

Summary
Human induced pluripotent stem cell (iPSC)-derived neurons are an attractive substrate for modeling disease, yet the heterogeneity of these cultures presents a challenge for functional characterization by manual patch-clamp electrophysiology. Here, we describe an optimized all-optical electrophysiology, “Optopatch,” pipeline for high-throughput functional characterization of human iPSC-derived neuronal cultures. We demonstrate the method in a human iPSC-derived motor neuron (iPSC-MN) model of amyotrophic lateral sclerosis (ALS). In a comparison of iPSC-MNs with an ALS-causing mutation (SOD1 A4V) with their genome-corrected controls, the mutants showed elevated spike rates under weak or no stimulus and greater likelihood of entering depolarization block under strong optogenetic stimulus. We compared these results with numerical simulations of simple conductance-based neuronal models and with literature results in this and other iPSC-based models of ALS. Our data and simulations suggest that deficits in slowly activating potassium channels may underlie the changes in electrophysiology in the SOD1 A4V mutation.
Keywords: all-optical electrophysiology, iPSC, motor neurons, ALS
Graphical Abstract
Highlights
-
•
All-optical electrophysiology enables high-throughput assays in hiPSC-derived neurons
-
•
Neurons derived from ALS patients fire differently from genome-corrected controls
-
•
A deficit in the Kv7 potassium current can explain the difference in firing
In this article, Kiskinis and coworkers use all-optical electrophysiology to characterize an iPSC-based model of ALS. By performing high-throughput optical measurements of excitability in motor neurons derived from patients with a SOD1 (A4V) mutations and genome-corrected controls, the authors identify differences in firing consistent with a deficit in KV7 current in the mutants.
Introduction
Cell-reprogramming technologies have created an unprecedented opportunity to study human neurons in vitro, probing disease mechanisms under each patient's unique genetic constellation (Han et al., 2011, Pankevich et al., 2014). Many studies have used induced pluripotent stem cell (iPSC)-based and direct conversion methods to model neurological, neuropsychiatric, and neurodegenerative diseases, effectively describing disease-related phenotypes in multiple neuronal subtypes (Ichida and Kiskinis, 2015). Here we present methodology for the design and analysis of optical electrophysiology experiments on iPSC-based disease models, with application to a model of amyotrophic lateral sclerosis (ALS).
Electrical spiking is the dominant function of every neuron. The spiking patterns, the action potential waveforms, and the subthreshold voltages under different stimulus waveforms represent an integrative phenotype that reflects the activity of a large number of ion channels, transporters, and pumps, as well as the underlying cellular metabolism. While it is not, in general, possible to deduce the complete ion channel composition of a cell from its spiking patterns (Brookings et al., 2014), differences in spiking patterns between disease-model and control states can point to likely differences in ion channel function, and pharmacological rescue of disease-associated functional phenotypes can support efficacy of a candidate therapeutic.
Electrophysiology data have been traditionally difficult to attain. Manual patch-clamp measurements can be highly accurate but are labor-intensive and slow. Multi-electrode arrays and calcium imaging probe overall spontaneous activity of a culture, but do not probe details of action potential (AP) waveforms, nor are these techniques typically combined with precisely targeted stimulation. The large effort required to record manually from many neurons, combined with the intrinsic variability of iPSC-derived cultures, presents a major obstacle to systematic exploration of patient populations or experimental conditions.
A recently developed system for all-optical electrophysiology (“Optopatch”) addresses this bottleneck (Hochbaum et al., 2014). Optogenetic actuation occurs through a blue light-activated channelrhodopsin, called CheRiff. Voltage imaging occurs through a spectrally orthogonal near-infrared genetically encoded fluorescent voltage indicator called QuasAr2. Specialized optics and software allow simultaneous stimulation and recording from multiple single cells embedded in a complex network (Werley et al., 2017, Zhang and Cohen, 2017). However, low expression levels of the Optopatch construct limited its application to highly robust primary neuron cultures and to commercially produced iPSC iCell neurons (Hochbaum et al., 2014). Furthermore, limitations in data handling and analysis constrained previous applications to relatively small numbers of well-separated neurons.
Scaling up the Optopatch platform for iPSC-based disease modeling posed a number of challenges in automated data analysis and statistical interpretation. We developed image segmentation techniques to extract the fluorescence traces and morphology of individual neurons, even when they were clumped and overlapping. We developed a suite of filtering and fitting techniques robust to the dominant noise sources in our dataset to extract spike times and AP waveform parameters (Cunningham and Yu, 2014, Druckmann et al., 2013). We then employed systematic regression techniques to determine population- and subpopulation-level differences between the mutant and control cell lines while controlling for significant sources of cell-to-cell variability.
Here we apply Optopatch assays to study the electrical properties of human iPSC-derived motor neurons (iPSC-MNs) in a model of ALS. We developed improved expression constructs and cell-culture protocols to measure spontaneous and optogenetically induced spiking in human iPSC-MNs. We applied these tools to a previously validated model of ALS with the SOD1 A4V mutation, and its gene-corrected but otherwise isogenic control. We measured 1,771 single cells across six differentiations, for mutant and control, in two independent isogenic pairs. We found that SOD1 A4V mutant cells had higher spontaneous activity than isogenic controls and greater firing rate at low stimulation, but lower firing rate under strong stimulation due to an increased likelihood of entering depolarization block. Mutant cells also had smaller-amplitude APs. Mutant and genome-corrected cells had indistinguishable maximum firing rates and intra-stimulus accommodation behavior.
To gain mechanistic insight into this array of seemingly distinct functional comparisons, we explored simplified conductance-based Hodgkin-Huxley-type models. Variation of a delayed rectifier potassium channel was sufficient to account for the bulk of our findings. The relative ease of acquiring Optopatch data creates an opportunity to explore electrophysiology in cell-based models of neurological disease in detail and at a population scale, and to make quantitative comparisons with theory.
Results
Expression and Characterization of Optopatch in Human iPSC-Derived Motor Neurons
We developed an experimental pipeline to apply Optopatch to an established (Kiskinis et al., 2014, Wainger et al., 2014) human iPSC-based model of ALS (Figure 1A). The major steps were (1) differentiation of iPSCs into MNs, (2) delivery of Optopatch genes, (3) optical stimulation and recording, (4) image segmentation, (5) voltage trace parameterization, (6) statistical analysis of population differences, and (7) comparison with numerical simulations. We applied the pipeline to two iPSC lines: one derived from an ALS patient (39b) harboring the A4V mutation in the SOD1 gene, the other an isogenic control cell line (39b-Cor), generated by correcting the mutation in SOD1 through zinc finger nuclease (ZFN)-mediated gene editing. Both lines have been extensively characterized and validated for pluripotency markers, developmental potency, and genomic integrity described previously (Kiskinis et al., 2014, Wainger et al., 2014). We validated the key results in a second patient-derived line with the same mutation in SOD1 (RB9d), and a corresponding isogenic control line (RB9d-Cor) (Figures S1A and S1B).
