

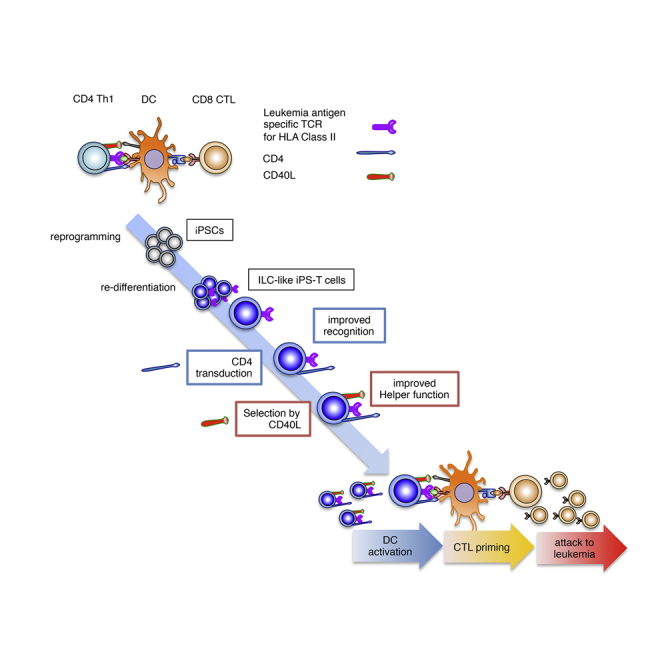
Summary
CD4+ T helper (Th) cell activation is essential for inducing cytotoxic T lymphocyte (CTL) responses against malignancy. We reprogrammed a Th clone specific for chronic myelogenous leukemia (CML)-derived b3a2 peptide to pluripotency and re-differentiated the cells into original TCR-expressing T-lineage cells (iPS-T cells) with gene expression patterns resembling those of group 1 innate lymphoid cells. CD4 gene transduction into iPS-T cells enhanced b3a2 peptide-specific responses via b3a2 peptide-specific TCR. iPS-T cells upregulated CD40 ligand (CD40L) expression in response to interleukin-2 and interleukin-15. In the presence of Wilms tumor 1 (WT1) peptide, antigen-specific dendritic cells (DCs) conditioned by CD4-modified CD40Lhigh iPS-T cells stimulated WT1-specific CTL priming, which eliminated WT1 peptide-expressing CML cells in vitro and in vivo. Thus, CD4 modification of CD40Lhigh iPS-T cells generates innate lymphoid helper-like cells inducing bcr-abl-specific TCR signaling that mediates effectiveanti-leukemic CTL responses via DC maturation, showing potential for adjuvant immunotherapy against leukemia.
Keywords: iPSCs, T cell differentiation, innate lymphoid cells, immuno-adjuvant function, bcr-abl, CD40L, DC activation, chronic myeloid leukemia, immunotherapy
Graphical Abstract
Highlights
-
•
iPSC-derived T cells have molecular similarity to group 1 innate lymphoid cells
-
•
iPSC-derived CD40Lhigh T cell-adjuvants induce leukemia-specific CTLs via DCs
Kaneko and colleagues describe the generation of CD4+ T helper clone-derived iPSCs and differentiation of the cells into T-lineage cells, which had molecular signatures and functional properties more consistent with group 1 innate lymphoid cells. CD4 transduction and CD40 ligand high population purification of the regenerated cells enhanced the antigen-specific adjuvant responses via dendritic cells in an antigen-specific manner.
Introduction
Tumor antigen-specific CD4+ T helper (Th) cells can induce a wide range of tumor antigen-specific cytotoxic T lymphocyte (CTL) responses via the maturation of dendritic cells (DCs), which avoid tumor evasion because of a mutation in a single target epitope (Kreiter et al., 2015). In addition, Th cells can attenuate the immunosuppressive properties of the tumor microenvironment by altering the cytokine milieu (Kim and Cantor, 2014). Therefore, adoptive T cell therapy using ex vivo expanded antigen-specific CD4+ Th cells may be a promising therapeutic strategy for refractory malignant tumors including hematological malignancies. However, clinical application is limited by the difficult isolation of CD4+ Th cells specific for relevant antigens and limited proliferative potential of these cells.
This problem may be solved by using induced pluripotent stem cell (iPSC) technology. We and others have reported methods for establishing iPSCs from mature antigen-specific T cells and re-differentiating the iPSCs into CD8+ T cells or invariant T cells with the same T cell antigen receptor (TCR) as the original T cells (Kitayama et al., 2016, Nishimura et al., 2013, Vizcardo et al., 2013, Wakao et al., 2013). The proliferative potential of iPSCs may provide a sufficient number of CD4+ Th cells for cancer treatment.
CD40 ligand (CD40L), which is expressed on activated CD4+ Th cells, is critical for inducing DC maturation via the CD40-CD40L interaction (Bennett et al., 1997, Bennett et al., 1998, Boise et al., 1995, Ridge et al., 1998, Schoenberger et al., 1998, Summers deLuca and Gommerman, 2012, Wiesel and Oxenius, 2012). Recently, the expression of CD40L on other types of immune cells known as innate lymphoid cells (ILCs) was reported (Magri et al., 2014, McKenzie et al., 2014, Summers deLuca and Gommerman, 2012). ILCs play a fundamental role in the immune system not only by initiating, regulating, and resolving inflammation, but also by modulating adaptive immunity (Sonnenberg and Artis, 2015). Although they lack TCRs, ILCs show T helper properties similar to Th1, Th2, Th17, and Th22 cells in terms of their cytokine profiles and transcription factors, which determine their development (McKenzie et al., 2014). The contribution of ILCs to pathogen control and pathogenesis, along with their similarity and redundancy to acquired immune cells, are current of interest in immunology research (Cording et al., 2016).
In the present study, we established iPSCs from a CD4+ Th1 clone specific for the junction region of BCR-ABL p210 (b3a2), a leukemia antigen, which is restricted by HLA class II (HLA-DR9) (Ueda et al., 2016). We induced re-differentiation of iPSCs to T-lineage cells expressing HLA class II-restricted TCR (iPS-T cells). The gene expression profile of iPS-T cells differed from that of αβTCR+ T cells and resembled a subset of ILCs. By transferring CD4 molecule to iPS-T cells and optimizing the ex vivo culture conditions to induce iPS-T cells with high CD40L expression, we successfully generated innate lymphoid helper-like cells that activated leukemic antigen-specific CTLs via DC maturation in a TCR-dependent antigen-specific manner. The activated CTLs showed effective anti-leukemic activity.
