

Abstract
Integral membrane proteins are prone to aggregation and misfolding in aqueous environments and therefore require binding by molecular chaperones during their biogenesis. Chloroplast signal recognition particle 43 (cpSRP43) is an ATP-independent chaperone required for the biogenesis of the most abundant class of membrane proteins, the light-harvesting chlorophyll a/b-binding proteins (LHCPs). Previous work has shown that cpSRP43 specifically recognizes an L18 loop sequence conserved among LHCP paralogs. However, how cpSRP43 protects the transmembrane domains (TMDs) of LHCP from aggregation was unclear. In this work, alkylation-protection and site-specific cross-linking experiments found that cpSRP43 makes extensive contacts with all the TMDs in LHCP. Site-directed mutagenesis identified a class of cpSRP43 mutants that bind tightly to the L18 sequence but are defective in chaperoning full-length LHCP. These mutations mapped to hydrophobic surfaces on or near the bridging helix and the β-hairpins lining the ankyrin repeat motifs of cpSRP43, suggesting that these regions are potential sites for interaction with the client TMDs. Our results suggest a working model for client protein interactions in this membrane protein chaperone.
Keywords: protein targeting, signal recognition particle (SRP), chloroplast, chaperone, membrane protein
Introduction
Proper protein folding and localization are critical for cellular protein homeostasis. The post-translational targeting of integral membrane proteins poses an acute challenge to protein homeostasis. Before arrival at the target membrane, nascent membrane proteins are highly prone to aggregation in the cytosol and other aqueous cellular compartments. Thus, effective molecular chaperones or chaperone networks are required to minimize improper exposure of the transmembrane domains (TMDs) on newly synthesized membrane proteins and to maintain them in a soluble, translocation-competent conformation. Many examples illustrate the intimate link between chaperone function and membrane protein biogenesis, including SecB, Skp, and SurA that protect bacterial outer membrane proteins, and Hsp70 homologues implicated in the import of precursor proteins to the endoplasmic reticulum, mitochondria, or chloroplast (1–7).
The light-harvesting chlorophyll a/b-binding proteins (LHCP)6 comprise over 50% of the protein content on the thylakoid membrane of green plants and form the most abundant family of membrane proteins on earth (8). LHCPs are nuclear encoded, initially synthesized in the cytosol, and imported across the chloroplast envelope in a largely unfolded state (8). In the chloroplast stroma, LHCPs are protected in a soluble “transit complex” by the chloroplast signal recognition particle (cpSRP), comprised of the cpSRP43 and cpSRP54 protein subunits (9–12). Via interactions between the GTPase domains of cpSRP54 and its receptor cpFtsY, LHCPs are delivered to the Alb3 translocase and inserted into the thylakoid membrane (11, 13–20). Previous work showed that the cpSRP43 subunit binds tightly to and quantitatively prevents the aggregation of multiple members of the LHCP family, and that it is necessary and sufficient to chaperone LHCPs (21, 22). Although the chaperone activity of cpSRP43 is allosterically regulated by additional components in the cpSRP pathway, such as cpSRP54 and Alb3 (19, 22–24), the simple composition of the cpSRP43–LHCP chaperone–client pair and the robustness of cpSRP43's chaperone activity make this pair an excellent system to understand the interaction and regulation of membrane protein chaperones.
A long-standing question about the cpSRP43 chaperone is the mechanism by which it prevents the hydrophobic TMDs on its substrate proteins from aggregation. The substrate-binding domain (SBD) of cpSRP43 is comprised of ankyrin repeat motifs 1–4, capped at the N terminus by a chromodomain (CD1) and at the C terminus by a bridging helix (BH) (21, 22, 25). Biochemical and crystallographic analyses showed that a conserved Tyr-204 in the third ankyrin repeat motif recognizes an FDPLGL motif in L18, a conserved 18-amino acid sequence between the second and third TMDs of LHCP (12, 22, 25–27). However, interaction with a soluble loop sequence is unlikely to be sufficient to protect LHCPs, which contain three TMDs, from aggregation. The ability of cpSRP43 to quantitatively prevent full-length LHCPs from aggregation is highly suggestive of additional interactions between cpSRP43 and the substrate TMDs. Moreover, a recent study showed that cpSRP43 also protects aggregation-prone regions of glutamyl-tRNA reductase to enhance the stability of this enzyme (28), indicating that cpSRP43 can contact hydrophobic segments on client proteins independently of L18 recognition. Nevertheless, deletion of individual TMDs in LHCP or replacement with the TMDs from unrelated membrane proteins did not severely disrupt the cpSRP43–LHCP interaction (26); this lack of specificity rendered the putative TMD interactions of cpSRP43 particularly challenging to demonstrate and identify. Although a cross-linking study identified three additional residues in TM3 of LHCP that can cross-link to cpSRP43 (27), the study did not identify additional possible interactions with the remainder of LHCP, nor the sites on cpSRP43 that interact with the substrate protein. Furthermore, conformational rearrangements occur in the cpSRP43 SBD upon substrate binding (22), making it particularly challenging to define the interaction of this chaperone with the substrate TMDs.
In this work, we used a combination of chemical modification-protection, cross-linking, and site-directed mutagenesis studies to understand the interaction between cpSRP43 and its client protein. The results showed that cpSRP43 can interact with LHCP across all three TMDs, and identified a set of mutant cpSRP43s that are specifically disrupted in their ability to chaperone LHCP without affecting recognition of the L18 motif. These observations suggest potential sites for TMD interactions on this membrane protein chaperone.
Results
Mapping the interaction sites of cpSRP43 on Lhcb5 through alkylation protection and site-specific cross-linking
Lhcb5 is a member of the LHCP family that strongly depends on cpSRP for its biogenesis. Previous work showed that Lhcb5 forms a tight complex with cpSRP43, with an apparent Kd value of ∼10 nm (26). To define the sites on Lhcb5 involved in complex formation with cpSRP43, we tested the ability of cpSRP43 to protect individual residues in Lhcb5 from alkylation by N-ethylmaleimide (NEM). To this end, we purified a set of Lhcb5 variants in which single cysteines were engineered at every 5–10 residues across the entire sequence of Lhcb5 (29). To ensure that all Lhcb5 are bound by the chaperone, we tested the efficiency of complex formation between each single cysteine variant of Lhcb5 and cpSRP43 (Table 1) and used cpSRP43 concentrations that are saturating for each Lhcb5 mutant during assembly of the respective cpSRP43·Lhcb5 complexes. The previously identified FDPLGL interaction motif in the L18 sequence was not tested, as point mutations at any of these residues severely impaired complex formation with cpSRP43 (25, 26).
Table 1.
Chaperone activity of cpSRP43 toward individual Lhcb5 single-cysteine mutants (second and third columns), and NEM alkylation efficiency at each cysteine in the presence of WT cpSRP43 (fourth and fifth columns)
| Lhcb5 construct | 1 μm cpSRP43 activitya | 5 μm cpSRP43 activitya | Alkylationb at 2′ | Alkylationb at 10′ |
|---|---|---|---|---|
| Ile-40 | NDc | ND | 1.0 | 1.0 |
| Gly-50 | ND | ND | 1.0 | 1.0 |
| Gln-70 | ND | ND | 0.55 ± 0.02 | 0.67 ± 0.10 |
| Ile-75 | 84.48 | ND | 0.53 ± 0.05 | 0.66 ± 0.10 |
| Ala-85 | 102.15 ± 0.66 | ND | 0.33 ± 0.00 | 0.39 ± 0.02 |
| Pro-90 | 57.06 ± 31.69 | ND | 0.19 | 0.37 ± 0.14 |
| Cys-100 (WT) | 95.59 | ND | 0.62 ± 0.16 | 0.66 ± 0.17 |
| Gly-110 | ND | ND | 0.29 ± 0.05 | 0.35 ± 0.03 |
| Asn-120 | ND | ND | 0.47 ± 0.04 | 0.52 ± 0.09 |
| Asn-125 | 74.62 | ND | 0.47 ± 0.00 | 0.56 ± 0.11 |
| Leu-130 | 68.18 ± 0.24 | ND | 0.28 ± 0.00 | 0.32 ± 0.01 |
| Val-135 | 89.89 ± 14.78 | ND | 0.13 | 0.18 ± 0.01 |
| Gly-143 | 89.20 | ND | 0.34 | 0.45 ± 0.01 |
| Thr-150 | 95.84 | ND | 0.80 | 0.88 ± 0.03 |
| Glu-156 | 80.74 | ND | 1.0 | 1.0 |
| Asp-157 | 86.34 | ND | 1.0 | 1.0 |
| Gly-162 | 95.39 | ND | 1.0 | 1.0 |
| Ala-171 | 86.33 | ND | 1.0 | 1.0 |
| Leu-180 | 89.85 | ND | 0.91 ± 0.12 | 0.91 ± 0.13 |
| Ile-185 | 82.86 ± 5.02 | ND | 0.91 ± 0.12 | 0.86 ± 0.02 |
| Leu-190 | 59.79 | 95.04 | 0.44 ± 0.10 | 0.48 ± 0.11 |
| Met-195 | 74.87 ± 23.58 | 95.50 | 0.17 ± 0.02 | 0.24 ± 0.05 |
| Ile-200 | 77.24 | ND | ND | 0.25 ± 0.10 |
| Val-210 | 92.27 | ND | 0.81 | 0.91 ± 0.13 |
| Pro-220 | 88.82 | ND | 0.44 | 0.50 ± 0.17 |
| Ala-230 | 87.60 | ND | 0.56 | 0.64 ± 0.14 |
a cpSRP43 chaperone activity was measured using 1 μm Lhcb5 in the light scattering assay. % soluble Lhcb5 at indicated cpSRP43 concentrations are reported.