Figure 1.

Optopatch Reports Firing Patterns of iPSC-Derived Motor Neurons in a Model of ALS
(A) Pipeline for disease modeling with optical electrophysiology.
(B) Timeline of motor neuron differentiation, gene transduction, maturation, and measurement.
(C) Top: domain structure of Optopatch constructs. Bottom: images of an iPSC-derived motor neuron expressing both CheRiff-EGFP and QuasAr2-mOrange2.
(D) Simultaneous fluorescence and patch-clamp recordings of spiking in iPSC-derived motor neurons under optical stimulation. Left: images from mutant and genome-corrected controls. Right: fluorescence (red, black) and voltage (blue). Illumination protocols are shown above.
All scale bars, 10 μm. See also Figure S1.
We differentiated the iPSC lines into post-mitotic, spinal MNs using a previously described protocol based on formation of embryoid bodies and subsequent neuralization through dual-SMAD inhibition (Figure 1B). MN specification was achieved through addition of retinoic acid and a Smoothened agonist (Kiskinis et al., 2014, Boulting et al., 2011). We and others have previously shown that the majority of MNs generated through this protocol are FOXP1/HOXA5 positive, indicative of a lateral motor column identity with a rostral phenotype, and are able to form neuromuscular junctions (Kiskinis et al., 2014, Amoroso et al., 2013). This 24-day protocol resulted in highly neuralized cultures (>95% MAP2/TUJ1+ cells) and significant numbers of spinal MNs (>30% of MAP2/TUJ1+ were ISL1/2 [ISL]+) (Figures S1A and S1B). At the end of the differentiation, MN cultures were plated onto poly-D-lysine/laminin-coated glass-bottomed dishes for subsequent maturation and electrophysiological analysis.
We tested the calcium-calmodulin-dependent kinase II type α (CamKIIα) promoter as a means to achieve selective and specific expression in iPSC-MNs. Previously published RNA-sequencing data acquired from fluorescence-activated cell sorting-isolated HB9+ MNs differentiated through this protocol (Kiskinis et al., 2014) revealed strong expression of CAMK2A (Figure S1C). The CaMKIIα promoter is known to be active in mature excitatory neurons (Lund and McQuarrie, 1997). To validate the specificity of the CamKIIα promoter for MNs, we infected iPSC-derived MN cultures with a CamKIIα-EGFP lentiviral construct and performed immunocytochemistry for EGFP and ISL (Figure S1D). Of the ISL+ MNs, 75% expressed EGFP. Of the EGFP+ cells, 89% were also ISL+ MNs (n = 1,147 ISL+ MNs and 1,289 EGFP+ cells; Figure S1E).
The previously published Optopatch construct (Hochbaum et al., 2014) contained the CheRiff and QuasAr2 genes joined by a self-cleaving 2A peptide. We found that this construct did not express highly enough for robust functional recordings in iPSC-MNs. The expression level was considerably higher when the two genes were packaged in separate lentiviruses. We generated low-titer lentiviruses (see Experimental Procedures) for QuasAr2-mOrange2 (Addgene #51692) and CheRiff-EGFP (Addgene #51693), both under control of the CamKIIα promoter (Figure 1C). We delivered the Optopatch genes via lentiviral transduction of the MN cultures 10 days before each recording.
Neurons were imaged 22–28 days post plating. Neurons showed robust expression of EGFP, indicative of CheRiff expression, as well as near-infrared fluorescence indicative of QuasAr2 expression (Figure 1C). Both proteins showed extensive membrane trafficking in the soma and in distal processes, although QuasAr2 also showed some intracellular puncta.
Optopatch expression was reported not to have significant effects on electrical properties of primary or iPSC-derived neurons (Hochbaum et al., 2014), but those measurements did not include MNs. To test for effects of expression on MN electrophysiology, we performed manual patch-clamp measurements in iPSC-MNs expressing either CaMKIIα-driven Optopatch or a CaMKIIα-driven EGFP control (see Experimental Procedures). Optopatch expression did not significantly perturb resting voltage, membrane resistance, membrane capacitance, rheobase, or AP threshold voltage relative to control cultures expressing EGFP (Figures S2A–S2E). Both Optopatch and EGFP-transduced cells had slightly lower membrane resistance and higher membrane capacitance than non-transduced controls, indicating that within the heterogeneous in vitro neuronal population, the CaMKIIα promoter targeted expression to neurons with larger surface area, a marker of greater maturity. We further tested for differences in AP properties between EGFP- and Optopatch-expressing MNs in both SOD1 A4V and genome-corrected controls. Individual cells showed widely varying firing patterns, particularly in the vicinity of depolarization block, ranging from single spikes to tonic firing. Many cells showed subthreshold ringing oscillations that gradually diminished as the cell entered depolarization block. In neither genotype did we observe significant differences between EGFP- and Optopatch-expressing MNs in AP amplitude, maximum firing rate, or width of the first spike following stimulus onset (Figures S2F–S2H).
To test whether the optical measurements were a faithful reporter of AP waveforms, we acquired simultaneous manual patch-clamp and optical recordings, with optogenetic stimulation (Experimental Procedures and Figure 1D). Fluorescence traces were extracted from single-cell recordings using a previously described pixel-weighting algorithm that automatically identified pixels whose fluorescence correlated with the whole-image mean (Kralj et al., 2012). APs were resolved optically on a single-trial basis and tagged with an automatic spike-finding algorithm (Supplemental Experimental Procedures). Of the optically identified spikes, 4% were not automatically identified in the patch-clamp recordings; of the electrically identified spikes, 5% were not automatically identified in the optical recordings (n = 148 spikes, 4 cells). These discrepancies came not from shot noise (which contributed an error rate of <10−6) but from low-amplitude oscillations near depolarization block whose classification as spike or not-spike was ambiguous even to human observers (see Figure 6B for an example).
Figure 6.

Numerical Simulation of Firing Patterns with Variable Channel Levels
(A) Simulation (blue) and fluorescence trace (black) of a neuron showing tonic firing.
(B) Simulation (blue) and fluorescence trace (black) of the same neuron shown in (A) approaching the transition to depolarization block under strong stimulus.
(C–E) Each trace shows the number of APs in a 500-ms interval as a function of the optogenetic stimulus strength (gm). Maximum conductances of the indicated channels were varied from 0.67- to 1.5-fold basal level. (C) Variation in NaV level. (D) Variation in KV (delayed rectifier) level. (E) Variation in KV7 level. The changes in the F-I curve that came with lower KV7 conductance phenotypically matched the differences and similarities between SOD1 A4V and control neurons.
