

Abstract
The radula is the central foraging organ and apomorphy of the Mollusca. However, in contrast to other innovations, including the mollusk shell, genetic underpinnings of radula formation remain virtually unknown. Here, we present the first radula formative tissue transcriptome using the viviparous freshwater snail Tylomelania sarasinorum and compare it to foot tissue and the shell-building mantle of the same species. We combine differential expression, functional enrichment, and phylostratigraphic analyses to identify both specific and shared genetic underpinnings of the three tissues as well as their dominant functions and evolutionary origins. Gene expression of radula formative tissue is very distinct, but nevertheless more similar to mantle than to foot. Generally, the genetic bases of both radula and shell formation were shaped by novel orchestration of preexisting genes and continuous evolution of novel genes. A significantly increased proportion of radula-specific genes originated since the origin of stem-mollusks, indicating that novel genes were especially important for radula evolution. Genes with radula-specific expression in our study are frequently also expressed during the formation of other lophotrochozoan hard structures, like chaetae (hes1, arx), spicules (gbx), and shells of mollusks (gbx, heph) and brachiopods (heph), suggesting gene co-option for hard structure formation. Finally, a Lophotrochozoa-specific chitin synthase with a myosin motor domain (CS-MD), which is expressed during mollusk and brachiopod shell formation, had radula-specific expression in our study. CS-MD potentially facilitated the construction of complex chitinous structures and points at the potential of molecular novelties to promote the evolution of different morphological innovations.
Keywords: chitin synthase, novelty, radula, RNAseq, shell, Tylomelania sarasinorum
Introduction
Evolutionary innovations can promote the origin and subsequent diversification of emerging lineages, and thus represent a central subject of evolutionary biology (Hunter 1998; Galis 2001; Shubin et al. 2009; Wagner and Lynch 2010; Erwin 2015). Understanding the genetic underpinnings of evolutionary novelties is essential for understanding the origin and evolutionary trajectories of both novelties and the respective lineages (Shubin et al. 2009; Hall 2012; Erwin 2015). The radula (rasping tongue) is the central foraging organ and innovation of the Mollusca, which make up one of the most specious animal phyla (Ponder and Lindberg 2008; Rosenberg 2014) and have successfully colonized habitats ranging from mountain peaks to deep sea vents (Brusca and Brusca 2003). Although mollusks exhibit an outstanding diversity of different body plans (Sigwart 2017), the radula has only been lost in one major lineage, the filter-feeding Bivalvia. Adaptation of the radula allowed mollusks to develop a multitude of foraging strategies including predation, herbivory, scavenging, detritivory, and filter-feeding. Especially within gastropods, the radula underwent remarkable adaptive evolution leading, for example, to the toxoglossan harpoon-like teeth of predatory cone snails (Shimek and Kohn 1981; Kantor and Puillandre 2012). Diversification of the radula was further suggested as a key character of trophic specialization in the course of adaptive radiations of the freshwater snail Tylomelania (von Rintelen et al. 2004, 2010; Glaubrecht and von Rintelen 2008). The radula has sparked the interest of not only evolutionary biologists but also of material scientists (Brooker and Shaw 2012; Ukmar-Godec et al. 2015), since the self-sharpening chiton radula teeth represent the hardest biomineralized structure known to date (Weaver et al. 2010). Nonetheless, the genetic basis of radula formation remains poorly characterized (but see Samadi and Steiner 2010; Nemoto et al. 2012).
The radula develops as a ventral outpocketing of the foregut (Page 2002; Page and Hookham 2017) and is usually made up of numerous rows of teeth, which are situated on a cuticular ribbon, the radular membrane (Runham 1963; Kerth 1973; Wiesel and Peters 1978; Mackenstedt and Märkel 1987). Since the radula is worn down anteriorly, it is continuously produced by the odontoblast cell group, which is situated at the caudal end of the radular sack (Runham 1963; Kerth 1973; Wiesel and Peters 1978; Mackenstedt and Märkel 1987). Coordinated cyclical secretion of tooth matrix by groups of odontoblasts defines size and shape of the developing teeth (Kerth 1973; Mackenstedt and Märkel 1987). The tooth matrix mainly consists of densely packed chitin fibers and so far unexplored nonchitinous macromolecules (Peters 1972; Sone et al. 2007). Mineralizing cells of the superior epithelium integrate organic and inorganic compounds into the matrix to harden and, in some cases, mineralize the teeth as they migrate toward the buccal cavity (Mackenstedt and Märkel 1987; Cruz et al. 1998; Sone et al. 2007; Weaver et al. 2010; Brooker and Shaw 2012). Thus far, very little is known about the genetic underpinnings of radula formation, but alkaline phosphatase and the ParaHox gene Gsx were reported to be expressed during radula development (Samadi and Steiner 2010; Hohagen and Jackson 2013) and Nemoto et al. (2012) identified six proteins in tooth cusps of the giant pacific chiton.
Both the radula and the shell, which represents another prominent molluscan innovation, are based on a chitinous matrix. However, in contrast to the radula, considerable effort was put into investigating the genetic basis of shell formation (Jackson et al. 2006; Bai et al. 2013; Harney et al. 2016; Vendrami et al. 2016). The mollusk shell is continuously produced by the mantle and composed of the periostracum and two to five calcified layers (Marin et al. 2012). Although the organic matrix comprises only 0.1–4% of the total shell weight, it is essential for mechanical properties and controlled mineralization (Lowenstam and Weiner 1989; Marin et al. 2008, 2012, 2013). It is mainly composed of chitin and a large variety of proteins and proteoglycans (Peters 1972; Marin et al. 2008, 2013; Fernández et al. 2016) that are rich in repetitive low complexity domains (Kocot et al. 2016; Aguilera et al. 2017). Mantle secretomes evolve rapidly, and can differ tremendously, even between closely related species (Jackson et al. 2006, 2010; Aguilera et al. 2014, 2017; Kocot et al. 2016). Genes involved in shell formation include both deeply conserved components and a large number of lineage-specific genes (Jackson et al. 2006; Jackson and Degnan 2016; Aguilera et al. 2017; Feng et al. 2017). The peripheral position of secreted proteins in gene regulatory networks (GRN), which allowed co-option and loss of genes, as well as gain, loss and shuffling of domains in shell proteins were suggested to promote the rapid evolution of mantle secretomes (Jackson and Degnan 2016; Kocot et al. 2016; Aguilera et al. 2017). Phylostratigraphic analyses further indicate that both novel orchestration of ancient genes, that is, changes in expression level or composition of pre-existing genes within a GRN, and ongoing evolution of novel genes contributed to the genetic basis of shell formation (Aguilera et al. 2017).
In summary, although very different in detail, shell and radula are both molluscan innovations based on partly chitinous matrices that can be mineralized. Both first evolved in early mollusks and subsequently underwent remarkable diversification. Here, we present the first radula building tissue transcriptome and compare it to transcriptomes of mantle edge and foot muscle of the same species to investigate the genetic basis of radula formation. Foot muscle was chosen as reference tissue, because it enables comparisons between mollusk innovations and an older, not mollusk-specific tissue. Additionally, it allows exclusion of genes, which are not related to shell formation but expressed by muscle cells in the mantle. The foot further contains a collagenous extracellular matrix, which allows excluding genes that are not specifically employed for radula and shell formation, but generally needed to produce any extracellular matrix. We identify specific and potentially shared genetic underpinnings of shell and radula formation and compare patterns of their evolutionary origin using the viviparous freshwater snail Tylomelania sarasinorum (Kruimel 1913). Tylomelania sarasinorum was chosen because it is a representative member of the adaptive radiations in the ancient lakes of Sulawesi, Indonesia (von Rintelen et al. 2012), which gave rise to impressive radula and shell diversities (von Rintelen et al. 2004, 2010). Our analyses indicate that the genetic underpinnings of both innovations were based on similar patterns of ongoing evolution of novel genes and novel orchestration of ancient gene sets. However, evolution of novel genes since the last common ancestor of mollusks likely played an especially important role for the evolution of the radula. Despite very distinct overall gene expression, some central transcription factors were employed for both radula and shell formation. Additionally, some genes with radula-specific expression are known to be involved in the formation of other lophotrochozoan hard structures, suggesting gene co-option for tissue patterning and hard structure formation. Thus, genes underlying radula formation in mollusks may have played a significant role for the evolution of other lophotrochozoan innovations as well.
Results
Sequencing and Transcriptome Assembly
Tissue samples from mantle, radula, and foot of 19 adult T. sarasinorum were pooled into four biological replicates (Pool1-4) for each tissue, and RNAseq was conducted to gain insight into gene sets expressed in these tissues. Sequencing generated 561 million 150-bp paired-end reads, and the initial assembly with Trinity (Grabherr et al. 2011; Haas et al. 2013) consisted of 315,700 sequences classified as genes and 93,017 isoforms (table 1), which is in line with previously published deep-sequenced mollusk transcriptomes (De Oliveira et al. 2016; Harney et al. 2016). The transcriptome N50 was 729 bp regarding only the longest isoform per gene and 1,300 bp when considering all contigs. BUSCO v1.1b1 (Simão et al. 2015), a program that searches for single-copy orthologs that are conserved among metazoans, indicated high completeness of the assembly. In a data set with only the longest isoform per gene, 90% of the conserved single-copy orthologs were present in full length. Despite the high number of transcripts, BUSCO suggested low redundancy of the assembly (table 1). Pool1 was excluded from further analyses, because hierarchical clustering and PCA identified mantle and radula of pool1 as outliers. For the three remaining pools, transcripts were filtered by expression level, using a threshold of FPKM ≥ 1 (one mapped fragment per kilobase of transcript per million mapped reads). Remaining transcripts were clustered based on sequence similarity, reads remapped, and transcripts again filtered by expression (FPKM ≥ 1). The final assembly consisted of 105,718 genes with an N50 of 1,740, and BUSCO (Simão et al. 2015) indicated both high completeness (89%) and low redundancy (8.4%) (table 1).
