–20 pairs of oligonucleotides referred to as a probe set. Each probe pair consists of a Perfect Match (PM) and a Mismatch (MM) oligonucleotide sequence, which differs only by the central base. Following hybridization and washing, scan images were produced and analyzed to calculate a fluorescence intensity value for each probe using the MicroArray Suite 5.0 Suite (Affymetrix). Our analysis began with the normalization of arrays, which is aimed at minimizing variations between different experiments. We used a nonlinear normalization method to generate comparable distributions of PM and MM quantiles of the arrays of interest. Each treatment and control array was first normalized against its replicate and then each treatment array was normalized against its corresponding control. We then calculated the fluorescence intensity of each probe set. Different methods have been previously proposed to combine probe intensities of a given probe set to get a measure of the expression of the corresponding gene at the mRNA level. Instead of using Affymetrix algorithms, we calculated expression levels following the method described by Efron et al. (16), which is defined by: intensity = (1/N){Σ[log2(PM) − 0.5*log2(MM)]}, where N indicates the number of probe pairs for a given probe set and PM and MM are the fluorescence intensities of the perfect match and mismatch probes of each pair, respectively. Following intensity calculation, genes that were differentially expressed between a treated and a control sample were identified. A commonly used approach is the simple-minded fold-change approach, in which a gene is referred as modulated if its expression level in a sample varies from more than a constant factor when compared with the corresponding control condition. Alternatively, statistical scores like t-test and regularized t-statistics (3,14,30,66) have been recently used to identify such genes. We combined several methods to statistically identify regulated genes using scores associated with each genes: 1) the fold change, 2) the Welch statistic, and 3) the regularized t-statistic, and 4) the entropy is defined by H = H (treated) − H (control) with H (condition) = Σ{p(x)log2 [p(x)]}, where p(x) is the expression intensity in the treated or control sample divided by the sum of the treated and control intensities and where the sum is performed on replicates, and a measure of correlation as described by Staunton et al. (62) as Stat = (μ1 − μ2)/(σ1 + σ2), where (μ1,σ1) and (μ2,σ2) indicate the means and standard deviations of the expression level of a given gene in treatment and control conditions, respectively. Those scores make it possible to integrate the effect of the gene population size and the intersample variability. Different scores were ranked and candidate modulated genes were selected given some randomly fixed cut-offs as the intersection of the top genes for each calculated score. Finally, selected genes for each treatment versus control comparison were retained for hierarchical cluster analysis. Entropy scores of the selected genes for each replicated comparison were used to perform two-dimensional hierarchical clustering using Cosine correlation as similarity measure and grouping following the average linkage algorithm (UPGMA) to cluster genes, and using Pearson correlation and WARD agglomeration to cluster drugs. Cluster analysis was performed using the GeneMaths software package (Applied Maths Inc.).
Protein Extract Preparation and Western Blot Analysis
Following treatment with different drugs, cells were harvested with trypsin-EDTA and centrifuged at 1400 rpm min−1. Cells were lysed in NP40 buffer (NaCl 150 mM, EDTA 1 mM, DTT 1 mM, Tris pH 7.5 at 50 mM, NP40 0.2%, SDS 0.1%, and Boeringher antiprotease mix) for 15 min. Total cell lysates were then briefly sonicated and, after centrifugation, the protein concentration in the supernatant was quantified using the Bio-Rad assay kit (Promega). After boiling in Laemmli buffer, 30 μg of cellular proteins was separated on 10% SDS-PAGE. Then proteins were transferred onto PVDF membranes (Hoeffer). To ensure an equivalent protein loading, PVDF membranes were stained with Amido Black solution (Sigma). Following blocking with 5% milk/PBS/0.1% Tween 20, blots were incubated for 1 h at room temperature with the anti-thymidine kinase 1 serum (1:1000) and visualized using an enhanced chemiluminescent detection system (Super-Signal; Pierce Chemical Company, Rockford, IL).
RESULTS
Selection of the Drug Doses
The transcriptional profiles of standard antineo-plastic drugs and SR31747A were determined in two prostate cancer cell lines, PC-3 and DU-145, and a breast cancer cell line, MDA-MB-231. These cell lines were selected as they were shown to express SR31747A receptors and to be sensitive to the anti-proliferative effect of SR31747A (2). They are hormone independent and mutated in the p53 gene and they weakly express the MDR gene product (1,49). The standard anticancer agents we used included two spindle inhibitors, Taxol and vincristine; two antimetabolites, methotrexate and 5-FU; an alkylating agent, melphalan; and two topoisomerase inhibitors, doxorubicin and etoposide.
Different criteria must be defined to assess the transcriptional signatures of drugs (i.e., the doses and treatment time to be used for each cell line). We first performed proliferation studies because drug sensitivity varies between cell lines. For each drug and for comparative purposes, the corresponding IC70 dose at 72 h was chosen as an endpoint. IC70 was selected as this concentration is elevated enough to guarantee a marked drug effect on cells, whereas beyond this threshold drugs may induce responses unrelated to the pharmacology under investigation (Table 1). The concentrations ranging from 50 nM to 30 μM are in accordance with those previously reported at the NCI (58). Even at the highest dose used, MDA-MB-231 and PC3 cells appeared to be resistant to the antimetabolites used and to etoposide, respectively. Therefore, those cell/treatment points were omitted for the transcriptional analysis. Considering SR31747A, a treatment with doses ranging from 0.1 to 100 μM produced a dose-dependent inhibition of cell growth in each cell line (data not shown). Although MDA-MB-231 were the most sensitive cells to low SR31747A concentrations, 30 μM SR31747A similarly inhibited cell proliferation in the three cell lines by approximately 50–60%. Therefore, we chose this dose for further transcriptional studies.
TABLE 1.
SR31747A AND STANDARD ANTICANCER DRUGS DOSES USED FOR TRANSCRIPTIONAL PROFILING IN THE DIFFERENT CELL LINES STUDIED
| PC-3 | DU-145 | MDA-MB-231 | |
|---|---|---|---|
| Taxol | 200 nM | 100 nM | 100 nM |
| Vincristine | 50 nM | 50 nM | 50 nM |
| 5-FU | 50 μM | 20 μM | not active |
| MTX | 100 nM | 200 nM | not active |
| Melphalan | 30 μM | 30 μM | 30 μM |
| Etoposide | not active | 5 μM | 10 μM |
| Doxorubicin | 2 μM | 2 μM | 5 μM |
| SR31747A | 30 μM | 30 μM | 30 μM |
IC70 doses were defined in the antiproliferative assays as described in Materials and Methods.
Kinetics of Gene Modulations in Response to Standard Anticancer Drugs or SR31747A
Anticancer drugs display a specific kinetics of action, and as a result one may expect specific kinetics of gene modulations both in terms of number and identity. Therefore, responses to the different anticancer drugs were first evaluated by analyzing the number of genes modulated in the three cell lines as a function of time using the Affymetrix DNA microarrays containing 1700 cancer-associated genes. Global gene expression patterns in treated cells at a given time were compared with that of control (vehicle-treated cells), and each experiment was performed in duplicate. Modulated genes (either induced or repressed) are numbered. This evaluation was aimed at determining the time period that would be the most appropriate for the clustering analysis of all drugs simultaneously, whatever their mode of action. It would be the time period in which all drugs gave a significant response in terms of modulated gene numbers.