Our findings indicate that functional helper-like cells can be acquired from iPS-T cells through genetic modification and purification of the population. Therefore, CD40Lhigh CD4+ iPS-T cells are a potential platform for novel adjuvant cell therapy against malignant tumors.
Results
ILC-like Properties of T-Lineage Cells Differentiated from CD4+ Th1 Clone-Derived iPSCs
We previously established an HLA-DR9-restricted leukemia antigen (b3a2)-specific CD4+ Th1 clone (SK). Using our T cell regeneration protocol with slight modifications (Figure S2A), we obtained CD3+ CD45+ CD5dim+ CD7+ CD8αdim+ CD8β− cells from CD4+ Th1 clone (SK)-derived iPSCs (Figure 1A, left panel). The cells did not express CD4 throughout cell processing and heterogeneously expressed several ILC markers including CD56, CD161, NKG2D, c-Kit, NKp30, NKp44, NKp46, and DNAM-1 (Figure 1A, right panel). Despite their heterogeneity, the cells consistently expressed the same TCR as the original CD4+ Th1 clone (SK) (Figure S2B). Based on the expression of CD161 and c-Kit, iPS-T cells were divided into four subpopulations (Figure S2C), and their global RNA expression patterns were compared with those of natural killer (NK) cells, type 1 ILCs (ILC1s), type 2 ILCs (ILC2s), type 3 ILCs (ILC3s), αβT cells, and γδT cells isolated from peripheral blood (Figure S2D). iPS-T cells had genetic properties more consistent with those of ILC1s, NK cells, and γδT cells than those of peripheral αβT cells (Figure S2E; Table S2). The expression of genes related to T cell and ILC functions in iPS-T cells were similar to those in NK cells or ILC1s (Figures 1B and S2F; Table S3). Gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed enrichment of genes related to “NK cell-related cytotoxicity” in iPS-T cells, NK cells, and ILC1s (Table S4). All subpopulations of iPS-T cells expressed relatively low levels of BCL11B, an essential transcription factor for T cell differentiation, compared with αβT cells, but relatively high levels of ID2 and ZBTB16, which are transcription factors for ILCs (Figures S2G and S4F) (Naito et al., 2011, Spits et al., 2013). All subpopulations commonly expressed ILC1-related genes, such as NCAM1, NCR1, NCR2, ICOS, and IL12RB2, but low levels of IL7R and IL1R, which are expressed on all ILCs except NK cells (Figure 1D) (Spits et al., 2013). The ILC1/NK-like genetic properties of iPS-T cells, indicated by the relatively high expression of TBX21 and relatively low expression of GATA3 and RORC, were confirmed by the reads per kilobase million value and qRT-PCR analysis (Figure 1C, and data not shown). As reported previously, our iPS-T cells exhibited TCR-independent NK cell-like cytotoxicity (Figure S2H) (Kitayama et al., 2016, Yamada et al., 2016). The iPS-T cells produced high levels of interferon-γ (IFN-γ), comparatively low levels of interleukin-4 (IL-4), and no IL-17, which are similar to the original CD4+ Th1 clone (SK) (Figure 1E). In addition, iPS-T cells were expanded by up to several thousand-fold by two rounds of phytohemagglutinin (PHA)-P stimulation (Figure S2I). These data suggest that iPS-T cells generated from the CD4+ Th1 clone have group 1 ILC-like properties despite their expression of TCR.
Figure 1.

Re-differentiation and CD4 Modification of T-Lineage Cells from CD4+ Th1 Clone-Derived iPSCs Exert HLA Class II-Restricted Responses
(A) Representative flow cytometry profiles of the indicated molecules on the original CD4+ Th clone (SK) and regenerated T cells (iPS-T cells) after 14 days of phytohemagglutinin (PHA)-P stimulation.
(B) Principal component analysis of expression profiles of 146 selected T cell/ILC-related genes (Table S3).
(C) GATA3, RORC, and TBX21 expression in the indicated population. mRNA expression levels were determined by RNA sequencing.
(D) Hierarchical clustering of expressions of 22 selected genes related to ILC subsets.
(E) Cytokine production of the original CD4+ Th1 clone (SK) and iPS-T cells. T cells were stimulated with plate-bound control immunoglobulin (immunoglobulin G [IgG]) or anti-CD3 mAb (10 μg/mL) for 24 hr. The indicated cytokines in the culture supernatant were measured by ELISA. Data shown are the means ± SD of triplicate cultures and are representative of two independent experiments.
(F) Representative flow cytometry profiles of CD4 and TCR-Vβ22 expression on the original CD4+ Th1 clone (SK), Mock transduced iPS-T cells (Mock iPS-T cells), and CD4-transduced iPS-T cells (CD4+ iPS-T cells).
(G) Proliferative responses of Mock iPS-T cells and CD4+ iPS-T cells to antigenic peptide. T cells were co-cultured with autologous PBMCs in the presence of b3a2 peptide (10 μM) and measured by the [3H]thymidine incorporation assay. Data shown are the means and are representative of two independent triplicate experiments.
(H) b3a2 peptide-specific IFN-γ production by Mock iPS-T cells and CD4+ iPS-T cells. Mock iPS-T cells and CD4+ iPS-T cells (1 × 105) were co-cultured for 24 hr with autologous DCs (5 × 104) that had been prepulsed with b3a2 peptide (10 μM). IFN-γ in the culture supernatant (24 hr) was measured by ELISA. Data shown are the means ± SD of triplicate cultures and are representative of two independent experiments.
CD4 Modification Augments b3a2-Specific Responses in iPSC-Derived T Cells
During T cell activation, the binding of CD4 to HLA class II increases TCR signaling 30- to 300-fold (Janeway, 1992), suggesting that CD4 is essential for the complete activation of Th cells. Because our iPS-T cells expressed HLA class II-restricted TCR from the original CD4+ Th1 clone (SK), we hypothesized that transduction of the CD4 gene in iPS-T cells results in enhanced TCR-dependent responses by peptide stimulation. We transduced the CD4 gene using a retroviral vector (Figure 1F). CD4-transduced iPS-T cells (CD4+ iPS-T cells) showed an antigen-dependent proliferative response and cytokine production restricted by HLA-DR9 (Figures 1G, 1H, and S3A), which was consistent with the response of the original CD4+ Th1 clone (SK) (Figures S3B–S3D). In contrast, iPS-T cells without CD4 transduction (Mock iPS-T cells) showed impaired proliferation and IFN-γ production (Figures 1G and 1H). Moreover, we confirmed that the acquired functions of CD4+ iPS-T cells did not depend on the specific positional effect of integrated retroviral vectors (Figure S3E). The effect of CD4 modification was also validated in iPS-T cells derived from an HLA-DR53-restricted GAD65113-131 peptide-specific CD4+ Th clone (SA32.5) (Figures S4A–S4E) (Tabata et al., 1998). Next, we analyzed the global gene expression profiles of Mock iPS-T cells and CD4+ iPS-T cells stimulated with b3a2 peptide- or vehicle-loaded THP-1-expressing HLA-DR9 (THP-1-DR9) cells. The gene expression profile of CD4+ iPS-T cells stimulated by b3a2 peptide differed from that of cells without b3a2 peptide and/or CD4 transduction (Figures S5A and S5B, Table S5). Gene ontology analysis revealed that categories related to cell proliferation were significantly upregulated in b3a2-stimulated CD4+ iPS-T cells compared with b3a2-stimulated Mock iPS-T cells (Figure S5C). These data collectively indicate that CD4 gene modification gives iPS-T cells an enhanced ability to exert b3a2 peptide-specific and HLA class II-restricted responses.
