

Abstract
Therapeutic efficacy of gemcitabine (GEM) is severely limited due to its rapid metabolism by enzymatic deamination in vivo. We recently determined its therapeutic efficacy before (F-GEM) and after conjugation to poly(ethylene glycol)-block-poly(2-methyl-2-carboxyl-propylene carbonate) (mPEG-b-PCC-g-GEM-g-DC, abbreviated as P-GEM) in subcutaneous and orthotopic pancreatic tumor bearing mice. In this study, pharmacokinetic (PK) parameters and biodistribution profiles of F-GEM and P-GEM were determined after intravenous injection into orthotopic pancreatic tumor bearing NSG mice. To assess the short-term toxicity, the levels of hematological, hepatic, and renal injury markers were measured after 24 h postadministration into these mice. P-GEM was distributed to all the major organs, with higher accumulation in the liver, spleen, and tumor compared to F-GEM. Area under the curve (AUC), elimination half-life (t1/2), and mean residence time (MRT) of P-GEM treated group were significantly higher compared to those of F-GEM treated group: 246,425 ± 1605 vs 83,591 ± 1844 ng/mL × h as AUC, 5.77 ± 2.02 vs 1.99 ± 0.09 h as t1/2, and 4.45 ± 0.15 vs 1.12 ± 0.13 h as MRT. Further, P-GEM exhibited negligible systemic toxicity as evidenced by almost similar alanine aminotransferase (ALT) and aspartate aminotransferase (AST) values for both P-GEM and F-GEM. These results suggest that P-GEM protects GEM from degradation and provides sustained drug release, resulting in enhanced GEM delivery to the tumor by more than 2.5-fold compared to F-GEM. Hence, P-GEM is a promising gemcitabine conjugated polymeric micelle for treating pancreatic cancer.
Keywords: gemcitabine, polymeric micelles, pancreatic cancer, pharmacokinetics, biodistribution
Graphical abstract
Introduction
Pancreatic ductal adenocarcinoma (PDCA) is the fourth leading cause of cancer related deaths in the United States.1 Currently, surgical resection is the only treatment, although 9% to 15% of patients are suitable for surgery and the median survival rate for all stages of pancreatic cancer is 3 to 5 months after diagnosis;2,3 however, the major challenges are the potential metastatic spread, high local recurrence, and chemoresistance induced by cancer stem cells.4,5 2,2′-Difluorodeoxyribofuranosylcytosine (commonly known as gemcitabine or GEM) is the first line therapy for pancreatic cancer. However, this drug has narrow therapeutic index due to its rapid enzymatic deamination leading to the formation of its inactive metabolite 2,2′-difluorodeoxyuridine (dFdU) with a short half-life of 13.7 min.6 Further, GEM induces drug resistance due to the downregulation of deoxycytidine kinase enzyme responsible for the primary phosphorylation of GEM.7 To enhance the in vivo stability of GEM and control its pharmacokinetic profiles, various formulations, and drug delivery strategies have been reported.
Drug delivery approaches to increase the overall intracellular GEM concentration via improved pharmacokinetics have been widely applied.8,9 Nanoformulations have been extensively applied to address the plasma instability, metabolic inactivation, and subsequent deamination of GEM into its inactive uracil derivatives. These include squalenoyl GEM conjugate,10 GEM loaded PEGylated unilamellar liposomes,11 incorporation into polyaspartylhydrazide copolymer-based supramolecular vesicular aggregates,12 and encapsulation of GEM lipophilic derivatives into polycyanoacrylate nanospheres and nano-capsules.13 Squalene (SQ) conjugated GEM encapsulated into liposomes has shown promising anticancer efficacy in leukemia cancer model.14 For elevated intracellular drug fate, Bildstein et al. demonstrated that SQdFdC application lead to better pharmacokinetic profiles and increased tumor accumulation in resistant cancer cells. However, these formulations were taken up by the cells of reticuloendothelial system (RES), leading to their high accumulation in the liver and spleen.15 Consequent increased toxicity demands new delivery approaches.
We recently conjugated GEM to the carboxyl pendant groups of methoxy poly(ethylene glycol)-block-poly(2-methyl-2-carboxyl-propylene carbonate) (mPEG-b-PCC), which self-assembled into micelles and resulted in its improved plasma stability, sustained drug release, enhanced internalization, and apoptosis of pancreatic cancer cells.16 Further, this GEM conjugated polymeric (mPEG-b-PCC-g-GEM-g-DC, abbreviated as P-GEM) micelles significantly inhibited subcutaneous as well as orthotopic xenograft pancreatic tumor after intravenous administration compared to free gemcitabine (F-GEM).16,17 These findings further encouraged us to determine the in vivo fate of this polymer–drug conjugate. This polymer was chosen, since polycarbonates degrade into carbon dioxide and alcohol, resulting in little local inflammation. Since this amphiphilic copolymer self-assembles into micelles, P-GEM is expected to have enhanced circulation half-life and enhanced permeability and retention (EPR) effect, which should facilitate extravasation of these micelles to pancreatic tumor tissues.18
In this study, we determined the biodistribution and PK profiles of P-GEM after systemic administration into orthotopic pancreatic tumor bearing NSG mice and compared with free GEM by simultaneous quantification of drug and metabolite concentrations in plasma, tumor, and other major organs. The in vivo hepatic, renal, and hematological toxicities were also determined and compared with the control animals.
Materials and Methods
Chemicals
Gemcitabine hydrochloride was purchased from AK Scientific (Union City, CA). Dodecanol (DC), triethyl-amine (TEA), 1-ethyl-3-(3- (dimethylamino)propyl) carbodiimide (EDC), hydroxybenzotriazole (HOBT), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), benzyl bromide, 2,2-bis(hydroxymethyl) propionic acid, and methoxy poly(ethylene glycol) (mPEG, Mn = 5000, PDI = 1.03) were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification. Matrigel matrix basement membrane was procured from Corning (Chicago, IL). All other chemicals were of analytical grade and purchased from Sigma-Aldrich (St. Louis, MO).
Synthesis and Characterization of Polymers
The monomer 2-methyl-2-benzyloxycarbonyl-propylene carbonate (MBC) was synthesized in two-step reaction, as reported by our group.16,19 Briefly, PEG-MBC was synthesized by ring-opening polymerization of MBC and mPEG in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) catalyst at room temperature for 3 h under nitrogen atmosphere. mPEG-MBC was in tetrahydrofuran (THF): methanol (1:1, v/v)) hydrogenated in the presence of 10 wt % palladium on charcoal (Pd/C) to obtain the copolymer containing carboxyl pendant groups (mPEG-b-PCC). Finally, GEM and DC were conjugated to the polymer by carbodiimide coupling reaction to obtain P-GEM using EDC/HOBt. The product was purified by precipitation in excess chilled diethyl ether and then with isopropyl alcohol. The final precipitate was dissolved in acetone and dialyzed against water and lyophilized to obtain pure P-GEM.