Table 1.
Assembly Statistics of the Raw and Expression Filtered Assembly.
| Trinity Genes | Trinity Transcripts | GC (%) | “Gene” N50 | Completea(%) | Duplicateda(%) | |
|---|---|---|---|---|---|---|
| Raw assembly | 315,700 | 408,717 | 45.4 | 729 | 90 | 9.8 |
| Expression filteredb | 105,718 | NA | 45 | 1,740 | 89 | 8.4 |
According to BUSCO.
FPKM ≥ 1.
Expression Analyses
All biological replicates clustered tightly together in both PCA and hierarchical clustering performed on expression data of all genes in the final assembly, that is, without any a priori assumptions concerning group affiliation (supplementary figs. 2 and 3, Supplementary Material online). While 32% (n = 33,928) of all genes in the assembly were expressed (FPKM ≥ 1) in all tissues, 44% (n = 46,568) were expressed only in a single tissue. Housekeeping genes, which are expressed across all tissues, can contribute differentially to transcriptomes of different tissues, because they make up larger proportions of transcriptomes with fewer expressed genes. To avoid confusion of transcriptome size (number of expressed genes) with transcriptome specificity when comparing transcriptomes, we focus on genes that are not universally expressed (NUE), that is, not expressed in all tissues investigated. Figure 1 illustrates the expression of NUE genes across the three tissues. In contrast to the foot muscle, which shared a considerable proportion of its NUE genes with the mantle (59%), radula forming tissue had a very distinct expression pattern (fig. 1). Although the radula sac expressed a lower total number of genes than the other tissues (n = 49,114), it had high specificity, with 57% (n = 8,628) of its NUE genes expressed in no other tissue. Radula shared more than twice as many NUE genes with mantle (n = 4,769) than with foot (n = 1,789). The mantle transcriptome had the largest total number of transcribed genes (n = 84,319), which included both a large number of genes that were not expressed in other tissues (n = 26,958), as well as a large overlap of NUE genes with foot tissue (n = 18,664). Foot had fewer NUE genes (n = 31,435) than mantle (n = 50,418), but expressed more genes (n = 65,363) than radula tissue. Differential expression analyses revealed differentially expressed (P ≤ 10−5; fold change ≥ 4) gene sets between tissues, and cutting the hierarchical clustering tree at 50% of its height resulted in 5 clusters named I–V, based on overall expression patterns (fig. 2). Figure 2 depicts differentially expressed genes that were clustered based on their expression across the three tissues, while figure 3 shows cluster-wise gene expression across all samples. One cluster (I) was made up of genes that were primarily overexpressed in foot tissue, while genes overexpressed in radula (IV, V) and mantle (II, III) were split into one moderately and one extremely differentially expressed cluster each (figs. 2 and 3).
Fig. 1.
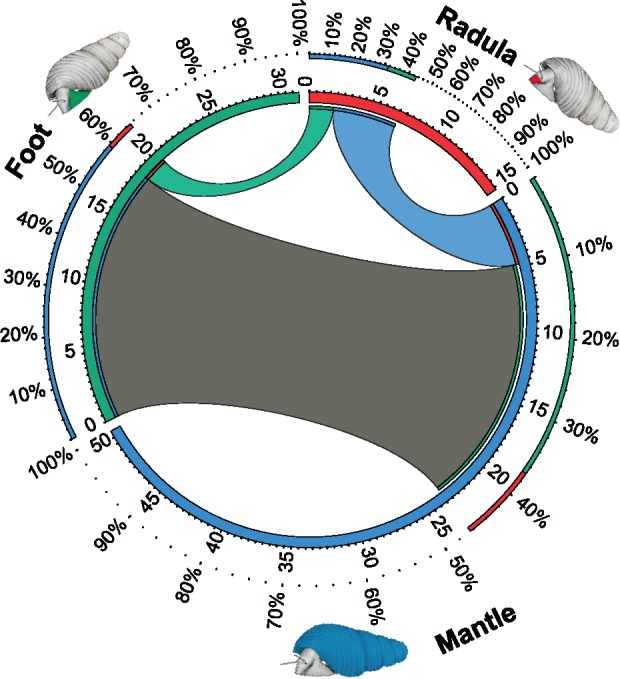
Shared and uniquely expressed transcripts across the three tissues. For each tissue, that is, radula (red) mantle (blue) and foot (green), the number of transcripts (in thousand) expressed at FPKM ≥ 1 is shown as a partition of the inner circle. Only transcripts that are not expressed at FPKM ≥ 1 in all tissues are displayed. Transcripts present in two tissues at FPKM ≥ 1 are connected with bands between tissues. Stretches of the circle unconnected to any of the other tissues represent transcripts uniquely expressed in this tissue at FPKM ≥ 1. This figure was generated with CIRCOS (Krzywinski et al. 2009).
Fig. 2.

Heatmap of hierarchically clustered differentially expressed (P ≤ 10−5) genes. Genes are displayed as horizontal lines across samples (columns). Genes with more similar expression cluster together. Overexpressed genes in a sample are colored yellow, while underexpressed genes are displayed in purple. Clusters generated by cutting the hierarchical clustering tree (left) at 50% of its height are indicated by roman numerals (I–V) on tree branches and black and white boxes (right).
Fig. 3.

Cluster-wise (I–V) expression of differentially expressed (P ≤ 10−5) genes across samples. For each cluster gene expression patterns across all samples are shown. Gray lines depict expression levels of all genes, while the blue line indicates average gene expression of all genes in each cluster. Genes overexpressed in the foot were primarily grouped in cluster I. Cluster II and III are moderately and extremely differentially expressed mantle clusters, respectively. Similarly, genes overexpressed in the radula were split into one moderately overexpressed cluster IV and one extremely overexpressed cluster V.
Annotation
Transcripts were functionally annotated with the Trinotate annotation pipeline (https://trinotate.github.io/). Most sequences remained without functional annotations, which has also been reported in previous mollusk studies (Harney et al. 2016). In our case, 29% (n = 31,128) of all genes could be annotated, while gene ontologies could be assigned to 15% (n = 16,248) of all genes based on Pfam domains and BLAST hits.
Gene Ontology Enrichment
To assess overrepresented molecular functions (MF), biological processes (BP), and cellular components (CC) of gene products in each differentially expressed gene cluster (I–V), gene ontology enrichment analyses were carried out with GOseq (Young et al. 2010), and similar terms were collapsed with Revigo (Supek et al. 2011). Numbers and functions, as well as P values of enriched GO-terms differed substantially between clusters (supplementary table 2 and fig. 4, Supplementary Material online). Generally, many more functions and processes were significantly enriched in clusters of radula and mantle than of foot tissue.
Foot Transcriptome Is Dominated by Muscle Functions
Cluster I, which primarily consisted of transcripts overexpressed in foot tissue, was enriched in comparatively few BP (n = 7), which were frequently related to muscle function like “sarcomere organization” and “cholinergic synaptic transmission” (supplementary table 2 and fig. 4, Supplementary Material online). Cellular components enriched in transcripts expressed in cluster I could mostly be attributed to the collagen matrix (“extracellular region,” “collagen trimer”) and other fundamental muscle cell components (“ion channel complex,” “postsynaptic membrane”). Enriched MF matched these observations (supplementary table 2, Supplementary Material online).
Mantle Transcriptome Is Dominated by Shell-Related Binding, Transport, and Regulation
The moderately differentially expressed mantle cluster II was enriched in diverse terms (n = 60) including “ion transport” and other likely shell formation related processes like “extracellular matrix organization” (supplementary table 2 and fig. 4, Supplementary Material online). Cellular components assigned to cluster II that were significantly enriched included “extracellular region” and “extracellular space,” while enriched MF included transporter related functions and metal-, carbohydrate-, and chitin binding. The extremely differentially expressed mantle cluster III was not significantly enriched in any GO-term.
Radula Transcriptome Is Dominated by Vesicular Secretion, Chitin, and Aminoglycan Metabolism
The moderately differentially expressed radula cluster IV had many and diverse enriched BP (n = 88) including “vesicle-mediated transport,” “chitin metabolic process,” and “growth” (supplementary table 2 and fig. 4, Supplementary Material online). While enriched CC were frequently linked to vesicular secretion (“membrane-enclosed lumen,” “brush border”), enriched MF included chitin-, protein-, and ion binding. Genes of the extremely overexpressed radula cluster V had few (n = 6), but at the same time the most significantly enriched BP. Enriched BP and MF were related to amino-sugar metabolism like “carbohydrate derivative metabolic process” and “aminoglycan metabolic process.” The only enriched CC was “extracellular region.”
Tissue-Specific Genes
Tissue-specific genes were determined, based on their expression across the three tissues in this study. The number of genes that met our criteria for “tissue-specific” (FPKM ≥ 1 in all replicates, fold change ≥ 32, and P ≤ 10−10 against both other tissues) and “strictly tissue-specific” (FPKM ≥ 2 in all replicates, fold change ≥ 32, and P ≤ 10−100 against both other tissues) differed widely between tissues. Radula had 606 and 99, Mantle 576 and 22, and foot tissue 169 and 0 tissue-specific and strictly tissue-specific genes, respectively. The shared genetic basis of radula and shell formation consisted of 66 genes that were overexpressed in both radula and mantle tissue (fold change ≥ 4 and P ≤ 10−5) in comparison to foot muscle. As was expected, well-known muscle genes had foot-specific expression. Foot-specific genes encoded, for example, myosin heavy chain, myosin essential light chain, paramyosin, and troponin T. Genes found in the remaining gene sets are presented in more detail in the following sections.
