

Abstract
Phosphorylation is a key post-translational modification in cell signaling, which is regulated by the equilibrium activities of kinases and phosphatases. The biological significance of many phosphorylation events remains poorly characterized due to the scarcity of tools to discover phosphatases substrates. In prior work, we established kinase-catalyzed biotinylation where kinases accept the γ-modified ATP analog, ATP-biotin, to label phosphoproteins. Here, we developed a novel method to study substrates of phosphatases using kinase-catalyzed biotinylation termed K-BIPS (Kinase-catalyzed Biotinylation to Identify Phosphatase Substrates). In a proof-of-concept experiment, K-BIPS was initially used to explore the substrates of phosphatases inhibited by okadaic acid. Many known phosphatase substrates were observed, confirming K-BIPS as a valid phosphatase substrate identification tool. Then, as a further application, K-BIPS was used to discover the substrates of the PP1-Gadd34 phosphatase complex in the context of unfolded protein response (UPR). In addition to the known substrate eIF2α, K-BIPS revealed several novel substrates, suggesting a more prominent role for the PP1-Gadd34 complex in UPR than previously appreciated. Overall, the two studies establish K-BIPS as a powerful tool to discover the cellular substrates of phosphatases.
Graphical Abstract
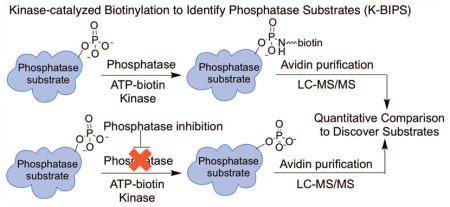
Introduction
Protein phosphorylation is a key post-translational modification involved in signal transduction.1 Two families of enzymes, kinases and phosphatases, tightly regulate phosphorylation to ensure proper cell signaling.2 While kinases mediate the transfer of a phosphoryl group from ATP (adenosine 5′-triphosphate) to cellular proteins, phosphatases remove the phosphoryl group (Figure 1A). The malfunction of either kinases or phosphatases results in various human diseases, such as cancer.3, 4 Despite the disease relevance, the substrates of many phosphatases remain elusive due to the scarcity of discovery tools. The challenge of discovering substrates may be partially explained by the structural complexity of protein phosphatases.
Figure 1.

Protein phosphorylation and the K-BIPS method. (A) Kinases transfer a phosphoryl group from ATP to cellular proteins. Phosphatases reverse the reaction. (B) Kinases utilize γ-modified ATP analogs, such as ATP-biotin, to covalently modify substrate proteins. The γ-modified phosphoryl group once attached is resistant to phosphatase degradation. (C) The K-BIPS method. Phosphatases present in lysates remove phosphoryl groups from already phosphorylated substrates (i, left path). However, when a phosphatase is inactivated by inhibitor treatment, the substrates are maintained in the phosphorylated form (ii, right path). (i) Phosphatase substrates in the untreated lysates are biotinylated by ATP-biotin and active kinases. After avidin purification of biotinylation proteins, LC-MS/MS analysis will identify the captured substrates. (ii) Phosphorylated substrates in the phosphatase-inhibited sample are immune to biotinylation by ATP-biotin. After avidin purification and LC-MS/MS analysis, quantitative comparison of proteins present at elevated levels in the untreated sample versus the phosphatase inhibitor-treated sample will reveal possible substrates.
Protein phosphatases are broadly classified into two families: Protein Tyr phosphatases (PTPs) and Ser/Thr phosphatases.5 PTPs act on phosphorylated Tyr substrates and comprise a single subunit containing both catalytic and regulatory domains. The catalytic domain is well conserved throughout different members of PTP family, while the regulatory domains dictate the substrate specificity of PTPs.6, 7 Ser/Thr phosphatases, on the other hand, act on phosphorylated Ser/Thr residues and are further classified into two families: PPP and PPM.5 The PPP subfamily consists of PP1, PP2A, PP2B, PP4, PP5, PP6 and PP7 phosphatases, whereas the PPM subfamily includes PP2C phosphatase. Similar to Tyr phosphatases, PP2C phosphatases carry both catalytic and regulatory domains on a single subunit.8 In contrast, PPP phosphatases contain only the catalytic domain. A separate regulatory protein controls the catalytic domain. Regulatory and catalytic proteins assemble into a protein phosphatase complex, which maintains distinct substrate specificity.8, 9 For example, interaction with regulatory protein Gadd34 directs the PP1 catalytic protein to dephosphorylate eIF2α during the unfolded protein response (UPR),10 whereas interaction with the regulatory protein MYPT1 leads to the dephosphorylation of MLC20 protein by PP1.11 Adding to the complexity of PPP phosphatase-mediated signaling, more than a hundred regulatory subunits exist for PP1 alone. The diverse nature of each subfamily of phosphatases has made the development of a general phosphatase substrate identification method challenging.
Only a few methods are currently available to discover phosphatase substrates. Among existing methods, the substrate trapping approach employs a catalytically inactive mutant to stabilize phosphatase-substrate interactions in cellular lysates for subsequent pull down.12, 13 In a second in vitro method, synthesized phosphopeptide libraries were arrayed and screened for dephosphorylation by recombinant phosphatases.14, 15 While these methods have been successful in identifying substrates of Tyr phosphatases, they have been inadequate for Ser/Thr phosphatase substrate discovery. As discussed earlier, the substrate specificities of the PPP family of Ser/Thr phosphatases are governed by associated regulatory protein. Because the substrate trapping and in vitro peptide screening strategies rely on mutated or recombinant phosphatases alone, both approaches fail to discover substrates of phosphatase-regulatory complexes. Beyond these two approaches, phosphoproteomics studies coupled with phosphatase inactivation and IMAC (Immobilized metal affinity chromatography) purification have revealed some Ser/Thr phosphatase substrates,16 although not widely and with bias towards highly abundant proteins. Substrates have also been discovered by co-immunoprecipitation and yeast-two-hybrid, although these methods are low throughput.17 As a result, many Ser/Thr phosphatase substrates remain unknown. For example, despite the complexity of the UPR in cells, the only UPR-involved PP1-Gadd34 substrate known is eIF2α.10 Alternative, unbiased methods applicable to both Tyr and Ser/Thr phosphatases are needed to thoroughly study phosphatase biology. Here, we developed a novel chemical method for phosphatase substrate discovery based on our work on kinase-catalyzed labeling.
In previous work, we demonstrated that kinases across the kinome accept γ-modified ATP analogs to modify proteins (Figure 1B).18–20 For example, ATP-biotin was used by cellular kinases to phosphorylbiotinylate proteins in lysates.18 Further studies revealed that phosphorylbiotinylated residues were resistant to phosphatase degradation (Figure 1B), indicating that the phosphorylbiotin group is a stable tag.21 The cellular stability of the phosphorylbiotin group makes kinase-catalyzed biotinylation ideal for identification of phosphoproteins. Interestingly, those same studies also revealed that robust kinase-catalyzed biotinylation required the presence of active phosphatases; biotinylation of cellular proteins was reduced when phosphatases were inactivated.21 The phosphatase dependency of kinase-catalyzed biotinylation is due to the presence of already existing phosphorylation, which prevents biotin labeling. In other words, phosphatase activity in lysates promotes removal of phosphoryl groups to allow subsequent ATP-biotin labeling by kinases.
Based on the phosphatase dependency of kinase-catalyzed biotinylation, we developed a new method to identify phosphatase substrates called K-BIPS (Kinase-catalyzed Biotinylation to Identify Phosphatase Substrates). In K-BIPS, biotinylation was carried out with ATP-biotin in lysates either with or without phosphatase inactivation (Figure 1C). In samples containing an active phosphatase (Figure 1C, i), substrates were dephosphorylated by the phosphatase, resulting in ATP-biotin labeling by kinases. In contrast, in samples where the phosphatase was inactivated (Figure 1C, ii), phosphatase substrates were trapped in the phosphorylated state, making them inaccessible to ATP-biotin labeling. Therefore, phosphatase substrates were less biotinylated in the phosphatase-inactive sample compared to the untreated sample (Figure 1C, i compared to ii). To identify the proteins differentially labeled in the two samples, biotinylated proteins were purified using avidin resin and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Candidate phosphatase substrates were discovered by observing proteins present at elevated levels in untreated samples compared to the phosphatase-inactivated samples.