As shown in Figure 1, 6-h treatments lead to contrasting drug responses. For example, spindle inhibitors (Taxol and vincristine) and antimetabolites (methotrexate and 5-FU) modulated no or very few genes. The rapid cellular response to doxorubicin is consistent with DNA being its primary target, contrasting with the indirect action of tubulin inhibitors or antimetabolites. By contrast, more genes are modulated thereafter. At 24 h, spindle inhibitors and antimetabolites modulated the expression of about 30 genes while treatments with topoisomerase inhibitors dramatically affected gene expression as evidenced by 40 to 182 modulated genes. At 48 h we observed that drug treatments affected twice to threefold more genes on average compared to 24 h. This change was even more pronounced for antimetabolites in the prostate cancer cell lines with a change by a factor of 2 to 8. At 48 h, 20 to 100 genes were modulated by antimetabolites and melphalan while spindle inhibitors and topoisomerase inhibitors modulated from 46 to 230 genes. Although adequate for some drugs with rapid action, a 6-h treatment is too short to compare all the drug responses. Conversely, considering that cells are doomed to die irrespective of the drug they were treated with, a long treatment may hamper dissecting drug primary responses. Indeed, it would be too long to characterize early events that are specific for a given drug. Prominent modulations observed at a long-time treatment may only reflect in their majority the activation of common cellular pathways that lead to cell death, precluding any drug distinction. Therefore, considering that substantial numbers of genes were modulated with all the drugs tested at 24 h, this time was chosen for clustering experiments. At 24 h, compared with the standard anticancer drugs, the numbers of genes modulated by SR31747A were intermediary. The molecule affected the expression of approximately 50 genes in DU-145 and 90 genes in PC-3 or MDA-MB-231 cell lines.
Figure 1.
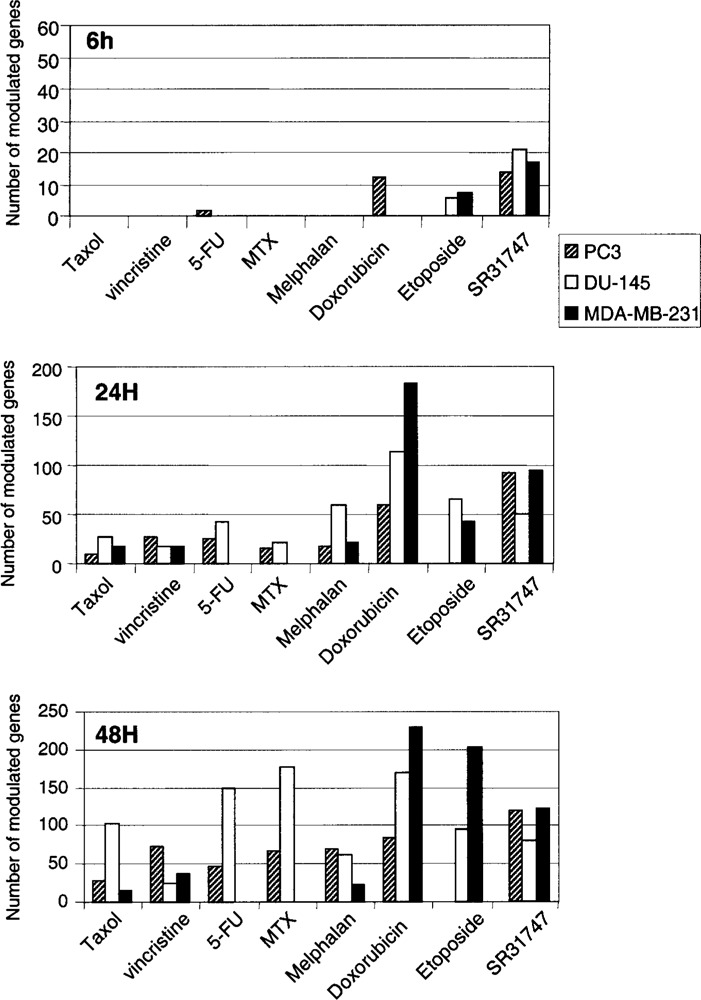
Numbers of genes modulated following a 24-h treatment with the indicated drug. PC-3, DU-145, and MDA-MB-231 cell lines were treated for 24 h at doses described in Table 1 and the number of modulated genes was evaluated as described in Materials and Methods.
Standard Anticancer Drug Profiles
To compare the gene expression signatures of the different molecules, the entropy scores for selected modulated genes were used to establish the two-dimensional hierarchical clustering using Pearson correlation and Ward agglomeration to classify sample and cosine distance with UPGMA to cluster genes as described in the Materials and Methods section. The clustering obtained for each cell line independently is shown in Figure 2.
Figure 2.
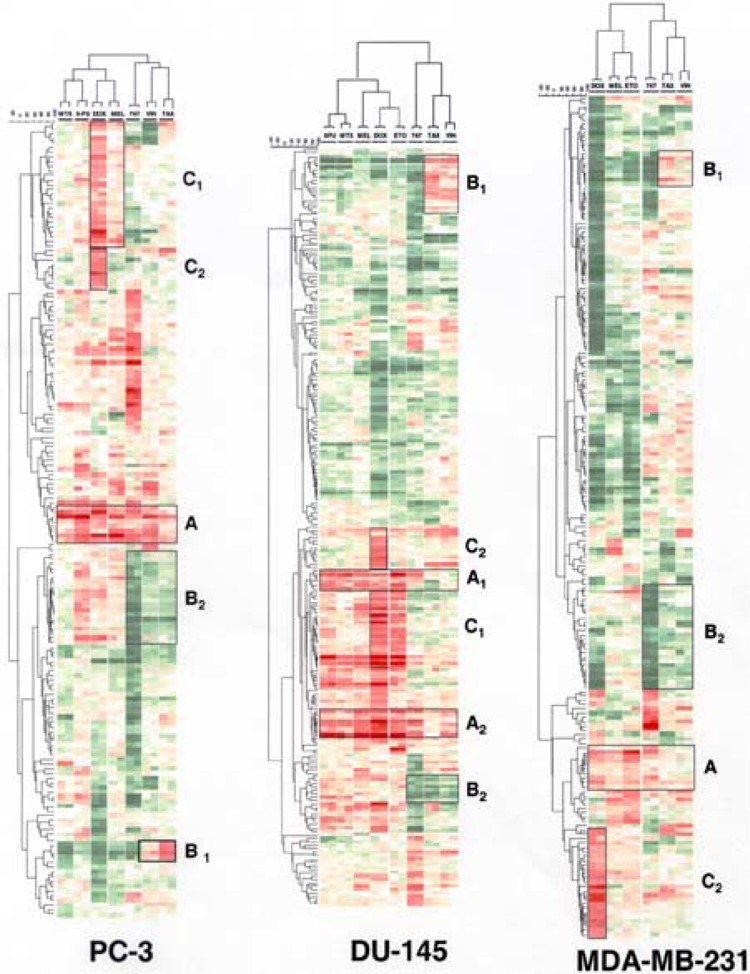
Two-dimensional hierarchical cluster analysis of gene expression profiles following treatments with anticancer drugs. PC-3, DU-145, and MDA-MB-231 cells were treated for 24 h at doses described in Table 1. Unsupervised hierarchical clustering (GeneMaths software) was applied to the list of modulated genes as described in Materials and Methods. Increases in mRNA expression are indicated by shades of red while decreases are shown by shades of green. Cluster A: genes involved in the stress/apoptosis responses in PC-3 and MDA-MB-231; this cluster is divided into clusters A1 and A2 in DU-145. Cluster B1: spindle inhibitors biomarkers; genes involved in cell cycle progression and mitosis. Cluster B2: spindle inhibitors biomarkers; genes involved in replication process. Cluster C1: topoisomerase inhibitors biomarkers; genes involved in response to interferon. Cluster C2: doxorubicin biomarkers; doxorubicin-induced genes.