Identification of a CD40Lhigh Population that Efficiently Exerts Adjuvant Function
CD40L expression on activated CD4+ Th cells is critical for the maturation of DCs, which provide costimulatory signals for effective activation and enhanced survival of antigen-specific CD8+ T cells (Bennett et al., 1997, Bennett et al., 1998, Boise et al., 1995, Ridge et al., 1998, Schoenberger et al., 1998, Wiesel and Oxenius, 2012). In addition to CD4+ Th cells, IL-7 plus IL-1β primed murine ILC3 cells (Magri et al., 2014) and IL-2-activated human NK cells (Carbone et al., 1997) were reported to express functional levels of CD40L. We hypothesized that iPS-T cells express CD40L under stimulation by IL-2 receptor subunit γ (common γ chain), which binds to other ligand (IL-2, 4, 7, 9, 15, 21)-specific receptors. We detected preferential expression of CD40LG and simultaneous expression of partner cytokine receptors for common γ chain including IL2RA, IL2RB, IL2RG, and IL15RA in the c-Kit+ CD161+ subpopulation (Figure S5D and data not shown). We cultured iPS-T cells in several combinations of common γ chain cytokines and found that IL-2 in combination with IL-15 upregulated CD40L expression (Figures 2A and 2B).
Figure 2.

CD40Lhigh Population in iPS-T Cells Shows High Responsiveness to TCR Stimulation
(A and B) CD40L expression of indicated iPS-T cells on day 13 after PHA-P stimulation. Mock iPS-T cells or CD4+ iPS-T cells were stimulated with PHA-P and cultured in the absence (A) or presence (B) of IL-2. The frequency of CD40L-positive cells is shown in the upper right corner of each panel.
(C and D) Expression of CD40L, CD4, and TCR-Vβ22 on the subpopulations are shown. CD40L high and low populations under IL-2 (100 U/mL) and IL-15 (5 ng/mL) were separated from Mock iPS-T cells (C) and CD4+ iPS-T cells (D) by flow cytometry sorting and expanded by PHA-P stimulation.
(E and F) Surface CD40L expression on different subpopulations (E; CD40L high and F; CD40L low) stimulated with plate-bound control IgG or anti-CD3 mAb (10 μg/mL). The original CD4+ Th1 clone (SK) served as a control. CD40L (red) and isotype-matched controls (gray) are shown.
(G) Cytokine production by the indicated population stimulated with plate-bound control IgG or anti-CD3 mAb (10 μg/mL). The original CD4+ Th1 clone (SK) served as control. (H) Cytokine production by CD40Lhigh CD4+ iPS-T cells (1 × 105) co-cultured with THP1 cells (5 × 104) expressing HLA-DR9 and BCR-ABL p210 gene. (G and H) The indicated cytokines in the culture supernatant (24 h) were measured by a bead-based multiplex immunoassay. Data shown are the means ± SD of triplicate cultures and are representative of two independent triplicate experiments.
The IL-2/IL-15-induced CD40Lhigh and CD40Llow populations were separated and expanded by PHA-P stimulation. Each CD40Lhigh and CD40Llow population from Mock iPS-T cells and CD4+ iPS-T cells expressed TCR-Vβ22 and retained their CD40L expression levels in the presence of IL-2 and IL-15 (Figures 2C and 2D). Upon stimulation with an anti-CD3 antibody, only CD40Lhigh iPS-T cells showed upregulated CD40L expression (Figures 2E and 2F). CD40Lhigh CD4+ iPS-T cells produced higher levels of IFN-γ and tumor necrosis factor alpha (TNF-α) than did CD40Llow CD4+ iPS-T cells, but the two populations consistently produced low levels of IL-2, IL-4, IL-6, IL-10, and IL-17 (Figures 2G and S6A). When stimulated with b3a2-peptide-loaded DCs, co-expression of CD4 and CD40L synergistically enhanced the production of IFN-γ and TNF-α, indicating a Th1-biased cytokine profile (Figure S6B). In addition, when CD40Lhigh CD4+ iPS-T cells were co-cultured with THP-1-DR9 cells expressing BCR-ABL p210 protein, they produced IFN-γ and TNF-α, indicating their ability to respond to the naturally processed BCR-ABL p210 epitope (Figures 2H and 2I). CD40Lhigh CD4+ iPS-T cells can expand against repeated stimulation without affecting CD3, CD5dim, CD7, and CD8α expression (Figures S6C and S6D). Similar findings associated with CD40L expression were obtained in iPS-T cells from an HLA-DR53-restricted GAD65113-131 peptide-specific CD4+ Th clone (SA32.5) (Figures S7A–S7E). Collectively, we identified a population of CD4+ iPS-T cells with upregulated CD40L expression in response to TCR stimulation and possessed a superior ability to respond to antigenic peptide stimulation.
Next, we analyzed the cellular adjuvant function of CD4+ iPS-T cells to induce DC maturation. When CD40Lhigh CD4+ iPS-T cells were co-cultured with immature DCs prepulsed with b3a2 peptide, they induced maturation of DCs (Figures 3A and S7F). In contrast, DC maturation by CD40Llow CD4+ iPS-T cells was impaired (Figures 3A and S7F). Furthermore, CD40Llow Mock iPS-T cells failed to induce DC maturation, possibly via impaired recognition of the HLA class II/peptide complex because of the absence of CD4 (Figures 3A and S7F). CD40Lhigh CD4+ iPS-T cells also enhanced the production of IL-12p70, CXCL9, and CXCL11, which are important soluble factors in the activation and migration of NK cells, Th1 cells, and CTLs (Figure 3B). These data indicate that CD40Lhigh CD4+ iPS-T cells have a superior ability to induce DC maturation.