b Fraction of NEM modified Lhcb5 in the cpSRP43·Lhcb5 complex after indicated times of the alkylation reaction. All values represent mean ± S.E., with n = 2.
c ND, not done.
The efficiency of NEM alkylation was quantified by intact MS and provides a direct measure for the solvent accessibility of individual cysteine residues in Lhcb5 (29). Previous work showed that LHCPs are imported into the chloroplast in a largely unfolded state (8), and its folding requires the lipids and binding of photosynthetic pigments in the thylakoid membrane (30–32). This loosely folded state of Lhcb5 in the stroma is mimicked by denaturation in 6 m GdmCl or 8 m urea, and the NEM alkylation of the engineered single cysteines in denaturant-solubilized Lhcb5 were used as a control to correct for the intrinsic differences in the reactivity of cysteines at different positions (29). Comparison of the alkylation efficiency in the cpSRP43·Lhcb5 complex to that of chemically denatured Lhcb5 provides a measure for the degree to which individual residues in Lhcb5 are protected by interaction with cpSRP43.
Representative data for the complexes of two Lhcb5 variants, V135C and E156C, are shown in Fig. 1, A and B, respectively. Deconvolution and quantification of the m/z spectrum showed that for Lhcb5(E156C), a single alkylated species was present after a 10-min alkylation reaction, indicating that this site was fully alkylated and thus solvent exposed in the complex (Fig. 1B). By contrast, the m/z spectrum of Lhcb5(V135C) contained both the unalkylated and alkylated species (Fig. 1A), indicating that this site was protected by cpSRP43.
Figure 1.

Alkylation pattern of Lhcb5 in the cpSRP43·Lhcb5 complex show cpSRP43-induced protection on the substrate protein. A and B, mass spectrum (upper left), deconvolution (lower panels), and component analysis (upper right) for a partially alkylated Lhcb5 residue, Cys-135 (A) and a completely alkylated Lhcb5 residue, Cys-156 (B). C, summary of the NEM alkylation efficiencies at individual sites in Lhcb5. Alkylation reactions were carried out for 10 min. For each engineered cysteine, “fraction accessible” was calculated from the ratio of the fraction of alkylation in the cpSRP43·Lhcb5 complex relative to that of Lhcb5 dissolved in 6 m GdmHCl. Error bars indicate S.E., with n = 2. D, the alkylation protection pattern of Lhcb5 in complex with cpSRP43 is mapped onto the sequence of Lhcb5. Colored triangles denote the extent of protection, with white denoting the least protection (0% protection, or 100% alkylation) and blue denoting the highest observed protection (>50% protection or <50% alkylation).
The results of the alkylation-protection experiments for all the Lhcb5 variants are shown in Fig. S1 and summarized in Fig. 1C. Mapping of the alkylation-protection efficiencies at 10 min onto the sequence of Lhcb5 (Fig. 1D) revealed several patterns. Residues 70–143, which span the first two TMDs of Lhcb5 and their intervening loop, were modestly to heavily protected, suggesting that they were contacted by cpSRP43 in the cpSRP43·Lhcb5 complex. Residues 190–200, which form the C-terminal part of TM3, were also extensively protected, consistent with the results of a previous cross-linking analysis (27). The C-terminal loop of Lhcb5 was also modestly protected. In contrast, residues in the N-terminal loop of Lhcb5 were highly accessible. In addition, residues in the loop connecting TM2 and TM3 of Lhcb5 were accessible, consistent with crystallographic analysis showing that the L18 peptide is bound at a solvent-accessible site on the surface of cpSRP43 (25). Together, these results show that cpSRP43 induced protections of Lhcb5 including all its TMDs, the TM1-TM2 loop, and the C terminus.
To independently probe for the interaction of the LHCP TMDs with cpSRP43, we incorporated a photoinducible cross-linker, p-benzoyl-l-phenylalanine (pBpa), into specific positions of Lhcb5 via amber suppression coupled to the S30 in vitro translation system (33–35). Cross-linking occurs when the ketone oxygen of the incorporated pBpa is within 3.1 Å of an interaction partner (36). To specifically examine the interactions between cpSRP43 and Lhcb5, we used a superactive mutant of cpSRP43 (intein–cpSRP43) that has been shown to mimic the conformation and activity of cpSRP54-activated cpSRP43 in both NMR spectroscopy and biochemical assays (22). We observed UV-induced, cpSRP43-dependent high molecular weight cross-linking products for pBpa incorporated at all three TMDs as well as the L18 motif of Lhcb5 (Fig. 2). Western blotting analyses with anti-Strep (for Lhcb5) and anti-cpSRP43 antibodies of both the in vitro translation reaction and the affinity-purified cpSRP43·Lhcb5 complex confirmed that the high-molecular weight band(s) contained both cpSRP43 and Lhcb5 (Fig. 3, A–C). MS analysis of the cross-linked bands for two Lhcb5 variants, with Bpa incorporated at residues 162 and 180, further confirmed that both bands contained cpSRP43 and Lhcb5 at a roughly 1:1 molar ratio (Fig. 3, D–F). These results indicate that all three of the TMDs of Lhcb5 can come into close contact with cpSRP43 in the complex. Cross-linking efficiency was highest with pBpa incorporated near the DPLG motif in the L18 sequence (Figs. 2, residues 162 and 164), consistent with specific recognition of this motif by cpSRP43. In comparison, the cross-linked bands between cpSRP43 and pBpa incorporated in the TMDs of Lhcb5 were weaker and more diffuse. The observation of two cross-linked bands, both containing equimolar cpSRP43 and Lhcb5 (Fig. 3, D–F), further suggest the presence of alternative conformations in the cpSRP43·Lhcb5 complex. These observations suggest that the interactions of Lhcb5 TMDs with cpSRP43 are potentially dynamic. Collectively, the combination of cross-linking and alkylation-protection experiments in this section provides strong evidence that, in addition to the L18 motif, cpSRP43 can make contacts with and induce protection of all three of the TMDs in its client protein.
Figure 2.

Site-specific cross-linking suggests extensive contacts between Lhcb5 and cpSRP43. A, SDS-PAGE analysis of 35S-labeled Lhcb5 containing a photocross-linker, pBpa, at the indicated positions. Purified superactive cpSRP43 was present (+ lanes) or absent (− lanes) during translation, and samples were protected from light (− lanes) or exposed to UV light to induce pBpa cross-links (+ lanes). Marked bands indicate the cross-linked cpSRP43·Lhcb5 complex (red), free Lhcb5 (green), and Lhcb5 cross-linked to an unknown protein in the translation extract (yellow). B, summary of cross-linking efficiencies between cpSRP43 and Lhcb5 with pBpa incorporated at different sites. Cross-linking efficiency was calculated from the ratio of the cpSRP43·Lhcb5 band to total Lhcb5 (after subtraction of background from the corresponding locations in the +UV, −cpSRP43 lane) for the SDS-PAGE analysis in A and replicates (not shown). Data are reported as mean ± S.E., with n = 2.
Figure 3.

