

Abstract
The study of developmental trajectories is where epigenetics truly shines. The “epi” in epigenetics captures the fact that although epigenetic processes also preside over the maintenance and termination of gene expression, the unfolding and remodeling of chromatin architecture are especially critical to prepare genes for regulated transcription. These properties imply being on a path, a trajectory to events that will occur later on thanks to epigenetic programming. Thus, epigenetics is about timed and timely events. In this article we discuss epigenetic and genetic evidence from several independent studies of asthma, COPD and lung function which converge to highlight a potential role of the TGFB pathway in these processes. These results raise the possibility that at least in a subset of individuals, these conditions may be functionally connected in ways that need to be further defined, but that likely reflect the uniquely pleiotropic nature of TGFB pathway genes – particularly, their ability to control both lung development and immune responses essential for regulation and inflammation. Further characterization of this pathway in longitudinally phenotyped populations may unmask novel trajectories to lung disease that begin in utero and unfold into old age
The study of developmental trajectories is where epigenetics truly shines. The “epi” in epigenetics captures the fact that although epigenetic processes also preside over the maintenance and termination of gene expression, the unfolding and remodeling of chromatin architecture (the stuff epigenetics is made of) are especially critical to prepare genes for regulated transcription. Indeed, epigenetic studies typically (albeit not exclusively) focus on what needs to happen for a cell or a tissue to become “poised” to express a given set of genes – those genes are still silent, but they are getting ready to go. This “getting ready to go” implies being on a path, a trajectory to events that will occur later on thanks to epigenetic programming. Thus, epigenetics is very much about timed and timely events, and epigenetic processes are the (almost invisible) puppeteer behind gene transcription.
Epigenetic Predictors of Childhood Asthma at Birth
The elegant work on perfectly timed waves of epigenetic events that sweep through the Th2 cytokine locus in order to allow regulated expression of IL-5, IL-13 and IL-4 in T cells (1) eloquently illustrates the power of basic epigenetic research to highlight fundamental biological trajectories. Unfortunately, leveraging that power for population studies in the asthma and allergy field is difficult for both logistical and technical reasons. Most population studies are cross-sectional. Bringing epigenetics into such studies may provide interesting insight into the pathogenesis of concurrent disease but may also fall into the cause/effect trap (if epigenetic differences are detected between cases and controls, are they a cause or an effect of the disease?). Much preferable are longitudinal approaches that, relying on the unique ability of epigenetic processes to shape temporal trajectories, allow exploring the relationship between a given pattern of epigenetic remodeling (DNA methylation or histone modifications) in early life and the subsequent emergence of the phenotype of interest. Our recent work on childhood asthma epigenetics in the Infant Immune Study (IIS), an unselected birth cohort closely monitored for asthma for over 10 years, testified to the power of this approach by establishing a nexus between the immune epigenome at birth and asthma in the first decade of life. Indeed, we could show that neonates who developed asthma by 9 years of age carried DNA methylation profiles in their cord blood mononuclear cells that were distinct from those of neonates who did not become asthmatic (2).
Some of these results were especially significant. Our initial epigenome-wide analysis found almost 600 regions that were differentially methylated in IIS children who did and did not develop asthma by age 9 years. When we constructed networks to elucidate the functional connections among these differentially methylated genomic regions, we found that a region in the SMAD3 promoter was the most connected node within the network. Bisulfite sequencing of cord blood cell DNA from our IIS neonates and two replication cohorts (The Manchester Asthma and Allergy Study, and the Childhood Origins of ASThma Study) showed that SMAD3 promoter methylation at birth was selectively and significantly increased in asthmatic children of asthmatic mothers and was associated with childhood asthma risk. These findings provided the first evidence that the trajectory to childhood asthma may begin at birth with epigenetic modifications that cluster primarily in the SMAD3 pathway and are influenced by asthma in the child’s mother. It is noteworthy that SMAD3 is a well-replicated asthma-associated gene from genome-wide association studies (GWAS) (3, 4) and a master regulator of TGF-β-dependent signaling. In this capacity, SMAD3 is uniquely positioned to affect the trajectory to, and the pathogenesis of childhood asthma. Indeed, TGF-β controls the differentiation of Treg and Th17 cells; altered Treg and Th17 activities have been reported in childhood asthma (5) and conversely, maternal exposure to asthma-protective environments such as farming has been shown to activate the Treg compartment (6) and influence the expression of Th17 markers (7). On the other hand, SMAD3 and all three TGF-β isoforms are expressed at high levels during early lung development and are critical to ensure normal branching morphogenesis, epithelial cell differentiation and maturation of surfactant synthesis (8). The identification of SMAD3 as an epigenetic predictor of childhood asthma at birth suggests that the early epigenetic trajectory to asthma involves processes at the intersection between immune regulation and lung development.