Quantification of GEM Payload in mPEG-b-PCC-g-GEM-g-DC
The amount of GEM conjugated to the copolymer was determined by alkaline hydrolysis as described previously.16 Briefly, 5 mg of copolymeric micelles were hydrolyzed in 1 M NaOH at 40 °C for 1 h, and the samples were analyzed by HPLC-UV using the following experimental conditions: a 20 μL volume of the standard and sample was injected into an autosampler through Inertsil ODS-3 column (4.5 × 250 mm) at an isocratic flow rate of 1 mL/min with sodium acetate buffer (20 mM, pH 5.6)/methanol (93:07, v/v) as a mobile phase.
Preparation of Micelles
GEM conjugated micelles were prepared by film hydration by dissolving 10 mg of P-GEM in 200 μL of chloroform and evaporating on a rotary evaporator (Heidolph Instruments GmbH and Co., Germany). Following vacuum desiccation overnight, the film was rehydrated with phosphate buffered saline (PBS; pH 7.4), vortexed, sonicated, centrifuged, and filtered through Acrodisc Syringe Filter (13 mm × 0.2 μm) (Waters, Milford, MA) to prepare micelles.
Stable Transfection of MIA PaCa-2 cells with Lentiviral Particles
MIA PaCa-2 cells were kindly gifted by Dr. Rakesh K. Singh (UNMC, Omaha, NE) and maintained in Dulbecco's Modified Eagle Medium (DMEM) containing 10% FBS and 1% antibiotic in an incubator at 37 °C/5% CO2. The cells were stably transfected with lentiviral vector encoding luciferase and green fluorescent protein (GFP) (hLUC-Lv201-0200) using Polybrene according to the manufacturer's instructions. Briefly, 2 × 105 MIA PaCa-2 cells were seeded in each well in a six-well plate for 24 h. For each well, 2 μL of lentiviral particles was mixed with 1 μL of Polybrene (10 mg/mL), and the final volume was adjusted to 2 mL using DMEM. The transfection mixture was then incubated for 15 min at RT with occasional agitating, and MIA PaCa-2 cells were washed with PBS and incubated with the transfection mixture overnight. The medium was replaced with 2 mL of fresh DMEM for an additional 48 h. The cells were trypsinized and cultured in T-75 flasks for 15 days in 10 mL of DMEM containing 2 μg/mL puromycin. The medium was removed on every alternate day with a fresh DMEM containing 2 μg/mL puromycin. MIA PaCa-2 cells were then grown for another 4 days with fresh DMEM containing a lower concentration of puromycin (1 μg/mL). Luciferase and GFP expression from stably transfected cells were confirmed by in vivo IVIS and epifluorescence microscopy, respectively. Finally, cells were sorted to obtain luciferase and GFP reporter gene-expressing cell population using fluorescence-activated cell sorting (FACS). In vitro and in vivo experiments were performed with stable luciferase and GFP-transfected cells culturing in DMEM without puromycin.
Generation of Orthotopic Pancreatic Tumor
Animal experiments were carried out as per the NIH animal use guidelines and protocol approved by the Institutional Animal Care and Use Committee (IACUC), University of Nebraska Medical Center, Omaha, NE. The orthotopic pancreatic cancer mouse model was generated by implantation of MIA PaCa-2 cells using PBS/matrigel (1:1 v/v) into the pancreas of 6–8 week-old immunodeficient NSG mice. The mice were anesthetized with isoflurane; a left lateral abdominal incision was made and opened peritoneum. MIA PaCa-2 cells (∼2 × 106) were implanted to the pancreas through 27-gauge needle. The pancreas was returned back to the peritoneal cavity, and the abdominal wall and muscles was stitched by suturing. Tumor growth was monitored by using the IVIS Living Image System (Caliper Life Sciences). After 15 d postimplantation of luciferase and GFP stably expressing cells into mice, 100 mg/kg of d-luciferin was injected intraperitoneally into each mouse and bioluminescent was recorded.
Pharmacokinetics and Biodistribution
When the tumor volume reached to 400–500 mm3, each group was further divided into 13 subgroups (each subgroup contained four mice). Mice were anesthetized by inhalation of isoflurane, and P-GEM or F-GEM was injected via the tail vein at a GEM equivalent dose of 40 mg/kg. At 15 min, 30 min, 1 h, 4 h, 12 h, and 24 h, blood was collected by cardiac puncture in heparinized tubes containing 10 μL tetrahydrouridine (10 μg/mL in water) and stored on ice immediately. The animals were then sacrificed, and major organs (liver, kidney, spleen, heart, lung, and tumor) were collected, washed, blotted dry, weighed, and stored on dry ice and then at −80 °C. Extraction and quantification of GEM and its dFdU metabolite in collected samples were carried out using a validated HPLC/UV method.
Determination of Pharmacokinetic Profiles
Non-compartmental analysis of GEM plasma and tissue concentration vs time was performed using WinNonlin Professional (version 6.4, Pharsight, Sunnyvale, CA) to obtain pharmacokinetic (PK) parameters including the area under the curve (AUC), mean resident time (MRT), elimination half-life (t1/2), and apparent volume of distribution at steady state (Vss).
HPLC Methodology
Quantification of GEM and its dFdU metabolite concentrations was performed using a Waters HPLC system consisting of a 2695 pump, an autosampler, and a 996 photodiode array detector (Milford, MA) by using 5-methylcytidine as an internal standard (IS).16,20 Chromato-graphic separation was obtained by isocratic conditions on an analytical InertsilODS-3 column (4.5 mm × 250 mm × 5 μm) at room temperature. The mobile phase consisted of 20 mM acetate buffer/methanol (93:7, v/v) at pH 5.6 and flow rate was 1 mL/min. Standard curves were generated for every analytical run. Stock solutions of GEM, its dFdU metabolite, and 5-methylcytidine were prepared by dissolving separately weighed amounts in 2 mL of glass vials in water, and the solutions were stored at −20 °C. The dynamic calibration concentration range for both GEM and dFdU was 0.2–80 μg/mL for both mouse plasma and control tissue homogenates. To 100 μL of plasma, 20 μL of IS (10 μg/mL) was added. Plasma and tissue homogenate samples were treated with 25 μL glacial acetic acid to terminate possible deamination and precipitated with acetonitrile for plasma and mixture of acetonitrile and ethyl acetate (85:15, v/v) for tissue homogenates. The drug and metabolite extracts were centrifuged at 10,000 rpm for 10 min. The supernatants were evaporated in a nitrogen evaporator (Zymark Turbovap Evaporator, East Lyme, CT) at 40 °C. The dried samples were reconstituted in mobile phase, vortexed, centrifuged at 12000 rpm at 4 °C, syringe filtered, and injected into the HPLC system. The assay performance was assessed by running a triplicate standard curve, and extraction recoveries were determined at each calibration concentration. The average recoveries of GEM and dFdU from plasma were 89% and 91%, respectively. The retention time for IS, GEM, and dFdU metabolite were 5.8, 11.1, and 21.7 min, respectively. The recovery rate of GEM in tissue homogenates were in the range of 85%–93%. The total time for the analysis of each samples was 25 min.