Analysis of the high molecular weight cross-linked bands between cpSRP43 and Lhcb5. The color-marked bands in A–D indicate the cross-linked cpSRP43·Lhcb5 complex (red), free Strep-Lhcb5 (green), free cpSRP43 (blue), and Strep-Lhcb5 cross-linked to an unknown protein (yellow). A and B, representative Western blotting analyses of the cross-linking reactions and their controls from Fig. 2A using anti-Strep (for Strep-tagged Lhcb5; A) or anti-cpSRP43 (B) antibodies. The lower Mr band for Lhcb5 is likely a C terminally proteolyzed product of full-length Lhcb5. C, representative Western blot of affinity purification of the cross-linked cpSRP43·Lhcb5 complex based on Strep-tagged Lhcb5. The final wash (W) and elution (E) from Strep-Tactin resin were shown for reactions with (+ lanes) and without (− lanes) intein–cpSRP43 and exposure to UV light. D, representative Coomassie-stained SDS-PAGE gels for the same cross-linking reactions purified based on His6-tagged cpSRP43, with Bpa incorporated at the indicated residues of Lhcb5. E, NuPAGE gels showing the purified (as in C and D) cpSRP43·Lhcb5(162Bpa) and cpSRP43·Lhcb5(180Bpa) cross-linking reactions. The two labeled bands (A and B) were digested and sent for MS analysis. F, results for MS analysis of the abundance of cpSRP43 and Lhcb5 in bands A and B excised the gel in E for Lhcb5(162Bpa).
cpSRP43's substrate-binding domain is highly sensitive to point mutations
Previous work established that CD1, the ankyrin repeat motifs, and the BH together form a structural and functional unit that comprises the SBD of cpSRP43, which is sufficient to chaperone LHCPs (22). To establish which sites of cpSRP43 are involved in complex formation with LHCP, we mutated all solvent-exposed hydrophobic residues (Leu, Ile, Val, and Trp) in the SBD, as well as additional residues on the β-hairpins of the ankyrin repeat motifs and on the BH (highlighted in blue in Fig. 4A). Each residue was mutated to cysteine in an otherwise cysteine-less cpSRP43 (C118A, C240S). Cys-less cpSRP43 is 5-fold reduced in binding and chaperoning LHCP compared with WT cpSRP43 because it is shifted to a less active conformation (22), but otherwise behaves analogously to WT cpSRP43.
Figure 4.

Single-cysteine mutants across the cpSRP43 SBD exhibit defects in chaperone activity in the light scattering assay. A, structure of cpSRP43 indicating all sites where cysteine mutations were made (blue). B and C, representative data showing the chaperone activity of neutral (B) and defective (C) cpSRP43 mutants. Light scattering time traces are shown for LHCP diluted into aqueous buffer (green), into a solution containing Cys-less WT (black), and into solutions containing the indicated cpSRP43 mutants. D, summary of the chaperone activity for all the single cysteine mutants of cpSRP43 measured by light scattering. Mutants exhibiting chaperone activity within 3-fold of that of Cys-less cpSRP43 are considered neutral (above dashed line), whereas mutants with lower activity are considered defective (below dashed line). Error bars indicate S.E., with n = 3–13.
We tested each single cysteine mutant of cpSRP43 for its ability to bind and protect LHCP from aggregation using a well-established light scattering assay (Fig. 4, B and C). In this assay, LHCP denatured and solubilized in 8 m urea was added to a solution containing either buffer, Cys-less cpSRP43 (referred to as WT), or the mutant cpSRP43 of interest, and the turbidity of the solution was monitored in real time. In the absence of cpSRP43, LHCP aggregated extensively in aqueous solution (Fig. 4, B and C, green lines). The presence of 2.5 μm Cys-less cpSRP43 prevented the aggregation of ∼55% LHCP (Fig. 4, B and C, black lines); this cpSRP43 concentration thus provides the most sensitive condition to screen for mutant cpSRP43s defective in chaperone activity.
We found that single point mutations of a surprisingly large number of residues in the cpSRP43 SBD compromised its chaperone activity. Of the 33 single cysteine mutants tested, only 10 mutants exhibited chaperone activities within 3-fold of that of Cys-less cpSRP43 (Fig. 4, B and D). Six mutants exhibited 3–5-fold reductions in the solubilization of LHCP compared with Cys-less cpSRP43, and chaperone activity was undetectable for 17 mutants (Fig. 4, C and D). The sites of mutations that induced modest or severe defects in chaperone activity span almost an entire surface of the cpSRP43 SBD (see Fig. 8). Thus, cpSRP43 is highly sensitive to conservative perturbations in its SBD.
Figure 8.

Mapping two classes of cpSRP43 mutants onto the crystal structure of the cpSRP43 SBD (Protein Data Bank code 3dep). A, residues whose mutations led to defective chaperone activity for LHCP but did not disrupt L18 binding are categorized as Class I and colored in orange. B, residues whose mutations disrupted both cpSRP43's chaperone activity and its interaction with the L18 motif are categorized as Class II and colored in magenta. C, a putative model for the interaction surfaces of cpSRP43 with LHCP, with Tyr-204 (blue) interacting with the L18 sequence, and the hydrophobic surfaces formed by Ank4, BH, and the β-hairpins along the ankyrin repeat motifs involved in protection of the TMDs of LHCP. The electrostatic surface potential of the cpSRP43 SBD was generated using Adaptive Poisson-Boltzmann Solver (52) and visualized in PyMOL.
To independently test the chaperone activity of cpSRP43 mutants, we used an alternative sedimentation assay (21). LHCP denatured in 8 m urea was added to either Cys-less (WT) or mutant cpSRP43, and the mixture was separated into soluble and insoluble fractions by sedimentation followed by analysis on SDS-PAGE (Fig. 5A). Qualitatively, most mutants displayed changes in chaperone activity in the sedimentation assay (Fig. 5B) that are consistent with the results of the turbidity assay. Nevertheless, a smaller mutational defect was observed in the sedimentation assay compared with the turbidity assay (cf. Fig. 5B versus 4D). Control experiments with a number of mutants for which this discrepancy is most pronounced revealed two major contributing factors: (i) the higher protein concentration used in the sedimentation than the turbidity assay, which provided a more favorable binding equilibrium for cpSRP43 variants with binding defects (Fig. 5C); (ii) even at the same protein concentrations, the sedimentation assay showed a smaller mutational defect compared with the light scattering assay (Fig. 5, D and E). This is likely due to less accurate quantification in Western blot analysis, especially for mutants with large defects (the band in either the soluble or pellet fraction is outside the linear range of quantification). Thus, the sedimentation assay corroborated the defects of many cpSRP43 variants in chaperone activity, but the light scattering assay allowed a more sensitive and accurate detection of mutational defects.
Figure 5.

Analysis of the chaperone activity of mutant cpSRP43 using the sedimentation assay and comparison with the results of light scattering assay. A, representative Coomassie-stained gels for analysis of the chaperone activity of cpSRP43 mutants using the sedimentation assay. C denotes lanes with cpSRP43 only; S denotes the soluble fraction; P denotes pellet. B, summary of the relative chaperone activity of all cpSRP43 mutants measured by the sedimentation assay. Values are reported relative to Cys-less cpSRP43 (WT). C, comparison of the chaperone activity of cpSRP43 mutants measured at high (green) and low (blue) protein concentrations using the light scattering assay. D, representative Western blot images for analysis of the chaperone activity of cpSRP43 mutants using the sedimentation assay at low protein concentrations. C denotes lanes with cpSRP43 only; S denotes the soluble fraction; P denotes pellet. LHCP denotes controls where indicated concentrations of purified LHCP were loaded to assess the dynamic range of Western blotting. E, comparison of the chaperone activity of cpSRP43 variants measured by the sedimentation (red; data from D and replicates) and light scattering (blue) assays at the same concentration. Data were reported as mean ± S.E., with n = 2–9.