In the three cell lines, when the reference molecules were classified according the gene modulations they induced, we observed two major distinct groups: one group consisted of spindle inhibitors (Taxol and vincristine) while the other group included antimetabolites, topoisomerase inhibitors, and the alkylating agent melphalan (Fig. 2). Those two groups were evidenced in the prostate cell lines and in the breast cancer cell line as well, indicating that this classification is not cell line dependent. The second group could be further subdivided: the antimetabolites (5-FU and methotrexate) were on the same branch, distinct from the topoisomerase inhibitors, which were grouped together. The alkylating agent defined its own branch; its signature appeared to be closer to that of the topoisomerase inhibitors than that of the antimetabolites—so close that in the MDA-MB-231 cell line, melphalan was grouped with etoposide within the group of topoisomerase inhibitors.
Such a classification made it possible to identify drug biomarkers easily, which are genes specifically and reproducibly modulated by a drug whatever the cell line. A group of genes was specifically induced by Taxol and vincristine while being repressed by other drugs. These genes are the spindle inhibitor drug biomarkers. They are involved in cell cycle progression and mitosis (cluster B1, Fig. 2) and included, for example, cyclin B, cyclin A, the centromere protein A (CENP-AP), which is an essential component of centromeres (16); E2-EPF, which is required for ubiquitin–protein conjugation (31); or CIP2, a tyrosine-serine phosphatase that interacts with cyclin-dependent kinases and inhibits progression through the cell cycle (18). Cluster B2 consisted of other spindle inhibitor biomarkers that were repressed by Taxol and vincristine while induced by other drugs. These are genes essential for the initiation of DNA replication (such as DNA primase, Cdc6, Cdc7-related kinase, mcm4, or mcm2) (34) or for progression of the DNA replication process (replication factors A and C, or PCNA) (24) (cluster B2, Fig. 2). The transcriptional signatures of topoisomerase inhibitors, doxorubicin and etoposide, in PC-3 and DU-145 revealed that those molecules specifically induced a cluster of genes involved in the response to interferon (cluster C1 included HUMII56KD, ISG54, HUMI-FN15K) (40,43,60). Cluster C2 (histone H2A.2, H2A.1B, H2, H4, CRABP-II, MK) was mainly induced following treatment with doxorubicin in the three cell lines. This marked cluster of upregulated genes likely reflects that this molecule specifically induces histone gene regulation in response to the retinoic acid and interferon regulatory factor pathways (71,73,75). Contrasting with specific modulations, we observed that a group of genes was induced by all the drugs in each cell line (clusters A in PC-3 and MDA-MB-231; A1 and A2 in DU-145). Those genes were previously described to be involved in stress/apoptosis responses: interleukin 8, Gadd153/Chop, ATF3, CL100, Ets-2, MAD-3, and A20 factor (13,37,38,52,56,57). CHOP/gadd153 is a transcription factor expressed in response to genotoxic, oxidative, UV, or MMS treatment (38). ATF-3 is a stress-inducible gene with a putative role in apoptosis (37). IL-8 does not kill cells per se but can be activated in response to a genotoxic stress; its expression correlates with induction of apoptosis (7) (clusters A in PC-3 and MDA-MB-231; A1 and A2 in DU-145). Interestingly, this cluster also contained genes involved in cell cycle progression (WAF-1/p21 and gadd45) (17,75). Their induction in response to DNA-damaging or antimitotic agents has been previously described. Because the three cell lines we used are deficient in p53, these results suggest a p21-mediated proliferation arrest that is independent of p53.
Validating our method, the hierarchical clustering we obtained correlated with the presumed mechanism of action of the reference anticancer molecules in each cell line tested, because reference drugs with similar mode of action clustered together.
Molecular Classification of SR31747A
We used the SR31747A transcriptional signature to classify and compare the molecule with other drugs. As seen in Figure 2, we observed that the molecule did not belong sensu stricto to any group defined by the reference anticancer drugs. The SR31747A signature appeared to differ markedly from that of antimetabolites, topoisomerase inhibitors, or the alkylating agent. The molecule tended to cluster with spindle inhibitors, indicating that SR31747A shared substantial gene modulations with that class. Specifically, the clustering of SR31747A with spindle inhibitors resulted from a similar repression of genes of cluster B2, but the molecule does not belong to this class as it did not induce cluster B1; even more, SR31747A strongly inhibited the expression of these genes.
Dissection of SR31747A Molecular Impact
Unsurprisingly, like other anticancer drugs and irrespective of the cell line, SR31747A induced genes of cluster A, indicating the establishment of apoptotic and stress responses (Figs. 2 and 3). For example, genes such as IL-8, GADD153/Chop, ATF3, and MAD3, as well as the A20 factor, the cysteine protease ICErel-II (45), and p57(Kip2), a member of the Cip/Kip family of CDK inhibitors (29), were induced following a treatment with SR31747A. Among the other genes that were highly affected by the drug, two prominent functional groups emerged: genes involved in cell cycle progression and DNA metabolism (Fig. 3).
Figure 3.
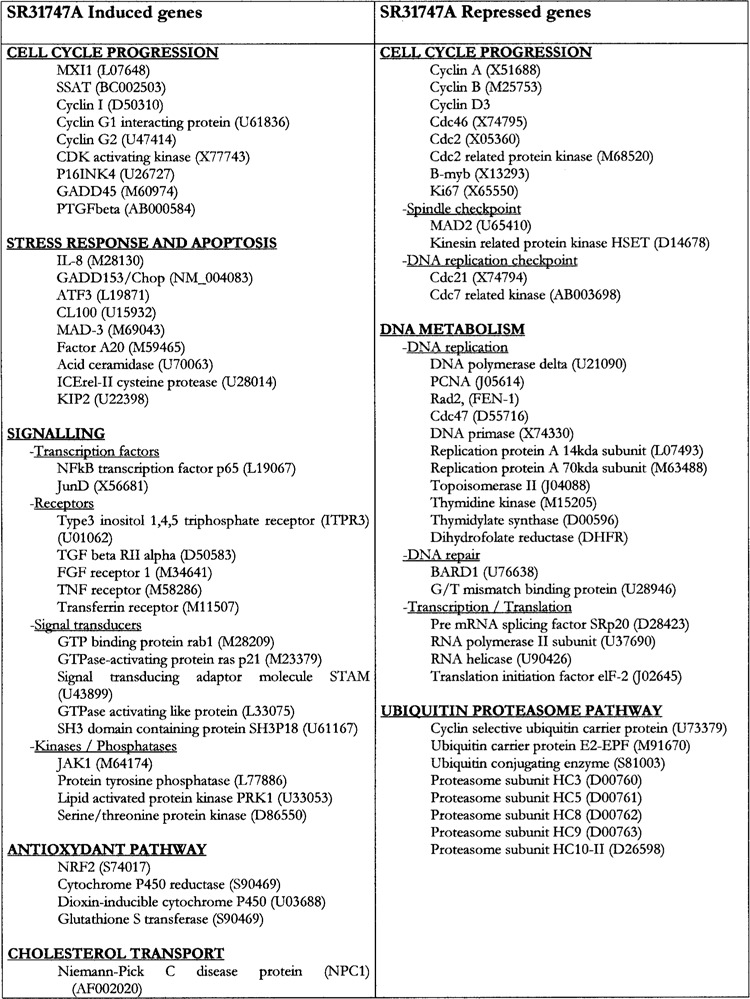
Functional assignment of genes that are modulated by SR31747A. The accession number of each gene is indicated.