Going One Step Backwards: Maternal Influences on Childhood Asthma Risk
The detection of asthma-associated differential epigenetic programming already at birth is consistent with the possibility that asthma might have its origins in utero, a hypothesis supported by the finding that maternal history of asthma is the strongest risk factor for asthma in the child (9). Further evidence for the in utero origins of asthma comes from associations of childhood asthma with other maternal prenatal conditions and exposures such as maternal age, smoking, infectious illness, stress, weight gain, and exposure to farm animals (10–12). To better understand maternal prenatal influences on the child’s trajectory to asthma, we recently relied again on the IIS population to explore the relation between maternal immune status during pregnancy and asthma risk in the child. We found that the maternal profiles of cytokine production during the third trimester of pregnancy predict asthma in the child. More specifically, there was a strong inverse association between maternal production of IFN-γ relative to IL-13 and IL-4 and asthma risk in the child’s first decade of life. Interestingly, this relationship was found for pre-natal but not post-natal maternal cytokine levels, was evident in children of non-asthmatic but not asthmatic mothers, and held for maternal but not paternal cytokines. Moreover, this relationship was independent of childhood allergy (13).
While, to our knowledge, our study provided the first identification of a maternal pre-natal immune indicator of subsequent asthma risk for children born to non-asthmatic mothers, the underlying mechanisms remained to be determined. Therefore, as a logical follow-up to our previous study of neonates (2), we asked whether the relationship between maternal immune profiles during pregnancy and childhood asthma involves the neonatal epigenome and if so, whether it differs in children of non-asthmatic and asthmatic mothers. We performed a genome-wide survey of DNA methylation in cord blood mononuclear cells from 32 IIS neonates (19 with non-asthmatic mothers, 13 with asthmatic mothers). The maternal prenatal immune profile was defined as the ratio of IFN-γ and IL-13 released in the supernatants of maternal mitogen-stimulated peripheral blood mononuclear cells isolated during the third trimester of pregnancy. Because of our previous findings (2), associations between the log10-transformed prenatal IFN-γ/IL-13 ratio and DNA methylation at individual neonatal CpG sites were assessed using linear regression separately in children of asthmatic and non-asthmatic mothers. We found that levels of DNA methylation at 553 CpG sites were significantly associated with the prenatal maternal cytokine ratio in children of non-asthmatic mothers, while no significant association was detected in children born to asthmatic mothers. Of particular interest was the finding that the neonatal CpG sites associated with the maternal IFN-γ/IL-13 ratio clustered predominantly in the retinoic acid receptor and aryl hydrocarbon receptor pathways and had TGFB1 as a prominent upstream regulator (14).
While not definitive, these data suggest that the pre-natal immune milieu influences epigenetic profiles at birth in ways that depend on maternal asthma status, and primarily affects genes involved in the response to environmental cues. Moreover, our results point to an intriguing convergence among the epigenetic pathways that link maternal immune status, neonatal methylome and development of asthma during childhood. Indeed, TGFB1 and SMAD3 are part of a single signaling pathway that regulates both lung development and immune responses, and in this capacity may modulate the inception and unfolding of the trajectory to childhood asthma. Finally, we note that RUNX3, another gene in the TGFB pathway, was also found to be differentially methylated in peripheral blood immune cells when comparing inner-city children with persistent atopic asthma and healthy controls (15).