We compared AP parameters of spikes recorded simultaneously optically and electrically. Optically recorded APs had a root-mean-square error of 1.2 ms in time of peak depolarization relative to the electrical signal (n = 148 spikes, 4 cells), and a systematic overestimate of spike full-width at half-maximum (FWHM) by 1.8 ± 1.1 ms (mean ± SD) compared with an electrically recorded mean spike FWHM of 5.6 ms (Figure S2I). These mean widths included exceptionally broad spikes near onset of depolarization block. The average width for the first electrically recorded AP after stimulus onset was 3.9 ms, comparable with the literature on developing motor neurons (in rat, AP duration is 9.3 ms at embryonic day 15–16 and 3.4 ms at postnatal day 1–3, at 27°C–29°C [Ziskind-Conhaim, 1988]). In the electrical recordings, the first AP after stimulus onset was narrower in width (3.9 ms versus 6.7 ms FWHM) and higher in amplitude (74 mV versus 60 mV) than subsequent APs. In the optical recordings the first AP was also narrower than subsequent APs (5.7 ms versus 8.7 ms), but appeared smaller in amplitude (2.5% ΔF/F versus 2.8% ΔF/F) than subsequent APs. The differing trends in apparent spike amplitude are explained by low-pass filtering of the optical signal due to the 2-ms exposure time of the camera and the 1.2-ms response time of QuasAr2. This level of time resolution enabled robust spike counting and coarse parameterization of AP waveforms but not detailed analysis of submillisecond dynamics.
The fluorescence signal showed a slow increase in baseline during each optical stimulus epoch (Figure 1D), which we traced to blue light photoproduction of a red-fluorescent product, as has been reported previously for other Arch-derived voltage indicators (Venkatachalam et al., 2014, Hou et al., 2014) (see Supplemental Discussion). This effect had been negligible in previous experiments in primary rodent neurons (Hochbaum et al., 2014) on account of higher CheRiff expression (necessitating lower blue stimulus intensity) in the primary cells. We did not include gradual changes in baseline in the analysis, focusing instead on spike timing and shape parameters.
Probing Neuronal Excitability with Optopatch
Figure 2A shows the illumination protocol used to probe the cell-autonomous excitability of human iPSC-MNs. Recordings were acquired at 500-Hz frame rate for 9,000 frames. Illumination with red light (635 nm, 800 W/cm2) induced near-infrared voltage-dependent fluorescence of QuasAr2. Cells were monitored for 10 s without stimulation to quantify spontaneous activity. Cells were then stimulated with eight 500-ms pulses of blue light of linearly increasing intensity from 6 mW/cm2 to 100 mW/cm2. After each blue stimulus pulse, cells were recorded for another 500 ms without blue stimulus and then given 5 s of rest with neither red nor blue illumination. Mutant SOD1 A4V and genome-corrected control MN cultures were differentiated in parallel, and recordings from paired cultures were performed on the same day.
Figure 2.

Optopatch Measurement and Analysis Pipeline
(A) Cells were subject to 10 s of unstimulated recording to measure spontaneous activity (red), and then to eight stimulation pulses of 500 ms duration and increasing intensity (blue).
(B) Activity-based movie segmentation. Image stacks were filtered spatially and temporally, then processed via principal components analysis (PCA) followed by independent components analysis (ICA) to identify clusters of pixels whose fluorescence values co-varied in synchrony. The movie was decomposed into a sum of overlapping neuron images, each with its own spiking pattern.
(C) Parameterization pipeline. Spikes were identified in the fluorescence traces. Spiking patterns were analyzed within stimuli, between stimuli, and between populations. AP waveforms were also parameterized, enabling comparison of width, height, and after-polarization within and between cells. The results of the segmentation in the spatial domain enabled measurement of morphological features (soma versus dendrite) and of CheRiff-EGFP expression level. Finally, all of this information was integrated to build a coherent picture of phenotypic differences between mutant and control cell lines.
All scale bars, 10 μm. See also Figures S2–S4.
Image Segmentation and Data Processing
To accommodate the large quantities of Optopatch data (1,039 movies, 200 gigabytes in total), we developed a pipeline for analysis in a parallel computing environment (Figure S3). The first stage comprised image segmentation. Cells often clustered, with overlapping somas and intertwined processes. On average, 60% of each cell body area overlapped with other cells in the controls, and 63% in the mutants (distribution difference p = 0.07 by Mann-Whitney U test, not significant). The large degree of overlap implied a need to un-mix the fluorescence signals from overlapping cells.
Our segmentation approach was derived from an independent components analysis (ICA) algorithm, originally developed for calcium imaging (Mukamel et al., 2009), with modifications to accommodate the differing morphological, statistical, and noise properties of voltage imaging data (see Supplemental Experimental Procedures). In brief, movies were high-pass filtered in time to accentuate the signals from spikes, and low-pass filtered in space to suppress spatially uncorrelated shot noise. Movies were then subjected to principal components analysis (PCA) to reduce the dimensionality of the dataset, then time-domain ICA to identify linear combinations of principal components that maximized statistical independence between intensity traces. The spatial filters produced from ICA were then applied to the original (unfiltered) movie to extract the underlying intensity traces (Supplemental Experimental Procedures and Figure 2B).
Complex images of up to six overlapping neurons were readily decomposed into single-cell traces. No information about cell morphology was used in the image segmentation, so emergence of neuron-shaped objects with corresponding stereotyped AP waveforms confirms the effectiveness of the algorithm. Inspection of the firing patterns revealed negligible crosstalk between signals derived from overlapping cells. Single-cell fluorescence traces were then processed with a spike-finding algorithm that used a dynamically adjusted threshold to accommodate different signal-to-noise ratios in different cells (Supplemental Experimental Procedures and Figure 2C). Sources were classified as active cells if they showed five or more spikes during the experiment and had a signal-to-noise ratio greater than 5, ensuring a shot noise contribution to errors in spike calling of <10−6.
The second stage of the pipeline comprised parameterization of the spike waveforms. A standard set of parameters has been proposed to describe AP waveforms recorded via conventional electrophysiology (Druckmann et al., 2013). Fluorescence differs from patch clamp in that fluorescence has a lower signal-to-noise ratio, does not have an absolute voltage scale, is subject to baseline drift, and has lower time resolution. To determine which parameters we could use robustly, we first described spikes with a large set of parameters and then used an information-theoretic approach to eliminate redundancies (Supplemental Discussion and Figure S4). Our final set of parameters described the upstroke duration, downstroke duration, initiation threshold relative to baseline, spike amplitude relative to baseline, and after-hyperpolarization relative to baseline.
Our image segmentation method also enabled quantitative description of cellular morphology. We used filters derived from the activity-based segmentation to identify the two-dimensional footprint of each cell. We then employed morphological image processing to identify cell soma and dendrites (Supplemental Experimental Procedures and Figure S4).