Figure 3.

DC Activation Induced by CD40Lhigh CD4+ iPS-T Cells
(A) Flow cytometry profiles of the surface molecules on DCs. Vehicle- or b3a2-peptide-pulsed DCs were cultured for 24 hr with the CD40Lhigh CD4+ and CD40Llow CD4+ population at a DC/CD4+ iPS-T cell ratio of 5:1. Relative fluorescence intensity (RFI) (%) of indicated molecule is calculated as follows: RFI (%) = 100 × (RFI of b3a2 peptide-treated cells)/(RFI of vehicle-treated cells).
(B) Cytokine production by DCs co-cultured with the indicated population. Cytokines in the culture supernatant were measured by a bead-based multiplex immunoassay. The indicated population (1 × 104) was co-cultured for 24 hr with autologous DCs (2.5 × 104) that had been prepulsed with b3a2 peptide (10 μM). The original CD4+ Th1 clone (SK) served as a control.
(A and B) Data shown are the means ± SD of triplicate cultures and are representative of two independent experiments.
It was previously reported that the CD40L-CD40-mediated ILC3-B cell interactions induce regulatory B cells to secrete the T cell-suppressing cytokine IL-10 (Komlosi et al., 2017); thus, we examined whether CD40Lhigh CD4+ iPS-T cells induce the immune inhibitory cytokine IL-10 by interacting with DCs or B cells. Both CD40Lhigh- and CD40Llow-CD4+ iPS-T cells alone did not produce IL-10 upon CD3 stimulation (Figure S6A). When DCs or B cells were co-cultured with CD40Lhigh CD4+ iPS-T cells in the presence of the b3a2 peptide, they did not produce IL-10, whereas DCs co-cultured with CD40Llow CD4+ iPS-T cells produced IL-10 (Figure S7G). These data indicate that CD40Lhigh CD4+ iPS-T cells do not stimulate IL-10 production, which is an appropriate property for eliciting anti-leukemic immune responses.
CD40Lhigh CD4+ iPSC-Derived T Cells Reduced TCR-Independent Cytotoxicity
CD4+ Th1 clone (SK)-derived iPS-T cells exhibited antigen-independent cytotoxicity against THP-1, and the cytotoxicity against THP-1 was partially dependent on perforin and DNAM-1 (Figure 4A). iPS-T cells exert DNAM-1- and NKG2D-dependent cytotoxicity against K562 cells expressing both DNAM-1 ligand (PVR, nectin-2) and NKG2D ligands (MICA/B) (Kitayama et al., 2016). Despite the expression of both NK receptors, the iPS-T cells used in this study did not show NKG2D-dependent cytotoxicity (Figure 4A). This may be because of the reduced expression of NKG2D ligands (MICA/B) on THP-1 target cells (Figure S7H). CD40Lhigh CD4+ iPS-T cells conditioned by IL-2/IL-15 exhibited reduced expression of DNAM-1 and NKG2D compared with CD40Llow CD4+ iPS-T cells (Figure 4B, left panel). Consistent with the reduced DNAM-1 and NKG2D expression, CD40Lhigh CD4+ iPS-T cells reduced NK cell-like cytotoxicity against THP-1-DR9 while retaining b3a2-specific cytotoxicity (Figure 4B, right panel). These data indicate that CD40Lhigh CD4+ iPS-T cells reduced antigen-independent cytotoxicity by downregulating DNAM-1 expression.
Figure 4.

CD40Lhigh CD4+ iPS-T Cells Reduced NK Cell-like Cytotoxicity
(A) Left panel: cytotoxic activity of iPS-T cells to THP-1 cells. Center panel: Mock iPS-T cells were treated with 10 nM concanamycin A (CMA) to block perforin. Right panel: Indicated Abs blocking receptor/ligand interactions were added. Cytotoxicity of iPS-T cells to THP-1 cells at an effector/target (E:T) ratio of 2.5:1.
(B) Left panel: representative flow cytometry profiles of TCR-Vβ22, CD4, CD40L, DNAM-1, and NKG2D on the indicated population. Right panel: cytotoxic activities of the indicated population to HLA-DR9-expressing THP-1 cells loaded with vehicle or b3a2 peptide (5 μM).
(A and B) Cytotoxicity was measured by 51Cr-release assay for 4 hr at the indicated E:T ratios. Data are representative of two independent triplicate experiments.
CD40Lhigh CD4+ iPSC-Derived T Cells Efficiently Induce Primary Expansion of Leukemia Ag-Specific CTLs
CD4+ Th cells help in the priming of CD8+ CTLs via DC activation (Bennett et al., 1997, Ridge et al., 1998, Schoenberger et al., 1998, Ueda et al., 2016). To determine whether CD40Lhigh CD4+ iPS-T cells can induce leukemia antigen-specific CTL responses, vehicle- or b3a2 peptide-loaded DCs were cultured with CD40Lhigh CD4+ iPS-T cells, and differentially conditioned DCs were loaded with Wilms tumor 1 (WT1235-243) peptide, irradiated, and cultured with CD8+ T cells (Figure S8A). After 7 days of culture, the proliferation of CD8+ T cells was evaluated. We observed markedly enhanced proliferation of CD8+ T cells upon stimulation with WT1235-243 peptide when CD40Lhigh CD4+ iPS-T cell/b3a2 peptide-conditioned DCs were used as antigen-presenting cells (Figure 5A, left panel). In contrast, neither CD40Llow CD4+ iPS-T cell/b3a2-conditioned DCs nor CD40Llow Mock iPS-T cell/b3a2-conditioned DCs induced the proliferation of CD8+ T cells (Figure 5A, center and right panel). CTLs stimulated by CD40Lhigh CD4+ iPS-T cell/b3a2 peptide-conditioned DCs contained a high frequency of WT1-tetramer-positive T cells (Figure 5B, left panel). WT1 peptide-specific CTLs were further expanded by repeated stimulation with WT1 peptide three times (Figure 5B, center panel). The expanded WT1-specific CTLs exhibited cytotoxic activity against WT1 peptide-loaded HLA-A24-expressing K562, but not against vehicle-loaded cells (Figure 5B, right panel). These data indicate that the activation of CD40Lhigh CD4+ iPS-T cells by the b3a2 peptide induces DC-mediated cellular adjuvant properties that can increase leukemic antigen-specific CTL responses, which is consistent with the T helper function of the original CD4+ Th1 clone (SK) (Ueda et al., 2016).
Figure 5.