In Vivo Toxicity Analysis
For evaluation of short-term toxicity, orthotopic pancreatic tumor bearing NSG mice were randomly divided into three groups: nontreated tumor bearing mice (control group) and F-GEM and P-GEM treated groups. Mice received F-GEM and P-GEM intravenously at a GEM equivalent dose of 40 mg/kg. Mice were euthanized after 24 h to collect blood, liver, kidney, and spleen to determine toxicity. Blood was collected in potassium EDTA microtainer tube (KE/1.3) via cardiac puncture. Approximately, ∼50 μL of blood was used and kept at room temperature to avoid hemolysis. Complete blood count (CBC) was performed with the Abaxis VetScan HM5 (Union City, CA). Blood chemistry markers (hepatic and renal) for each group were quantified on using VetScan liver and kidney profile rotors with the Abaxis VetScan VS2 analyzer. Approximately, 100 μL of blood was collected in lithium heparin tubes (LH/1.3).
Statistical Analysis
All data were presented as the mean ± SD. Data from different groups were compared using the Student's t test. A p value less than 0.01 was considered to be statistically significant.
Results
Synthesis and Characterization of P-GEM
We have synthesized P-GEM using mPEG-b-PCC, GEM, and dodecanol (DC) by carbodiimide coupling reaction using the same synthetic route as previous report (Figure S1).16,21 However, there were slight batch to batch variations in the number of PCC units grafted to the polymer backbone and the number of GEM molecules conjugated per PEG–PCC. The final purified P-GEM was characterized by 1HNMR (Figure S2). mPEG-b-PCC showed copolymer backbone peaks corresponding to PEG (–CH2–CH2–O) at δ 3.4–3.6 and –CH2 unit of PEG–PCC at δ 4.2–4.4. After the removal of pendant benzyl group by hydrogenation process, the characteristic peak of phenyl ring at δ 7.3 disappeared and –COOH were observed at δ 12–13.5. Average molecular weight of mPEG-b-PCC was 10 196 Da with 24 PCC units, as determined from integral value corresponding to protons of –CH2 of PEG and –CH2 of PCC units. The presence of amide proton at δ 8.2–8.4 confirmed the chemical conjugation of GEM through the formation of an amine bond with the pendant carboxyl group of mPEG-b-PCC, which is in agreement with the literatures.22,23 From the integration of –CONH– proton peak at δ 8.2–8.4, GEM loading in P-GEM was estimated to be 13.5% (Figure S2). However, 1H NMR can only confirm the conjugation of GEM to mPEG-b-PCC backbone but cannot predict accurate drug loading. Therefore, we also determined GEM loading in P-GEM by alkaline hydrolysis and then analyzed by HPLC-UV as reported previously.16 The mean particle size of these micelles was 30 nm with PDI = 0.14 (Figure S3). GEM payload was ∼16.2% (w/w) confirmed with the calculation of corresponding HPLC peak at 10.8 min after alkaline hydrolysis of GEM conjugated micelles (Figure S4).
Biodistribution and Pharmacokinetics
We previously demonstrated the enhanced antitumor activity of P-GEM in subcutaneous and orthotopic xenograft pancreatic tumor models compared to F-GEM. This encouraging finding prompted us to determine the biodistribution and pharmacokinetic profiles after intravenous administration of F-GEM and P-GEM into orthotopic pancreatic tumor bearing NSG mice. At day 14 postimplantation of luciferase expressing MIA PaCa-2 cells into the pancreas, NSG mice were bioimaged for luciferase expression and photon counts were calculated (Figure 1). After 3 weeks when the tumor size reached to 200–300 mm3 and photon count to ∼9 × 107 p/sec/cm2/Sr, F-GEM or P-GEM was injected intravenously into these tumor bearing mice at a GEM equivalent dose of 40 mg/kg; we selected this dose based on our previous studies.16
Figure 1.
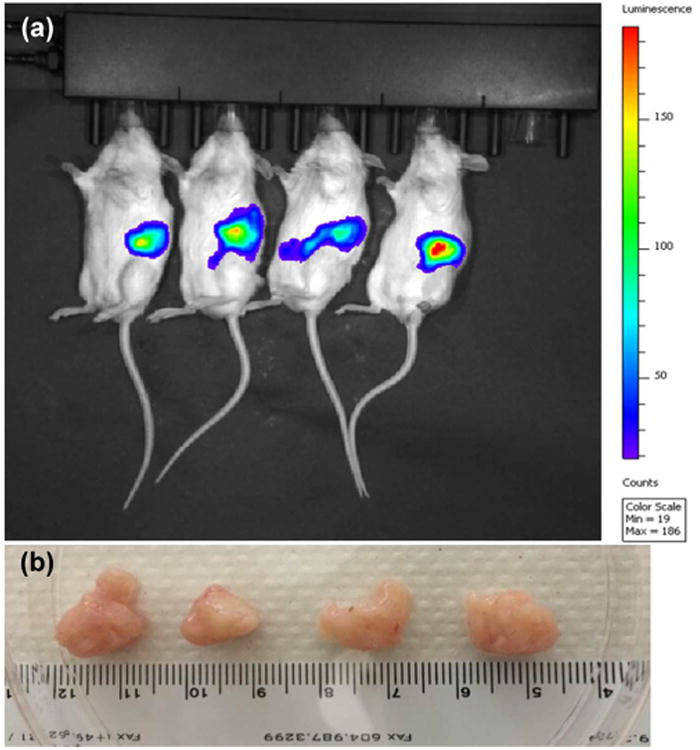
(a) Luciferase expression from orthotopic pancreatic tumors after imaging of pancreas in NSG mice. Each mouse received ∼2 million MIA PaCa-2 cells. (b) Tumor morphology.
The mean plasma concentration-time profiles of GEM and its metabolite dFdU after intravenous bolus administration of F-GEM and P-GEM into orthotopic pancreatic tumor bearing NSG mice are shown in Figures 2 and 3. There is rapid decline of GEM after systemic administration of F-GEM, but significant decline after systemic administration of P-GEM. In contrast, dFdU concentration for F-GEM treated group first increased but rapidly declined with time. For P-GEM treated group, there was also increase in plasma dFdU concentration but decreased slowly with time. These findings are in good agreement with the previous findings in mice24 and human plasma analysis.25,26
Figure 2.
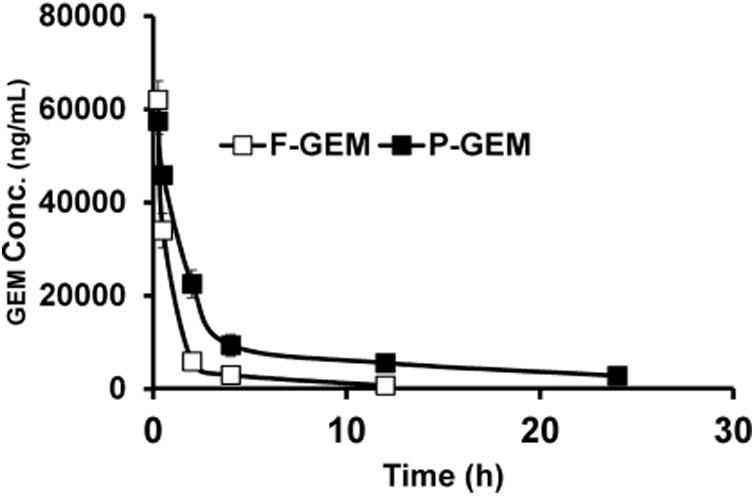
Plasma concentration/of gemcitabine after single injection of free and polymer conjugated gemcitabine (F-GEM and P-GEM) in the tail vein of NSG mice at GEM equivalent dose of 40 mg/kg. Results are represented as the mean ± SD of four mice.