Previous work showed that the ability of cpSRP43 to bind LHCP and generate a soluble transit complex is integral to the subsequent targeting and insertion of LHCP (10–14, 37); this was the case for both in vitro translated as well as chemically denatured LHCP (21, 38). To further assess the relationship between the ability of cpSRP43 to solubilize LHCP and the efficiency of LHCP targeting and integration, we measured and compared the two activities in parallel. LHCP unfolded in 8 m urea were either prevented from aggregation by dilution into a solution containing cpSRP43 (Fig. 6A, lane 1), or allowed to aggregate by dilution into buffer (Fig. 6A, lane 2). As cpSRP43 is also able to reverse LHCP aggregation (21, 26, 39), preformed LHCP aggregates were also incubated with increasing concentrations of cpSRP to allow re-solubilization by the chaperone (Fig. 6A, lanes 3–5). These samples were tested for the extent of LHCP solubilization (by the sedimentation assay) and for the efficiency of LHCP integration into thylakoid membrane (Fig. 6). Successful integration leads to protection of LHCP from thermolysin, giving rise to two protease-protected bands (Fig. 6A, DP1 and DP2). We observed a strong correlation between the degree of LHCP solubilization by cpSRP43 and the efficiency of LHCP insertion (Fig. 6B). As a constant amount of targeting factors (cpSRP43, cpSRP54, and cpFtsY) was present in all the integration reactions regardless of the conditions of pre-incubation, the observed differences in LHCP integration efficiency do not arise from the differences in the concentration of targeting factors, but rather from differences in the conformation of LHCP prior to initiation of the integration reaction. Together with previous work (10–14, 37), these results strongly suggest that the ability of cpSRP43 to solubilize LHCP is required for its proper targeting and integration into the thylakoid membrane.
Figure 6.
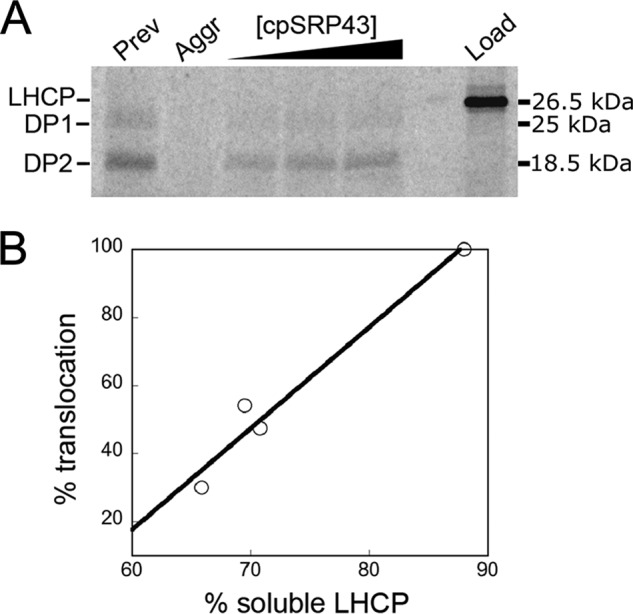
Solubilization of LHCP by cpSRP43 correlates with LHCP targeting and integration. A, [35S]methionine-labeled LHCP (lane 6, Load) were preincubated under different conditions with cpSRP43 and tested for targeting and insertion into thylakoid membrane in the presence of 3 μm cpSRP43, cpSRP54, and cpFtsY. Lanes 1 and 2, 2 μl of 35S-LHCP in 8 m urea was added to 40 μl of Buffer D with (lane 1) or without (lane 2) 3 μm cpSRP43/54 and incubated for 60 min. Lanes 3–5, 2 μl of 35S-LHCP in 8 m urea was added to 33.6 μl of Buffer D and allowed to aggregate at room temperature for 60 s, followed by addition of an equimolar ratio of cpSRP43/54 to final concentrations of 5, 15, and 30 μm in a final volume of 40 μl. 20 μl of the preincubated sample was used for the LHCP integration assay. DP1 and DP2 (25 and 18.5 kDa) are the protease-protected fragments of integrated LHCP (51). The remaining 20 μl was subjected to the sedimentation assay as described under “Experimental procedures,” except that LHCP bands were quantified by autoradiography using Storm 840 (Molecular Dynamics) and ImageQuant (GE Healthcare). Details of the LHCP integration assay are described under “Experimental procedures.” B, correlation of the translocation efficiency of LHCP with the degree to which LHCP is solubilized by cpSRP43.
Two distinct classes of defective cpSRP43 SBD mutants
The large number of surface residues that exhibit a mutational defect in substrate binding could arise from an extensive interaction surface of cpSRP43 with LHCP, or from perturbations of the global conformation of the SBD by the mutations. Recent NMR studies showed that the SBD of apo-cpSRP43 intrinsically samples active and inactive conformations with equal probability (22), supporting the possibility that the activity of cpSRP43 could be susceptible to mutations that shift the conformational equilibrium. To control for mutational effects on the global conformation of the SBD, we tested the ability of cpSRP43 mutants to bind the L18 recognition motif of LHCP. All the chaperone-defective mutations examined here are located away from the crystallographically identified L18-binding site of cpSRP43 (Tyr-204 highlighted in Fig. 8 below (25)); thus, a defect in L18 binding caused by these mutations most likely arises from a global structural defect of the SBD, rather than disruption of a direct interaction with L18.
The binding affinity of cpSRP43 for L18 was measured based on the cpSRP43-induced increase in the fluorescence anisotropy of a HiLyte-Fluor 488-conjugated L11 peptide, which represents the minimal sequence in L18 required for high affinity binding to cpSRP43 (26, 29). Representative equilibrium titrations for L11–cpSRP43 binding are shown in Fig. 7, A and B. The equilibrium dissociation constants (Kd) for L11 binding to WT and mutant cpSRP43's derived from the equilibrium titrations are summarized in Table 2. The anisotropy change of L11 induced by a subsaturating concentration (0.19 μm) of each mutant cpSRP43 relative to that of WT cpSRP43 are summarized in Fig. 7C.
Figure 7.
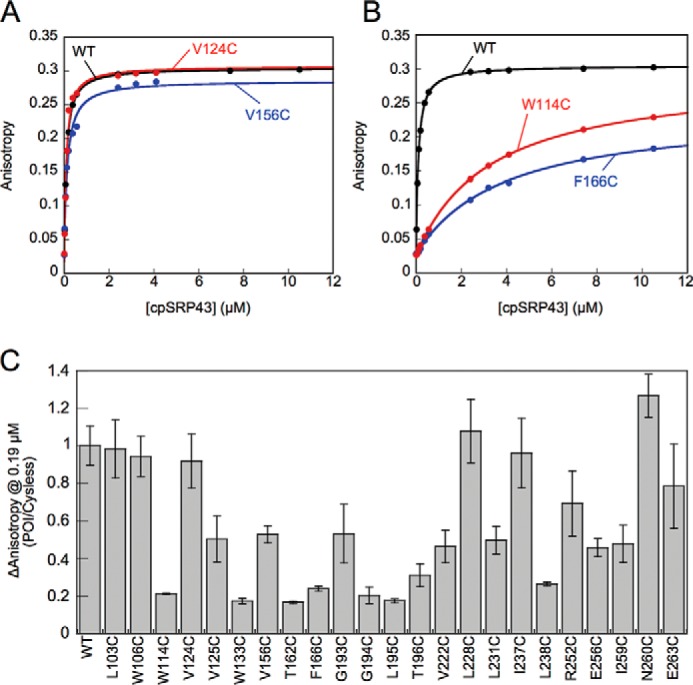
Characterization of the interaction of mutant cpSRP43s with the L18 motif. A and B, representative equilibrium titrations for the binding of WT and mutant cpSRP43s to HiLyte-Fluor 488-labeled L11. Representative data for cpSRP43 mutants that can bind L11 with high affinity are shown in A, and those for mutants exhibiting weakened L11 binding are shown in B. C, summary of the cpSRP43-induced changes in the fluorescence anisotropy of L11 at 0.19 μm, which is subsaturating for binding of Cys-less cpSRP43 to L11. The data for all mutants are normalized to that of Cys-less cpSRP43 (denoted as WT). All data are reported as mean ± S.E., with n ≥ 2.
Table 2.
Kd values for binding of HiLyte-conjugated L11 to individual cpSRP43 mutants
All cpSRP43 mutants shown in this table are derived from Cys-less cpSRP43 (denoted as WT). Green highlights mutants that exhibit Kd values within 3-fold of WT cpSRP43; yellow highlights mutants exhibiting 3–5-fold defects in L11 binding; and red highlights mutants that are severely defective in L11 binding.
