

In response to oxidative stress, cells must choose either to live or to die. Here we show that the E3 ligase SCFGrr1 mediates the destruction of Med13, which releases cyclin C into the cytoplasm and results in cell death. The Med13 SCF degron is most likely primed by the Cdk8 kinase and marked for destruction by the MAPK Slt2.
Abstract
In response to oxidative stress, cells decide whether to mount a survival or cell death response. The conserved cyclin C and its kinase partner Cdk8 play a key role in this decision. Both are members of the Cdk8 kinase module, which, with Med12 and Med13, associate with the core mediator complex of RNA polymerase II. In Saccharomyces cerevisiae, oxidative stress triggers Med13 destruction, which thereafter releases cyclin C into the cytoplasm. Cytoplasmic cyclin C associates with mitochondria, where it induces hyperfragmentation and regulated cell death. In this report, we show that residues 742–844 of Med13’s 600–amino acid intrinsic disordered region (IDR) both directs cyclin C-Cdk8 association and serves as the degron that mediates ubiquitin ligase SCFGrr1-dependent destruction of Med13 following oxidative stress. Here, cyclin C-Cdk8 phosphorylation of Med13 most likely primes the phosphodegron for destruction. Next, pro-oxidant stimulation of the cell wall integrity pathway MAP kinase Slt2 initially phosphorylates cyclin C to trigger its release from Med13. Thereafter, Med13 itself is modified by Slt2 to stimulate SCFGrr1-mediated destruction. Taken together, these results support a model in which this IDR of Med13 plays a key role in controlling a molecular switch that dictates cell fate following exposure to adverse environments.
INTRODUCTION
Protein–protein interactions are at the heart of nearly all facets of cell physiology, including transducing exogenous signals necessary for proper cell fate decisions to be adopted. For example, following exposure to cytotoxic compounds, the cell must assess the level of damage and decide whether to arrest cell division and repair the damage or execute programmed cell death. Recently, it has become apparent that intrinsic-disordered regions (IDRs), defined by a continuous stretch of disordered promoting residues, play key roles in directing protein−protein interaction networks, especially those involved in macromolecular decisions including signaling and control pathways (reviewed in Dyson and Wright, 2005; Fuxreiter et al., 2014). This hotspot for communication is achieved in part by the ability of IDRs to contribute to the formation of large, malleable interfaces that can interact with multiple partners. These protein interactions can occur via short segments termed molecular recognition features (MoRFs) that undergo disorder-to-order transition upon binding to their cognate ligands (Fuxreiter et al., 2007). Consistent with these domains being communication hubs, the binding of IDRs to their targets is often regulated by covalent modifications including phosphorylation, which can serve as simple biological switches (Vuzman et al., 2012).
In budding yeast, cyclin C and its kinase partner Cdk8 are predominantly negative regulators of a diverse set of stress response genes (Surosky et al., 1994; Cooper et al., 1997; Holstege et al., 1998; Chi et al., 2001; van de Peppel et al., 2005). Together with Med13 and Med12, they form the Cdk8 kinase module (CKM) of the multisubunit Mediator complex, which acts as an interface between DNA bound transcription factors and RNA polymerase (Bourbon, 2008; Yin and Wang, 2014; Allen and Taatjes, 2015). In addition to its transcriptional role, cyclin C possesses another function following oxidative stress that is found in both yeast and mammalian cells (Cooper et al., 2012, 2014; Strich and Cooper, 2014; Wang et al., 2015). Specifically, nuclear release of cyclin C, but not Cdk8, allows its relocalization to the mitochondrial outer membrane. At this new subcellular address, cyclin C is necessary and sufficient for stress-induced mitochondrial fission and is required for MOMP-dependent normal regulated cell death (RCD) execution (Cooper et al., 2012, 2014; Wang et al., 2015). This type of cell death is the reclassification of programmed cell death (PCD) passed by the international Nomenclature Committee on Cell Death in 2015 (Galluzzi et al., 2015). In budding yeast, cyclin C release is dependent on its direct phosphorylation by Slt2 MAPK (Jin et al., 2014) and the ubiquitin-mediated degradation of Med13 (Khakhina et al., 2014, and see Figure 1) In the CKM, Med13 contains a large centrally located IDR, the extent and placement of which is conserved across metazoans, plants, and fungi (Nagulapalli et al., 2016). As Med13 also bridges the CKM to the Mediator complex (Knuesel et al., 2009a) this centrally placed IDR may provide structural plasticity, allowing the CKM to interface with different mediator components according to the environment. Consistent with Med13 IDR being a potential communication hub, computer predictions suggest the presence of several MoRFs that are biased toward an alpha helical confirmation (Toth-Petroczy et al., 2008).
FIGURE 1:
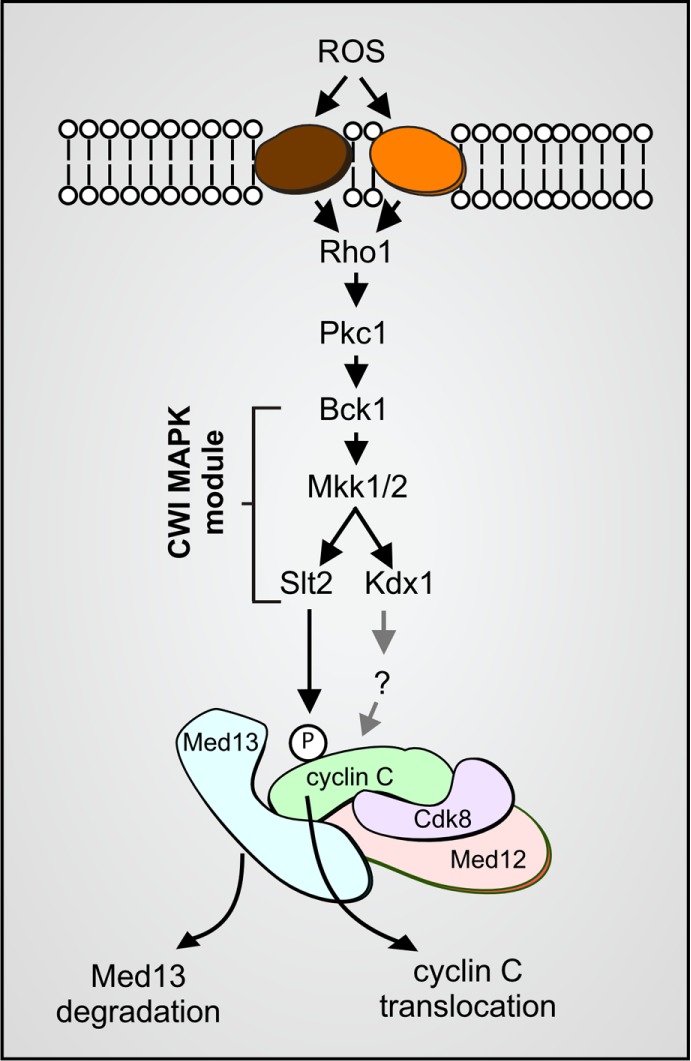
Cdk8 module (Cdk8, cyclin C, Med12 and Med13) regulation by the CWI MAPK pathway. H2O2 stimulates cell wall sensors, leading to activation of the CWI MAP kinase module. Slt2 phosphorylation triggers cyclin C translocation to the mitochondria and Med13 degradation (Khakhina et al., 2014). The pseudokinase Kdx1 is also required for cyclin C nuclear release via an unknown mechanism (Jin et al., 2014). Med12 is not required for cyclin C nuclear release (Khakhina et al., 2014).
In this report, we provide mechanistic details on how Med13 is targeted for destruction following oxidative stress in the budding yeast Saccharomyces cerevisiae. We show that the cell walI integrity (CWI) pathway MAPK Slt2 directly phosphorylates Med13, which is required for its recognition by the SCFGrr1 E3 ligase complex. Consistent with other SCF targets in yeast and higher eukaryotes (reviewed in Ang and Wade Harper, 2005), degradation of Med13 by SCFGrr1 also requires phosphorylation by cyclin C-Cdk8, which most likely primes this phosphodegron. Furthermore, this phosphodegron partly overlaps with the predicted Med13 IDR (Toth-Petroczy et al., 2008). Consistent with this region serving as an interaction hub (Uversky, 2013), this IDR directs both Cdk8 and Grr1 binding and is sufficient to retain cyclin C in the nucleus. Taken together, these results define a multistep switch required for the cellular decision to release cyclin C from the nucleus and identify a Med13 IDR as the communication hub that coordinates this prodeath decision.
RESULTS
SCFGrr1 is necessary for Med13 H2O2-mediated degradation
We have previously shown that Med13 degradation is dependent on a functional 26S proteasome, indicating that a ubiquitin ligase is required (Khakhina et al., 2014). In unstressed human cells, Med13 is turned over by the conserved SCF ubiquitin ligase utilizing the Fbw7 F-box recognition protein (Davis et al., 2013). Grr1, a yeast homologue of Fbw7, is a nonessential F-box protein that utilizes a leucine-rich region (LRR; Figure 2A) for substrate recognition (Flick and Johnston, 1991; Hsiung et al., 2001). SCFGrr1 is necessary for degradation of the mediator component Med3 (Gonzalez et al., 2014), as well as proteins regulating the glycolytic–gluconeogenic switch (Benanti et al., 2007). More recently, it has also been shown to regulate Whi7, a repressor of Start transcriptional gene expression (Gomar-Alba et al., 2017). To determine whether SCFGrr1 plays a role in H2O2-induced Med13 degradation, we examined Med13 protein levels following 0.4 mM H2O2 treatment in wild-type and grr1∆ cells harboring functional Med13-HA on a single copy plasmid. This concentration of H2O2 has long been established to induce a MOMP-dependent RCD response in yeast (Madeo et al., 1999; Carmona-Gutierrez et al., 2010; Galluzzi et al., 2011), as well as triggering cyclin C nuclear release (Cooper et al., 2012, 2014; Jin et al., 2014; Khakhina et al., 2014). The results (Figure 2B) show that Med13-HA is significantly more stable in grr1∆ cells. We next repeated these experiments using endogenous MED13 tagged with the myc epitope in wild-type (RSY1798) and grr1∆ mutant strains (RSY1771) harboring a plasmid expressing either wild-type GRR1, a vector control, or a GRR1 derivative deleted for the LRR domain (grr1∆L) (Hsiung et al., 2001). Following H2O2 treatment, Med13-myc was again significantly more stable in grr1∆ cells harboring either the vector control or grr1∆L than in wild-type cells (Figure 2C and Supplemental Figure S1A). These results suggest that SCFGrr1 is the E3 ligase responsible for mediating Med13 degradation following H2O2 stress.