Comparison of Mutant SOD1 A4V with Isogenic Control MNs
We compared the firing patterns between iPSC-MNs with the SOD1 A4V mutation (n = 331) and genome-corrected controls (n = 843). Figure 3A shows a raster plot of the spike timing for a subset of the cells (not including the recording of spontaneous activity at the start of each trace). Figure 3B shows that, on average, SOD1 A4V cells had higher spontaneous activity than genome-corrected controls (mean spontaneous rates 1.50 ± 0.18 Hz in mutant, 0.98 ± 0.12 Hz in control; p = 0.003, Wilcoxon signed-rank test used because of non-normal distribution). Figure 3C shows the population-average spike count in a 500-ms stimulus as a function of stimulus intensity for the two genotypes. The curves for mutant and control crossed: in the most strongly stimulated epoch, mutant cells fired less on average than controls (mean rates 12.6 ± 0.5 Hz in mutant, 14.6 ± 0.3 Hz in control; p = 0.0012, unpaired t test). We then performed matched experiments in a second patient-derived SOD1 A4V cell line (RB9d) and its isogenic control (RB9d-Cor). As with the 39b line, the RB9d mutant cells showed, on average, enhanced spontaneous activity, hyperexcitability at weak stimulus, and hypoexcitability at strong stimulus (Figure S5).
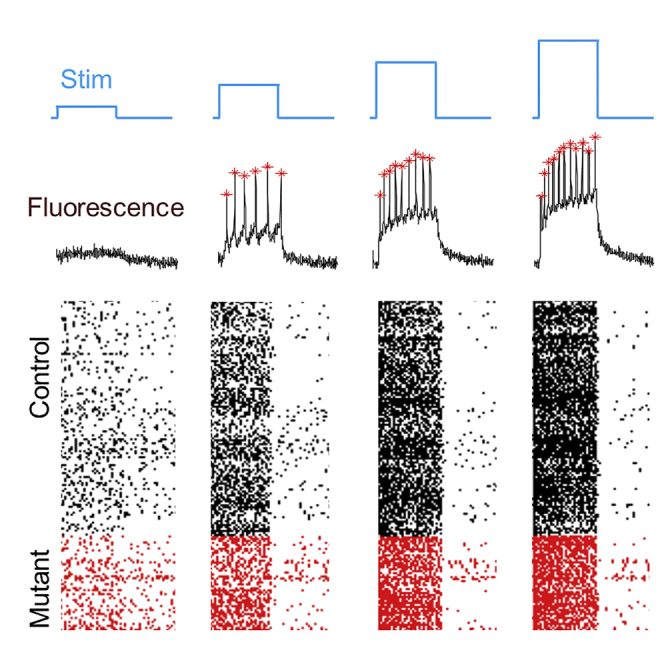
Figure 3.

Comparison of Spiking Patterns in SOD1 A4V and Control Motor Neurons
(A) Top: representative optical trace with stimulus protocol (blue) and identified spikes (red stars). Bottom: raster plot showing spike timing for a subset of the recorded cells. Black are controls, red are SOD1 A4V.
(B) Spontaneous activity.
(C) Population-average number of APs as a function of optogenetic stimulus strength.
(D and F) Histograms of number of APs as a function of stimulus intensity for (D) controls and (F) SOD1 A4V mutants.
(E) Left: spike train in a control neuron showing monotonically increasing number of APs as a function of stimulus strength. Right: image of the cell.
(G) Left: spike train in an SOD1 A4V mutant showing depolarization block upon strong stimulus. Right: image of the cell.
(H) Number of APs as a function of stimulus strength among the subpopulation of cells that did not enter depolarization block. No significant differences were observed between mutant and control.
(I) Fraction of cells in depolarization block as a function of stimulus number.
Error bars represent SEM. Asterisks indicate significance differences between mutant and control, ∗p < 0.01. Data in all panels based on n = 834 control neurons, n = 331 SOD1 A4V, and six rounds of differentiation. Scale bars, 10 μm. See also Figures S5 and S6.
Population-level differences in activity could arise from uniform shifts in all cells or from redistribution of cells among subpopulations with different firing patterns. The single-cell Optopatch data allowed us to examine the underlying distributions of single-cell behavior that led to the population-average differences (Figure 3D for controls and Figure 3F for mutants).
Under strong stimulus, two clear subpopulations emerged: cells that fired rapidly and tonically (Figure 3E) and cells that generated just one or two spikes before going quiet (Figure 3G). We presumed that these quiet cells were constitutively inactive. However, a majority of these cells (64% in the control, 72% in the mutant) fired four or more times during a stimulus of intermediate intensity. These results established that a subpopulation of neurons showed a non-monotonic dependence of firing rate on stimulus strength, with a maximum in firing rate at intermediate stimulus strength.
We then examined the fluorescence waveforms of the cells that inactivated under strong stimulus. These cells were depolarized but not firing, a signature of depolarization block (Pontiggia et al., 1993). To quantify the populations in depolarization block, we defined the onset of depolarization block as a decrease in number of spikes upon an increase in stimulus strength. Cells that did not show depolarization block had statistically indistinguishable firing rates in mutant and control populations at high intensity (p = 0.11, unpaired t test at the strongest stimulus; Figure 3H). However, the proportion of cells that entered depolarization block differed significantly between mutant and control: at the strongest stimulus, the SOD1 A4V cells were 32% more likely to be in depolarization block than the controls (p = 0.008, binomial model t test with Holm-Bonferroni correction, two hypotheses; Figure 3I). This difference in propensity to enter depolarization block under strong stimulus was the most dramatic difference between mutant and control.
Aligning all cells' F-I curves by the stimulus pulse at which they reached their maximum firing rate revealed a stereotyped F-I curve (Figure 4). Cells showed linear dependence of firing rate on stimulus strength up to a maximum, maintenance of the maximum firing rate in a plateau phase, and then a rapid collapse into depolarization block. The mutant population tended to have a narrower plateau phase and a greater propensity to enter depolarization block than the controls. For cells with matched firing rates at a given stimulus strength, the odds of a cell going into depolarization block in the next stronger stimulus were 88% higher in the mutants than in the controls (p = 1.7 × 10−4, logistic regression coefficient t test). The mutant and control neurons reached the same maximum firing rate (mean rates 8.7 ± 0.4 Hz in mutant, 9.2 ± 0.3 Hz in control; p = 0.51, linear model coefficient t test).
Figure 4.

Characteristic Firing Pattern of iPSC-MNs as a Function of Stimulus Strength
(A and B) Heatmaps showing universal shape of the F-I curves for control and SOD1 A4V mutant iPSC-MNs. F-I curves from each cell were rescaled along the x and y axes as follows. Firing rate was expressed as a percentage of the cell's maximum firing rate. Stimulus intensities were aligned to the lowest intensity stimulus at which this maximum firing rate was achieved. This rescaling revealed typical F-I trajectory shapes in a manner that was independent of changes in CheRiff expression level. Control (A) and SOD1 A4V mutant iPSC-MNs (B) showed a linear increase in firing rate versus stimulus strength for weak stimuli, a plateau in firing rate for moderate stimuli, and a collapse in firing under strong stimuli. Data based on n = 834 control neurons, n = 331 SOD1 A4V, and six rounds of differentiation.