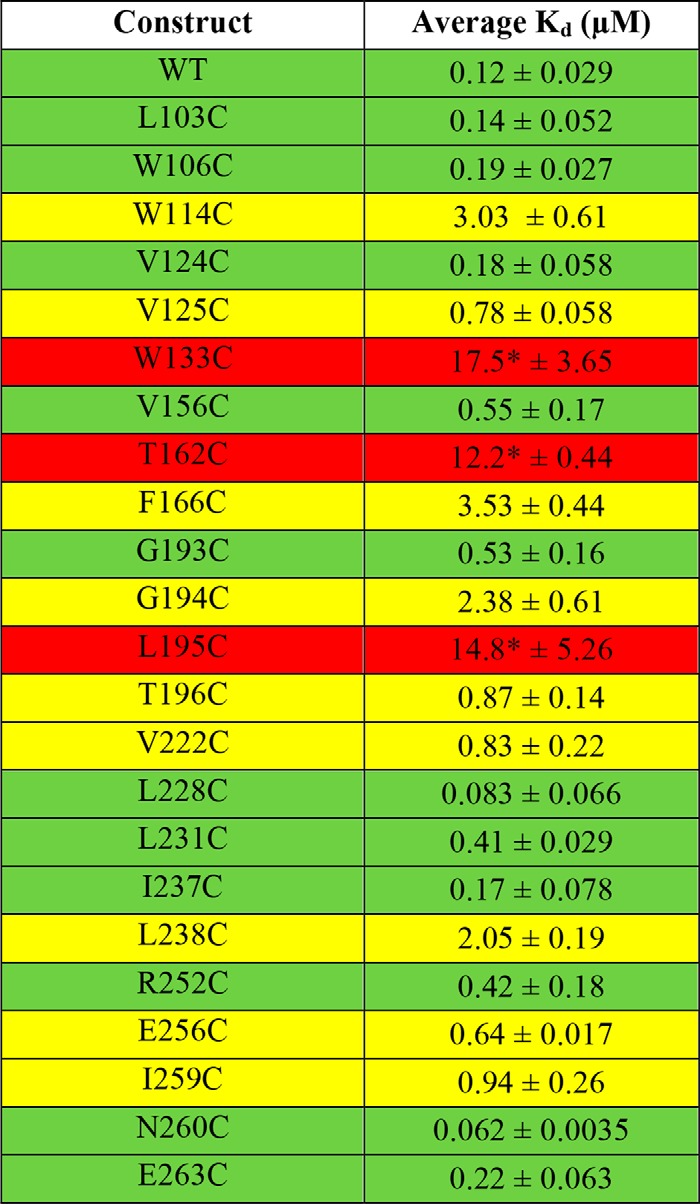
* indicates that saturation could not be reached with the mutant during equilibrium titrations, and their Kd values for L11 were estimated assuming the same end point in the titration curve as cys-less cpSRP43.
We found that mutation of a large number of residues affected the interaction of cpSRP43 with the L18 motif. Eight mutants bound the L11 peptide an order of magnitude more weakly than WT cpSRP43 (Kd ∼ 0.8–3.5 μm; Fig. 7B and Table 2, yellow), and three mutants exhibited ∼100-fold weakened binding to L11 (Kd >10 μm; Table 2, red). In contrast, 11 mutant cpSRP43s bound the L11 peptide with Kd values within 5-fold of that of WT cpSRP43 (Kd <0.6 μm; Fig. 7A and Table 2, green). We designated 10 of these mutants as Class I: they either bind L11 with similar affinity compared with WT cpSRP43 but are defective in chaperoning LHCP (L103C, W106C, V124C, L228C, I237C, N260C, and E263C), or the modest reductions in L11 binding observed with these mutants were insufficient to account for their complete loss of chaperone activity toward LHCP (V156C, G193C, L231C). Thus, Class I mutants specifically disrupt the ability of cpSRP43 to protect LHCP from aggregation. The remainder of the chaperone-defective mutants were designated as Class II. Although a direct involvement in TMD binding cannot be excluded, much of the defects of these mutants can be attributed to disruptions in interaction with the L18 motif. Because these mutations are located away from the vicinity of the L18-binding site, Class II mutants disrupt L18 binding allosterically by altering the conformation of the SBD.
Discussion
cpSRP43 is a small, ATP-independent chaperone with an SBD comprised mostly of ankyrin repeat motifs. At a size of 25 kDa, the cpSRP43 SBD is able to effectively chaperone multipass membrane protein substrates comparable with its own size, providing an attractive system to understand how a small protein scaffold interacts with and provides protection for large client proteins. Previous understanding of the cpSRP43–LHCP interaction was limited to recognition of the L18 loop sequence in LHCP by cpSRP43–Tyr-204 (21, 25, 26, 37). In this work, the results of alkylation-protection and cross-linking experiments showed that LHCP interacts more extensively with cpSRP43; the regions of interaction and/or cpSRP43-induced protection span all three TMDs of LHCP as well as the TM1–TM2 loop and the C terminus. Furthermore, site-directed mutagenesis of the cpSRP43 SBD identified two classes of mutant cpSRP43's: Class I, which disrupts cpSRP43's ability to protect LHCP from aggregation without affecting high-affinity recognition of L18; and Class II, which allosterically disrupts binding of the L18 motif. Together, these results provide evidence for much more extensive cpSRP43–client interactions than previously recognized, and suggest potential sites of cpSRP43 that bind and protect the TMDs of LHCP.
When mapped onto the crystal structure of the cpSRP43 SBD, the two classes of mutants are enriched in different regions of the cpSRP43 SBD, suggesting that different surfaces in the SBD mediate distinct functions. The residues that give rise to Class II mutants are primarily located on the helices in the ankyrin repeat motifs (Fig. 8B, magenta). As the sites of Class II mutations are away from the previously identified L18-binding site (Tyr-204; highlighted in blue), we attribute their defects to disruption of the active conformation of the cpSRP43 SBD (see the next paragraph). In contrast, residues that give rise to Class I mutants are enriched in the bridging helix, the β-hairpins in the ankyrin repeat motifs, and a hydrophobic surface in CD1 (Fig. 8A, orange), suggesting that these regions may either form or are in close vicinity of the TMD-binding sites in cpSRP43. In support of this notion, Class I mutations cluster on or near major hydrophobic surfaces on the cpSRP43 SBD (Fig. 8C). This model is also consistent with the general structural and functional features of ankyrin repeat proteins, which are formed by individual repeats of helix-loop-helix folds connected by β-hairpins. Structural, computational, and protein engineering studies showed that intra- and inter-repeat interactions between the helices allow ankyrin repeat proteins to cooperatively fold into concave L-shaped structures; in contrast, the loops and β-hairpins, which project outward from the helices, often form the recognition site for interaction partners (40–43). We therefore propose that client recognition by cpSRP43 may occur analogously, with the L18 sequence specifically recognized by the loop in Ank3, whereas the TMDs in LHCP are bound and protected by the hydrophobic surfaces on Ank4 and BH, and on or near the β-hairpins.
The large number of residues in the cpSRP43 SBD, at which a single conservative mutation away from the direct L18 interaction site severely disrupts substrate binding and chaperone activity, is extraordinary. This behavior is characteristic of molecular systems that sample inactive conformations with a high probability, such that small perturbations are sufficient to drive the molecule or complex into the inactive state (44–47). Likewise, the sensitivity of cpSRP43 to point mutations suggests that its SBD is at the threshold of a cooperative conformational change required to attain a chaperone-active conformation. This model is consistent with recent NMR data that detected distinct conformational states in the cpSRP43 SBD that are equally populated in apo-cpSRP43 (22). As observed previously, this property of cpSRP43 may be particularly useful in enabling regulation, allowing cpSRP43 to be readily switched “on” and “off” by its regulators in the stroma and at the target membrane, respectively (22). The precise nature of the conformational changes in cpSRP43 remains an important question for future investigations.
Experimental procedures
Protein expression and purification
Single cysteine mutants of Lhcb5 and cpSRP43 were constructed using the QuikChange Mutagenesis procedure (Stratagene) according to the manufacturer's instructions. WT and mutant cpSRP43, LHCP, and WT and mutant Lhcb5 were overexpressed and purified as previously described (20, 21).