FIGURE 2:

SCFGrr1 mediates Med13 degradation following H2O2 stress. (A) Model of the SCFGrr1. (B) Wild-type (RSY10) and grr1∆ cells (RSY1770) harboring Med13-HA (pKC801) were treated with 0.4 mM H2O2 for the time points indicated and Med13 levels were analyzed by Western blot. (C) Top panel: RSY1798 (MED13-myc::KAN) was treated with 0.4 mM H2O2 for the time points indicated and Med13 levels were analyzed by Western blot. Tub1 levels were used as loading controls. Bottom panel: RSY1771 (grr1∆::HIS3 MED13-myc::KAN) harboring ADH1PRO-grr1∆L was analyzed as for RSY1798. Tub1 levels were used as loading controls. (D) Yeast two-hybrid analysis of Med13 and Grr1 derivatives. Y69a cells harboring Med13-activating domain plasmid (pKC800) and either pAS2, pAS-Grr1, or pAS2-Grr1∆L binding domain plasmids were grown on –LEU, –TRP dropout medium to select for both plasmids (left panel) and on –TRP, –LEU, –HIS –ADE (middle panel), and –TRP, –LEU, –HIS, –ADE +3-AT (right panel) to test for Med13-Grr1 interaction.
If SCFGrr1 is the ubiquitin ligase directing Med13 proteolysis, then Grr1 should interact with Med13. To test this possibility, Grr1 and Med13 association was assayed using a two-hybrid strategy. This approach has been used previously both to identify and confirm Grr1 substrates (Wang and Solomon, 2012; Gonzalez et al., 2014). We expressed either the wild-type or the grr1∆L mutant GRR1 allele fused to the Gal4 DNA-binding domain bait with the full-length Med13 fused to the activator domain (AD) prey. These studies revealed that Grr1 interacts with Med13, and this interaction survives addition of the histidine analogue 3-amino-1,2,4-triazole (3-AT), suggesting that the HIS3 reporter gene induction is robust (Figure 2D). With the grr1∆L mutant bait, an interaction is detected selecting for the dual HIS3 and ADE2 reporter genes, but colony formation is uneven (middle panel) or absent in the presence of 3-AT (right panel). Taken together with the increased stability of Med13 observed in grr1∆ cells, these results argue that Med13 is an SCFGrr1 substrate.
The intrinsic disordered region of Med13 interacts with Grr1
Many SCF targets require phosphorylated substrates for F-box recognition (Skowyra et al., 1997). Consistent with this model, the LRR of Grr1 specifically recognizes phosphorylated serine/threonine–proline motifs within its substrate degron (Hsiung et al., 2001). In human cells, phosphorylation of T326 is required for Med13 destruction by SCFFbw7 (Davis et al., 2013). To address whether a similar mechanism directs H2O2-induced Med13 degradation in yeast, the requirement of the analogous threonine in yeast (T210; Supplemental Figure S1B) was tested. For these studies, we expressed Med13-HA from a single-copy plasmid in med13∆ cells. These studies revealed that both the wild type and the Med13T210A derivative were still degraded following 0.4 mM H2O2 stress (Supplemental Figure S1, C, D, and quantitated in E), indicating that T210 phosphorylation is not necessary for oxidative stress–induced destruction of Med13. Furthermore, consistent with our previously published results with endogenously tagged Med13-myc (Khakhina et al., 2014), Med13-HA degradation is not dependent on new protein synthesis, as it is still observed following cycloheximide treatment (Supplemental Figure S1C).
We next employed Y2H assays to identify the Med13 region able to interact with Grr1. Med13 has structured head and tail domains flanking a large IDR (Toth-Petroczy et al., 2008; Figure 3A). A series of Gal4AD-Med13 constructs that span the length of Med13 were built and tested for their ability to interact with Gal4BD-Grr1. The results revealed that the Grr1 interaction domain lies within the unstructured domain, between amino acids 571 and 906 (Figure 3B, summarized in Figure 3A). To confirm that the Med13 degron lies within this region, we performed a classical degron assay. The levels of the prey Gal4AD-Med13571-906 fusion protein were monitored following 0.4 mM H2O2 stress in wild-type and grr1∆ cells. The results indicated that Med13571-906 was destroyed following oxidative stress in a Grr1-dependent manner (Figure 3C). Taken together, these results indicate that the SCFGrr1 degron lies within the IDR.
FIGURE 3:

The unstructured domain of Med13 binds Grr1. (A) ProteinPredict (Yachdav et al., 2014) analysis of yeast Med13. Med13 contains amino- and carboxyl-terminal structured domains separated by an intrinsic disordered domain (IDR). Grr1 and Med13 interaction regions are indicated below the schematic. ± = qualitatively reduced binding (B). Med13-Grr1 Y2H analysis. Y69a cells harboring pAS-Grr1 and the indicated Gal4AD-Med13 subclone were streaked on media selecting for plasmid maintenance (left) or induction of the ADE2 and HIS3 reporter genes (right) by Y2H interaction. (C) Wild-type (RSY10) and grr1∆ (RSY1770) cells harboring the minimal Med13 interaction domain expression plasmid (Gal4AD-Med13571–906) were treated with 0.4 mM H2O2 for the time points indicated and Med13571–906 levels were analyzed by Western blot. Pgk1 levels were used as loading controls.
The Med13 intrinsic disordered region binds cyclin C
As cyclin C nuclear release represents an important step toward entering the cell death pathway, we next sought to identify the Med13 region that binds cyclin C, using two-hybrid strategies. However, the yeast cyclin C self-activates when tethered to a yeast two-hybrid (Y2H) bait protein (Cooper et al., 1997). Therefore, we used the human cyclin C, as it does not self-activate two-hybrid reporter genes (Krasley et al., 2006). Using this approach, we found that the same region associating with Grr1 (amino acid residues 571–906) also bound cyclin C (Figure 4A; summarized in Figure 3A). To confirm these results, as well as to address if this interaction is direct, we used a pull-down approach. We expressed a central region of the Med13 interaction domain (amino acid residues 742–844) as a GST fusion protein. GST-Med13742-844 was passed over a cobalt column on which Escherichia coli–prepared human His6-cyclin C was bound. Following extensive washing, bound proteins were eluted with imidazole and the presence of GST-Med13742-844 was determined by Western blot analysis using antibodies to His6 or human cyclin C. These results show that human cyclin C interacts with Med13742–844 (Figure 4B). The experiments were repeated using yeast cyclin C (Figure 4C). Although the residual cleaved GST that was present in the GST-Med13 construct showed some interaction with yeast cyclin C, the full-length construct showed a significantly stronger interaction. Confidence was gained in this interaction being specific, as no interaction was observed when GST alone was used in Figure 4, B or C. Interestingly, although amino acid residues 571–650 of Med13’s IDR can associate with Grr1 (Figure 3B), this region does not associate with cyclin C (Figure 4D). Thus, taken together, these results indicate that cyclin C binds the same region of Med13’s large IDR that is recognized by SCFGrr1.
FIGURE 4:

The Med13 IDR binds cyclin C. (A) Y2H analysis of the human cyclin C DNA–binding domain plasmid (pSW108) and the indicated Gal4AD-Med13 subclone derivatives. The numbers indicate the amino acids remaining in the activation domain plasmids. Transformants were patched onto either –LEU, –TRP (–L–T) or –HIS –ADE –LEU –TRP (–H–A–L–T) to select for plasmid maintenance or Y2H interaction, respectively. (B) Western blot analysis of pull-down assays with His6-human cyclin C and GST-Med13742–844 (DS30). The load control contains 1/10 of the input. Single and double asterisks represent cleaved GST and cross-reaction between anti-GST antibody and human cyclin C, respectively. Molecular weight markers (kDa) are indicated. (C) As in B except that yeast cyclin C was used. (D) Western blot analysis of pull-down assays with GST yeast cyclin C and His6-Med13571–650 (DS22). The load control contains 1/50 of the input. Molecular weight markers (kDa) are indicated.
The Med13 intrinsic disordered region is sufficient to retain cyclin C in the nucleus
To determine whether yeast cyclin C also associates with the same region of Med13’s IDR in vivo, we asked whether this region (Med13571–906) is sufficient to retain cyclin C in the nucleus in the absence of stress. To address this question, mitochondrial morphology and subcellular localization of a functional cyclin C-YFP (Cooper et al., 2012) were monitored in unstressed med13∆ cells harboring either a vector control or one of four Gal4AD-Med13 fusion proteins. As expected, cyclin C was predominantly cytoplasmic and colocalized with fragmented mitochondria in the vector control (Figure 5A, top row, quantitated in Figure 5B, and see Supplemental Figure S2 for DAPI stained images). Similarly, expression of Gal4AD-Med13571–650 was unable to retain cyclin C in the nucleus or prevent mitochondrial fragmentation (second row). However, cells expressing either Gal4AD-Med13571–906, Gal4AD-Med13651–906, or Gal4AD-Med13742–844 exhibited both nuclear retention of cyclin C and fused mitochondria. Taken together with the pull-down assays, these results suggest that Med13742–844 is sufficient to retain yeast cyclin C in the nucleus.