Mantle-Specific Genes Include Conserved Genes Involved in Biomineralization
Mantle-specific genes included genes that were already known to be essential for mollusk shell formation, which supports the validity of our approach. The four strictly mantle-specific genes that could be annotated encoded putative ferric-chelate reductase 1, a peroxidase, and a secreted tyrosinase-like protein. Another tissue-specific mantle transcript encoded carbonic anhydrase, and two were annotated as alkaline phosphatases. Additionally, two mantle-specific transcripts produced significant BLAST hits against the nacre protein perlucin, which nucleates growth of calcium carbonate crystals (Blank et al. 2003). Mantle-specific transcription factors included the homeobox protein goosecoid, which is also involved in larval shell field patterning (Lartillot, Le Gouar, et al. 2002), and pax6, which has conserved expression in the developing and adult nervous system and eyes of mollusks (Scherholz et al. 2017).
Many Radula-Specific Genes Are Known from Foregut Development or Hard Structure Formation
Radula-specific genes primarily comprised genes that are also known to be involved in foregut development or hard structure formation. Tooth matrix forming genes included a Lophotrochozoa-specific chitin synthase (PF03142.12) with a myosin head motor domain (PF00063.18) that is also involved in mollusk and brachiopod shell formation (Weiss et al. 2006; Marin et al. 2008; Luo et al. 2015). Hephaestin (heph), a ferroxidase needed for efficient iron transport in vertebrates (Petrak and Vyoral 2005), which is also employed for skeletogenesis in corals (Ramos-Silva et al. 2014), and mollusk as well as brachiopod shell formation (Liao et al. 2015; Luo et al. 2015) also had radula-specific expression. Radula-specific transcripts further encoded the classic notch responsive gene hes1 and aristaless-related homeobox protein arx, which are both involved in morphogenesis of chitinous bristles, referred to as chaetae, in brachiopods and annelids (Schiemann et al. 2017). Furthermore, two fork head box transcription factors that were most similar to foxA and foxL1, which are involved in lophotrochozoan foregut development (Boyle and Seaver 2008; Shimeld et al. 2010; Perry et al. 2015), as well as gbx, which is involved in brain regionalization and shell field patterning in mollusks (Wollesen et al. 2017), had radula-specific expression. Finally, otx, which is known to be involved in eye maturation and brain development in mollusks and annelids (Steinmetz et al. 2011; Buresi et al. 2012) and has conserved expression in larval mouth regions across bilaterians (Arendt et al. 2001), had radula-specific expression in our study.
Shared Expression of Genes during Radula and Shell Formation
The T-box gene brachyury (bra) is expressed during larval foregut development (Arendt et al. 2001; Perry et al. 2015) and shell field patterning in gastropods (Lartillot, Lespinet, et al. 2002; Jackson and Degnan 2016). Although bra was expressed during both radula and shell formation, it was highly significantly overexpressed in the radula. Commonly expressed genes during radula and shell formation further included the lophotrochozoan paired box gene paxβ, which has only recently been discovered and has primarily been associated with CNS development (Schmerer et al. 2009; Scherholz et al. 2017). Additionally, ets1, a conserved transcription factor with a variety of functions, including a role in gene regulatory networks upstream of skeletogenesis in echinoderms and vertebrates (Raouf and Seth 2000; Sharma and Ettensohn 2010; Rafiq et al. 2014), was expressed during radula and shell formation. Both radula and shell further shared expression of a member of the solute carrier family 22, the actin binding and brush border cytoskeleton organizing villin, and a chitinase. Finally, some genes involved in cell–cell recognition, adhesion, and signaling were shared between both tissues, including two protocadherins, an innexin, a molluscan insulin related peptide, and a Nemo-like kinase.
Origin of Tissue-Specific Genes
To investigate and compare the evolutionary origin of important genes in each tissue, genes that were identified as tissue-specific were searched in a database of predicted proteins of 21 genomes across the tree of life. We focused our analyses on genes for which at least one significant BLAST hit (E-value ≤ 10−10) was returned. The percentage of tissue-specific genes for which putative homologs were found in other species differed between tissues and ranged from 29% (radula) to 35% (foot). In contrast, significant BLAST hits were found for only 18% of all expressed genes. Figure 4 shows gene origin (in %) of tissue-specific gene sets across nine phylostrata in comparison to the background gene origin. The origin of most tissue-specific genes that were found in at least one other genome predates the evolution of the Mollusca (fig. 4). Nonetheless, irrespective of the method applied to correct for multiple testing, a two-tailed hypergeometric test indicated that a significantly higher than average proportion of radula-specific genes originated in all phylostrata since the emergence of stem-mollusks. Mantle-specific genes showed a trend of increased gene origin in multiple phylostrata, which was, however, only significant in stem-gastropods. Additionally, a significantly lower than average proportion of radula-specific and mantle-specific genes originated between the emergence of cellular life and the origin of the eukaryotes. Foot-specific genes showed a trend of higher than average gene origin in stem-Metazoa, and less pronounced following the origin of stem-Protostomia, but never deviated significantly from background gene evolution.
Fig. 4.

Evolutionary origin of tissue-specific genes. Phylostratigraphic analyses reveal evolutionary origin (in percent) of radula-specific (red), mantle-specific (blue), and foot-specific (green) genes in comparison to the origin of all genes included in the final assembly (black) across nine phylostrata. Only genes for which at least one significant BLAST hit was returned were included in this analysis. Asterisks indicate in which phylostrata gene origin of tissue-specific gene sets differed significantly (* ≤ 0.05; ** ≤ 0.01; *** ≤ 0.001) from background gene origin according to a two-tailed hypergeometric test with correction for multiple testing using either Bonferroni or the false discovery rate (FDR). Asterisks have similar coloration to the graph of the gene sets they refer to (radula-specific, red; mantle-specific, blue; foot-specific, green).
Genes with radula-specific expression that were predicted to have originated in stem-Mollusca and stem-Gastropoda predominantly encoded chitin binding proteins, which occurred less frequently in other phylostrata and tissues (fig. 5 and supplementary fig. 5, Supplementary Material online). Additionally, two radula-specific genes that originated in stem-mollusks had EGF-like and delta serrate ligand domains (supplementary table 3, Supplementary Material online), suggesting that they likely encode notch ligands.
Fig. 5.

Annotation of genes predicted to have originated before and after the origin of stem-Mollusca. Proportion of genes with the functional annotations chitin binding, other gene ontology, and no predicted gene ontology are displayed for genes predicted to have originated before (inner circle) and after (outer circle) the origin of stem-Mollusca, based on phylostratigraphic analyses. Functional annotations of radula-specific (right) and all other genes included in the phylostratigraphic analyses (left) are based on Pfam annotations.
Discussion
Understanding the genetic underpinnings of evolutionary novelties is a central, yet difficult to achieve, goal in evolutionary biology. However, in recent years, it has been aided tremendously by massively parallelized sequencing, and RNAseq has successfully been employed to investigate gene expression underlying various complex innovations (Manousaki et al. 2013; Whittington et al. 2015; Babonis et al. 2016; Santos et al. 2016; Aguilera et al. 2017). Here, we used RNAseq to investigate the genetic basis of radula formation, a central molluscan innovation, and compare it to the genetic basis of a second prominent molluscan innovation, the shell.
Expressed Genes and Enriched Functions Match Previously Known Tissue Functions
We first investigated dominant functions in clusters of genes that were differentially expressed between tissues. In general, enriched biological processes and molecular functions corresponded to known tissue functions. Muscle genes dominated in the foot cluster, ion transport, and extracellular structure development were enriched in the shell building mantle, and radula clusters were dominated by genes associated with vesicle mediated secretion, chitin, carbohydrate, and aminoglycan processing. Enrichment of GO-terms related to developmental processes like “developmental process” in the mantle and “growth” in the radula could be explained by the presence of more proliferative tissue in radula and shell building tissues (Mackenstedt and Märkel 1987; Fang et al. 2008). In contrast to the radula, the mantle fulfills diverse functions, which is also apparent on the gene expression level, as it includes the largest number of expressed genes and a variety of enriched functions. The larger number of commonly expressed genes expressed in both mantle and foot in comparison to the transcribed gene sets that either tissue shares with the radula is likely due to nervous tissue and muscle cells in mantle and foot, both of which are absent from radula forming tissue. Specialization on functions not present in multiple tissues of our data set (muscle dominates foot, but is also present in mantle) is reflected by more enriched functions and a higher percentage of tissue-specific genes among NUE genes in mantle and radula than in the foot muscle transcriptome. Mantle-specific genes encode proteins involved in biomineralization like tyrosinases, alkaline phosphatases, carbonic anhydrases, and peroxidases, which supports previous studies that identified these genes as essential for shell formation (Aguilera et al. 2014; Feng et al. 2017) and discussed them in the context of an ancestral biomineralization toolkit (Jackson and Degnan 2016; Karakostis et al. 2016; Marin et al. 2016).