Induction of Leukemia Antigen-Specific CTLs By CD40Lhigh CD4+ iPS-T Cells
(A) Proliferative response of CD8+ T cells. Indicated iPS-T cells (5 × 103) and DCs (1 × 104) ± b3a2 peptide (5 μM) were initially co-cultured for 5 hr to mature the DCs, after which DCs and iPS-T cells were irradiated and cultured with autologous CD8+ T cells (5 × 104) in the presence of WT1 peptide (5 μM). The proliferative response (day 7) was measured as the amount of [3H]thymidine incorporated. Data shown are the means ± SD of triplicate cultures and are representative of two independent experiments.
(B) Left panels: frequency of WT1/HLA-A24 tetramer-positive CD8+ T cells primed by CD40Lhigh CD4+ iPS-T cell-conditioned DCs. CD40Lhigh CD4+ iPS-T cells (5 × 103) and DCs (1 × 104) prepulsed with b3a2 peptide (5 μM) were initially co-cultured for 5 hr to mature the DCs, after which DCs and CD4+ iPS-T cells were irradiated and cultured with autologous CD8+ T cells (5 × 104) in the presence of WT1 peptide (5 μM). Tetramer staining at day 10 after stimulation is shown. Center panel: frequency of WT1/HLA-A24 tetramer-positive CD8+ T cells after a third stimulation with WT1 peptide. Representative flow cytometry profiles of two independent experiments. HIV-env/HLA-A24 tetramer was used as a control. Right panel: cytotoxic activities of expanded WT1-specific CD8+ T cells against K562-A24 loaded with vehicle or WT1 peptide. Cytotoxicity was measured by 51Cr-release assay for 4 hr at the indicated E:T ratios. Data are representative of two independent triplicate experiments.
WT1-Specific CTLs Primed By CD40Lhigh CD4+ iPSC-Derived T Cell/DC Interaction Exert Anti-leukemic Effects In Vivo
To examine whether primed WT1 peptide-specific CTLs exert anti-leukemic effects in vivo, WT1 epitope-expressing K562 cells were subcutaneously injected into NSG mice with or without WT1 peptide-specific CTLs. Tumor growth was monitored each week by bioluminescence imaging and external caliper measurements. In the presence of WT1 peptide-specific CTLs, tumor growth was significantly inhibited (Figure 6A) and the survival of mice was significantly prolonged (Figure 6B). Collectively, leukemic antigen-specific CTLs primed by the interaction of CD40Lhigh CD4+ iPS-T cells and DCs exert effective anti-leukemic effects.
Figure 6.

Anti-leukemic Activity of CTLs Primed By CD40Lhigh CD4+ iPS-T Cell-Conditioned DCs
(A) In vivo anti-leukemia effect. NSG mice were subcutaneously injected with mixtures of K562-A24-Luc-WT1 minigene and either saline or WT1-specific CTLs. Upper panel: tumor burden was measured weekly by bioluminescence imaging. Lower panel: the average tumor size for each group from day 0 to 42 is shown. Error bars represent ± SD. ∗p < 0.05 by unpaired Student's t test (two-tailed).
(B) Kaplan-Meier survival curves for treated and control mice. ∗p < 0.05 by the log rank (Mantel-Cox) test. WT1-CTL, n = 10; no treatment, n = 5.
Discussion
Although tumor antigen-specific CD4+ Th cells are essential for inducing downstream activation of diverse tumor antigen-specific CTLs, it is difficult to obtain clinically sufficient numbers of Th cells for adoptive cell therapy. The unlimited supply of Th cells from iPSCs may resolve this issue. To date, studies have not reported the successful generation of HLA class II-restricted CD4+ Th cells from iPSCs. We established iPSCs from b3a2-specific CD4+ Th cells (SK) and induced T-lineage cells (iPS-T cells) from these cells. Using the current differentiation protocol, the iPS-T cells spontaneously failed to express CD4 molecules, and their gene expression profiles were closer to those of ILC1s/NK cells than to those of αβT cells, which may be partially explained by insufficient control of the transcription factor BCL11B for specification to the T cell-lineage (Kitayama et al., 2016). Bcl11b insufficiency is known to cause biased lineage-reprogramming from T cells to NK cells (a lineage of type 1 ILCs) in conditional knockout mice (Li et al., 2010). Our iPS-T cells from multiple donors expressed ID2 and ZBTB16, but not BCL11B (Figure S4F). These features may partially explain the lineage-shift from T cells to type 1 ILCs during in vitro differentiation.
Modification of CD4 expression in the regenerated cells and purification of the CD40Lhigh population were performed to acquire ILC1-like cells that can stimulate DCs, thereby, promoting leukemia antigen-specific CTL responses. Although the detailed mechanisms of this artificial TCR signaling in group 1 ILC-like T cells are presently not known, TCR ligation via antigenic peptide/HLA molecule complexes induced proliferation and cytokine production in a manner consistent with that in αβT cells.
The interaction between CD40Lhigh CD4+ iPS-T cells and DCs facilitates the production of chemokines to recruit CXCR3-expressing effector cells (Figure 3B). IFN-γ production from these recruited effector cells activates DCs to amplify the production of chemokines (Groom and Luster, 2011). This positive feedback loop pathway may further facilitate the recruitment and activation of effector cells in tumor tissues. In the actual leukemia environment, CD40Lhigh CD4+ iPS-T cells may condition DCs to take up leukemia cells and cross-present leukemia-derived antigens, resulting in the activation of diverse CTLs that specifically recognize multiple leukemia antigens, a phenomenon known as epitope spreading. Chronic myelogenous leukemia (CML) cells carry a number of well-defined leukemia antigens including BCR-ABL, PR1, and WT1 (Molldrem et al., 2000, Rezvani et al., 2003). Moreover, TKI treatment of CML induces various mutations in the BCR-ABL kinase domain because of resistance. The mutated antigens may then generate highly immunogenic neoepitopes (Cai et al., 2012), and these neoepitopes are potential targets of epitope spreading by CD40Lhigh CD4+ iPS-T cells. However, few suitable animal models are available that reproduce general reactions of human immune cells, particularly in HLA class II-restricted helper T cell/DC-mediated CTL priming and subsequent tumor elimination. Animal models reconstituted with the human hematopoietic system, immune system, tumor xenograft, and iPS-T cells derived from the same patient may overcome this limitation in the future.