FIGURE 5:

The unstructured domain of Med13 retains cyclin C in the nucleus. (A) Fluorescence microscopy of mid–log phase med13∆ cells (RSY1701) harboring the Gal4AD-Med13 construct indicated or a vector control (pACT2) and both cyclin C-YFP and DsRed mitochondrial targeting (mt-DsRed) plasmids. Blue boxes indicated enlarged regions of the image. Bar = 10 µM. (B) The percentage of the population of cells in A displaying at least three cyclin C-YFP foci in the cytoplasm (top panel) or fragmented mitochondria (bottom panel) is quantified (mean ± SEM). At least 200 cells were counted per time point from three individual isolates. *p < 0.05 difference from vector control.
Cdk8 phosphorylation primes Med13 for destruction
SCF substrates are typically recognized following phosphorylation of the degron. However, many substrates require two phosphorylation marks: one to “prime” the substrate and another that represents the trigger for ubiquitylation (reviewed in Ang and Wade Harper, 2005). Previous work revealed that protein kinase A (PKA) phosphorylates Med13 on Serine 608 and 1236 (Chang et al., 2004). However, a derivative mutated for both phosphor acceptor sites (Med13S608A,S1236A-HA) was still destroyed with kinetics similar to wild type (Supplemental Figure S3A), indicating that PKA phosphorylation is not required for Med13 degradation following H2O2 treatment. Consistent with this result, Med13S608A,S1236A-HA complements the aberrant mitochondrial morphology exhibit by med13∆ cells (Supplemental Figure S3B).
In addition to PKA, cyclin C-Cdk8 phosphorylates Med13 in human cells (Knuesel et al., 2009b; Poss et al., 2016) suggesting the possibility that Cdk8 represents the priming kinase. To test this model, Med13 destruction kinetics were monitored in a cnc1∆ null strain transformed with either the wild-type CNC1 gene or the vector control. These experiments indicated that cyclin C-Cdk8 kinase is necessary for Med13 destruction (Figure 6A and quantified in Supplemental Figure S4A). Our previous studies showed that Slt2 phosphorylates cyclin C on Ser266 following H2O2, and this modification is required for its release from Med13 and translocation to the cytoplasm (Jin et al., 2014). Therefore, one possibility is that cyclin C release is a prerequisite for Med13 destruction. Consistent with this possibility, Med13 was protected from destruction in a cnc1∆ mutant harboring a plasmid expressing cyclin CS266A (Figure 6A, right panel). These results indicate that cyclin C phosphorylation by Slt2 is also required for Med13-HA destruction (see below).
FIGURE 6:
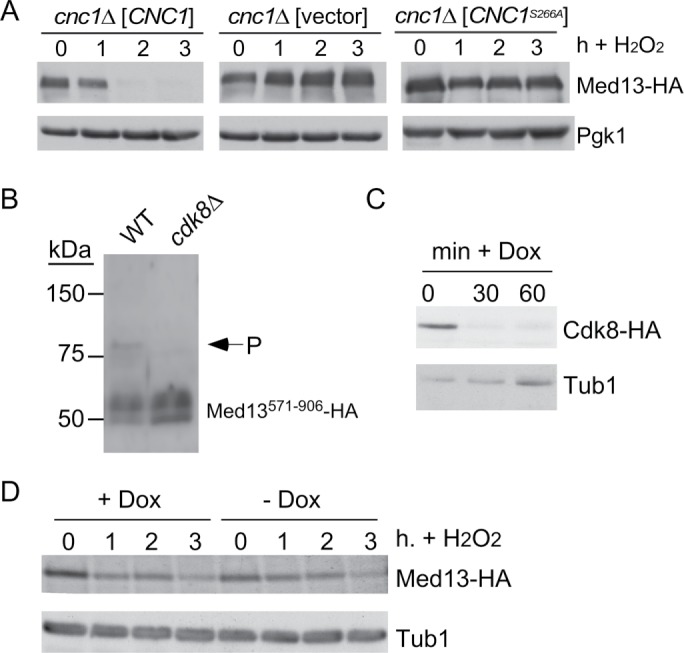
Cyclin C-Cdk8 directly stimulates Med13 proteolysis. (A) Mid-log cnc1∆ cultures (RSY391) harboring either wild-type CNC1 (pKC337), vector (pRS314), or cnc1S266A (pLR166) expression plasmids and pKC803 (CEN, Med13-HA) were subjected to an H2O2 time-course experiment. Pgk1 levels were used as loading controls. (B) Phos-tag gel analysis of pDS8 (Gal4AD-Med13571–906) in unstressed wild-type (RSY10) or cdk8∆ (RSY1796) cultures. Arrow indicates phosphorylated Med13571–906 species. Molecular weight markers (kDa) are indicated. (C) RSY2066 harboring the Ubi-Ile3-Cdk8-HA expression plasmid (pCM1888) were grown to mid–log phase and doxycycline added for the indicated times. Cdk8-HA levels were monitored by Western blot analysis. Tub1 levels were monitored for loading control. (D) RSY2066 harboring pCM1888 and Med13-HA (pKC803) were grown to mid–log phase, the culture split, and doxycycline added to one for 1 h. Both cultures were then treated with 0.8 mM H2O2 and Med13-HA degradation monitored as previously described. Tub1p was monitored for loading control.
The results just presented indicated that the cyclin C-Cdk8 kinase is required for Med13 destruction in response to oxidative stress. Consistent with this, Med13 is protected from degradation in cells harboring kinase dead cdk8 as the only source of this enzyme (Khakhina et al., 2014). As cyclin C-Cdk8 is active in unstressed cells, these results suggest that this kinase could prime the degron for recognition by SCFGrr1. To test this model, we attempted to perform in vitro kinase assays with cyclin C-Cdk8 and Med13571–906 as the substrate. Unfortunately, we were unable to get these assays to work. Thus we decided to examine the phosphorylation state of the Med13 degron derivative (GalADMed13571–906) using the Phos-Tag SDS–PAGE system, which severely retards the movement of a phosphorylated peptide (Kinoshita et al., 2006). Protein extracts prepared from unstressed wild-type or cdk8∆ cultures harboring GalADMed13571–906 were subjected to Phos-Tag Western blot analysis. The results revealed the presence of a slower migrating band in the wild-type extract that was missing in the cdk8∆ sample (Figure 6B). This indicates that Cdk8 is required for Med13 phosphorylation in unstressed cells.
To test whether cyclin C-Cdk8 activity was required before or after oxidative stress, a Cdk8-N-end rule degron was constructed. This system takes advantage of the rapid turnover of proteins that contain arginine as the amino terminal residue (Bachmair et al., 1986; Varshavsky, 1992; Gnanasundram and Kos, 2015). The Cdk8-N-end rule construct is under the control of a doxycycline-repressible promoter, so that the administration of doxycycline coupled with the rapid turnover of the Cdk8-N-end rule fusion protein results in rapid depletion of Cdk8 from yeast cells (Figure 6C). To test the execution point for Cdk8 phosphorylation, a mid–log culture expressing Med13-HA and Cdk8-N-end degron was untreated or treated with doxycycline for 1 h prior to H2O2 addition to ensure Cdk8 depletion. A time course was conducted and samples were collected, extracts prepared, and Med13-HA levels monitored by Western blot analysis. These studies revealed that Med13-HA levels were reduced similarly in both cultures (Figure 6D), indicating that Cdk8 function was required before oxidative stress. Taken together, these results are consistent with a model in which Cdk8 phosphorylation primes Med13 for degradation.
The CWI pathway is required for reactive oxygen species–mediated destruction of Med13
The CWI signal transduction pathway is the major MAPK pathway in yeast that transmits the reactive oxygen species (ROS) stress signal to transcription factors (Staleva et al., 2004). To facilitate cyclin C nuclear release, the CWI employs two MAPKs, Slt2 and its pseudokinase paralogue Kdx1. Slt2 phosphorylates cyclin C on Serine 266 to induce cytoplasmic relocalization (Cooper et al., 2012; Jin et al., 2013, 2014), while Kdx1 functions through an unknown mechanism (Jin et al., 2015). To address the question of whether Kdx1 and/or Slt2 is required for Med13 degradation, we examined Med13-HA degradation kinetics in wild-type and slt2∆ or kdx1∆ single-mutant cultures following 0.4 mM H2O2 stress. As slt2∆ mutants are temperature-sensitive, these experiments were conducted at 23°C. The results show that Med13-HA was degraded in wild-type or kdx1∆ cells but stable in the slt2∆ mutant (Figure 7A, quantified in Supplemental Figure S4B). These results indicate that unlike cyclin C, only Slt2, but not Kdx1, is required for Med13 destruction. Repeating this experiment with cells expressing a kinase-dead version of SLT2 (slt2K54R) produced similar results (Figure 7B), indicating that Slt2 kinase activity is required for Med13 proteolysis. To investigate whether the role of Slt2 in Med13 degradation was direct, in vitro kinase assays were performed using activated Slt2-HA or the kinase-dead derivative (Slt2K54R-HA; Kim et al., 2008) immunoprecipitated from yeast extracts. The immunoprecipitate was incubated with the E. coli prepared Med13 degron (amino acids 571–906) described above. These experiments revealed enhanced phosphorylation of Med13571–906 above background that was dependent on a functional kinase (Figure 7C), indicating that Slt2 directly regulates Med13 stability. Finally, we asked whether Slt2 activation was sufficient to induce Med13 destruction or whether another stress signal was required. To address this question, Med13-HA levels were monitored in an unstressed culture expressing a constitutively active allele of the upstream MAPKKK BCK1 (BCK1-20). Mid-log cultures expressing BCK1-20 exhibited dramatically reduced Med13-HA levels compared with vector controls (Figure 7D). These results indicate that the CWI pathway is both necessary and sufficient to induce Med13 destruction. Finally, to test the epistatic relationship between BCK1-20 and cdk8∆ alleles, the experiment just described was repeated in a cdk8∆ background. Western blot analysis revealed that Cdk8 was required for BCK1-20-induced Med13 destruction in unstressed cells (Figure 6D). Taken together, these results indicate that Slt2 activation is necessary for H2O2-mediated Med13-HA degradation and that this signal requires Cdk8 activity. These results are consistent with a three-step model for initiating the Med13 destruction pathway (see the Discussion).