Alkylation
Single-cysteine mutants of Lhcb5 solubilized in 8 m urea was treated with 4 mm tris(2-carboxyethyl)phosphine overnight. Lhcb5 was rapidly diluted to a final concentration of 1 μm in Buffer D (200 mm NaCl, 50 mm KHEPES, pH 7.5) containing 4 μm cpSRP43 and incubated for 10 min to allow complex formation. The sample was centrifuged at 13,000 rpm for 30 min to remove aggregated proteins, and the soluble fraction was subject to NEM alkylation as described (29). Briefly, the pre-formed Lhcb5·cpSRP43 complex was incubated with 100 μm NEM for 2 or 10 min and quenched with 50 mm DTT. Quenched samples were treated with 0.2% formic acid and analyzed on an LC-MSD SL 1100 series (Agilent) using a 2.1 × 150-mm Zorbax 300SB-C3 column (Agilent) and a gradient consisting of 0.2% formic acid as solvent A and 0.2% formic acid in acetonitrile (89.8%) and methanol (10%) as solvent B. Intact masses were determined in the single quadrupole. Chemstation software (Agilent) was used to deconvolute the masses and quantify the proteins. The accessibility of each site (1 − protection) was calculated from the alkylation efficiency of each cysteine mutant observed in the cpSRP43·Lhcb5 complex after subtracting that of the same mutant denatured in 6 m GdmCl.
Photoinducible cross-linking
Amber stop codons were introduced into Lhcb5-coding plasmids at the indicated sites using the QuikChange mutagenesis procedure (Stratagene). To incorporate pBpa, Lhcb5 harboring the amber codon at specific positions were in vitro translated using an S30 translation extract coupled to amber suppression, as previously described (35), except that an evolved aminoacyl-tRNA synthetase specific for pBpa was used in place of that for coumarin (34). Translation reactions were incubated for 90 min at 30 °C in the presence or absence of 20 μm intein–cpSRP43. Cross-links were induced by exposure to 365 nm light for 2 h at 4 °C. 10-μl reactions were mixed with an equal volume of 2× SDS and 4 m urea and analyzed by SDS-PAGE. Gels with 35S-labeled Lhcb5 were visualized by autoradiography using a Typhoon scanner. Western blots with 1× Strep-tagged Lhcb5 were immunoblotted with either anti-Strep or anti-cpSRP43 antibodies and visualized on a LI-COR Odyssey imaging system at 800 nm. Bands were quantified using the GelAnalyzer software. Cross-linking efficiency was determined by the ratio of the high-molecular weight cross-linked Lhcb5 bands to total Lhcb5 after subtracting the cpSRP43-independent cross-links in the corresponding area in control reactions lacking cpSRP43.
To purify cpSRP43·Lhcb5 after cross-linking, translation reactions were scaled up to 1 ml and incubated for 60 min at 30 °C in the presence or absence of 5 μm intein–cpSRP43. Cross-links were induced by exposure to 365 nm light for 45 min at 4 °C. After centrifugation at 16,000 × g for 15 min, the supernatant was loaded onto 100 μl of Strep-Tactin resin pre-equilibrated with Strep Wash Buffer (20 mm HEPES, 500 mm NaCl, pH 7.5), or 100 μl of Ni-NTA resin pre-equilibrated with Ni-NTA Wash Buffer (20 mm HEPES, 300 mm KCl, 20 mm imidazole, pH 7.5). The resin was washed five times with 500 μl of Strep or Ni-NTA Wash Buffer and eluted with 500 μl of Strep Elution Buffer (20 mm HEPES, 500 mm NaCl, 20 mm Biotin, pH 7.5) or Ni-NTA Elution Buffer (20 mm HEPES, 300 mm KCl, 200 mm imidazole, pH 7.5). The final wash and elution fractions from Strep-Tactin purification were immunoblotted with anti-cpSRP43 antibody as previously described. The wash and elution fractions from Ni-NTA purification were stained with Coomassie Blue and sent for MS analysis.
Mass spectrometry analysis of cpSRP43·Lhcb5 cross-links
Samples were separated by SDS-PAGE using a 4–12% gradient NuPAGE gel (Invitrogen) with NuPAGE MES running buffer (Invitrogen) for 35 min at 200 V. The gel was stained with colloidal Coomassie stain (Invitrogen) and de-stained with water and ammonium bicarbonate. The desired bands were excised and digested by trypsin overnight at 37 °C. Digested peptides were extracted from gel and lyophilized. Digested samples were subjected to LC-MS/MS analysis on a nanoflow LC system, EASY-nLC 1200 (Thermo Fisher Scientific) coupled to a QExactive HF Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with a Nanospray Flex ion source. Samples were directly loaded onto a 20 cm × 50-μm ID PicoFrit column (New Objective, Woburn, MA) packed in house with ReproSil-Pur C18AQ 1.9 μm resin (120A° pore size, Dr. Maisch, Ammerbuch, Germany) and heated to 60 °C. Peptides were separated with the following gradient at a flow rate of 220 nl/min: 2–6% Solvent B (3 min), 6–25% B (40 min), 25–40% B (17 min), and 100% B (9 min). Solvent A consisted of 97.8% H2O, 2% acetonitrile, and 0.2% formic acid, and solvent B consisted of 19.8% H2O, 80% acetonitrile, and 0.2% formic acid. The QExactive HF Orbitrap was operated in data-dependent mode with the Tune (version 2.7 SP1build 2659) instrument control software. Spray voltage was set to 2.5 kV, S-lens RF level at 50, and heated capillary at 275 °C.
Raw files were searched against the protein sequences using MaxQuant (48, 49) (version 1.6.0.16) assuming trypsin digestion with up to two missed cleavages. Precursor mass tolerances were less than 4.5 ppm after mass recalibration and fragment ion tolerances were 20 ppm. Protein abundances were estimated by iBAQ (50).
Chaperone activity of cpSRP43
The ability of cpSRP43 to prevent LHCP aggregation was measured as described (21). cpSRP43 were ultracentrifuged in a TLA-100 rotor (Beckman Coulter) at 100,000 rpm for 30 min at 4 °C prior to the experiment. Light scattering experiments were performed by addition of 3 μl of 50 μm LHCP denatured in 8 m urea to 150 μl of buffer D (50 mm KHEPES, pH 7.5, 200 mm NaCl) or 2.5 μm cpSRP43 in buffer D. Assays at higher protein concentrations were performed by addition of 6 μl of 125 μm LHCP to 150 μl of 15 μm cpSRP43. Light scattering was monitored at 360 nm on a UV-visible spectrometer (Beckman Coulter) over time until equilibrium was reached. The percentage of soluble LHCP (% soluble) at 300 s was calculated from Equation 1,
| (Eq. 1) |
in which A0 and Aobsd are the optical density readings in the absence and presence of cpSRP43, respectively. cpSRP43 itself does not contribute to the optical density reading (21).
Sedimentation experiments were performed by addition of 1 μl of 100 μm LHCP denatured in 8 m urea to 19 μl of 15 μm cpSRP43 in buffer D. Sedimentation assays at lower concentrations were performed by adding 1 μl of 20 μm LHCP to 19 μl of 5 μm cpSRP43. The mixtures were incubated at room temperature for 30 min, and the soluble and pellet fractions were separated by centrifugation at 18,000 × g for 30 min at 4 °C. The pellet was resuspended in 8 m urea, and both the pellet and soluble fractions were analyzed by SDS-PAGE. Gels were stained with Coomassie Blue (when using 5 μm LHCP) or Western blotting using an anti-LHCP antibody (when using 1 μm LHCP), and were imaged on a LI-COR Odyssey imaging system at 700 nm (for Coomassie stain) or 800 nm (for Western blots). The intensity of LHCP bands was quantified using the GelAnalyzer software.
Measurement of L18 binding
The L18 binding affinity of cpSRP43 was measured using L11 (GSFDPLGLADD), the minimal binding motif in L18, conjugated to HiLyte-Fluor 488. Anisotropy measurements were conducted in Buffer D on a Fluorolog 3–22 spectrofluorometer (Jobin Yvon), using 100 nm HiLyte-Fluor 488-labeled L11 and varying concentrations of cpSRP43. Samples were excited at 500 nm and fluorescence anisotropy was recorded at 527 nm, as previously described (21, 22). The data were fit to Equation 2,
| (Eq. 2) |
in which [pro] is cpSRP43 concentration, Aobsd is the observed anisotropy value, A0 is the anisotropy value without cpSRP43, ΔA is the change in anisotropy at saturating cpSRP43 concentrations, and Kd is the equilibrium dissociation constant for the interaction of cpSRP43 with L11–HiLyte-Fluor 488.