Our study is an initial step toward clinically applicable iPSC-derived T helper-like cell therapy. However, iPSC-based regenerative medicine is accompanied by challenges, such as the fact that these strategies are time-consuming and expensive. Compared with an autologous setting, the use of HLA-matched or homozygous HLA-type allogeneic iPSCs transduced with desired TCR (i.e., obtained TCR sequences from this research) may resolve this issue in the preparation of CD40Lhigh CD4+ iPS-T cells (Neofytou et al., 2015). In addition, reproducible production of CD4-expressing iPS-T cells is a prerequisite for their clinical application. In this study, CD4 gene transduction was performed after re-differentiation of iPSCs to T-lineage cells. Genome editing of iPSCs enables targeted insertion of the CD4 transgene into the downstream region of T cell-specific promoters, such as Lck promoter. Using such a system, it may be possible to establish an ideal iPSC clone expressing transgenes only at an ideal differentiation step when the promoter is activated. This may provide reproducible CD4-expressing iPS-T cells with safe profiles for clinical use. Moreover, establishing a good manufacturing practice-compliant manufacturing protocol using xenogeneic-free and feeder cell-free material is essential and currently under development (Fransen et al., 2011, Neofytou et al., 2015).
In conclusion, we showed that iPS-T cells from CD4+ Th clone-derived iPSCs constitute a heterogeneous population of TCR-expressing group 1 ILC-like lymphoid cells. Gene modification and cell purification resulted in the acquisition of HLA class II-restricted TCR-expressing adjuvant cells that induce anti-leukemia effects via DC maturation. These results support iPS-T cells as a potential platform for novel adjuvant therapy for leukemia.
Experimental Procedures
Peptide, Cytokines, and Chemicals
HLA-DR9 (DRB1∗09:01)-restricted BCR-ABL b3a2-junctional peptide (ATGFKQSSKALQRPVAS), HLA-A24 (A∗24:02)-restricted modified WT1235–243 epitope peptide (CYTWNQMNL), and HLA-DR53 (DRB4∗01:03)-restricted glutamic acid decarboxylase 65 (GAD65)113-131 peptide (DVMNILLQYVVKSFDRSTK) were commercially synthesized and supplied at >90% purity (Toray Research Center, Kamakura, Japan, Scrum, Tokyo, Japan). In the modified WT1235-243 peptide, Y was substituted for M at amino acid position 2 of the natural WT1235-243 peptide (CYTWNQMNL). The following reagents were from commercial sources: recombinant human (rh) IL-2, rhIL-4, and rh granulocyte macrophage colony-stimulating factor (Primmune, Osaka, Japan); rhIL-7, rhIL-15, and rh fms-related tyrosine kinase 3 ligand (Flt-3L) (PeproTech, Rocky Hill, NJ, USA); rh basic fibroblast growth factor and PHA-P (Wako Pure Chemicals Industries, Osaka, Japan); rh vascular endothelial growth factor and rh stem cell factor (R&D Systems, Minneapolis, MN, USA); and penicillin-killed Streptococcus pyogenes (OK432) (Chugai Pharmaceutical, Tokyo, Japan).
Generation of Retrovirus for CD4 Transduction
The human CD4 gene was inserted into pDON-AI2 (Takara Bio, Shiga, Japan). Transient retroviral particles were produced in G3T-hi cells (Takara Bio), followed by transduction of PG13 cells to generate GaLV-pseudotyped retrovirus producer cells (Kaneko et al., 2001).
Generation of iPSCs Derived from HLA Class II-Restricted Antigen-Specific CD4+ T Cell Clone
HLA-DR9-restricted b3a2-specific CD4+ Th1 clone (SK) (Ueda et al., 2016) and HLA-DR53-restricted GAD65113-131 peptide-specific CD4+ Th clone (SA32.5) (Tabata et al., 1998) were reprogrammed to iPSCs by transduction of reprogramming factors by the Sendai viral system using pSeV[KOSM302L] (kindly provided by Dr. Nakanishi, AIST, Tsukuba, Japan) as described previously (Kitayama et al., 2016, Nishimura et al., 2013). The iPSC clones were negative for residual transgenes, showed pluripotency characterized by the expression of pluripotency-related molecules and teratoma formation in immunodeficient mice, and were confirmed to have a normal karyotype (Figure S1). The use of cells isolated from healthy adults was approved by the Ethics Committee of Kyoto University, and informed consent was obtained from all donors in accordance with the Declaration of Helsinki.
T Cell Differentiation from iPSCs and CD4 Transduction
We differentiated iPSCs into T cells using a previously described method (Nishimura et al., 2013). In brief, clumps of iPSCs were transferred onto C3H10T1/2 feeder cells and cultured in EB medium containing rh vascular endothelial growth factor. On day 7, rh stem cell factor and rhFlt-3L were added to the culture. On day 14, hematopoietic progenitor cells were collected and transferred onto OP9-DL1 cells and co-cultured in OP9 medium in the presence of rhIL-7 and rhFLT-3L. On day 35, regenerated T cells were stimulated by PHA-P in the presence of rhIL-7 and rhIL-15 using allogeneic peripheral blood mononuclear cells (PBMCs) as antigen-presenting cells at 14-day intervals. Expanded iPSC-T cells (1 × 106) were transduced with human CD4 using the RetroNectin-bound virus infection method, in which retroviral solution was preloaded onto RetroNectin-coated plates, centrifuged at 2,000 ×g for 2 hr at 32°C, and rinsed with 1.5% human serum albumin PBS, after which the cells were applied to the virus-preloaded plate for 20 hr. After 10 days of culture, a CD4-positive population (15%–35% of total cells) was sorted by flow cytometry and restimulated for expansion.
Functional Assay of T Cells
Cell proliferation was evaluated by the [3H]thymidine incorporation assay, as described previously (Zhang et al., 2015). Cytotoxic activity was measured using the 51Cr-release assay, as described previously (Zhang et al., 2015). Cytokine levels in the culture supernatants were evaluated by ELISA (hIFN-γ: eBioscience, San Diego, CA, USA) or a bead-based multiplex immunoassay (BD Cytomertic Beads Array; BD Biosciences, Franklin Lakes, NJ, USA).
CTL Priming and Cytotoxicity Assays
CD8+ T cells and DCs were obtained from the same donor from whom the b3a2-specific T cell clone (SK) was established. The cells were used to induce antigen-specific CTLs to avoid alloreactive responses. CD8+ T cells were isolated from PBMCs by negative magnetic cell sorting using a CD8+ T cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). DCs were cultured for 3 hr in 96-well round plates in the presence or absence of b3a2 peptide, and then the original CD4+ Th1 clone (SK) or re-differentiated SK was added to the culture and incubated for 5 hr. After irradiation at 30 Gy, CD8+ T cells were added together with WT1235–243 peptide. On day 7, we added 1 μCi of [3H]thymidine to the cultures; after 16-hr incubation, we assessed the proliferative response of CD8+ T cells in a [3H]thymidine incorporation assay. Another experiment was performed in the same manner on day 10. The frequencies of WT1-peptide-specific CTLs were determined by staining with the HLA-A∗2402/WT1235–243 tetramer. We restimulated the resulting CD8+ T cells with WT1235–243 peptide in the presence of 35-Gy-irradiated autologous PBMCs. The cells were later used in cytotoxicity assays and in vivo experiments.