To establish K-BIPS as a phosphatase substrate discovery tool, two studies were performed here focused on Ser/Thr phosphatases. We selected Ser/Thr phosphatases for these studies given the challenge to identify the substrates of Ser/Thr phosphatase-regulatory complexes. As an initial proof-of-concept study, K-BIPS was used after the inactivation of a majority of PPP family Ser/Thr phosphatases with the broad phosphatase inhibitor, okadaic acid (OA). LC-MS/MS analysis after K-BIPS with OA identified many known substrates of PPP family members, confirming that K-BIPS is a viable discovery tool. In a second discovery-oriented study, K-BIPS was used to identify substrates of the PP1-Gadd34 phosphatase complex in the context of UPR. In this case, selective inactivation of the PP1-Gadd34 complex was achieved with the small molecule guanabenz (Gb), which was previously documented to reduce activity of the PP1-Gadd34 complex.22 K-BIPS with Gb identified eIF2α, the known PP1-Gadd34 substrate, confirming that K-BIPS is a useful tool for substrate discovery. Importantly, many other UPR-related candidate substrates were also identified, and secondary validation studies indicated that several K-BIPS hits are PP1-Gadd34 substrates. The novel substrates identified by K-BIPS implicate the PP1-Gadd34 complex in several unprecedented functions in UPR regulation. Altogether, these studies document that K-BIPS is a valuable discovery tool to identify phosphatase substrates and to discover unanticipated biological functions of phosphatases.
Materials and methods
Synthesis of ATP-biotin
The synthesis and characterization of ATP-biotin has been previously described.21
OA treatment of HeLa cells
HeLa cells (20X106) were grown in F12 media (45 mL, ThermoFisher) containing 10% FBS (fetal bovine serum, ThermoFisher) and 1X antibiotic/antimycotic solution (ThermoFisher) at 37°C in a growth environment supplied with 5% CO2. When cells reached 80% confluency, they were serum starved overnight in F12 media (45 mL) without FBS or antibiotic/antimycotic in the same growth environment. The next day cells were treated with OA (1 μM or 10nM in ethanol, 45 μL, Santa Cruz Biotechnology) or without OA (0.1% ethanol, EMD) in F12 media without FBS or antibiotic/antimycotic for 30 minutes in the same growth environment. Next, the cells were harvested using the following protocol. Media was removed and the cells were washed with DPBS (Dulbecco’s Phosphate Buffered Saline, 10 mL, ThermoFisher). The cells were then incubated with trypsin-EDTA (0.25%, 12 mL, ThermoFisher) for 5 minutes at 37°C. Next, cold DPBS (15 mL) was added and the released cells were collected by centrifugation at 1000 rpm, 4°C for 5 minutes. The supernatant was discarded and the cells were resuspended in cold DPBS (2 mL). The cells were again collected by centrifugation at 1000 rpm, at 4°C for 5 minutes. The collected cell pellet was either stored at −80°C or immediately lysed.
Induction of UPR and Gb treatment of HeLa cells
HeLa cells (20X106) plated in F12 media (45 mL) supplemented with 10% FBS and 1X antibiotic/antimycotic solution were allowed to grow to at least 80% confluence, as previously described. Then, all cells were treated with tunicamycin (Tm, 2.5 μg/mL in DMSO, 45 μL, Sigma, catalog number T7765). At the same time, cells were also treated with guanabenz (Gb, 50 μM in ethanol, 45 μL, Sigma-Aldrich, catalog number G110) or without Gb (0.1 % ethanol (EMD) and 0.1% DMSO (ATCC)). The cells were then incubated at 37°C for 6 hours in a growth environment. After removing the media, cells were harvested, as described.
Gadd34 knock down in HeLa cells
HeLa cells (0.3X106) plated in F12 media (2 mL) supplemented with 10% FBS in 6 well plates (Sigma Aldrich) were grown to 70% confluency. Next, cells were treated with a pool of Gadd3-targeting siRNA (25 nM, Dharmacon, catalog number M-004442-01-0005) or a control non-targeting pool of siRNA (25 nm, Dharmacon, D-001206-14-05), along with a transfection reagent (6 μL/well, Dharmacon, T-2001-02). As another control, cells were treated with the transfection reagent alone. After 60 hours, cells were treated with tunicamycin (Tm, 2.5 μg/mL in DMSO, 45 μL, Sigma, catalog number T7765) and incubated for 6 hours at 37°C in a growth environment. Then cells were harvested and lysed, as described.
Cell lysis
Cell pellets after treatment, as described above, were resuspended in lysis buffer (300 μL; 50 mM Tris pH 7.5, 150 mM NaCl, 0.5% Triton X-100 and 10% glycerol), and rocked at 4°C for 20 minutes. Then, cell debris was separated from the soluble fraction by centrifugation at 13.2 rpm for 20 minutes. The supernatant was stored in single reaction aliquots and saved at −80°C. Protein concentration was determined by Bradford assay (BioRad).
ATP-biotin labeling of OA-treated lysates for K-BIPS
Lysates from OA-treated cells (500 μg total protein) were pre-incubated with OA (1 μM or 10 nM in water) for 10 minutes at room temperature. Lysates from untreated cells were also incubated for 10 minutes at room temperature with the same volume of water. Biotinylation was initiated by adding ATP-biotin (2 mM) to the lysates in a final reaction volume of 60 μL. Reactions were incubated for 2 hours at 31°C. Then, biotinylated proteins were purified by streptavidin affinity chromatography, as described below.
Assessment of Gadd34 expression in Gb-treated and Gadd34 siRNA-treated lysates
Lysates from Tm and Gb-treated cells or Gadd34 siRNA-treated cells (100 μg total protein) were boiled at 95°C for 1 min in 1X Laemmli sample buffer (60 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 0.0005% bromophenol blue, and 2% beta-mercaptoethanol). Then, proteins in each sample were separated by 10% SDS-PAGE. Total proteins were visualized using a Typhoon imager (GE Healthcare Life Sciences) after staining with Sypro®Ruby (ThermoFisher). For the Gadd34 Western blot, proteins were transferred onto a PVDF membrane (Milipore Immobilon-P) and then probed with a Gadd34 specific antibody (Santa Cruz Biotechnology, SC-8327).
Co-immunoprecipitation of PPI and Gadd34 from HeLa lysates
Lysates (3.5 mg total protein) obtained from Tm and Gb-treated cells were pre-incubated with Gb (50 μm in 0.1 % ethanol) at room temperature for 15 minutes. Lysates (3.5 mg total protein) from cells treated only with Tm were pre-incubated with vehicle (0.1% ethanol) for 15 minutes at room temperature. A fraction of lysates (100 μg) was saved as an input control for gel analysis. The remaining lysates were incubated for 1 hour at 4°C with a PP1 antibody (14 μg, Santa Cruz Biotechnology, SC-7482) with rotation. Next, the lysate-antibody mixture was incubated with Protein A/G agarose beads (20 μL, Santa Cruz Biotechnology, SC-2003) for 1 hour at 4°C. Then, bead-bound proteins were eluted by boiling at 95°C for 5 minutes in 2X Laemmli sample buffer. After separation of eluted proteins by 10% SDS-PAGE, proteins were transferred onto a PVDF membrane (Milipore Immobilon-P) and the levels of PP1 and Gadd34 were monitored by Western blot using specific antibodies (Santa Cruz Biotechnology, SC-7482 for PP1 and SC-8327 for Gadd34).
Analysis of eIF2α phosphorylation
Lysates (500 μg total protein) from Tm and Gb-treated cells were pre-incubated with Gb (50 μm in 0.1 % ethanol) at room temperature for 15 minutes. Lysates from cells treated only with Tm (500 μg total protein) were pre-incubated with vehicle (0.1% ethanol) for 15 minutes at room temperature. A portion of the lysates (100 μg) was saved as an input control for gel analysis. The remaining lysates from the Gb-treated and untreated samples were incubated at 31°C for 2 hours, which simulated the conditions of the ATP-biotin labeling reaction. Portions of lysates (100 μg) were removed at the 1 hour and 2 hour time points. Proteins in each lysate sample were separated by 10% SDS-PAGE, transferred onto a PVDF membrane (Milipore Immobilon-P), and analyzed by Western blot using a specific antibody recognizing eIF2α phosphorylation at Ser52 (Cell Signaling Technology, catalog number 9721). As a control, total eIF2α levels were also probed with an antibody to eIF2α (Santa Cruz Biotechnology, SC-133132).