Signs of Gene Co-Option for Chitinous Hard Structure Formation in Lophotrochozoa
Although gene expression of radula formative tissue is very distinct, radula expression patterns are much more similar to mantle than to foot expression (fig. 1). Genes uniquely shared between mantle and radula are involved in central processes of shell and radula formation like chitin processing, cell–cell recognition, regulation of gene expression and organization of cell protrusions of the secretory epithelium. Generally, similarities in gene expression between tissues can arise from evolutionarily related cell lineages in both organs (Arendt et al. 2016) or independent co-option of genes into gene regulatory networks. Although the former cannot be entirely excluded, gene co-option appears much more likely in our case given the different developmental origins of mantle and radula, very distinct overall gene expression of the two tissues, and reportedly extensive gene co-option during shell evolution (Aguilera et al. 2017). Alternatively, it could be argued that some similarity in gene expression is caused by more cell proliferation or cell migration in radula and mantle in comparison to foot muscle. However, these processes alone are unlikely to account for the observed similarities between mantle and radula. Instead, genes overexpressed in both mantle and radula in comparison to foot muscle included central transcription factors such as brachyury (bra) and ets1, which are known for their important roles in cell differentiation (Raouf and Seth 2000; Livingston et al. 2006; Sharma and Ettensohn 2010; Rafiq et al. 2014; Zhu et al. 2016). Although bra is reportedly involved in larval shell field patterning (Lartillot, Lespinet, et al. 2002; Jackson and Degnan 2016), it is also expressed in multiple other tissues during mollusk development (Perry et al. 2015) and is thought to play a role in major developmental processes like anterior–posterior axis development (Arendt et al. 2001; Lartillot, Lespinet, et al. 2002). Little is known about the role of ets1 in mollusks, but in other organisms it is primarily known for its role in cell differentiation including that of cells involved in skeletogenesis in vertebrates and echinoderms (Raouf and Seth 2000; Livingston et al. 2006; Sharma and Ettensohn 2010; Rafiq et al. 2014). Additionally, paxβ, which is a lophotrochozoan paired box gene of the recently discovered bilaterian paxα/β subfamily (Schmerer et al. 2009; Franke et al. 2015), was expressed during both radula and shell formation. Although paxβ has only been studied in a few organisms, where it is primarily associated with nervous system development (Schmerer et al. 2009; Franke et al. 2015; Scherholz et al. 2017), it was apparently also employed for the formation of different molluscan innovations and might thus have played a significant role in mollusk evolution. Additional data concerning the role of these genes in mollusks is needed and will help to disentangle the role these genes play in radula and shell formation.
Radula-specific genes include transcription factors that most likely reflect the radula’s developmental origin from the ventral foregut (Crofts 1937; Ghose 1962). These transcription factors include foxA, foxL1, and otx, which are involved in lophotrochozoan foregut development (Arendt et al. 2001; Boyle and Seaver 2008; Shimeld et al. 2010; Perry et al. 2015). In contrast, radula-specific genes that were potentially co-opted for hard structure formation include regulatory genes such as aristaless related protein (arx), the notch responsive gene hes1, and structural genes like chitinases, chitin synthases, and hephaestin. All of these are reportedly involved in lophotrochozoan hard structure formation and were employed in brachiopod shell formation, mollusk shell formation and/or annelid chaetae formation (table 2) (Liao et al. 2015; Luo et al. 2015; Schiemann et al. 2017). Further, gbx, which is involved in feather bud formation in birds (Obinata and Akimoto 2012), was recently found to be co-opted from brain regionalization into shell field patterning in mollusks (Wollesen et al. 2017). Based on our expression analyses, gbx was also co-opted into GRNs for radula formation. Additionally, a Lopohotrochozoa-specific chitin synthase with a myosin motor domain (CS-MD) (Zakrzewski et al. 2014), which is expressed in brachiopod lophophores (Luo et al. 2015) and employed for both brachiopod (Luo et al. 2015) and mollusk shell building (Weiss et al. 2006; Schönitzer and Weiss 2007) is also involved in radula formation. CS-MD evolved independently in fungi and Lophotrochozoa (Zakrzewski et al. 2014), two lineages for which chitinous structures play a pivotal role. Especially within Lophotrochozoa, chitin is employed in a variety of complex structures that include chaetae and spicules, lophophores, jaws of rotifers and annelids, mollusk radulae, tubes of horseshoe worms, and mollusk and brachiopod shells (Peters 1972; Klusemann et al. 1990; Hausen 2005; Weiss et al. 2006; Luo et al. 2015). It seems compelling that evolution of CS-MD facilitated precise spatial control of chitin synthesis via actin filaments. This molecular innovation may have promoted the evolution of more sophisticated chitin matrices and thus contributed to the evolution of multiple innovations based on such matrices in Lophotrochozoa. Additional studies are needed to further investigate this possibility, but rapidly declining sequencing costs and recent advances in nonmodel species gene editing using CRISPR/cas-9 (Perry and Henry 2015) are making this a testable hypothesis.
Table 2.
Expressed Genes in the Radula and Other Hard Structures As Known from Literature (blue) or As Indicated by This Study (green).
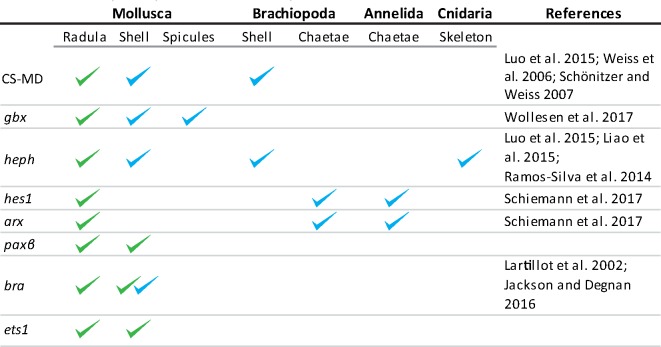 |
Note.—Clades, tissues, and references included in this table are nonexhaustive.
Novel Orchestration of Preexisting Genes and Evolution of Novel Genes Gave Rise to the Genetic Basis of Radula Formation
The role of novel genes in generating evolutionary innovation has been the subject of recent discussion, and the relative contribution of the former to the latter remains to be investigated (McLysaght and Guerzoni 2015; Babonis et al. 2016; McLysaght and Hurst 2016). We used phylostratigraphic analyses to investigate when tissue-specific genes first evolved (Domazet-Lošo et al. 2007). It was previously argued that phylostratigraphic inferences were prone to various biases (McLysaght and Hurst 2016; Moyers and Zhang 2016), especially underestimating ages of small and fast evolving genes. However, general patterns were recently suggested to be consistent despite the bias introduced by error-prone genes (Domazet-Lošo et al. 2017). It should additionally be pointed out that none of the genes in this analysis are young genes, because lineage specific genes were excluded and the last common ancestor of Tylomelania and Biomphalaria/Aplysia diverged between 310 and 496 Ma (Kumar et al. 2017).
Our results indicate that many tissue-specific genes originated long before the evolution of the Mollusca and thus, the radula. As seen in figure 4, a significantly higher than average proportion of radula-specific genes originated during and after the evolution of stem-mollusks. Additionally, mantle-specific and radula-specific genes originated at a significantly lower than average rate during early organismal evolution until the origin of the Eukaryota. Mantle-specific gene origin was only significantly above average in stem-gastropods. In contrast, foot-specific genes never deviated significantly from average gene origin. It should be taken into consideration that foot had fewer specific genes, which decreases the statistical power to detect significant deviation from average gene origin. Nonetheless, origin of foot-specific genes was always closer to background gene origin than at least one of the two other gene sets in all phylostrata except stem-Metazoa. Foot-specific genes were closest to the average whenever both radula-specific and mantle-specific gene origin deviated significantly from the average. Taken together, this indicates that novel orchestration of already existing gene sets in combination with the ongoing evolution of novel genes contributed to the evolution of both the radula and the shell. Here, novel orchestration refers to evolution leading to changes in expression level or combination of pre-existing genes within a GRN, independent of the underlying molecular mechanism. Our results further suggest that novel gene evolution since the last common ancestor of mollusks has played an especially important role for the evolution of the radula.
Proteins encoded by genes with radula-specific expression that were predicted to have originated in stem-Mollusca and stem-Gastropoda are dominated by chitin binding proteins, which occur much less frequently in other gene sets and phylostrata (fig. 5 and supplementary table 3, Supplementary Material online). Additionally, EGF-like and delta serrate ligand domains of two radula-specific genes that originated in stem-mollusks indicate that their products bind to notch receptors (supplementary table 3, Supplementary Material online). An important role of the notch signaling pathway in the radula is further supported by the radula-specific expression of the classic notch-responsive gene hes1. Notch signaling regulates cell-fate decisions and tissue patterning in various morphogenic processes including dental development and dental regeneration in vertebrates (Mumm and Kopan 2000; Lai 2004; Gazave et al. 2009; Fraser et al. 2013; Corson et al. 2017; Thiery et al. 2017). In contrast to comparing overall patterns of gene origin, conclusions based on the origin of single genes should be treated with caution, because predicted gene origin of single sequences may depend on the cutoff E-value or the genomes included in our database. Nonetheless, potentially novel ligands of a likely important morphogenic signaling pathway in the radula represent promising candidates for future in-depth analyses.
Taken together, these patterns underline the contribution of novel genes to evolutionary innovations and are in general concordance with results reported for the shell-building mantle secretome (Jackson et al. 2006; Aguilera et al. 2017). A combination of novel genes and novel orchestration of ancestral gene sets was previously found in other major innovations, including the evolution of muscles, where a core set of muscle proteins already occurred in unicellular organisms (Steinmetz et al. 2012).
Conclusions
This study provides a first insight into the genetic basis of radula formation and enlarges the available resources to target molluscan shell formation. Our results support previous findings that novel orchestration of ancient gene sets and evolution of novel genes gave rise to the genetic basis of the shell (Aguilera et al. 2017) and suggest similar patterns for the evolution of the molecular underpinnings of the radula. Novel genes, which evolved in and after the last common ancestor of the Mollusca likely played an especially important role for radula evolution. Finally, some genes, like the Lophotrochozoa-specific chitin synthase with a myosin motor domain, were likely co-opted into different GRN for lophotrochozoan hard structure formation. Gene co-option of this molecular innovation potentially contributed to the evolution of multiple morphological innovations across the Lophotrochozoa, including the molluscan radula and shell. In the future, in situ hybridization or similar approaches could be used to localize candidate gene expression in Mollusca and other Lophotrochozoa, and gene editing or knockdown could be employed to examine phenotypic consequences of distorted expression of candidate genes. Additional radula transcriptomes would allow interspecific comparisons, while radula transcriptomes from limpets or polyplacophorans would further allow investigating whether genes involved in shell biomineralization are also employed for radula biomineralization in these clades. Last but not least, our results can be used as a valuable resource to target the genetic changes that underlie rapid radula and shell diversification during the adaptive radiation of Tylomelania. Comparative transcriptomic analyses between the radula-morphs of Tylomelania sarasinorum appear especially promising for gaining insight into radula shape diversification.