Figure 3.
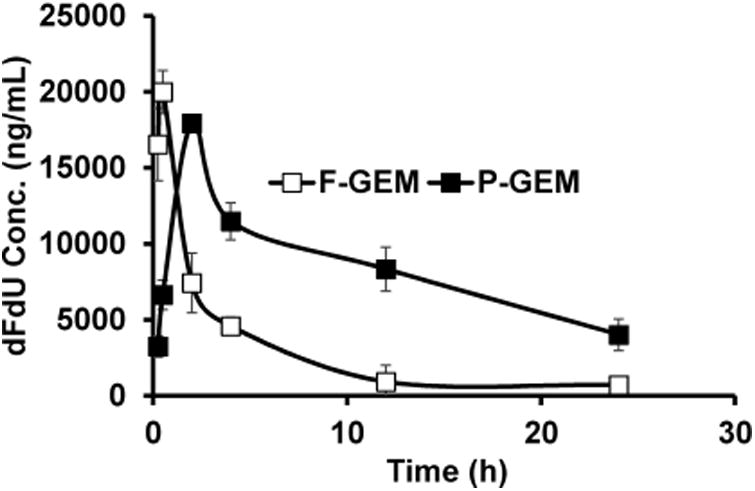
Plasma dFdU concentration after single injection of free and polymer conjugated gemcitabine (F-GEM and P-GEM) in the tail vein of NSG mice at GEM equivalent dose of 40 mg/kg. The values are the mean ± SD of four mice.
Table 1 summarizes PK parameters such as Tmax, Cmax, AUC, t1/2, and MRT. Calculation of plasma PK parameters showed that t1/2 was 1.99 ± 0.09 h for F-GEM injected mouse group, but increased to 5.77 ± 0.49 h for P-GEM injected mouse group. GEM concentration was too low to be detected in F-GEM treated groups at 12 h postadministration, possibly due to the quantification limit from our developed method. In contrast, GEM was detected in P-GEM treated group for up to 24 h, leading to higher MRT compared to F-GEM treated group: 4.45 ± 0.15 h vs 1.12 ± 0.13 h. As expected, the AUC of P-GEM injected mouse group was 2.9-fold higher compared to F-GEM injected mice (246,425 ± 1,605 vs 83,591 ± 1,844 ng/ mL × h).
Table 1. Plasma Pharmacokinetic Parameters of Gemcitabine (GEM) and Its Inactive Metabolite 2′,2′-Difluorodeoxyuridine (dFdU) after Single Intravenous Injection of Free GEM and mPEG-b-PCC-g-GEM-g-DC (Abbreviated as F-GEM and P-GEM) at GEM Equivalent Dose of 40 mg/kg in NSG Mice Bearing Orthotopic Pancreatic Tumors.
| samples | t1/2 (h) | Cmax (ng/mL) | Tmax (h) | AUC (ng/mL × h) | MRT (h) |
|---|---|---|---|---|---|
| F-GEM | 1.99 ± 0.09 | 62923 ± 3479 | 0.25 | 83591 ± 1844 | 1.12 ± 0.13 |
| P-GEM | 5.77 ± 0.49 | 57462 ± 2403 | 0.25 | 246425 ± 1605 | 4.45 ± 0.15 |
| dFdU from F-GEM | 5.9 ± 2.02 | 19988 ± 1223 | 0.5 | 77555 ± 3570 | 6.58 ± 1.31 |
| dFdU from P-GEM | 10.03 ± 1.11 | 18287 ± 976 | 2 | 245708 ± 16552 | 14.93 ± 2.09 |
As shown in Figure 3, in F-GEM treated group, the highest plasma dFdU (Cmax, 19,988 ng/mL) was measured at 0.5 h postinjection (Tmax, 0.5h), indicating the rapid metabolism of GEM. However, comparable value of Cmax (18,287 ng/mL) was achieved at 2 h postinjection (Tmax, 2 h). Interestingly, the AUC of dFdU from P-GEM treated group was found to be 3.2-fold higher (245,708 vs 77,555 ng/mL × h) compared to F-GEM treated group. To understand the fate of biotransformation of dFdU, the metabolic ratio of plasma concentrations of dFdU to GEM was calculated and are shown in Figure 4.
Figure 4.
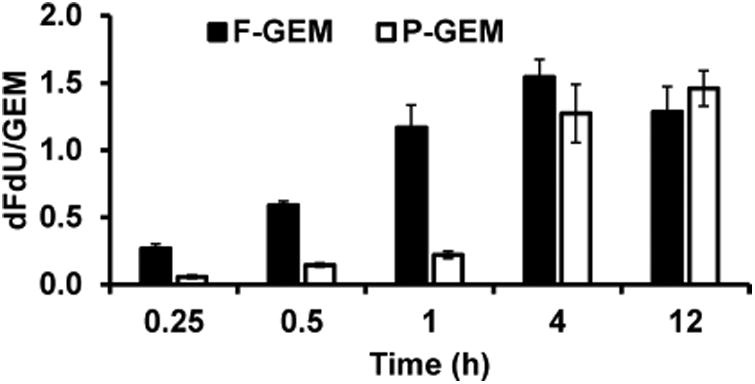
Ratio of the plasma concentration of dFdU to gemcitabine (GEM) in mice after single injection of free and polymer conjugated gemcitabine (F-GEM and P-GEM) in the tail vein of NSG mice at GEM equivalent dose of 40 mg/kg.
Biodistribution of GEM in major organs like liver, spleen, tumor, kidneys, lungs, and heart was also determined. The distribution of GEM after intravenous injection of P-GEM injected mice was higher in the liver, heart, spleen, kidney, and tumor than F-GEM treated mice, especially after 1 h postinjection (Figures 5 and 6). Interestingly, the distribution and retention of GEM resulting from P-GEM in the reticuloendothelial system (RES) were higher than that of F-GEM from 4 to 24 h postinjection (Figures 5 and 6). The accumulation of GEM in liver and spleen from P-GEM treated mice increased up to 4 h postinjection then slowly decreased in the later time point. The highest concentration of GEM resulting from P-GEM was found in liver and spleen at 4 h postinjection. GEM concentration in lung was higher up to 4 h postinjection in F-GEM treated mice compared to P-GEM treated mice, but reverse trend was found in the lung at later time point in P-GEM treated mice. Similarly, GEM concentration in heart was higher up to 1 h postinjection in F-GEM treated mice compared to P-GEM treated mice, but reverse trend was found in lung at later time point in P-GEM treated mice (Figures 5 and 6).27,28 Interestingly, the distribution and retention of GEM resulting from P-GEM in tumor were higher than that of F-GEM at all time points from 15 min to 24 h postinjection (Figures 5 and 6). Additionally, calculation of PK parameters in organs revealed comparable Cmax and higher AUC and t1/2 values in the liver, heart, lung, spleen, tumor, and kidney for P-GEM treated mice compared to F-GEM treated mice as presented in Table 2.