A treatment with SR31747A modulated many genes that are known to control cell cycle progression. They included the induction of cyclin G2, a negative regulator of cell cycle progression (20); p16INK4, a tumor suppressor gene encoding an inhibitor of cdk4 that blocks entry into the S phase (39); MXI1, a transcriptional repressor that antagonizes myc activity (63); the spermidine/spermine N1 acetyltransferase, SSAT, that belongs to the polyamine metabolism and whose enhanced activity resulted in accumulation of cells in the G2/M phases (60). Concomitantly, SR31747A inhibited the expression of genes controlling mitosis. Those genes are cyclin A, cyclin B, cyclin D3, and cdc2, which is required for entry into S phase and mitosis. Consistent with these modulations, the expression of Ki67, a proliferation marker, was repressed (4). Finally, two genes that belong to the spindle checkpoint were reduced: MAD2 and HSET, the kinesin-related protein kinase; they regulate the microtubule organization at mitotic spindle poles (8,44). Among the genes that were specifically modulated by SR31747A (and not by other drugs), we observed that the molecule significantly induced PTGF-β. PTGF-β is a distantly related member of the TGF-β superfamily, designated placental TGF-β that is upregulated in response to both p53-dependent and -independent apoptotic signaling events arising from DNA damage. PTGF-β has been described to inhibit tumor cell growth via the TGF-β signaling pathway (65).
Besides the modulation of genes involved in cell cycle progression, altered expression of many other genes is consistent with a general shutdown of the DNA metabolism. This was noted at different levels. First, DNA replication was dramatically affected with the inhibition of the expression of key actors as well as regulators of that process (35). They included the DNA polymerase delta and its auxiliary protein, PCNA; the replication protein A 14- and 70-kDa sub-units (24); the DNA primase, which is the polymerase synthesizing small RNA primers for the okazaki fragments (34); thymidine kinase (72), thymidylate synthase (6), and dihydrofolate reductase (59), three key enzymes of the purine synthesis pathway; and topoisomerase II, which makes double strand breaks (5). The transcription/translation process was also repressed as shown by the inhibition of the expression of the RNA polymerase II subunit, the RNA helicase, the translational initiation factor 2 (elF-2) (68), and the pre-mRNA splicing factor SRp20 (22). DNA repair was also affected by the inhibition of the expression of BARD1, which plays a regulatory role during transcription (27), and the G/T mismatch binding protein (23), rad2 (FEN-1), which is required for chromosome segregation and recovery from DNA damage (21). Genes of the DNA replication checkpoint (P1-cdc21 and cdc7 related kinase) were also repressed (36,46). Some of those genes were included in cluster B2, which defined the spindle inhibitor biomarkers.
Contrasting with the blockade of the DNA replication process, cellular signaling was boosted as indicated by the induction of many actors of the signaling machinery. For instance, SR31747A triggered the TGF-β and TNF signaling pathways by inducing the expression of the TGF-β type II receptor and the TNF receptor, respectively. JAK1, which is one of the early components of TNF signaling, was also induced (42). Signal transducers such as rab1, ras, STAM, and the GTPase activating like protein showed enhanced expression (32,48,55). Two transcription factors, NFKB and JunB, which are known to cooperate, were also induced. In addition, an antioxidant pathway was induced as evidenced by the enhanced expression of cytochrome P450 reductase, dioxin-inducible cytochrome P450, the transcription factor, NRF2, and one of its targets, glutathione-S-transferase (47). A gene encoding the Niemann-Pick disease type C1 protein (NPC1) was significantly induced by SR31747A. NPC1 is a lysosomal sterol transporter whose overexpression is associated with abnormal regulation of cellular cholesterol content and distribution (41). This modulation is of interest considering the role played by HSI, one of the SR31747A receptors, in cholesterol metabolism (61). Finally, a cluster of genes involved in the ubiquitin proteasome pathway was repressed: they included the cyclin selective ubiquitin carrier protein, the ubiquitin carrier protein E2-EPF, the ubiquitin conjugating enzyme, and numerous proteasome subunits (HC3, HC5, HC8, HC9, and HsC10-II), suggesting that the ubiquitin/proteasome-dependent protein degradation pathway is impaired (9).
Taken together, these results showed that the molecule had a marked impact on cells: it affected different pathways, mainly cell cycle progression and DNA metabolism. The modulations induced by the drug likely account for a blockade of critical cellular pathways, ultimately leading to cell death.
SR31747A Inhibits TK Expression Both at the mRNA and Protein Levels
SR31747A strongly reduced the expression of TK, thymidylate synthase (TS), and dihydrofolate reductase (DHFR). These repressions were SR31747A specific compared with the reference molecules. Only SR31747A and 5-FU modulated these genes but in opposite directions (Fig. 4A), as 5-FU induced their expression. To confirm the results obtained using the DNA microarrays, we focused our interest on one target (i.e., TK) and analyzed the impact of SR31747A on its expression both at the mRNA and protein levels. As shown in Figure 4B, Northern blot experiments indicated a strong decrease in the TK transcript level observed between 6 and 24 h when cells were treated with SR31747A. Finally, to determine whether this transcriptional downregulation of TK by SR31747 also occurred at the protein level, we performed Western blot experiments with total protein extracts from cells treated with SR31747A using a polyclonal TK antibody we produced. As shown in Figure 4C, SR31747A led to a complete inhibition of the TK protein expression.
Figure 4.
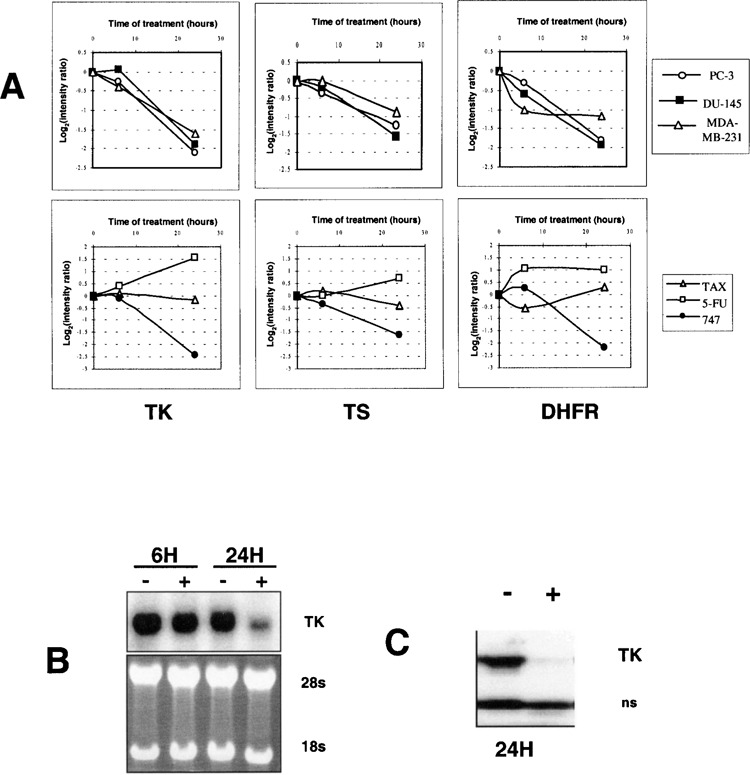
Effect of SR31747A on the expression of genes involved in nucleotide synthesis. (A) PC-3, DU-145, and MDA-MB-231 cells were treated with SR31747A 30 μM for 6 and 24 h. Transcript levels of TK (thymidine kinase), TS (thymidylate synthase), and DHFR (dihydrofolate reductase) were evaluated using cDNA microarrays as described in Materials and Methods. The effect of SR31747A was compared with that of Taxol and 5-FU on TK, TS, and DHFR gene expression. The average modulation in gene expression compared with untreated cells was calculated from duplicate hybridizations. Representative results in the PC-3 cell line are shown. (B) Northern blot validation of SR31747A-dependent inhibition of the expression of the thymidine kinase gene expression. Log-phase cells were treated (+) or not (−) with SR31747A, for 6 and 24 h. Total RNA was prepared and analyzed by Northern blot. Representative results in the DU-145 cell line are shown. (C) Western blot analysis of the thymidine kinase protein expression level in response to SR31747A in PC-3 cells treated (+) or not (−) for 24 h with SR31747A (30 μM). TK protein expression level was evaluated from total protein extracts by Western blot analysis using an anti-TK serum.