Materials and Methods
Animal and Tissue Collection
Adult specimens of Tylomelania sarasinorum were collected from submerged wood by a snorkeler in March 2015 at the northern shore of Loeha Island (Lake Towuti, South Sulawesi, Indonesia; 2.76075 S 121.5586 E). Animals were dissected in the field, and tissue samples of distal radular sack (supplementary fig. 1, Supplementary Material online), mantle edge, and foot muscle were directly stored in RNAlater to ensure efficient RNA preservation. T. sarasinorum occurs on wood and rock surfaces, and exhibits a pronounced substrate correlated radula polymorphism (von Rintelen et al. 2007; Glaubrecht and von Rintelen 2008). To avoid confounding factors caused by this radula polymorphism, radula morphs of all individuals were inspected, and only wood morph individuals were chosen for further experiments.
Sample Preparation and Sequencing
To increase the amount of tissue for RNA extraction and to reduce noise in expression analysis due to anatomically heterogeneous sampling and/or different state of individual tooth production, 19 individuals of T. sarasinorum were grouped into three pools of five and one pool of four individuals. Tissue samples of individuals in each pool were weighed (Mettler AT 261), and equal amounts of tissue for each individual were pooled, resulting in four biological replicates of each tissue (Tissue [Average tissue weight; Average SD in each pool], radula [0.46 mg; 0.11 mg] mantle [0.63 mg; 0.08 mg] foot [5.31 mg; 0.41 mg]). In other words, each of the four pools was made up of similar amounts of tissue from a defined group of specimens, irrespective of which tissue was sampled. Samples were homogenized with a Precellys Minilys, and total RNA was extracted from mantle edge and radula formative tissue using a customized protocol of the RNeasy Plus Micro Kit (Qiagen). Specifically, to increase total RNA yield from minute amounts of tissue, homogenization was conducted in 150 µl lysis buffer, which was subsequently diluted to enable further digestion with proteinase K. After proteinase digestion, 450 µl lysis buffer was added to allow efficient DNA removal with gDNA spin columns. Since larger amounts of foot tissue were available, RNA was extracted from foot muscle with a TRIzol extraction according to the manufacturer’s protocol. Amount and quality of extracted total RNA was inspected using Agilent’s 2100 Bioanalyzer. Samples showed no signs of degradation or DNA contamination. RNA integrity (RIN) estimates were not applicable due to the presence of a so called “hidden break,” which led to a sharp 18S band, but a much reduced or lacking 28S rRNA peak in our samples. This effect is known from many protostomes (Gayral et al. 2011) and was recently also observed in the cone snail Conus episcopatus (Lavergne et al. 2015). Messenger RNA was enriched with poly (A) capture using NEXTflex Poly (A) Beads, and strand-specific libraries were built using NEXTflex Rapid Illumina Directional RNA-Seq Library Prep Kit (Bioo Scientific) with modifications suggested in Sultan et al. (2012). Quality and concentrations of libraries were evaluated using Agilent’s 2100 Bioanalyzer and qPCR (Kapa qPCR High Sensitivity Kit). Libraries had average fragment sizes between 450 and 500 bp and were sequenced (150 bp, paired end) on an illumina NextSeq sequencing platform at the Berlin Center for Genomics in Biodiversity research (BeGenDiv).
Sequence Preprocessing, Assembly, and Quality Assessment
Raw sequences were quality trimmed with a quality threshold of 30, minimum read length of 25 bp, and removing all N using sickle (Joshi and Fass 2011). Subsequent removal of adapter sequences with cutadapt (Martin 2011) generated 493 million paired end reads (supplementary table 1, Supplementary Material online). Trinity v2.1.1 (Grabherr et al. 2011; Haas et al. 2013) was run in strand-specific mode with a minimal transcript length of 250 bp, in silico read normalization (max. read coverage = 50), and 2-fold minimal kmer coverage to generate a single assembly of all tissues. General assembly statistics, including number and length distribution of contigs, were assessed with the script included in the trinity pipeline. BUSCO v1.1b1 (Simão et al. 2015) was employed to search for a set of single copy orthologs that are conserved among metazoans to get an estimate of transcriptome completeness and redundancy.
Expression Analysis and Assembly Filtering
Gene expression analysis was performed using the pipeline included in Trinity v2.1.1 (Grabherr et al. 2011; Haas et al. 2013). Briefly, quality-filtered adapter trimmed reads of each sample were mapped to the transcriptome using bowtie2 (Langmead et al. 2009), followed by abundance estimation with RSEM (Li and Dewey 2011). Ribosomal RNA (rRNA) was identified using a BLAST search of 28S rRNA (Brotia pagodula; HM229688.1) and 18S rRNA (Stenomelania crenulata; AB920318.1), and the best hits with the highest number of mapped reads were removed from the read count matrices. Abundance of rRNA mostly reflected polyA capture success, and sample correlation between biological replicates increased when rRNA was removed from the expression data set. A principal component analysis (PCA) and hierarchical clustering was performed with the log2 transformed counts per million mapped reads (cpm) for all samples to inspect data integrity. Pool1 radula and pool1 mantle were identified as outliers using the above-mentioned methods (supplementary fig. 3, Supplementary Material online). This was most likely due to a combination of deeper sequencing of pool1 (supplementary table 1, Supplementary Material online) and lower yield of total RNA, which led to a decrease in library complexity. Since pool1 was further sequenced separately, a batch effect could also not be excluded. Thus, all tissue samples of pool1 were removed from expression analyses. Transcripts were subsequently filtered by expression (FPKM ≥ 1, i.e., one mapped fragment per kilobase of transcript per million mapped reads) based on the nine remaining samples using the script provided in the Trinity pipeline. To reduce redundancy within our data set, the longest isoforms of all “trinity genes” (sequences with gene level assigned by Trinity) were clustered based on sequence similarity (97% sequence identity threshold; 90% minimum alignment coverage of the shorter sequence) with CD-HIT version 4.6 (Li and Godzik 2006), and the longest transcript of each cluster was retained. Quality filtered, adapter trimmed reads were remapped to the remaining transcripts. Following this reduction of redundancy, transcripts with very low expression (FPKM ≥ 1) were again removed to create a new final assembly. Differentially expressed genes (P ≤ 10−5; FC ≥ 4) were determined using edgeR (Robinson et al. 2010). Hierarchical clustering was performed to group differentially expressed genes based on their expression pattern. The hierarchical clustering tree was cut at 50% of its height to group differentially expressed genes into clusters based on expression patterns across all tissues.
Annotation
The Trinotate annotation pipeline (v3.0.1) was used to functionally annotate the longest isoform of each gene in the assembly. Specifically, the custom Swissprot and Pfam database files were downloaded from the Trinotate website (https://trinotate.github.io/) and used for homology searches. In addition, transmembrane regions and signal peptides were predicted using TmHmm and Signalp, respectively. All results were imported into the Trinotate-SQLite database, and the annotation report for the transcriptome was generated using default parameters.
To manually verify T. sarasinorum genes mentioned by name in this manuscript, T. sarasinorum open reading frames were compared against the UniProt database (fungi, human, invertebrate, mammals, rodents, and vertebrate divisions of Swiss-Prot and TrEMBL) using BLASTP. Up to ten best hits with an E-value of 10−10 or lower for which the alignment covered at least 60% of the database sequence were collected. Query- and database sequences fulfiling these requirements were aligned using MAFFT and used to confirm gene identities.
Gene Ontology Enrichment
GO (Gene Ontology) enrichment analyses were carried out to determine dominant functions in differentially expressed gene clusters. Gene ontology assignments and parental terms were extracted for all genes expressed at FPKM ≥ 1 from the Trinotate annotation report using a script included in the Trinotate-2.0.2 pipeline. Enriched gene ontologies were identified for each cluster using GOseq (Young et al. 2010). Significantly enriched gene ontologies of each cluster with a false discovery rate (FDR) ≤ 0.05 were summarized and redundant terms removed (allowed similarity: 0.5) with REVIGO (Supek et al. 2011).
Tissue-Specific Gene Identification and Evolution
Tissue-specific genes were determined, based on their expression across the three tissues included in this study. Genes with a minimal expression of FPKM ≥ 1 in all biological replicates of one tissue that were overexpressed with a fold change (FC) ≥ 32 and P ≤ 10−10 against both other tissues were termed “tissue-specific.” A subset of these genes was named “strictly tissue-specific,” if they were differentially expressed with a FC ≥ 32 and P ≤ 10−100 against both other tissues, while their expression was FPKM ≥ 2 in all biological replicates of one tissue and FPKM < 1 in all other samples. Genes with a minimal expression of FPKM = 1 in radula and mantle that were overexpressed in both radula and mantle tissue in comparison to foot muscle with a FDR < 10−5 and a FC of at least 4, were identified as likely employed for both radula and shell formation.