FIGURE 7:

Slt2 directly stimulates Med13 destruction. (A) Wild-type (RSY10), kdx1∆ (RSY1736) and slt2∆ (RSY1006) cultures expressing pKC801 (CEN, Med13-HA) were grown to mid–log phase (0 h) and then treated with 0.4 mM H2O2 for the indicated times. Med13-HA levels were determined by Western blot analysis. (B) As in A except that the slt2∆ cells expressed a kinase dead SLT2 mutant (slt2K54R) plasmid. (C) Top panel: Slt2-HA or Slt2K54R-HA (kinase dead) was immunoprecipitated from extracts prepared from cells treated for 60 min with 5 mM sodium orthovanadate was mixed with Med13571–906 and radioactive ATP as indicated. All reactions were conducted in duplicate and then separated by SDS PAGE and subject to autoradiography. Med13571–906 is indicated by the arrowhead. Bottom panel: Coomassie stained gel showing the Med13571–906 input used in the kinase assays. (D) Wild-type (RSY10) or cdk8∆ (RSY1796) mutant cells expressing either an empty vector control or the hyperactive allele of BCK1 (BCK1-20) and MED13-HA (pKC803) were grown to midlog and Med13-HA levels determined by Western blot analysis. Tub1 served as a loading control.
Slt2 and Cdk8 phosphorylation of the intrinsic disordered region is required for Med13 destruction
IDRs confer conformational flexibility on proteins that are often targets of posttranslational modifications (Diella et al., 2008). Notably, IDRs are enriched in phosphorylation sites, raising the possibility that they are substrates for multiple kinases (Collins et al., 2008). As our data indicate that Cdk8 and Slt2 are required for Med13 degradation, we next asked if the sites they phosphorylate lie within the same IDR of Med13 that associates with Grr1 and cyclin C (amino acids 651–906). Slt2 and Cdk8 are both proline-directed kinases that preferentially phosphorylate the consensus sequences PX(S/T)P and S/T-P-X-K/R (where X is any amino acid), respectively. They also can phosphorylate the minimal consensus sequence S/T-P (Nigg, 1993). Med13651–906 contains a conserved Slt2 consensus sequence at position 748 that has been identified as potential phosphorylation site in two genomic screens (Albuquerque et al., 2008; Holt et al., 2009) as well as four additional TP sites (Figure 8A). Thus, to address whether these kinases phosphorylate the five S/T-P sites in Gal4AD-Med13651–906 fusion protein, its degradation was monitored following H2O2 stress in wild-type, slt2∆ and cdk8∆ cells. The results show that Gal4AD-Med13651–906 fusion protein is stable in either slt2∆ or cdk8∆ cells (Figure 8B). These results are consistent with a model where the Med13 phosphodegron is targeted by both Cdk8 and Slt2. Consistent with the Y2H data presented in Figure 3B, the phosphodegron is stable in grr1∆ cells (Figure 8C). If the Med13 phosphodegron is regulated by two kinases, then two separate S/T-P sites within the degron should stabilize the protein. To test this, wild-type cells harboring either a wild-type Gal4AD-Med13742–844 fusion protein or mutant derivatives were subjected to 0.4 mM H2O2 stress and analyzed as described above. As anticipated, the smaller Gal4AD-Med13742–844 fusion protein that contains all five S/T-P sites was still degraded in wild-type cells (Figure 8C). Interestingly, the S748A, T781A, and T803A mutants were also degraded, with the same kinetics as wild type (Supplemental Figure S4C). In addition, the double mutants (T781A, T803A and S748A, T781A) were also degraded with wild-type kinetics (Figure 8, D and E). However, the T835A, T837A double mutant was significantly more stable than the wild type (Figure 8, C and E), suggesting that these sites are needed for Med13 degradation. Taken together, these results suggest that both kinases have to be active for SCFGrr1 to recognize this 100–amino acid phosphodegron at positions T835 and T837.
FIGURE 8:

The Med13 IDR contains the phosphodegron. (A) Map of Med13 showing the location of potential Cdk and MAPK sites within the IDR. (B) Wild-type (RSY10), slt2∆ (RSY1737), and cdk8∆ (RSY1796) cultures expressing pDS16 (Gal4AD-Med13651–906) were grown to mid–log phase (0 h) and then treated with 0.4 mM H2O2 for the indicated times. Med13651–906-HA levels were determined by Western blot analysis. Pgk1 levels were used as a loading control. (C, D) Wild-type (RSY10) cultures harboring wild-type Gal4AD-Med13742–844 (pDS32) or various point mutations as indicated were grown to mid–log phase (0 h) then treated with 0.4 mM H2O2 for the indicated times. Med13-HA levels were determined by Western blot analysis. Pgk1 levels were used as a loading control. The Gal4AD-Med13742–844 levels were also monitored in the grr1∆ strain. (E) Degradation kinetics of the Gal4AD-Med13742–844 constructs shown in C and D.
DISCUSSION
Communication between organelles is critical to coordinate the cellular response to a variety of external or internal signals, including oxidative stress. The data presented here and in previous publications (Cooper et al., 1997, 2014; Krasley et al., 2006; Jin et al., 2013, 2014, 2015; Khakhina et al., 2014; Wang et al., 2015) have revealed that the CKM plays a key role in both yeast and mammalian cells. Previously, we reported that cyclin C, but not Cdk8, translocates from the nucleus to the mitochondria following oxidative stress in both yeast and mammalian cells. At the mitochondria, cyclin C associates with the fission machinery to induce fragmentation and directs programmed cell death (Cooper et al., 2012, 2014; Wang et al., 2015). Therefore, releasing cyclin C from the nucleus represents an important step determining whether the cell initiates the RCD pathway or not. Importantly, we found that the nuclear anchor for cyclin C, Med13, is destroyed in response to oxidative stress, providing a mechanism for orchestration of cyclin C nuclear release (Khakhina et al., 2014). Interestingly, Med12 is neither required for this response (Khakhina et al., 2014) nor destroyed following H2O2 stress (Supplemental Figure S4D). In this report, we provide the molecular details on how Med13 destruction is triggered to operate this important molecular switch in yeast. First, Med13 proteolysis is mediated by the ubiquitin ligase SCF and the specificity factor Grr1. Several studies in yeast and higher eukaryotes found that SCF substrates require a complex interplay between multiple kinases to generate the requisite phosphodegron on the substrate (reviewed in Ang and Wade Harper, 2005). For example, Cdk1 acts as a priming kinase for Cdc5 (Asano et al., 2005; Yoshida et al., 2006), whereas Cdc5 acts as a priming kinase for Wee1 (Watanabe et al., 2004). In all cases, the priming kinase first phosphorylates the substrate, while the second kinase completes the formation of the phosphodegron by modifying residues that are recognized by the SCF. This has led to the current model, in which priming phosphorylations create docking sites for downstream kinases. Moreover, local structural disorder most likely facilitates multiple kinases regulating a protein within a small region. We identified a small region (amino acids 742–844) within the IDR of Med13 that acts as a communication hub. This region is sufficient to retain cyclin C in the nucleus, associates with Grr1, and contains the Med13 phosphodegron that is primed by Cdk8 and activated by Slt2 following oxidative stress exposure. This dual kinase requirement provides the cell with a mechanism to integrate multiple input signals into a single readout.
Med13 phosphodegron activation requires a three-step process
Our results indicate that cyclin C-Cdk8 provides the priming signal for Med13 destruction (Figure 9, Step 1). Although technical reasons prevented us from addressing whether this activity is direct or indirect, four independent experiments support a model of this as a direct phosphorylation event: 1) The observation that Med13651–906 was not destroyed following H2O2 stress in cdk8∆ cells (Figure 8B). 2) The observation of a slower migrating band seen in Phos-Tag SDS–PAGE analysis of unstressed in wild-type extracts that was missing in the cdk8∆ sample (Figure 6B). 3) The end-N rule experiments conducted in Figure 6, C and D, also revealed that Med13-HA levels were reduced regardless of the presence of Cdk8 after stress. 4) Last, in vitro evidence revealed that cyclin C interacts directly with this region of Med13 (Figure 4, B and C), which would position the kinase with direct access to the Med13 degron. Thus, taken together, these results suggest that Cdk8 phosphorylation primes Med13 for degradation, most likely by a direct event. Consistent with this, human Cdk8 directly phosphorylates human Med13 on S749, which lies in the intrinsic disorder domain in what appears to be a nonconserved residue compared with yeast (Poss et al., 2016). This requirement may be necessary to inform the cell that Med13 is in the Cdk8 module and its destruction will lead to cyclin C release and Cdk8 inactivation. Slt2 activation by oxidative stress provides the trigger mechanism that commits the cell to destroying Med13 and releasing cyclin C from the nucleus (Step 3). This event accomplishes two goals. First, removing cyclin C and destroying Med13 inhibits the repressor activity of Cdk8 for several stress-responsive genes, including chaperones (e.g., SSA1), anti-oxidants such as catalase (CTT1), and the multi-stress-responsive gene DDR2 (Cooper et al., 1997, 2012; Holstege et al., 1998). In addition, Slt2 activation stimulates several transcription factors, such as Rlm1, which is required for the transcriptional arm of the stress response (Dodou and Treisman, 1997; Watanabe et al., 1997; Jung et al., 2002). Therefore, Slt2 activation both stimulates transcriptional activators and inhibits repressors. By releasing cyclin C, the cell is able to transmit the stress signal to the mitochondria. We have previously demonstrated that cyclin C–induced fission alone is not sufficient to induce cell death, but does make cells hypersensitive to oxidative stress (Khakhina et al., 2014). Therefore, cyclin C release poises the cell to initiate RCD, but additional regulatory layers exist. Unlike other priming/trigger two-kinase degron phosphorylation switches, we have identified an additional step in the system regulating Med13. Specifically, Slt2 also modifies cyclin C on S266, which promotes its dissociation from Med13 (Jin et al., 2014). Our finding that the S266A mutation protects Med13 from destruction is consistent with a model in which S266 modification disrupts cyclin C–Med13 interaction (Step 2), allowing Slt2 access to the phosphodegron. Taken together, our model predicts a three-step system that controls Med13 stability and cyclin C nuclear release.