DISCUSSION
Monitoring global changes in gene expression using DNA microarrays is now routinely used for different purposes. For example, gene expression profiles are defined to analyze the impact of the expression/deletion of a gene of interest, during a physiological or a pathological process to follow with time changes in gene expression, in response to the treatment with a drug, or to compare and classify pathological samples according their respective gene expression patterns. In oncology, the latter has been exemplified by numerous recent reports where gene expression profiles were used to classify tumors to generate tools for diagnostic or therapeutic purposes.
The group of Weinstein et al. at the NCI developed an original strategy whereby gene expression profiles are used to determine the molecular basis of differential chemosensitivity responses (58,70). With the ultimate aim of producing predictive tools that would be used clinically to define potential responses of patients before initiating the treatment, their approach is devoted to correlate drug sensitivity with gene expression patterns of a panel of 60 human cancer cell lines. Their procedures focus on untreated cells.
In contrast with this approach, our strategy was to profile gene expression of cells treated with different anticancer drugs, to classify reference molecules according to the modulations they induced at the mRNA level and to apply this classification as a tool to assign an investigational drug into classes of reference anticancer compounds to obtain information on its mode of action. To compare drug effects at the transcriptional level, a careful experimental design is warranted considering cells, doses, and duration of treatment. We selected three different cell lines (PC-3, DU-145, and MDA-MB-231), which were previously documented to be sensitive to SR31747A and to most anticancer drugs used in this study. The doses we used were the IC70 of the molecules as defined in preliminary proliferation experiments. Those doses are sufficient to induce a marked cellular response. Upon treatment, alterations in gene expression occurred in a time-dependent manner, exhibiting gradual changes over time. We decided to perform 24-h treatments because this time appeared to be the most appropriate to analyze all the drugs included in our study simultaneously. Shorter treatments would limit the impact of molecules with a slow mechanism of action while longer treatments would favor the appearance of drug secondary or tertiary responses. Considering that cell death is the ultimate end point for all the compounds, common pathways leading to cell death would prevail. This would impair the molecule classification.
Validating our approach, the seven reference anti-cancer compounds included in this study were grouped into classes that reflected the families of molecules based on their known mechanism of action. The two spindle inhibitors (Taxol and vincristine) were grouped together in a branch distinct from other molecules that directly or indirectly target DNA. The two topoisomerase inhibitors (doxorubicin and etoposide) were classified on the same branch near melphalan, which binds DNA. Finally, the two antimetabolites (5-FU and methotrexate) were grouped together. This classification made it possible to identify clusters of genes that are either common between drug families or specific to a given anticancer drug class. As expected, shared modulations included genes involved in apoptosis and stress responses, indicative of a common cell death induction. Contrasting with these common modulated genes, several clusters were specifically modulated in response to a class of anti-cancer drugs, thus reflecting drug signatures. For example, the induction of cluster B1 comprising many genes regulating cell cycle progression such as cyclin B or CENP-A was a hallmark of spindle inhibitors after a 24-h treatment, while the modulation of genes involved in the retinoic acid or interferon pathways was associated with topoisomerase inhibitors.
To characterize the SR31747A antiproliferative effect, we first compared its transcriptional signature with that of reference anticancer compounds. We observed that the molecule signature was significantly different from that of topoisomerase inhibitors, alkylating agents, or antimetabolites. Even though the closest signatures appeared to be those of spindle inhibitors, we found that the molecule exhibited specific modulations (e.g., contrasting with the spindle inhibitors, SR31747A inhibited the expression of genes included in cluster B2). This classification suggested that SR31747A modulated different pathways that conferred to the molecule an original mechanism of action.
The analysis of the impact of SR31747A on tumor cells revealed that the molecule dramatically affected two key cellular pathways: the molecule interfered with cell cycle progression and impaired the DNA metabolism process at different levels. A treatment with the molecule resulted in the induction of negative regulators of the cell cycle and dramatically reduced the expression of numerous genes involved in DNA synthesis and repair. Besides its direct impact on cell cycle, SR31747A may affect its progression indirectly though the reduction of the expression of many elements of the proteasome/ubiquitin pathway. Indeed, many of the short-lived regulatory proteins that govern cell division, growth, activation, signaling, and transcription are substrates that are temporally degraded by the proteasome. Thus, the impaired activity of the proteasome may interfere with the temporal control of the cell cycle and thereby contribute to the antiproliferative effect of SR31747A. All these modulations may account for the blockade of tumor cell growth and are consistent with the G1/S arrest induced by the drug. Similar to conventional anticancer drugs, SR31747A induces cell death as shown by a fast and high upregulation of genes involved in apoptosis and stress responses.
Among the genes that are modulated by SR31747A, we gave special emphasis on the inhibition of TK and TS, because they are being considered as potential key targets for cancer chemotherapy (64). For example, 5-FU is an inhibitor of TS activity. One of the main barriers to its chemotherapeutic efficacy in the clinic is the emergence of cellular resistance, which was shown to correlate with increased TS expression. High TK expression levels have also been proposed to predict a poor response to 5-FU chemotherapy (11,51,53,54). Strategies aimed at preventing the emergence of such cellular resistances are the focus of considerable attention. The blockage of TS protein induction together with the repression of TK gene expression, which would limit the activity of the alternative salvage pathway of pyrimidine synthesis, may be one of these strategies. In this setting, we propose that the combination of 5-FU with SR31747A would be interesting in regard to the concomitant inhibition of TK and TS gene expression obtained in response to a treatment with that molecule. In vivo studies would be warranted to test this hypothesis and this is currently explored in our laboratory.
Notably, our approach of the mechanism of action of SR31747A was based upon the characterization of the global gene modulations induced by the molecule in three different cell lines. Such an approach made it possible to visualize the overall impact of the molecule. However, when a cell is treated by a drug, transcriptional changes can be either primary responses to the treatment or secondary to cellular processes responding to the treatment. Therefore, one can infer that among all the genes that were modulated following SR31747A treatment, only some or a few modulations may be crucial and mediate SR31747A’s effect. Dissecting the molecule’s mode of action warranted the identification of its primary targets. The answer to this could help understand mechanistically how SR31747A blocks cell proliferation. This issue could be assessed by subtracting genes that are modulated by SR31747A in cells that have been selected for their resistance to the antiproliferative activity of the molecule. An additional important question would be to define the role played by each receptor in mediating SR31747A’s antiproliferative effect. This can be performed on different cells expressing one receptor exclusively or a combination of different SR31747A receptors. These cells could be identified or produced through an expression screening, the use of receptor killing by RNAi, and/or the characterization of new molecules with a specific selectivity spectrum towards HSI/SRBP1/SRBP2/sigma2.