In Vivo Experiments
All in vivo animal studies were approved by the Animal Research Committee of Kyoto University. Six-week-old female NOD-SCID IL2Rγcnull (NSG) mice were purchased from Charles River (Yokohama, Japan) and inoculated subcutaneously in the left shaved shank with mixtures of K562-Luc-A24-WT1 minigene (1.0 × 105) cells and either saline or WT1-specific CTLs (1.0 × 106). The mice were monitored for tumor growth and survival. Tumor growth was monitored weekly by bioluminescence imaging for 4 weeks and external caliper measurements until the mice died or were sacrificed when tumors exceeded 25 mm in diameter.
Retroviral Integration Site Analysis by Linear Amplification-Mediated PCR
Retroviral integration site analysis was performed using the Retro-X Integration Site Analysis Kit (Clontech, Mountain View, CA, USA) according to the manufacturer's instructions.
Statistical Analysis
STATA version 13.0 (StataCorp LP, College Station, TX, USA) was used for all statistical analyses. To compare multiple experimental groups, one-way ANOVA with Bonferroni post hoc test was used to assess significance; to compare two experimental groups, unpaired t tests (two-tailed) were used. For statistical analysis of Kaplan-Meier survival curves, a log rank (Mantel-Cox) test was used to calculate p values; p values < 0.05 were considered statistically significant and are indicated in the figures by asterisks.
Author Contributions
N.U., Y.U., and S. Kaneko designed the study. N.U., R.Z., S. Kitayama, Y.Y., S.I., T.-Y.L., Y.M., C.O., A.W., S.S., Y.N., K.K., H.K., Y.U., and S. Kaneko interpreted the data. N.U., Y.U., R.Z., S. Kitayama, Y.Y., M.T., and Y.M. performed the experiments. M.N. provided critical materials. N.U., R.Z., Y.M., and A.W. analyzed the data. S.I., Y.K., T.U., T.-Y.L., S.S., K.K., Y.N., H.K., and T.N. contributed to analyzing and discussing the data. Y.U. and S. Kaneko supervised the study. N.U., Y.U., and S. Kaneko wrote the manuscript.
Acknowledgments
CSII-EF, pCMV-VSV-G-RSV-Rev, and pCAG-HIVgp were kindly provided by Dr. H. Miyoshi (RIKEN BioResource Center, Tsukuba, Japan). cDNA encoding HLA-DRB1∗09:01 (DR9) was kindly provided by Dr. H. Kobayashi (Asahikawa Medical College, Asahikawa, Japan). We thank Dr. P. Karagiannis (Kyoto University, Kyoto, Japan) for reading the manuscript. This study was performed as a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P-DIRECT) and Core Center for iPS Cell Research of Research Center Network for Realization of Regenerative Medicine, Japan Agency for Medical Research and Development. This study was supported by grants from Grants-in-Aid “Carcinogenic Spiral,” Nagono Medical Foundation, the National Cancer Center Research and Development Fund (28-A-8) and the Takeda Science Foundation. Shin Kaneko is a founder, shareholder, and scientific advisor at Thya and received research funding from Kyowa Hakko Kirin, Takeda Pharmaceutical, Sumitomo Chemical, and Thyas. Hitoshi Kiyoi received research funding from Chugai Pharmaceutical, Bristol-Myers Squibb, Kyowa Hakko Kirin, Zenyaku Kogyo, FUJIFILM Corporation, Nippon Boehringer Ingelheim, Astellas Pharma, and Celgene Corporation, consulting fees from Astellas Pharma and Daiichi Sankyo, and honoraria from Bristol-Myers Squibb. The remaining authors declare no competing financial interests.
Published: May 24, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, eight figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.04.025.
Contributor Information
Yasushi Uemura, Email: yuemura@east.ncc.go.jp.
Shin Kaneko, Email: kaneko.shin@cira.kyoto-u.ac.jp.
Supplemental Information
References
- Bennett S.R., Carbone F.R., Karamalis F., Flavell R.A., Miller J.F., Heath W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Bennett S.R., Carbone F.R., Karamalis F., Miller J.F., Heath W.R. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J. Exp. Med. 1997;186:65–70. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise L.H., Minn A.J., Noel P.J., June C.H., Accavitti M.A., Lindsten T., Thompson C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- Cai A., Keskin D.B., DeLuca D.S., Alonso A., Zhang W., Zhang G.L., Hammond N.N., Nardi V., Stone R.M., Neuberg D. Mutated BCR-ABL generates immunogenic T-cell epitopes in CML patients. Clin. Cancer Res. 2012;18:5761–5772. doi: 10.1158/1078-0432.CCR-12-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone E., Ruggiero G., Terrazzano G., Palomba C., Manzo C., Fontana S., Spits H., Karre K., Zappacosta S. A new mechanism of NK cell cytotoxicity activation: the CD40-CD40 ligand interaction. J. Exp. Med. 1997;185:2053–2060. doi: 10.1084/jem.185.12.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cording S., Medvedovic J., Aychek T., Eberl G. Innate lymphoid cells in defense, immunopathology and immunotherapy. Nat. Immunol. 2016;17:755–757. doi: 10.1038/ni.3448. [DOI] [PubMed] [Google Scholar]
- Fransen M.F., Sluijter M., Morreau H., Arens R., Melief C.J. Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin. Cancer Res. 2011;17:2270–2280. doi: 10.1158/1078-0432.CCR-10-2888. [DOI] [PubMed] [Google Scholar]
- Groom J.R., Luster A.D. CXCR3 in T cell function. Exp. Cell Res. 2011;317:620–631. doi: 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway C.A., Jr. The T cell receptor as a multicomponent signalling machine: CD4/CD8 coreceptors and CD45 in T cell activation. Annu. Rev. Immunol. 1992;10:645–674. doi: 10.1146/annurev.iy.10.040192.003241. [DOI] [PubMed] [Google Scholar]
- Kaneko S., Onodera M., Fujiki Y., Nagasawa T., Nakauchi H. Simplified retroviral vector gcsap with murine stem cell virus long terminal repeat allows high and continued expression of enhanced green fluorescent protein by human hematopoietic progenitors engrafted in nonobese diabetic/severe combined immunodeficient mice. Hum. Gene Ther. 2001;12:35–44. doi: 10.1089/104303401450942. [DOI] [PubMed] [Google Scholar]