(C) Top: fluorescence traces from a single representative cell passed through three distinct stages of firing in response to monotonically increasing optogenetic stimulus strength. Bottom: fit of piecewise-continuous F-I curves to the data in (A) and (B) for (black) genome-corrected and (red) SOD1 A4V mutant cell lines. Curves were constructed from measurements of spontaneous rate, average slope of the F-I curve (controlling for expression level), maximum firing rate, and the number of stimulus steps spent at the maximum.
We next studied the timing of the APs within each stimulus epoch. After the first two APs, cells showed nearly constant firing rate throughout the stimulus, provided that the cell was not in depolarization block (Figure 5). To quantify the degree of firing rate adaptation, we defined the nth inter-spike interval by ISIn = tn+1 – tn, where tj represents the time of peak of spike j. We then defined the degree of firing rate adaptation by , where the average is over all spikes during a stimulus epoch. This quantity did not differ significantly between SOD1 A4V and control neurons (8.7% in control, 8.9% in mutants; p = 0.87, unpaired t test). We also detected no differences between mutant and control in the ratio of first inter-spike interval to the average inter-spike interval during a stimulus epoch, (33% in control, 34% in mutant; p = 0.45, unpaired t test).
Figure 5.

Comparison of AP Waveforms in Mutant and Control
(A and B) Average phase plots, , from steps with nine action potentials, for (A) control and (B) SOD1 A4V cell lines. The first two spikes are highlighted in blue, after which the cells converged to a stable limit cycle.
(C and D) Average AP waveforms from all stimuli, which produced (C) two and (D) nine APs during a single stimulus epoch. APs from stimuli during which the cell entered depolarization block were excluded. Spikes were aligned in time by their peak and in by their pre-peak minimum. Average waveforms from stimulus epochs with different numbers of APs are quantified in Table S2.
Finally, we studied the waveforms of individual APs. Within each 500-ms stimulus epoch, the AP waveform of each single cell was consistent from spike to spike, after the first two spikes, provided that the cell was not in depolarization block (Figure 5 and Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6). AP waveforms did vary with firing rate and with stimulus intensity. After controlling for these parameters, average AP waveforms still differed between mutant and control iPSC-MNs. In the weak stimulus regime, where firing rate was proportional to stimulus strength, mutants had 10% smaller AP peak amplitude than controls (p = 3.1 × 10−8, linear model coefficient t test, significant after Holm-Bonferroni multiple comparison correction; Figure 5 and Table S2).
In accord with previous findings (Kiskinis et al., 2014), we observed statistically significant morphological differences between the mutant and control MNs. The mutants had a smaller soma and fewer projections than the controls (Figure S6 and Supplemental Experimental Procedures). This observation suggested that the changes in excitability seen in the mutant cells might be a by-product of the mutant line's differences in morphology. We included terms for soma area and ratio of soma area to whole-cell area (“soma fraction”) in a logistic regression model for the probability of depolarization block as a function of firing rate. When trained on control data, the coefficients on these morphological parameters were not significant (p = 0.06 for soma area, p = 0.47 for soma fraction, separate models, logistic regression coefficient t test). When mutant data were controlled for these parameters, the difference between mutant and control remained significant (p = 1.4 × 10−4 for soma area, p = 1.2 × 10−4 for soma fraction, logistic regression coefficient t test). Thus morphological parameters (soma area and soma fraction) did not predict the probability of depolarization block either within or between genotypes. While this analysis cannot rule out possible contributions from other morphological parameters, we focused subsequent analysis on electrophysiological variables.
Simulations
We sought to relate the phenotypic differences and similarities between SOD1 A4V mutant and control neurons to hypotheses about underlying disease mechanisms. While complex multi-compartment models of MNs have been developed (Powers et al., 2012, Powers and Heckman, 2015), it is well established that voltage recordings alone are insufficient to constrain the parameters of such models (Brookings et al., 2014). Furthermore, considering the large cell-to-cell variability in the observed firing patterns, a morphologically and molecularly detailed model was deemed inappropriate. Instead, we sought a parsimonious model that could account for multiple population-level observations with a small number of parameters.
Previous studies have singled out potassium channels in general (Kanai et al., 2006) and KV7 (KCNQ) channels in particular (Wainger et al., 2014) as a target of investigation in both SOD1 and C9ORF72 models of ALS, but it is not clear how these mutations affect ion channel expression or function. We asked whether changes in Kv7 current alone could account for some or all of the observed functional effects. We performed numerical simulations of a minimal Hodgkin-Huxley-type model, containing only a NaV channel, a fast KV channel, a slow KV7 channel, and a channelrhodopsin (Powers et al., 2012, Powers and Heckman, 2015). We varied the model parameters systematically and studied the resulting simulated firing patterns using the same parameters as for the experimental data.
The starting ion channel parameters were taken from previous numerical simulations of a human MN (Powers et al., 2012, Powers and Heckman, 2015), and the channelrhodopsin was modeled as a conductance with a reversal potential of 0 mV and an opening time constant of 1 ms. Following our experimental illumination protocol, simulations were run with steps of increasing maximal channelrhodopsin conductance. To account for the capacitive load from passive membrane structures and to match simulated firing rates to the range observed experimentally, we increased the membrane capacitance beyond the literature value (see Supplemental Experimental Procedures). The simulated cells showed stimulus-dependent firing and depolarization block, clearly recapitulating the main qualitative features of the data (Figure 6A). The experimentally recorded waveforms varied considerably from cell to cell, so we focused on studying the dependence of spiking properties on channel conductances rather than on trying to match simulated and experimental waveforms precisely.
We varied each conductance in the model to determine its effect on low-stimulus excitability, threshold for depolarization block, maximum firing rate, and AP waveform. Figure 6 shows simulated firing rate as a function of optogenetic stimulus strength, for a range of channel conductances bracketing the original model parameters. Decreases in KV7 conductance increased low-stimulus excitability, decreased threshold for depolarization block, decreased spike height, and had no effect on maximum firing rate (Table S3). Thus, remarkably, changes only in the KV7 conductance were sufficient to reproduce all the major functional phenotypes and null results cataloged in our Optopatch experiments. Neither variation in the NaV conductance nor in the fast KV channel showed the correct qualitative trends (Figure 6).
Discussion
Our demonstration of optical electrophysiology recordings in a delicate and complex human cellular preparation opens the prospect to record large quantities of functional data in this and other human models of neuronal disease. Key to extracting meaning from these data is a statistically robust analysis pipeline and comparisons with numerical simulations at an appropriate level of detail.