LHCP integration assay
Thylakoids were collected from chloroplasts of 9–12-day-old pea leaves (Laxton Progressive 9) hypotonically lysed in lysis buffer (10 mm KHEPES, pH 7.5, 10 mm MgCl2) as described (51). Thylakoids were further salt-washed and resuspended to a concentration of 1 mg/ml of chlorophyll (1 time). Each 150-μl LHCP integration reaction contained 20 μl of [35S]methionine-labeled LHCP preincubated with cpSRP43 (generated as described in legends to Fig. 6), 50 ml of 1× salt-washed thylakoid, 2 mm ATP, 2 mm GTP, 3 μm cpFtsY, and cpSRP43/54 supplemented to a final concentration of 3 μm. Integration reactions were incubated at 25 °C for 30 min and quenched on ice. The reaction mixtures were thermolysin-treated for 40 min and centrifuged to isolate the thylakoid membrane as described (51). The resulting pellets were resuspended in 2× SDS and analyzed by SDS-PAGE and quantified using Storm 840 (Molecular Dynamics) and ImageQuant (GE Healthcare).
Author contributions
C. Z. M., A. S., S. P., E. M., T. N., A. M., M. J. S., and S. H. data curation; C. Z. M., A. S., S. P., E. M., M. Y., T. N., A. M., S. H., and S.-o. S. formal analysis; C. Z. M., A. S., S. P., E. M., M. Y., T. N., and S. H. investigation; C. M. writing-original draft; A. S. and S.-o. S. validation; A. S., S. H., and S.-o. S. writing-review and editing; M. J. S. software; S. H. and S.-o. S. supervision; S.-o. S. conceptualization; S.-o. S. funding acquisition; S.-o. S. project administration.
Supplementary Material
Acknowledgments
We thank members of the Shan group for helpful comments on the manuscript. The Proteome Exploration Laboratory was supported by Betty and Gordon Moore Foundation Grant GBMF775 and the Beckman Institute at Caltech.
This work was supported by Betty and Gordon Moore Foundation Grant 94-3397785 and National Institutes of Health Grant R01 GM114390 (to S.-o. S.) and by National Institutes of Health Training Grant 2 T32 GM 7616-36 (to C. Z. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- LHCP
- chlorophyll a/b-binding protein
- cpSRP
- chloroplast signal recognition particle
- SBD
- substrate-binding domain
- BH
- bridging helix
- TMD
- transmembrane domain
- NEM
- N-ethylmaleimide
- GdmCl
- guanidinium chloride
- pBpa
- p-benzoyl-l-phenylalanine
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Randall L. L., and Hardy S. J. (2002) SecB, one small chaperone in the complex milieu of the cell. Cell. Mol. Life Sci. 59, 1617–1623 10.1007/PL00012488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walton T. A., Sandoval C. M., Fowler C. A., Pardi A., and Sousa M. C. (2009) The cavity chaperone Skp protects its substrates from aggregation but allows independent folding of substrate domains. Proc. Natl. Acad. Sci. U.S.A. 106, 1772–1777 10.1073/pnas.0809275106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Deshaies R. J., Koch B. D., Werner-Washburne M., Craig E. A., and Schekman R. (1988) 70 kD stress protein homologues facilitate translocation of secretory and mitochondrial precursor polypeptides. Nature 332, 800–805 10.1038/332800a0 [DOI] [PubMed] [Google Scholar]
- 4. Deshaies R. J., Koch B. D., and Schekman R. (1988) The role of stress proteins in membrane biogenesis. Trends Biochem. Sci. 13, 384–388 10.1016/0968-0004(88)90180-6 [DOI] [PubMed] [Google Scholar]
- 5. Thoma J., Burmann B. M., Hiller S., and Müller D. J. (2015) Impact of holdase chaperones Skp and SurA on the folding of β-barrel outer-membrane proteins. Nat. Struct. Mol. Biol. 22, 795–802 10.1038/nsmb.3087 [DOI] [PubMed] [Google Scholar]
- 6. Hennecke G., Nolte J., Volkmer-Engert R., Schneider-Mergener J., and Behrens S. (2005) The periplasmic chaperone SurA exploits two features characteristic of integral outer membrane proteins for selective substrate recognition. J. Biol. Chem. 280, 23540–23548 10.1074/jbc.M413742200 [DOI] [PubMed] [Google Scholar]
- 7. Mihara K., and Omura T. (1996) Cytoplasmic chaperones in precursor targeting to mitochondria: the role of MSF and hsp 70. Trends Cell Biol. 6, 104–108 10.1016/0962-8924(96)81000-2 [DOI] [PubMed] [Google Scholar]
- 8. Schunemann D. (2004) Structure and function of the chloroplast signal recognition particle. Curr. Genet. 44, 295–304 10.1007/s00294-003-0450-z [DOI] [PubMed] [Google Scholar]
- 9. Klimyuk V. I., Persello-Cartieaux F., Havaux M., Contard-David P., Schuenemann D., Meiherhoff K., Gouet P., Jones J. D., Hoffman N. E., and Nussaume L. (1999) A chromodomain protein encoded by the Arabidopsis CAO gene is a plant-specfic compoment of the chloroplast signal recognition particle pathway that is involved in LHCP targeting. Plant Cell 11, 87–99 10.1105/tpc.11.1.87,10.2307/3870840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li X., Henry R., Yuan J., Cline K., and Hoffman N. (1995) A chloroplast homologue of the signal recognition particle subunit SRP54 is involved in the posttranslational integration of a protein into thylakoid membranes. Proc. Natl. Acad. Sci. U.S.A. 92, 3789–3793 10.1073/pnas.92.9.3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schuenemann D., Gupta S., Persello-Cartieaux F., Klimyuk V. I., Jones J. D., Nussaume L., and Hoffman N. E. (1998) A novel signal recognition particle targets light-harvesting proteins to the thylakoid membranes. Proc. Natl. Acad. Sci. U.S.A. 95, 10312–10316 10.1073/pnas.95.17.10312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tu C. J., Peterson E. C., Henry R., and Hoffman N. E. (2000) The L18 domain of light-harvesting chlorophyll proteins binds to chloroplast signal recognition particle 43. J. Biol. Chem. 275, 13187–13190 10.1074/jbc.C000108200 [DOI] [PubMed] [Google Scholar]
- 13. Tu C.-J., Schuenemann D., and Hoffman N. E. (1999) Chloroplast FtsY, chloroplast signal recognition particle, and GTP are required to reconstitute the soluble phase of light-harvesting chlorophyll protein transport into thylakoid membranes. J. Biol. Chem. 274, 27219–27224 10.1074/jbc.274.38.27219 [DOI] [PubMed] [Google Scholar]
- 14. Eichacker L. A., and Henry R. (2001) Function of a chloroplast SRP in thylakoid protein export. Biochim. Biophys. Acta 1541, 120–134 10.1016/S0167-4889(01)00151-3 [DOI] [PubMed] [Google Scholar]
- 15. Kuhn A., Stuart R., Henry R., and Dalbey R. E. (2003) The Alb3/Oxa1/YidC protein family: membrane-localized chaperones facilitating membrane protein insertion? Trends Cell Biol. 13, 510–516 10.1016/j.tcb.2003.08.005 [DOI] [PubMed] [Google Scholar]
- 16. Lewis N. E., Marty N. J., Kathir K. M., Rajalingam D., Kight A. D., Daily A., Kumar T. K., Henry R. L., and Goforth R. L. (2010) A dynamic cpSRP43-Albino3 interaction mediates translocase regulation of cpSRP targeting components. J. Biol. Chem. 285, 34220–34230 10.1074/jbc.M110.160093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moore M., Harrison M. S., Peterson E. C., and Henry R. (2000) Chloroplast Oxa1p homolog Albino3 is required for post-translational integration of the light harvesting chlorophyll-binding protein into thylakoid membranes. J. Biol. Chem. 275, 1529–1532 [DOI] [PubMed] [Google Scholar]