FIGURE 9:
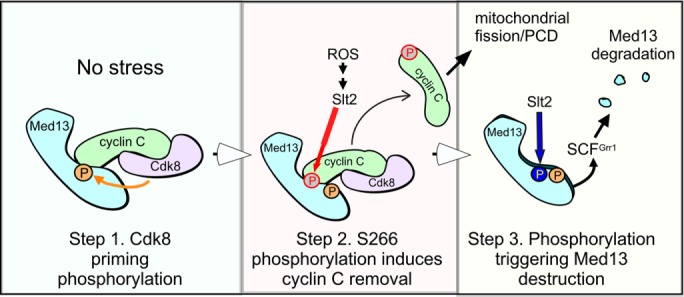
Three-step model of cyclin C release from Med13 following H2O2 stress. Step 1: Cdk8-cyclin C kinase phosphorylates Med13 in unstressed cells to provide the priming modification for this degron. Step 2 requires stress-activated Slt2 that phosphorylates cyclin C on S266 leading to dissociation of cyclin C from Med13. This event allows Slt2 access to the phosphodegron to deliver the trigger modification leading to Med13 recognition by SCFGrr1 and its destruction (Step 3).
If cyclin C phosphorylation is sufficient to initiate cyclin C nuclear release, then why would it be important to completely destroy Med13? First, our results indicate that S266 phosphorylation is capable of stimulating only partial release of cyclin C in unstressed cells (Jin et al., 2014). Therefore, Med13 destruction may be necessary for complete cyclin C release. Not exclusive of this model, another possibility is that Med13 destruction prevents the reaccumulation of cyclin C in the nucleus after its cytoplasmic translocation. In this model, Med13 proteolysis fixes the decision of cyclin C release thus directing a complete transcriptional and mitochondrial response to oxidative stress.
The Med13 intrinsic disordered region is a communication hub
It has recently been suggested that IDRs play important roles in protein:protein communication, as they provide a flexible interaction surface to multiple partners (Wright and Dyson, 2015). Using two different protein structure prediction algorithms, IUPred/ANCHOR and Phyre2 (Dosztanyi et al., 2005, 2009; Kelley et al., 2015), Med13 has a large IDR spanning the middle of the protein (amino acids residues 307–906 and Supplemental Figure S5, A and B). Although not conserved in amino acid sequence, it is notable that the position and presence of Med13’s IDR is conserved throughout three kingdoms (Nagulapalli et al., 2016). Also conserved is the observation that Med13 is the CKM member that brings this kinase module in contact with the Mediator complex (Knuesel et al., 2009a). This suggests a model in which the IDR of Med13 provides conformational plasticity to the CKM, which is important for bringing the kinase and other proteins into contact with different mediator members on a pro re nata basis. This idea is consistent with the results presented here, in which we show that the IDR of Med13 can bind to cyclin C before stress and Grr1 after. Although speculative, taken together, this suggests a model in which Med13 IDR is conserved functionally and may contain signature motifs that are important for protein binding. Consistent with this idea is the observation that two other conserved proteins that regulate transcription (CCR4-NOT4 and TFIIS) also bind to the IDR of yeast Med13, but in a different location (Supplemental Figure S5; Liu et al., 2001; Wery et al., 2004). Such a signature could be a MoRF, which is defined as a short binding region located within a longer intrinsically disordered region that binds to protein partners via disorder-to-order transitions (Kotta-Loizou et al., 2013). ANCHOR analysis (Dosztanyi et al., 2009) predicts that the IDR of Med13 has many such domains, with two lying within the cyclin C binding region (residues 742–844, Supplemental Figure S5A). As additional factors are identified that bind the Med13 IDR, the rules that govern these interactions may become better understood.
The need to understand the role that Med13 IDR plays in protein communication has become important in the past decade, as in humans truncating or loss-of-function mutations in MED13L, a paralogue of MED13 (Muncke et al., 2003), results in MED13L haploinsufficiency syndrome. This syndrome is complex and presents with a range of symptoms including congenital heart and neurodevelopmental defects, severe neurocognitive deficiencies, and facial dysmorphism (Adegbola et al., 2015; van Haelst et al., 2015; Asadollahi et al., 2017; Yamamoto et al., 2017). Interestingly many of these mutations lie in the IDR of MED13L (Asadollahi et al., 2017). In yeast, a hallmark of med13∆ cells is that they are respiratory deficient, which is dependent on cyclin C nuclear release (Khakhina et al., 2014). Thus, it would be anticipated that a strain deleted for Med13 residues 742–844 would have a similar phenotype. Taken together, these studies emphasize the need for further analysis of both yeast and human Med13 IDRs to understand the molecular details of how this region contributes to Med13 function.
Role of SCFFbw7 and Med13 in higher eukaryotes following stress
Our previous studies described the conserved role of cyclin C in stressed and unstressed cells (Cooper et al., 2014; Wang et al., 2015). However, differences are observed between yeast and humans in cyclin C regulation. In yeast, all of cyclin C is released into the cytoplasm, followed by its destruction by the Not1 ubiquitin ligase. Conversely, only ∼10% of total cyclin C is released from the nucleus in mammalian cells following oxidative stress, and we do not detect any significant changes in its overall protein levels (Wang et al., 2015). Steady state turnover of the human Med13 is directed by the analogous SCF ubiquitin ligase by a homologue of Grr1, called Fbw1 (Davis et al., 2013). As Med13 tethers the Cdk8 module to the Mediator complex (Knuesel et al., 2009a), Med13 turnover determines the promoter residency of the module and therefore impacts transcriptional regulation. However, we found that the phosphodegron directing the turnover of Med13 in human cells (T326) is not required for stress-induced destruction of Med13 in yeast. These results suggest either that a different degron system is employed in these two systems or that stress-induced destruction is regulated differently than that observed under normal growth conditions. Consistent with this is the observation that Cdk substrates are often clustered in regions of intrinsic disorder (reviewed in Enserink and Kolodner, 2010). However, similar to other IDRs, their exact position in the protein is often poorly conserved in evolution, indicating that precise positioning of phosphorylation may not be required to permit a shared function (reviewed in Enserink and Kolodner, 2010). Thus, it is possible that following oxidative stress in human cells, a similar two-kinase mechanism could regulate Med13 degradation. In support of this model, phosphorylation by two kinases has been observed for other SCFFbw7 substrates (Akhoondi et al., 2007) including oncogenic transcription factors. This has led to Fbw7 being classified as a tumor suppressor (reviewed in Welcker and Clurman, 2008). Fbw7 degrades numerous oncogenic transcription factors (e.g., Myc, Notch, and Jun), as well as other proteins that contribute to carcinogenesis. As cyclin C has also recently been given this tumor suppressor designation (Li et al., 2014), it is imperative to understand the relationship between these proteins following stress in higher eukaryotes.
MATERIALS AND METHODS
Yeast strains and plasmids
All experiments, except the Y2H assays, were performed on the S. cerevisiae W303 strain RSY10 (Strich et al., 1989) and are listed in Supplemental Table S1. The Y2H assays were performed on PJ69-4a (James et al., 1996). In accordance with the Mediator nomenclature unification effort (Bourbon et al., 2004), cyclin C (SSN8/UME3/SRB11) and Cdk8 (SSN3/UME5/SRB10) will use the CNC1 and CDK8 gene designations, respectively. For reference, the human cyclin C gene name is CCNC. The cnc1∆, cdk8∆, med13∆, slt2∆, kdx1∆, and kdx1 slt2∆ strains have been previously described (Cooper et al., 1997, 2014; Krasley et al., 2006; Jin et al., 2014). The grr1∆ strain RSY1770 and the myc-,tagged Med12 strain (RSY1787) were constructed using gene replacement methodology as described (Longtine et al., 1998). The MED13-13myc allele was generated in RSY1770 to create RSY1771 using the same methodology. RSY2066 was made by integrating pMK634 (Gnanasundram and Kos, 2015) into the CDK8 locus of RSY10. The strain was then transformed with pCM188 (Gari et al., 1997), which contains the TET activator on a plasmid. The yeast hybrid strain PJ69-4a was obtained from the Yeast Resource center, courtesy of a gift from S. Fields (University of Washington). All cells were grown at 30°C, apart from slt2∆ and slt2∆ kdx1∆, which were grown at 23°C.
All plasmids used in this study are listed in Supplemental Table S2. The wild-type epitope tagged plasmids pHY1066 (MED13-HA, 2 µ), pKC337 (ADH1Pro-cyclin C-myc), pUM511 (GPD1Pro-CDK8-HA), and pBK38 (ADH1Pro-CNC1-YFP) are functional and have been described previously (Cooper et al., 1997, 2012; Cooper and Strich, 1999; Chang et al., 2004; Jin et al., 2013). pLR141 was made by cloning the ADH1Pro-CNC1-myc, CYCTerm from pKC337 into pRS426. The 2 µ MED13-HA plasmids (PHY1066 and PHY1089) and were a gift from P. Herman (Ohio State University) (Chang et al., 2004). The wild-type MED13 centromere–based plasmids pKC801 and pKC803 were made by PCR cloning the ADH1 promoter MED13-3HA ORF and the MED13 terminator from PHY1022 into pRS316 and pRS315, respectively. Site-directed mutagenesis (New England Bio-Rad Q5) was used to create plasmids harboring amino acid mutations, except for DS36 which was made using the Change-IT kit by Affymetrix. The CNC1S266A and the SLT2K54R (a gift from D. Levin, Boston University) have been described previously (Kim et al., 2008; Jin et al., 2014). The MED13 Y2H plasmids were constructed by PCR cloning from pHY1022 into the Xho1 site of the Gal4 activating domain plasmid pACT2. Likewise, the human cyclin C Y2H Gal4 binding domain plasmid pSW108 was made by PCR cloning of cyclin C cDNA amplified from pEGFP-cyclin C (unpublished data) into Nco1-digested pAS2. The GST-MED13 fusion plasmid was made by PCR cloning from PHY1022 into pGEX4T-1. Oligonucleotide sequences were used to make plasmids, and strains are available upon request. In short, all constructs were amplified from plasmid DNA using Phusion Taq (Thermo) digested using NEB fast digest restriction enzymes and ligated using Thermo fast ligase into their respective vectors. All protein fusion constructs were sequenced (Eurofins Genomics). Other plasmids that were used in this study that have been previously described are listed in Supplemental Table S2.