Patch versus Optopatch
The optical and manual patch-clamp techniques offer different tradeoffs in resolution and throughput, and thus should be seen as complementary rather than competing techniques. One must exercise caution in applying concepts from conventional electrophysiology to Optopatch. Principally, Optopatch and patch deliver different kinds of stimulation. CheRiff is a conductance while electrical stimulation is via a current source. CheRiff current reverses direction when the membrane voltage crosses 0 mV, while a current clamp maintains constant current irrespective of membrane voltage. In our MN simulation, the dependence of firing rate on stimulus strength and channel levels had the same qualitative features for CheRiff stimulation as for current injection. In other parameter regimes, however, the two modes of stimulation induced strikingly different firing patterns. Spike trains induced optogenetically may be a more faithful indicator of in vivo firing because AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors are a conductance with reversal potential of ∼0 mV, not a current source. One can simulate membrane conductances with dynamic patch clamp (Prinz et al., 2004), but this technique is not widely used.
Optical and electrical recordings also are subject to different types of noise and artifacts (Cohen and Venkatachalam, 2014). Patch-clamp recordings remain the gold standard for accuracy and time resolution: one can record submillivolt changes in membrane voltage on a submillisecond timescale. One can also use a voltage clamp to dissect the contributions of distinct conductances to membrane currents. However, patch-clamp measurements are slow and laborious (four cells per hour in the present experiments), increasing the risk of statistical artifacts from small sample sizes in highly heterogeneous stem cell-derived cultures. Furthermore, in the most commonly used whole-cell configuration, manual patch clamp risks dialyzing cytoplasmic contents. Manual patch-clamp measurements also lack spatial resolution and are exceedingly difficult to apply to a single cell on successive days.
Optopatch has lower temporal resolution (1–2 ms) and lower precision than patch clamp. The signal-to-noise ratio in the optical recordings was 13.0 ± 6.7 (mean ± SD), corresponding to a noise level of approximately 5.2 mV in a 500-Hz bandwidth. The lower signal-to-noise ratio relative to published results in primary rodent neurons (Hochbaum et al., 2014) was due to a smaller soma size and lower Optopatch expression in the iPSC-derived neurons. Cell-to-cell variations in QuasAr2 expression level prevented assignment of absolute voltage values to fluorescence measurements. Thus, Optopatch measurements are most useful for determining spiking statistics and for examining AP waveforms, and at present less so for quantifying subthreshold events or absolute voltage values.
Optopatch has considerably higher throughput than a manual patch clamp (34 cells per hour in the present experiments). Wide-field imaging systems could potentially increase this throughput considerably (Hochbaum et al., 2014). Although not used here, Optopatch measurements can, in principle, be readily combined with genetic or immunohistochemical targeting with cell-type-specific markers. Measurements targeted to MNs (e.g., via HB9-Cre [Peviani et al., 2012] or post-measurement HB9 staining) are a natural extension of the work. Optopatch can also probe spatial relations of electrical activity, both within and between cells.
Neuronal Excitability in ALS
Our simple computational models showed non-monotonic dependence of firing rate on stimulus strength in all cases, consistent with our data. Variations in KV7 currents led to firing rate curves that crossed each other. Together, these observations show that “excitability” is not a well-defined attribute of a neuron, but rather depends on the magnitude of the stimulus strength. In our data, in both the mutant and controls cells with a higher spontaneous rate were more likely to show a decrease in firing under strong stimulus: the odds of entering depolarization block increased by 34% with every 1-Hz increase in spontaneous rate (p = 0.005, logistic regression coefficient t test) in control and 49% per Hz in mutant (p = 0.0004). Thus neurons that appeared hyperexcitable under weak or zero stimulus tended to appear hypoexcitable under strong stimulus. One should therefore use caution in speaking of hyper- or hypoexcitability as intrinsic neuronal properties.
These observations also highlight the importance of analyzing neuronal recordings at the single-cell level rather than simply looking at aggregate population-level statistics. The population-average curves of firing frequency versus stimulus strength may be strikingly different from the curves for every individual neuron. Due to the non-linear dependence of firing frequency on ion channel levels, efforts to fit the population-average data may lead to incorrect mechanistic conclusions.
By recording data in two pairs of mutant and genome-corrected lines, we can compare the effect size due to the SOD1 A4V mutation versus the effect size due to line-to-line differences (39b-Cor versus RB9d-Cor). In our recordings, the line-to-line differences in firing rate were comparable in magnitude with the difference between mutant and corrected within each line (Figure S5). At present it is not known whether these line-to-line differences reflect underlying genetic heterogeneity or epigenetic effects reflecting the different histories of the iPSC lines. Resolving that question would require comparisons of multiple lines derived from the same individuals, as well as lines derived from different individuals. Nonetheless, these results highlight the importance of making matched isogenic comparisons when looking for mutation-associated phenotypes at the level of individual lines.
The pathways by which mutations in ALS-causing genes lead to a change in neuronal excitability remain unknown. The KV7 potassium channel has recently emerged as a potential therapeutic target and is the subject of an ongoing clinical trial of retigabine for ALS (McNeish et al., 2015). While expression profiling did not identify effects of the SOD1 A4V mutation at the transcriptional level (Kiskinis et al., 2014), there are many post-translational mechanisms for regulating the KV7 current. This channel has multiple interaction partners (Delmas and Brown, 2005), is regulated by PtdIns(4,5)P2 (Suh and Hille, 2008), and is redox sensitive (Gamper et al., 2006). Thus defects in any of these interaction partners, in lipid metabolism, or in redox homeostasis could contribute to excitability defects. Recently, deficits in trafficking of RNA granules have been found in a TDP43 model of ALS (Alami et al., 2014). These findings provide a plausible mechanism for proteostatic deficits in ALS, including deficits in ion channels.
In a mutant C9ORF72 model of ALS, Sareen et al. (2013) reported hypoexcitability of the mutants relative to controls. However, the previously published plots of firing frequency as a function of stimulus strength (Figures 3H and S12 of Sareen et al.) resemble our data showing depolarization block at strong stimulus (Figures 3B and 3C). Although we did not probe the C9ORF72 model, these observations raise the intriguing possibility that loss of KV7 conductance could provide a common mechanism across genotypes. The sufficiency of a KV7 deficit to account for both hyper- and hypoexcitability phenotypes cannot rule out other possible contributions to the electrophysiology, including other ion channels, shifts in channel kinetics or gating thresholds, and shifts in cellular morphology, resting potential, or ion channel spatial distribution.
The iPSC-MNs studied here represent an immature developmental state, while ALS typically strikes in adulthood. Whereas the disease-causing mutation is present throughout the life of the patient, its effect is clearly age dependent. It is not known whether time in vitro is a realistic proxy for chronological age in vivo and, if so, the relative scaling of these timelines. Thus, while time-course data in vitro may yield interesting changes in function or physiology, it is not clear whether such data provide information relevant to age-related processes. Despite these limitations, functional optogenetic screening has potential uses in identifying disease mechanisms, testing prospective therapeutics, and stratifying patients.