To determine the time of origin of tissue-specific genes, putative homologs were identified using a BLASTX search (E-value = 10−10) against predicted proteins of annotated genomes (species [phylum; phylostratum; GenBank/NCBI accession number]) of Biomphalaria glabrata (Mollusca; Gastropoda; APKA00000000.1), Aplysia californica (Mollusca; Gastropoda; AASC00000000.3), Lottia gigantea (Mollusca; Gastropoda; NZ_AMQO00000000.1), Crassostrea gigas (Mollusca; Mollusca; AFTI00000000.1), Octopus bimaculoides (Mollusca; Mollusca; LGKD00000000.1), Capitella teleta (Annelida, Lophotrochozoa; AMQN00000000.1), Lingula anatina (Brachiopoda; Lophotrochozoa; LFEI00000000.1), Drosophila melanogaster (Arthropoda; Protostomia; GCA_000001215.4); Daphnia pulex (Arthropoda; Protostomia; ACJG00000000.1), Takifugu rubripes (Chordata; Bilateria; CAAB00000000.2), Branchiostoma floridae (Chordata; Bilateria; ABEP00000000.2), Ciona intestinalis (Chordata; Bilateria; GCA_000224145.2), Strongylocentrotus purpuratus (Echinodermata; Bilateria; AAGJ00000000.5), Amphimedon queenslandica (Porifera; Metazoa; ACUQ00000000.1), Agaricus bisporus (Basidiomycota; Opisthokonta; GCA_000300575.2), Saccharomyces cerevisiae (Ascomycota; Opisthokonta; GCA_000146045.2), Paramecium tetraurelia (Ciliophora; Eukaryota; CAAL00000000.1), Arabidopsis thaliana (Tracheophyta; Eukaryota; GCA_000001735.1), Phaeodactylum tricornutum (Stramenopiles; Eukaryota; ABQD00000000.1), Methanobrevibacter smithii (Euryarchaeota; Cellular life; GCA_000016525.1), and Escherichia coli (Proteobacteria; Cellular life; GCA_000005845.2). If putative homologs were identified in any of these species, it was assumed that the last common ancestor of Tylomelania and the respective species already possessed a copy of this gene. The same procedure was carried out with all genes in our final assembly to obtain a measure of background gene origin. A two-tailed hypergeometric test with Bonferroni correction was employed to test whether gene evolution of each tissue-specific gene set differed significantly from background gene origin in each of the phylostrata. Since Bonferroni correction is a relatively strict correction for multiple testing, we also calculated the false discovery rate as measure of significance. The two-tailed hypergeometric test with both corrections for multiple testing was calculated using the R script provided by Domazet-Lošo et al. (2017). We chose to focus our analyses on genes for which at least one significant BLASTX hit was returned, since our transcriptome included too many sequences that were assigned gene status, and assembly errors like fragmented or chimeric transcripts together with transcription of intergenic regions would falsely inflate the number of lineage-specific genes. The likelihood of such errors is dependent on expression level, repetitive regions and other features that complicate correct assembly, and which are further likely to differ between tissue-specific gene sets. Thus, although the differing rates of putative homologs found for genes of different gene sets indicated different rates of gene origin leading to Tylomelania, there was no reliable measure to assess to what degree this was a true signal. Genomes or comparable transcriptomes of more closely related species will allow insight into gene origin of younger genes in future studies.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank David Garfield for helpful discussions and comments to a previous version of this manuscript. We also thank Jobst Pfaender for collecting specimens, Isabelle Waurick and the BeGenDiv (Berlin Center for Genomics in Biodiversity Research) for assistance in the lab, and all members of the Hofreiter Lab and von Rintelen Lab for useful discussions. LIPI (Indonesian Institute of Sciences) and RISTEK (Indonesian State Ministry of Research and Technology) kindly issued the permits to conduct research in Indonesia. We would in particular like to thank Ristiyanti M. Marwoto (Museum Zoologicum Bogoriense, LIPI, Cibinong) for her continuous support of the project. Last but not least, we would like to express our gratitude to two anonymous reviewers, who helped improve this manuscript. This work was supported by the German Research Council (DFG) (grant number: Ri 1738/9-1). The authors declare no conflict of Interest.
References
- Aguilera F, McDougall C, Degnan BM.. 2014. Evolution of the tyrosinase gene family in bivalve molluscs: independent expansion of the mantle gene repertoire. Acta Biomater. 109:3855–3865. [DOI] [PubMed] [Google Scholar]
- Aguilera F, McDougall C, Degnan BM.. 2017. Co-option and de novo gene evolution underlie molluscan shell diversity. Mol Biol Evol. 344:779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt D, Musser JM, Baker CVH, Bergman A, Cepko C, Erwin DH, Pavlicev M, Schlosser G, Widder S, Laubichler MD.. 2016. The origin and evolution of cell types. Nat Rev Genet. 1712:744–757. [DOI] [PubMed] [Google Scholar]
- Arendt D, Technau U, Wittbrodt J.. 2001. Evolution of the bilaterian larval foregut. Nature 4096816:81–85. [DOI] [PubMed] [Google Scholar]
- Babonis LS, Martindale MQ, Ryan JF.. 2016. Do novel genes drive morphological novelty? An investigation of the nematosomes in the sea anemone Nematostella vectensis. BMC Evol Biol. 161:114.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Z, Zheng H, Lin J, Wang G, Li J.. 2013. Comparative analysis of the transcriptome in tissues secreting purple and white nacre in the pearl mussel Hyriopsis cumingii. PLoS One 81:e53617.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank S, Arnoldi M, Khoshnavaz S, Treccani L, Kuntz M, Mann K, Grathwohl G, Fritz M.. 2003. The nacre protein perlucin nucleates growth of calcium carbonate crystals. J Microsc. 212(Pt 3):280–291. [DOI] [PubMed] [Google Scholar]
- Boyle MJ, Seaver EC.. 2008. Developmental expression of FoxA and Gata genes during gut formation in the polychaete annelid Capitella sp. Evol Dev. 101:89–105. [DOI] [PubMed] [Google Scholar]
- Brooker L, Shaw J.. 2012. The chiton radula: a unique model for biomineralization studies In: Seto J, editor. Advanced topics in biomineralization. Rijeka: InTech; p. 65–84. [Google Scholar]
- Brusca RC, Brusca GJ.. 2003. Invertebrates. 2nd ed.Sunderland (MA: ): Sinauer Associates, Inc. [Google Scholar]
- Buresi A, Baratte S, Da Silva C, Bonnaud L.. 2012. Orthodenticle/otx ortholog expression in the anterior brain and eyes of Sepia officinalis (Mollusca, Cephalopoda). Gene Expr Patterns 12(3–4):109–116. [DOI] [PubMed] [Google Scholar]
- Corson F, Couturier L, Rouault H, Mazouni K, Schweisguth F.. 2017. Self-organized Notch dynamics generate stereotyped sensory organ patterns in Drosophila. Science 356:eaai7407. [DOI] [PubMed] [Google Scholar]
- Crofts DR. 1937. The Development of Haliotis tuberculata, with special reference to organogenesis during torsion. Philos Trans R Soc Lond B Biol Sci. 228552:219–268. [Google Scholar]
- Cruz R, Lins U, Farina M.. 1998. Minerals of the radular apparatus of Falcidens sp. (Caudofoveata) and the evolutionary implications for the phylum mollusca. Biol Bull. 1942:224–230. [DOI] [PubMed] [Google Scholar]
- De Oliveira AL, Wollesen T, Kristof A, Scherholz M, Redl E, Todt C, Bleidorn C, Wanninger A.. 2016. Comparative transcriptomics enlarges the toolkit of known developmental genes in mollusks. BMC Genomics 171:905.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domazet-Lošo T, Brajkovic J, Tautz D.. 2007. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2311:533–533. [DOI] [PubMed] [Google Scholar]
- Domazet-Lošo T, Carvunis A-R, Albà MM, Šestak MS, Bakarić R, Neme R, Tautz D.. 2017. No evidence for phylostratigraphic bias impacting inferences on patterns of gene emergence and evolution. Mol Biol Evol. 344:843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin DH. 2015. Novelty and innovation in the history of life. Curr Biol. 2519:R930–R940. [DOI] [PubMed] [Google Scholar]
- Fang Z, Feng Q, Chi Y, Xie L, Zhang R.. 2008. Investigation of cell proliferation and differentiation in the mantle of Pinctada fucata (Bivalve, Mollusca). Mar Biol. 1534:745–754. [Google Scholar]
- Feng D, Li Q, Yu H, Kong L, Du S.. 2017. Identification of conserved proteins from diverse shell matrix proteome in Crassostrea gigas: characterization of genetic bases regulating shell formation. Sci Rep. 7:45754–45712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández MS, Valenzuela F, Arias JI, Neira-Carrillo A, Arias JL.. 2016. Is the snail shell repair process really influenced by eggshell membrane as a template of foreign scaffold? J Struct Biol. 1962:187–196. [DOI] [PubMed] [Google Scholar]
- Franke FA, Schumann I, Hering L, Mayer G.. 2015. Phylogenetic analysis and expression patterns of Pax genes in the onychophoran Euperipatoides rowelli reveal a novel bilaterian Pax subfamily. Evol Dev. 171:3–20. [DOI] [PubMed] [Google Scholar]
- Fraser GJ, Bloomquist RF, Streelman JT.. 2013. Common developmental pathways link tooth shape to regeneration. Dev Biol. 3772:399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galis F. 2001. Key innovations and radiations In: Wagner GP, editor. The character concept in evolutionary biology. San Diego: Academic Press; p. 581–605. [Google Scholar]
- Gayral P, Weinert L, Chiari Y, Tsagkogeorga G, Ballenghien M, Galtier N.. 2011. Next-generation sequencing of transcriptomes: a guide to RNA isolation in nonmodel animals. Mol Ecol Resour. 114:650–661. [DOI] [PubMed] [Google Scholar]
- Gazave E, Lapebie P, Richards GS, Brunet F, Ereskovsky AV, Degnan BM, Borchiellini C, Vervoort M, Renard E.. 2009. Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC Evol Biol. 9:249.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose KC. 1962. Origin and development of the digestive system of the giant land snail Achatina fulica Bowdich. Proc R Soc Edinb B Biol. 6803:186–207. [Google Scholar]
- Glaubrecht M, von Rintelen T.. 2008. The species flocks of lacustrine gastropods: Tylomelania on Sulawesi as models in speciation and adaptive radiation. Hydrobiologia 6151:181–199. [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson D, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q.. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 297:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M.. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 88:1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BK. 2012. Parallelism, deep homology, and evo-devo. Evol Dev. 141:29–33. [DOI] [PubMed] [Google Scholar]
- Harney E, Dubief B, Boudry P, Basuyaux O, Schilhabel MB, Huchette S, Paillard C, Nunes FLD.. 2016. De novo assembly and annotation of the European abalone Haliotis tuberculata transcriptome. Mar Genomics 28:11–16. [DOI] [PubMed] [Google Scholar]
- Hausen H. 2005. Chaetae and chaetogenesis in polychaetes (Annelida). Hydrobiologia 535-5361:37–52. [Google Scholar]
- Hohagen J, Jackson DDJ.. 2013. An ancient process in a modern mollusc: early development of the shell in Lymnaea stagnalis. BMC Dev Biol. 131:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JP. 1998. Key innovations and the ecology of macroevolution. Trends Ecol Evol. 131:31–36. [DOI] [PubMed] [Google Scholar]
- Jackson DJ, Degnan BM.. 2016. The importance of evo-devo to an integrated understanding of molluscan biomineralisation. J Struct Biol. 1962:67–74. [DOI] [PubMed] [Google Scholar]
- Jackson DJ, McDougall C, Green K, Simpson F, Wörheide G, Degnan BM.. 2006. A rapidly evolving secretome builds and patterns a sea shell. BMC Biol. 4:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DJ, McDougall C, Woodcroft B, Moase P, Rose RA, Kube M, Reinhardt R, Rokhsar DS, Montagnani C, Joubert C et al. , . 2010. Parallel evolution of nacre building gene sets in molluscs. Mol Biol Evol. 273:591–608. [DOI] [PubMed] [Google Scholar]
- Joshi NA, Fass JN.. 2011. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. [Software]. Available at https://github.com/najoshi/sickle.