Figure 5.
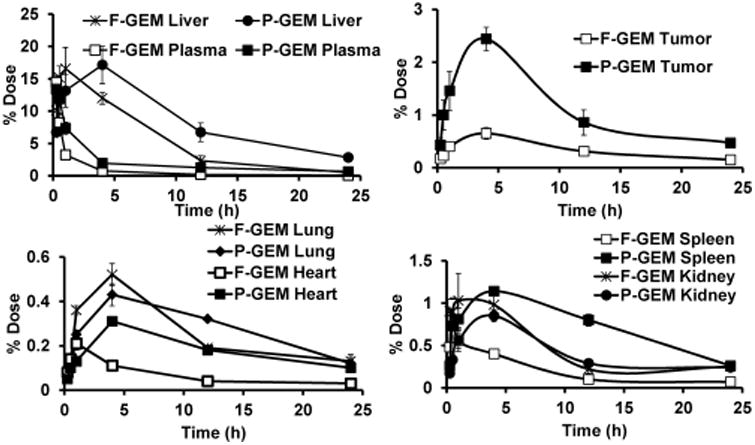
Tissue accumulation of free and polymer conjugated gemcitabine (F-GEM and P-GEM) after injection into the tail vein of NSG mice at a dose of 40 mg/kg GEM equivalent dose. Results are presented as the mean ± SD of four mice.
Figure 6.
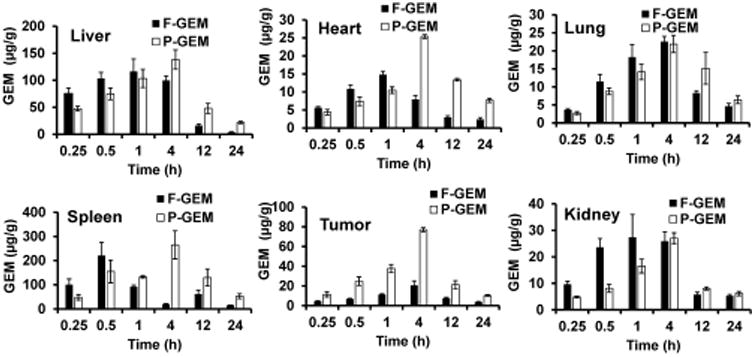
Gemcitabine concentration in major organs after single injection of free and polymer conjugated gemcitabine (F-GEM and P-GEM) at GEM equivalent dose of 40 mg/kg. Results are represented as the mean ± SD of four mice.
Table 2. Organ Pharmacokinetic Parameters of Gemcitabine (GEM) and Its Inactive Metabolite 2′,2′-Difluorodeoxyuridine (dFdU) after Single Intravenous Injection of Free GEM and mPEG-b-PCC-g-GEM-g-DC (Abbreviated as F-GEM and P-GEM) at GEM Equivalent Dose of 40 mg/kg in NSG Mice Bearing Orthotopic Pancreatic Tumors.
| pharmacokinetic parameters | ||||
|---|---|---|---|---|
|
| ||||
| organs | samples | t1/2 (h) | Cmax (ng/mL) | AUC (ng/mL × h) |
| liver | F-GEM | 4.5 ± 0.3 | 43,406 ± 6249 | 347,329 ± 26,578 |
| P-GEM | 12.6 ± 0.9 | 31,598 ± 2636 | 564,977 ± 37,638 | |
| heart | F-GEM | 9.3 ± 0.7 | 3,879 ± 113 | 38,237 ± 3,494 |
| P-GEM | 11.7 ± 0.4 | 3,449 ± 129 | 121,475 ± 1,378 | |
| spleen | F-GEM | 8.2 ± 0.5 | 25,635 ± 1204 | 20,5084 ± 9,533 |
| P-GEM | 8.7 ± 0.3 | 25,366 ± 5435 | 59,3119 ± 8,122 | |
| lung | F-GEM | 10.3 ± 0.8 | 4,593 ± 395 | 80,216 ± 1,378 |
| P-GEM | 14.4 ± 1.7 | 3,720 ± 264 | 131,612 ± 6,115 | |
| tumor | F-GEM | 7.8 ± 0.05 | 3,009 ± 125 | 79,970 ± 7,715 |
| P-GEM | 8.5 ± 0.4 | 9,765 ± 1025 | 263,251 ± 9,148 | |
| kidney | F-GEM | 9.6 ± 0.3 | 6,811 ± 497 | 102,983 ± 6,557 |
| P-GEM | 11.8 ± 1.1 | 4,069 ± 226 | 105,975 ± 5,451 | |
In Vivo Toxicity
To investigate whether P-GEM possesses any short-term systemic toxicity, hematological parameters of mice after systemic administration of F-GEM and P-GEM were determined. The estimation of blood biochemical parameters of both groups at the dose of 40 mg/kg showed no statistically significant differences between the control and treated groups (Table 3). Toxic effects on renal function were evaluated by blood urea nitrogen (BUN), glucose (GLU), creatinine (CRE), calcium (Ca), phosphorus (PHOS), sodium (Na+), potassium (K+), chlorine (Cl−), and total carbon dioxide (tCO2), as depicted in Table 4. Changes of alanine aminotransferase (ALT), alkaline phosphate (ALP), total bilirubin (TBIL), bile acid (BA), gamma glutamyl transferase (GGT), cholesterol (CHOL), and albumin (ALB) were used to identify acute hepatic injury. We did not observe any significant changes in biochemical parameters such as ALT, ALP, TBIL, BA, GGT, CHOL, or ALB in GEM formulation-treated animals compared to the control group (Table 5).
Table 3. Hematological Parameters after Single Intravenous Injection of Free GEM and mPEG-b-PCC-g-GEM-g-DC (Abbreviated as F-GEM and P-GEM) at GEM Equivalent Dose of 40 mg/kg in NSG Mice Bearing Orthotopic Pancreatic Tumors.