Experimental Procedures
Cell Culture
All cell cultures were maintained at 37°C and 5% CO2. Cells tested negative for mycoplasma contamination. Pluripotent stem cells were grown on Matrigel (BD Biosciences) with mTeSR1 medium (STEMCELL Technologies). Culture medium was changed every 24 hr and cells were passaged by Dispase (Gibco) or Accutase (Innovative Cell Technologies) as required.
Motor Neuron Differentiation
Stem cell cultures were differentiated into motor neurons as previously described (Kiskinis et al., 2014). In brief, iPSCs were dissociated to single cells and plated in suspension in low-adherence flasks (400 k/mL), in mTeSR medium with 10 μM ROCK inhibitor. Medium was gradually diluted (50% on day 3 and 100% on day 4) to knockout serum replacement (KOSR) (DMEM/F12, 10% KOSR) between days 1 and 4 and to a neural induction medium (DMEM/F12 with L-glutamine, non-essential amino acids [NEAA], 2 μg/mL heparin, N2 supplement [Invitrogen]) for days 5–24. From days 1–6 cells were cultured in the presence of 10 μM SB431542 (Sigma-Aldrich) and 1 μM dorsomorphin (Stemgent), and from days 5–24 with 10 ng/mL brain-derived neurotrophic factor (BDNF; R&D Systems), 0.4 μg/mL ascorbic acid (Sigma), 1 μM retinoic acid (Sigma), and 1 μM Smoothened agonist 1.3 (Calbiochem). On day 24 floating cell aggregates were dissociated to single cells with Papain/DNase (Worthington Bio) and plated onto poly-D-lysine/laminin-coated dishes for electrophysiological analysis. Once dissociated, MN cultures were fed every 2–3 days with complete neurobasal medium (neurobasal with L-glutamine, NEAA, Glutamax, N2, and B27), with 10 ng/mL BDNF/ciliary neurotrophic factor/glial cell-derived neurotrophic factor (R&D) and 0.2 μg/mL ascorbic acid (Sigma).
Gene Editing
Correction of the SOD1 A4V mutation in the ALS patient iPSC line RB9d was performed using ZFN-mediated targeting as described previously (Kiskinis et al., 2014).
Electrophysiology
Measurements were conducted in Tyrode's solution containing 125 mM NaCl, 2.5 mM KCl, 3 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 30 mM glucose (pH 7.3) and adjusted to 305–310 mOsm with sucrose. Prior to imaging, neurons were incubated with 5 μM all-trans retinoic acid for 30 min and then washed with Tyrode's solution.
Synaptic blockers were added to the imaging medium for measurements of single-cell electrophysiology. The blockers comprised 10 μM NBQX (Tocris), 25 μM D(−)-2-amino-5-phosphonovaleric acid (Tocris), and 20 μM gabazine (SR-95531; Tocris). Patch-clamp data were used if and only if access resistance was <25 MΩ, and did not vary over the experiment. Recordings were terminated if membrane resistance changed by >10%. Experiments were performed at 23°C under ambient atmosphere.
Optopatch Recordings
Cells were imaged on a custom-built epifluorescence inverted microscope. Imaging experiments were conducted in Tyrode's buffer (pH 7.3). Excitation of QuasAr2 was via a 500-mW 640-nm diode laser (Dragon Lasers), which provided a field of view of 31 × 37 μm, with an intensity at the sample of 800 W/cm2. Blue illumination from a 50-mW 488-nm solid-state laser (Coherent OBIS) was modulated by an acousto-optical tunable filter (Gooch and Housego) to control timing and amplitude of the optogenetic stimulation. The blue and red beams were then combined and imaged onto the sample through the objective lens. Images were collected with a 60× water objective (Olympus, numerical aperture 1.2) and imaged onto a scientific CMOS camera (Flash 4.0, Hamamatsu). Data were collected at a frame rate of 500 Hz. Custom code written in LabView (National Instruments) controlled the hardware.
The stimulation protocol consisted of:
-
1.
10 s of continuous red light to measure spontaneous firing
-
2.
50 ms with red light only
-
3.
500 ms with red and blue light to stimulate firing
-
4.
500 ms with red light only
-
5.
5 s with no light for cell recovery
Steps 2–5 were repeated 8–10 times with increasing intensities of blue light.
Procedures for immunocytochemistry, virus production, gene delivery, data analysis, and simulation are described in detail in Supplemental Experimental Procedures.
Author Contributions
E.K., J.M.K., K.E., and A.E.C. designed the project. E.K. performed iPSC differentiation and characterization. J.M.K., P.Z., and H.Z. performed Optopatch and manual patch-clamp recordings. J.A.O., O.W., and K.T. performed differentiations and neuronal characterization. J.M.K., P.Z., H.Z., E.N.W., and A.E.C. analyzed the data. E.K., J.M.K., E.N.W., and A.E.C. wrote the manuscript with input from all other authors. E.K., K.E., and A.E.C. oversaw the research.
Acknowledgments
Scott Linderman provided an optimization algorithm for fitting piecewise linear functions. Vicente Parot helped with PCA-ICA segmentation. Samouil Farhi performed patch-clamp electrophysiology measurements. This work was supported by US NIH grants 1-R01-EB012498 and 1-DP2-OD007428 and support from the Howard Hughes Medical Institute. The Kiskinis lab is supported by grants from the Les Turner ALS Foundation and the Muscular Dystrophy Association. E.K. is a Les Turner ALS Research and Patient Center Investigator. K.E. acknowledges support from Project ALS, Target ALS, and National Institute of Neurological Disorders and Stroke of the NIH awards R01NS089742 and RC2NS069395. J.M.K. and E.K. own stock in Q-State Biosciences. E.N.W. has worked as a consultant for Q-State Biosciences. K.E. and A.E.C. are co-founders of Q-State Biosciences.
Published: May 17, 2018
Footnotes
Supplemental Information includes Supplemental Discussion, Supplemental Experimental Procedures, six figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.04.020.
Contributor Information
Kevin Eggan, Email: keggan@scrb.harvard.edu.
Adam E. Cohen, Email: cohen@chemistry.harvard.edu.