Epigenetics, Genetics and the Link between childhood Asthma and Chronic Obstructive Pulmonary Disease (COPD)
The trajectory to lung disease may not end in early life and may not stop at asthma. More and more attention focuses on the possibility that COPD, a condition that currently claims more victims than any other disease except cancer and cardiovascular illnesses, may have its origins in early life thanks to its complex relationships with reduced lung function and childhood asthma (16). Children with persistent asthma have been consistently shown to reach a lower forced expiratory volume in 1 second (FEV1) plateau during the third decade of life than children without asthma (17). Only a fraction of children with asthma go on to have fixed airflow limitation, but those who do typically proceed through three phases (5, 16). Children with persistent asthma have a slightly but significantly lower maximal expiratory flow and respiratory system compliance shortly after birth than do those without asthma (18), suggesting that impaired lung growth in utero predisposes for the development of asthma. By the time they reach early school age, these children already have a large percentage of the deficits in FEV1 that they will show during the plateau phase of lung function (19), which suggests that the period between birth and age 6 years is critical for the development of airflow limitation in persistent asthma. Further declines in FEV1 occur during the school years (20) and in adult life as part of the natural history of asthma, but these declines seem to account for a much smaller fraction of the total impairment than those observed in early life (16). As a result of these deficits in FEV1 growth, by the third decade of life approximately 17% of all patients with mild or moderate asthma reach stage 1 COPD [post-bronchodilator FEV1/forced vital capacity (FVC) ratio <0.70 and FEV1 ≥80% of the predicted value], and 5% reach stage 2 COPD (post-bronchodilator FEV1/FVC ratio <0.70 and FEV1<80% of the predicted value). Overall, multi-decade follow ups in several cohorts around the world demonstrate that a history of asthma confers a 10-30 fold risk of COPD, with individuals who experience reduced maximum growth of FEV1 in early adulthood being at risk for early or more severe disease (21).
The possibility that, at least in some patients, asthma and COPD may represent temporal stages within a continuum of disease has led some to propose a new entity, the Asthma-COPD Overlap Syndrome (ACOS) (22). While the criteria to diagnose ACOS and even its existence as a separate entity are still debated (23, 24), the intertwining of asthma in childhood, deficits in lung function growth and COPD later in life is increasingly recognized. The underlying mechanisms are not understood. Airway biopsies from infants with recurrent wheezing show no evidence of airway remodeling, but incipient thickening of the reticular basement membrane in medium-size bronchi can be observed in preschool children with wheezing at approximately the age at which the largest deficits in airway growth appear in children with subsequent persistent asthma (16).
Not much is currently known about the role of altered airway development and immune function in these trajectories, but it is tempting to speculate that thanks to its unique positioning at the intersection of lung development and immune regulation, epigenetic (or genetic) modifications in the TGFB pathway may provide a mechanistic link between asthma in early life and COPD in later years. Unfortunately, because patients with a COPD phenotype are typically excluded from asthma cohorts, and few if any populations have been monitored long enough to cover the temporal unfolding of the childhood asthma-to-COPD trajectory, we cannot assess whether peculiar DNA methylation profiles characterize the neonatal epigenome in individuals who develop asthma in the first years of life and are diagnosed with COPD later on.
If epigenetics cannot currently illuminate the asthma-COPD trajectory, genetics might. COPD and lung function GWAS, which rely on large populations and well powered meta-analyses, are providing tantalizing clues for a contribution of the TGFB pathway to these phenotypes. In a 2011 study, TGFB2 was identified as a locus strongly associated with pulmonary function in a study of FEV1 and FEV1/FVC ratio in 48,201 individuals of European ancestry, with follow-up of top associations in 46,411 additional individuals (25). Soon thereafter, TGFB2 was found to be significantly associated with quantitative measures of emphysema and airway imaging phenotypes in the COPDGene, ECLIPSE (Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints), NETT (National Emphysema Treatment Trial), and GenKOLS (Genetics of COPD) studies, and with percentage gas trapping in COPDGene (26).
The year 2017 saw the publication of three distinct studies all converging on the TGFB pathway. The first one reported COPD-targeted analyses in 15,256 cases and 47,936 controls followed by replication in 9,498 cases and 9,748 controls from the International COPD Genetics Consortium, and identified 22 genome-wide significant loci, including TGFB2. Thirteen novel genome-wide significant loci emerged from these analyses, and one of them was RARB (retinoic acid receptor-β), also a member of the TGFB pathway (27) (Figure 1). The second study also focused on COPD and included 2,588 cases and 1,782 controls from four cohorts, combining results with existing data from 6,633 cases and 5,704 controls. The analysis of moderate-to-severe COPD confirmed signals in a number of previously identified loci, including TGFB2, and the analysis of severe COPD again found genome-wide significant associations for TGFB2. The locus was also found to be associated with lung function. A variant in TGFB2 (rs4846480) was also part of a 7-gene genetic risk score constructed using genome-wide significant single nucleotide polymorphisms associated with COPD. This risk score was associated with a 1.86% reduction in FEV1 percent predicted for each additional risk allele (28). Finally, a study of 48,943 individuals selected from the extremes of the lung function distribution in UK Biobank, with follow-up in 95,375 individuals, found 97 independent signals for lung function. These signals tended to cluster in pathways related to development, elastic fibers and epigenetic regulation, and included several genes (TGFB2, GLIS3, BMP2 and TGFBR3) specifically belonging to the TGFB pathway (29). Finally, a recent GWAS examined current or former smoking non-Hispanic whites with COPD. Overlap subjects reported a history of physician-diagnosed asthma before the age of 40 years. A variant in SOX5 (rs59569785), which also related to TGFB pathway genes, was found to be associated with COPD and asthma overlap (30) (Figure 1).