ATP-biotin labeling of GB-treated lysates
Lysates from Tm and Gb-treated cells were pre-incubated with Gb (50 μm in 0.1 % ethanol) at room temperature for 15 minutes. Lysates from Tm treated cells were pre-incubated with vehicle (0.1% ethanol) at room temperature for 15 minutes. For biotinylation reactions that did not include streptavidin purification (Figure 3A), lysates (100 μg) were used in a final volume of 20 μL with ATP-biotin (2 mM) and incubated for 2 hours at 31°C. After reaction, total protein samples were boiled at 95°C for 1 minute in 1X Laemmli buffer and separated by 10% SDS-PAGE. Total proteins were visualized by Sypro®Ruby gel stain. For visualizing biotinylated proteins, proteins were transferred onto a PVDF membrane (Milipore Immobilon-P) and probed with a Streptavidin-Cy5 conjugate (Life Technologies). For K-BIPS reactions (Figure S7), lysates (500 μg) were used in a volume of 30 μL with ATP-biotin (2 mM). Biotinylation reactions were carried out for 2 hours at 31°C. Then biotinylated proteins were purified by streptavidin resin, as described below.
Figure 3.

K-BIPS study with Gb treatment. (A) Biotinylation of proteins after Gb treatment. Untreated or Gb-treated HeLa lysates were labeled by ATP-biotin. Proteins were separated by SDS-PAGE and visualized with Streptavidin-Cy5 (top) and Sypro®Ruby (bottom). The arrows and brackets indicate proteins that are reduced in the GB-treated sample (lane 3) compared to the untreated samples (lanes 1–2). The full gel image is shown in Figure S6. (B) Known physical protein-protein interactions among the 130 K-BIPS hits and the PP1-Gadd34 complex were mapped using GeneMANIA in Cytoscape. The inner circles of hexagons include known direct interacting proteins of PP1 catalytic subunits (PPP1CA, PPP1CB, PPP1CC, red rectangles) or Gadd34 (PPP1R15A, red rectangle), including the known PP1-Gadd34 substrate, EIF2S1 (eIF2α, red star). The outer circles represent indirect interacting proteins of PP1-Gadd34. Protein colors indicate the biological function of the proteins: yellow - translation initiation; pink – ribosome; dark blue – UPR; orange - ER stress; light blue – protein ubiquitination; purple - stress granules; grey - oxidative stress. Except when cited (Table S2), functional information was obtained from Uniprot database. Grey lines indicate interactions among the enriched proteins. The other line colors indicate the proteins that directly interact with PPP1CA (blue lines), PPP1CB (green lines), PPP1CC (black lines) or Gadd34 (PPP1R15A, red lines). Line thickness and length were adjusted arbitrary to improve clarity. The black star highlights the proteins validated in Figure 4. An enlarged version of this figure is available as Figure S9. The Cytoscape file is also available as a supplementary document. (C) The interaction of COPS5 with stress granule proteins. (D) K-BIPS hits not reported to interact with PP1-Gadd34 are also shown.
Streptavidin purification of biotinylated proteins for K-BIPS
After ATP-biotin labeling as described above, a fraction of lysates (80 μg) was saved to be analyzed as the input. The rest of the lysates were filtered using 3 kDa centiprep spin columns (Millipore) per manufacturer’s instructions to remove excess ATP-biotin and endogenous biotin. Streptavidin resin (200 μL of packed beads, Genscript) was washed three times with phosphate binding buffer (200 μL; 0.1 M phosphate pH 7.2, 0.15 M NaCl). The filtered samples were then allowed to bind the streptavidin resin by rotating for 1 hour at room temperature. The flow through was collected and the streptavidin beads were washed with phosphate binding buffer (200 μL) ten times and four times with water (200 μL). The final wash was collected. The bound, biotinylated proteins were eluted by boiling the beads in 2% SDS in water (200 μL) for 8 minutes. The eluate was then concentrated by lyophilization. The input, flow through, last wash, and concentrated eluate were boiled at 95°C for 1 min in 1X Laemmli sample buffer and separated by 10% SDS-PAGE gels. SYPRO®Ruby stain was used to visualize total proteins.
LC-MS/MS analysis of K-BIPS with OA treatment
The proteins in eluate lanes from gels described above were excised and in gel digested as previously described.23 Digested peptides were then labeled with TMT (Tandem Mass Tag™, ThermoFisher Scientific) per manufacturer’s instructions. Peptides were separated by reverse phase chromatography over a 90 min gradient (5% – 28% acetonitrile in 0.1% formic acid) followed by a 20 min gradient (28% – 40% acetonitrile in 0.1% formic acid) using an Easy-nLC pump (Thermo) at 300 nL/min. Peptides were analyzed with an Orbitrap Fusion Tribrid mass spectrometer (Thermo). MS1 scans were performed within the orbitrap at a 120,000 resolution and 350–1600 m/z scan range. The top 10 ions with a charge of +2 to +7 were isolated in the ion trap and fragmented with CID (30% collision energy; activation Q = 0.25). Dynamic exclusion was turned on (after 1 isolation the ion was excluded for analysis for 30 s). Quantitation of isobaric TMT tags was performed with MS3 scans. The top 10 fragment ions were isolated, re-fragmented by HCD (65% collision energy) and sent to the orbitrap for analysis (60,000 resolution over a scan range of 100–500 m/z).
LC-MS/MS analysis of K-BIPS with Gb treatment
Biotinylated proteins from eluate lanes from gels described above were excised from the gel and trypsin digested as previously described.23 Digested peptides were separated on an EASY nLC-1000 UHPLC system (Thermo) by reverse phase chromatography under acidic conditions (0.1% formic acid) and analyzed by a Q-Exactive mass spectrometer (Thermo). MS1 profiling was performed over a 375–1600 m/z range at a resolution of 70,000. MS2 fragmentation was done using higher energy collision induced dissociation (HCD) on the top 15 ions using a 1.6 m/z window and normalized collision energy of 29. Dynamic exclusion was turned on (15 s).
MS data analysis
MS raw data was analyzed by MaxQuant (version 1.5.2.8). A human protein database from UniProt (downloaded 2016.04.07, 20159 entries) was used. Searches allowed 2 missed tryptic cleavages. The iodoacetamide derivative of cysteine was set as a fixed modification while oxidation of methionine and acetylation of protein N-termini were set as variable modifications. Mass tolerances for parent ions were 20 ppm for the first search and 4.5 ppm for the second search and 20 ppm for fragment ions. Minimum protein and peptide identification probabilities were specified at ≤1% false discovery rate (FDR) as determined by a reversed database search, and proteins required 1 unique peptide. All other parameters were used at their default settings. Fold enrichment in the OA-mediated K-BIPS study was calculated by dividing the TMT reporter intensity observed for the OA-untreated sample by that of the OA-treated sample. Proteins showing fold enrichment of at least 1.5 in both replicates were considered as K-BIPS hits (Table S1). For the PP1-Gadd34 K-BIPS study, fold enrichment was calculated by dividing the peptide intensity observed in the PP1-Gadd34-active sample by that observed for the PP1-Gadd34-inactive sample. Proteins showing fold enrichment of at least 1.3 in both replicates in the PP1-Gadd34 active sample compared to the PP1-Gadd34 inactive sample were considered hits (Table S2).
Interactome and functional analysis
The known physical protein-protein interactions among the K-BIPS hits (Table S2), Gadd34, and different isoforms of PP1 catalytic subunit were mapped using the GeneMANIA application in Cytoscape 3.3.0.24 The functions of the K-BIPS hits were discovered using a literature search and the Uniprot database. K-BIPS hits were then manually colored in the Cytoscape map according to function.
Secondary validation of COPS5, WDR5, CAPRIN1 and G3BP1
Biotinylated proteins from K-BIPS experiments using either Gb (500 μg total protein) or Gadd34 siRNA (350 μg total protein) treated lysates were streptavidin enriched, as detailed earlier. Eluted proteins (30% of eluate for COPS5 or 100% of eluate for WDR5, CAPRIN1 and G3BP1 from Gb-treated lysates; 100% of eluate for COPS5 from Gadd34 siRNA-treated lysates), along with input lysates, flow through, and the wash from streptavidin enrichment, were separated by 10% SDS-PAGE. The proteins were then transferred onto a PVDF membrane (Milipore Immobilon-P) and probed with specific antibodies: COPS5 (Santa Cruz-SC-9074), WDR5 (Bethyl Laboratories, A302-429), CAPRIN1 (Bethyl Laboratories, A303-881), G3BP1 (Bethyl Laboratories, A302-033).