Altogether our results demonstrated the feasibility of drug classification based solely on gene expression monitoring in treated cells and suggested a general strategy for characterizing investigational drugs independently of a biological knowledge. The assessment of gene expression patterns in different cell types and under diverse treatments with anticancer agents would lead to increasingly detailed maps of gene expression profiles associated with drugs.
REFERENCES
- 1. Alvarez M.; Paull K.; Monks A.; Hose C.; Lee J. S.; Weinstein J.; Grever M.; Bates S.; Fojo T. Generation of a drug resistance profile by quantitation of mdr-1/P-glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. J. Clin. Invest. 95:2205–2214; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berthois Y.; Bourrie B.; Galiegue S.; Vidal H.; Carayon P.; Martin P. M.; Casellas P. SR31747A is a sigma receptor ligand exhibiting antitumoural activity both in vitro and in vivo. Br. J. Cancer 88:438–446; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Broberg P. Statistical methods for ranking differentially expressed genes. Genome Biol. 4:R41; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown D. C.; Gatter K. C. Ki67 protein: The immaculate deception? Histopathology 40:2–11; 2002. [DOI] [PubMed] [Google Scholar]
- 5. Burden D. A.; Osheroff N. Mechanism of action of eukaryotic topoisomerase II and drugs targeted to the enzyme. Biochim. Biophys. Acta 1400:139–154; 1998. [DOI] [PubMed] [Google Scholar]
- 6. Carreras C. W.; Santi D. V. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev. Biochem. 64:721–762; 1995. [DOI] [PubMed] [Google Scholar]
- 7. Choi C.; Kutsch O.; Park J.; Zhou T.; Seol D. W.; Benveniste E. N. Tumor necrosis factor-related apoptosis-inducing ligand induces caspase-dependent interleukin-8 expression and apoptosis in human astroglioma cells. Mol. Cell. Biol. 22:724–736; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chung E.; Chen R. H. Spindle checkpoint requires Mad1-bound and Mad1-free Mad2. Mol. Biol. Cell 13: 1501–1511; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ciechanover A. The ubiquitin-mediated proteolytic pathway: Mechanisms of action and cellular physiology. Biol. Chem. Hoppe Seyler 375:565–581; 1994. [DOI] [PubMed] [Google Scholar]
- 10. Cinato E.; Peleraux A.; Silve S.; Galiegue S.; Dhers C.; Jbilo O.; Loison G.; Casellas P. A DNA micro-array-based approach to elucidate the effects of the immunosuppressant SR 31747A on gene expression in Saccharomyces cerevisiae . Gene Expr. 10:213–230; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Copur S.; Aiba K.; Drake J. C.; Allegra C. J.; Chu E. Thymidylate synthase gene amplification in human colon cancer cell lines resistant to 5-fluorouracil. Biochem. Pharmacol. 49:1419–1426; 1995. [DOI] [PubMed] [Google Scholar]
- 12. Dan S.; Tsunoda T.; Kitahara O.; Yanagawa R.; Zembutsu H.; Katagiri T.; Yamazaki K.; Nakamura Y.; Yamori T. An integrated database of chemosensitivity to 55 anticancer drugs and gene expression profiles of 39 human cancer cell lines. Cancer Res. 62: 1139–1147; 2002. [PubMed] [Google Scholar]
- 13. De Valck D.; Jin D. Y.; Heyninck K.; Van de C. M.; Contreras R.; Fiers W.; Jeang K. T.; Beyaert R. The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene 18:4182–4190; 1999. [DOI] [PubMed] [Google Scholar]
- 14. Dudoit S.; Yang H. Y.; Callow M. J.; Speed T. P. Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Technical report; 2000. http://www.stat.berkeley.edu/users/terry/zarray/TechReport/578.pdf
- 15. Earnshaw W. C.; Cooke C. A. Proteins of the inner and outer centromere of mitotic chromosomes. Genome 31:541–552; 1989. [DOI] [PubMed] [Google Scholar]
- 16. Efron B.; Tibshirani R.; Goss V.; Chu G. Micro-arrays and their use in a comparative experiment. Technical report; 2000. http://www-stat.stanford.edu/∼tibs/ftp/microarrays.pdf
- 17. El Deiry W. S.; Tokino T.; Velculescu V. E.; Levy D. B.; Parsons R.; Trent J. M.; Lin D.; Mercer W. E.; Kinzler K. W.; Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell 19(75): 817–825; 1993. [DOI] [PubMed] [Google Scholar]
- 18. Gyuris J.; Golemis E.; Chertkov H.; Brent R. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell 19(75):791–803; 1993. [DOI] [PubMed] [Google Scholar]
- 19. Hanner M.; Moebius F. F.; Weber F.; Grabner M.; Striessnig J.; Glossmann H. Phenylalkylamine Ca2+ antagonist binding protein. Molecular cloning, tissue distribution, and heterologous expression. J. Biol. Chem. 270:7551–7557; 1995. [DOI] [PubMed] [Google Scholar]
- 20. Horne M. C.; Donaldson K. L.; Goolsby G. L.; Tran D.; Mulheisen M.; Hell J. W.; Wahl A. F. Cyclin G2 is up-regulated during growth inhibition and B cell antigen receptor-mediated cell cycle arrest. J. Biol. Chem. 272:12650–12661; 1997. [DOI] [PubMed] [Google Scholar]
- 21. Hosfield D. J.; Mol C. D.; Shen B.; Tainer J. A. Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: Coupling DNA and PCNA binding to FEN-1 activity. Cell 95:135–146; 1998. [DOI] [PubMed] [Google Scholar]
- 22. Huang Y.; Steitz J. A. Splicing factors SRp20 and 9G8 promote the nucleocytoplasmic export of mRNA. Mol. Cell 7:899–905; 2001. [DOI] [PubMed] [Google Scholar]
- 23. Hughes M. J.; Jiricny J. The purification of a human mismatch-binding protein and identification of its associated ATPase and helicase activities. J. Biol. Chem. 267:23876–23882; 1992. [PubMed] [Google Scholar]
- 24. Iftode C.; Daniely Y.; Borowiec J. A. Replication protein A (RPA): The eukaryotic SSB. Crit. Rev. Biochem. Mol. Biol. 34:141–180; 1999. [DOI] [PubMed] [Google Scholar]
- 25. Jbilo O.; Vidal H.; Paul R.; De Nys N.; Bensaid M.; Silve S.; Carayon P.; Davi D.; Galiegue S.; Bourrie B.; Guillemot J. C.; Ferrara P.; Loison G.; Maffrand J. P.; Le Fur G.; Casellas P. Purification and characterization of the human SR 31747A-binding protein. A nuclear membrane protein related to yeast sterol isomerase. J. Biol. Chem. 272:27107–27115; 1997. [DOI] [PubMed] [Google Scholar]
- 26. Kekuda R.; Prasad P. D.; Fei Y. J.; Leibach F. H.; Ganapathy V. Cloning and functional expression of the human type 1 sigma receptor (hSigmaR1). Biochem. Biophys. Res. Commun. 229:553–558; 1996. [DOI] [PubMed] [Google Scholar]