Figure 1. Genes associated with asthma, COPD and lung function within the TGFB signaling pathway.
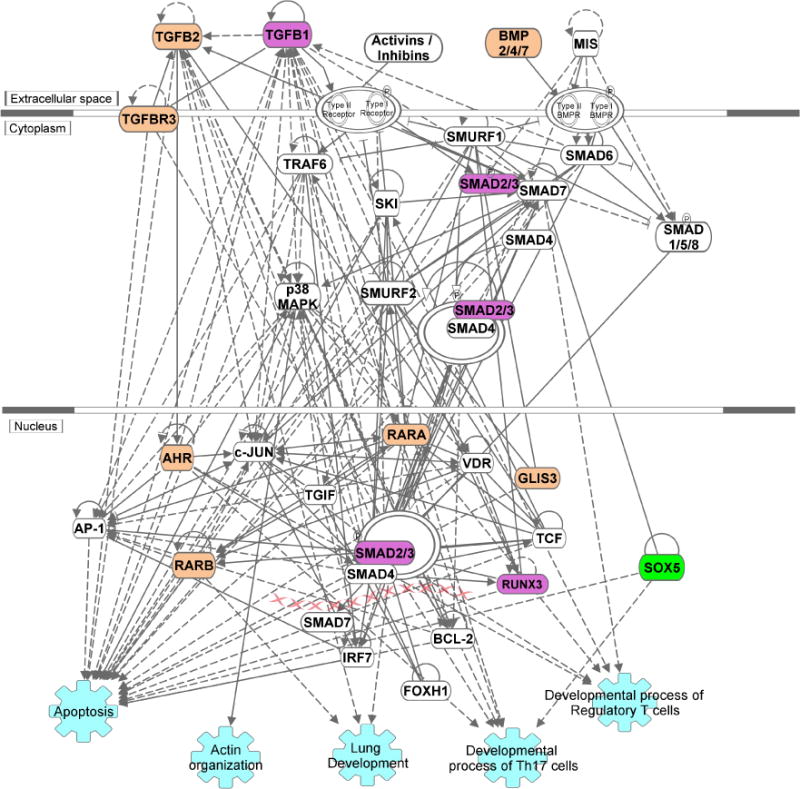
Ingenuity Pathway Analysis was used to depict the pathway and highlight the position of genes that are associated with COPD (tan) or asthma/COPD overlap (green) in GWAS and/or harbor asthma-associated differential methylation (purple). Selected biological functions (shown in light blue) were added to the network. Direct and indirect interactions are shown as solid and dashed lines, respectively. The primary data are found in Refs. 2, 14, 15, 25–30.
Conclusions
Unbiased epigenetic and genetic studies have the potential to identify biological pathways with a previously unrecognized role in underpinning the phenotypes of interest. The GWAS of asthma that pointed to genes in the epithelium/ILC2 axis (3) provides an eloquent case in point. In this context it is noteworthy that epigenetic and genetic evidence from several independent studies of asthma, COPD and lung function converges to highlight a potential role of the TGFB pathway in these processes. These results raise the possibility that at least in a subset of individuals, these conditions may be functionally connected in ways that need to be further defined, but that likely reflect the uniquely pleiotropic nature of TGFB pathway genes – particularly, their ability to control both lung development and immune responses essential for regulation and inflammation. Therefore, further characterization of this pathway in longitudinally phenotyped populations may unmask novel trajectories to lung disease that begin in utero and unfold into old age (Figure 2).
Figure 2.