- Kantor YI, Puillandre N.. 2012. Evolution of the radular apparatus in Conoidea (Gastropoda: neogastropoda) as inferred from a molecular phylogeny. Malacologia 551:55–90. [Google Scholar]
- Karakostis K, Zanella-Cléon I, Immel F, Guichard N, Dru P, Lepage T, Plasseraud L, Matranga V, Marin F.. 2016. A minimal molecular toolkit for mineral deposition? Biochemistry and proteomics of the test matrix of adult specimens of the sea urchin Paracentrotus lividus. J Proteomics 136:133–144. [DOI] [PubMed] [Google Scholar]
- Kerth K. 1973. Radulaersatz und Zellproliferation in der röntgenbestrahlten Radulascheide der Nacktschnecke Limax flavus L. Ergebnisse zur Arbeitsteilung der Scheidengewebe. Wilhelm Roux’ Arch. 1724:317–348. [DOI] [PubMed] [Google Scholar]
- Klusemann J, Kleinow W, Peters W.. 1990. The hard parts (trophi) of the rotifer mastax do contain chitin: evidence from studies on Brachionus plicatilis. Histochemistry 943:277–283. [DOI] [PubMed] [Google Scholar]
- Kocot KM, Aguilera F, McDougall C, Jackson DJ, Degnan BM.. 2016. Sea shell diversity and rapidly evolving secretomes: insights into the evolution of biomineralization. Front Zool. 13:23.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruimel JH. 1913. Verzeichnis der von Herrn E.C. Abendanon in Celebes gesammelten Süsswasser-Mollusken. Bijdr tot Dierkd. 19:217–235. [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA.. 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 199:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Suleski M, Hedges SB.. 2017. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Bio Evol. 347:1812–1819. [DOI] [PubMed] [Google Scholar]
- Lai EC. 2004. Notch signaling: control of cell communication and cell fate. Development 1315:965–973. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL.. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 103:R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Le Gouar M, Adoutte A.. 2002. Expression patterns of fork head and goosecoid homologues in the mollusc Patella vulgata supports the ancestry of the anterior mesendoderm across Bilateria. Dev Genes Evol. 21211:551–561. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Lespinet O, Vervoort M, Adoutte A.. 2002. Expression pattern of Brachyury in the mollusc Patella vulgata suggests a conserved role in the establishment of the AP axis in Bilateria. Development 129:1411–1421. [DOI] [PubMed] [Google Scholar]
- Lavergne V, Harliwong I, Jones A, Miller D, Taft RJ, Alewood PF, Lavergne V, Harliwong I, Jones A, Miller D.. 2015. Optimized deep-targeted proteotranscriptomic profiling reveals unexplored Conus toxin diversity and novel cysteine frameworks. Proc Natl Acad Sci U S A. 11229:E3782–E6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN.. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A.. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2213:1658–1659. [DOI] [PubMed] [Google Scholar]
- Liao Z, Bao LF, Fan MH, Gao P, Wang XX, Qin CL, Li XM.. 2015. In-depth proteomic analysis of nacre, prism, and myostracum of Mytilus shell. J. Proteomics 122:26–40. [DOI] [PubMed] [Google Scholar]
- Livingston BT, Killian CE, Wilt F, Cameron A, Landrum MJ, Ermolaeva O, Sapojnikov V, Maglott DR, Buchanan AM, Ettensohn CA.. 2006. A genome-wide analysis of biomineralization-related proteins in the sea urchin Strongylocentrotus purpuratus. Dev Biol. 3001:335–348. [DOI] [PubMed] [Google Scholar]
- Lowenstam HA, Weiner S.. 1989. On biomineralization. Oxford: Oxford University Press. [Google Scholar]
- Luo Y-J, Takeuchi T, Koyanagi R, Yamada L, Kanda M, Khalturina M, Fujie M, Yamasaki S-I, Endo K, Satoh N.. 2015. The Lingula genome provides insights into brachiopod evolution and the origin of phosphate biomineralization. Nat Commun. 61:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenstedt U, Märkel K.. 1987. Experimental and comparative morphology of radula renewal in pulmonates (Mollusca, Gastropoda). Zoomorphology 1074:209–239. [Google Scholar]
- Manousaki T, Hull PM, Kusche H, MacHado-Schiaffino G, Franchini P, Harrod C, Elmer KR, Meyer A.. 2013. Parsing parallel evolution: ecological divergence and differential gene expression in the adaptive radiations of thick-lipped Midas cichlid fishes from Nicaragua. Mol Ecol. 223:650–669. [DOI] [PubMed] [Google Scholar]
- Marin F, Bundeleva I, Takeuchi T, Immel F, Medakovic D.. 2016. Organic matrices in metazoan calcium carbonate skeletons: composition, functions, evolution. J Struct Biol. 1962:98–106. [DOI] [PubMed] [Google Scholar]
- Marin F, Le Roy N, Marie B.. 2012. The formation and mineralization of mollusk shell. Front Biosci. 4:1099–1125. [DOI] [PubMed] [Google Scholar]
- Marin F, Luquet G, Marie B, Medakovic D.. 2008. Molluscan shell proteins: primary structure, origin, and evolution In: Schatten GP, editor. Current topics in developmental biology. Vol. 80 Elsevier Inc; p. 209–276. [DOI] [PubMed] [Google Scholar]
- Marin F, Marie B, Hamada SB, Silva P, Roy NL, Wolf SE, Montagnani C, Joubert C, Piquemal D.. 2013. “Shellome”: proteins involved in mollusc shell biomineralization – diversity, functions. In: Watabe S, Maeyama K, Nagasawa H, editors. Recent Advances in Pearl Research. Tokyo: Terrapub; p. 149–166. [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 171:10–12. [Google Scholar]
- McLysaght A, Guerzoni D.. 2015. New genes from non-coding sequence: the role of de novo protein-coding genes in eukaryotic evolutionary innovation. Philos Trans R Soc Lond B Biol Sci. 3701678:20140332.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLysaght A, Hurst LD.. 2016. Open questions in the study of de novo genes: what, how and why. Nat Rev Genet. 179:567–578. [DOI] [PubMed] [Google Scholar]
- Moyers BA, Zhang J.. 2016. Evaluating phylostratigraphic evidence for widespread de novo gene birth in genome evolution. Mol Biol Evol. 335:1245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumm JS, Kopan R.. 2000. Notch signaling: from the outside in. Dev Biol. 2282:151–165. [DOI] [PubMed] [Google Scholar]
- Nemoto M, Wang Q, Li D, Pan S, Matsunaga T, Kisailus D.. 2012. Proteomic analysis from the mineralized radular teeth of the giant Pacific chiton, Cryptochiton stelleri (Mollusca). Proteomics 1218:2890–2894. [DOI] [PubMed] [Google Scholar]
- Obinata A, Akimoto Y.. 2012. Effects of retinoic acid and Gbx1 on feather-bud formation and epidermal transdifferentiation in chick embryonic cultured dorsal skin. Dev Dyn. 2419:1405–1412. [DOI] [PubMed] [Google Scholar]
- Page LR. 2002. Larval and metamorphic development of the foregut and proboscis in the caenogastropod Marsenina (Lamellaria) stearnsii. J Morphol. 2522:202–217. [DOI] [PubMed] [Google Scholar]
- Page LR, Hookham B.. 2017. The gastropod foregut—evolution viewed through a developmental lens. Can J Zool. 954:227–238. [Google Scholar]
- Perry KJ, Henry JQ.. 2015. CRISPR/Cas9-mediated genome modification in the mollusc, Crepidula fornicata. Genesis 532:237–244. [DOI] [PubMed] [Google Scholar]
- Perry KJ, Lyons DC, Truchado-Garcia M, Fischer AHL, Helfrich LW, Johansson KB, Diamond JC, Grande C, Henry JQ.. 2015. Deployment of regulatory genes during gastrulation and germ layer specification in a model spiralian mollusc Crepidula. Dev Dyn. 24410:1215–1248. [DOI] [PubMed] [Google Scholar]
- Peters W. 1972. Occurrence of chitin in mollusca. Comp Biochem Physiol B Biochem Mol Biol. 413:541–550. [Google Scholar]
- Petrak J, Vyoral D.. 2005. Hephaestin – a ferroxidase of cellular iron export. Int J Biochem Cell Biol. 376:1173–1178. [DOI] [PubMed] [Google Scholar]
- Ponder WF, Lindberg DR.. 2008. Molluscan evolution and phylogeny: an introduction In: Ponder WF, Lindberg DR, editors. Phylogeny and evolution of the Mollusca. Berkeley: University of California Press; p. 1–17. [Google Scholar]
- Rafiq K, Shashikant T, McManus CJ, Ettensohn C. a.. 2014. Genome-wide analysis of the skeletogenic gene regulatory network of sea urchins. Development 1414:950–961. [DOI] [PubMed] [Google Scholar]