| samples | |||
|---|---|---|---|
|
|
|||
| parametersa | control | F-GEM | P-GEM |
| WBC (109/L) | 0.27 ± 0.1 | 0.69 ± 0.25 | 0.69 ± 0.19 |
| LYM (109/L) | 0.30 ± 0.09 | 0.10 ± 0.03 | 0.19 ± 0.09 |
| MON (109/L) | 0.22 ± 0.17 | 0.15 ± 0.14 | 0.23 ± 0.15 |
| NEU (109/L) | 0.82 ± 0.11 | 0.54 ± 0.22 | 1.87 ± 1.2 |
| LYM (%) | 7.63 ± 0.78 | 13.6 ± 5.3 | 8.95 ± 1.7 |
| MON (%) | 8.47 ± 2.15 | 8.5 ± 2.84 | 9.88 ± 0.9 |
| NEU (%) | 83.93 ± 2.03 | 76.8 ± 2.81 | 81.2 ± 1.48 |
| RBC (1012/L) | 8.12 ± 0.14 | 9.1 ± 2.41 | 7.4 ± 1.6 |
| HGB (g/dL) | 14.03 ± 0.49 | 13.3 ± 1.57 | 13.6 ± 1.1 |
| HCT (%) | 36.9 ± 0.86 | 38.3 ± 4.09 | 31.7 ± 5.7 |
| MCV (fl) | 45.3 ± 0.58 | 46 ± 2.65 | 45.5 ± 1 |
| MCH (pg) | 16.8 ± 0.93 | 16.8 ± 1.31 | 18.4 ± 0.7 |
| MCHC (g/dL) | 37.8 ± 2.08 | 35.7 ± 2.54 | 39.5 ± 1.73 |
| RDW (%) | 17.6 ± 0.81 | 18.7 ± 0.75 | 18.2 ± 0.6 |
| PLT (109/L) | 426.6 ± 61.72 | 371.6 ± 29.02 | 343 ± 53.8 |
| MPV (fl) | 6.13 ± 0.25 | 6.63 ± 0.95 | 6.28 ± 0.23 |
| PCT (%) | 0.24 ± 0.18 | 0.41 ± 0.26 | 0.47 ± 0.27 |
| PDW (%) | 28.7 ± 1.63 | 30.8 ± 2.02 | 32.1 ± 3.34 |
WBC, white blood cells; LYM, lymphocytes; MON, monocytes; NEU, neutrophils; RBC, red blood cells; HCB, hemoglobin; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; RDW, red cell distribution width; PLT, platelets; MPV, mean platelet volume; PCT, procalcitonin; PDW, platelets distribution width.
Table 4. Quantitative Determination of Kidney Profile Markers in Lithium-Heparinized Whole Blood from NSG Mice Bearing Orthotopic Pancreatic Tumors after Single Intravenous Injection of Free GEM and mPEG-b-PCC-g-GEM-g-DC (Abbreviated as F-GEM and P-GEM) at GEM Equivalent Dose of 40 mg/kg.
| samples | |||
|---|---|---|---|
|
|
|||
| parametersa | control | F-GEM | P-GEM |
| BUN (mg/dL) | 25 ± 0.8 | 23 ± 3.5 | 21.7 ± 2.8 |
| GLU (mg/dL) | 233 ± 22 | 209 ± 48 | 224 ± 21 |
| CRE (mg/dL) | 0.2 ± 0.1 | 0.25 ± 0.1 | 0.2 ± 0.1 |
| Ca (mg/dL) | 10.1 ± 0.2 | 9.9 ± 0.7 | 9.6 ± 0.3 |
| PHOS (mg/dL) | 8.6 ± 0.3 | 8.3 ± 1.1 | 8.9 ± 0.8 |
| Na+ (mmol/L) | 144 ± 2.2 | 150 ± 3 | 150 ± 1.8 |
| Cl−(mmol/L) | 105 ± 5.2 | 109 ± 4.4 | 107 ± 2.4 |
| K+(mmol/L) | 7.4 ± 0.7 | 8.7 ± 0.4 | 6.9 ± 0.5 |
| tCO2(mmol/L) | 18.7 ± 2.5 | 20.3 ± 4 | 17.2 ± 2.5 |
F-GEM, free gemcitabine; P-GEM, mPEG-b-PCC-g-GEM-g-DC; BUN, blood urea nitrogen; GLU, blood glucose; CRE, creatinine; Ca, calcium; PHOS, phosphorus; ALB, albumin; Na+, sodium ion; Cl−,chloride ion; K+, potassium ion; tCO2, total carbondioxide. Data are presented as the mean ± SD (n = 4).
Table 5. Quantitative Determination of Liver Profile Markers in Lithium-Heparinized Whole Blood after Single Intravenous Injection of Free GEM and mPEG-b-PCC-g-GEM-g-DC (Abbreviated as F-GEM and P-GEM) at GEM Equivalent Dose of 40 mg/kg in NSG Mice Bearing Orthotopic Pancreatic Tumors.
| samples | |||
|---|---|---|---|
| parametersa | control | F-GEM | P-GEM |
| ALT (U/L) | 37 ± 4.2 | 34 ± 6.5 | 34 ± 5.2 |
| ALP (U/L) | 35.5 ± 7.1 | 37.3 ± 5.8 | 42.4 ± 6.2 |
| TBIL (mg/dL) | 0.2 ± 0.09 | 0.2 ± 0.06 | 0.2 ± 0.05 |
| BA (μmol/L) | <1 | <1 | <1 |
| GGT (U/L) | <5 | <5 | <5 |
| CHOL (mg/dL) | 87.7 ± 9 | 95 ± 23 | 85 ± 8.2 |
| ALB (g/dL) | 3.2 ± 0.2 | 2.9 ± 0.1 | 2.7 ± 0.3 |
F-GEM, free gemcitabine; P-GEM, mPEG-b-PCC-g-GEM-g-DC; control mice refers the mice bearing pancreatic orthotopic tumor without treatment; ALT, alanine aminotransferase; ALP, alkaline phosphatase; TBIL, total bilirubin; BA, bile acid; GGT, gamma glutamyl transferase; CHOL, cholesterol; BUN, blood urea nitrogen; ALB, albumin. Data are presented as the mean ± SD (n = 4).
Discussion
GEM has significant antitumor activity against several solid tumors, and its pharmacodynamics features made this drug an important regimen for chemotherapy. The narrow PK parameters and the loss of therapeutic activity after systemic administration due to its rapid metabolism into dFdU is a major challenge. Various strategies have been reported to overcome the short half-life and to increase the bioavailability of GEM. For example, the delivery vehicles such as liposomes and nanoparticles improve the in vivo fate of the drug by controlling PK properties, improving biodistribution profiles, and accumulating the drug at the tumor site.14,27 After intravenous administration, GEM is rapidly distributed throughout the body, as clearly evidenced by its fast concentration decay in plasma, reaching the highest concentration at 15 min after administration (Figure 2). This may be due to high lipophilicity of P-GEM and its ability to permeate membrane barrier.29
Our results clearly suggest that the chemical conjugation provides metabolic protection to GEM, leading to significant delay in its metabolism compared to the free drug after systemic administration. Lower CMC value (1.56 μg/mL) of P-GEM micelles compared to nonionic surfactants, Pluronic block copolymers and other PEGylated amphiphilic polymers, increases formulation stability and sustained drug release prevent GEM metabolism, resulting in enhanced therapeutic efficacy of GEM at target tissue.16 It is further supported by the prolonged terminal elimination t1/2 of the formulation where it has been enhanced by ∼2.9-fold (Table 1). This is also supported by the fact that very low concentration (722 ng/mL) was observed at 12 h postinjection of such high GEM dose (40 mg/kg), and no GEM was detected after 12 h, which was below the detection limit of our HPLC analytical method.
One of the interesting findings of this study is the desirable modifications on PK profiles of GEM through its conjugation to our amphiphilic polymer, which can form micelles. For example, GEM release from the conjugate not only displayed prolonged t1/2 but also increased drug exposure (AUC) by ∼2.95-fold and MRT by ∼3.97-fold, illustrating that chemical conjugation significantly altered the PK profiles of GEM. However, the deamination of GEM by deoxycytidine deaminase was significantly delayed because of the stealth properties of P-GEM, which significantly decreases GEM's interaction with the plasma proteins. For better understanding, we have assigned the plasmatic ratio of dFdU metabolite and GEM, where the data clearly showed that the metabolic ratio is constantly lower for P-GEM injected mouse group at least up to 4 h postinjection, suggesting that GEM conjugation to our copolymer slows down its biotransformation (Figure 4).