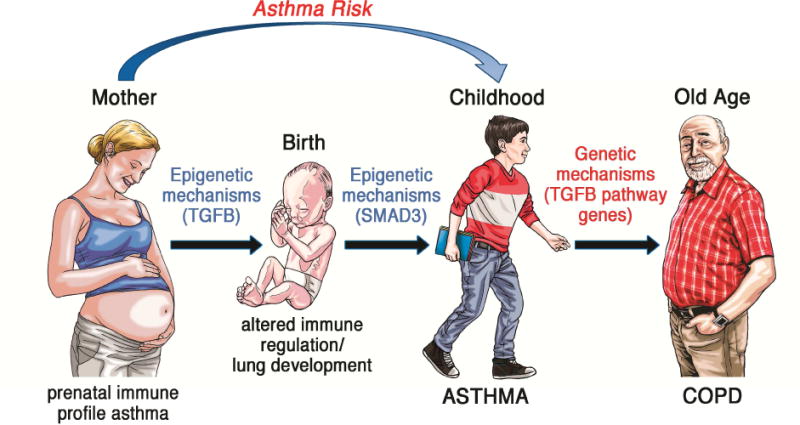
A potential trajectory to asthma and COPD that begins during gestation and involves the TGFB pathway and its members
Acknowledgments
This work was supported by a Pre-doctoral Fellowship from HL007249 T32 (to ADV) and R21AI133765 (to DV).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol. 2006;24:607–56. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- 2.DeVries A, Wlasiuk G, Miller SJ, Bosco A, Stern DA, Lohman IC, et al. Epigenome-wide analysis links SMAD3 methylation at birth to asthma in children of asthmatic mothers. J Allergy Clin Immunol. 2017;140(2):534–42. doi: 10.1016/j.jaci.2016.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211–21. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinds DA, McMahon G, Kiefer AK, Do CB, Eriksson N, Evans DM, et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat Genet. 2013;45(8):907–11. doi: 10.1038/ng.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez FD, Vercelli D. Asthma. Lancet. 2013;382(9901):1360–72. doi: 10.1016/S0140-6736(13)61536-6. [DOI] [PubMed] [Google Scholar]
- 6.Schaub B, Liu J, Hoppler S, Schleich I, Huehn J, Olek S, et al. Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J Allergy Clin Immunol. 2009;123(4):774–82. doi: 10.1016/j.jaci.2009.01.056. [DOI] [PubMed] [Google Scholar]
- 7.Lluis A, Ballenberger N, Illi S, Schieck M, Kabesch M, Illig T, et al. Regulation of TH17 markers early in life through maternal farm exposure. J Allergy Clin Immunol. 2014;133(3):864–71. doi: 10.1016/j.jaci.2013.09.030. [DOI] [PubMed] [Google Scholar]
- 8.Bartram U, Speer CP. The role of transforming growth factor beta in lung development and disease. Chest. 2004;125(2):754–65. doi: 10.1378/chest.125.2.754. [DOI] [PubMed] [Google Scholar]
- 9.Lim RH, Kobzik L, Dahl M. Risk for asthma in offspring of asthmatic mothers versus fathers: a meta-analysis. PLoS One. 2010;5(4):e10134. doi: 10.1371/journal.pone.0010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neuman A, Hohmann C, Orsini N, Pershagen G, Eller E, Kjaer HF, et al. Maternal smoking in pregnancy and asthma in preschool children: a pooled analysis of eight birth cohorts. Am J Respir Crit Care Med. 2012;186(10):1037–43. doi: 10.1164/rccm.201203-0501OC. [DOI] [PubMed] [Google Scholar]
- 11.Halonen M, Lohman IC, Stern DA, Ellis WL, Rothers J, Wright AL. Perinatal tumor necrosis factor-alpha production, influenced by maternal pregnancy weight gain, predicts childhood asthma. Am J Respir Crit Care Med. 2013;188(1):35–41. doi: 10.1164/rccm.201207-1265OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harpsoe MC, Basit S, Bager P, Wohlfahrt J, Benn CS, Nohr EA, et al. Maternal obesity, gestational weight gain, and risk of asthma and atopic disease in offspring: a study within the Danish National Birth Cohort. J Allergy Clin Immunol. 2013;131(4):1033–40. doi: 10.1016/j.jaci.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Rothers J, Stern DA, Lohman IC, Wright AL, DeVries A, Vercelli D, et al. Maternal cytokine profiles during pregnancy predict asthma in children of nonasthmatic mothers. Am J Resp Cell Mol Biol. 2018 doi: 10.1165/rcmb.2017-0410OC. under revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeVries A, Stern DA, Wright AL, Halonen M, Vercelli D. Neonatal DNA methylation profiles are associated with the maternal prenatal immune milieu selectively in children with non-asthmatic mothers. ATS International Conference. 2017 [Google Scholar]
- 15.Yang IV, Pedersen BS, Liu A, O’Connor GT, Teach SJ, Kattan M, et al. DNA methylation and childhood asthma in the inner city. J Allergy Clin Immunol. 2015;136(1):69–80. doi: 10.1016/j.jaci.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez FD. Early-life origins of chronic obstructive pulmonary disease. N Engl J Med. 2016;375(9):871–8. doi: 10.1056/NEJMra1603287. [DOI] [PubMed] [Google Scholar]