Effect of Guanabenz on COPS5 and G3BP1 protein levels
HeLa cells (80% confluent, 7X106 cells) in F12 media (30 mL, ThermoFisher), 10% FBS, and 1X antibiotic/antimycotic were treated with tunicamycin (Tm, 2.5 μg/mL in DMSO, 30 μL). To some samples were added Gb (50 μM in ethanol, 30 μL, Sigma-Aldrich, catalog number G110), Sephin1 (10 mg/mL in 0.1% DMSO, Sigma, catalog number SML 1356), and/or ISRIB (1 mg/mL in 0.1% DMSO, Sigma, catalog number SML 0843). After 6 hours incubation at 37°C, cells were harvested and lysed as described earlier. Each lysate (100 μg total protein) was treated with 1X Laemmli sample buffer and heated at 95°C for 1 min. The proteins in the lysates were separated by 10% SDS-PAGE gels, followed by visualization with Sypro®Ruby total protein stain or immunoblotting after transfer to PVDF membrane (Milipore Immobilon-P) using antibodies to COPS5 (Santa Cruz-SC-9074) or G3BP1 (Santa Cruz-365338). The gels images were obtained using a Typhoon imager (GE Healthcare Life Sciences).
Secondary validation of COPS5 with Phos-tag™ SDS-PAGE
Proteins in lysates (100 μg total protein) from HeLa cells treated with Tm only or Tm and Gb were separated using 10% SDS-PAGE containing 25 μM Phos-tag™ and ZnCl2 (10 μM). Total proteins in the lysates were visualized with Sypro®Ruby stain. The various phosphorylated forms of COPS5 proteins were visualized with a COPS5 primary antibody (Santa Cruz-SC-9074) after electrotransfer to PVDF membrane (Milipore Immobilon-P). The gels images were obtained using a Typhoon imager (GE Healthcare Life Sciences).
Results
K-BIPS with Okadaic Acid
Okadaic acid (OA) is a broad-spectrum phosphatase inhibitor that targets PP2A, PP4, PP5 and PP6 at a concentration of 1–2 nM and PP1 at around 1 uM.25, 26 Given that PP1 and PP2A carry out roughly 90% of Ser/Thr dephosphorylation in the cell and have a large number of known substrates,27 K-BIPS with OA will serve to test the feasibility of K-BIPS for phosphatase substrate identification. Specifically, by comparing known PPP phosphatase substrates with the proteins identified by K-BIPS, the viability of the method will be established. In addition, unknown substrates of the PPP phosphatase family can also be identified.
We carried out K-BIPS with OA treatment in HeLa lysates. HeLa cells were untreated or treated with OA at either 10 nM or 1 μM concentrations to differentially influence PP2A and PP1. After cell lysis, biotinylation was carried out by incubating each lysate sample with ATP-biotin either in the absence or the presence of OA. The biotinylated proteins were then purified with avidin resin and analyzed by SDS-PAGE. As expected, reduced protein levels were observed in the elution of OA-treated compared to untreated samples (Figure 2A, compare lanes 4 to 5 and 6), confirming that inactivation of phosphatases results in less biotinylation. As anticipated, the sample treated with 1 μM OA showed the most reduction (Figure 2A, compare lane 5 to 4 and 6). The results confirmed that efficient kinase-catalyzed biotinylation requires the activity of phosphatases.
Figure 2.

K-BIPS study with OA. (A) Avidin purification of biotinylated proteins after kinase-catalyzed biotinylation. Biotinylation was carried out with lysates from HeLa cells untreated (ethanol only; lane 1) or treated with OA (1 or 0.01 μM in ethanol; lanes 2–3). After reaction, biotinylated proteins were enriched with avidin resin (elution in lanes 4–6). Proteins in each sample were separated by SDS-PAGE and stained for total proteins using SYPRO® Ruby stain. Full gel image shown in Figure S1. (B) Known substrates identified by K-BIPS (Substrate), along with the corresponding PPP family member. Refer to Table S1 for primary literature. (C) Phosphatase interacting proteins (Gene name) from the K-BIPS hits. Interaction information was obtained from Uniprot and BioGrid databases, or literature (see Table S1). The catalytic and regulatory subunits known to interact with the hit are indicated. For example, PPP1CA refers to PP1 alpha isoform of the catalytic subunit, whereas PPP1R8 refers to the regulatory subunit 8 of PP1 phosphatase.
To identify the substrates of phosphatases inhibited by OA, we performed LC-MS/MS analysis after in-gel digestion of proteins eluted from untreated and 1 μM OA-treated samples (Figure 2, lanes 4 and 5). TMT (Tandem mass tag™)-based quantitation of the LC-MS/MS data was used to select proteins enriched by at least 1.5-fold in the untreated versus OA-treated samples in two independent trials. K-BIPS identified 71 proteins as hits (Table S1). A literature search revealed that 15 of the K-BIPS hit proteins (21% of the total hits, Figure 2B or Table S1, green colored rows) are known PP1 or PP2A substrates, showing that K-BIPS is capable of discovering phosphatase substrates. Interestingly, most of the known substrates were reported to also interact directly with the catalytic or regulatory subunits of the inactivated phosphatases (Table S1). Therefore, we analyzed if any of the other K-BIPS hits have documented interactions with PP1, PP2A, PP4, PP5, or PP6. An additional 18 proteins (25% of the total hits, Figure 2C or Table S1, blue colored rows) are known interactors of PP1, PP2A, PP4, PP5 and PP6 catalytic or regulatory subunits. These additional 18 K-BIPS hits (Figure 2C) are also likely substrates. Overall, 46% of the K-BIPS hits were either known substrates or known interactors of the inhibited phosphatase complexes.
To further analyze the K-BIPS hits, we assessed the functional roles and the abundance levels of the proteins. Functional analysis using Gene Ontology28 revealed that K-BIPS hits belonged to diverse functions, such as metabolism, cellular processes and localization (Figure S2), consistent with the wide spectrum of biological roles associated with phosphatases.9 To assess the possibility that K-BIPS hits were identified due to their high abundance, the abundances of the K-BIPS hits were also analyzed by using previously reported values.29 The results showed that proteins with a range of abundance (0.01 to 8,785 ppm, Figure S3) were identified by K-BIPS, which is similar to the abundance range of all HeLa cell proteins (0.01 to 10,000 ppm). The presence of proteins at all abundance levels demonstrated that K-BIPS is not biased toward proteins with high abundance.
In summary, K-BIPS was carried out after inactivating phosphatases with OA. While gel based experiments confirmed that phosphatase activity is necessary for efficient biotinylation, proteomics studies identified both known and novel phosphatase substrates. Altogether, the study with OA verified that K-BIPS is capable of phosphatase substrates identification.
K-BIPS control studies with the PP1-Gadd34 complex
Having established that K-BIPS is a viable strategy for phosphatase substrate identification, we used K-BIPS to discover potential substrates of the PP1-Gadd34 phosphatase complex. Gadd34 (PPP1R15A) is a regulatory subunit of PP1 that plays a key role during the unfolded protein response (UPR).10, 30 The UPR is induced when cells are faced with various insults that disrupt correct protein folding. Misfolded proteins accumulate in the endoplasmic reticulum (ER), causing ER stress. Consequently, ER stress triggers the activation of three main stress sensors in the cell (IRE1α, PERK and ATF6) to induce a cascade of downstream pathways in the UPR. In a bid to restore cellular protein homeostasis and save the cell from apoptosis during UPR, expression is elevated for a series of proteins involved in various restorative processes, such as protein folding, protein degradation, and apoptosis suppression. In addition, through PERK-mediated phosphorylation of the translation initiation factor eIF2α, the synthesis of new proteins is blocked during UPR by stalling translation and allowing the cell to recover from stress. As cells rebound from stress, Gadd34 is induced in a feedback loop. The PP1-Gadd34 complex acts to dephosphorylate eIF2α and remove the block on translation.30–32 Thus, PP1-Gadd34 functions a key modulator of UPR and has been implicated in various neurodegenerative diseases, cancer and viral infections.32–34 Despite being a feedback regulator of the UPR, surprisingly, the only known UPR-involved PP1-Gadd34 substrate is eIF2α. Given the disease relevance of the PP1-Gadd34 phosphatase complex, we aimed to use K-BIPS to explore unknown substrates of PP1-Gadd34 in the context of the UPR.