The rate at which the various tissues uptake a drug greatly depends on the drug's lipophilicity, the tissue blood flow, drug molecular weight, and binding affinity of the drug to the plasma proteins.29 After systemic administration of F-GEM, the drug was rapidly distributed throughout the body as evidenced by its fast concentration decay in the plasma. The relatively lower drug concentration in kidneys was observed throughout the studied time points, suggesting slower excretion of the drug. The highest drug accumulation was observed in the liver and then in the spleen (Figures 5 and 6) after systemic administration of P-GEM: 17.14% and 1.14% of dose for the liver and spleen, respectively, at 4 h postinjection compared to F-GEM. In contrast, the drug accumulation in the kidney was very low (0.85% of the dose) at 4 h postinjection.30 Our findings are in accordance with the previous studies where the nanoparticles were preferentially taken up by the liver due to opsonization.31 Tumor distribution and retention of GEM from P-GEM treated mice was significantly higher than F-GEM treated mice at all time points (Figures 5 and 6). Controlled release of GEM from P-GEM in the vascular compartment results in increased t1/2 and improved exposer of drug to target tissue. This tumor distribution pattern of GEM from P-GEM confirmed our previous observation of pancreatic tumor growth inhibition potential of P-GEM in both subcutaneous and orthotopic tumor models.
To establish the clinical relevance of our GEM formulation, we investigated in vivo toxicity by measuring the levels of hematological, renal, and hepatic markers. Changes in hematologic parameters are a common indication of drug toxicity and inflammation. There were no significant differences in the erythrocytic parameters such as hemoglobin concentration, red blood cell (RBC), and mean cell volume (MCV) for both free GEM treated groups compared to that of control animals (Table 3). However, there were different white blood cell (WBC) and platelet values. Since the used NSG mice are immune compromised, high WBC count indicates an immune system disorder, possibly due to the reaction to a drug.32 From our examination of blood biochemical marker of kidney function (Table 4), we observed that the kidney function was not affected after systemic administration of F-GEM and P-GEM at a GEM equivalent dose of 40 mg/kg in orthotopic pancreatic tumor bearing NSG mice. Table 5 shows the value of different hepatic enzymes assayed under different groups. Liver is the primary organ responsible for metabolism and excretion of drugs and molecules through its well-established RES. In the event of damage to hepatic cells, there is a consequent increase in hepatic enzymes in pancreatic cancer.33,34 From our evaluation of liver enzyme markers (ALT, ALP, TBIL, BA, GGT, CHOL, and ALB) up to 24 h postinjection, no significant differences in enzyme levels were observed between the drug treated groups and control, suggesting that P-GEM conjugate did not induce any hepatic toxicities.
Conclusion
The present study clearly demonstrated that chemical conjugation dramatically modified the circulation residency, metabolism, and biodistribution of GEM. P-GEM had prolonged circulation in the vascular compartment and greater distribution to reticuloendothelial organs with significant drug accumulation at the tumor site. Investigation of single dose toxicity up to 24 h clearly suggested that the formulation is safe for in vivo application. To sum up, our results clearly suggest that delivery of P-GEM to the tumor is increased and toxicity is reduced upon conjugation to an amphiphilic polymer, which forms micelles, allowing further evaluation of P-GEM in nonclinical and clinical studies.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (1R01EB017853), the Fred and Pamela Buffet Cancer Center, and a Faculty Start-up fund to R.I.M. from the University of Nebraska Medical Center.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.6b00929.
Synthesis scheme, NMR spectra, particle size distribution, and gemcitabine payload (PDF)
Notes: The authors declare no competing financial interest.
References
- 1.Berger AC, Meszoely IM, Ross EA, Watson JC, Hoffman JP. Undetectable preoperative levels of serum CA 19–9 correlate with improved survival for patients with resectable pancreatic adenocarcinoma. Ann Surg Oncol. 2004;11:644–649. doi: 10.1245/ASO.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 2.Shore S, Vimalachandran D, Raraty MG, Ghaneh P. Cancer in the elderly: pancreatic cancer. Surg Oncol. 2004;13:201–210. doi: 10.1016/j.suronc.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Kattel K, Evande R, Tan C, Mondal G, Grem JL, Mahato RI. Impact of CYP2C19 polymorphism on the pharmacokinetics of nelfinavir in patients with pancreatic cancer. Br J Clin Pharmacol. 2015;80:267–275. doi: 10.1111/bcp.12620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D, Sarkar FH. Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol. 2011;8:27–33. doi: 10.1038/nrgastro.2010.188. [DOI] [PubMed] [Google Scholar]
- 6.Reid JM, Qu W, Safgren SL, Ames MM, Krailo MD, Seibel NL, Kuttesch J, Holcenberg J. Phase I trial and pharmacokinetics of gemcitabine in children with advanced solid tumors. J Clin Oncol. 2004;22:2445–2451. doi: 10.1200/JCO.2004.10.142. [DOI] [PubMed] [Google Scholar]
- 7.Bergman AM, Pinedo HM, Peters GJ. Determinants of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine) Drug Resist Updates. 2002;5:19–33. doi: 10.1016/s1368-7646(02)00002-x. [DOI] [PubMed] [Google Scholar]
- 8.Cavallaro G, Licciardi M, Salmaso S, Caliceti P, Gaetano G. Folate-mediated targeting of polymeric conjugates of gemcitabine. Int J Pharm. 2006;307:258–269. doi: 10.1016/j.ijpharm.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 9.Bornmann C, Graeser R, Esser N, Ziroli V, Jantscheff P, Keck T, Unger C, Hopt UT, Adam U, Schaechtele C, Massing U, von Dobschuetz E. A new liposomal formulation of Gemcitabine is active in an orthotopic mouse model of pancreatic cancer accessible to bioluminescence imaging. Cancer Chemother Pharmacol. 2008;61:395–405. doi: 10.1007/s00280-007-0482-z. [DOI] [PubMed] [Google Scholar]
- 10.Couvreur P, Stella B, Reddy LH, Hillaireau H, Dubernet C, Desmaele D, Lepetre-Mouelhi S, Rocco F, Dereuddre-Bosquet N, Clayette P, Rosilio V, Marsaud V, Renoir JM, Cattel L. Squalenoyl nanomedicines as potential therapeutics. Nano Lett. 2006;6:2544–2548. doi: 10.1021/nl061942q. [DOI] [PubMed] [Google Scholar]