Supplemental Information
References
- Alami N.H., Smith R.B., Carrasco M.A., Williams L.A., Winborn C.S., Han S.S.W., Kiskinis E., Winborn B., Freibaum B.D., Kanagaraj A. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoroso M.W., Croft G.F., Williams D.J., O'Keeffe S., Carrasco M.A., Davis A.R., Roybon L., Oakley D.H., Maniatis T., Henderson C.E., Wichterle H. Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J. Neurosci. 2013;33:574–586. doi: 10.1523/JNEUROSCI.0906-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulting G.L., Kiskinis E., Croft G.F., Amoroso M.W., Oakley D.H., Wainger B.J., Williams D.J., Kahler D.J., Yamaki M., Davidow L. A functionally characterized test set of human induced pluripotent stem cells. Nat. Biotechnol. 2011;29:279–286. doi: 10.1038/nbt.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookings T., Goeritz M.L., Marder E. Automatic parameter estimation of multicompartmental neuron models via minimization of trace error with control adjustment. J. Neurophysiol. 2014;112:2332–2348. doi: 10.1152/jn.00007.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A.E., Venkatachalam V. Bringing bioelectricity to light. Annu. Rev. Biophys. 2014;43:211–232. doi: 10.1146/annurev-biophys-051013-022717. [DOI] [PubMed] [Google Scholar]
- Cunningham J.P., Yu B.M. Dimensionality reduction for large-scale neural recordings. Nat. Neurosci. 2014;17:1500–1509. doi: 10.1038/nn.3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P., Brown D.A. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Druckmann S., Hill S., Schürmann F., Markram H., Segev I. A hierarchical structure of cortical interneuron electrical diversity revealed by automated statistical analysis. Cereb. Cortex. 2013;23:2994–3006. doi: 10.1093/cercor/bhs290. [DOI] [PubMed] [Google Scholar]
- Gamper N., Zaika O., Li Y., Martin P., Hernandez C.C., Perez M.R., Wang A.Y., Jaffe D.B., Shapiro M.S. Oxidative modification of M-type K(+) channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 2006;25:4996–5004. doi: 10.1038/sj.emboj.7601374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.S., Williams L.A., Eggan K.C. Constructing and deconstructing stem cell models of neurological disease. Neuron. 2011;70:626–644. doi: 10.1016/j.neuron.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Hochbaum D.R., Zhao Y., Farhi S.L., Klapoetke N., Werley C.A., Kapoor V., Zou P., Kralj J.M., Maclaurin D., Smedemark-Margulies N. All-optical electrophysiology in mammalian neurons using engineered microbialrhodopsins. Nat. Methods. 2014;11:825–833. doi: 10.1038/nmeth.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J.H., Venkatachalam V., Cohen A.E. Temporal dynamics of microbialrhodopsin fluorescence reports absolute membrane voltage. Biophys. J. 2014;106:639–648. doi: 10.1016/j.bpj.2013.11.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichida J.K., Kiskinis E. Probing disorders of the nervous system using reprogramming approaches. EMBO J. 2015;34:1456–1477. doi: 10.15252/embj.201591267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai K., Kuwabara S., Misawa S., Tamura N., Ogawara K., Nakata M., Sawai S., Hattori T., Bostock H. Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage. Brain. 2006;129:953–962. doi: 10.1093/brain/awl024. [DOI] [PubMed] [Google Scholar]
- Kiskinis E., Sandoe J., Williams L.A., Boulting G.L., Moccia R., Wainger B.J., Han S., Peng T., Thams S., Mikkilineni S. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell. 2014;14:781–795. doi: 10.1016/j.stem.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralj J.M., Douglass A.D., Hochbaum D.R., Maclaurin D., Cohen A.E. Optical recording of action potentials in mammalian neurons using a microbialrhodopsin. Nat. Methods. 2012;9:90–95. doi: 10.1038/nmeth.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund L.M., McQuarrie I.G. Calcium/calmodulin-dependent protein kinase II expression in motor neurons: effect of axotomy. J. Neurobiol. 1997;33:796–810. doi: 10.1002/(sici)1097-4695(19971120)33:6<796::aid-neu7>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- McNeish J., Gardner J.P., Wainger B.J., Woolf C.J., Eggan K. From dish to bedside: lessons learned while translating findings from a stem cell model of disease to a clinical trial. Cell Stem Cell. 2015;17:8–10. doi: 10.1016/j.stem.2015.06.013. [DOI] [PubMed] [Google Scholar]
- Mukamel E.A., Nimmerjahn A., Schnitzer M.J. Automated analysis of cellular signals from large-scale calcium imaging data. Neuron. 2009;63:747–760. doi: 10.1016/j.neuron.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankevich D.E., Altevogt B.M., Dunlop J., Gage F.H., Hyman S.E. Improving and accelerating drug development for nervous system disorders. Neuron. 2014;84:546–553. doi: 10.1016/j.neuron.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peviani M., Kurosaki M., Terao M., Lidonnici D., Gensano F., Battaglia E., Tortarolo M., Piva R., Bendotti C. Lentiviral vectors carrying enhancer elements of Hb9 promoter drive selective transgene expression in mouse spinal cord motor neurons. J. Neurosci. Methods. 2012;205:139–147. doi: 10.1016/j.jneumeth.2011.12.024. [DOI] [PubMed] [Google Scholar]
- Pontiggia A., Negri A., Beltrame M., Bianchi M.E. Protein HU binds specifically to kinked DNA. Mol. Microbiol. 1993;7:343–350. doi: 10.1111/j.1365-2958.1993.tb01126.x. [DOI] [PubMed] [Google Scholar]
- Powers R.K., Heckman C.J. Contribution of intrinsic motoneuron properties to discharge hysteresis and its estimation based on paired motor unit recordings: a simulation study. J. Neurophysiol. 2015;114:184–198. doi: 10.1152/jn.00019.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers R.K., Elbasiouny S.M., Rymer W.Z., Heckman C.J. Contribution of intrinsic properties and synaptic inputs to motoneuron discharge patterns: a simulation study. J. Neurophysiol. 2012;107:808–823. doi: 10.1152/jn.00510.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz A.A., Abbott L., Marder E. The dynamic clamp comes of age. Trends Neurosci. 2004;27:218–224. doi: 10.1016/j.tins.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Sareen D., O'Rourke J.G., Meera P., Muhammad A.K., Grant S., Simpkinson M., Bell S., Carmona S., Ornelas L., Sahabian A. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B.C., Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu. Rev. Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam V., Brinks D., Maclaurin D., Hochbaum D., Kralj J., Cohen A.E. Flash memory: photochemical imprinting of neuronal action potentials onto a microbialrhodopsin. J. Am. Chem. Soc. 2014;136:2529–2537. doi: 10.1021/ja411338t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger B.J., Kiskinis E., Mellin C., Wiskow O., Han S.S., Sandoe J., Perez N.P., Williams L.A., Lee S., Boulting G. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werley C.A., Chien M.P., Cohen A.E. An ultrawidefield microscope for high-speed fluorescence imaging and targeted optogenetic stimulation. Biomed. Opt. Express. 2017;8:5794–5813. doi: 10.1364/BOE.8.005794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Cohen A.E. Optogenetic approaches to drug discovery in neuroscience and beyond. Trends Biotechnol. 2017;35:625–639. doi: 10.1016/j.tibtech.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziskind-Conhaim L. Electrical properties of motoneurons in the spinal cord of rat embryos. Dev. Biol. 1988;128:21–29. doi: 10.1016/0012-1606(88)90262-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.