Cell growth
Yeast cells were grown in either rich, nonselective medium (YPDA) or synthetic minimal medium (SC), allowing plasmid selection as previously described (Cooper et al., 1997). For all experiments, the cells were grown to mid–log phase (∼6 × 106) before treatment with low concentrations of 0.4 mM H2O2, as previously described (Jin et al., 2013). Samples of 25 ml of cells were collected per time point and washed in water; then the pellets were flash frozen in liquid nitrogen. Y2H experiments were executed as described (Wang and Solomon, 2012). A sample of 10 mM 3-amino-1,2,4-triazole (3-AT) was added to the selective plates to increase the stringency of the interactions. E. coli cells were grown in LB medium with selective antibiotics. The degron experiments described in Figure 5, D and E, were executed as follows. In Figure 5D, cells (RSY2066) were grown to midlog and samples removed at the time points shown for analysis of Ubi-isoleucine::3HA-Cdk8 degradation after the addition of 2 µg/ml doxycycline. In Figure 5E, RSY2066 was transformed with Med13-3HA plasmid (pKC803), grown to midlog, and split and into one flask 2 µg/ml doxycycline was added. After 60 min, 0.8 mM H2O2 was added to both sets of cells and samples taken for NaOH lysis and Western analysis as described below.
Western blot analysis
Tagged Med13 constructs were detected using NaOH lysis of cell pellets as described by Zhang et al. (2011). In short, the frozen cell pellets were defrosted on ice, resuspended in 2 M LiOAc on ice for 5 min, centrifuged, and then resuspended in 0.4 M NaOH for a further 5 min on ice. Thereafter, the pellets were resuspended in 100 µl 2X SDS loading dye (Cooper et al., 1997) and boiled for 5 min, and 15 µl was loaded onto a 6% SDS–PAGE gel (Novagen). For the phos-tag gel (Figure 9B), proteins were prepared as just described and analyzed on a 7.5% polyacrylamide gel containing 7.5 µM phos-tag (Wako Laboratory Chemicals) and 15 µM MnCl2. The proteins were transferred to a nylon membrane (Millipore) using 10% methanol, 0.02% SDS, and 10X running buffer (Cooper et al., 1997). To detect Med13-myc, Med12-myc, and Med13-HA, 1-in-5000 dilutions of either anti-myc (Roche) or anti-HA antibodies (Clontech) were used. Tub1 and Pgk1 were visualized as previously described (Tan et al., 2011), except that anti–alpha tubulin antibodies (12G10) were obtained from the Developmental Studies Hybridoma Bank, University of Iowa. Western blot signals were detected using either goat anti-mouse or goat anti-rabbit secondary antibodies conjugated to alkaline phosphatase (Sigma) and the CDP-Star chemiluminescence kit (Tropix). Signals were quantitated by CCD camera imaging (Kodak). All degradation assays were performed more than once. SEM were generated for each point and error bars are indicated on the graphs, which were generated using the GraphPad Prism 7 program.
Fluorescence microscopy
YFP-cyclin C subcellular localization and mitochondrial morphology were monitored as described previously (Cooper et al., 2012, 2014). For all experiments, the cells were grown to midlog (6 × 106 cells/ml), treated with 0.4 mM H2O2 for the time points indicated, and then analyzed by fluorescence microscopy. Cyclin C-YFP export analysis was performed on cells fixed with 4% paraformaldehyde/3.4% sucrose for 1 h at room temperature. The cells were washed three times in water and prepared for fluorescence microscopy as described previously (Guacci et al., 1997) using mounting medium (10 mg/ml p-phenylenediamine, 50 ng/ml 4′,6-diamidino-2-phenylindole [DAPI]) to visualize nuclei and prevent photo bleaching. Images were obtained using a Nikon microscope (Model E800) with a 100× objective with 1.2× camera magnification (Plan Fluor Oil, NA 1.3) and a CCD camera (Hamamatsu Model C4742). Data were collected using NIS software and processed using Image Pro software. All images of individual cells were optically sectioned (0.2 µM slices at 0.3 µM spacing) and deconvolved, and the slices were collapsed to visualize the entire fluorescent signal within the cell. Cyclin C-YFP foci were scored as being cytoplasmic when three or more foci were observed outside the nucleus. Mitochondrial fission assays were performed on live cells as described (Cooper et al., 2014). In brief, mitochondrial fission was scored positive if no reticular mitochondria were observed that transversed half the cell diameter. Fusion was scored when cells exhibited one or more reticular mitochondria the diameter of the cell. Fission and fusion was scored for 200 cells from three independent isolates. Statistical analysis was performed using Student’s t test, with p < 0.05 used to indicate significant differences.
Pull-down assays and kinase assays
Purified proteins that were used in the pull-down assay (Figure 4B) were made as follows. The GST constructs (DS30, GST hCCNC, and GST alone) were transformed into the BL21 DE3 E. coli strain and expression was induced with 0.5 M IPTG at 37°C for 3 h. Cell pellets were resuspended in 50 ml GST lysis buffer (150 mM KCl, 50 mM HEPES, pH 7.5, 1 mM βME) and sonicated (25 × 10 s bursts), and the supernatant was recovered by centrifugation at 40,000 × g for 30 min. The supernatant was then added to 1 ml of washed glutathione beads (LifeTech) rotated at 4°C for 1 h. The unbound fraction was removed from the beads by gravity flow. The beads were washed twice with lysis buffer and the bound protein was eluted in five 1 ml fractions with the elution buffer (lysis buffer supplemented with 10 mM reduced glutathione). The protein-containing fractions were pooled and then desalted using 4 ml Zebra7K desalting columns that had been preequilibrated with the lysis buffer. The protein fraction was supplemented with glycerol to a final concentration of 10% and stored at –80°C in small aliquots. His6 tagged human or yeast cyclin C was purified the same way with the following exceptions. The lysis buffer (buffer A) was 500 mM KCl, 50 mM HEPES, pH 7.5, 1 mM βME, and 20 mM imidazole. Talon beads (Clontech) were used and washed with buffer B (150 mM KCl, 50 mM HEPES, pH 7.5, 1 mM βME , and 20 mM imidazole) and the bound protein was eluted in elution buffer (150 mM KCl, 50 mM HEPES, pH 7.5, 1 mM βME, and 500 mM imidazole). The desalting columns were preequilibrated with the B buffer without imidazole. The concentrations of all the recombinant proteins were determined by Bradford assay. The pull-down assays in Figure 4, B and C, were performed using His6-tagged human or yeast cyclin C, respectively, as bait and GST-tagged Med13742–844 (DS30) as target. A quantity of 500 nM cyclin C was mixed with 1 μM of Med13742–844 in a final reaction volume of 250 µl with B buffer and incubated for 1 h at room temperature. Bound protein was precipitated with 100 µl of washed Talon beads and eluted using 500 mM imidazole. The protein mixture was resolved using SDS–PAGE and Western blotting with either anti-GST (Abcam), anti-His (Bethal), or anti–human cyclin C antibodies (Bethal). Controls with only the target protein and no bait protein were included in all the experiments. The pull-down assays in Figure 4D were performed using 500 nM of either GST or GST-tagged yeast cyclin C and 1 µM His6-Med13571–660 (DS22). The proteins were incubated together in GST lysis buffer and incubated at RT for 1 h. Then 100 μl of GSH beads (preequilibrated with GST wash/lysis buffer) was added to the protein mixture and incubated with rotation for 1 h. The beads were centrifuged for 30 s washed twice with 500 μl of GST wash/lysis buffer. The protein mixture was resolved using SDS–PAGE and Western blotting with either anti-GST or anti-His antibodies.
The in vitro kinase assays were executed basically as described by DeMille et al. (2015). Slt2-HA and Slt2K54R-HA kinases were immunoprecipitated from cells treated with 5 mM sodium orthovanadate for 60 min, which activates Slt2 (Martin et al., 2000). In short, yeast extracts harboring the respective plasmids were made by resuspending cells in lysis buffer (20 mM HEPES, 10 mM KCl, 1 mM EDTA, 1 mM ethylene glycol tetraacetic acid [EGTA], 50 mM NaCl, 10% glycerol, 1 mM β-mercaptoethanol, and cOmplete Protease Inhibitor Cocktail Tablet, pH 7.4, with phosphatase inhibitors for Cdk8-HA purification) and protein immunoprecipitation was conducted as previously described (DeMille et al., 2015), with anti-HA magnetic beads (PierceTM)/anti-myc magnetic beads (Cell Signaling). Med13571–906 was purified from an E. coli BL21 DE3 strain induced with 0.5 mM IPTG for 5 h at 37°C using the 6X His-tagged purification method previously described (DeMille et al., 2015). The Slt2 in vitro kinase assay was conducted as previously described (Carmody et al., 2010) by resuspending Slt2 beads in kinase buffer (20 mM HEPES, 10 mM MgCl2, 100 μM Na3VO4, pH 7.5), with 20 μM ATP, 5 μCi of 32P-ATP, and purified Med13571–907. Reactions were incubated at 30°C for 30 min.