- 17.Sears MR, Greene JM, Willan AR, Wiecek EM, Taylor DR, Flannery EM, et al. A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. N Engl J Med. 2003;349(15):1414–22. doi: 10.1056/NEJMoa022363. [DOI] [PubMed] [Google Scholar]
- 18.Haland G, Carlsen KC, Sandvik L, Devulapalli CS, Munthe-Kaas MC, Pettersen M, et al. Reduced lung function at birth and the risk of asthma at 10 years of age. N Engl J Med. 2006;355(16):1682–9. doi: 10.1056/NEJMoa052885. [DOI] [PubMed] [Google Scholar]
- 19.Morgan WJ, Stern DA, Sherrill DL, Guerra S, Holberg CJ, Guilbert TW, et al. Outcome of asthma and wheezing in the first 6 years of life: follow-up through adolescence. Am J Respir Crit Care Med. 2005;172(10):1253–8. doi: 10.1164/rccm.200504-525OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strunk RC, Weiss ST, Yates KP, Tonascia J, Zeiger RS, Szefler SJ. Mild to moderate asthma affects lung growth in children and adolescents. J Allergy Clin Immunol. 2006;118(5):1040–7. doi: 10.1016/j.jaci.2006.07.053. [DOI] [PubMed] [Google Scholar]
- 21.McGeachie MJ. Childhood asthma is a risk factor for the development of chronic obstructive pulmonary disease. Curr Opin Allergy Clin Immunol. 2017;17:104–9. doi: 10.1097/ACI.0000000000000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung JM, Sin DD. Asthma-COPD overlap syndrome: pathogenesis, clinical features, and therapeutic targets. BMJ. 2017;358:j3772. doi: 10.1136/bmj.j3772. [DOI] [PubMed] [Google Scholar]
- 23.Woodruff PG, van den Berge M, Boucher RC, Brightling C, Burchard EG, Christenson SA, et al. American Thoracic Society/National Heart, Lung, and Blood Institute asthma-ohronic obstructive pulmonary disease overlap workshop report. Am J Respir Crit Care Med. 2017;196(3):375–81. doi: 10.1164/rccm.201705-0973WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sin DD, Miravitlles M, Mannino DM, Soriano JB, Price D, Celli BR, et al. What is asthma-COPD overlap syndrome? Towards a consensus definition from a round table discussion. Eur Respir J. 2016;48(3):664–73. doi: 10.1183/13993003.00436-2016. [DOI] [PubMed] [Google Scholar]
- 25.Soler Artigas M, Loth DW, Wain LV, Gharib SA, Obeidat M, Tang W, et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat Genet. 2011;43(11):1082–90. doi: 10.1038/ng.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho MH, Castaldi PJ, Hersh CP, Hobbs BD, Barr RG, Tal-Singer R, et al. A genome-wide association study of emphysema and airway quantitative imaging phenotypes. Am J Respir Crit Care Med. 2015;192(5):559–69. doi: 10.1164/rccm.201501-0148OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hobbs BD, de Jong K, Lamontagne M, Bosse Y, Shrine N, Artigas MS, et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet. 2017;49(3):426–32. doi: 10.1038/ng.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Busch R, Hobbs BD, Zhou J, Castaldi PJ, McGeachie MJ, Hardin ME, et al. Genetic association and risk scores in a chronic obstructive pulmonary disease meta-analysis of 16,707 subjects. Am J Respir Cell Mol Biol. 2017;57(1):35–46. doi: 10.1165/rcmb.2016-0331OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wain LV, Shrine N, Artigas MS, Erzurumluoglu AM, Noyvert B, Bossini-Castillo L, et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet. 2017;49(3):416–25. doi: 10.1038/ng.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardin M, Cho M, McDonald ML, Beaty T, Ramsdell J, Bhatt S, et al. The clinical and genetic features of COPD-asthma overlap syndrome. Eur Respir J. 2014;44(2):341–50. doi: 10.1183/09031936.00216013. [DOI] [PMC free article] [PubMed] [Google Scholar]