To perform K-BIPS to identify PP1-GADD34 substrates, we created conditions to promote UPR by using the known ER stress-inducing compound, tunicamycin (Tm).22 To inhibit the PP1-Gadd34 complex for K-BIPS, we used the small molecule guanabenz (Gb). Prior work documented increased phosphorylation of eIF2α, a Gadd34/PP1 substrate, after treatment of cells with Gb.22, 35 As an initial control study, we confirmed that Gb treatment altered phosphorylation of eIF2α under K-BIPS experimental conditions.30–32 HeLa cells were treated with Tm alone or both Tm and Gb before lysis and incubation for 2 hours either with or without Gb, which simulated the K-BIPS experiment (Figure 1C). After incubation, cellular proteins were separated by SDS-PAGE, transferred to a membrane, and then probed by Western blot using antibodies to eIF2α or Ser52-phosphorylated eIF2α. The expectation was that Gb treatment would result in increased eIF2α phosphorylation compared to untreated samples. As expected, the level of phosphorylated eIF2α was greater in the Gb-treated compared to the untreated sample at all tested time points (Figure S4, compare lanes 1 and 2, or 3 and 4, or 5 and 6), which is consistent with decreased PP1-Gadd34 activity in Gb-treated sample. This control study with the known PP1-Gadd34 substrate, eIF2α, confirmed that K-BIPS experimental conditions are suitable for monitoring changes in substrate phosphorylation.
Previous studies accounted for the increase in eIF2α phosphorylation by Gb due to disruption of PP1-Gadd34 interaction, which resulted in reduced phosphatase activity.22 However, recent in vitro studies questioned the influence of Gb on PP1-Gadd34 interaction.36 As an second control experiment, we confirmed that Gb treatment of HeLa cells disrupted the PP1-Gadd34 complex under the K-BIPS experimental conditions. PP1 was immunoprecipitated from Tm-treated lysates either with or without co-treatment with Gb, and the levels of co-immunoprecipitated Gadd34 were monitored by Western blotting. Gb treatment resulted in reduced co-immunoprecipitation of Gadd34 by PP1 (Figure S5A, compare lanes 3 to 4), consistent with Gb-mediated disruption of the PP1-Gaddd34 complex. To assure that the reduced co-immunoprecipitation of Gadd34 by PPI with Gb treatment was a result of complex disruption and not reduced expression, we monitored the expression of Gadd34 after Gb treatment by Western blot. Gadd34 expression was similar with and without Gb treatment (Figure S5A, compare lanes 1 and 2, and Figure S5B, compare lanes 2 and 3). These initial control studies document that Gb treatment disrupted the Gadd34-PPI complex under conditions of the K-BIPS study.
Finally, as a first step towards the K-BIPS method, we tested if Gb treatment, and the resulting loss of PPI-Gadd34 activity, reduced biotinylation of cellular proteins using gel analysis. Using Tm alone or Tm and Gb treated lysates, biotinylation was initiated by incubating with ATP-biotin. For the Gb-treated samples, Gb was also added to the lysates to maintain the PP1-Gadd34 complex in an inactive state during biotinylation. After reaction, cellular proteins were separated by SDS-PAGE, transferred to a membrane, and then probed with streptavidin-Cy5 to detect biotinylation. A reduction in biotinylation was observed in the Gb-treated compared to untreated lysates (Figure 3A, compare lane 2 to 3), consistent with the expectation that Gb treatment leads to inactivation of PP1-Gadd34 leads and reduced biotinylation (Figure 1C).
K-BIPS with Guanabenz
To perform a full K-BIPS experiment and identify UPR-implicated PP1-Gadd34 substrates using mass spectrometric analysis, biotinylated proteins obtained from UPR-induced lysates treated with or without Gb were purified with avidin resin. The purified proteins were then separated by SDS-PAGE (Figure S7) and analyzed by LC-MS/MS. After analysis by label free quantitation, 130 proteins enriched by at least 1.3-fold in the untreated versus the Gb-treated sample in two trials were identified as PP1-Gadd34 candidate substrates (Table S2). Importantly, the known PP1-Gadd34 substrate, eIF2α (EIF2S1, Figure 3B, red star, or Table S2, green color) was among the enriched proteins, confirming that K-BIPS is able to discover PP1-Gadd34 substrates.
To test if the identification of possible substrates was abundance dependent, we performed an abundance analysis of the 130 K-BIPS hits using reported values.29 The analysis showed that the K-BIPS hits were in the abundance range of 0.02 to 5,188 ppm (Figure S8), which is similar to the abundance of the full proteome of HeLa cells (0.01 to 10,000 ppm). The analysis indicated that abundance was not a factor for K-BIPS identification.
To determine if K-BIPS hits are known interacting proteins of PP1-Gadd34 phosphatase, we next performed an interactome analysis. Specifically, previously reported protein-protein interactions among the 130 K-BIPS hits and PP1-Gadd34 complex were explored using the GeneMania24 application in Cytoscape.37 The interactome analysis revealed that several K-BIPS hits are direct interacting proteins of either PP1 or Gadd34 (Figure 3B, hexagons, direct interacting proteins in inner circle). In addition, many other K-BIPS hits interact indirectly with PP1 or Gadd34 through the direct binding proteins (Figure 3B, circles, indirect interacting proteins in outer circle). Overall, 68% of the K-BIPS hits were either direct or indirect interacting proteins of the PP1-Gadd34 complex, suggesting that K-BIPS revealed likely candidate PP1-Gadd34 substrates.
Next, we carried out a literature search to assess if the K-BIPS hits have biological functions related to PP1-Gadd34. Besides eIF2α, three other proteins involved in translation initiation38 (EIF4G3, EIF3M and EIF3G, Figure 3B, yellow) and four ribosomal proteins (RPS12, RPL10A, RPL26L1 and RPLP0, Figure 3B, pink) were observed among the enriched proteins, consistent with the role of PP1-Gadd34 in translation. In addition, several K-BIPS hits also have known roles in UPR (Figure 3B, dark blue), ER stress (Figure 3B, orange), protein ubiquitination (Figure 3B, light blue), oxidative stress (Figure 3B, grey) and stress granules (Figure 3B, purple), in agreement with the role of PP1-Gadd34 in modulating stress response.
Secondary validation of K-BIPS hits as PP1-Gadd34 substrates
To validate the K-BIPS results, we selected a few hits that are known to have cellular functions related to UPR, but have not been previously identified as PP1-Gadd34 substrates. Specifically, the K-BIPS hits COPS5 and WDR5 represented interesting candidate substrates (Figure 3B, hexagons indicated by a black star). Besides being known interacting proteins of PP1 (Figure 3B, hexagons), COPS5 and WDR5 have known roles in UPR (Figure 3B, dark blue).39, 40 In addition, COPS5 associates directly with Gadd34 (Figure 3B, hexagons). Therefore, COPS5 and WDR5 were picked for further validation. Interestingly, COPS5 interacts with all stress granule proteins observed in K-BIPS (Figure 3C), including CAPRIN1 and G3BP1. The association of COPS5 with stress granules suggests a previously unknown role of PP1-Gaddd34 in stress granules formation, possibly through COPS5. Therefore, the stress granule proteins CAPRIN1 and G3BP1 were also selected for validation.
Before performing validation experiments, we first performed a series of control experiments to confirm that enrichment in K-BIPS studies is independent of changes in protein expression levels. The fact is that the PP1-Gadd34 complex influences protein expression during UPR through dephosphorylation of eIF2α.30–32 To assure that changes in proteins levels do not explain the enrichment of the candidate proteins with K-BIPS, the levels of COPS5, WDR5, Caprin, and G3BP1 were monitored after treatment with Tm alone or Tm and Gb. SDS-PAGE separation of proteins in the treated lysates and immunoblotting with primary antibodies showed that all four candidate substrates maintained similar protein levels after GB treatment in a Tm background (Figures 4A and S10-S13, compare lane 1 to lane 2). As a further control, the expression of COPS5 and G3BP1 was monitored after treatment with Gb, the Gb analog Sephin1, or the PERK signaling inhibitor ISRIB to prevent translational arrest. The results showed no significant difference in protein expression with any treatment condition (Figure S14, compare lane 1 to lanes 2–6). These control studies confirmed that COPS5, WDR5, Caprin, and G3BP1 maintained similar protein expression levels during the K-BIPS study, making them candidate PP1-Gadd34 substrates.