- 11.Cosco D, Bulotta A, Ventura M, Celia C, Calimeri T, Perri G, Paolino D, Costa N, Neri P, Tagliaferri P, Tassone P, Fresta M. In vivo activity of gemcitabine-loaded PEGylated small unilamellar liposomes against pancreatic cancer. Cancer Chemother Pharmacol. 2009;64:1009–1020. doi: 10.1007/s00280-009-0957-1. [DOI] [PubMed] [Google Scholar]
- 12.Paolino D, Cosco D, Licciardi M, Giammona G, Fresta M, Cavallaro G. Polyaspartylhydrazide copolymer-based supramolecular vesicular aggregates as delivery devices for anticancer drugs. Biomacromolecules. 2008;9:1117–1130. doi: 10.1021/bm700964a. [DOI] [PubMed] [Google Scholar]
- 13.Stella B, Arpicco S, Rocco F, Marsaud V, Renoir JM, Cattel L, Couvreur P. Encapsulation of gemcitabine lipophilic derivatives into polycyanoacrylate nanospheres and nanocapsules. Int J Pharm. 2007;344:71–77. doi: 10.1016/j.ijpharm.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 14.Pili B, Reddy LH, Bourgaux C, Lepetre-Mouelhi S, Desmaele D, Couvreur P. Liposomal squalenoyl-gemcitabine: formulation, characterization and anticancer activity evaluation. Nanoscale. 2010;2:1521–1526. doi: 10.1039/c0nr00132e. [DOI] [PubMed] [Google Scholar]
- 15.Bildstein L, Dubernet C, Marsaud V, Chacun H, Nicolas V, Gueutin C, Sarasin A, Benech H, Lepetre-Mouelhi S, Desmaele D, Couvreur P. Transmembrane diffusion of gemcitabine by a nanoparticulate squalenoyl prodrug: an original drug delivery pathway. J Controlled Release. 2010;147:163–170. doi: 10.1016/j.jconrel.2010.07.120. [DOI] [PubMed] [Google Scholar]
- 16.Chitkara D, Mittal A, Behrman SW, Kumar N, Mahato RI. Self-assembling, amphiphilic polymer-gemcitabine conjugate shows enhanced antitumor efficacy against human pancreatic adenocarcinoma. Bioconjugate Chem. 2013;24:1161–1173. doi: 10.1021/bc400032x. [DOI] [PubMed] [Google Scholar]
- 17.Mondal G, Kumar V, Shukla SK, Singh PK, Mahato RI. EGFR-Targeted Polymeric Mixed Micelles Carrying Gemcitabine for Treating Pancreatic Cancer. Biomacromolecules. 2016;17:301–313. doi: 10.1021/acs.biomac.5b01419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larson N, Ghandehari H. Polymeric conjugates for drug delivery. Chem Mater. 2012;24:840–853. doi: 10.1021/cm2031569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li F, Danquah M, Mahato RI. Synthesis and characterization of amphiphilic lipopolymers for micellar drug delivery. Biomacromolecules. 2010;11:2610–2620. doi: 10.1021/bm100561v. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Fan W, Dai X, Katragadda U, Mckinley D, Teng Q, Tan C. Enhanced tumor delivery of gemcitabine via PEG-DSPE/TPGS mixed micelles. Mol Pharmaceutics. 2014;11:1140–1150. doi: 10.1021/mp4005904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karaca M, Dutta R, Ozsoy Y, Mahato RI. Micelle Mixtures for Coadministration of Gemcitabine and GDC-0449 To Treat Pancreatic Cancer. Mol Pharmaceutics. 2016;13:1822–1832. doi: 10.1021/acs.molpharmaceut.5b00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pasut G, Canal F, Dalla Via L, Arpicco S, Veronese FM, Schiavon O. Antitumoral activity of PEG-gemcitabine prodrugs targeted by folic acid. J Controlled Release. 2008;127:239–248. doi: 10.1016/j.jconrel.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Khare V, Kour S, Alam N, Dubey RD, Saneja A, Koul M, Gupta AP, Singh D, Singh SK, Saxena AK, Gupta PN. Synthesis, characterization and mechanistic-insight into the anti-proliferative potential of PLGA-gemcitabine conjugate. Int J Pharm. 2014;470:51–62. doi: 10.1016/j.ijpharm.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Controlled Release. 2004;100:331–346. doi: 10.1016/j.jconrel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Fogli S, Danesi R, Gennari A, Donati S, Conte PF, Del Tacca M. Gemcitabine, epirubicin and paclitaxel: pharmacokinetic and pharmacodynamic interactions in advanced breast cancer. Ann Oncol. 2002;13:919–927. doi: 10.1093/annonc/mdf164. [DOI] [PubMed] [Google Scholar]
- 26.Wang LR, Huang MZ, Xu N, Shentu JZ, Liu J, Cai J. Pharmacokinetics of gemcitabine in Chinese patients with non-small-cell lung cancer. J Zhejiang Univ Sci. 2005;6:446–450. doi: 10.1631/jzus.2005.B0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reddy LH, Khoury H, Paci A, Deroussent A, Ferreira H, Dubernet C, Decleves X, Besnard M, Chacun H, Lepetre-Mouelhi S, Desmaele D, Rousseau B, Laugier C, Cintrat JC, Vassal G, Couvreur P. Squalenoylation favorably modifies the in vivo pharmacokinetics and biodistribution of gemcitabine in mice. Drug Metab Dispos. 2008;36:1570–1577. doi: 10.1124/dmd.108.020735. [DOI] [PubMed] [Google Scholar]
- 28.Reddy LH, Dubernet C, Mouelhi SL, Marque PE, Desmaele D, Couvreur P. A new nanomedicine of gemcitabine displays enhanced anticancer activity in sensitive and resistant leukemia types. J Controlled Release. 2007;124:20–27. doi: 10.1016/j.jconrel.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 29.Castelli F, Sarpietro MG, Ceruti M, Rocco F, Cattel L. Characterization of lipophilic gemcitabine prodrug-liposomal membrane interaction by differential scanning calorimetry. Mol Pharmaceutics. 2006;3:737–744. doi: 10.1021/mp060059y. [DOI] [PubMed] [Google Scholar]
- 30.Chen F, Zhang J, He Y, Fang X, Wang Y, Chen M. Glycyrrhetinic acid-decorated and reduction-sensitive micelles to enhance the bioavailability and anti-hepatocellular carcinoma efficacy of tanshinone IIA. Biomater Sci. 2016;4:167–182. doi: 10.1039/c5bm00224a. [DOI] [PubMed] [Google Scholar]
- 31.Paolino D, Cosco D, Racanicchi L, Trapasso E, Celia C, Iannone M, Puxeddu E, Costante G, Filetti S, Russo D, Fresta M. Gemcitabine-loaded PEGylated unilamellar liposomes vs GEMZAR: biodistribution, pharmacokinetic features and in vivo antitumor activity. J Controlled Release. 2010;144:144–150. doi: 10.1016/j.jconrel.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 32.Vozarova B, Weyer C, Lindsay RS, Pratley RE, Bogardus C, Tataranni PA. High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes. 2002;51:455–461. doi: 10.2337/diabetes.51.2.455. [DOI] [PubMed] [Google Scholar]
- 33.Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med. 2010;77:195–204. doi: 10.3949/ccjm.77a.09064. [DOI] [PubMed] [Google Scholar]
- 34.Nyblom H, Berggren U, Balldin J, Olsson R. High AST/ALT ratio may indicate advanced alcoholic liver disease rather than heavy drinking. Alcohol Alcohol. 2004;39:336–339. doi: 10.1093/alcalc/agh074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.