- 27. Kleiman F. E.; Manley J. L. Functional interaction of BRCA1-associated BARD1 with polyadenylation factor CstF-50. Science 285:1576–1579; 1999. [DOI] [PubMed] [Google Scholar]
- 28. Labit-Le Bouteiller C.; Jamme M. F.; David M.; Silve S.; Lanau C.; Dhers C.; Picard C.; Rahier A.; Taton M.; Loison G.; Caput D.; Ferrara P.; Lupker J. Antiproliferative effects of SR31747A in animal cell lines are mediated by inhibition of cholesterol biosynthesis at the sterol isomerase step. Eur. J. Biochem. 256:342–349; 1998. [DOI] [PubMed] [Google Scholar]
- 29. Lee M. H.; Yang H. Y. Negative regulators of cyclin-dependent kinases and their roles in cancers. Cell Mol. Life Sci. 58:1907–1922; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li C.; Wong W. H. Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. Proc. Natl. Acad. Sci. USA 98:31–36; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Z.; Diaz L. A.; Haas A. L.; Giudice G. J. cDNA cloning of a novel human ubiquitin carrier protein. An antigenic domain specifically recognized by endemic pemphigus foliaceus autoantibodies is encoded in a secondary reading frame of this human epidermal transcript. J. Biol. Chem. 267:15829–15835; 1992. [PubMed] [Google Scholar]
- 32. Lohi O.; Lehto V. P. STAM/EAST/Hbp adapter proteins—integrators of signalling pathways. FEBS Lett. 508:287–290; 2001. [DOI] [PubMed] [Google Scholar]
- 33. Mach R. H.; Smith C. R.; al Nabulsi I.; Whirrett B. R.; Childers S. R.; Wheeler K. T. Sigma 2 receptors as potential biomarkers of proliferation in breast cancer. Cancer Res. 57:156–161; 1997. [PubMed] [Google Scholar]
- 34. MacNeill S. A. DNA replication: Partners in the Okazaki two-step. Curr. Biol. 11:R842–R844; 2001. [DOI] [PubMed] [Google Scholar]
- 35. Masai H.; Arai K. Regulation of DNA replication during the cell cycle: Roles of Cdc7 kinase and coupling of replication, recombination, and repair in response to replication fork arrest. IUBMB Life 49:353–364; 2000. [DOI] [PubMed] [Google Scholar]
- 36. Masai H.; Sato N.; Takeda T.; Arai K. CDC7 kinase complex as a molecular switch for DNA replication. Front. Biosci. 4:D834–840; 1999. [DOI] [PubMed] [Google Scholar]
- 37. Mashima T.; Udagawa S.; Tsuruo T. Involvement of transcriptional repressor ATF3 in acceleration of caspase protease activation during DNA damaging agent-induced apoptosis. J. Cell Physiol. 188:352–358; 2001. [DOI] [PubMed] [Google Scholar]
- 38. Maytin E. V.; Ubeda M.; Lin J. C.; Habener J. F. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp. Cell Res. 267:193–204; 2001. [DOI] [PubMed] [Google Scholar]
- 39. Medema R. H.; Herrera R. E.; Lam F.; Weinberg R. A. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc. Natl. Acad. Sci. USA 92:6289–6293; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meraro D.; Gleit-Kielmanowicz M.; Hauser H.; Levi B. Z. IFN-stimulated gene 15 is synergistically activated through interactions between the myelocyte/lymphocyte-specific transcription factors, PU.1, IFN regulatory factor-8/IFN consensus sequence binding protein, and IFN regulatory factor-4: Characterization of a new subtype of IFN-stimulated response element. J. Immunol. 168:6224–6231; 2002. [DOI] [PubMed] [Google Scholar]
- 41. Millard E. E.; Srivastava K.; Traub L. M.; Schaffer J. E.; Ory D. S. Niemann-pick type C1 (NPC1) over-expression alters cellular cholesterol homeostasis. J. Biol. Chem. 275:38445–38451; 2000. [DOI] [PubMed] [Google Scholar]
- 42. Miscia S.; Marchisio M.; Grilli A.; Di V. V.; Centurione L.; Sabatino G.; Garaci F.; Zauli G.; Bonvini E.; Di Baldassarre A. Tumor necrosis factor alpha (TNF-alpha) activates Jak1/Stat3-Stat5B signaling through TNFR-1 in human B cells. Cell Growth Differ. 13:13–18; 2002. [PubMed] [Google Scholar]
- 43. Mitsiadis T. A.; Muramatsu T.; Muramatsu H.; Thesleff I. Midkine (MK), a heparin-binding growth/differentiation factor, is regulated by retinoic acid and epithelial-mesenchymal interactions in the developing mouse tooth, and affects cell proliferation and morphogenesis. J. Cell Biol. 129:267–281; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mountain V.; Simerly C.; Howard L.; Ando A.; Schatten G.; Compton D. A. The kinesin-related protein, HSET, opposes the activity of Eg5 and cross-links microtubules in the mammalian mitotic spindle. J. Cell Biol. 147:351–366; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Munday N. A.; Vaillancourt J. P.; Ali A.; Casano F. J.; Miller D. K.; Molineaux S. M.; Yamin T. T.; Yu V. L.; Nicholson D. W. Molecular cloning and pro-apoptotic activity of ICErelII and ICErelIII, members of the ICE/CED-3 family of cysteine proteases. J. Biol. Chem. 270:15870–15876; 1995. [DOI] [PubMed] [Google Scholar]
- 46. Musahl C.; Schulte D.; Burkhart R.; Knippers R. A human homologue of the yeast replication protein Cdc21. Interactions with other Mcm proteins. Eur. J. Biochem. 230:1096–1101; 1995. [DOI] [PubMed] [Google Scholar]
- 47. Nguyen T.; Sherratt P. J.; Pickett C. B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 43:233–260; 2003. [DOI] [PubMed] [Google Scholar]
- 48. Nuoffer C.; Davidson H. W.; Matteson J.; Meinkoth J.; Balch W. E. A GDP-bound of rab1 inhibits protein export from the endoplasmic reticulum and transport between Golgi compartments. J. Cell Biol. 125:225–237; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. O’Connor P. M.; Jackman J.; Bae I.; Myers T. G.; Fan S.; Mutoh M.; Scudiero D. A.; Monks A.; Sausville E. A.; Weinstein J. N.; Friend S.; Fornace A. J. Jr.; Kohn K. W. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anti-cancer agents. Cancer Res. 57:4285–4300; 1997. [PubMed] [Google Scholar]
- 50. Paul R.; Lavastre S.; Floutard D.; Floutard R.; Canat X.; Casellas P.; Le Fur G.; Breliere J. C. Allosteric modulation of peripheral sigma binding sites by a new selective ligand: SR 31747. J. Neuroimmunol. 52: 183–192; 1994. [DOI] [PubMed] [Google Scholar]
- 51. Pestalozzi B. C.; Peterson H. F.; Gelber R. D.; Goldhirsch A.; Gusterson B. A.; Trihia H.; Lindtner J.; Cortes-Funes H.; Simmoncini E.; Byrne M. J.; Golouh R.; Rudenstam C. M.; Castiglione-Gertsch M.; Allegra C. J.; Johnston P. G. Prognostic importance of thymidylate synthase expression in early breast cancer. J. Clin. Oncol. 15:1923–1931; 1997. [DOI] [PubMed] [Google Scholar]