- 18. Dünschede B., Bals T., Funke S., and Schünemann D. (2011) Interaction studies between the chloroplast signal recognition particle subunit cpSRP43 and the full-length translocase Alb3 reveal a membrane-embedded binding region in Alb3. J. Biol. Chem. 286, 35187–35195 10.1074/jbc.M111.250746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Falk S., Ravaud S., Koch J., and Sinning I. (2010) The C-terminus of the Alb3 membrane insertase recruits cpSRP43 to the thylakoid membrane. J. Biol. Chem. 285, 5954–5962 10.1074/jbc.M109.084996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jaru-Ampornpan P., Chandrasekar S., and Shan S. (2007) Efficient interaction between two GTPases allows the chloroplast SRP pathway to bypass the requirement for an SRP RNA. Mol. Biol. Cell 18, 2636–2645 10.1091/mbc.E07-01-0037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jaru-Ampornpan P., Shen K., Lam V. Q., Ali M., Doniach S., Jia T. Z., and Shan S. (2010) ATP-independent reversal of a membrane protein aggregate by a chloroplast SRP subunit. Nat. Struct. Mol. Biol. 17, 696–702 10.1038/nsmb.1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liang F. C., Kroon G., McAvoy C. Z., Chi C., Wright P. E., and Shan S. O. (2016) Conformational dynamics of a membrane protein chaperone enables spatially regulated substrate capture and release. Proc. Natl. Acad. Sci. U.S.A. 113, E1615–E1624 10.1073/pnas.1524777113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao F., Kight A. D., Henderson R., Jayanthi S., Patel P., Murchison M., Sharma P., Goforth R. L., Kumar T. K., Henry R. L., and Heyes C. D. (2015) Regulation of structural dynamics within a signal recognition particle promotes binding of protein targeting substrates. J. Biol. Chem. 290, 15462–15474 10.1074/jbc.M114.624346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Falk S., and Sinning I. (2010) The C terminus of Alb3 interacts with the chromodomains 2 and 3 of cpSRP43. J. Biol. Chem. 285, le25–le26; author reply le26–le28 10.1074/jbc.L110.160093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stengel K. F., Holdermann I., Cain P., Robinson C., Wild K., and Sinning I. (2008) Structural basis for specific substrate recognition by the chloroplast signal recognition particle protein cpSRP43. Science 321, 253–256 10.1126/science.1158640 [DOI] [PubMed] [Google Scholar]
- 26. Jaru-Ampornpan P., Liang F. C., Nisthal A., Nguyen T. X., Wang P., Shen K., Mayo S. L., and Shan S. O. (2013) Mechanism of an ATP-independent protein disaggregase: II. distinct molecular interactions drive multiple steps during aggregate disassembly. J. Biol. Chem. 288, 13431–13445 10.1074/jbc.M113.462861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cain P., Holdermann I., Sinning I., Johnson A. E., and Robinson C. (2011) Binding of chloroplast signal recognition particle to a thylakoid membrane protein substrate in aqueous solution and delineation of the cpSRP43-substrate interaction domain. Biochem. J. 437, 149–155 10.1042/BJ20110270 [DOI] [PubMed] [Google Scholar]
- 28. Wang P., Liang F. C., Wittmann D., Siegel A., Shan S. O., and Grimm B. (2018) Chloroplast SRP43 acts as a chaperone for glutamyl-tRNA reductase, the rate-limiting enzyme in tetrapyrrole biosynthesis. Proc. Natl. Acad. Sci. U.S.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nguyen T. X., Jaru-Ampornpan P., Lam V. Q., Cao P., Piszkiewicz S., Hess S., and Shan S. (2013) Mechanism of an ATP-independent protein disaggregase: I. structure of a membrane protein aggregate reveals a mechanism of recognition by its chaperone. J. Biol. Chem. 288, 13420–13430 10.1074/jbc.M113.462812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Horn R., and Paulsen H. (2002) Folding in vitro of light-harvesting chlorophyll a/b protein is coupled with pigment binding. J. Mol. Biol. 318, 547–556 10.1016/S0022-2836(02)00115-8 [DOI] [PubMed] [Google Scholar]
- 31. Paulsen H., Finkenzeller B., and Kühlein N. (1993) Pigments induce folding of light-harvesting chlorophyll a/b-binding protein. Eur. J. Biochem. 215, 809–816 10.1111/j.1432-1033.1993.tb18096.x [DOI] [PubMed] [Google Scholar]
- 32. Reinsberg D., Booth P. J., Jegerschöld C., Khoo B. J., and Paulsen H. (2000) Folding, assembly, and stability of the major light-harvesting complex of higher plants, LHCII, in the presence of native lipids. Biochemistry 39, 14305–14313 10.1021/bi001365z [DOI] [PubMed] [Google Scholar]
- 33. Ryu Y., and Schultz P. G. (2006) Efficient incorporation of unnatural amino acids into proteins in Escherichia coli. Nat. Methods 3, 263–265 10.1038/nmeth864 [DOI] [PubMed] [Google Scholar]
- 34. Young T. S., Ahmad I., Yin J. A., and Schultz P. G. (2010) An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374 10.1016/j.jmb.2009.10.030 [DOI] [PubMed] [Google Scholar]
- 35. Saraogi I., Zhang D., Chandrasekaran S., and Shan S. (2011) Site-specific fluorescent labeling of nascent proteins on the translating ribosome. J. Am. Chem. Soc. 133, 14936–14939 10.1021/ja206626g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wittelsberger A., Mierke D. F., and Rosenblatt M. (2008) Mapping ligand-receptor interfaces: approaching the resolution limit of benzophenone-based photoaffinity scanning. Chem. Biol. Drug Des. 71, 380–383 10.1111/j.1747-0285.2008.00646.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jonas-Straube E., Hutin C., Hoffman N. E., and Schünemann D. (2001) Functional analysis of the protein-interacting domains of chloroplast SRP43. J. Biol. Chem. 276, 24654–24660 10.1074/jbc.M100153200 [DOI] [PubMed] [Google Scholar]
- 38. Yuan J., Kight A., Goforth R. L., Moore M., Peterson E. C., Sakon J., and Henry R. (2002) ATP stimulates signal recognition particle (SRP)/FtsY-supported protein integration in chloroplasts. J. Biol. Chem. 277, 32400–32404 10.1074/jbc.M206192200 [DOI] [PubMed] [Google Scholar]
- 39. Falk S., and Sinning I. (2010) cpSRP43 is a novel chaperone specific for light-harvesting chlorophyll a,b-binding proteins. J. Biol. Chem. 285, 21655–21661 10.1074/jbc.C110.132746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Barrick D., Ferreiro D. U., and Komives E. A. (2008) Folding landscapes of ankyrin repeat proteins: experiments meet theory. Curr. Opin. Struct. Biol. 18, 27–34 10.1016/j.sbi.2007.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Binz H. K., Stumpp M. T., Forrer P., Amstutz P., and Plückthun A. (2003) Designing repeat proteins: well-expressed, soluble, and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 332, 489–503 10.1016/S0022-2836(03)00896-9 [DOI] [PubMed] [Google Scholar]
- 42. Binz H. K., Amstutz P., Kohl A., Stumpp M. T., Briand C., Forrer P., et al. (2004) High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 22, 575–582 10.1038/nbt962 [DOI] [PubMed] [Google Scholar]
- 43. Zahnd C., Wyler E., Schwenk J. M., Steiner D., Lawrence M. C., McKern N. M., Pecorari F., Ward C. W., Joos T. O., and Plückthun A. (2007) A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J. Mol. Biol. 369, 1015–1028 10.1016/j.jmb.2007.03.028 [DOI] [PubMed] [Google Scholar]
- 44. Peracchi A., Karpeisky A., Maloney L., Beigelman L., and Herschlag D. (1998) A core folding model for catalysis by the hammerhead ribozyme accounts for its extraordinary sensitivity to abasic mutations. Biochemistry 37, 14765–14775 10.1021/bi980867y [DOI] [PubMed] [Google Scholar]
- 45. Peracchi A., Matulic-Adamic J., Wang S., Beigelman L., and Herschlag D. (1998) Structure-function relationships in the hammerhead ribozyme probed by base rescue. RNA 4, 1332–1346 10.1017/S1355838298980979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marcos E., Crehuet R., and Bahar I. (2010) On the conservation of the slow conformational dynamics within the amino acid kinase family: NAGK the paradigm. PLoS Comput. Biol. 6, e1000738 10.1371/journal.pcbi.1000738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kikani C. K., Antonysamy S. A., Bonanno J. B., Romero R., Zhang F. F., Russell M., Gheyi T., Iizuka M., Emtage S., Sauder J. M., Turk B. E., Burley S. K., and Rutter J. (2010) Structural bases of PAS domain-regulated kinase (PASK) activation in the absence of activation loop phosphorylation. J. Biol. Chem. 285, 41034–41043 10.1074/jbc.M110.157594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- 49. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 10.1021/pr101065j [DOI] [PubMed] [Google Scholar]
- 50. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- 51. Nguyen T. X., Chandrasekar S., Neher S., Walter P., and Shan S. O. (2011) Concerted complex assembly and GTPase activation in the chloroplast signal recognition particle. Biochemistry 50, 7208–7217 10.1021/bi200742a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.