Figure 4.

Validation of K-BIPS hits. (A) Enrichment of K-BIPS hits with Gb treatment. HeLa cells were treated with tunicamycin (Tm) alone, or Tm + guanabenz (Gb) before lysis and kinase-catalyzed labeling with ATP-biotin. After avidin enrichment of biotinylated proteins, the presence of the four candidate substrates (COPS5, WDR5, CAPRIN, and G3BP1) was assessed by Western blot analysis with specific antibodies. Full gel images and repetitive trials are shown in Figures S10–S13. (B) COPS5 enrichment after Gadd34 knockdown. HeLa cells were treated with a pool of siRNA targeting Gadd34 or a pool of control siRNA (Figure S15), followed by treatment with (Tm). Cells were lysed and labeled with ATP-biotin. COPS5 enrichment was monitored by Western blot after avdin enrichment. Full gel image and repetitive trials are shown in Figure S16. (C) Phos-tag™ SDS-PAGE of COPS5 phosphorylation. HeLa cells were treated with Tm and Gb, or Tm alone. After lysis, proteins were separated using Phos-tag™ SDS-PAGE. COPS5 was visualized by Western blot analhysis (anti-COPS5). Total proteins were visualized as a load control by Sypro®Ruby total protein stain. Bands affected by Gb treatment are indicated with a bracket. Full image and repetitive trials was shown in Figure S17.
For secondary validation, we used K-BIPS to monitor the differential biotinylation of COPS5, WDR5, CAPRIN1 and G3BP1 with Gb treatment by gel methods. Specifically, proteins from untreated and Gb-treated lysates were biotinylated with ATP-biotin. Biotin-labeled proteins were purified with avidin resin, separated by SDS-PAGE, and analyzed by Western blot analysis. The expectation was that substrates of the PP1-Gadd34 complex would show elevated phosphorylation, and hence reduced biotinylation, in Gb-treated versus untreated samples (Figure 1C). As expected, after avidin enrichment, reduced levels of all proteins were observed in the Gb-treated sample compared to the untreated sample (Figure 4A, compare lanes 3 and 4). These studies are consistent with COPS5, WDR5, Caprin, and G3BP1 being PP1-Gadd34 substrates.
As further validation, expression of Gadd34 was knocked down using siRNA treatment in HeLa cells (Figure S15), and K-BIPS was carried out with monitoring of COPS5. Gel analysis following ATP-biotin labeling and avidin purification showed decreased levels of COPS5 in Gadd34 knockdown compared to the control samples (Figure 4B, compare lanes 3 and 4). Thus, the data confirmed that both Gb treatment and knockdown affected biotinylation of the selected substrate proteins, consistent with the role of the PP1-Gadd34 complex in regulating phosphorylation.
As a final validation study, the levels of phosphorylated COPS5 as a function of Gb treatment were directly monitored using Phos-tag™ SDS-PAGE. If COPS5 is a PP1-Gadd34 substrate, the expectation was that more intense phosphorylated COPS5 bands would be observed in Gb-treated compared to untreated cells due to inactivation of the phosphatase activity. Cells were treated with Tm alone or Tm and Gb before lysis, separation of proteins by Phos-tag™ SDS-PAGE, and visualization of COPS5 using Western blot analysis. As expected, more intense COPS5 bands were observed in cells treated with Tm and Gb compared to Tm alone (Figure 4C and S17, lanes 4 versus lanes 3). This data is consistent with increased COPS5 phosphorylation in the presence of Gb due to reduced PP1-Gadd34 activity.
Looking back at the K-BIPS LC-MS/MS data, COPS5 and WDR5 showed only modest enrichment in the proteomics experiment (Table S2, 1.3- and 1.4-fold enrichment, respectively). These secondary studies, therefore, experimentally confirmed that the 1.3-fold enrichment threshold for selection of K-BIPS hit was appropriate. In addition, these studies validated that K-BIPS can be used to monitor the activity of a specific phosphatase-regulatory subunit activity in cells. Taken together, K-BIPS was able to not only discover COPS5, WDR5, CAPRIN1 and G3BP1 as previously unknown PP1-Gadd34 substrates, but also validate candidate substrates by monitoring phosphatase activity in lysates.
Discussion
Despite being key players in cell signaling and human diseases, the biological functions of protein phosphatases have remained elusive due to the lack of tools to identify substrates. Here we developed K-BIPS as a chemical method for discovering phosphatase substrates (Figure 1C). As an initial proof-of-concept, K-BIPS with OA-mediated phosphatase inactivation led to the identification of already known phosphatase substrates, which established K-BIPS as a tool for phosphatase substrate discovery. Further, K-BIPS identified many candidate phosphatase substrates, indicating previously unexplored functions of the PPP family phosphatases. For an example, the K-BIPS hit PCNA (Proliferating Cell Nuclear Antigen) interacts with PP2A, and nuclear transport of PCNA was inhibited by OA treatment.41 The presence of PCNA among the K-BIPS hits, combined with the prior work, suggests that PCNA is dephosphorylated by PP2A to effect nuclear transport. As a second example, LDHA was among the OA K-BIPS hits, and previous studies reported that the expression and enzymatic activity of LDHA was reduced after knock down of CIP2A, the endogenous inhibitor of PP2A.42 Taken together, prior work and the K-BIPS study suggest that PP2A-mediated dephosphorylation regulates LDHA expression and activity. In total, the study with OA established K-BIPS as a viable tool to identify substrates of phosphatases.
With the success of K-BIPS with OA treatment, we next studied the PP1-Gadd34 complex in the context of the UPR. Despite the clear role of PP1-Gadd34 in UPR regulation, the only known UPR-related substrate is eIF2α. Identification of PP1-Gadd34 substrates is complicated by the presence of many other PP1-regulatory subunit complexes in the cell; distinguishing substrates of the PP1-Gadd34 complex from the substrates of the many other PP1-regulatory complexes is challenging with current methods. To achieve selectivity for only the PP1-Gadd34 complex, K-BIPS was performed after inactivation of the PP1-Gadd34 complex using the small molecule guanabenz (Gb). By specifically targeting interaction of PP1 and Gadd34 and incorporating a biotin pull-down enrichment step, K-BIPS was expected to identify candidate substrates of PP1-Gadd34 in the UPR, which has the potential to reveal new functions of Gadd34.
K-BIPS was performed after incubation of cells with Gb and the UPR-inducing compound, tunicamycin (Tm). As expected, K-BIPS with Gb identified the known PP1-Gadd34 substrate eIF2α. Consistent with the earlier OA experiment, the presence of the only known UPR-related substrate of the PP1-Gadd34 complex confirmed the utility of K-BIPS to identify phosphatase substrates. In addition to eIF2α, many other proteins related to ER stress and UPR were observed among the K-BIPS hits (Figure 3B). For example, K-BIPS identified the chaperone proteins PDIA6, FKBP10, DNABJ11 and DNAJA1, which have been implicated in protein homeostasis during UPR.43–46 Although not previously implicated in the UPR, the E3 ligases, RNF213 and UBE3A, and the E2 ubiquitin-conjugating enzyme, UBE2Z, were enriched in the K-BIPS study, suggesting a role for these ubiquitin-modifying enzymes in degrading misfolded proteins during UPR. Among the K-BIPS-enriched proteins was also the ER stress-associated protein UBQLN2, which mediates the movement of ubiquitin-linked proteins from the ER to the cytosolic proteasome complex for degradation.47 The discovery of many ER stress and UPR-associated proteins in the K-BIPS study is consistent with the role of the PP1-Gadd34 complex in protein homeostasis. Importantly, the data suggest that PP1-Gadd34 is involved in other UPR-related functions in addition to eIF2α dephosphorylation and translation regulation.