Supplementary Material
Acknowledgments
We thank C. Wittenburg, S. Fields, P. Herman, D. Levin, and M. Solomon for strains and plasmids. We also thank A. Sardallah and S. Khakhina for help in making Med13 plasmids and the Med12-myc strain, respectively. We thank members of the Cooper, Strich, and Grose laboratories for critical reading of the manuscript. This work was supported by grants from the National Institutes of Health (NIH) awarded to K.F.C. (GM113196), R.S. (GM113052), and J.G. (GM100376) and by the New Jersey Cancer Commission (DHFS16PPC067) to V.G. The Y2H strain was made by work supported by NIH Grant P41 RR11823, awarded to T.N. Davis.
Abbreviations used:
- 3-AT
3-amino-1,2,4-triazole
- βME
β-mercaptoethanol
- Cdk
cyclin-dependent kinase
- CKM
cyclin C/Cdk8 kinase module
- IDR
intrinsic disordered region
- MAPK
MAP kinase
- MAPKKK
MAP kinase kinase kinase
- MoRF
molecular recognition feature
- ORF
open reading frame
- PKA
protein kinase A
- RCD
regulated cell death
- ROS
reactive oxygen species
- SCF
Skip1, Cullin, F box E3 ligase
- Y2H
yeast two-hybrid.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-08-0493) on December 6, 2017.
REFERENCES
- Adegbola A, Musante L, Callewaert B, Maciel P, Hu H, Isidor B, Picker-Minh S, Le Caignec C, Delle Chiaie B, Vanakker O, et al. Redefining the MED13L syndrome. Eur J Hum Genet. 2015;23:1308–1317. doi: 10.1038/ejhg.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteom. 2008;7:1389–1396. doi: 10.1074/mcp.M700468-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Taatjes DJ. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16:155–166. doi: 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang XL, Wade Harper J. SCF-mediated protein degradation and cell cycle control. Oncogene. 2005;24:2860–2870. doi: 10.1038/sj.onc.1208614. [DOI] [PubMed] [Google Scholar]
- Asadollahi R, Zweier M, Gogoll L, Schiffmann R, Sticht H, Steindl K, Rauch A. Genotype-phenotype evaluation of MED13L defects in the light of a novel truncating and a recurrent missense mutation. Eur J Med Genet. 2017;60:451–464. doi: 10.1016/j.ejmg.2017.06.004. [DOI] [PubMed] [Google Scholar]
- Asano S, Park JE, Sakchaisri K, Yu LR, Song S, Supavilai P, Veenstra TD, Lee KS. Concerted mechanism of Swe1/Wee1 regulation by multiple kinases in budding yeast. EMBO J. 2005;24:2194–2204. doi: 10.1038/sj.emboj.7600683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- Benanti JA, Cheung SK, Brady MC, Toczyski DP. A proteomic screen reveals SCFGrr1 targets that regulate the glycolytic–gluconeogenic switch. Nat Cell Biol. 2007;9:1184–1191. doi: 10.1038/ncb1639. [DOI] [PubMed] [Google Scholar]
- Bourbon HM. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 2008;36:3993–4008. doi: 10.1093/nar/gkn349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourbon HM, Aguilera A, Ansari AZ, Asturias FJ, Berk AJ, Bjorklund S, Blackwell TK, Borggrefe T, Carey M, Carlson M, et al. A unified nomenclature for protein subunits of mediator complexes linking transcriptional regulators to RNA polymerase II. Mol Cell. 2004;14:553–557. doi: 10.1016/j.molcel.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Carmody SR, Tran EJ, Apponi LH, Corbett AH, Wente SR. The mitogen-activated protein kinase Slt2 regulates nuclear retention of non–heat shock mRNAs during heat shock–induced stress. Mol Cell Biol. 2010;30:5168–5179. doi: 10.1128/MCB.00735-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Gutierrez D, Eisenberg T, Buttner S, Meisinger C, Kroemer G, Madeo F. Apoptosis in yeast: triggers, pathways, subroutines. Cell Death Differ. 2010;17:763–773. doi: 10.1038/cdd.2009.219. [DOI] [PubMed] [Google Scholar]
- Chang YW, Howard SC, Herman PK. The Ras/PKA signaling pathway directly targets the Srb9 protein, a component of the general RNA polymerase II transcription apparatus. Mol Cell. 2004;15:107–116. doi: 10.1016/j.molcel.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Chi Y, Huddleston MJ, Zhang X, Young RA, Annan RS, Carr SA, Deshaies RJ. Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev. 2001;15:1078–1092. doi: 10.1101/gad.867501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MO, Yu L, Campuzano I, Grant SG, Choudhary JS. Phosphoproteomic analysis of the mouse brain cytosol reveals a predominance of protein phosphorylation in regions of intrinsic sequence disorder. Mol Cell Proteom. 2008;7:1331–1348. doi: 10.1074/mcp.M700564-MCP200. [DOI] [PubMed] [Google Scholar]
- Cooper KF, Khakhina S, Kim SK, Strich R. Stress-induced nuclear-to-cytoplasmic translocation of cyclin C promotes mitochondrial fission in yeast. Dev Cell. 2014;28:161–173. doi: 10.1016/j.devcel.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KF, Mallory MJ, Smith JB, Strich R. Stress and developmental regulation of the yeast C-type cyclin Ume3p (Srb11p/Ssn8p) EMBO J. 1997;16:4665–4675. doi: 10.1093/emboj/16.15.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KF, Scarnati MS, Krasley E, Mallory MJ, Jin C, Law MJ, Strich R. Oxidative-stress-induced nuclear to cytoplasmic relocalization is required for Not4-dependent cyclin C destruction. J Cell Sci. 2012;125:1015–1026. doi: 10.1242/jcs.096479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KF, Strich R. Functional analysis of the Ume3p/ Srb11p-RNA polymerase II holoenzyme interaction. Gene Expr. 1999;8:43–57. [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Larimore EA, Fissel BM, Swanger J, Taatjes DJ, Clurman BE. The SCF-Fbw7 ubiquitin ligase degrades MED13 and MED13L and regulates CDK8 module association with Mediator. Genes Dev. 2013;27:151–156. doi: 10.1101/gad.207720.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMille D, Badal BD, Evans JB, Mathis AD, Anderson JF, Grose JH. PAS kinase is activated by direct SNF1-dependent phosphorylation and mediates inhibition of TORC1 through the phosphorylation and activation of Pbp1. Mol Biol Cell. 2015;26:569–582. doi: 10.1091/mbc.E14-06-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diella F, Haslam N, Chica C, Budd A, Michael S, Brown NP, Trave G, Gibson TJ. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci. 2008;13:6580–6603. doi: 10.2741/3175. [DOI] [PubMed] [Google Scholar]
- Dodou E, Treisman R. The Saccharomyces cerevisiae MADS-box transcription factor Rlm1 is a target for the Mpk1 mitogen-activated protein kinase pathway. Mol Cell Biol. 1997;17:1848–1859. doi: 10.1128/mcb.17.4.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- Dosztanyi Z, Meszaros B, Simon I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–2746. doi: 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Kolodner RD. An overview of Cdk1-controlled targets and processes. Cell Div. 2010;5:11. doi: 10.1186/1747-1028-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick JS, Johnston M. GRR1 of Saccharomyces cerevisiae is required for glucose repression and encodes a protein with leucine-rich repeats. Mol Cell Biol. 1991;11:5101–5112. doi: 10.1128/mcb.11.10.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M, Tompa P, Simon I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics. 2007;23:950–956. doi: 10.1093/bioinformatics/btm035. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M, Toth-Petroczy A, Kraut DA, Matouschek A, Lim RY, Xue B, Kurgan L, Uversky VN. Disordered proteinaceous machines. Chem Rev. 2014;114:6806–6843. doi: 10.1021/cr4007329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58–73. doi: 10.1038/cdd.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vanden Berghe T, Vanlangenakker N, Buettner S, Eisenberg T, Vandenabeele P, Madeo F, Kroemer G. Programmed necrosis from molecules to health and disease. Int Rev Cell Mol Biol. 2011;289:1–35. doi: 10.1016/B978-0-12-386039-2.00001-8. [DOI] [PubMed] [Google Scholar]
- Gari E, Piedrafita L, Aldea M, Herrero E. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast. 1997;13:837–848. doi: 10.1002/(SICI)1097-0061(199707)13:9<837::AID-YEA145>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Gnanasundram SV, Kos M. Fast protein-depletion system utilizing tetracycline repressible promoter and N-end rule in yeast. Mol Biol Cell. 2015;26:762–768. doi: 10.1091/mbc.E14-07-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomar-Alba M, Mendez E, Quilis I, Bano MC, Igual JC. Whi7 is an unstable cell-cycle repressor of the Start transcriptional program. Nat Commun. 2017;8:329. doi: 10.1038/s41467-017-00374-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez D, Hamidi N, Del Sol R, Benschop JJ, Nancy T, Li C, Francis L, Tzouros M, Krijgsveld J, Holstege FC, Conlan RS. Suppression of Mediator is regulated by Cdk8-dependent Grr1 turnover of the Med3 coactivator. Proc Natl Acad Sci USA. 2014;111:2500–2505. doi: 10.1073/pnas.1307525111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci V, Hogan E, Koshland D. Centromere position in budding yeast: evidence for anaphase A. Mol Biol Cell. 1997;8:957–972. doi: 10.1091/mbc.8.6.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiung YG, Chang HC, Pellequer JL, La Valle R, Lanker S, Wittenberg C. F-box protein Grr1 interacts with phosphorylated targets via the cationic surface of its leucine-rich repeat. Mol Cell Biol. 2001;21:2506–2520. doi: 10.1128/MCB.21.7.2506-2520.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Kim KK, Willis SD, Cooper KF. The MAPKKKs Ste11 and Bck1 jointly transduce the high oxidative stress signal through the cell wall integrity MAP kinase pathway. Microb Cell. 2015;2:239–242. doi: 10.15698/mic2015.09.