- Ramos-Silva P, Kaandorp J, Herbst F, Plasseraud L, Alcaraz G, Stern C, Corneillat M, Guichard N, Durlet C, Luquet G, Marin F.. 2014. The skeleton of the staghorn coral Acropora millepora: molecular and structural characterization. PLoS One 96:e97454.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raouf A, Seth A.. 2000. Ets transcription factors and targets in osteogenesis. Oncogene 1955:6455–6463. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK.. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 261:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg G. 2014. A new critical estimate of named species-level diversity of the recent Mollusca. Am Malacol Bull. 322:308–322. [Google Scholar]
- Runham NW. 1963. A study of the replacement mechanism of the pulmonate radula. Q J Microsc Sci. 104:271–277. [Google Scholar]
- Samadi L, Steiner G.. 2010. Conservation of ParaHox genes’ function in patterning of the digestive tract of the marine gastropod Gibbula varia. BMC Dev Biol. 101:74–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos ME, Baldo L, Gu L, Boileau N, Musilova Z, Salzburger W.. 2016. Comparative transcriptomics of anal fin pigmentation patterns in cichlid fishes. BMC Genomics 17:712.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherholz M, Redl E, Wollesen T, de Oliveira AL, Todt C, Wanninger A.. 2017. Ancestral and novel roles of Pax family genes in mollusks. BMC Evol Biol. 171:81.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann SM, Martín-durán JM, Børve A, Vellutini BC, Passamaneck YJ, Hejnol A.. 2017. Clustered brachiopod Hox genes are not expressed collinearly and are associated with lophotrochozoan novelties. Proc Natl Acad Sci U S A. 11410:E1913–E1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmerer M, Savage RM, Shankland M.. 2009. Paxβ: a novel family of lophotrochozoan Pax genes. Evol Dev. 116:689–696. [DOI] [PubMed] [Google Scholar]
- Schönitzer V, Weiss IM.. 2007. The structure of mollusc larval shells formed in the presence of the chitin synthase inhibitor Nikkomycin Z. BMC Struct Biol. 7:71.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma T, Ettensohn CA.. 2010. Activation of the skeletogenic gene regulatory network in the early sea urchin embryo. Development 137:1149–1157. [DOI] [PubMed] [Google Scholar]
- Shimek RL, Kohn AJ.. 1981. Functional morphology and evolution of the toxoglossan radula. Malacologia 20:423–438. [Google Scholar]
- Shimeld SM, Boyle MJ, Brunet T, Luke GN, Seaver EC.. 2010. Clustered Fox genes in lophotrochozoans and the evolution of the bilaterian Fox gene cluster. Dev Biol. 3402:234–248. [DOI] [PubMed] [Google Scholar]
- Shubin N, Tabin C, Carroll S.. 2009. Deep homology and the origins of evolutionary novelty. Nature 4577231:818–823. [DOI] [PubMed] [Google Scholar]
- Sigwart JD. 2017. Zoology: molluscs all beneath the sun, one shell, two Shells, more, or none. Curr Biol. 2714:R708–R710. [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 3119:3210–3212. [DOI] [PubMed] [Google Scholar]
- Sone ED, Weiner S, Addadi L.. 2007. Biomineralization of limpet teeth: a cryo-TEM study of the organic matrix and the onset of mineral deposition. J Struct Biol. 1583:428–444. [DOI] [PubMed] [Google Scholar]
- Steinmetz PRH, Kostyuchenko RP, Fischer A, Arendt D.. 2011. The segmental pattern of otx, gbx, and Hox genes in the annelid Platynereis dumerilii. Evol Dev. 131:72–79. [DOI] [PubMed] [Google Scholar]
- Steinmetz PRH, Kraus JEM, Larroux C, Hammel JU, Amon-Hassenzahl A, Houliston E, Wörheide G, Nickel M, Degnan BM, Technau U.. 2012. Independent evolution of striated muscles in cnidarians and bilaterians. Nature 4877406:231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan M, Dökel S, Amstislavskiy V, Wuttig D, Sültmann H, Lehrach H, Yaspo ML.. 2012. A simple strand-specific RNA-Seq library preparation protocol combining the Illumina TruSeq RNA and the dUTP methods. Biochem Biophys Res Commun. 4224:643–646. [DOI] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T.. 2011. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One 67:e21800.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery AP, Shono T, Kurokawa D, Britz R, Johanson Z, Fraser GJ.. 2017. Spatially restricted dental regeneration drives pufferfish beak development. Proc Natl Acad Sci U S A. 11422:E4425–E4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukmar-Godec T, Kapun G, Zaslansky P, Faivre D.. 2015. The giant keyhole limpet radular teeth: a naturally-grown harvest machine. J Struct Biol. 1923:392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendrami DLJ, Shah A, Telesca L, Hoffman JI.. 2016. Mining the transcriptomes of four commercially important shellfish species for single nucleotide polymorphisms within biomineralization genes. Mar Genomics 27:17–23. [DOI] [PubMed] [Google Scholar]
- von Rintelen T, Bouchet P, Glaubrecht M.. 2007. Ancient lakes as hotspots of diversity: a morphological review of an endemic species flock of Tylomelania (Gastropoda: cerithioidea: pachychilidae) in the Malili lake system on Sulawesi, Indonesia. Hydrobiologia 5921:11–94. [Google Scholar]
- von Rintelen T, von Rintelen K, Glaubrecht M.. 2010. The species flocks of the viviparous freshwater gastropod Tylomelania (Mollusca: cerithioidea: pachychilidae) in the ancient lakes of Sulawesi, Indonesia: the role of geography, trophic morphology and color as driving forces in adaptive radiation. In: Glaubrecht M, editor. Evolution in action. Berlin: Springer; p. 485–512. [Google Scholar]
- von Rintelen T, von Rintelen K, Glaubrecht M, Schubart CD, Herder F.. 2012. Aquatic biodiversity hotspots in Wallacea: the species flocks in the ancient lakes of Sulawesi, Indonesia In: Gower DJ, Johnson KG, Richardson JE, Rosen BR, Rüber L, Williams ST, editors. Biotic evolution and environmental change in Southeast Asia. Cambridge: Cambridge University Press; p. 290–315. [Google Scholar]
- von Rintelen T, Wilson AB, Meyer A, Glaubrecht M.. 2004. Escalation and trophic specialization drive adaptive radiation of freshwater gastropods in ancient lakes on Sulawesi, Indonesia. Proc R Soc Lond B Biol Sci. 2711557:2541–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GP, Lynch VJ.. 2010. Evolutionary novelties. Curr Biol. 202:R48–R52. [DOI] [PubMed] [Google Scholar]
- Weaver JC, Wang Q, Miserez A, Tantuccio A, Stromberg R, Bozhilov KN, Maxwell P, Nay R, Heier ST, DiMasi E, Kisailus D.. 2010. Analysis of an ultra hard magnetic biomineral in chiton radular teeth. Mater Today 13(1–2):42–52. [Google Scholar]
- Weiss IM, Schönitzer V, Eichner N, Sumper M.. 2006. The chitin synthase involved in marine bivalve mollusk shell formation contains a myosin domain. FEBS Lett. 5807:1846–1852. [DOI] [PubMed] [Google Scholar]
- Whittington CM, Griffith OW, Qi W, Thompson MB, Wilson AB.. 2015. Seahorse brood pouch transcriptome reveals common genes associated with vertebrate pregnancy. Mol Biol Evol. 3212:3114–3131. [DOI] [PubMed] [Google Scholar]
- Wiesel R, Peters W.. 1978. Licht- und elektronenmikroskopische Untersuchungen am Radulakomplex und zur Radulabildung von Biomphalaria glabrata Say (= Australorbis gl.) Gastropoda, Basommatophora). Zoomorphologie 891:73–92. [Google Scholar]
- Wollesen T, Scherholz M, Rodríguez Monje SV, Redl E, Todt C, Wanninger A.. 2017. Brain regionalization genes are co-opted into shell field patterning in Mollusca. Sci Rep. 71:5486.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, Oshlack A.. 2010. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 112:R14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewski AC, Weigert A, Helm C, Adamski M, Adamska M, Bleidorn C, Raible F, Hausen H.. 2014. Early divergence, broad distribution, and high diversity of animal chitin synthases. Genome Biol Evol. 62:316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Kwan KM, Mackem S.. 2016. Putative oncogene Brachyury (T) is essential to specify cell fate but dispensable for notochord progenitor proliferation and EMT. Proc Natl Acad Sci U S A. 11314:3820–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.