Cellular stress conditions that stall translation, such as Tm treatment, also result in formation of cytoplasmic bodies called stress granules. Stress granules contain transient aggregates of mRNA and translation pre-initiation complexes and allow the rapid recovery of translation once the stress is resolved.48, 49 Overexpression of Gadd34 was previously shown to reduce the formation of stress granules under conditions that otherwise promote stress granule formation, suggesting a role for Gadd34 in dissociating stress granules.35, 50 Related to the PP1-Gadd34 complex, prior work documented that Gb treatment increased the formation of stress granules.35 Building on these prior connections between PP1, Gadd34, and stress granules, K-BIPS identified several stress granule proteins (Figure 3C), including TIAL1,49 G3BP1, CAPRIN1,51 FMR1, FXR149 and IGF2BP3.52 Importantly, both CAPRIN1 and G3BP1 were validated as PP1-Gadd34 substrates in secondary studies (Figure 4). Overall, the combined K-BIPS data and prior studies suggest a model where the PP1-Gadd34 complex regulates phosphorylation of stress granule proteins to influence stress granule assembly in the UPR.
The Gadd34-interacting protein COPS5 (JAB1) was also identified by K-BIPS, and K-BIPS validation experiments confirmed that PP1-Gadd34 affects COPS5 phosphorylation (Figure 4). COPS5 is both a transcriptional coactivator and a component of the COP9 signalosome, with a role in key cellular processes, such as cell proliferation, apoptosis and cell cycle arrest.53 Related to the UPR, COPS5 interacts with ER stress-modulating kinase IRE1α under normal conditions, but dissociates from IRE1α when ER stress is induced. Disruption of the COPS5-IRE1α complex results in the downstream activation of IRE1α during UPR.40 The identification of COPS5 as a substrate of PP1-Gadd34 hints at a model where reversible phosphorylation regulates the various functions of COPS5 during UPR. According to the model, phosphorylation disrupts IRE1α binding to COPS5 under ER stress conditions, controlling both activation of IRE1α and COPS5 activities in UPR. Once the UPR is resolved, PP1-Gadd34 dephosphorylates COPS5 to promote re-association of COPS5 with IRE1α, restoring regular cellular processes. Beyond the IRE1α interaction, COPS5 interacts with stress granule proteins, which were also pulled down in the K-BIPS study (Figure 3C). Therefore, the K-BIPS study also suggests a connection between COPS5 activity and regulation of stress granules in UPR.
UPR is a tightly regulated process that culminates in either cell survival or cell death, depending on the severity of the stress.54 A recent study reported crosstalk between the UPR and the pro-apoptotic Hippo pathway. The Hippo pathway induces phosphorylation of the transcriptional coactivator YAP1 to block its nuclear translocation, which suppresses the proliferative activities of YAP1 and promotes apoptosis.55, 56 Expression of Gadd34 was previously correlated with increased phosphorylation and cytoplasmic retention of YAP1 with Tm treatment, suggesting a link between UPR and Hippo pathway.56 Notably, the Hippo pathway kinase STK3 (MST2), which promotes phosphorylation of Yap1 through LATS kinase,55 was one of the K-BIPS hits. Consistent with a model where Gadd34 activity influences the Hippo pathway to promote apoptosis, the activity of STK3 is reduced upon phosphorylation.57 Therefore, the identification of STK3 by K-BIPS suggests a possible mechanism where PP1-Gadd34 indirectly affects the phosphorylation of YAP1 through dephosphorylation of STK3. The crosstalk between UPR and Hippo pathway may represent a regulatory mechanism through which the balance between cell survival and cell death under cell stress conditions is determined. Beyond the Hippo pathway, STK3 interacts with the scaffolding protein SLC9A3R1, which in turn associates with PKA through cytoplasmic proteins EZR and RDX.58–60 Interestingly, K-BIPS identified SLC9A3R1 and PKA regulatory subunits, PRKAR1A and PRKAR2A, suggesting connections between the Hippo pathway and PKA signaling during UPR. Altogether, the K-BIPS study implicates a greater role for PP1-Gadd34 in UPR than previously appreciated and validates the use of K-BIPS as a phosphatase substrate identification method.
In this work, kinase-catalyzed biotinylation was exploited to develop K-BIPS as a tool for discovering phosphatase substrates. Application to two Ser/Thr phosphatase systems established K-BIPS for identification of unanticipated substrates, which linked phosphatases to novel biological functions. K-BIPS requires a method to inactivate the phosphatase of interest, such as small molecule perturbation. Although not explored directly here, cells with knockout or knockdown of a phosphatase or regulatory protein would also be compatible with K-BIPS. Because small molecule inhibition or knockdown/knockout strategies are available for both Try and Ser/Thr phosphatases, we anticipate application of K-BIPS to both Tyr and Ser/Thr phosphatases. Compared to currently available phosphoproteomic methods, K-BIPS has the advantage of detecting dynamically changing phosphorylation events due to the use of kinase-catalyzed biotinylation. In contrast, the IMAC purification used widely in phosphoproteomics enriches all phosphoproteins, regardless of dynamics. K-BIPS has the added advantage of attaching a stable biotin tag to phosphoproteins for subsequent enrichment.21 By focusing on dynamically changing phosphoproteins and relying on enrichment of biotinylated proteins, K-BIPS has the potential to identify substrates independent of cellular abundance. In fact, both K-BIPS studies reported here observed proteins with a range of cellular abundances (see Figures S3 and S8), suggesting that K-BIPS is an effective tool to discover even low abundance proteins.
A similar challenge with both K-BIPS and phosphoproteomics methods is that they discover both direct phosphatase substrates and proteins indirectly affected by the fed phosphatase. Therefore, validation studies are necessary to confirm that discovered proteins are substrates of a particular phosphatase. Fortunately, K-BIPS is also a validation tool for substrate confirmation when coupled with gel analysis. Current validation methods typically involve in vitro incubation of candidate substrates with purified phosphatases.10 In contrast, K-BIPS validation is compatible with complex mixtures, including lysates, and only requires phosphatase inactivation. With application to substrate discovery and validation, K-BIPS provides an enabling tool to explore the biological role of phosphatases in cells. Given the importance of protein phosphatases in cell signaling, K-BIPS will assist in unraveling the complex regulatory network in the cell to reveal mechanisms behind human diseases.