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Parshin AV, Daly I, Strich R, Cooper KF. The cell wall sensors Mtl1, Wsc1, and Mid2 are required for stress-induced nuclear to cytoplasmic translocation of cyclin C and programmed cell death in yeast. Oxid Med Cell Longev. 2013;2013:320823. doi: 10.1155/2013/320823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Strich R, Cooper KF. Slt2p phosphorylation induces cyclin C nuclear-to-cytoplasmic translocation in response to oxidative stress. Mol Biol Cell. 2014;25:1396–1407. doi: 10.1091/mbc.E13-09-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung US, Sobering AK, Romeo MJ, Levin DE. Regulation of the yeast Rlm1 transcription factor by the Mpk1 cell wall integrity MAP kinase. Mol Microbiol. 2002;46:781–789. doi: 10.1046/j.1365-2958.2002.03198.x. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakhina S, Cooper KF, Strich R. Med13p prevents mitochondrial fission and programmed cell death in yeast through nuclear retention of cyclin C. Mol Biol Cell. 2014;25:2807–2816. doi: 10.1091/mbc.E14-05-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Truman AW, Levin DE. Yeast Mpk1 mitogen-activated protein kinase activates transcription through Swi4/Swi6 by a noncatalytic mechanism that requires upstream signal. Mol Cell Biol. 2008;28:2579–2589. doi: 10.1128/MCB.01795-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteom. 2006;5:749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 2009a;23:439–451. doi: 10.1101/gad.1767009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Donner AJ, Espinosa JM, Taatjes DJ. The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol Cell Biol. 2009b;29:650–661. doi: 10.1128/MCB.00993-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotta-Loizou I, Tsaousis GN, Hamodrakas SJ. Analysis of molecular recognition features (MoRFs) in membrane proteins. Biochim Biophys Acta. 2013;1834:798–807. doi: 10.1016/j.bbapap.2013.01.006. [DOI] [PubMed] [Google Scholar]
- Krasley E, Cooper KF, Mallory MJ, Dunbrack R, Strich R. Regulation of the oxidative stress response through Slt2p-dependent destruction of cyclin C in Saccharomyces cerevisiae. Genetics. 2006;172:1477–1486. doi: 10.1534/genetics.105.052266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Fassl A, Chick J, Inuzuka H, Li X, Mansour MR, Liu L, Wang H, King B, Shaik S, et al. Cyclin C is a haploinsufficient tumour suppressor. Nat Cell Biol. 2014;16:1080–1091. doi: 10.1038/ncb3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HY, Chiang YC, Pan J, Chen J, Salvadore C, Audino DC, Badarinarayana V, Palaniswamy V, Anderson B, Denis CL. Characterization of CAF4 and CAF16 reveals a functional connection between the CCR4-NOT complex and a subset of SRB proteins of the RNA polymerase II holoenzyme. J Biol Chem. 2001;276:7541–7548. doi: 10.1074/jbc.M009112200. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Madeo F, Frohlich E, Ligr M, Grey M, Sigrist SJ, Wolf DH, Frohlich KU. Oxygen stress: a regulator of apoptosis in yeast. J Cell Biol. 1999;145:757–767. doi: 10.1083/jcb.145.4.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin H, Rodriguez-Pachon JM, Ruiz C, Nombela C, Molina M. Regulatory mechanisms for modulation of signaling through the cell integrity Slt2-mediated pathway in Saccharomyces cerevisiae. J Biol Chem. 2000;275:1511–1519. doi: 10.1074/jbc.275.2.1511. [DOI] [PubMed] [Google Scholar]
- Muncke N, Jung C, Rudiger H, Ulmer H, Roeth R, Hubert A, Goldmuntz E, Driscoll D, Goodship J, Schon K, Rappold G. Missense mutations and gene interruption in PROSIT240, a novel TRAP240-like gene, in patients with congenital heart defect (transposition of the great arteries) Circulation. 2003;108:2843–2850. doi: 10.1161/01.CIR.0000103684.77636.CD. [DOI] [PubMed] [Google Scholar]
- Nagulapalli M, Maji S, Dwivedi N, Dahiya P, Thakur JK. Evolution of disorder in Mediator complex and its functional relevance. Nucleic Acids Res. 2016;44:1591–1612. doi: 10.1093/nar/gkv1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA. Cellular substrates of p34(cdc2) and its companion cyclin-dependent kinases. Trends Cell Biol. 1993;3:296–301. doi: 10.1016/0962-8924(93)90011-o. [DOI] [PubMed] [Google Scholar]
- Poss ZC, Ebmeier CC, Odell AT, Tangpeerachaikul A, Lee T, Pelish HE, Shair MD, Dowell RD, Old WM, Taatjes DJ. Identification of mediator kinase substrates in human cells using cortistatin A and quantitative phosphoproteomics. Cell Rep. 2016;15:436–450. doi: 10.1016/j.celrep.2016.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin–ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Staleva L, Hall A, Orlow SJ. Oxidative stress activates FUS1 and RLM1 transcription in the yeast Saccharomyces cerevisiae in an oxidant-dependent Manner. Mol Biol Cell. 2004;15:5574–5582. doi: 10.1091/mbc.E04-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strich R, Cooper KF. The dual role of cyclin C connects stress regulated gene expression to mitochondrial dynamics. Microb Cell. 2014;1:318–324. doi: 10.15698/mic2014.10.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strich R, Slater MR, Esposito RE. Identification of negative regulatory genes that govern the expression of early meiotic genes in yeast. Proc Natl Acad Sci USA. 1989;86:10018–10022. doi: 10.1073/pnas.86.24.10018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surosky RT, Strich R, Esposito RE. The yeast UME5 gene regulates the stability of meiotic mRNAs in response to glucose. Mol Cell Biol. 1994;14:3446–3458. doi: 10.1128/mcb.14.5.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan GS, Magurno J, Cooper KF. Ama1p-activated anaphase-promoting complex regulates the destruction of Cdc20p during meiosis II. Mol Biol Cell. 2011;22:315–326. doi: 10.1091/mbc.E10-04-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth-Petroczy A, Oldfield CJ, Simon I, Takagi Y, Dunker AK, Uversky VN, Fuxreiter M. Malleable machines in transcription regulation: the mediator complex. PLoS Comput Biol. 2008;4:e1000243. doi: 10.1371/journal.pcbi.1000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN. The most important thing is the tail: multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 2013;587:1891–1901. doi: 10.1016/j.febslet.2013.04.042. [DOI] [PubMed] [Google Scholar]
- van de Peppel J, Kettelarij N, van Bakel H, Kockelkorn TT, van Leenen D, Holstege FC. Mediator expression profiling epistasis reveals a signal transduction pathway with antagonistic submodules and highly specific downstream targets. Mol Cell. 2005;19:511–522. doi: 10.1016/j.molcel.2005.06.033. [DOI] [PubMed] [Google Scholar]
- van Haelst MM, Monroe GR, Duran K, van Binsbergen E, Breur JM, Giltay JC, van Haaften G. Further confirmation of the MED13L haploinsufficiency syndrome. Eur J Hum Genet. 2015;23:135–138. doi: 10.1038/ejhg.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. The N-end rule. Cell. 1992;69:725–735. doi: 10.1016/0092-8674(92)90285-k. [DOI] [PubMed] [Google Scholar]
- Vuzman D, Hoffman Y, Levy Y. Modulating protein-DNA interactions by post-translational modifications at disordered regions. Pac Symp Biocomput. 2012:188–199. [PubMed] [Google Scholar]
- Wang K, Yan R, Cooper KF, Strich R. Cyclin C mediates stress-induced mitochondrial fission and apoptosis. Mol Biol Cell. 2015;26:1030–1043. doi: 10.1091/mbc.E14-08-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Solomon MJ. Identification of She3 as an SCF(Grr1) substrate in budding yeast. PloS One. 2012;7:e48020. doi: 10.1371/journal.pone.0048020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci USA. 2004;101:4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Takaesu G, Hagiwara M, Irie K, Matsumoto K. Characterization of a serum response factor-like protein in Saccharomyces cerevisiae, Rlm1, which has transcriptional activity regulated by the Mpk1 (Slt2) mitogen-activated protein kinase pathway. Mol Cell Biol. 1997;17:2615–2623. doi: 10.1128/mcb.17.5.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- Wery M, Shematorova E, Van Driessche B, Vandenhaute J, Thuriaux P, Van Mullem V. Members of the SAGA and Mediator complexes are partners of the transcription elongation factor TFIIS. EMBO J. 2004;23:4232–4242. doi: 10.1038/sj.emboj.7600326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachdav G, Kloppmann E, Kajan L, Hecht M, Goldberg T, Hamp T, Honigschmid P, Schafferhans A, Roos M, Bernhofer M, et al. PredictProtein—an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014;42:W337–W343. doi: 10.1093/nar/gku366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Shimojima K, Ondo Y, Shimakawa S, Okamoto N. MED13L haploinsufficiency syndrome: a de novo frameshift and recurrent intragenic deletions due to parental mosaicism. Am J Med Genet A. 2017;173:1264–1269. doi: 10.1002/ajmg.a.38168. [DOI] [PubMed] [Google Scholar]
- Yin JW, Wang G. The Mediator complex: a master coordinator of transcription and cell lineage development. Development. 2014;141:977–987. doi: 10.1242/dev.098392. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Kono K, Lowery DM, Bartolini S, Yaffe MB, Ohya Y, Pellman D. Polo-like kinase Cdc5 controls the local activation of Rho1 to promote cytokinesis. Science. 2006;313:108–111. doi: 10.1126/science.1126747. [DOI] [PubMed] [Google Scholar]
- Zhang T, Lei J, Yang H, Xu K, Wang R, Zhang Z. An improved method for whole protein extraction from yeast Saccharomyces cerevisiae. Yeast. 2011;28:795–798. doi: 10.1002/yea.1905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.