- Kim H.J., Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol. Res. 2014;2:91–98. doi: 10.1158/2326-6066.CIR-13-0216. [DOI] [PubMed] [Google Scholar]
- Kitayama S., Zhang R., Liu T.Y., Ueda N., Iriguchi S., Yasui Y., Kawai Y., Tatsumi M., Hirai N., Mizoro Y. Cellular adjuvant properties, direct cytotoxicity of re-differentiated Valpha24 invariant NKT-like cells from human induced pluripotent stem cells. Stem Cell Rep. 2016;6:213–227. doi: 10.1016/j.stemcr.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlosi Z.I., Kovacs N., van de Veen W., Kirsch A.I., Fahrner H.B., Wawrzyniak M., Rebane A., Stanic B., Palomares O., Ruckert B. Human CD40 ligand-expressing type 3 innate lymphoid cells induce IL-10-producing immature transitional regulatory B cells. J. Allergy Clin. Immunol. 2017 doi: 10.1016/j.jaci.2017.07.046. [DOI] [PubMed] [Google Scholar]
- Kreiter S., Vormehr M., van de Roemer N., Diken M., Lower M., Diekmann J., Boegel S., Schrors B., Vascotto F., Castle J.C. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Leid M., Rothenberg E.V. An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science. 2010;329:89–93. doi: 10.1126/science.1188989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magri G., Miyajima M., Bascones S., Mortha A., Puga I., Cassis L., Barra C.M., Comerma L., Chudnovskiy A., Gentile M. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat. Immunol. 2014;15:354–364. doi: 10.1038/ni.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie A.N., Spits H., Eberl G. Innate lymphoid cells in inflammation and immunity. Immunity. 2014;41:366–374. doi: 10.1016/j.immuni.2014.09.006. [DOI] [PubMed] [Google Scholar]
- Molldrem J.J., Lee P.P., Wang C., Felio K., Kantarjian H.M., Champlin R.E., Davis M.M. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat. Med. 2000;6:1018–1023. doi: 10.1038/79526. [DOI] [PubMed] [Google Scholar]
- Naito T., Tanaka H., Naoe Y., Taniuchi I. Transcriptional control of T-cell development. Int. Immunol. 2011;23:661–668. doi: 10.1093/intimm/dxr078. [DOI] [PubMed] [Google Scholar]
- Neofytou E., O'Brien C.G., Couture L.A., Wu J.C. Hurdles to clinical translation of human induced pluripotent stem cells. J. Clin. Invest. 2015;125:2551–2557. doi: 10.1172/JCI80575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T., Kaneko S., Kawana-Tachikawa A., Tajima Y., Goto H., Zhu D., Nakayama-Hosoya K., Iriguchi S., Uemura Y., Shimizu T. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Rezvani K., Grube M., Brenchley J.M., Sconocchia G., Fujiwara H., Price D.A., Gostick E., Yamada K., Melenhorst J., Childs R. Functional leukemia-associated antigen-specific memory CD8+ T cells exist in healthy individuals and in patients with chronic myelogenous leukemia before and after stem cell transplantation. Blood. 2003;102:2892–2900. doi: 10.1182/blood-2003-01-0150. [DOI] [PubMed] [Google Scholar]
- Ridge J.P., Di Rosa F., Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Schoenberger S.P., Toes R.E., van der Voort E.I., Offringa R., Melief C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Sonnenberg G.F., Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med. 2015;21:698–708. doi: 10.1038/nm.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H., Artis D., Colonna M., Diefenbach A., Di Santo J.P., Eberl G., Koyasu S., Locksley R.M., McKenzie A.N., Mebius R.E. Innate lymphoid cells – a proposal for uniform nomenclature. Nat. Rev. Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- Summers deLuca L., Gommerman J.L. Fine-tuning of dendritic cell biology by the TNF superfamily. Nat. Rev. Immunol. 2012;12:339–351. doi: 10.1038/nri3193. [DOI] [PubMed] [Google Scholar]
- Tabata H., Kanai T., Yoshizumi H., Nishiyama S., Fujimoto S., Matsuda I., Yasukawa M., Matsushita S., Nishimura Y. Characterization of self-glutamic acid decarboxylase 65-reactive CD4+ T-cell clones established from Japanese patients with insulin-dependent diabetes mellitus. Hum. Immunol. 1998;59:549–560. doi: 10.1016/s0198-8859(98)00050-0. [DOI] [PubMed] [Google Scholar]
- Ueda N., Zhang R., Tatsumi M., Liu T.Y., Kitayama S., Yasui Y., Sugai S., Iwama T., Senju S., Okada S. BCR-ABL-specific CD4+ T-helper cells promote the priming of antigen-specific cytotoxic T cells via dendritic cells. Cell. Mol. Immunol. 2016;15:15–26. doi: 10.1038/cmi.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcardo R., Masuda K., Yamada D., Ikawa T., Shimizu K., Fujii S., Koseki H., Kawamoto H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 2013;12:31–36. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Wakao H., Yoshikiyo K., Koshimizu U., Furukawa T., Enomoto K., Matsunaga T., Tanaka T., Yasutomi Y., Yamada T., Minakami H. Expansion of functional human mucosal-associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:546–558. doi: 10.1016/j.stem.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Wiesel M., Oxenius A. From crucial to negligible: functional CD8(+) T-cell responses and their dependence on CD4(+) T-cell help. Eur. J. Immunol. 2012;42:1080–1088. doi: 10.1002/eji.201142205. [DOI] [PubMed] [Google Scholar]
- Yamada D., Iyoda T., Vizcardo R., Shimizu K., Sato Y., Endo T.A., Kitahara G., Okoshi M., Kobayashi M., Sakurai M. Efficient regeneration of human Valpha24+ invariant natural killer T cells and their anti-tumor activity in vivo. Stem Cells. 2016;34:2852–2860. doi: 10.1002/stem.2465. [DOI] [PubMed] [Google Scholar]
- Zhang R., Liu T., Senju S., Haruta M., Hirosawa N., Suzuki M., Tatsumi M., Ueda N., Maki H., Nakatsuka R. Generation of mouse pluripotent stem cell-derived proliferating myeloid cells as an unlimited source of functional antigen-presenting cells. Cancer Immunol. Res. 2015;3:668–677. doi: 10.1158/2326-6066.CIR-14-0117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.