Supplementary Material
Acknowledgments
We would like to thank the National Institutes of Health (GM079529) and Wayne State University for funding, the Wayne State University and Karmanos Cancer Center Proteomics Core, which is supported by NIH Grants P30 ES020957, P30 CA022453, and S10 OD010700, J. Caruso and N. Carruthers for technical support, and V. Ramanayake-Mudiyanselage and A. Gamage for comments on the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions
P.M.D.A. performed all experiments, except the Gadd34 expression study (Figure S5B), repetitive trials of secondary substrate validation studies (Figure S10–S13), and Phos-tag™ SDS-PAGE studies (Figure 4C and S17) completed by N.P.N.A. M.K.H.P. conceived of the project. M.K.H.P and P.M.D.A carried out experimental design, interpretation, and wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Graves JD, Krebs EG. Pharmacol Ther. 1999;82:111–121. doi: 10.1016/s0163-7258(98)00056-4. [DOI] [PubMed] [Google Scholar]
- 2.Hunter T. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 3.Tsatsanis C, Spandidos DA. Int J Mol Med. 2000;5:583–590. doi: 10.3892/ijmm.5.6.583. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Q, Claret FX. Enzyme Res. 2012;2012:659649. doi: 10.1155/2012/659649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson MD, Denu JM. Chem Rev. 2001;101:2313–2340. doi: 10.1021/cr000247e. [DOI] [PubMed] [Google Scholar]
- 6.Soulsby M, Bennett AM. Physiology (Bethesda) 2009;24:281–289. doi: 10.1152/physiol.00017.2009. [DOI] [PubMed] [Google Scholar]
- 7.Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Ceulemans H, Bollen M. Physiol Rev. 2004;84:1–39. doi: 10.1152/physrev.00013.2003. [DOI] [PubMed] [Google Scholar]
- 10.Novoa I, Zeng H, Harding HP, Ron D. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinheiro AS, Marsh JA, Forman-Kay JD, Peti W. J Am Chem Soc. 2011;133:73–80. doi: 10.1021/ja107810r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flint AJ, Tiganis T, Barford D, Tonks NK. Proc Natl Acad Sci U S A. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanchetot C, Chagnon M, Dube N, Halle M, Tremblay ML. Methods. 2005;35:44–53. doi: 10.1016/j.ymeth.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Garaud M, Pei D. J Am Chem Soc. 2007;129:5366–5367. doi: 10.1021/ja071275i. [DOI] [PubMed] [Google Scholar]
- 15.Ren L, Chen X, Luechapanichkul R, Selner NG, Meyer TM, Wavreille AS, Chan R, Iorio C, Zhou X, Neel BG, Pei D. Biochemistry. 2011;50:2339–2356. doi: 10.1021/bi1014453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Ma D, Caruso M, Lewis M, Qi Y, Yi Z. Journal of proteomics. 2014;109C:63–75. doi: 10.1016/j.jprot.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Virshup DM, Shenolikar S. Mol Cell. 2009;33:537–545. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 18.Green KD, Pflum MK. J Am Chem Soc. 2007;129:10–11. doi: 10.1021/ja066828o. [DOI] [PubMed] [Google Scholar]
- 19.Senevirathne C, Embogama DM, Anthony TA, Fouda AE, Pflum MK. Bioorg Med Chem. 2016;24:12–19. doi: 10.1016/j.bmc.2015.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dedigama-Arachchige PM, Pflum MK. ACS Chem Biol. 2016 doi: 10.1021/acschembio.6b00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Senevirathne C, Pflum MK. ChemBioChem. 2013;14:381–387. doi: 10.1002/cbic.201200626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsaytler P, Harding HP, Ron D, Bertolotti A. Science. 2011;332:91–94. doi: 10.1126/science.1201396. [DOI] [PubMed] [Google Scholar]
- 23.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 24.Montojo J, Zuberi K, Rodriguez H, Kazi F, Wright G, Donaldson SL, Morris Q, Bader GD. Bioinformatics. 2010;26:2927–2928. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen P, Klumpp S, Schelling DL. FEBS Lett. 1989;250:596–600. doi: 10.1016/0014-5793(89)80803-8. [DOI] [PubMed] [Google Scholar]
- 26.Swingle M, Ni L, Honkanen RE. Methods Mol Biol. 2007;365:23–38. doi: 10.1385/1-59745-267-X:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castermans D, Somers I, Kriel J, Louwet W, Wera S, Versele M, Janssens V, Thevelein JM. Cell Res. 2012;22:1058–1077. doi: 10.1038/cr.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.C. Gene Ontology. Nucleic Acids Res. 2015;43:D1049–1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geiger T, Wehner A, Schaab C, Cox J, Mann M. Mol Cell Proteomics. 2012;11:M111 014050. doi: 10.1074/mcp.M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brush MH, Weiser DC, Shenolikar S. Mol Cell Biol. 2003;23:1292–1303. doi: 10.1128/MCB.23.4.1292-1303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hetz C. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 32.Wang M, Kaufman RJ. Nat Rev Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 33.Hetz C, Chevet E, Harding HP. Nat Rev Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- 34.Minami K, Tambe Y, Watanabe R, Isono T, Haneda M, Isobe K, Kobayashi T, Hino O, Okabe H, Chano T, Inoue H. J Virol. 2007;81:11106–11115. doi: 10.1128/JVI.01063-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruggieri A, Dazert E, Metz P, Hofmann S, Bergeest JP, Mazur J, Bankhead P, Hiet MS, Kallis S, Alvisi G, Samuel CE, Lohmann V, Kaderali L, Rohr K, Frese M, Stoecklin G, Bartenschlager R. Cell Host Microbe. 2012;12:71–85. doi: 10.1016/j.chom.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crespillo-Casado A, Chambers JE, Fischer PM, Marciniak SJ, Ron D. eLife. 2017:6. doi: 10.7554/eLife.26109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, Hanspers K, Isserlin R, Kelley R, Killcoyne S, Lotia S, Maere S, Morris J, Ono K, Pavlovic V, Pico AR, Vailaya A, Wang PL, Adler A, Conklin BR, Hood L, Kuiper M, Sander C, Schmulevich I, Schwikowski B, Warner GJ, Ideker T, Bader GD. Nature protocols. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jackson RJ, Hellen CU, Pestova TV. Nat Rev Mol Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song ZT, Sun L, Lu SJ, Tian Y, Ding Y, Liu JX. Proc Natl Acad Sci U S A. 2015;112:2900–2905. doi: 10.1073/pnas.1419703112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oono K, Yoneda T, Manabe T, Yamagishi S, Matsuda S, Hitomi J, Miyata S, Mizuno T, Imaizumi K, Katayama T, Tohyama M. Neurochem Int. 2004;45:765–772. doi: 10.1016/j.neuint.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Morrow PW, Tung HY, Hemmings HC., Jr Biochem Biophys Res Commun. 2004;323:645–651. doi: 10.1016/j.bbrc.2004.08.147. [DOI] [PubMed] [Google Scholar]
- 42.Peng B, Lei N, Chai Y, Chan EK, Zhang JY. Mol Biosyst. 2015;11:105–114. doi: 10.1039/c4mb00513a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sano R, Reed JC. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy LA, Ramirez EA, Trinh VT, Herman AM, Anderson VC, Brewster JL. Cell Stress Chaperones. 2011;16:607–619. doi: 10.1007/s12192-011-0270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Genereux JC, Qu S, Zhou M, Ryno LM, Wang S, Shoulders MD, Kaufman RJ, Lasmezas CI, Kelly JW, Wiseman RL. EMBO J. 2015;34:4–19. doi: 10.15252/embj.201488896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gotoh T, Terada K, Oyadomari S, Mori M. Cell Death Differ. 2004;11:390–402. doi: 10.1038/sj.cdd.4401369. [DOI] [PubMed] [Google Scholar]
- 47.Xia Y, Yan LH, Huang B, Liu M, Liu X, Huang C. J Neurochem. 2014;129:99–106. doi: 10.1111/jnc.12606. [DOI] [PubMed] [Google Scholar]
- 48.Kawai T, Lal A, Yang X, Galban S, Mazan-Mamczarz K, Gorospe M. Mol Cell Biol. 2006;26:3295–3307. doi: 10.1128/MCB.26.8.3295-3307.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson P, Kedersha N. Trends Biochem Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 50.Dalet A, Arguello RJ, Combes A, Spinelli L, Jaeger S, Fallet M, Vu Manh TP, Mendes A, Perego J, Reverendo M, Camosseto V, Dalod M, Weil T, Santos MA, Gatti E, Pierre P. EMBO J. 2017;36:761–782. doi: 10.15252/embj.201695000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, Thomas M, Lieberman J, McInerney GM, Ivanov P, Anderson P. J Cell Biol. 2016;212:845–860. doi: 10.1083/jcb.201508028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kobayashi T, Winslow S, Sunesson L, Hellman U, Larsson C. PLoS One. 2012;7:e35820. doi: 10.1371/journal.pone.0035820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shackleford TJ, Claret FX. Cell Div. 2010;5:26. doi: 10.1186/1747-1028-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szegezdi E, Logue SE, Gorman AM, Samali A. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hao Y, Chun A, Cheung K, Rashidi B, Yang X. J Biol Chem. 2008;283:5496–5509. doi: 10.1074/jbc.M709037200. [DOI] [PubMed] [Google Scholar]
- 56.Wu H, Wei L, Fan F, Ji S, Zhang S, Geng J, Hong L, Fan X, Chen Q, Tian J, Jiang M, Sun X, Jin C, Yin ZY, Liu Q, Zhang J, Qin F, Lin KH, Yu JS, Deng X, Wang HR, Zhao B, Johnson RL, Chen L, Zhou D. Nat Commun. 2015;6:6239. doi: 10.1038/ncomms7239. [DOI] [PubMed] [Google Scholar]
- 57.Kim D, Shu S, Coppola MD, Kaneko S, Yuan ZQ, Cheng JQ. PLoS One. 2010;5:e9616. doi: 10.1371/journal.pone.0009616. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58.Varjosalo M, Sacco R, Stukalov A, van Drogen A, Planyavsky M, Hauri S, Aebersold R, Bennett KL, Colinge J, Gstaiger M, Superti-Furga G. Nat Methods. 2013;10:307–314. doi: 10.1038/nmeth.2400. [DOI] [PubMed] [Google Scholar]
- 59.Wang B, Means CK, Yang Y, Mamonova T, Bisello A, Altschuler DL, Scott JD, Friedman PA. J Biol Chem. 2012;287:24148–24163. doi: 10.1074/jbc.M112.369405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, Cai Z, Chen JH, Ju M, Xu Z, Sheppard DN. Sheng Li Xue Bao. 2007;59:416–430. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.