- 52. Queva C.; McArthur G. A.; Iritani B. M.; Eisenman R. N. Targeted deletion of the S-phase-specific Myc antagonist Mad3 sensitizes neuronal and lymphoid cells to radiation-induced apoptosis. Mol. Cell. Biol. 21:703–712; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Romain S.; Christensen I. J.; Chinot O.; Balslev I.; Rose C.; Martin P. M.; Thorpe S. M. Prognostic value of cytosolic thymidine kinase activity as a marker of proliferation in breast cancer. Int. J. Cancer 61:7–12; 1995. [DOI] [PubMed] [Google Scholar]
- 54. Romain S.; Martin P. M.; Klijn J. G.; van Putten W. L.; Look M. P.; Guirou O.; Foekens J. A. DNA-synthesis enzyme activity: A biological tool useful for predicting anti-metabolic drug sensitivity in breast cancer? Int. J. Cancer 74:156–161; 1997. [DOI] [PubMed] [Google Scholar]
- 55. Ross E. M.; Wilkie T. M. GTPase-activating proteins for heterotrimeric G proteins: Regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 69:795–827; 2000. [DOI] [PubMed] [Google Scholar]
- 56. Sanchez-Perez I.; Martinez-Gomariz M.; Williams D.; Keyse S. M.; Perona R. CL100/MKP-1 modulates JNK activation and apoptosis in response to cisplatin. Oncogene 19:5142–5152; 2000. [DOI] [PubMed] [Google Scholar]
- 57. Sanij E.; Hatzistavrou T.; Hertzog P.; Kola I.; Wolvetang E. J. Ets-2 is induced by oxidative stress and sensitizes cells to H(2)O(2)-induced apoptosis: Implications for Down’s syndrome. Biochem. Biophys. Res. Commun. 287:1003–1008; 2001. [DOI] [PubMed] [Google Scholar]
- 58. Scherf U.; Ross D. T.; Waltham M.; Smith L. H.; Lee J. K.; Tanabe L.; Kohn K. W.; Reinhold W. C.; Myers T. G.; Andrews D. T.; Scudiero D. A.; Eisen M. B.; Sausville E. A.; Pommier Y.; Botstein D.; Brown P. O.; Weinstein J. N. A gene expression database for the molecular pharmacology of cancer. Nat. Genet. 24:236–244; 2000. [DOI] [PubMed] [Google Scholar]
- 59. Schweitzer B. I.; Dicker A. P.; Bertino J. R. Dihydrofolate reductase as a therapeutic target. FASEB J. 4:2441–2452; 1990. [DOI] [PubMed] [Google Scholar]
- 60. Scorcioni F.; Corti A.; Davalli P.; Astancolle S.; Bettuzzi S. Manipulation of the expression of regulatory genes of polyamine metabolism results in specific alterations of the cell-cycle progression. Biochem. J. 354:217–223; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Silve S.; Dupuy P. H.; Labit-Lebouteiller C.; Kaghad M.; Chalon P.; Rahier A.; Taton M.; Lupker J.; Shire D.; Loison G. Emopamil-binding protein, a mammalian protein that binds a series of structurally diverse neuroprotective agents, exhibits delta8-delta7 sterol isomerase activity in yeast. J. Biol. Chem. 271:22434–22440; 1996. [DOI] [PubMed] [Google Scholar]
- 62. Staunton J. E.; Slonim D. K.; Coller H. A.; Tamayo P.; Angelo M. J.; Park J.; Scherf U.; Lee J. K.; Reinhold W. O.; Weinstein J. N.; Mesirov J. P.; Lander E. S.; Golub T. R. Chemosensitivity prediction by transcriptional profiling. Proc. Natl. Acad. Sci. USA 98:10787–10792; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Taj M. M.; Tawil R. J.; Engstrom L. D.; Zeng Z.; Hwang C.; Sanda M. G.; Wechsler D. S. Mxi1, a Myc antagonist, suppresses proliferation of DU145 human prostate cells. Prostate 47:194–204; 2001. [DOI] [PubMed] [Google Scholar]
- 64. Takemura Y.; Jackman A. L. Folate-based thymidylate synthase inhibitors in cancer chemotherapy. Anti-cancer Drugs 8:3–16; 1997. [DOI] [PubMed] [Google Scholar]
- 65. Tan M.; Wang Y.; Guan K.; Sun Y. PTGF-beta, a type beta transforming growth factor (TGF-beta) superfamily member, is a p53 target gene that inhibits tumor cell growth via TGF-beta signaling pathway. Proc. Natl. Acad. Sci. USA 97:109–114; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tusher V. G.; Tibshirani R.; Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98:5116–5121; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vidal H.; Mondesert G.; Galiègue S.; Carrière D.; Dupuy P.-H.; Carayon P.; Combes T.; Bribes E.; Simony-Lafontaine J.; Kramar J.; Loison G.; Casellas P. Identification and pharmacological characterization of SRBP-2: A novel SR31747A-binding protein. Cancer Res. 63:4809–4818; 2003. [PubMed] [Google Scholar]
- 68. Voorma H. O. Regulatory steps in the initiation of protein synthesis. Horiz. Biochem. Biophys. 7:139–153; 1983. [PubMed] [Google Scholar]
- 69. Wathelet M. G.; Clauss I. M.; Content J.; Huez G. A. The IFI-56K and IFI-54K interferon-inducible human genes belong to the same gene family. FEBS Lett. 231:164–171; 1988. [DOI] [PubMed] [Google Scholar]
- 70. Weinstein J. N.; Myers T. G.; O’Connor P. M.; Friend S. H.; Fornace A. J. Jr.; Kohn K. W.; Fojo T.; Bates S. E.; Rubinstein L. V.; Anderson N. L.; Buolamwini J. K.; van Osdol W. W.; Monks A. P.; Scudiero D. A.; Sausville E. A.; Zaharevitz D. W.; Bunow B.; Viswanadhan V. N.; Johnson G. S.; Wittes R. E.; Paull K. D. An information-intensive approach to the molecular pharmacology of cancer. Science 275:343–349; 1997. [DOI] [PubMed] [Google Scholar]
- 71. Widschwendter M.; Daxenbichler G.; Dapunt O.; Marth C. Effects of retinoic acid and gamma-interferon on expression of retinoic acid receptor and cellular retinoic acid-binding protein in breast cancer cells. Cancer Res. 55:2135–2139; 1995. [PubMed] [Google Scholar]
- 72. Wintersberger E. Regulation and biological function of thymidine kinase. Biochem. Soc. Trans. 25:303–308; 1997. [DOI] [PubMed] [Google Scholar]
- 73. Xie R.; van Wijnen A. J.; van Der M. C.; Luong M. X.; Stein J. L.; Stein G. S. The cell cycle control element of histone H4 gene transcription is maximally responsive to interferon regulatory factor pairs IRF-1/IRF-3 and IRF-1/IRF-7. J. Biol. Chem. 276:18624–18632; 2001. [DOI] [PubMed] [Google Scholar]
- 74. Yeung T. K.; Germond C.; Chen X.; Wang Z. The mode of action of taxol: Apoptosis at low concentration and necrosis at high concentration. Biochem. Biophys. Res. Commun. 263:398–404; 1999. [DOI] [PubMed] [Google Scholar]
- 75. Zhao H.; Jin S.; Antinore M. J.; Lung F. D.; Fan F.; Blanck P.; Roller P.; Fornace A. J. Jr.; Zhan Q. The central region of Gadd45 is required for its interaction with p21/WAF1. Exp. Cell Res. 258:92–100; 2000. [DOI] [PubMed] [Google Scholar]
- 76. Zhou B. B.; Elledge S. J. The DNA damage response: Putting checkpoints in perspective. Nature 408:433–439; 2000. [DOI] [PubMed] [Google Scholar]